

Abstract
Background
Mismatch repair deficiency (dMMR) is a characteristic feature of cancers linked to Lynch syndrome. However, in most cases, it results from sporadic somatic events rather than hereditary factors. The term ‘Lynch-like syndrome’ (LLS) has been used to guide colorectal cancer surveillance for relatives of individuals with a dMMR tumour when somatic and germline genomic testing is uninformative. As the assessment of mismatch repair through immunohistochemistry and/or microsatellite instability is increasingly applied across various tumour types for treatment planning, dMMR is increasingly detected in tumours where suspicion of hereditary aetiology is low. Our objective was to establish current practices and develop national guidance for investigating, and managing relatives of, patients with cancers demonstrating unexplained dMMR.
Methods
This was achieved through a virtual consensus meeting involving key stakeholders from the UK, through premeeting surveys, structured discussions and in-meeting polling to formulate best practice guidance.
Results
We identified variability in the availability of diagnostic technologies across specialist centres. It was agreed that equitable access to baseline testing is required, acknowledging the need for a pragmatic approach to investigating dMMR cancers not traditionally associated with Lynch syndrome. Factors such as family history, age, tumour type, protein loss pattern and extent of the investigation were deemed crucial in guiding family management. The term ‘unexplained dMMR’ was recommended over LLS.
Conclusion
Decisions regarding investigations and future cancer risk management in patients and relatives should be nuanced, considering factors like clinical suspicion of hereditary predisposition to allocate limited resources efficiently and avoid unnecessary investigations in low-suspicion families.
Keywords: Clinical Decision-Making
WHAT IS ALREADY KNOWN ON THIS TOPIC
Mismatch repair deficiency (dMMR) may be due to an underlying heritable variant impacting mismatch repair gene function or may be due to a sporadic, somatic aetiology. Even in cases where germline and somatic testing is undertaken, dMMR in a minority of cases remains unexplained. The investigative pathway and clinical management of patients with tumours demonstrating unexplained dMMR, and their relatives, pose challenges.
WHAT THIS STUDY ADDS
Following a national, multidisciplinary consensus meeting, we have outlined a pragmatic approach to investigation of patients with dMMR cancers, guided by clinical suspicion of hereditary predisposition as well as considering available resources in a publicly funded health system.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
We have identified a number of areas where consensus could not be achieved, which warrant further research and long-term prospective data collection and interrogation. Further data are necessary to elucidate the underlying aetiology of dMMR in tumours not classically associated with Lynch syndrome, but undertaking necessary investigations in the National Health Service(NHS) will require further investment in existing services, as well as addressing inequities in access to key technologies.
Introduction
Lynch syndrome is defined by the identification of constitutional pathogenic variants in four mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS2). Although functional MMR deficiency (dMMR) is a hallmark of cancers associated with Lynch syndrome, dMMR may also be a feature of sporadic cancers as a consequence of somatic aberrations impacting MMR gene function.1 dMMR, as evidenced by abnormal expression of one or more MMR proteins, and/or microsatellite instability (MSI) is most commonly detected in colorectal (12%–15%) and endometrial (20%–30%) cancers but can be a feature of multiple other cancer types, including upper gastrointestinal2 (~10%), ovarian (~10%), hepatobiliary (10%) and adrenocortical cancers.3 4 Given that dMMR cancers typically demonstrate favourable response to immune checkpoint inhibitors (ICIs),5 the expanding availability of such therapeutic agents has fuelled increasing testing for, and detection of, dMMR in a host of cancer types, including common and rare Lynch syndrome-associated malignancies, as well as cancers not usually considered part of the Lynch syndrome phenotypic spectrum.6
The majority of dMMR detected in colorectal and endometrial cancers is attributable to sporadic somatic hypermethylation of the MLH1 promoter,7 8 and, usually, detection of hypermethylation of the MLH1 promoter in tumour-derived DNA (or, in colorectal cancer, detection of a pathogenic somatic BRAF variant as a surrogate marker of same) will obviate the need for constitutional genetic testing.9 Testing for constitutional hypermethylation of the MLH1 promoter, if not undertaken at the time of initial tumour-based analysis, and/or constitutional MMR gene testing may still be warranted if clinical suspicion for Lynch syndrome, based on patient age and/or family history, persists.10
At present, in the UK, constitutional and tumour-based genetic testing is undertaken, for the most part, in an unsynchronised, unpaired manner, driven, in part, by increasing awareness of Lynch syndrome as an underlying aetiology for a small but significant proportion of cancer risk but also by increasing application of and access to ICIs. The National Institute for Health and Care Excellence (NICE) has published guidance for assessment of MMR in endometrial and colorectal cancers, with investigative pathway guided by tumour type and, where available, pattern of MMR protein loss (figure 1).11 12 Detection of a pathogenic activating BRAF variant in colorectal cancer demonstrating loss of MLH1 (usually with concomitant loss of PMS2) can be used as a surrogate marker of MLH1 promoter hypermethylation, but 18%–20% of cancers demonstrating MLH1 promoter hypermethylation will not harbour a detectable BRAF variant.9 13 BRAF variants are rare in endometrial cancers, such that BRAF testing is not usually informative for such cancers.14 The utility of such testing in other extracolonic cancers is uncertain, as is the contribution of MLH1 promoter hypermethylation to dMMR in other tumour types.
Figure 1.
