

Summary
Chemotherapy remains the first-line treatment for advanced esophageal cancer. However, durable benefits are achieved by only a limited subset of individuals due to the elusive chemoresistance. Here, we utilize patient-derived xenografts (PDXs) from esophageal squamous-cell carcinoma to investigate chemoresistance mechanisms in preclinical settings. We observe that activated cancer-associated fibroblasts (CAFs) are enriched in the tumor microenvironment of PDXs resistant to chemotherapy. Mechanistically, we reveal that cancer-cell-derived S100A8 triggers the intracellular RhoA-ROCK-MLC2-MRTF-A pathway by binding to the CD147 receptor of CAFs, inducing CAF polarization and leading to chemoresistance. Therapeutically, we demonstrate that blocking the S100A8-CD147 pathway can improve chemotherapy efficiency. Prognostically, we found the S100A8 levels in peripheral blood can serve as an indicator of chemotherapy responsiveness. Collectively, our study offers a comprehensive understanding of the molecular mechanisms underlying chemoresistance in esophageal cancer and highlights the potential value of S100A8 in the clinical management of esophageal cancer.
Keywords: esophageal squamous-cell carcinoma, chemotherapy, patient-derived xenografts, the tumor microenvironment, cancer-associated fibroblasts, S100A8
Graphical abstract
Highlights
-
•
Estimation of responsiveness to chemotherapy using ESCC PDX models
-
•
S100A8 imparts chemoresistance by reprogramming CAFs
-
•
Inhibition of S100A8-CD147 pathway improves chemotherapy efficiency
-
•
S100A8 serves as a biomarker for predicting chemotherapy responsiveness
Chen et al. investigate the mechanisms of chemoresistance using well-established ESCC PDX models. They present new chemoresistance mechanisms conferred by S100A8-mediated cancer cell-fibroblast crosstalk and identify S100A8 as a potential non-invasive biomarker for evaluating chemosensitivity.
Introduction
Esophageal squamous-cell carcinoma (ESCC) emerges as a prominent and lethal malignancy within the digestive system, presenting a dismal 5-year survival rate of only 20%.1,2 Fluorouracil (5-FU)/cisplatin (CDDP)-based chemotherapy is a standard treatment for localized ESCC.3,4 However, the current standard of care offers only modest relief for patients with ESCC. This is exacerbated by the fact that most patients eventually suffer tumor recurrence due to primary or acquired multidrug resistance, and the underlying mechanisms of chemoresistance are not fully understood.
The intractable tumor microenvironment (TME) remains one of the key issues that impedes effective cancer management in clinical practice. Mounting advances on the TME have revealed its importance in driving cancer heterogeneity and treatment resistance.5,6,7,8 Within the TME, various nonmalignant cells coexist with malignant cells, primarily including cancer-associated fibroblasts (CAFs), endothelial cells, tumor-infiltrating lymphocytes, and myeloid cells. Among all types of stromal cells, CAFs are the abundant and heterogeneous components that support diverse critical cancer-promoting functions.9,10,11 For example, CAFs have the capacity to remodel the extracellular matrix (ECM), creating physical barriers that hinder drug delivery and impede immune cells infiltration.12,13 Due to the tumor-supporting functions exerted by CAFs in cancer development, the modulation of CAFs presents a tempting and promising prospect for enhancing anti-cancer efficacy.14 Nevertheless, directly eliminating CAFs may promote the dissemination and invasion of cancer cells, leading to decreased survival rates.15,16 Furthermore, clinical trials focused on targeting CAFs have encountered limited progress.17 Therefore, it is imperative to develop innovative strategies to reprogram the pro-tumor CAFs into a quiescent state without disrupting the tumor-stroma architecture, which will hold potential for enhancing therapeutic outcomes in a safe and effective manner.
Devising effective approaches targeting CAFs relies on understanding their interactions with cancer cells. Accumulating evidence suggests that the symbiotic interactions between cancer cells and CAFs are crucial in the tumorigenesis,18,19 progression,20,21 and resistance to anti-cancer therapies of ESCC.22,23,24,25 Our prior work has demonstrated that cancer cell-secreted CXCL1 activates myofibroblastic CAFs (myCAFs), triggering collagen deposition and ECM remodeling. The deposited collagen supports the survival of ESCC cells after ionizing radiation exposure, ultimately leading to radioresistance.25 Additionally, CAFs are typically polarized toward an immunosuppressive phenotype, thus promoting immune evasion and tumor progression in ESCC.23,26 These findings expand our understanding of how cancer cell-stromal cell interactions contribute to cancer development and propose a number of strategies for targeting CAFs to improve outcomes in patients with ESCC.
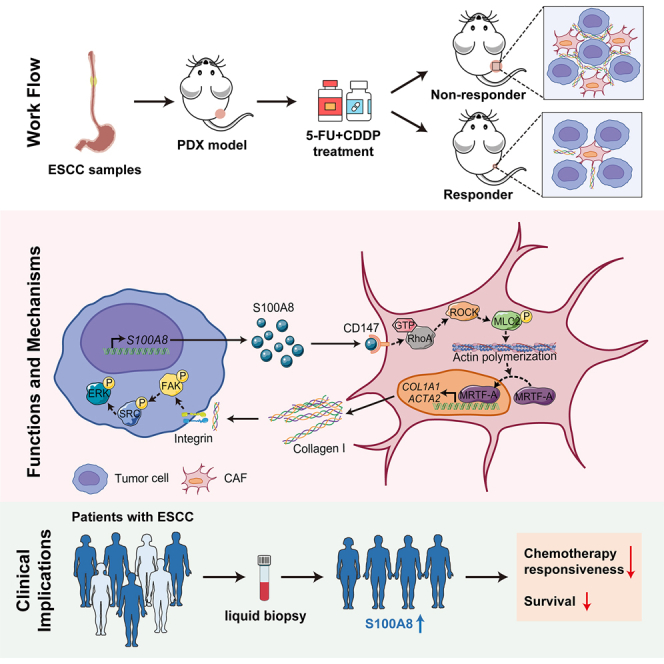
The utilization of preclinical animal models that preserve the intact characteristics of interactions between cancer cells and stromal cells is of utmost importance for investigating the impact of CAF polarization on treatment resistance in ESCC. Patient-derived xenograft (PDX) models offer a sophisticated platform for comprehending the disease mechanisms of ESCC. PDXs can mimic the genetic and composition diversity of the clinical settings due to the high histological and pathological relevance between donor tumors and established tumor xenografts.27 In the present study, we comprehensively investigate the mechanisms underlying chemoresistance of ESCC utilizing well-established PDX models. We demonstrate the enrichment of myCAFs, the primary source of tumor-associated ECM, in the non-responsive PDXs’ TME. Importantly, we illustrate that the cancer-cell-derived alarmin S100A8 triggers myCAF accumulation and imparts chemoresistance. Analysis of human ESCC samples aligns with our PDX findings. Our findings highlight alarmin S100A8 as a promising target for circumventing ESCC chemoresistance and as a valuable marker for predicting chemotherapy responsiveness.
Results
5-FU/CDDP-based chemotherapeutics screening reveals the enrichment of myCAF-associated signatures in non-responsive PDXs
To comprehensively investigate the molecular signatures that determine chemotherapy responsiveness in ESCC, we expanded the ESCC PDX cohort based on our prior studies.25,28 The enlarged cohort comprised 20 PDX cases from surgical specimens of distinct donors (detailed clinical information in Table S1). Subsequently, in vivo screening for 5-FU- and CDDP-based chemotherapy was carried out. All PDXs were subjected to three cycles of treatment with 5-FU/CDDP or vehicle reagents once the xenografts reached approximately 150 mm3 in size (Figure 1A). The mice in the experiments exhibited a good tolerance to the chemotherapy dosage. Although some experienced mild weight loss during treatment, there were no significant differences in body weight between the groups receiving chemotherapeutics and those receiving vehicle reagents (Figure S1A). The chemotherapy responsiveness was assessed by applying two criteria to compare the changes in relative tumor volume (RTV) between the drug-treated and vehicle-treated groups: (1) determining if drug treatment significantly restrained tumor growth (p < 0.05 for responders), and (2) calculating the percentage of tumor growth inhibition (TGI [%], as described in STAR Methods) at the endpoint (TGI > 50% for responders). According to the defined criteria, PDXs were classified into 12 non-responders (NRs) and eight responders (Rs) (Figure 1B). The average TGI was 63.1% for Rs and 12.2% for NRs, with a significant disparity between the two groups (Figure 1C). Additionally, the tumor growth rates of vehicle-treated mice were not significantly different between the R and NR groups (Figure S1B), suggesting that the inherent tumor growth capacity had minimal impact on the classification of responsiveness.
Figure 1.

Generation of ESCC PDXs chemotherapy cohort and identification of myCAF-associated gene signatures in non-responsive PDXs
(A) Graphical overview of the study.
(B) RTV curves of tumor xenografts from 20 distinct ESCC PDXs that were treated with chemotherapeutics or vehicle (n = 3–5 mice per group). Data are presented as mean ± SD. p values are determined using two-tailed Student’s t test.
(C) Boxplot exhibiting the percentage of tumor growth inhibition (TGI) of PDXs between the R (n = 8) and NR (n = 12) groups. Boxes represent the interquartile range, and whiskers represent the minimum and maximum values. p value is determined using two-tailed Student’s t test.
(D) Principal-component analysis (PCA) plot showing overall patterns of gene expression of 20 PDX donors’ tumor tissues. Circles indicate two separate clusters (R cluster [n = 8], blue; NR cluster [n = 12], red).
(E) Gene set enrichment analysis (GSEA) of pathways enriched in the NR group, using RNA-seq data of PDX donors’ tumor tissues.
(F) Spearman correlation between the TGI (%) and the collagen-associated genes score. The gray area represents 95% CI (n = 20).
(G) Representative images of H&E staining (top) and MTS (bottom) in the PDX donors’ biopsies of the R (n = 8) and NR (n = 12) groups. Scale bar, 100 μm.
(H) Quantification of (G). Data are presented as mean ± SEM. ∗p < 0.05 of two-tailed Student’s t test.
(I) Spearman correlation between the TGI (%) and the MTS-positive area (%). The gray area represents 95% CI (n = 20). See also Figure S1 and Table S1.
Next, we investigated the transcriptomic signatures of the R and NR groups by analyzing the RNA sequencing (RNA-seq) data of the 20 PDX donors’ tumor tissues. Initially, we assessed the expression variances of two well-known cell proliferation markers, MKI67 and PCNA,29 and found no correlation with chemosensitivity (Figure S1C). This finding reinforces our earlier conclusion that the inherent tumor growth rate was not responsible for chemoresistance. The principal-component analysis (PCA) revealed a clear separation between the R and NR groups (Figure 1D), indicating that variations in gene expression patterns accounted for the discrepancies in chemotherapy responsiveness. The gene set enrichment analysis (GSEA) revealed a significant enrichment of gene sets associated with cancer cells-stromal cells interactions, such as “focal adhesion” and “ECM-receptor interaction,” in the NR group (Figure 1E). Additionally, the activation of the “smooth muscle contraction” pathway, regulated by Rho-kinase signaling and responsible for actomyosin contractility in the stroma,30 was observed in the NR group (Figure 1E). These findings suggest that the myofibroblastic phenotypes of CAFs, including ECM remodeling, deposition, and their interactions with cancer cells, may play a crucial role in determining the responsiveness to chemotherapy in ESCC. We then calculated the Z scores of expression values for the pathway-associated genes and established signature scores for pathways enriched in the NR group. Correlation analysis revealed a significant and negative correlation between these pathway activities and chemotherapy responsiveness (Figure S1D).
To identify more robust representative tumor expression marker associated with responsiveness to chemotherapy, we analyzed the differentially expressed genes and observed significant upregulation of genes encoding collagen, fibronectin, and other ECM proteins, such as COL1A1, COL3A1, COL5A1, FN1, and POSTN, in the NR group (Figure S1E). Subsequently, we selected the upregulated collagen-associated genes in the NR group and calculated the signature score. Correlation analysis showed the signature score of collagen-associated genes was negatively correlated with chemotherapy responsiveness (Figure 1F). Masson’s trichrome staining (MTS) further confirmed a significant deposition of collagen in the NR group (Figures 1G and 1H), with the collagen content serving as a representation of chemoresistance severity (Figure 1I). Given the capability of myCAFs for depositing and remodeling ECM in the TME,9 these findings collectively indicate that stromal components, particularly myCAFs and the abnormally deposited ECM, and their interactions with cancer cells in the TME, play vital roles in determining the responsiveness to 5-FU/CDDP-based chemotherapy.
