

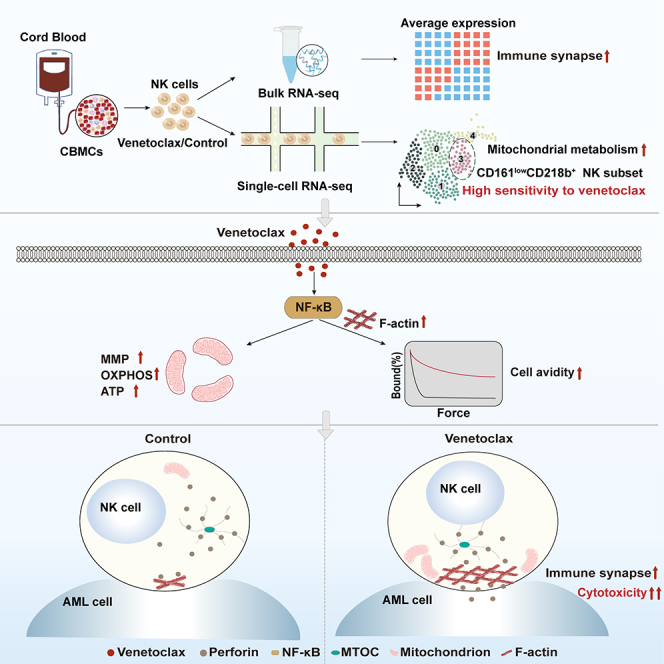
Summary
Natural killer (NK) cell-based immunotherapy holds promise for cancer treatment; however, its efficacy remains limited, necessitating the development of alternative strategies. Here, we report that venetoclax, an FDA-approved BCL-2 inhibitor, directly activates NK cells, enhancing their cytotoxicity against acute myeloid leukemia (AML) both in vitro and in vivo, likely independent of BCL-2 inhibition. Through comprehensive approaches, including bulk and single-cell RNA sequencing, avidity measurement, and functional assays, we demonstrate that venetoclax increases the avidity of NK cells to AML cells and promotes lytic granule polarization during immunological synapse (IS) formation. Notably, we identify a distinct CD161lowCD218b+ NK cell subpopulation that exhibits remarkable sensitivity to venetoclax treatment. Furthermore, venetoclax promotes mitochondrial respiration and ATP synthesis via the NF-κB pathway, thereby facilitating IS formation in NK cells. Collectively, our findings establish venetoclax as a multifaceted immunometabolic modulator of NK cell function and provide a promising strategy for augmenting NK cell-based cancer immunotherapy.
Keywords: venetoclax, natural killer cells, immunotherapy, acute myeloid leukemia, cytotoxicity, immunological synapse, avidity, mitochondrial respiration, NF-κB, RNA sequencing
Graphical abstract
Highlights
-
•
Venetoclax enhances NK cell cytotoxicity, likely independent of BCL-2 inhibition
-
•
Venetoclax increases NK cell avidity for AML cells and improves IS formation
-
•
Venetoclax boosts mitochondrial metabolism via NF-κB, enhancing IS formation
-
•
The CD161lowCD218b+ NK cell subpopulation exhibits high sensitivity to venetoclax
Wang et al. reveal that venetoclax activates NK cells, enhancing their cytotoxicity against leukemia through NF-κB-mediated metabolic and cytoskeletal changes. This improves NK cell avidity and IS formation in leukemic cells, with CD161lowCD218b+ NK cells showing heightened sensitivity, indicating potential targets for NK cell-based immunotherapy.
Introduction
Cell-based anticancer immunotherapies have revolutionized cancer treatment. Diverse immune cell types, particularly those that destroy target cells through direct contact or release of cytotoxic molecules, have been used for this purpose.1,2,3 Natural killer (NK) cells are cytotoxic innate lymphoid cells that protect the host against viral infections and cancer. Unlike T cells, NK cells can spontaneously detect and eliminate infected and malignant cells without prior specific activation.4 Allogeneic NK cells are particularly well suited for use in adoptive transfer, as they alleviate the risk of cytokine release syndrome occurrence.5 These properties of NK cells have rendered them highly appealing for use in cell-based cancer immunotherapy.