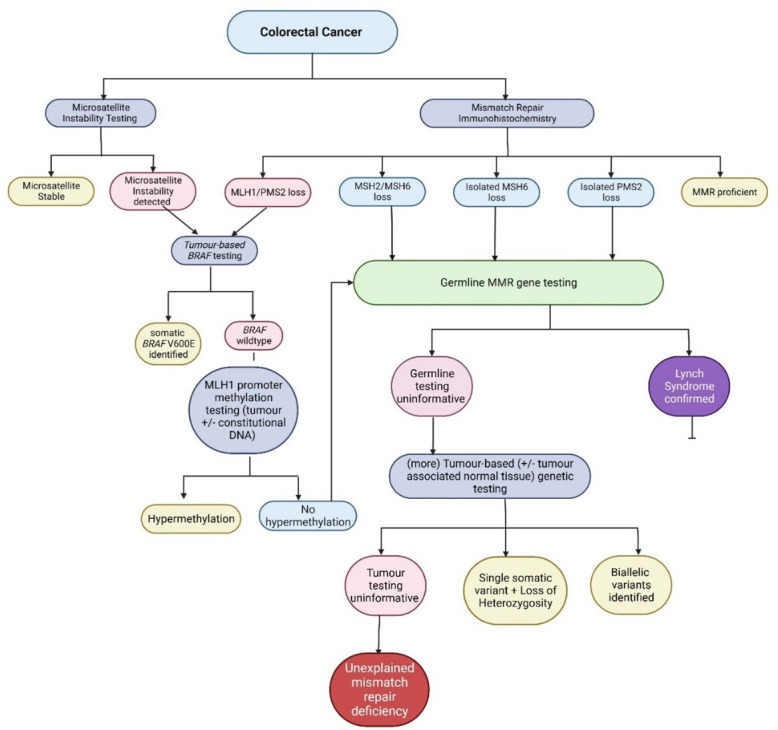
Pathway for investigation of mismatch repair (MMR) and/or microsatellite instability in colorectal cancers. Decisions about the extent of investigation along this pathway are guided by clinical suspicion and the availability of relevant investigations. If investigation is stopped following germline MMR gene testing, the proportion of unexplained MMR deficiency will be higher.
For patients with cancers demonstrating combined MLH1/PMS2 deficiency and/or microsatellite instability where MLH1 promoter hypermethylation is not detected, or for patients with cancers demonstrating isolated loss of PMS2 or MSH6, or combined loss of MSH2 and MSH6, or where clinical suspicion for Lynch syndrome persists based on age and/or family history, constitutional MMR gene testing is indicated.11 12 Type and extent of constitutional testing should be guided by the pattern of protein loss and the a priori suspicion that a constitutional pathogenic variant explains the patient phenotype and in line with criteria outlined in the NHS England Genomic Testing Directory15 or equivalent in Wales, Northern Ireland and Scotland.16 In patients with tumours demonstrating isolated PMS2 deficiency, testing should include long-range PCR of PMS2 (or equivalent assessment of region impacted by pseudogene interference), while for tumours demonstrating loss of MSH2 and/or MSH6, testing should include dosage analysis of 3′ end of EPCAM, and testing for the recurrent paracentric inversion of exons 1–7 in MSH2.17 Where constitutional testing is uninformative, testing on tumour-derived DNA (‘somatic testing’) may then be undertaken to determine if an underlying somatic aetiology accounts for dMMR.18 In those cases where biallelic somatic events (biallelic variants or a single variant with evidence of loss of heterozygosity) impacting an MMR gene are not identified, the dMMR is classified as ‘unexplained’ (henceforth abbreviated as u-dMMR), and, historically, affected individuals were said to have ‘Lynch-like syndrome’ (LLS). The proportion of cases deemed ‘unexplained’ is influenced by the extent of investigation—after somatic BRAF sequencing and MLH1 promoter hypermethylation and, where indicated, constitutional MMR gene testing is undertaken, approximately 4.24% of colorectal cancers have u-dMMR; when somatic MMR analysis is undertaken, this reduces to 0.61%.19
Possible explanations for u-dMMR are undetectable somatic or constitutional pathogenic variants.20 Other explanations may include inaccuracies in the tests performed. For example, the performance of immunohistochemistry (IHC) is dependent on platform (including antibodies) and interpretation of results is observer-dependent and may be influenced by tumour fixation. Therefore, repeating an apparently abnormal IHC result may be useful as a first step in cases where an aetiology cannot be identified, particularly if IHC results are discordant with MSI results, and if clinical suspicion of Lynch syndrome is low.21 22 Testing an alternative well-fixed specimen, for example, diagnostic biopsy may be helpful to avoid the reduction/loss of staining noted with poorly fixed resection specimens. Consideration should also be given to the possibility of BRAF variants and/or MLH1 hypermethylation being missed by testing a sample with inadequate tumour cellularity.23
Published recommendations for bowel screening in relatives of affected probands deemed to have LLS are equivalent to that offered to patients with Lynch syndrome.24 However, while a proportion may be due to an unidentified heritable event, where clinical suspicion of a hereditary predisposition is low, it is more likely that dMMR will be due to an unidentified somatic aetiology.25 26 This is evidenced by the fact that relatives of probands with LLS have a lower cancer risk than in those families with molecularly confirmed Lynch syndrome, although higher than in those families with sporadic colorectal cancer.27
In a publicly funded health system, it could be argued that it is most appropriate that limited resources are best focused on detection of rare, atypical mechanisms of heritable dMMR20 and screening of relatives of probands where suspicion of hereditable aetiology remains high after uninformative investigations. However, conversely, as screening recommendations can be ‘downgraded’ once an aetiology for MMR is established, it could be argued that further investigations could be warranted where an explanation for MMR deficiency is not immediately obvious but where clinical suspicion of constitutional mechanism is low.
National multidisciplinary team meeting
Every two months, a virtual multidisciplinary meeting involving clinical geneticists, genetic counsellors and other healthcare professionals working in Cancer Genetics convenes as part of a national collaborative initiative between the Cancer Research UK-funded CanGene-CanVar Programme and the UK Cancer Genetics Group (UKCGG), a constituent group of the British Society of Genomic Medicine.28 Each meeting has a specific theme, and clinicians are invited to submit challenging cases aligned with that theme. The session in November 2022 was on the theme of u-dMMR. A number of cases were presented (table 1), all of whom were patients with dMMR cancers where aetiology was not established. The audience was asked to comment on bowel screening recommendations based on clinical features (eg, age of cancer diagnoses, cancer type, family history and results of tumour and constitutional testing). Recommendations ranged from population bowel cancer screening for relatives to Lynch-equivalent bowel screening.
Table 1.