Cancer-cell-derived S100A8 contributes to chemoresistance in a CAF-dependent manner
Growing evidence suggests that cancer cells can induce the transformation of CAFs into diverse oncogenic phenotypes by secreting various cytokines, leading to the acquisition of distinct cancer-supporting capacities.31,32,33 In the PDX model, the TME components, primarily comprising stromal cells and non-cellular matrix proteins, will progressively be replaced by murine components, resulting in the coexistence of human-originating cancer cells and mouse-originating stromal cells.34 Therefore, to investigate the culprits responsible for ECM remodeling and chemoresistance, we conducted RNA-seq of PDX tumors and then focused on analyzing the human-originating components of the TME (Figure 1A). Mouse-originating RNA reads were filtered out using Disambiguate software,35 and the resulting reads were used for subsequent analysis. Differential expression analysis using human-originating reads revealed that the expression of S100A8 was robustly upregulated in non-responsive PDXs (Figure 2A). S100A8 belongs to the calcium-binding S100 alarmin family and typically complexes with S100A9 to form a heterodimer.36 Immunohistochemistry (IHC) analysis of PDX tumors showed increased S100A8 staining in the cancer cells of the NR group (Figures 2B and 2C). Moreover, the expression levels of S100A8 were negatively correlated with chemotherapy responsiveness (Figure 2D), suggesting a potential role for cancer-cell-derived S100A8 in promoting chemoresistance.
Figure 2.

Cancer-cell-derived S100A8 imparts chemoresistance in a CAF-dependent manner
(A) Volcano plots exhibiting differentially expressed genes between the R and NR groups, using RNA-seq data of the PDX mice’s tumor tissues.
(B) Representative images of H&E staining (top) and S100A8 IHC staining (bottom) in the PDX tumor tissues of the R (n = 8) and NR (n = 12) groups. Scale bar, 100 μm.
(C) Quantification of (B). Data are presented as mean ± SEM. p value is determined using two-tailed Student’s t test.
(D) Spearman correlation between the TGI (%) and the S100A8 RNA level (left) and the S100A8 IHC intensity (right). The gray areas represent 95% CI (n = 20).
(E) ELISA analysis of S100A8 in the CM (top) and western blot analysis of S100A8 in cell lysate (bottom) of KYSE510 and KYSE180 cells stably transfected with S100A8 shRNA or control nontargeting shRNA (n = 3 biological replicates). Data are presented as mean ± SD. p values are determined using two-tailed Student’s t test.
(F) The left panel shows the schematic of co-culture system and cell viability assay. The right panel shows the quantification of cell viability in KYSE510 and KYSE180 cell lines (n = 3 biological replicates). Data are presented as mean ± SD. p values are determined using two-tailed Student’s t test.
(G) Tumor growth curves of control and S100A8-knockdown xenografts treated with chemotherapeutics or vehicle reagents (n = 4 per group). Data are presented as mean ± SD. p values are determined using two-tailed Student’s t test.
(H) Representative images of H&E, S100A8, MTS, αSMA, and CD31 staining of tumor tissues in (G). Scale bar, 100 μm.
(I) Quantification of (H). Data are presented as mean ± SEM. p values are determined using two-tailed Student’s t test.
(J) Representative images of αSMA and CD31 staining in the PDX tumors of the R (n = 8) and NR (n = 12) groups. Scale bar, 100 μm.
(K) Quantification of (J). Data are presented as mean ± SEM. p values are determined using two-tailed Student’s t test.
For all panels, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, and n.s., not significant. Each assay for western blot had three biological repeats. See also Figure S2.
S100A8 is frequently secreted into the extracellular space where it binds to TLR4, RAGE, CD147, and other receptors.37 As S100A8 was predominantly expressed in cancer cells in ESCC PDX tumors (Figure 2B), we initially hypothesized that modulating its expression in cancer cells could affect cell growth and response to chemotherapy. To this end, we used two sequence-independent short hairpin RNAs (shRNAs) to knock down S100A8 expression in ESCC cell lines (Figure S2A), and we confirmed that this inhibition reduced both intracellular and extracellular S100A8 protein levels (Figure 2E). However, this manipulation had limited effects on cell proliferation and chemosensitivity in vitro (Figure S2B). These findings indicate that the autocrine pathway of S100A8 does not directly influence the malignant behavior of cancer cells but may instead play a cancer-promoting role by influencing other cellular components within the TME.
We subsequently analyzed cell-cell interactions between S100A8-positive malignant epithelial cells and stromal cells using our previously published ESCC single-cell RNA-seq (scRNA-seq) data (GSE160269).20 Given that PDX models were established in NOD/SCID/IL-2Rγnull (NSG) mice, which lacked functional immune cells, we focused on non-immune stromal cells. A robust interaction was observed between S100A8-positive cancer cells and BSG (encoding CD147)-positive CAFs through ligand-receptor interaction analysis (Figures S2C–S2E), suggesting that S100A8 may drive tumor progression and chemoresistance by polarizing CAFs. Next, we assessed phenotypic changes of S100A8-knockdown ESCC cells upon the interactions with CAFs (Figure 2F). Esophageal CAFs derived from primary ESCC tissues were used in the following assays. Compared with ESCC cells expressing nonspecific shRNA (NC) cocultured with CAFs, the S100A8-knockdown ESCC cells cocultured with CAFs displayed markedly reduced proliferation and enhanced sensitivity to chemotherapeutic treatment (Figures 2F and S2F). Additionally, we observed that S100A8 knockdown suppressed the growth of ESCC xenografts in vivo, irrespective of chemotherapeutic application (Figures 2G and S2G). In the context of xenografts, S100A8 ablation in ESCC cells resulted in reduced activation of CAFs and decreased collagen deposition in the TME, as confirmed by alpha-smooth muscle actin (αSMA) and MTS staining, respectively (Figures 2H and 2I). Conversely, there was no disparity in the quantity of vascular endothelial cells between the groups, as revealed by CD31 staining (Figures 2H and 2I). Similarly, non-responsive PDX tumors consistently exhibited higher αSMA staining compared to responsive ones, with no discrepancy in CD31 levels (Figures 2J and 2K). Taken together, these findings suggest that cancer-derived S100A8 contributes to chemoresistance, necessitating the involvement of stromal components in the TME, particularly CAFs.
S100A8 triggers the activation and accumulation of myCAFs in the TME
Based on the role of CAFs in S100A8-mediated chemoresistance, we monitored the possible interplay between S100A8 and CAF polarization. CAFs can be skewed toward myCAFs, inflammatory CAFs (iCAFs), or antigen-presenting CAFs (apCAFs) under diverse stimuli to promote cancer progression and invasion.10 Consequently, we profiled CAFs to investigate the intricate and dynamic cell-cell interactions relationship in the ESCC TME using scRNA-seq data.20 Our analysis revealed that CAFs could be classified into five distinct subtypes (Figure S3A), with each subtype exhibiting unique patterns of marker genes expression (Figure S3B). Notably, ligand-receptor interaction analysis demonstrated that S100A8-positive cancer cells exhibited the strongest interactions with myCAFs (Figure S3C). We then calculated the CAF signature scores of marker genes expressed in myCAFs and iCAFs to quantify their biological functions and conducted correlation analysis with the expression level of S100A8 in cancer cells. The results revealed a positive correlation between the S100A8 RNA level and the myCAF signature score, while no such correlation was observed with the iCAF signature score (Figure 3A). Additionally, the S100A8 RNA level showed a positive correlation with myCAF marker genes (Figure S3D), but not with iCAF marker genes (Figure S3E). These findings indicate that S100A8 produced by ESCC cells may significantly influence the cancer-stroma crosstalk, thereby governing the polarization of microenvironmental CAFs.
Figure 3.

S100A8 polarizes CAFs toward myCAFs
(A) Spearman correlations between the S100A8 RNA level of epithelial cells and the myCAF (left) and iCAF (right) signature scores of fibroblasts. The gray areas represent 95% CIs (n = 60).
(B) Representative multiple immunofluorescence images of KRT6A, S100A8, and αSMA in the PDX donors’ primary tumor tissues of the R (n = 8) and NR (n = 12) groups. Scale bar, 100 μm.
(C) Quantification of S100A8 and αSMA fluorescence intensity in (B). Data are presented as mean ± SEM. p values are determined using two-tailed Student’s t test.
(D) Spearman correlation between the S100A8 and the αSMA fluorescence intensity. The gray area represents 95% CI (n = 20).
(E) Western blot analysis of the myCAF-related marker proteins collagen type 1 and αSMA, the iCAF-related marker CXCL1 in CAFs cocultured with KYSE510 and KYSE180 cells with or without S100A8 knockdown.
(F) Representative images of CAF migration induced by CM derived of KYSE510 and KYSE180 cells with or without S100A8 knockdown. Scale bar, 100 μm.
(G) Quantification of (F) (n = 3 biological replicates). Data are presented as mean ± SD. p values are determined using two-tailed Student’s t test.
(H) Western blot analysis of collagen type 1, αSMA, and CXCL1 in CAFs treated with indicated concentration of rS100A8 proteins.
(I) Representative images of CAF migration induced by indicated concentration of rS100A8 proteins. Scale bar, 100 μm.
(J) Quantification of (I) (n = 3 biological replicates). Data are presented as mean ± SD. p value is determined using two-tailed Student’s t test.
For all panels, ∗∗p < 0.01, ∗∗∗p < 0.001. Each assay for western blot had three biological repeats. See also Figure S3.
To examine the role of S100A8 in the polarization of CAFs, we deciphered the interactions between S100A8 and myCAFs in clinical samples. Multiple immunofluorescence analysis (mIF) of the PDX donors’ tumor tissues showed that S100A8 expression was predominantly observed in cancer cells, as shown by the esophageal epithelial-specific marker KRT6A (Figure 3B).38 Furthermore, the NR group exhibited elevated S100A8 levels in epithelial cells and increased αSMA levels in fibroblasts compared to the R group (Figures 3B and 3C). Notably, there was a significant and positive correlation between the intensity of S100A8 immunofluorescence and the activation level of myCAFs, as demonstrated by the αSMA intensity (Figure 3D). These results were consistent with scRNA-seq analysis (Figure 3A and S3D). Subsequently, we investigated the impact of endogenous S100A8 on CAFs. The CAFs cocultured with S100A8-knockdown ESCC cells displayed reduced expression of myCAF markers, including collagen type 1 and αSMA, compared to those cocultured with NC ESCC cells. However, there was no significant difference in the expression of the iCAF marker (CXCL1) (Figure 3E). Transwell-based chemotaxis assays demonstrated that CAFs incubated with the conditional medium (CM) derived from S100A8-knockdown ESCC cells exhibited significantly reduced migration capacity in comparison to those incubated with CM from NC ESCC cells (Figures 3F and 3G). Additionally, we investigated the impact of exogenous S100A8 on CAFs polarization by introducing recombinant human S100A8 (rS100A8) protein into the culture medium. Similarly, following rS100A8 protein treatment, there was a significant dose-dependent induction in the expression of myCAF markers but not the iCAF marker (Figure 3H), along with enhanced migration ability of CAFs (Figures 3I and 3J). Collectively, these findings indicate that S100A8 contributes to the conversion of CAFs into myCAFs, promoting the formation of a chemoresistant TME.
myCAF activation induced by S100A8 is triggered via CD147-RhoA-ROCK-MLC2-MRTF-A pathway
To better comprehend how S100A8 promotes the conversion of CAFs to myCAFs, we used scRNA-seq data to conduct pathway enrichment analysis for these CAF subtypes. The results revealed PI3K-Akt signaling and smooth muscle contraction pathways were enriched in myCAFs (Figure 4A). These two pathways regulate the actomyosin contraction of myofibroblasts and are responsible for myCAF activation.30,39 Additionally, correlation analysis demonstrated a positive correlation between the expression of S100A8 and the signature score of the smooth muscle contraction pathway (Figure 4B). These findings further support the role of S100A8 in inducing the polarization of CAFs toward myCAFs.
Figure 4.

myCAF activation induced by S100A8 is triggered via CD147-RhoA-ROCK-MLC2-MRTF-A pathway
(A) KEGG pathway enrichment analysis of distinct CAF subtypes.
(B) Spearman correlation between the S100A8 RNA level and the pathway signature score. The gray area represents 95% CI (n = 60).
(C) Western blot analysis of RhoA-associated protein markers in CAFs treated with indicated concentration of rS100A8 proteins.
(D) Western blot analysis of RhoA-associated and myCAF-related protein markers in CAFs transfected with siRNAs targeting RhoA, ROCK1, MLC2, or negative control and treated with rS100A8 proteins or PBS.
(E) Representative immunofluorescence images of MRTF-A nuclear translocation in CAFs treated with rS100A8 proteins or PBS. White arrows indicate MRTF-A nuclear translocation in cells. Scale bar, 100 μm.
(F) Quantification of the percentage of MRTF-A nuclear translocation in (E) (n = 6 biological replicates). Data are presented as mean ± SD. ∗∗∗∗p < 0.0001 of two-tailed Student’s t test.
(G) Western blot analysis of the indicated proteins in CAFs transfected with MRTF-A or control siRNA and treated with indicated concentration of rS100A8 proteins.