NK immunotherapy has shown immense promise as a feasible and effective treatment approach in preclinical and early clinical studies.4,5,6 However, NK-based cancer immunotherapy faces significant challenges, primarily centered on optimizing NK cell sources and enhancing their functional capabilities and long-term persistence in vivo.5,7 Extensive efforts are being made to overcome these hurdles and to maximize the therapeutic potential of NK cells. Diverse sources of therapeutic NK cells have been explored, including haploidentical, umbilical cord blood (CB), stem cell-derived, adaptive, cytokine-induced memory-like, chimeric antigen receptor, and NK cell lines.5,6,7 Cytokine-based agents, engager molecules, and immune checkpoint inhibitors have been employed to enhance NK cell function. Despite considerable progress in preclinical and clinical trials investigating NK cell-based immunotherapy, its efficacy in cancer treatment remains limited.7,8 Thus, there is an urgent need to explore strategies that can potentiate NK cell immunotherapy and improve its efficacy in cancer treatment.
Venetoclax is an FDA-approved small-molecule inhibitor of BCL-2, a key antiapoptotic protein that plays a crucial role in tumorigenesis, tumor survival, lymphocyte development, and immune system regulation.9,10,11 It is prescribed for adults with chronic lymphocytic leukemia/small lymphocytic leukemia and is also used in combination with a hypomethylating agent or low-dose cytarabine for adults with acute myeloid lymphoma (AML) aged over 75 or those with medical conditions preventing intensive induction chemotherapy.12,13,14,15 Despite its well-established apoptotic role, the broader impact of venetoclax on antitumor immune responses remains largely unexplored.
Our recent study revealed a significant enhancement in NK cell cytotoxicity in patients with AML receiving venetoclax treatment. While venetoclax is traditionally used for leukemia treatment by inhibiting BCL-2 and inducing apoptosis, this intriguing finding raises the possibility that venetoclax may directly enhance NK cell cytotoxicity, thereby boosting NK cell-mediated antitumor immune responses. Given the limited exploration of the complex interplay between venetoclax and its direct effects on NK cells, our present study delves into thoroughly investigating the impacts and underlying mechanisms of venetoclax on human NK cells. Additionally, we aim to explore the potential applications of venetoclax in NK cell-based immunotherapy.
Results
Venetoclax directly activates human NK cells and enhances their cytotoxicity against AML in vitro
NK cells in patients with AML exhibit impaired cytotoxicity, which is associated with disease development, progression, and relapse.16,17 Unexpectedly, we observed that venetoclax effectively restored NK cell cytotoxicity in patients with AML (Figures S1A and S1B), indicating its potential role in modulating NK cell activity. To investigate this phenomenon, we first evaluated the tolerance of NK cells to venetoclax-induced apoptosis. We observed that umbilical CB-derived NK (CB-NK) cells exhibited tolerance (Figures S1C and S1D), whereas resting peripheral blood-derived NK (PB-NK) cells were vulnerable due to elevated BCL-2 expression (Figures S1E and S1F), and this result was consistent with the reported study.18 Next, upon investigating the impact of venetoclax on CB-NK cell cytotoxicity against AML cells, we noted a significant increase, particularly at a dose of 400 nM for 18 h (Figures S1G and S1H), which was adopted for subsequent tests. Although venetoclax treatment did not significantly increase NK cell proliferation (Figures S1I and S1J), NK cells treated with venetoclax consistently induced apoptosis in target cells at significantly higher levels compared to control NK cells, even at low effector-to-target ratios (e.g., 1:2) (Figures 1A–1C). This was accompanied by increased interferon (IFN)-γ and CD107a expression (Figures 1D and S1K), indicating improved effector functions. Moreover, compared to untreated NK cells, venetoclax-treated NK cells more effectively reduced colony formation in the AML cell lines KG-1a, THP-1, HL60-Luc (HL60 cells stably expressing luciferase), and U937 (Figures 1E and S1L–S1N).
Figure 1.

Venetoclax enhances the cytotoxicity of cord blood and AML patient-derived NK cells against AML in vitro
(A) Schematic representation of the cytotoxicity assays using CB-NK cells.