Cases discussed at UKCGG/CanGene-CanVar Cancer Genetics MDT
| Summary: Clinical scenarios | Consensus |
| 1. Young patient (age 40–50 years) with MMR-deficient endometrial cancer from an Amsterdam-positive family history (unconfirmed). Germline genetic testing uninformative. Single MSH2 somatic variant detected. | Manage as Lynch-like unless further information obtained |
| 2. Deceased proband with MMR-deficient colorectal cancer (isolated loss of PMS2) at 40–50 years. Molecular testing on tissue-derived DNA identified PMS2 pathogenic variant—origin (germline/somatic) could not be determined based on available tissue. | Offer germline testing to FDRs for PMS2 variant identified in tumour |
| 3. MMR-deficient (loss of MSH2 and MSH6) endometrial cancer at 50–60 years, keratoacanthoma at 50–60 years. Constitutional testing uninformative. Somatic testing identified two somatic MSH2 variants at low VAF. Cousin with young onset endometrial and rectal cancer also MMR-deficient (loss of MSH2 and MSH6). Somatic testing failed. Amsterdam-positive family history. | Manage as Lynch-like unless further information obtained |
| 4. Man with MMR-deficient (loss of MLH1 and PMS2) colorectal at 30–40 years. Mother hysterectomy in late 30s. Brother polyps at 40–50 years. Uninformative constitutional testing. | Manage as Lynch-like unless further information obtained |
| 5. MMR-deficient (isolated loss PMS2) pancreatic cancer in a male aged 70–80 years; constitutional testing uninformative, testing on tumour-derived DNA unsuccessful. | Colonoscopic screening based on family history |
| 6. MMR-deficient (loss of MLH1 and PMS2) colorectal cancer in 60–70 year old male. Mother ovarian cancer in 80s. No MLH1 promoter hypermethylation or constitutional MMR gene variants identified. Single somatic MLH1 variant identified. | National bowel screening |
FDR, first-degree relative; MDT, multidisciplinary team; MMR, mismatch repair; UKCGG, UK Cancer Genetics Group; VAF, Variant Allele Frequency.
During the meeting, consensus was easily achieved for bowel screening recommendations for each case, although this was only informally recorded given the nature of the meeting. From the discussions, it was noted that most centres consider tumour type, age and family history when determining extent of testing to establish aetiology of dMMR and screening recommendations for relatives. However, it was noted that regional variability in access to certain technologies such as long-range PCR for PMS2 and loss of heterozygosity analysis was a limitation.
In cases where constitutional MMR testing is uninformative and clinical suspicion of Lynch syndrome low, there was discussion about whether the cumulative human resources and cost required in undertaking additional tumour-based testing to try to determine aetiology for dMMR is unacceptably high when considering diagnostic yield. It was questioned whether this would be cost-effective considering numbers needed to test/screen to prevent one cancer-related death. It was suggested that tumour-based testing to establish somatic aetiology of dMMR might be most appropriate in those cases where clinical suspicion of a heritable predisposition is at least moderate, in order to determine the most appropriate familial bowel screening recommendations; and ensure limited resources and highest intensity screening are directed to those families where suspicion for Lynch syndrome remains high. To more fully address these issues, the UKCGG convened a national consensus meeting on 25 April 2023.
National consensus meeting preparation and format
Key experts representing gastroenterology, pathology, gynaecology and clinical genetics from each genomic medicine service alliance and each of the four countries in the UK were invited to contribute to a national consensus meeting. Participants from the Republic of Ireland were also permitted to attend. In advance of the meeting, an electronic survey was circulated to cancer genetics leads in each regional clinical genetics service, to establish current practices across the UK.
Specifically, attendees were asked to comment on:
Their approach to different clinical scenarios regarding colorectal surveillance strategies
The extent to which testing was completed according to the algorithm in figure 1, according to patients of different ages with typical versus atypical Lynch syndrome spectrum tumours (online supplemental table 1)
Access to testing in their centres (online supplemental figure 1), and what testing they felt should be available.
The factors that should be considered in determining the likelihood of a heritable cause of dMMR cancer, for example, age and tumour type.
When molecular testing could be considered completed in order to make clinical recommendations (ie, was ‘exhaustive’ testing required as standard or could a more pragmatic approach be taken?)
jmg-2024-109886supp001.pdf (184.3KB, pdf)
Key expert speakers were invited to give an overview of current evidence on prevalence of, and approaches to testing for, dMMR across a host of tumour types. The agenda of the meeting is outlined in appendix 1. These talks provided the multidisciplinary audience with relevant overview of evidence on which to base their responses to proposed statements. Recordings of these talks and copies of speakers’ slides are available to download (https://www.ukcgg.org/information-education/ukcgg-consensus-meetings/). In-meeting polls were then run to achieve consensus on a number of statements generated based on thematic analysis of survey results. In line with standard Delphi processes, consensus was deemed to be achieved when at least 50 responses (approximately half of respondents to which relevant statements applied) were recorded, and when at least 80% of responses were ‘agree/strongly agree’ with the proposed statement.29 Open discussion was encouraged where minimum number of responses or consensus was not achieved, or if audience members indicated strong feelings in contravention of the proposed statement. This approach has been successfully implemented for several other UKCGG-led national consensus meetings to generate clinical guidelines.30–34
jmg-2024-109886supp002.pdf (120KB, pdf)
Results
Premeeting survey
The premeeting survey was completed by 44 respondents. A response to a question was considered ‘valid’ if not left blank. The largest single professional group of respondents were clinical geneticists (n=18) (figure 2).
Figure 2.
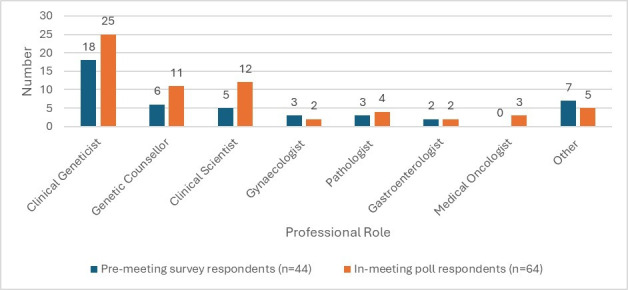
Professional roles of respondents to premeeting survey and in-meeting polls. Other: clinical nurse specialist, colorectal surgeon, oncologist, patient representative, not specified.
In most centres, MMR IHC was the preferred method of assessment of MMR for colorectal cancer and endometrial cancer. Most respondents indicated that reflex testing of MMR was being undertaken on all colorectal (35/36 valid responses) and endometrial cancers (33/36 valid responses), but some respondents indicated that such testing was also reflexively undertaken across a host of other tumour types (online supplemental table 1).
Most respondents indicated that they had routine access to tumour-based BRAF and MLH1 promoter hypermethylation testing, with a slightly lower proportion of respondents indicating routine access to constitutional or tumour-based MMR gene sequencing. Fewer than half of respondents indicated routine access to long-range PCR testing of PMS2 (13/37 valid responses) or loss of heterozygosity analysis on tumour-derived DNA (15/37 valid responses) (online supplemental figure 1).