(H) Representative immunofluorescence images of CD147 and αSMA in control and CD147-knockout CAFs. Scale bar, 100 μm.
(I) Western blot analysis of the indicated proteins in control and CD147-knockout CAFs treated with indicated concentration of rS100A8 proteins.
(J) Western blot analysis of the indicated proteins in control and CD147-knockout CAFs cocultured with control or S100A8-overexpression KYSE450 cells.
Each assay for western blot had three biological repeats. See also Figures S4 and S5.
Previous studies have indicated that RhoA has the ability to activate myosin light chain 2 (MLC2) through Rho-associated, coiled-coil-containing kinase (ROCK), thereby facilitating the activation of myCAFs and the deposition of ECM.40,41,42,43 Thus, we evaluated the expression levels of the GTP-bound form of RhoA, ROCK1, and phosphorylated MLC2 following treatment with rS100A8 protein. The findings demonstrated a significant induction of the three myCAF activation markers (Figure 4C), consistent with the increased expression level of collagen type 1 (Figure 3H). Notably, the suppression of RhoA, ROCK1, and MLC2 through small interfering RNA (siRNA) resulted in diminished activation of their downstream effectors, as well as reduced expression levels of collagen type 1 and αSMA (Figure 4D). Myocardin-related transcription factor-A (MRTF-A), a coactivator of the serum response factor, plays a role in regulating fibroblast motility, contractility, and ECM remodeling.44 The activity of MRTF-A is regulated by Rho signaling and G-actin, which forms a repressive complex by binding to MRTF-A.45 When activated by altered actin dynamics, MRTF-A is released from G-actin in the cytoplasm and translocated to the nucleus to promote the expression of profibrotic genes.46 Immunofluorescence staining depicted the increased nuclear translocation of MRTF-A in CAFs following treatment with rS100A8 protein (Figures 4E and 4F), and the downregulation of MRTF-A reversed the S100A8-induced upregulation of myCAF markers (Figures 4G and S4A).
It has been reported that S100A8 acts as a ligand that binds to the CD147 receptor, thereby regulating various cell fates.47,48,49 Likewise, ligand-receptor interaction analysis between cancer cells and myCAFs indicated stronger interaction of S100A8 with the CD147 receptor compared to other receptors (Figure S4B). In this study, we generated CD147-knockout CAFs using CRISPR-Cas9-mediated gene targeting, in which CD147 deletion significantly suppressed the expression of αSMA (Figure 4H). Notably, the deletion of CD147 in CAFs markedly eliminated S100A8-induced activation of RhoA signaling and CAF polarization (Figure 4I). Additionally, the inhibition of the CD147 receptor by its specific inhibitor, AC-73,50 markedly suppressed the S100A8-induced upregulation of collagen type 1 and αSMA (Figures S4C and S4D). To further validate the role of endogenous S100A8, S100A8-overexpressing (OE) ESCC cells were established (Figure S4E), and they were cocultured with either CD147-deficient or control CAFs. As anticipated, compared to the respective control, the activation markers of RhoA signaling and myCAFs showed consistent upregulation in CAFs cocultured with S100A8-OE cells; notably, this activation effect was significantly mitigated with the blockade of the CD147 receptor (Figure 4J). Collectively, these data suggest that cancer-cell-derived S100A8 triggers the CD147-RhoA-ROCK-MLC2-MRTF-A signaling pathway in CAFs, leading to their conversion into myCAFs.
The expression of S100A8 is regulated by the esophageal-specific transcription factor TFAP2A
We next interrogated how S100A8 expression is regulated in ESCC. Since the expression pattern of S100A8 is specific to the esophageal epithelium,51 we therefore searched for transcription factors (TFs) that might govern S100A8 expression within esophageal-specific regulatory networks. Utilizing the PROMO database52 and our previously identified esophageal-specific TFs,53 we identified three candidate TFs potentially involved in regulating S100A8 expression (Figure S5A). Correlation analysis using scRNA-seq data revealed a positive correlation exclusively with TFAP2A expression and S100A8 (Figure S5B). Silencing TFAP2A with siRNA notably suppressed S100A8 expression in ESCC cells (Figure S5C). Further in silico analysis indicated the potential presence of a TFAP2A cis element located between −64 and −56 bp upstream of the S100A8 transcriptional start site (Figure S5D), which was validated by chromatin immunoprecipitation (ChIP) assays (Figure S5E). These data reveal a TFAP2A-dependent positive regulation of S100A8 expression in ESCC.
We further explored the impact of TFAP2A on CAF polarization. For this purpose, we knocked down TFAP2A in ESCC cells and collected the CM. Immunoblot assays revealed reduced expression levels of collagen type 1 and αSMA in CAFs exposed to the CM from TFAP2A-knockdown ESCC cells compared to those from control ESCC cells. Furthermore, the addition of rS100A8 protein to the CM restored the expression of collagen type 1 and αSMA (Figure S5F). These results indicate that the specific expression of S100A8 in the esophageal epithelium is regulated by the esophageal-specific transcription factor TFAP2A.
S100A8-induced myCAFs endow ESCC cells to acquire chemoresistance by activating anti-apoptotic pathways
We aimed to investigate how activated myCAFs induce chemoresistance in ESCC cells. The GSEA of RNA-seq data from PDX tumors revealed an enrichment of the apoptosis pathway in the R group (Figure S6A). Furthermore, the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis demonstrated significant activation of pathways related to ECM-mediated integrin signaling and downstream effectors, as well as anti-apoptosis pathways in the NR group (Figures S6B and S6C). These data indicate that activated myCAFs and deposited ECM may enhance the anti-apoptotic capability by activating integrin-related pathways in cancer cells, thereby promoting cell survival post chemotherapy.
To further our conclusions, we developed a co-culture model in which ESCC cells and CAFs were cocultured for 24 h and then treated with chemotherapeutic or vehicle reagents for another 24 h. Subsequently, the ESCC cells were collected for subsequent assays (Figure 5A). When compared to ESCC cells expressing nonspecific shRNA and cultured alone, those cocultured with CAFs showed a significant increase in the phosphorylation levels of FAK, SRC, and ERK, regardless of chemotherapeutic treatment. Specifically, S100A8 ablation suppressed the phosphorylation of FAK, SRC, and ERK only under co-culture conditions. Additionally, ESCC cells cocultured with CAFs exhibited reduced expression of cell apoptosis markers, including cleaved-Caspase 3 and cleaved-PARP, compared to ESCC cells cultured alone. However, S100A8 knockdown reversed CAFs’ protective effect on ESCC cells, as shown by increased apoptosis levels under co-culture conditions (Figure 5A). Furthermore, we conducted in vitro co-culture assays using S100A8-OE ESCC cells and CD147-knockout CAFs (Figure S6D). Immunoblot assays revealed that ESCC cells cocultured with CAFs expressed higher levels of phosphorylated FAK, SRC, and ERK and lower levels of cleaved-Caspase 3 and cleaved-PARP than ESCC cells cultured alone, and S100A8 overexpression augmented these effects. Deletion of CD147 in CAFs significantly annulled the anti-apoptosis effects induced by S100A8 overexpression under co-culture conditions (Figure 5B). The S100A8-OE ESCC cells cocultured with CAFs demonstrated significantly increased proliferation and survival following chemotherapeutic treatment, while CD147 deletion in CAFs reversed this effect (Figures 5C and S6E).
Figure 5.

S100A8-induced myCAFs endow ESCC cells to acquire chemoresistance by activating anti-apoptotic pathways
(A) The left panel shows the schematic of co-culture system. The right panel shows western blot analysis of the indicated proteins in control and S100A8-knockdown KYSE510 and KYSE180 cells cocultured with or without CAFs and treated with chemotherapeutics or vehicle reagents.
(B) Western blot analysis of the indicated proteins in control and S100A8-overexpression KYSE450 cells cocultured with control or CD147-knockout CAFs and treated with chemotherapeutics or vehicle reagents.
(C) The quantification of cell viability of control and S100A8-overexpression KYSE450 cells cocultured with control or CD147-knockout CAFs and treated with chemotherapeutics or vehicle reagents (n = 3 biological replicates). Data are presented as mean ± SD. p values are determined using two-tailed Student’s t test.
(D) Tumor growth curves and excised tumor images of xenografts derived from co-injection of control and S100A8-overexpression KYSE450 cells with CAFs and treated with chemotherapeutics or vehicle reagents (n = 5 per group). Data are presented as mean ± SD. p values are determined using two-tailed Student’s t test.
(E) Representative images of H&E, S100A8, MTS, αSMA, and cleaved-Caspase 3 staining of tumor tissues in (D). Scale bar, 100 μm.
(F) Quantification of (E). Data are presented as mean ± SEM. p values are determined using two-tailed Student’s t test.
For all panels, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figure S5. Each assay for western blot had three biological repeats. See also Figure S6.
To validate the in vitro findings in an in vivo setting, mice were subcutaneously co-inoculated with S100A8-OE or control (vector) ESCC cells in conjunction with CAFs. The mice injected with S100A8-OE cells along with CAFs revealed greater tumor growth following chemotherapy in comparison to those injected with control cells and CAFs (Figure 5D). IHC analysis of xenografts demonstrated that, consistent with S100A8 staining, the activation of myCAFs and the deposition of collagen were markedly higher in the OE group than in the control group (Figures 5E and 5F). The apoptosis level in OE group was significantly lower than that in control group (Figure 5F), suggesting that overexpression of S100A8 can enhance the tolerance of cancer cells to chemotherapy in vivo. Taken together, these results imply that the interaction between cancer-cell-derived S100A8 and CAFs induced polarization of CAFs toward myCAFs, subsequently activating the FAK/SRC/ERK-mediated anti-apoptotic signaling pathways in cancer cells, ultimately leading to chemoresistance.
Blockade of S100A8-CD147 pathway improves chemotherapy efficiency in vivo
In light of the involvement of the S100A8-CD147 signaling pathway in myCAF activation and chemoresistance, we subsequently investigated the potential improvement in therapeutic responses by targeting S100A8-mediated polarization of CAFs in vivo. We isolated ESCC primary cells (PCs) from a treatment-naive patients and conducted in vivo chemosensitivity assays by co-injecting PCs with CD147-knockout CAFs and control CAFs (Figure S7A). As shown in Figure 6A and S7B, the ablation of CD147 receptor in CAFs significantly inhibited the growth of xenografts, and chemotherapeutics treatment further augmented the inhibitory effect. Additionally, the deletion of CD147 notably inhibited the polarization of CAFs and collagen deposition in vivo and no longer shielded cancer cells from chemotherapy-induced apoptosis (Figures 6B and 6C).
Figure 6.

Disruption of S100A8-CD147 pathway improves chemotherapy efficiency in vivo
(A) Tumor growth curves of xenografts derived from co-transplantation of control and CD147-knockout CAFs with PCs and treated with chemotherapeutics or vehicle reagents (n = 4 per group). Data are presented as mean ± SD. p values are determined using two-tailed Student’s t test.
(B) Representative images of H&E, MTS, αSMA, cleaved-Caspase 3, and S100A8 staining of tumor tissues in (A). Scale bar, 100 μm.
(C) Quantification of (B). Data are presented as mean ± SEM. p values are determined using two-tailed Student’s t test.
(D) Tumor growth curves of xenografts of PDX-19 treated with AC-73 and control solvent and with chemotherapeutics or vehicle reagents (n = 4 per group). Data are presented as mean ± SD. p values are determined using two-tailed Student’s t test.
(E) Representative images of H&E, MTS, αSMA, and cleaved-Caspase 3 staining of tumor tissues in (D). Scale bar, 100 μm.
(F) Quantification of (E). Data are presented as mean ± SEM. p values are determined using two-tailed Student’s t test. For all panels, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, and n.s., not significant. Each assay for western blot had three biological repeats. See also Figure S7.
To extrapolate these findings to a preclinical application, we then administered AC-73 or control reagent in conjunction with chemotherapy to a non-responsive PDX case. Notably, the combination of AC-73 and chemotherapy significantly inhibited tumor growth compared to chemotherapy alone (Figures 6D and S7C). Also, AC-73 treatment had negligible impact on the body weight of PDX mice (Figure S7D). The blockade of the CD147 receptor hindered the activation of myCAFs and diminished their protective effect on cancer cells (Figures 6E and 6F). These findings indicate that S100A8-mediated signaling pathways activate myCAFs in the TME, ultimately contributing to chemoresistance. Moreover, targeting the S100A8-CD147 pathway could reprogram the pro-tumor CAFs into a quiescent state and reduce the viability of ESCC cells, thereby improving the efficiency of chemotherapy. In light of these findings, we posit that S100A8 may be a promising target for overcoming chemoresistance in ESCC.