(B and C) Untreated CB-NK cells or those pretreated with 400 nM venetoclax for 18 h were incubated with KG-1a or THP-1 cells (2 × 104 cells per well) at different effector-to-target ratios for 4 h, followed by Annexin V/7-AAD assay (n = 3/4, biological replicates). The results represent three independent experiments using NK cells from different donors. The calculation of the specific killing is provided in the STAR Methods.
(D) Representative flow cytometry plots (left) and summary data (right) of CD107a (n = 7, biological replicates) and IFN-γ (n = 10, biological replicates) production by CB-NK cells treated with or without 400 nM venetoclax. The results represent three independent experiments.
(E) Colony formation assay using KG-1a cells co-cultured with venetoclax-treated or untreated CB-NK cells at a ratio of 2.5:1 (n = 3, biological replicates). Equal numbers of AML cells (2 × 103 cells per dish) were used in the colony formation assays, and the colonies were counted after 14 days. The results represent three independent experiments using NK cells from three different donors, with detailed experimental procedures in the STAR Methods.
(F) Cytotoxicity assay using venetoclax-treated or untreated CB-NK cells (5 × 104 cells per well) against primary AML cells from newly diagnosed (n = 6, biological replicates) or relapsed patients (n = 5, biological replicates) at a 2.5:1 ratio for 4 h. Primary AML cell viability was assessed using flow cytometry with the Annexin V/7-AAD assay. The results represent three independent experiments using NK cells from different donors.
(G) Colony formation assay using primary AML cells co-cultured with venetoclax-treated or untreated CB-NK cells at a ratio of 2.5:1 (n = 3, biological replicates). Equal numbers of primary AML cells (1 × 104 cells per dish) were used in the colony formation assays, and the colonies were counted after 14 days. The results represent three independent experiments using NK cells and primary AML cells from different donors.
(H) Experimental setup of co-culture assays using NK cells derived from fresh bone marrow (BM) of newly diagnosed patients with AML.
(I) Cytotoxicity assay using NK cells derived from fresh AML BM samples (5 × 104 cells per well) treated with or without 400 nM venetoclax against KG-1a cells (n = 3, biological replicates) or autologous AML cells (n = 6, biological replicates) at a 2.5:1 ratio for 4 h. The results for autologous AML cells are representative of four independent experiments using NK cells from six different donors.
Data were analyzed using two-tailed unpaired Student’s t test (B, C, F, and I), paired Student’s t test (D), or one-way ANOVA with Tukey’s multiple comparisons test (E and G). Data are presented as mean ± standard deviation (SD).
See also Figures S1 and S2.
Furthermore, venetoclax-treated CB-NK cells effectively killed primary AML cells from both newly diagnosed and relapsed/refractory patients with AML, even those resistant to CB-NK-mediated killing (Figures 1F and S1O). These observations were extended to a colony formation assay (Figure 1G). Moreover, venetoclax pretreatment significantly enhanced the cytotoxicity of NK cells derived from the bone marrow of patients with AML (AML-NK) and PB-NK cells against AML cells (Figures 1H, 1I, and S1P), indicating broader effectiveness.
In clinical settings, combining venetoclax with demethylating agents like decitabine or azacitidine is a common strategy for AML treatment. This prompted us to examine the effects of these agents, both individually and in combination, on NK cell cytotoxicity. Utilizing CB-NK cells, we found that venetoclax significantly enhanced NK cell cytotoxicity, an effect not observed with decitabine or azacitidine alone (Figure S1Q), emphasizing the unique role of venetoclax in boosting NK cell-mediated cytotoxicity within the context of combination therapy.
To determine whether the enhancement of NK cell cytotoxicity by venetoclax is directly linked to its canonical role as a BCL-2 inhibitor, we evaluated the effects of other BCL-2 inhibitors, including S55746 and lisaftoclax (potent and selective BCL-2 inhibitors), as well as navitoclax (a dual BCL-2 and BCL-xL inhibitor), on NK cell cytotoxicity. Surprisingly, none of these inhibitors mimicked venetoclax’s significant cytotoxicity enhancement in NK cells (Figures S2A–S2F). Furthermore, knockdown of BCL-2 in CB-NK cells did not alter cytotoxicity compared to controls (Figures S2G and S2H), indicating that venetoclax may enhance NK cell cytotoxicity in a BCL-2-independent manner.