Variability in extent of testing offered in different clinical scenarios was noted; with some centres offering the same extent of testing in all circumstances, and other services offering no further testing to older patients after uninformative germline results. Similar variability in screening recommendations was noted, with some centres recommending Lynch-equivalent screening for relatives of all probands with unexplained dMMR cancers irrespective of age at diagnosis.
Consensus meeting attendees
The consensus meeting was attended by 82 participants. Because of the multidisciplinary nature of the audience, not all statements were applicable to each attendee, for which reason complete participation for every poll was not expected. 64 participants participated in at least one poll (figure 2).
In-meeting polls
Statements for which consensus was achieved
There were a number of statements for which consensus was achieved after discussion (table 2).
Table 2.
Statements on which consensus was achieved
| Statement | Responses (n) | Proportion of respondents indicating ‘agree/ strongly agree’ or ‘yes’ (%) |
| The abbreviations that should be used to define MMR proficient and MMR deficient should be ‘pMMR’ and ‘dMMR’ as per the IARC ‘Blue Book’ volume on genetic tumour syndromes. | 58 | 96 |
| The term unexplained dMMR should be used instead of the term ‘Lynch-like syndrome’. | 60 | 88 |
| Long-range PCR analysis of PMS2 should routinely be included as part of constitutional testing of PMS2 when cancer demonstrates isolated loss of PMS2. | 59 | 85 |
| Loss of heterozygosity analysis should be routinely available when somatic next-generation sequencing of MMR genes is undertaken. | 59 | 95 |
| It is not mandatory to pursue additional investigations for patients with dMMR cancers beyond standard testing* and constitutional genetic testing in situations where prior probability of heritable cause for cancer is low. | 55 | 90 |
| The following factors should be considered in determining probability of heritable cause of dMMR cancer to guide further testing: family history, age, tumour type, pattern of protein loss, extent of investigation. | 54 | 94 |
| It is not mandatory to undertake Lynch-equivalent surveillance for probands or their first-degree relatives in situations where there is unexplained dMMR, particularly where clinical suspicion of a heritable cause is low. | 56 | 96 |
*Standard testing: somatic BRAF sequencing and/or MLH1 promoter hypermethylation testing where indicated; constitutional MMR gene testing.
IARC, International Agency for Research on Cancer; MMR, mismatch repair.
Terminology
It was agreed that the abbreviations that should be used to define MMR proficient and MMR deficient should be pMMR and dMMR, respectively, as per the International Agency for Research on Cancer ‘Blue Book’ volume on genetic tumour syndromes35 (responses: 58; agreement: 96%). It was also agreed that the term u-dMMR should be used instead of the term Lynch-like Syndrome (LSS) (responses: 60; agreement: 88%).
Extent of molecular testing for dMMR cancer
Participants were asked to comment on the extent of investigation for patients with dMMR cancers beyond ‘standard testing’ (that specified by NICE guidelines DG2711 /DG4212 or the NHSE Genomic Test Directory (or equivalent in devolved nations)). It was agreed that long-range PCR analysis of PMS2 should routinely be included as part of constitutional genetic testing if isolated loss of PMS2 was observed in the cancer (responses: 59; agreement: 85%), and that loss of heterozygosity analysis should be routinely available when somatic next-generation sequencing of MMR genes is undertaken on tumour-derived DNA (responses: 59; agreement: 95%). It was also agreed that it should not be considered mandatory to pursue additional investigations for patients with dMMR cancers beyond standard testing in situations where prior probability of heritable cause for cancer is low (responses: 55; agreement: 90%).
Factors affecting clinical interpretation of dMMR results and need for further testing or enhanced colorectal cancer screening
It was agreed by attendees that although a baseline level of testing as outlined in NICE guidelines and genomic test directories was clinically appropriate for evaluation of patients with dMMR cancers, a pragmatic approach could be taken with regard to the extent of testing for dMMR cancers beyond such testing where the index of suspicion of Lynch syndrome was low for example, for dMMR cancers in older patients without a family history of cancer.
In determining the likelihood of heritable cause of dMMR, and therefore to guide further testing, it was agreed that family history, age, tumour type, pattern of protein loss and extent of investigation (including availability of long-range PCR testing of PMS2) should be considered (responses: 54; agreement: 94%).
In determining screening recommendations for the proband with dMMR cancer and their first-degree relatives (FDRs), it was agreed that it was not always appropriate to recommend Lynch-equivalent surveillance where clinical suspicion of a heritable cause is low, and that such recommendations should not be considered mandatory (responses: 56; agreement: 96%).
Statements where consensus could not be achieved
Despite extensive debate and discussion, there were specific statements for which consensus could not be reached, a variance which reflects uncertainty in testing and clinical interpretation, and difficulty in making general recommendations given the wide variability in clinical scenarios in which dMMR will be encountered (table 3).
Table 3.
Statements for which minimum number of responses and/or consensus was not achieved
| Statement | Responses (n) (minimum required: 50) | Proportion of respondents indicating ‘agree/ strongly agree’ or ‘yes’ (%) (minimum required: 80%) |
| For all colorectal and endometrial cancers, comprehensive testing (BRAF/MLH1 promoter hypermethylation testing/germline testing/somatic MMR next-generation sequencing and loss of heterozygosity analysis) should be performed to rule out a diagnosis of Lynch syndrome prior to making surveillance recommendations. | 54 | 60 |
| Additional investigations for patients with dMMR cancers beyond standard testing as per DG2711/DG4212 and germline genetic testing depend on prior probability of heritable cause for cancer and are not necessary where clinical suspicion is low. | 45 | 60 |
| For proband with dMMR cancer diagnosed >65 years of age without a family history of Lynch syndrome-associated cancers, colorectal surveillance in the proband and their relatives should be guided by the clinical picture and specialist MDT discussion. | 24 | 83 |
| Lynch-equivalent screening (for probands and first-degree relatives) is only appropriate where clinical suspicion of a heritable cause for dMMR is high. | 47 | 75 |
| The extent of testing for unexplained dMMR prior to making surveillance recommendations may be adjusted for non-colorectal/non-endometrial cancers. | 7 | 100 |
| Age and family history should be considered when deciding to proceed to additional testing beyond BRAF/MLH1 hypermethylation and germline testing, for colorectal and endometrial cancers to rule out a diagnosis of Lynch syndrome prior to making surveillance recommendations. | 13 | 69 |
Where minimum number of responses have not been achieved, or where the proportion of respondents indicating "agree"/"strongly agree" is less than 80%, the values are in bold.