S100A8 serves as a prognostic biomarker for predicting chemotherapy responsiveness
To determine the clinical implications of S100A8, we collected peripheral blood samples from 152 patients with ESCC who had undergone neoadjuvant chemotherapy (ESCC cohort 1; detailed clinical information in Table S2) and measured the levels of S100A8 in their plasma (Figure 7A). Based on the response evaluation criteria in solid tumors (RECIST) guidelines,54 100 patients’ treatment responses met the criteria for complete response and partial response, thereby classifying them as Rs. Meanwhile, the treatment responses of 52 patients were categorized as stable disease and progressive disease, thus identifying them as NRs (Figure 7B). Strikingly, the circulating S100A8 levels in the NR group were significantly higher than those in the R group (Figure 7C). In univariate analysis of all patients, elevated plasma S100A8/S100A9 levels were significantly associated with higher odds ratio (OR) for chemoresistance (Figure 7D; OR, 2.23 [95% confidence interval (CI), 1.12–4.43; p = 0.023]). Other clinical variables that demonstrated a higher OR in univariate analysis included moderately differentiated histologic grade (G2 grade) (Figure 7D; OR, 2.33 [95% CI, 1.04–5.24; p = 0.04]). In the multivariate analysis for OR in all patients, which involved adjustments for age, gender, histologic grade, tumor-node-metastasis (TNM) stage, and treatment cycles, only elevated S100A8/S100A9 levels remained associated with higher OR (Figure 7D; OR, 2.21 [95% CI, 1.09–4.48; p = 0.029]). The data suggest that the levels of circulating S100A8 serve as an independent risk factor in evaluating the chemotherapy responsiveness of patients with ESCC. However, to comprehensively establish the clinical implications of S100A8, a larger prospective clinical study is required.
Figure 7.

S100A8 serves as a prognostic biomarker for predicting chemotherapy responsiveness
(A) Schematic overview of the experimental design of liquid biopsy.
(B) Representative images showing computed tomography of patients’ esophageal tumors of the R and NR groups before and after neoadjuvant chemotherapy. Scale bar, 5 cm.
(C) Histogram exhibiting plasma S100A8/S100A9 concentration of patients with ESCC before receiving neoadjuvant chemotherapy in the R (n = 100) and NR (n = 52) groups. Data are presented as mean ± SEM. p value is determined using two-tailed Student’s t test.
(D) Forest plot exhibiting odds ratio (OR) with 95% CI and p value calculated by univariate and multivariate logistic regression analysis for age, gender, histologic grade, pathologic TNM stage, treatment cycles, and S100A8/A9 level.
(E) Histogram exhibiting serum S100A8/S100A9 concentration of vehicle-treated PDX mice between the R (n = 8) and NR (n = 12) groups. Data are presented as mean ± SEM. p value is determined using two-tailed Student’s t test.
(F) Spearman correlation between the TGI and S100A8/A9 concentration. The gray area represents 95% CI (n = 20).
(G) Representative images of S100A8 IHC staining in clinical pre-treatment ESCC biopsies of the stable disease (n = 10) and partial response (n = 23) groups. Scale bar, 100 μm.
(H) Quantification of (G). Data are presented as mean ± SEM. p value is determined using two-tailed Student’s t test.
(I) Kaplan-Meier plot comparing the overall survival (OS) of patients with ESCC treated with chemotherapy with low or high S100A8 RNA level. Hazard ratio (HR) and 95% CI are calculated by Cox proportional hazards model with age, gender, and tumor stage as covariates.
(J) Dot plot showing normalized score of HRs of high or low S100A8 and S100A9 RNA level in patients with different cancers receiving chemotherapy from The Cancer Genome Atlas (TCGA). HR and p values are determined by Cox proportional hazards model with age, gender, and tumor stage as covariates. For all panels, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. See also Figure S8, Tables S2 and S3.
Furthermore, we measured S100A8/S100A9 levels in the plasma of vehicle-treated PDX mice. The enzyme-linked immunosorbent assay (ELISA) revealed higher plasma S100A8/S100A9 levels in the NR group compared to the R group (Figure 7E), and the circulating levels of S100A8/S100A9 exhibited a negative correlation with the responsiveness to chemotherapy (Figure 7F). We also examined S100A8 in pre-treatment biopsy samples from 33 cases of ESCC (cohort 2). These 33 cases were reported in our previous studies,25,28 with 10 classified as stable disease and 23 as partial response. IHC analysis revealed a higher level of S100A8 staining in the stable disease group compared to the partial response group (Figures 7G and 7H).
Finally, we analyzed the correlation between the expression of S100A8 and S100A9 in epithelial cells and the survival time of patients receiving chemotherapy based on our previously published scRNA-seq data.20 In this cohort, 21 individuals underwent postoperative chemotherapy based on 5-FU (ESCC cohort 3; detailed clinical information in Table S3). The survival analysis revealed that patients with high expression of S100A8 and S100A9 experienced a shorter survival time (S100A8, log rank p = 0.04; S100A9, log rank p = 0.02), with adjusted hazard ratios (HRs) of 6.58 for S100A8 (Figure 7I) and 8.07 for S100A9 (Figure S8A). However, due to the relatively small sample size, the accuracy of the HR is relatively insufficient (S100A8, 95% CI, 0.701–61.6; S100A9, 95% CI, 0.817–79.8). Collectively, these data support considering S100A8 as a biomarker for predicting the therapeutic response of ESCC to chemotherapy.
We endeavored to extend our findings to encompass other types of cancer. To this end, we investigated the survival association of S100A8 and S100A9 RNA levels in The Cancer Genome Atlas (TCGA) patients who underwent chemotherapy and had survival data (16 cancer types, excluding mesenchymal and hematologic tumors). The analysis revealed that the expression of S100A8 and S100A9 was significantly associated with lower overall survival (OS) in multiple cancers, including cervical cancer (CESC), liver cancer (LIHC), bladder cancer (BLCA), pancreatic cancer (PAAD), head and neck cancer (HNSC), glioblastoma (GBM), and kidney clear cell carcinoma (KIRC) (Figures 7J and S8B–S8I). Altogether, these findings suggest that elevated S100A8 expression in tumors is strongly associated with diminished chemotherapy effectiveness and unfavorable survival of patients, highlighting its potential as a prognostic biomarker for predicting chemotherapy responsiveness.
Discussion
Cancer progression and therapy resistance are critical outcomes of the symbiotic interactions between cancer cells and the TME. Comprehending the reciprocal interactions between cancer cells and TME components is essential for devising effective cancer treatment strategies. In this study, we comprehensively investigated the transcriptome characteristics associated with chemoresistance in ESCC from both cancer cell and TME perspectives using well-established PDX models, unmasking a mechanism involving bidirectional malignant cell-fibroblast crosstalk, which drives chemoresistance. We identified an enrichment of genetic signatures related to myCAF activation and ECM remodeling in the non-responsive PDXs. The myCAF-induced perturbation of collagen and other ECM within the TME has been shown to shield cancer cells and enable their survival following anti-cancer treatment.25,43,55,56,57 Importantly, our findings uncover a previously unrecognized mechanism in which cancer-cell-derived S100A8 reprograms CAFs to modulate chemosensitivity in ESCC, highlighting the potential of S100A8 as a biomarker for predicting chemotherapy responsiveness, as well as a promising target for overcoming chemoresistance.
Mechanistically, we have identified an important function of S100A8, which contributes to chemoresistance in a CAF-dependent manner. Recent studies have shed light on the important role of S100A8 in promoting cancer progression and determining anti-cancer effects.37 Functioning as a critical immunomodulatory molecule, S100A8 and S100A8/S100A9 heterodimers drive the accumulation and activation of myeloid-derived suppressor cells (MDSCs),58,59,60 promote macrophage M2 polarization,61 mediate the formation of neutrophil extracellular traps,62 and impede the maturation of dendritic cells.63 These actions collectively establish an immunosuppressive TME that fosters tumor survival. Additionally, S100A8 and its heterodimers are extensively implicated in numerous oncogenic signaling pathways within cancer cells. Through interactions with TLR4, RAGE, CD147, and other membrane receptors of tumor cells, S100A8 activates the nuclear factor κB (NF-κB), phosphatidylinositol 3-kinase (PI3K)-protein kinase B (Akt), and mitogen-activated protein kinase (MAPK)-extracellular signal-regulated kinase (ERK) pathways, promoting cell proliferation, migration, epithelial-mesenchymal transition, and metabolic reprogramming, thus driving cancer progression and conferring resistance to treatment.64,65,66
In contrast to previous findings, our in vitro experiments did not demonstrate significant impacts on cancer cell proliferation and therapeutic phenotypes with altered S100A8 expression. This led to an exploration of how S100A8 influences chemotherapy responsiveness from the TME perspective. Given the properties of PDX mice, our subsequent studies focused on fibroblasts. Although the connection between S100A8 and CAFs polarization has not been thoroughly investigated, prior research has highlighted the association between S100A8 and fibrosis in non-tumor conditions. Araki et al. revealed that S100A8 has the capacity to induce pulmonary fibrosis,67 an interstitial lung disease characterized by fibroblast activation and ECM remodeling. Similarly, Hou et al. demonstrated that hepatic macrophage-derived S100A8 activated hepatic stellate cells, leading to hepatic fibrosis.68 These studies provide clues for S100A8 to regulate CAF polarization.
Subsequent ligand-receptor interaction analysis and functional experiments revealed that S100A8 can trigger RhoA-related signaling pathways through its interaction with CD147 receptors on CAFs, resulting in the polarization of CAFs into myCAFs. The transmembrane glycoprotein CD147 is expressed in various tumors and is known as a critical tumor promoter by regulating tumor proliferation, invasion, angiogenesis, metabolism, and immune evasion properties.69,70,71,72,73 While there is currently no evidence linking CD147 to myCAF activation, Wu et al. demonstrated that CD147 contributes to progressive pulmonary fibrosis induced by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and the ablation of CD147 in fibroblasts reverses disease progression.74 This study, in conjunction with the research of Araki et al.67 and Hou et al.,68 and the findings of our ligand-receptor interaction analysis, prompted us to further investigate the potential role of S100A8 in polarizing CAFs by binding to the CD147 receptor.
Our study holds important clinical implications. From a therapeutic perspective, we have shown that genetic inhibition of S100A8 or CD147, or pharmacological blockade of the CD147 receptor, suppresses myCAF activation, diminishes collagen deposition in the TME, and overcomes chemoresistance. Prior clinical trials have demonstrated that using metuximab to block CD147 significantly enhances the survival of patients with CD147-expressing hepatocellular carcinoma.75,76,77,78 Several studies have established that cancer cells expressing high levels of CD147 exhibit significant chemoresistance, and targeting CD147 can enhance the efficacy of chemotherapy.79,80,81 This evidence, along with our in vivo results, suggests that targeting the S100A8-CD147 pathway could be an effective approach to overcome chemoresistance.
Prognostically, we have demonstrated that the levels of S100A8 in peripheral blood can serve as a non-invasive marker, known as alarmin, to evaluate the chemosensitivity of patients with ESCC. Although chemotherapy remains a frontline treatment for advanced ESCC, its position in cancer treatment is weakening. The survival benefits from chemotherapy are limited, not only due to the emergence of resistance but also because chemotherapy toxicity restricts the continuous application of drugs.3,4,38,82,83 In our study, we propose that S100A8 can be used as a biomarker for liquid biopsy. Therefore, in the clinical management of ESCC, we can stratify patients according to the levels of S100A8 in peripheral blood and decide whether patients need further chemotherapy. If possible, patients with higher circulating S100A8 levels may reduce the use of chemotherapy and choose other more appropriate treatment options or receive S100A8 inhibitors in combination with chemotherapy, which may reduce the side effects caused by chemotherapy to some extent.
In conclusion, our findings suggest that the acquisition of chemoresistance in ESCC is related to the initiation of cell plasticity mechanisms triggered by crosstalk between cancer cells and the TME. The reprogramming of stromal cells by cancer cells might foster the development of distinct cellular and functional phenotypes (that is, myCAFs), which are implicated in therapy resistance. Understanding this interaction mechanism between cancer cells and stromal cells might provide insights for novel strategies and advancements in personalized cancer treatment.