Overall, these results indicated that venetoclax activates human NK cells and enhances their cytotoxicity against AML cells without compromising their survival in vitro.
Venetoclax potentiates NK cell-mediated cytotoxicity against AML in vivo
To assess the therapeutic potential of venetoclax-treated NK cells for AML in vivo, we used three models of human AML in NOD-PrkdcscidIl2rgem1/Smoc (M-NSG) immunodeficient mice injected with KG-1a, HL60-Luc, or patient-derived primary AML cells. In the first model, we intravenously injected KG-1a cells into M-NSG mice. After the mice developed leukemia, they were randomly divided into three groups and treated according to the regimen shown in Figure 2A. Similar to the in vitro findings, venetoclax-treated CB-NK cells were more effective at killing leukemic cells, leading to a significant reduction in the leukemic cell burden in the bone marrow compared to that in the phosphate-buffered saline (PBS) or untreated CB-NK cell groups (Figure 2B).
Figure 2.

Venetoclax potentiates NK cell-mediated cytotoxicity against AML in vivo
(A) Schematic illustration of KG-1a mouse model construction (3 groups, n = 8/group).
(B) Representative flow cytometry plots (left) and quantification (right) illustrating BM engraftment of KG-1a cells. Two weeks post-injection of KG-1a cells, engraftment of KG-1a cells (human CD45+, gating from human CD3−CD56−) in the BM was determined by flow cytometry. NK cells used for in vivo studies were generated following the procedures described in the STAR Methods. The results were obtained from three independent experiments using NK cells from eight donors.
(C) Schematic outline of AML patient-derived xenograft model generation (3 groups, n = 5/group).
(D) Representative flow cytometry plots (left) and quantification illustrating BM engraftment of primary AML cells (right). Four weeks post-AML injection, the engraftment of primary AML cells (human CD45+ CD33+) in the BM was assessed through flow cytometry. NK cells used for in vivo studies were generated following the procedures described in the STAR Methods. The results were obtained from three independent experiments.
(E and F) Representative images of Wright-Giemsa BM smear staining of KG-1a (left) and primary AML cell (right) xenograft mice. Red arrowheads indicate leukemia cells.
(G) Schematic outline of HL60-Luc mouse model construction (3 groups, n = 10/group), with detailed experimental procedures in the STAR Methods. AML burden was monitored by bioluminescence imaging at the indicated time points.
(H) Kinetic analysis of AML burden in each group assessed by bioluminescence imaging. Statistical analysis was performed for the average radiance between venetoclax-pretreated and non-pretreated NK cell groups 14 days post-NK cell infusion.
(I) Kaplan-Meier survival analysis of HL60-Luc-engrafted mice.
(J) Imaging of AML burden using bioluminescence.
Statistical significance was determined using one-way ANOVA with Tukey’s multiple comparisons test (B and D), unpaired Student’s t test (H), or log rank Mantel-Cox test (I). For (B) and (D), the data are presented as mean ± SD. For (H), data are presented as mean ± SEM.
In the second mouse model, we investigated the therapeutic effects of venetoclax-treated CB-NK cells on the engraftment of primary AML samples. We established a leukemic animal model by intravenously injecting primary AML cells into the M-NSG mice (Figure 2C). Human AML cells expressing the cell surface markers CD45 and CD33 in M-NSG mice were identified by flow cytometry. The venetoclax-treated CB-NK cell group exhibited a significant reduction in the AML burden in the bone marrow compared to that in the PBS or untreated CB-NK cell groups (Figure 2D). Wright-Giemsa staining of bone marrow smears further confirmed the decreased leukemic cell burden in mice receiving venetoclax-treated CB-NK cells (Figures 2E and 2F).