Extent of molecular testing for dMMR cancer
It was suggested that comprehensive testing of dMMR colorectal and endometrial cancers should include BRAF and/or MLH1 promoter hypermethylation testing, constitutional MMR genetic testing, somatic MMR next-generation sequencing and loss of heterozygosity analysis and that these investigations should be performed to rule out a diagnosis of Lynch syndrome prior to making surveillance recommendations. Consensus could not be reached on this point (responses: 54; agreement: 60%), with participants citing logistic issues in accessing relevant samples and certain diagnostic technologies, available capacity and resources within clinical departments and lengthy time for full diagnostic workup as barriers to provision of timely, fully informed screening recommendations. Many clinicians provide interim screening recommendations with a view to reviewing the need for screening if updated information from testing becomes available.
It was suggested that additional investigations for patients with dMMR cancers beyond standard testing should be guided by the prior probability of heritable cause for cancer and is not necessary where clinical suspicion is low. Consensus could not be achieved for this statement (responses: 45; agreement: 60%), with many participants citing that such investigations may be most helpful where clinical suspicion of hereditary predisposition is low, in order to ‘downgrade’ screening recommendations. Ultimately, after discussion, consensus was reached that the likelihood of heritable predisposition could be considered in guiding further testing, as outlined above and may be best undertaken in a local MDT setting.
Factors affecting clinical interpretation of dMMR results and need for further testing or enhanced colorectal cancer screening
When attempting to determine the indications for, and extent of additional investigations (beyond germline genetic testing) for patients with dMMR cancers, there was a lack of consensus as to how a prior probability of diagnosing Lynch syndrome should be evaluated and implemented. Discussion around this focused on lack of suitable models to estimate this probability, a minority of participants indicating use of models such as PREMM5 algorithm in clinical practice, however, while this is a useful tool to estimate the probability of Lynch syndrome prior to constitutional testing, it does not currently consider residual likelihood post-testing.36 Consensus could not be achieved when it was proposed that Lynch equivalent screening be limited to those families where suspicion of Lynch syndrome remained high after uninformative constitutional testing (responses: 47; agreement: 75%), for the same reasons. However, consensus was achieved when the statement was reframed to state that such screening was not mandatory if clinical suspicion for Lynch syndrome was low, as outlined above. Participants were reluctant to use age at diagnosis in isolation, as a factor to determine further investigation/screening.
There was uncertainty as to whether surveillance recommendations would be modified based on tumour site as well as clinical features, with most participants abstaining from commenting on the proposed statement, although those participants who did respond agreed this would be acceptable (responses: 7; agreement: 100%).
Discussion
The term LLS refers to an intermediate state of testing, and not a distinct heritable syndrome per se, and has therefore never been clearly defined despite advances in molecular testing and evaluation. Thus, some authors have suggested that the term may cause confusion and is redundant and should not be used.35 37 38 The consensus in this meeting was that the term ‘u-dMMR’ more accurately reflected intermediate and/or uncertainty in testing for dMMR cancers.
There remain significant diagnostic and management challenges when considering patients with cancers demonstrating u-dMMR. Although it was agreed by participants that the term LLS was ill-defined and obsolete, this term has been used in clinical guidelines for colorectal cancer screening in relatives of probands with u-dMMR cancers. The redefinition of LLS to ‘u- dMMR’ by this consensus meeting therefore necessitates a reassessment of existing clinical guidelines24 to align with our consensus indicating that Lynch syndrome-equivalent surveillance is not always appropriate or necessary in families where one individual is affected by u-dMMR cancer.
Significant discussion was generated regarding the extent of testing required to ‘rule out’ Lynch syndrome before surveillance recommendations could be made, with participants citing issues related to access to certain technologies and resources required to perform such testing. Many participants also highlighted the different paradigms in arranging tests to ‘exclude’ a hereditary predisposition, compared with most clinical scenarios where resources are generally focused on diagnosis of heritable mechanisms of disease. The likelihood of Lynch syndrome can be significantly reduced in the majority of patients with dMMR cancers following constitutional testing, and therefore a pragmatic approach to additional testing and ongoing surveillance for family members may be justifiable in some situations. Limitations of genomic testing not unique to Lynch syndrome but to all inherited conditions are such that heritable risk factors can never be fully excluded, and the considerable resources required to investigate dMMR cancers over and above an uninformative germline test, where the prior probability of Lynch syndrome is low must be considered.
Much discussion and variance in opinion focused on lack of data regarding proportion of non-colorectal/non-endometrial dMMR cancers accounted for by Lynch syndrome. This was clearly an issue when considering statements related to extent of investigation/screening recommendations for probands with non-colorectal/non-endometrial cancers.
Although reconfiguration of Genomic Medicine Services in NHS England in 2019 was intended to standardise availability and application of genomic testing in different clinical scenarios across England,39 it is clear that variability in both access and use of different technologies exists between genomic laboratory hubs in England, not to mention between different nations within the UK. Lack of routine access to loss of heterozygosity analysis was flagged as one particular issue hampering comprehensive investigation of probands presenting with dMMR, such that individuals with dMMR tumours demonstrating a single somatic variant and uninformative germline genetic testing are managed in the same way as those individuals where no causative variants are identified in constitutional or tumour-derived DNA. This is likely inappropriate as a significant proportion of tumours in which a single relevant variant will also demonstrate loss of heterozygosity at the locus of interest. Inequitable access to, and application of, long-range PCR testing of PMS2 was also identified as an issue, although consensus was reached that this should be implemented as part of standard testing for those individuals with tumours demonstrating isolated PMS2 loss.
In 2023, the National Lynch Syndrome Registry was successfully implemented in England, where entry to the register requires a molecular confirmation of Lynch syndrome.40 We did not discuss how we can prospectively follow probands with u-dMMR, but it is clear that such data are required to inform guidelines in the future. Furthermore, data are required to determine proportion of dMMR in non-colorectal/non-endometrial cancers accounted for by various somatic aetiologies, not to mention clinical and analytical sensitivity and specificity of available assays to detect underlying mechanisms of disease.
Data so far indicate that the number of patients with dMMR colorectal cancer appropriately referred for germline genetic testing is suboptimal,40 41 and this issue may be even more pronounced for patients with those cancers less commonly recognised as a Lynch syndrome-associated malignancy. Further training and education are required to ensure timely diagnosis of Lynch syndrome, and, where resources are limited, this may represent a more acute clinical risk than scenarios related to u-dMMR where significant genomic testing has already been undertaken.