Limitations of the study
The establishment of PDX models in NSG mice, lacking functional immune cells including neutrophils and macrophages, led us to focus solely on cancer cells and stromal fibroblasts in the TME. However, as S100A8 also functions as a crucial immunomodulatory molecule targeting various immune cells, including macrophages, neutrophils, and MDSCs in the TME, potential effects of S100A8 on the immune microenvironment of ESCC require further investigation. To fully address the clinical significance of S100A8, the development of S100A8-specific genetically engineered mouse models is essential.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Polyclonal anti-KRT6A | Proteintech | Cat# 10590-1-AP; RRID: AB_2134306 |
| Rabbit Polyclonal anti-S100A8 | Proteintech | Cat# 15792-1-AP; RRID: AB_10666315 |
| Rabbit Monoclonal anti-αSMA | Abcam | Cat# ab124964; RRID: AB_11129103 |
| Rabbit Monoclonal anti-Collagen 1A1 | Cell Signaling Technology | Cat# 72026; RRID: AB_2904565 |
| Rabbit Monoclonal anti-FAK | Abcam | Cat# ab40794; RRID: AB_732300 |
| Rabbit Monoclonal anti-phospho-FAK (Tyr 397) | Cell Signaling Technology | Cat# 8556; RRID: AB_10891442 |
| Rabbit Monoclonal anti-Src | Cell Signaling Technology | Cat# 2123; RRID: AB_2106047 |
| Rabbit Monoclonal anti-phospho-Src (Tyr 416) | Cell Signaling Technology | Cat# 6943; RRID: AB_10013641 |
| Rabbit Polyclonal anti-CD31 | Servicebio | Cat# GB11063-2-100; RRID: AB_2922436 |
| Rabbit Polyclonal anti-CXCL1 | Proteintech | Cat# 12335-1-AP; RRID: AB_2087568 |
| Mouse Monoclonal anti-RhoA-GTP | NewEast Biosciences | Cat# 26904; RRID: AB_1961799 |
| Rabbit Monoclonal anti-RhoA | Cell Signaling Technology | Cat# 2117; RRID: AB_10693922 |
| Rabbit Monoclonal anti-ROCK1 | Cell Signaling Technology | Cat# 4035; RRID: AB_2238679 |
| Rabbit Monoclonal anti-phospho-MLC2 (Ser 19) | Cell Signaling Technology | Cat# 3671; RRID: AB_330248 |
| Rabbit Monoclonal anti-MLC2 | Cell Signaling Technology | Cat# 8505; RRID: AB_2728760 |
| Rabbit Polyclonal anti-MRTF-A | Proteintech | Cat# 21166-1-AP; RRID: AB_2878822 |
| Rabbit Polyclonal anti-CD147 | Proteintech | Cat# 11989-1-AP; RRID: AB_2290597 |
| Rabbit Monoclonal anti-phospho-ERK1/2 (Thr 202/Tyr 204) | Cell Signaling Technology | Cat# 4370; RRID: AB_2315112 |
| Rabbit Monoclonal anti-ERK1/2 | Cell Signaling Technology | Cat# 4695; RRID: AB_390779 |
| Rabbit Monoclonal anti-cleaved-PARP (Asp 214) | Cell Signaling Technology | Cat# 5625; RRID: AB_10699459 |
| Rabbit Monoclonal anti-cleaved-Caspase3 (Asp 175) | Cell Signaling Technology | Cat# 9661; RRID: AB_2341188 |
| Rabbit Polyclonal anti-TFAP2A | Proteintech | Cat# 13019-3-AP; RRID: AB_2199414 |
| Mouse Monoclonal anti-Vinculin | Proteintech | Cat# 66305-1-Ig; RRID: AB_2810300 |
| Mouse Monoclonal anti-GAPDH | Proteintech | Cat# 60004-1-Ig; RRID: AB_2107436 |
| Normal Rabbit IgG | Cell Signaling Technology | Cat# 2729; RRID: AB_1031062 |
| Rabbit monoclonal anti-Histone H3 | Cell Signaling Technology | Cat# 4620; RRID: AB_1904005 |
| Goat anti-rabbit IgG, HRP-linked | Cell Signaling Technology | Cat# 7074; RRID: AB_2099233 |
| Horse anti-mouse IgG, HRP-linked | Cell Signaling Technology | Cat# 7076; RRID: AB_330924 |
| Bacterial and virus strains | ||
| S100A8 shRNA lentivirus | Genechem | N/A |
| S100A8 overexpression lentivirus | Genechem | N/A |
| CD147 Cas9-sgRNA lentivirus | Genechem | N/A |
| Biological samples | ||
| ESCC tumor specimens | Linzhou Cancer Hospital | See Table S1 for details |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Corning | Cat# 10-013-CV |
| Advanced DMEM/F12 | Gibco | Cat# 12634028 |
| Fetal Bovine Serum | Cell Technologies | Cat# 30070 |
| 5-Fluorouracil | MCE | Cat# HY-90006 |
| Cisplatin | MCE | Cat# HY-17394 |
| Collagenase I | Gibco | Cat# 17100017 |
| Collagenase IV | Gibco | Cat# 17104019 |
| Hyaluronidase | Sigma-Aldrich | Cat# H1115000 |
| B-27 | Gibco | Cat# 17504044 |
| HEPES | Gibco | Cat# 15630080 |
| GlutaMAX™ | Gibco | Cat# 35050061 |
| EGF | Gibco | Cat# PMG8043 |
| Y-27632 | Selleck Chemicals | Cat# S1049 |
| A83-01 | Tocris Bioscience | Cat# 2939 |
| Puromycin | Gibco | Cat# A1113803 |
| Lipofectamine 2000 | Invitrogen | Cat# 11668027 |
| AC-73 | MCE | Cat# HY-122214 |
| Human recombinant S100A8 protein | Novoprotein | Cat# C794 |
| Triton X-100 | Solarbio | Cat# T8200 |
| Critical commercial assays | ||
| RNA-Quick Purification Kit | ES Science | Cat# RN001 |
| TB Green® Premix Ex Taq™II (Tli RNaseH Plus) | TaKaRa | Cat# RR820A |
| Masson’s Trichrome Stain Kit | Solarbio | Cat# G1340 |
| Meilunbio® FGSuper Sensitive ECL Luminescence Reagent | Meilunbio | Cat# MA0186-1 |
| Pierce™ Rapid Gold BCA | Thermo Fisher Scientific | Cat# A53226 |
| Human S100A8/S100A9 ELISA Kit | Proteintech | Cat# KE00177 |
| Opal 5-Color Manual IHC Kit | PANOVUE | Cat# 10144100100 |
| SimpleChIP® Plus Sonication Chromatin IP Kit | Cell Signaling Technology | Cat# 56383 |
| Deposited data | ||
| RNA-seq data of PDX mice | This paper | GSA: CRA013775 |
| RNA-seq data of PDX donors | Yang et al.25 | GSA: HRA004329 |
| ESCC scRNA-seq data | Zhang et al.20 | GEO: GSE160269 |
| Experimental models: Cell lines | ||
| KYSE450 | Dr. Y. Shimada | JCRB1430 |
| KYSE510 | Dr. Y. Shimada | JCRB1436 |
| KYSE180 | Dr. Y. Shimada | JCRB1083 |
| Human esophageal cancer associated fibroblasts (ECAFs) | This paper | N/A |
| PDX-derived primary tumor cells (PDCs) | This paper | N/A |
| Experimental models: Organisms/strains | ||
| NSG mouse | BEIJING IDMO Co., Ltd | N/A |
| Oligonucleotides | ||
| shRNA sequences, see Table S2 | Genechem | N/A |
| sgRNA sequences, see Table S2 | Genechem | N/A |
| siRNA, see Table S2 | JTSBIO Co.,Ltd (Wuhan, China) | N/A |
| qRT-PCR primers, see Table S3 | Tsingke Biotechnology | N/A |
| Software and algorithms | ||
| R 4.2.2 | R Core Team | https://www.R-project.org/ |
| Trim Galore (version 0.6.6) | Felix Krueger | https://github.com/FelixKrueger/TrimGalore |
| Disambiguate (version 1.0) | Ahdesmäki et al.35 | https://github.com/AstraZeneca-NGS/disambiguate |
| Salmon (version 1.2.0) | Patro et al.84 | https://combine-lab.github.io/salmon/ |
| DESeq2 (version 1.38.3) | Love et al.85 | https://bioconductor.org/packages/DESeq2/ |
| tximport (version 1.26.1) | Soneson et al.86 | https://bioconductor.org/packages/tximport/ |
| clusterProfiler (version 4.6.2) | Wu et al.87 | https://github.com/YuLab-SMU/clusterProfiler |
| Seurat (version 4.3.0) | Hao et al.88 | https://github.com/satijalab/seurat |
| Harmony (version 0.1.1) | Korsunsky et al.89 | https://github.com/immunogenomics/harmony |
| CellChat (version 1.6.1) | Jin et al.90 | https://github.com/sqjin/CellChat |
| survminer (version 0.4.9) | Kassambara et al. | https://github.com/kassambara/survminer |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Prism 8.0 | GraphPad | http://www.graphpad.com/ |
Resource availability
Lead contact
Further information and requests for reagents may be directed to and will be fulfilled by the lead contact, Chen Wu (chenwu@cicams.ac.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The raw RNA sequencing data of 20 PDX tumor tissues generated in this study have been deposited in GSA (Genome Sequence Archive: CRA013775). Other resources of published datasets we used are listed in the key resources table and as follows:
The raw RNA sequencing data of PDX donors25 can be accessed under HRA004329 in GSA-Human. The ESCC scRNA-seq data20 is available in GEO with accession number GSE160269. This paper does not report original code. Any additional information required to re-analyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Human tumor and blood samples
The ESCC tumor specimens used to establish PDXs were obtained from patients who underwent surgery at Linzhou Cancer Hospital (Henan Province, China) between 2015 and 2017. All enrolled patients had not received any anti-cancer treatment before the surgery, and their ESCC diagnoses were confirmed through histopathological examination. The comprehensive clinical information of each PDX donor is provided in Table S1.
We also recruited 152 patients with ESCC who underwent neoadjuvant chemotherapy using 5-fluorouracil (5-FU)/cisplatin (CDDP) or paclitaxel/CDDP at Chinese Academy of Medical Sciences Cancer Hospital (Beijing, China). Peripheral blood samples were collected prior to the initiation of chemotherapy. Tumor response to chemotherapy was assessed using computed tomography following the RECIST guidelines (version 1.1).54 We classified complete response and partial response as responders (R), while stable disease and progressive disease were identified as non-responders (NR). Detailed clinical information for each patient is available in Table S2.
The study protocol was approved by the ethics committee of Chinese Academy of Medical Sciences Cancer Hospital (NCC2022C-141) with written informed consent from all individuals.
Animals and xenografts
All animal experiments were conducted in strict accordance with the approved protocols and guidelines from the Institutional Animal Care and Use Committee of the Chinese Academy of Medical Science and adhered to established animal care standards. All protocols were approved by the Animal Care Committee of Chinese Academy of Medical Sciences under the reference code NCC2022A061.
PDXs were established using NSG (NOD/SCID/IL-2Rγnull) male mice aged 4 to 6 weeks. Tumor samples obtained from donor patients (F0 tumor) were subcutaneously implanted in NSG mice to generate tumor expansion (F1 tumor). Tumor volume was calculated using the formula (length × width × width)/2. Upon reaching a size of approximately 800 mm3, the F1 tumors were excised and dissected for the next-generation passage (F2 tumor) or subjected to cryopreservation. The mice bearing F2 and F3 tumors were randomly allocated into experimental groups for each study.
Xenografts of ESCC tumor cells were established via subcutaneous inoculation. For co-injection assays, KYSE450 cells (4×106) were mixed with CAFs (1×106) in 100 μL PBS, while PCs (ESCC primary cells) cells (2×106) were mixed with CAFs (5×105) in 100 μL PBS before subcutaneous injection into NSG mice. For single-injection experiments, KYSE180 cells (4×106) were directly injected subcutaneously into NSG mice.
Cell culture and treatment
The human ESCC cell lines KYSE180, KYSE450, and KYSE510, were generously provided by Dr. Y. Shimada from Hyogo College of Medicine, Japan. These cell lines were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin.
Human ESCC-associated fibroblasts (CAFs) were isolated from primary ESCC tissue samples. Following digestion of fresh ESCC tissue with 2 mg/mL collagenase I, 2 mg/mL collagenase IV, and 1 mg/mL hyaluronidase in DMEM medium with 10% FBS at 37°C for 40 min, the resulting cell suspension underwent sieving and centrifugation for 5 min. Subsequently, the cell pellet obtained was resuspended and cultured in advanced DMEM/F12 medium. After 60 min, non-adherent cells were removed by replacing the cell culture medium with a fresh one. CAFs were obtained after 3 passages for analysis and subsequently cultured in DMEM/F12 medium with 20% FBS and 1% penicillin-streptomycin.
Patient-derived ESCC primary cells (PCs) were isolated from human ESCC sample. Briefly, fresh ESCC tumor tissue was digested following the same steps as CAFs isolation. Then, cell suspension was filtered through a 70-μm cell strainer, briefly centrifuged for 5 min and the resultant cell pellet was resuspended in advanced DMEM/F12 medium supplemented with B-27, HEPES, GlutaMAX, epithelial growth factor (EGF), ROCK inhibitor Y-27632, and TGFβ inhibitor A83-01.