To visually demonstrate the efficacy of venetoclax-treated NK cells in eliminating AML cells in vivo, we created a leukemia xenograft model by injecting HL60-Luc cells into M-NSG mice via the tail vein. The biodistribution of leukemic cells was monitored using bioluminescence imaging (Figure 2G). Once engraftment was confirmed, the mice were injected with PBS, venetoclax-pretreated CB-NK cells, or untreated CB-NK cells, and the AML burden was monitored using bioluminescence imaging. Notably, the group of mice injected with venetoclax-pretreated CB-NK cells had a significantly lower AML burden and longer survival time than the groups that received untreated CB-NK cells or PBS (Figures 2H–2J). Taken together, these findings provide strong evidence that venetoclax enhances the cytotoxicity of NK cells in AML in vivo.
Venetoclax enhances NK cell-binding avidity and lytic granule polarization during IS formation
To explore the mechanisms underlying the enhanced killing activity of NK cells upon venetoclax treatment, we performed bulk RNA sequencing (RNA-seq) analysis of untreated and venetoclax-treated CB-NK cells. Venetoclax treatment upregulated genes involved in synapse formation, actin reorganization, and cell adhesion and downregulated genes suppressing NK cell function (Figures 3A, 3B, S3A, and S3B). These findings were further validated by real-time qPCR and flow cytometric analyses (Figures S3C and S3D). Gene Ontology (GO) enrichment analysis using Metascape19 confirmed enhanced cellular adhesion and synaptic assembly capabilities in venetoclax-treated NK cells (Figures 3C and S3E).
Figure 3.

Venetoclax treatment increases NK cell avidity for AML cells and promotes lytic granule polarization during immune synapse formation
(A) Volcano plot of differentially expressed genes (DEGs) between the control and venetoclax-treated groups, with selected DEGs labeled.
(B) The distribution of DEG values in each sample from RNA-seq while classifying genes with similar functions.
(C) GO term enrichment analysis of upregulated DEGs in the venetoclax-treated group.
(D) F-actin (red) and perforin (green) staining in cell conjugates was acquired at different time points after mixing CB-NK cells (1 × 105 cells per well) treated or untreated with 400 nM venetoclax with KG-1a cells at a ratio of 1:1.
(E) Granule-to-synapse distance was quantified for 15–55 conjugates per group. The results represent two independent experiments.
(F) Schematic diagram of the single-cell avidity binding experimental outline.
(G) Representative bright-field raw micrographs of a microfluidic chip loaded with KG-1a cells and exposed to venetoclax-treated or untreated NK cells for 10 min before gradually applying force up to 1,000 pN. Orange circles represent bound effectors, while green circles represent regions where effectors were bound at the start of force application but then dislodged.
(H) Evaluation of binding avidity between venetoclax-treated or untreated NK cells and KG-1a targets using acoustic force microfluidic microscopy.
(I) Normalized fold change in the binding of venetoclax-treated NK cells compared to that of untreated NK cells from (H), n = 3.
(J) Cell-binding avidity from (H) at 1,000 pN, n = 3.
Data are presented as mean ± SD for (H)–(J). Statistical significance was determined using unpaired Student’s t test (E and J).
See also Figure S3.
Cellular cytotoxicity relies on lytic immunological synapse (IS) formation between NK and target cells.20,21 This process involves the migration of lytic granules (such as perforin and granzymes) toward the microtubule-organizing center (MTOC) and subsequent MTOC polarization to the synapse. F-actin remodeling facilitates granule movement and fusion with the plasma membrane for cytotoxic exocytosis.21,22,23 We formed conjugates between NK cells and target cells and subsequently stained these conjugates with antibodies targeting γ-tubulin (to label the MTOC), perforin (to label lytic granules), and phalloidin (to label F-actin at the IS) (Figures 3D and S3F). We measured the distances between the cytotoxic granules/MTOC and IS to assess polarization. Our findings revealed that venetoclax treatment expedited the movement of lytic granules and the MTOC toward the IS (Figures 3E, S3G, and S3H), along with increased F-actin intensity (Figure S3I), indicating cytoskeletal remodeling and enhanced IS formation, potentially contributing to improved NK cell cytotoxicity.