Further work
As assessment of MMR function continues to expand across different tumour types and constitutional testing of MMR genes increases in patients with non-colorectal/non-endometrial cancers, we hope to generate robust data regarding proportion of dMMR in such cancers accounted for by Lynch syndrome or alternative genomic mechanisms, by continuing and building on previous collaborative work between NHS genomic laboratory hubs and National Disease Registration Service.42 We appreciate, however, that a lack of uniformity in investigation of u-dMMR where clinical suspicion is low will hamper data collection for such cases—further emphasising the need for national collaboration to enable collection of at least some meaningful data. This information about the outcomes of testing, especially for non-colorectal/non-endometrial cancers, will help to inform diagnostic strategies, for example, clarifying the diagnostic yield of somatic and/or germline testing in these tumours.
Limitations of time at this meeting precluded discussion of gynaecological cancer risk management in probands with dMMR cancers and their at-risk relatives. Furthermore, we did not consider indications for consideration of constitutional mosaicism.
Conclusion
While it is not possible to reach uniform advice for all u-dMMR cases, the authors hope that this article describes the difficulties and limitations encountered in clinical practice and provides the reader with support in clinical decision making for individual cases. Recommendations regarding extent of investigation and colorectal cancer surveillance for probands with dMMR cancers and their at-risk relatives should be made based on the best available evidence; but it is crucial that collection of evidence should be practicable in the face of limited resources (money, staff and time).
Despite efforts in standardising availability of comprehensive testing, inequity of access to the same is evident across the UK, hampering efforts to develop uniform guidance for best practice. As a compromise, if it is not reasonably feasible to perform comprehensive constitutional and tumour-based testing for every patient with dMMR cancer, bearing in mind local resources, identification of heritable risk factors in those patients where Mendelian cancer predisposition is most suspected should be prioritised. Consideration of further testing for cryptic constitutional MMR gene variants (currently only available on a research basis) should be considered where clinical suspicion of Lynch syndrome persists—for example, with those patients fulfilling Amsterdam II43/Revised Bethesda44 criteria, or based on a high PREMM (PREdiction Model for gene Mutations)36 score.20 In such cases, Lynch-equivalent colonoscopic screening may be appropriate for FDRs of affected individuals.
Clinical judgement should be used to inform extent to which testing beyond standard testing is performed in families where clinical suspicion of such predisposition is lower. Importantly, clinical judgement should be used to inform surveillance recommendations in probands and their family members, and Lynch-equivalent surveillance is not always indicated where aetiology of dMMR remains elusive after reasonable efforts to determine the same.
We recommend that, at a minimum, ‘reasonable effort’ should include (where relevant), somatic BRAF testing and/or MLH1 promoter hypermethylation testing and constitutional MMR gene testing (and additional genes if indicated as per national genomic testing directories), with further testing beyond that depending on clinical circumstances as outlined above.
Basic principles should also be considered, and it is important that performance and reporting of results from IHC and/or genomic testing be undertaken in line with best practice.45–47
Acknowledgments
The authors thank the attendees of the national multidisciplinary team meeting. The authors also wish to thank any of those participants at the national consensus meeting who logged into the meeting anonymously or with a colleague and therefore could not be explicitly listed as a collaborator.
Footnotes
@mcveighterri, @Helen_Hanson1
Collaborators: UKCGG dMMR consensus meeting attendees: Ruth Armstrong, Andrew Beggs, Cheryl Berlin, Adam Boyde, Angela Brady, Jeremy Bulmer, George Burghel, John Burn, Mark Catherwood, Joseph Christopher, Ruth Cleaver, Beth Coad, Hector Conti, Jacqueline Cook, Wei Cope, Gemma Corbett, Emma Crosbie, Rosemarie Davidson, Bianca DeSouza, Alan Donaldson, Miranda Durkie, Diana Eccles, Nicola Foot, Ian Frayling, Andrew George, Gareth Gerrard, Sarah Gibson, Andrew Green, Stephanie Greville-Heygate, Sarah Hamilton, Helen Hanson, Rachel Hart, Shirley Hodgson, Debbie Holliday, Jacqui Hoyle, Rosalyn Jewell, Zoe Kemp, Louise Kiely, Vicki Kiesel, Kelly Kohut, Rebecca Kristeleit, Ajith Kumar, Fiona Lalloo, Andrew Latchford, Natalie Lee, Donna Lobo, Maurice Loughrey, Suzanne MacMahon, Richard Martin, Sally Martin, Terri McVeigh, Zosia Miedzybrodzka, Eleanor Minshall, Kevin Monahan, Laura Monje-Garcia, Meleri Morgan, Hood Mugalaasi, Jennie Murray, Hannah Musgrave, Emily Nastali, Gail Norbury, Kai Ren Ong, Nicola Onyeador, Judith Pagan, Kezia Quigley, Elizabeth Ratsma, Gillian Rea, Dimitra Repana, Adam Rosenthal, Malcolm Scott, Claire Searle, Adam Shaw, Lucy Side, Kate Simon, Katherine Smith, Joyce Solomons, Avani Varde, Nick West, Jennifer Wiggins, Dorte Wren, Laura Yarram-Smith.
Contributors: The authors (TPMcV, KJM, JC, NW, MS, JM and HH) conceived of the idea, organised the consensus meeting, devised poll questions, collated and analysed the data and drafted the manuscript. Consensus meeting attendees, as collaborators, attended the meeting and answered the polls from which data were derived. TPMcV is the author responsible for the overall content as the guarantor.