ESCC primary cells (PCs) were isolated from fresh tumor tissue through a process similar to CAF isolation. Briefly, the cell suspension underwent filtration using a 70-μm cell strainer, a brief 5-min centrifugation, and the resulting cell pellet was resuspended in advanced DMEM/F12 medium. The resuspension was supplemented with B-27, HEPES, GlutaMAX, epithelial growth factor (EGF), ROCK inhibitor Y-27632, and TGFβ inhibitor A83-01.
All cells were maintained at 37°C in a humidified incubator with 5% CO2, routinely authenticated by short tandem repeat (STR) analysis, and regularly treated with a mycoplasma-removing agent.
For experiments involving stimulation with recombinant human S100A8 protein (rS100A8), a specific concentration of the rS100A8 was added to serum-free DMEM medium following the attainment of confluency by the CAFs; the cells were then incubated for the specified duration. In the CD147 receptor inhibitory assay, the CD147 inhibitor AC-73 was introduced to serum-free DMEM medium for 6 h after CAFs reached confluency, followed by the addition of the rS100A8 protein to the medium for an additional 18 h.
Method details
In vivo pharmacological studies
The drug treatments commenced when the xenografts reached approximately 150 mm3. Mice were randomly assigned to different groups (n = 3–5 per group) and received treatment with 5-FU (30 mg/kg, i.p., biw), CDDP (3 mg/kg, i.p., q5d), AC-73 (20 mg/kg, i.p., biw), and vehicle reagents. Tumor volume was measured every 2 or 3 days, and the mice were euthanized when the tumor volume reached 2000 mm3 or at the experimental endpoint.
The chemotherapy responsiveness was assessed by applying two criteria to compare the changes in relative tumor volume (RTV) between the drug-treated and vehicle-treated groups: (1) determining if drug treatment significantly restrained tumor growth (p < 0.05 for the R group, p > 0.05 for the NR group); and (2) calculating the percentage of tumor growth inhibition (TGI [%]) at the end point (TGI >50% for R, TGI <50% for NR). The calculation of TGI is derived using the following formula: × 100. In this formula, RTVvehicle represents the RTV for the vehicle-treated animals at the end point, RTVtreatment denotes the RTV of the drug treatment groups at the end point, and RTVinitial signifies the initial RTV at the start of the treatment.
RNA sequencing data analysis
RNA sequencing of PDX tumor tissues was conducted by Illumina NovaSeq 6000 (Annoroad Gene Company [Beiing, China]). The initial processing of the RNA-seq data involved adaptor trimming and quality read filtering using Trim Galore (v0.6.6). Subsequently, Disambiguate (v1.0)35 was utilized to eliminate mouse-derived reads, employing the mouse (GRCm38, GENCODE vM19) and human (GRCh38, GENCODE v29) reference genomes. The resulting clean sequences were quantified utilizing the pseudo-alignment software Salmon (v1.2.0).84 The transcript-level quantification results from Salmon were imported into DESeq2 (v1.38.3)85 using tximport (v1.26.1)86 package. Differentially expressed genes (DEGs) meeting the criteria of |log2 fold-change| > 1 and FDR <0.05 were further assessed with DESeq2. Furthermore, gene set enrichment analysis (GSEA), gene ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed using the R package clusterProfiler (v4.6.2).87 The signature scores of enriched pathways for each sample were computed based on the mean expression of pathway-associated genes, with the signature genes for each pathway sourced from the KEGG database.
scRNA-seq data analysis
The ESCC scRNA-seq data were reported by our previous study,20 and the epression matrix was obtained from the GEO with accession number GSE160269. Quality control and downstream analysis were conducted using the Seurat package (v4.3.0),88 and potential batch effects across samples were corrected using the Harmony package (v0.1.1).89 Cell types were annotated based on the expression of known markers: EPCAM, SFN, KRT5, and KRT14 for epithelial cell; FN1, DCN, COL1A1, COL1A2, COL3A1, and COL6A1 for fibroblast; VWF, PECAM1, ENG and CDH5 for endothelial cell. The signature scores for individual cells were calculated using the AddModuleScore function of the Seurat package. The signature genes were COL1A1, COL1A2, COL3A1, ACTA2, TAGLN, CTHRC1, POSTN, MMP11, MMP1, SPARC, MFAP2, SERPINH1, THY1, TPM2, and SFRP4 for myCAFs, CXCL1, CXCL3, CXCL8, CXCL12, IL6, IL24, CHI3L1, CCL11, IGFBP2, IGFBP3, PTGDS, IGF1, LUM, ASPN, and CCN1 for iCAFs. Cell-cell interactions were assessed using the CellChat package (v1.6.1)90 based on the expression of receptors in one cell type and ligands in another cell type.
RNA interference
To achieve transient gene expression knockdown, cells were transiently transfected with siRNA using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. For the establishment of stable S100A8-knockdown cell lines, ESCC cells were infected with S100A8-shRNA and the control lentivirus. To generate stable CD147-knockout CAFs, the CRISPR/Cas9 gene editing system was employed to induce CD147 deletion in the cells. The CAFs were transduced with CRISPR/Cas9 virus targeting genomic CD147 and control virus. Stable clones were obtained by selecting cells in culture medium containing 2 μg/mL puromycin for 7 days. The efficiency of siRNA, shRNA, or sgRNA-mediated knockdown and knockout was assessed utilizing Western blot assays. All the target sequences of the siRNAs, shRNAs, and sgRNAs can be found in Table S4.
Establishment of S100A8 ectopic overexpression cells
To establish cell lines with ectopic overexpression of S100A8, the S100A8 (GenBank: NM_001319196) coding sequence was cloned into the GV341 vector (Ubi-MCS-3FLAG-SV40-puromycin). Subsequently, clones were selected in culture medium containing 2 μg/mL puromycin for 7 days. The overexpression of S100A8 was confirmed through Western blot and qRT-PCR assays.
Immunohistochemistry and immunofluorescence staining
Formalin-fixed and paraffin-embedded tumor tissues were sectioned into 4-μm-thick sections. For immunohistochemistry (IHC), the deparaffinized sections underwent antigen retrieval by heating in a pressure cooker for 3 min in Tris-EDTA buffer (pH 9.0) and subsequent blocking with peroxidase blocker for 30 min at room temperature. Following the blockade, the sections were incubated overnight at 4°C with specific primary antibodies for S100A8 (1:200), αSMA (1:1500), CD31 (1:250), and cleaved-caspase3 (1:100). Then, the sections were washed thrice with PBS, followed by incubation with IHC secondary antibody. The staining was visualized using DAB substrate and counterstained with hematoxylin.
We conducted multiplex immunofluorescence (mIF) staining utilizing the Opal 5-Color Manual IHC Kit (PANOVUE). Following the manufacturer’s instructions, we used Opal 520, Opal 570, and Opal 650 for the αSMA (1:500), KRT6A (1:800), and S100A8 (1:200) antibodies, respectively, to generate unique immunofluorescent signals.
For the immunofluorescence staining of fibroblasts cultured in vitro, cells were cultured on tissue culture-treated coverslips until reaching 50% confluency and then treated with conditional medium or rS100A8 protein for 24 h. Subsequently, the cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.1% Triton X-100 for 5 min, and blocked with 3% bovine serum albumin in PBS for 1 h at room temperature. Following this, the cells were incubated overnight at 4°C with specific primary antibodies for αSMA (1:500), CD147 (1:100), or MRTF-A (1:100), and then with Alexa Fluor secondary antibodies. The coverslips were mounted with DAPI-containing mounting medium, and the images were captured using the Cytation5 (BioTek) microscopy.
For quantification, the ImageJ software was utilized for analyzing IHC and mIF images and quantifying the average intensity of specific protein. In brief, for each section, three non-overlapping visual fields were randomly selected and the intensity of protein expression in each field was evaluated. The mean value of the three visual fields was calculated and used for subsequent statistical analyses. All the aforementioned procedures for staining, imaging, and quantification were performed blinded to the sample identity and phenotype.
Western blot analysis
Total protein was extracted from cell lysate, and its concentration was determined using the BCA kit (Thermo Fisher Scientific). Lysates containing 10–20μg of protein underwent separation by SDS-PAGE and were transferred to the PVDF membrane (Millipore) for analysis. Following this, the membranes were blocked with 5% skim milk in TBST for 1 h at room temperature, and then incubated with primary antibodies overnight at 4°C, followed by secondary antibodies for 1 h at room temperature. The signal was visualized using the Chemiluminescent Substrate kit (Thermo Fisher) via the Amersham Imager 600. The primary and secondary antibodies utilized in this study are listed in the key resources table.
Chromatin immunoprecipitation (ChIP) assay and ChIP-qPCR analysis
The ChIP assay was conducted using the SimpleChIP Plus Sonication Chromatin IP Kit (#56383, Cell Signaling Technology). Initially, ESCC cells were crosslinked with formaldehyde (final concentration 1%) for 10 min, followed by a 5-min glycine incubation at room temperature. The cells were then harvested, resuspended using cell lysis and nuclear lysis buffers, and the chromatin was fragmented using the Covaris S220 Focused-ultrasonicator (peak incident power: 140 W, duty factor: 5%, cycles per burst: 200, treatment time: 600 s, temperature 4°C). After fragmentation, the chromatin was subjected to overnight incubation at 4°C with rotation using anti-TFAP2A (1:100), anti-Histone H3 (1:50), or IgG (1 μg). Subsequently, the final DNA was purified and analyzed via qPCR. The ChIP-qPCR primer of S100A8 promoter is listed in Table S5.
qRT-PCR analysis
Total RNA extraction was performed using the RNA-Quick Purification Kit (ES Science) and subsequently reverse-transcribed with PrimeScript RT reagent kit (Takara). Quantification of mRNA expression was conducted utilizing SYBR Premix Ex Taq II kits (Takara). The expression levels of the specified genes were determined using ΔΔCt relative to the expression of GAPDH as the housekeeping gene. The qRT-PCR primers utilized in this process are available in Table S5.
Conditional medium (CM) preparation
A certain number of ESCC cells were seeded in the 6-well plate to attain approximately 70% confluency within 24 h. Subsequently, the cells were twice washed with PBS and further incubated for 24 h in serum-free medium. The cell culture supernatant was then collected, centrifuged at 2000 g at 4°C for 10 min, and either utilized immediately or stored at −80°C for subsequent use.
Indirect co-culture assays
The indirect co-culture assays were conducted using the 6-well and 12-well Transwell inserts with a 0.4 μm pore (Corning). For CAFs polarization assays, a specific number of CAFs were seeded in the lower chamber, and an equivalent number of ESCC cells were seeded in the upper chamber. In the chemosensitivity analysis of ESCC cells, a specific number of ESCC cells were seeded in the lower chamber, and an equivalent number of CAFs were seeded in the upper chamber. Following a 24-h incubation period, chemotherapeutic agents or control reagents were introduced to the co-culture system and continued to culture for 72 h, or ESCC cells or CAFs were harvested for protein expression analysis.
Migration and chemotaxis assays
The migration and chemotaxis capabilities of CAFs were assessed using the 24-well Transwell insert with an 8.0 μm pore (Corning). The CM of ESCC cells served as the chemoattractant in the lower chamber. CAFs were seeded in the upper chamber and incubated for 18–20 h. Subsequently, the cells were fixed with 100% methanol and stained with a 0.5% crystal violet solution. Cell counting was performed on the bottom of the membrane under a microscope, with three randomly selected non-overlapping visual fields per group.
Detection of S100A8/S100A9 by ELISA
The S100A8/S100A9 level in the CM and plasma samples was analyzed using the human S100A8/S100A9 ELISA Kit (Proteintech) according to the manufacturer’s instructions. For the plasma analysis, the samples were diluted in the sample diluent at a ratio of 1:1000.
Survival analysis and logistic regression analysis
The overall survival time analysis was conducted by the Kaplan-Meier method and the log rank test. For the survival analysis of patients with ESCC, the S100A8 and S100A9 mean expression levels of epithelial cells in 21 patients who underwent chemotherapy were calculated using our previously published scRNA data.20 For the survival analysis of pan-cancer, the gene expression and clinical information were downloaded from the Cancer Genome Atlas (TCGA). The best group-dividing cutoffs were determined by the surv_cutpoint function in survminer package (v0.4.9) to obtain the most minimal p values of log rank test. Hazard ratio (HR) and 95% confidence interval (CI) were calculated by Cox proportional hazards model using age, gender, and tumor stage as covariates.
The odds ratios (ORs) with 95% confidence intervals (CIs) and p values for age, gender, histologic grade, pathological TNM stage, treatment cycles, and S100A8/A9 levels were calculated using univariate and multivariate logistic regression analysis performed with the R package autoReg (version 0.3.3).
Quantification and statistical analysis
The sample size, statistical details, and methods are outlined in the figure legends, text, or methods. The data are presented as means and standard error of measurement (SEM) or standard deviation (SD), with the error bars representing SD or SEM for a minimum of three independent experiments. Statistical significance was considered when p values were <0.05. All statistical analyses were carried out using R 4.2.2 and GraphPad Prism 8.