We conducted a more in-depth examination of cellular avidity, which encompasses synaptic processes such as binding, adhesion, and cytoskeleton remodeling. Using acoustic force microscopy (z-Movi), we assessed the binding force between AML cells and NK cells treated with venetoclax or left untreated (Figure 3F). Compared to untreated NK cells, venetoclax-treated NK cells exhibited significantly higher avidity for AML cells, with a 6-fold increase in binding force at 1,000 pN (Figures 3G–3J). This enhanced avidity resulted in faster conjugation of AML cells to venetoclax-treated NK cells than to untreated NK cells (Figure S3J). These changes in cellular avidity, along with cytoskeleton remodeling and improved lytic granule polarization, contributed to a more efficient IS, enhancing NK cell-mediated killing of AML cells.
CD161lowCD218b+ NK cells exhibit the most substantial expansion and transcriptomic modifications upon venetoclax treatment
To understand the effect of venetoclax on the gene expression profiles of individual NK cells and its role in enhancing NK cell cytotoxicity, we performed single-cell RNA-seq (scRNA-seq) analysis on CB-NK cells. (Figure 4A). We analyzed 15,046 high-quality single transcriptomes, with 6,050 from the control group and 8,996 from the venetoclax-treated group (Figures 4B, S4A, and S4B).
Figure 4.

scRNA-seq analysis reveals notable expansion and transcriptomic alterations in CD161lowCD218b+ NK cell subpopulation following venetoclax treatment
(A) scRNA-seq workflow chart. CB-NK cells were isolated from CB, treated with or without venetoclax (400 nM for 18 h), and then subjected to scRNA-seq using the 10× platform (control group n = 4; venetoclax-treated group n = 4).
(B) Uniform manifold approximation and projection (UMAP) visualization of major cell types from the scRNA-seq data after quality control.
(C) Heatmap showing the expression of marker genes in the five indicated clusters.
(D) Representative cluster GO terms.
(E) Pie charts displaying the percentage of cells assigned to each cluster within the indicated groups.
(F) Pie charts comparing the proportions of NK cell subclusters (left, log2 odds ratio) and the number of DEGs (right) in each subpopulation in the venetoclax-treated group vs. the control group.
(G) Violin plot displaying the expression of KLRB1 and IL-18RAP in each cell subcluster.
(H) Representative flow cytometry plots (left) and summary data (right) of C3 NK cells within total NK cell population from the control and venetoclax (400 nM for 18 h)-treated groups (n = 7, biological replicates). The results represent three independent experiments.
(I) Specific killing ability of NK cell subsets (sorted based on CD161 and CD218b, 2 × 104 cells per well) co-cultured with KG-1a cells at a 1:1 ratio for 4 h. The values in red indicate the fold change in the mean value in the venetoclax-treated group compared to the control group. The results represent three independent experiments.
(J) Flow cytometric analysis of IFN-γ (n = 7, biological replicates), CD107a (n = 3, biological replicates), perforin (n = 7, biological replicates), and granzyme B (n = 7, biological replicates) expression in the venetoclax (400 nM for 18 h)-treated or untreated C3 NK cells. The results represent three independent experiments. CD107a expression in C3 NK cells was assessed through flow cytometry following stimulation with KG-1a cells.
Statistical significance was determined using unpaired Student’s t test (I), paired Student’s t test (H and J), or hypergeometric test (D). Data are presented as mean ± SD.
See also Figure S4.
Due to limited gene coverage per cell, CD56 (NCAM1) expression was relatively low in the dataset.24 Instead, we used the well-established NK cell markers CD94 (KLRD1), NKG2A (KLRC1), and NKG7 to define the NK cell lineage25,26 (Figure S4C). All the clusters showed elevated expression of markers that are characteristic of NK cells. By integrating the control and venetoclax-treated samples using Seurat,27 we identified five NK cell subpopulations (C0, C1, C2, C3, and C4) using uniform manifold approximation and projection visualization (Figure 4B). Based on these signature genes, we performed GO analysis to uncover the functional characteristics of these NK cell subpopulations. Cluster C0 was associated with antioxidant activity and response to reactive oxygen species, whereas C1 and C2 were linked to cell activation and regulation of NK cell activation. C3 was related to ATP hydrolysis activity and positive regulation of NF-κB inducing kinase (NIK)/NF-κB signaling, and C4 was associated with the cellular response to cytokine stimulus and cadherin binding, resembling CD56bright NK cells (Figures 4C, 4D, and S4D).