Funding: HH is supported by the NIHR Exeter Biomedical Research Centre (NIHR203320). This study was supported by the National Institute for Health and Care Research Exeter Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Contributor Information
Collaborators: UKCGG dMMR Consensus Meeting Attendees, Ruth Armstrong, Andrew Beggs, Cheryl Berlin, Adam Boyde, Angela Brady, Jeremy Bulmer, George Burghel, John Burn, Mark Catherwood, Joseph Christopher, Ruth Cleaver, Beth Coad, Hector Conti, Jacqueline Cook, Wei Cope, Gemma Corbett, Emma Crosbie, Rosemarie Davidson, Bianca DeSouza, Alan Donaldson, Miranda Durkie, Diana Eccles, Nicola Foot, Ian Frayling, Andrew George, Gareth Gerrard, Sarah Gibson, Andrew Green, Stephanie Greville-Heygate, Sarah Hamilton, Helen Hanson, Rachel Hart, Shirley Hodgson, Debbie Holliday, Jacqui Hoyle, Rosalyn Jewell, Zoe Kemp, Louise Kiely, Vicki Kiesel, Kelly Kohut, Rebecca Kristeleit, Ajith Kumar, Fiona Lalloo, Andrew Latchford, Natalie Lee, Donna Lobo, Maurice Loughrey, Suzanne MacMahon, Richard Martin, Sally Martin, Terri McVeigh, Zosia Miedzybrodzka, Eleanor Minshall, Kevin Monahan, Laura Monje-Garcia, Meleri Morgan, Hood Mugalaasi, Jennie Murray, Hannah Musgrave, Emily Nastali, Gail Norbury, Kai Ren Ong, Nicola Onyeador, Judith Pagan, Kezia Quigley, Elizabeth Ratsma, Gillian Rea, Dimitra Repana, Adam Rosenthal, Malcolm Scott, Claire Searle, Adam Shaw, Lucy Side, Kate Simon, Katherine Smith, Joyce Solomons, Avani Varde, Nick West, Jennifer Wiggins, Dorte Wren, and Laura Yarram-Smith
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
Not applicable.
References
- 1. Eikenboom EL, Moen S, van Leeuwen L, et al. Unexplained mismatch repair deficiency: case closed. HGG Adv 2023;4:100167. 10.1016/j.xhgg.2022.100167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ratti M, Lampis A, Hahne JC, et al. Microsatellite instability in gastric cancer: molecular bases, clinical perspectives, and new treatment approaches. Cell Mol Life Sci 2018;75:4151–62. 10.1007/s00018-018-2906-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kang Y-J, O’Haire S, Franchini F, et al. A scoping review and meta-analysis on the prevalence of Pan-tumour biomarkers (dMMR, MSI, high TMB) in different solid tumours. Sci Rep 2022;12:20495. 10.1038/s41598-022-23319-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bonneville R, Krook MA, Kautto EA, et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol 2017;2017. 10.1200/PO.17.00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee V, Murphy A, Le DT, et al. Mismatch repair deficiency and response to immune checkpoint blockade. Oncologist 2016;21:1200–11. 10.1634/theoncologist.2016-0046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13. 10.1126/science.aan6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ryan NAJ, McMahon R, Tobi S, et al. The proportion of endometrial tumours associated with Lynch syndrome (PETALS): a prospective cross-sectional study. PLoS Med 2020;17:e1003263. 10.1371/journal.pmed.1003263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology 2010;138:2073–2087. 10.1053/j.gastro.2009.12.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gausachs M, Mur P, Corral J, et al. MLH1 promoter hypermethylation in the analytical algorithm of Lynch syndrome: a cost-effectiveness study. Eur J Hum Genet 2012;20:762–8. 10.1038/ejhg.2011.277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ward RL, Dobbins T, Lindor NM, et al. Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the colon cancer family registry. Genet Med 2013;15:25–35. 10.1038/gim.2012.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. NICE . National Institute for Health and Care Excellence Guidance: molecular testing strategies for Lynch syndrome in people with colorectal cancer. Diagnostics guidance [DG27]; 2017.
- 12. NICE . National Institute for Health and Care Excellence Guidance: testing strategies for Lynch syndrome in people with endometrial cancer. Diagnostics guidance [DG42]; 2020.
- 13. West NP, Gallop N, Kaye D, et al. Lynch syndrome screening in colorectal cancer: results of a prospective 2-year regional programme validating the NICE diagnostics guidance pathway throughout a 5.2-million population. Histopathology 2021;79:690–9. 10.1111/his.14390 [DOI] [PubMed] [Google Scholar]
- 14. Metcalf AM, Spurdle AB. Endometrial tumour BRAF mutations and MLH1 promoter methylation as predictors of Germline mismatch repair gene mutation status: a literature review. Fam Cancer 2014;13:1–12. 10.1007/s10689-013-9671-6 [DOI] [PubMed] [Google Scholar]
- 15. NHS England . National Genomic test directory - rare and inherited disease eligibility criteria; 2024. 6.
- 16. NHS Scotland . Scottish strategic network for genomic medicine genomic test directory rare & inherited disease; 2023. 2.
- 17. Leclerc J, Vermaut C, Buisine MP. Diagnosis of Lynch syndrome and strategies to distinguish Lynch-related tumors from sporadic MSI/dMMR tumors. Cancers (Basel) 2021;13:467. 10.3390/cancers13030467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pope BJ, Clendenning M, Rosty C, et al. Germline and tumor sequencing as a diagnostic tool to resolve suspected Lynch syndrome. J Mol Diagn 2021;23:358–71. 10.1016/j.jmoldx.2020.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eikenboom EL, van der Werf-’t Lam A-S, Rodríguez-Girondo M, et al. Universal immunohistochemistry for Lynch syndrome: a systematic review and meta-analysis of 58,580 colorectal carcinomas. Clin Gastroenterol Hepatol 2022;20:e496–507. 10.1016/j.cgh.2021.04.021 [DOI] [PubMed] [Google Scholar]
- 20. Te Paske I, Mensenkamp AR, Neveling K, et al. Noncoding aberrations in mismatch repair genes underlie a substantial part of the missing heritability in Lynch syndrome. Gastroenterology 2022;163:1691–4. 10.1053/j.gastro.2022.08.041 [DOI] [PubMed] [Google Scholar]
- 21. Walker R, Mahmood K, Joo JE, et al. A tumor focused approach to resolving the etiology of DNA mismatch repair deficient tumors classified as suspected Lynch syndrome. medRxiv 2023. 10.1101/2023.02.27.23285541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arends M, Ibrahim M, Happerfield L, et al. Interpretation of immunohistochemical analysis of mismatch repair (MMR) protein expression in tissue sections for investigation of suspected Lynch / hereditary non-polyposis colorectal cancer (HNPCC) syndrome. NEQAS ICC & ISH recommendations; 2008.