Acknowledgments
This project was funded by the National Natural Science Foundation of China (grant 81988101 to D.L. and to C.W.), the Medical and Health Technology Innovation Project of Chinese Academy of Medical Sciences (2021-I2M-1-013 and 2022-I2M-2-003 to D.L. and to C.W.), and the National Natural Science Foundation of China (grant 82203256 to S.Z.).
Author contributions
C.W. and D.L. conceptualized and supervised this study. X.C. designed most experiments. X.C., G.C., and L.Z. conducted most experiments. X.C. and T.L. conducted data analysis and visualization. X.Y. and S.Z. conducted PDX generation. R.L., Z.O., and W.T. conducted clinical data and sample collection. X.C., G.C., and L.Z. drafted and C.W. and D.L. reviewed and prepared the final manuscript. All authors approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 21, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101576.
Contributor Information
Dongxin Lin, Email: lindx@cicams.ac.cn.
Chen Wu, Email: chenwu@cicams.ac.cn.
Supplemental information
References
- 1.Abnet C.C., Arnold M., Wei W.Q. Epidemiology of Esophageal Squamous Cell Carcinoma. Gastroenterology. 2018;154:360–373. doi: 10.1053/j.gastro.2017.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matz M., Valkov M., Šekerija M., Luttman S., Caldarella A., Coleman M.P., Allemani C. Worldwide trends in esophageal cancer survival, by sub-site, morphology, and sex: an analysis of 696,974 adults diagnosed in 60 countries during 2000-2014 (CONCORD-3) Cancer Commun. (London, England) 2023;43:963–980. doi: 10.1002/cac2.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puhr H.C., Prager G.W., Ilhan-Mutlu A. How we treat esophageal squamous cell carcinoma. ESMO open. 2023;8 doi: 10.1016/j.esmoop.2023.100789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okines A., Sharma B., Cunningham D. Perioperative management of esophageal cancer. Nat. Rev. Clin. Oncol. 2010;7:231–238. doi: 10.1038/nrclinonc.2010.20. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D., Coussens L.M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 7.Quail D.F., Joyce J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pitt J.M., Marabelle A., Eggermont A., Soria J.C., Kroemer G., Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016;27:1482–1492. doi: 10.1093/annonc/mdw168. [DOI] [PubMed] [Google Scholar]
- 9.Sahai E., Astsaturov I., Cukierman E., DeNardo D.G., Egeblad M., Evans R.M., Fearon D., Greten F.R., Hingorani S.R., Hunter T., et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer. 2020;20:174–186. doi: 10.1038/s41568-019-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalluri R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer. 2016;16:582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y., McAndrews K.M., Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021;18:792–804. doi: 10.1038/s41571-021-00546-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi Y., Du L., Lin L., Wang Y. Tumour-associated mesenchymal stem/stromal cells: emerging therapeutic targets. Nat. Rev. Drug Discov. 2017;16:35–52. doi: 10.1038/nrd.2016.193. [DOI] [PubMed] [Google Scholar]
- 13.Sleeboom J.J.F., van Tienderen G.S., Schenke-Layland K., van der Laan L.J.W., Khalil A.A., Verstegen M.M.A. The extracellular matrix as hallmark of cancer and metastasis: From biomechanics to therapeutic targets. Sci. Transl. Med. 2024;16 doi: 10.1126/scitranslmed.adg3840. [DOI] [PubMed] [Google Scholar]
- 14.Chen X., Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019;18:99–115. doi: 10.1038/s41573-018-0004-1. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y., Kim J., Yang S., Wang H., Wu C.J., Sugimoto H., LeBleu V.S., Kalluri R. Type I collagen deletion in αSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell. 2021;39:548–565.e66. doi: 10.1016/j.ccell.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Özdemir B.C., Pentcheva-Hoang T., Carstens J.L., Zheng X., Wu C.C., Simpson T.R., Laklai H., Sugimoto H., Kahlert C., Novitskiy S.V., et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang D., Liu J., Qian H., Zhuang Q. Cancer-associated fibroblasts: from basic science to anticancer therapy. Exp. Mol. Med. 2023;55:1322–1332. doi: 10.1038/s12276-023-01013-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y., Zhu S., Liu T., Zhang S., Lu J., Fan W., Lin L., Xiang T., Yang J., Zhao X., et al. Epithelial cells activate fibroblasts to promote esophageal cancer development. Cancer Cell. 2023;41:903–918.e8. doi: 10.1016/j.ccell.2023.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Yao J., Cui Q., Fan W., Ma Y., Chen Y., Liu T., Zhang X., Xi Y., Wang C., Peng L., et al. Single-cell transcriptomic analysis in a mouse model deciphers cell transition states in the multistep development of esophageal cancer. Nat. Commun. 2020;11:3715. doi: 10.1038/s41467-020-17492-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X., Peng L., Luo Y., Zhang S., Pu Y., Chen Y., Guo W., Yao J., Shao M., Fan W., et al. Dissecting esophageal squamous-cell carcinoma ecosystem by single-cell transcriptomic analysis. Nat. Commun. 2021;12:5291. doi: 10.1038/s41467-021-25539-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dinh H.Q., Pan F., Wang G., Huang Q.F., Olingy C.E., Wu Z.Y., Wang S.H., Xu X., Xu X.E., He J.Z., et al. Integrated single-cell transcriptome analysis reveals heterogeneity of esophageal squamous cell carcinoma microenvironment. Nat. Commun. 2021;12:7335. doi: 10.1038/s41467-021-27599-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiu L., Yue J., Ding L., Yin Z., Zhang K., Zhang H. Cancer-associated fibroblasts: An emerging target against esophageal squamous cell carcinoma. Cancer Lett. 2022;546 doi: 10.1016/j.canlet.2022.215860. [DOI] [PubMed] [Google Scholar]
- 23.Huang T.X., Tan X.Y., Huang H.S., Li Y.T., Liu B.L., Liu K.S., Chen X., Chen Z., Guan X.Y., Zou C., Fu L. Targeting cancer-associated fibroblast-secreted WNT2 restores dendritic cell-mediated antitumour immunity. Gut. 2022;71:333–344. doi: 10.1136/gutjnl-2020-322924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H., Yue J., Jiang Z., Zhou R., Xie R., Xu Y., Wu S. CAF-secreted CXCL1 conferred radioresistance by regulating DNA damage response in a ROS-dependent manner in esophageal squamous cell carcinoma. Cell Death Dis. 2017;8:e2790. doi: 10.1038/cddis.2017.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang X., Chen X., Zhang S., Fan W., Zhong C., Liu T., Cheng G., Zhu L., Liu Q., Xi Y., et al. Collagen 1-mediated CXCL1 secretion in tumor cells activates fibroblasts to promote radioresistance of esophageal cancer. Cell Rep. 2023;42 doi: 10.1016/j.celrep.2023.113270. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Q., Huang L., Qin G., Qiao Y., Ren F., Shen C., Wang S., Liu S., Lian J., Wang D., et al. Cancer-associated fibroblasts induce monocytic myeloid-derived suppressor cell generation via IL-6/exosomal miR-21-activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett. 2021;518:35–48. doi: 10.1016/j.canlet.2021.06.009. [DOI] [PubMed] [Google Scholar]
- 27.Lee N.P., Chan C.M., Tung L.N., Wang H.K., Law S. Tumor xenograft animal models for esophageal squamous cell carcinoma. J. Biomed. Sci. 2018;25:66. doi: 10.1186/s12929-018-0468-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu W., Miao C., Zhang S., Liu Y., Niu X., Xi Y., Guo W., Chu J., Lin A., Liu H., et al. VAV2 is required for DNA repair and implicated in cancer radiotherapy resistance. Signal Transduct. Target. Ther. 2021;6:322. doi: 10.1038/s41392-021-00735-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerdes J. Ki-67 and other proliferation markers useful for immunohistological diagnostic and prognostic evaluations in human malignancies. Semin. Cancer Biol. 1990;1:199–206. [PubMed] [Google Scholar]
- 30.Sanz-Moreno V., Gaggioli C., Yeo M., Albrengues J., Wallberg F., Viros A., Hooper S., Mitter R., Féral C.C., Cook M., et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell. 2011;20:229–245. doi: 10.1016/j.ccr.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 31.Massagué J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erez N., Truitt M., Olson P., Arron S.T., Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell. 2010;17:135–147. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 33.Scherz-Shouval R., Santagata S., Mendillo M.L., Sholl L.M., Ben-Aharon I., Beck A.H., Dias-Santagata D., Koeva M., Stemmer S.M., Whitesell L., Lindquist S. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;158:564–578. doi: 10.1016/j.cell.2014.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshida G.J. Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 2020;13:4. doi: 10.1186/s13045-019-0829-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahdesmäki M.J., Gray S.R., Johnson J.H., Lai Z. Disambiguate: An open-source application for disambiguating two species in next generation sequencing data from grafted samples. F1000Res. 2016;5:2741. doi: 10.12688/f1000research.10082.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jukic A., Bakiri L., Wagner E.F., Tilg H., Adolph T.E. Calprotectin: from biomarker to biological function. Gut. 2021;70:1978–1988. doi: 10.1136/gutjnl-2021-324855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y., Ouyang Y., Li Z., Wang X., Ma J. S100A8 and S100A9 in Cancer. Biochim. Biophys. Acta. Rev. Cancer. 2023;1878 doi: 10.1016/j.bbcan.2023.188891. [DOI] [PubMed] [Google Scholar]
- 38.Smyth E.C., Lagergren J., Fitzgerald R.C., Lordick F., Shah M.A., Lagergren P., Cunningham D. Oesophageal cancer. Nat. Rev. Dis. Primers. 2017;3 doi: 10.1038/nrdp.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu F., Yang J., Liu J., Wang Y., Mu J., Zeng Q., Deng S., Zhou H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021;6:218. doi: 10.1038/s41392-021-00641-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukata Y., Amano M., Kaibuchi K. Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol. Sci. 2001;22:32–39. doi: 10.1016/s0165-6147(00)01596-0. [DOI] [PubMed] [Google Scholar]
- 41.Vicente-Manzanares M., Ma X., Adelstein R.S., Horwitz A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 2009;10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Totsukawa G., Yamakita Y., Yamashiro S., Hartshorne D.J., Sasaki Y., Matsumura F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J. Cell Biol. 2000;150:797–806. doi: 10.1083/jcb.150.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu X., Lu Y., Huang J., Xing Y., Dai H., Zhu L., Li S., Feng J., Zhou B., Li J., et al. CD16(+) fibroblasts foster a trastuzumab-refractory microenvironment that is reversed by VAV2 inhibition. Cancer Cell. 2022;40:1341–1357.e13. doi: 10.1016/j.ccell.2022.10.015. [DOI] [PubMed] [Google Scholar]
- 44.Foster C.T., Gualdrini F., Treisman R. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev. 2017;31:2361–2375. doi: 10.1101/gad.304501.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miralles F., Posern G., Zaromytidou A.I., Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–342. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 46.Yokota S., Chosa N., Kyakumoto S., Kimura H., Ibi M., Kamo M., Satoh K., Ishisaki A. ROCK/actin/MRTF signaling promotes the fibrogenic phenotype of fibroblast-like synoviocytes derived from the temporomandibular joint. Int. J. Mol. Med. 2017;39:799–808. doi: 10.3892/ijmm.2017.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Möller A., Jauch-Speer S.L., Gandhi S., Vogl T., Roth J., Fehler O. The roles of toll-like receptor 4, CD33, CD68, CD69, or CD147/EMMPRIN for monocyte activation by the DAMP S100A8/S100A9. Front. Immunol. 2023;14 doi: 10.3389/fimmu.2023.1110185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sakaguchi M., Yamamoto M., Miyai M., Maeda T., Hiruma J., Murata H., Kinoshita R., Winarsa Ruma I.M., Putranto E.W., Inoue Y., et al. Identification of an S100A8 Receptor Neuroplastin-β and its Heterodimer Formation with EMMPRIN. J. Invest. Dermatol. 2016;136:2240–2250. doi: 10.1016/j.jid.2016.06.617. [DOI] [PubMed] [Google Scholar]