- 23. Jennings LJ, Arcila ME, Corless C, et al. Guidelines for validation of next-generation sequencing–based oncology panels: a joint consensus recommendation of the association for molecular pathology and college of American pathologists. J Mol Diagn 2017;19:341–65. 10.1016/j.jmoldx.2017.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Monahan KJ, Bradshaw N, Dolwani S, et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut 2020;69:411–44. 10.1136/gutjnl-2019-319915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martínez-Roca A, Giner-Calabuig M, Murcia O, et al. Lynch-like syndrome: potential mechanisms and management. Cancers (Basel) 2022;14:1115. 10.3390/cancers14051115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pearlman R, Haraldsdottir S, de la Chapelle A, et al. Clinical characteristics of patients with colorectal cancer with double somatic mismatch repair mutations compared with Lynch syndrome. J Med Genet 2019;56:462–70. 10.1136/jmedgenet-2018-105698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rodríguez-Soler M, Pérez-Carbonell L, Guarinos C, et al. Risk of cancer in cases of suspected Lynch syndrome without Germline mutation. Gastroenterology 2013;144:926–32. 10.1053/j.gastro.2013.01.044 [DOI] [PubMed] [Google Scholar]
- 28. UKCGG information and education: National cancer Genetics MDT. 2023. Available: https://www.ukcgg.org/information-education [Accessed 09 Jan 2024].
- 29. Diamond IR, Grant RC, Feldman BM, et al. Defining consensus: a systematic review recommends methodologic criteria for reporting of Delphi studies. J Clin Epidemiol 2014;67:401–9. 10.1016/j.jclinepi.2013.12.002 [DOI] [PubMed] [Google Scholar]
- 30. Kohut K, Speight B, Young J, et al. Co-design of patient information leaflets for Germline predisposition to cancer: recommendations for clinical practice from the UK cancer Genetics group (UKCGG), cancer research UK (CRUK) funded CanGene-CanVar programme and the association of genetic nurse counsellors (AGNC). J Med Genet 2024;61:142–9. 10.1136/jmg-2023-109440 [DOI] [PubMed] [Google Scholar]
- 31. Clark A, Thomas S, Hamblin A, et al. Management of patients with Germline predisposition to haematological malignancies considered for allogeneic blood and marrow transplantation: best practice consensus guidelines from the UK cancer Genetics group (UKCGG), Cangene-Canvar, NHS England Genomic laboratory Hub (GLH) haematological malignancies working group and the British society of blood and marrow transplantation and cellular therapy (BSBMTCT). Br J Haematol 2023;201:35–44. 10.1111/bjh.18682 [DOI] [PubMed] [Google Scholar]
- 32. Speight B, Hanson H, Turnbull C, et al. Germline predisposition to haematological malignancies: best practice consensus guidelines from the UK cancer genetics group (UKCGG), Cangene-Canvar and the NHS England haematological oncology working group. Br J Haematol 2023;201:25–34. 10.1111/bjh.18675 [DOI] [PubMed] [Google Scholar]
- 33. Hanson H, Kulkarni A, Loong L, et al. UK consensus recommendations for clinical management of cancer risk for women with Germline pathogenic variants in cancer predisposition genes: RAD51C, RAD51D, BRIP1 and PALB2. J Med Genet 2023;60:417–29. 10.1136/jmg-2022-108898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hanson H, Brady AF, Crawford G, et al. UKCGG consensus group guidelines for the management of patients with constitutional TP53 pathogenic variants. J Med Genet 2020;58:135–9. 10.1136/jmedgenet-2020-106876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lokuhetty D, White VA, Watanabe R, et al. Genetic tumour syndromes of the digestive system, in digestive system tumours. Lyon: International Agency for Research on Cancer; 2019. 511. [Google Scholar]
- 36. Kastrinos F, Uno H, Ukaegbu C, et al. Development and validation of the PREMM(5) model for comprehensive risk assessment of Lynch syndrome. J Clin Oncol 2017;35:2165–72. 10.1200/JCO.2016.69.6120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ladabaum U. What is Lynch-like syndrome and how should we manage it. Clin Gastroenterol Hepatol 2020;18:294–6. 10.1016/j.cgh.2019.08.009 [DOI] [PubMed] [Google Scholar]
- 38. Holter S, Hall MJ, Hampel H, et al. Risk assessment and genetic counseling for Lynch syndrome - practice resource of the National society of genetic counselors and the collaborative group of the Americas on inherited gastrointestinal cancer. J Genet Couns 2022;31:568–83. 10.1002/jgc4.1546 [DOI] [PubMed] [Google Scholar]
- 39. Barwell J, Snape K, Wedderburn S. The new genomic medicine service and implications for patients. Clin Med (Lond) 2019;19:273–7. 10.7861/clinmedicine.19-4-273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Monahan KJ, Ryan N, Monje-Garcia L, et al. The English national Lynch syndrome transformation project: an NHS Genomic medicine service alliance (GMSA) programme. Bmjonc 2023;2:e000124. 10.1136/bmjonc-2023-000124 [DOI] [Google Scholar]
- 41. Monje-Garcia L, Bill T, Farthing L, et al. From diagnosis of colorectal cancer to diagnosis of Lynch syndrome: the RM partners quality improvement project. Colorectal Dis 2023;25:1844–51. 10.1111/codi.16707 [DOI] [PubMed] [Google Scholar]
- 42. Loong L, Huntley C, McRonald F, et al. Germline mismatch repair (MMR) gene analyses from English NHS regional molecular genomics laboratories 1996-2020: development of a national resource of patient-level genomics laboratory records. J Med Genet 2023;60:669–78. 10.1136/jmg-2022-108800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vasen HF, Watson P, Mecklin JP, et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International collaborative group on HNPCC. Gastroenterology 1999;116:1453–6. 10.1016/s0016-5085(99)70510-x [DOI] [PubMed] [Google Scholar]
- 44. Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004;96:261–8. 10.1093/jnci/djh034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Singh N, Wong R, Tchrakian N, et al. Interpretation and reporting terminology for mismatch repair protein immunohistochemistry in endometrial cancer. The British Association of Gynaecological Pathologists; 2020. [Google Scholar]
- 46. Garrett A, Allen S, Loong L, et al. CanVig-UK consensus specification for cancer susceptibility genes (CSGs) of ACGS best practice guidelines for variant classification; 2022.
- 47. Ellard S, Baple EL, Callaway A, et al. ACGS best practice guidelines for variant classification in rare disease 2020; 2020. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jmg-2024-109886supp001.pdf (184.3KB, pdf)
jmg-2024-109886supp002.pdf (120KB, pdf)
Data Availability Statement
Data are available upon reasonable request.