- 49.Hibino T., Sakaguchi M., Miyamoto S., Yamamoto M., Motoyama A., Hosoi J., Shimokata T., Ito T., Tsuboi R., Huh N.H. S100A9 is a novel ligand of EMMPRIN that promotes melanoma metastasis. Cancer Res. 2013;73:172–183. doi: 10.1158/0008-5472.Can-11-3843. [DOI] [PubMed] [Google Scholar]
- 50.Fu Z.G., Wang L., Cui H.Y., Peng J.L., Wang S.J., Geng J.J., Liu J.D., Feng F., Song F., Li L., et al. A novel small-molecule compound targeting CD147 inhibits the motility and invasion of hepatocellular carcinoma cells. Oncotarget. 2016;7:9429–9447. doi: 10.18632/oncotarget.6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., et al. Proteomics. Tissue-based map of the human proteome. Science (New York, N.Y.) 2015;347 doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 52.Messeguer X., Escudero R., Farré D., Núñez O., Martínez J., Albà M.M. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics. 2002;18:333–334. doi: 10.1093/bioinformatics/18.2.333. [DOI] [PubMed] [Google Scholar]
- 53.Liu T., Zhao X., Lin Y., Luo Q., Zhang S., Xi Y., Chen Y., Lin L., Fan W., Yang J., et al. Computational Identification of Preneoplastic Cells Displaying High Stemness and Risk of Cancer Progression. Cancer Res. 2022;82:2520–2537. doi: 10.1158/0008-5472.Can-22-0668. [DOI] [PubMed] [Google Scholar]
- 54.Watanabe H., Okada M., Kaji Y., Satouchi M., Sato Y., Yamabe Y., Onaya H., Endo M., Sone M., Arai Y. [New response evaluation criteria in solid tumours-revised RECIST guideline (version 1.1)]. Gan to kagaku ryoho. Gan To Kagaku Ryoho. 2009;36:2495–2501. [PubMed] [Google Scholar]
- 55.Li L., Wei J.R., Dong J., Lin Q.G., Tang H., Jia Y.X., Tan W., Chen Q.Y., Zeng T.T., Xing S., et al. Laminin γ2-mediating T cell exclusion attenuates response to anti-PD-1 therapy. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abc8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deng M., Lin J., Nowsheen S., Liu T., Zhao Y., Villalta P.W., Sicard D., Tschumperlin D.J., Lee S., Kim J., Lou Z. Extracellular matrix stiffness determines DNA repair efficiency and cellular sensitivity to genotoxic agents. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abb2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pietilä E.A., Gonzalez-Molina J., Moyano-Galceran L., Jamalzadeh S., Zhang K., Lehtinen L., Turunen S.P., Martins T.A., Gultekin O., Lamminen T., et al. Co-evolution of matrisome and adaptive adhesion dynamics drives ovarian cancer chemoresistance. Nat. Commun. 2021;12:3904. doi: 10.1038/s41467-021-24009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De Veirman K., De Beule N., Maes K., Menu E., De Bruyne E., De Raeve H., Fostier K., Moreaux J., Kassambara A., Hose D., et al. Extracellular S100A9 Protein in Bone Marrow Supports Multiple Myeloma Survival by Stimulating Angiogenesis and Cytokine Secretion. Cancer Immunol. Res. 2017;5:839–846. doi: 10.1158/2326-6066.Cir-17-0192. [DOI] [PubMed] [Google Scholar]
- 59.Wang C., Zheng X., Zhang J., Jiang X., Wang J., Li Y., Li X., Shen G., Peng J., Zheng P., et al. CD300ld on neutrophils is required for tumour-driven immune suppression. Nature. 2023;621:830–839. doi: 10.1038/s41586-023-06511-9. [DOI] [PubMed] [Google Scholar]
- 60.Chen L., Shi V., Wang S., Sun L., Freeman R., Yang J., Inkman M.J., Ghosh S., Ruiz F., Jayachandran K., et al. SCCA1/SERPINB3 suppresses antitumor immunity and blunts therapy-induced T cell responses via STAT-dependent chemokine production. J. Clin. Invest. 2023;133 doi: 10.1172/jci163841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kwak T., Wang F., Deng H., Condamine T., Kumar V., Perego M., Kossenkov A., Montaner L.J., Xu X., Xu W., et al. Distinct Populations of Immune-Suppressive Macrophages Differentiate from Monocytic Myeloid-Derived Suppressor Cells in Cancer. Cell Rep. 2020;33 doi: 10.1016/j.celrep.2020.108571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhan X., Wu R., Kong X.H., You Y., He K., Sun X.Y., Huang Y., Chen W.X., Duan L. Elevated neutrophil extracellular traps by HBV-mediated S100A9-TLR4/RAGE-ROS cascade facilitate the growth and metastasis of hepatocellular carcinoma. Cancer Commun. 2023;43:225–245. doi: 10.1002/cac2.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maus R.L.G., Jakub J.W., Nevala W.K., Christensen T.A., Noble-Orcutt K., Sachs Z., Hieken T.J., Markovic S.N. Human Melanoma-Derived Extracellular Vesicles Regulate Dendritic Cell Maturation. Front. Immunol. 2017;8:358. doi: 10.3389/fimmu.2017.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Monteiro C., Miarka L., Perea-García M., Priego N., García-Gómez P., Álvaro-Espinosa L., de Pablos-Aragoneses A., Yebra N., Retana D., Baena P., et al. Stratification of radiosensitive brain metastases based on an actionable S100A9/RAGE resistance mechanism. Nat. Med. 2022;28:752–765. doi: 10.1038/s41591-022-01749-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang N., Zhao G., Yang Q., Tan J., Tan Y., Zhang J., Cheng Y., Chen J. Intracellular and extracellular S100A9 trigger epithelial-mesenchymal transition and promote the invasive phenotype of pituitary adenoma through activation of AKT1. Aging. 2020;12:23114–23128. doi: 10.18632/aging.104072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu N., Zhang B.B., Huang X.N., Yi X., Yan X.M., Cai Y., He Q., Han Z.J., Huang Y.J., Liu W., Jiao A.J. S100A8/A9 Molecular Complexes Promote Cancer Migration and Invasion via the p38 MAPK Pathway in Nasopharyngeal Carcinoma. Bioinorg. Chem. Appl. 2021;2021 doi: 10.1155/2021/9913794. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Araki K., Kinoshita R., Tomonobu N., Gohara Y., Tomida S., Takahashi Y., Senoo S., Taniguchi A., Itano J., Yamamoto K.I., et al. The heterodimer S100A8/A9 is a potent therapeutic target for idiopathic pulmonary fibrosis. J. Mol. Med. 2021;99:131–145. doi: 10.1007/s00109-020-02001-x. [DOI] [PubMed] [Google Scholar]
- 68.Hou C., Wang D., Zhao M., Ballar P., Zhang X., Mei Q., Wang W., Li X., Sheng Q., Liu J., et al. MANF brakes TLR4 signaling by competitively binding S100A8 with S100A9 to regulate macrophage phenotypes in hepatic fibrosis. Acta Pharm. Sin. B. 2023;13:4234–4252. doi: 10.1016/j.apsb.2023.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao P., Zhang W., Wang S.J., Yu X.L., Tang J., Huang W., Li Y., Cui H.Y., Guo Y.S., Tavernier J., et al. HAb18G/CD147 promotes cell motility by regulating annexin II-activated RhoA and Rac1 signaling pathways in hepatocellular carcinoma cells. Hepatology (Baltimore, Md.) 2011;54:2012–2024. doi: 10.1002/hep.24592. [DOI] [PubMed] [Google Scholar]
- 70.Wang K., Huang W., Chen R., Lin P., Zhang T., Ni Y.F., Li H., Wu J., Sun X.X., Geng J.J., et al. Di-methylation of CD147-K234 Promotes the Progression of NSCLC by Enhancing Lactate Export. Cell Metab. 2021;33:160–173.e6. doi: 10.1016/j.cmet.2020.12.010. [DOI] [PubMed] [Google Scholar]
- 71.Simanovich E., Brod V., Rahat M.A. Active Vaccination With EMMPRIN-Derived Multiple Antigenic Peptide (161-MAP) Reduces Angiogenesis in a Dextran Sodium Sulfate (DSS)-Induced Colitis Model. Front. Immunol. 2018;9:2919. doi: 10.3389/fimmu.2018.02919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cui H.Y., Wang S.J., Song F., Cheng X., Nan G., Zhao Y., Qian M.R., Chen X., Li J.Y., Liu F.L., et al. CD147 receptor is essential for TFF3-mediated signaling regulating colorectal cancer progression. Signal Transduct. Target. Ther. 2021;6:268. doi: 10.1038/s41392-021-00677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen Y., Xu J., Wu X., Yao H., Yan Z., Guo T., Wang W., Wang P., Li Y., Yang X., et al. CD147 regulates antitumor CD8(+) T-cell responses to facilitate tumor-immune escape. Cell. Mol. Immunol. 2021;18:1995–2009. doi: 10.1038/s41423-020-00570-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu J., Chen L., Qin C., Huo F., Liang X., Yang X., Zhang K., Lin P., Liu J., Feng Z., et al. CD147 contributes to SARS-CoV-2-induced pulmonary fibrosis. Signal Transduct. Target. Ther. 2022;7:382. doi: 10.1038/s41392-022-01230-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li J., Xing J., Yang Y., Liu J., Wang W., Xia Y., Yan Z., Wang K., Wu D., Wu L., et al. Adjuvant (131)I-metuximab for hepatocellular carcinoma after liver resection: a randomised, controlled, multicentre, open-label, phase 2 trial. The lancet. Lancet. Gastroenterol. Hepatol. 2020;5:548–560. doi: 10.1016/s2468-1253(19)30422-4. [DOI] [PubMed] [Google Scholar]
- 76.Bian H., Zheng J.S., Nan G., Li R., Chen C., Hu C.X., Zhang Y., Sun B., Wang X.L., Cui S.C., et al. Randomized trial of [131I] metuximab in treatment of hepatocellular carcinoma after percutaneous radiofrequency ablation. J. Natl. Cancer Inst. 2014;106 doi: 10.1093/jnci/dju239. [DOI] [PubMed] [Google Scholar]
- 77.Xu J., Shen Z.Y., Chen X.G., Zhang Q., Bian H.J., Zhu P., Xu H.Y., Song F., Yang X.M., Mi L., et al. A randomized controlled trial of Licartin for preventing hepatoma recurrence after liver transplantation. Hepatology (Baltimore, Md.) 2007;45:269–276. doi: 10.1002/hep.21465. [DOI] [PubMed] [Google Scholar]
- 78.Chen Z.N., Mi L., Xu J., Song F., Zhang Q., Zhang Z., Xing J.L., Bian H.J., Jiang J.L., Wang X.H., et al. Targeting radioimmunotherapy of hepatocellular carcinoma with iodine (131I) metuximab injection: clinical phase I/II trials. Int. J. Radiat. Oncol. Biol. Phys. 2006;65:435–444. doi: 10.1016/j.ijrobp.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 79.Nan G., Zhao S.H., Wang T., Chao D., Tian R.F., Wang W.J., Fu X., Lin P., Guo T., Wang B., et al. CD147 supports paclitaxel resistance via interacting with RanBP1. Oncogene. 2022;41:983–996. doi: 10.1038/s41388-021-02143-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bu X., Qu X., Guo K., Meng X., Yang X., Huang Q., Dou W., Feng L., Wei X., Gao J., et al. CD147 confers temozolomide resistance of glioma cells via the regulation of β-TrCP/Nrf2 pathway. Int. J. Biol. Sci. 2021;17:3013–3023. doi: 10.7150/ijbs.60894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Peng L., Jiang J., Chen H.N., Zhou L., Huang Z., Qin S., Jin P., Luo M., Li B., Shi J., et al. Redox-sensitive cyclophilin A elicits chemoresistance through realigning cellular oxidative status in colorectal cancer. Cell Rep. 2021;37 doi: 10.1016/j.celrep.2021.110069. [DOI] [PubMed] [Google Scholar]
- 82.Higuchi T., Shoji Y., Koyanagi K., Tajima K., Kanamori K., Ogimi M., Yatabe K., Ninomiya Y., Yamamoto M., Kazuno A., et al. Multimodal Treatment Strategies to Improve the Prognosis of Locally Advanced Thoracic Esophageal Squamous Cell Carcinoma: A Narrative Review. Cancers. 2022;15 doi: 10.3390/cancers15010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hirao M., Ando N., Tsujinaka T., Udagawa H., Yano M., Yamana H., Nagai K., Mizusawa J., Nakamura K., Japan Esophageal Oncology Group/Japan Clinical Oncology Group Influence of preoperative chemotherapy for advanced thoracic oesophageal squamous cell carcinoma on perioperative complications. Br. J. Surg. 2011;98:1735–1741. doi: 10.1002/bjs.7683. [DOI] [PubMed] [Google Scholar]
- 84.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soneson C., Love M.I., Robinson M.D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. doi: 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jin S., Guerrero-Juarez C.F., Zhang L., Chang I., Ramos R., Kuan C.H., Myung P., Plikus M.V., Nie Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021;12:1088. doi: 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw RNA sequencing data of 20 PDX tumor tissues generated in this study have been deposited in GSA (Genome Sequence Archive: CRA013775). Other resources of published datasets we used are listed in the key resources table and as follows:
The raw RNA sequencing data of PDX donors25 can be accessed under HRA004329 in GSA-Human. The ESCC scRNA-seq data20 is available in GEO with accession number GSE160269. This paper does not report original code. Any additional information required to re-analyze the data reported in this paper is available from the lead contact upon request.