Of particular interest, subpopulation C3, showing a 1.76-fold increase in the venetoclax-treated group, exhibited the most extensive transcriptomic changes during venetoclax treatment, characterized by the highest number of differentially expressed genes (Figures 4E and 4F). Two surface marker genes, IL18RAP (CD218b) and KLRB1 (CD161), distinguished C3 from other subpopulations (Figure 4G). Flow cytometry confirmed the presence of the CD161lowCD218b+ NK cell subset (C3), which increased in percentage after venetoclax treatment (Figure 4H).
We further conducted in vitro assays to assess the cytotoxic potential of various NK cell subpopulations, including the C3 subset, following venetoclax pretreatment. Our findings indicate that venetoclax pretreatment enhances the cytotoxicity of various NK cell subpopulations, with the C3 subpopulation showing the most significant increase, notably exhibiting a 25.7-fold increase in cytotoxicity (Figure 4I). This enhancement in cytotoxicity was accompanied by increased IFN-γ and CD107a secretion and upregulated expression of cytotoxic granule molecules following venetoclax treatment (Figure 4J). However, it is crucial to note that the baseline cytotoxicity of the C3 subpopulation against AML cells was initially lower than that of other NK cell subpopulations (Figure 4I), likely due to inherent functional limitations, including diminished expression of key cytotoxic molecules (Figure S4E). These data suggest that venetoclax treatment notably enhances the cytotoxic potential of the C3 NK cell subpopulation, transforming a less active C3 NK subpopulation into one with significantly enhanced cytotoxic activity.
Venetoclax treatment upregulates mitochondrial energy metabolism and NF-κB activity in both CD161lowCD218b+ NK cells and the total NK cell population
scRNA-seq analysis revealed significant transcriptional changes in the C3 NK cell subpopulation following venetoclax treatment. Venetoclax-treated C3 NK cells exhibited upregulation of genes associated with oxidative phosphorylation (OXPHOS) and ATP-synthesis-coupled electron transport pathways, which are crucial for cellular energy production (Figures 5A and 5B). Additionally, these cells showed enhanced expression of genes linked to OXPHOS, ATP synthesis, and the respiratory electron transport chain (ETC) (Figures S5A–S5C). Gene set enrichment analysis also supported the enrichment of OXPHOS-related pathways in the venetoclax-treated group (Figures S5D–S5F). Mitochondrial activity was significantly increased in terms of both the mitochondrial membrane potential (MMP) and mitochondrial mass within the C3 subpopulation after venetoclax treatment (Figures 5C, 5D, and S5G). These findings suggest that venetoclax treatment enhances OXPHOS and ATP synthesis in the C3 NK subpopulation, potentially contributing to improved functionality.
Figure 5.

Venetoclax treatment upregulates mitochondrial energy metabolism and increases NF-κB activity in NK cells
(A) Enriched GO terms of upregulated DEGs in the C3 subpopulation between the control and venetoclax-treated groups.
(B) Expression of OXPHOS-related genes in C3 NK cells.
(C and D) Representative images (C) and quantification (D) of the mitochondrial phenotype in C3 NK cells (control group, n = 27; venetoclax-treated group, n = 54; green, MitoTracker Green, an indicator of mitochondrial mass; red, MitoTracker Red CMXRos, an indicator of mitochondrial membrane potential; blue, Hoechst, a dye for cell nuclei). The results represent two independent experiments.
(E) Violin plot showing the NF-κB activation signal score and expression levels of representative NF-κB target genes in C3 NK cells.
(F and G) Expression of p-p65 (F, n = 7, biological replicates) and p-IKKα (G, n = 4, biological replicates) in venetoclax (400 nM for 18 h)-treated or untreated C3 NK cells analyzed by flow cytometry. The results represent three independent experiments.
(H) Confocal microscopy images (left) and quantification (right) of intranuclear p-p65 signal intensity in venetoclax (400 nM for 18 h)-treated or untreated C3 NK cells (control group, n = 76; venetoclax-treated group, n = 104). The results represent three independent experiments using NK cells from different donors.
(I) GO terms of genes upregulated following venetoclax treatment of total NK cells.
(J) Oxygen consumption rate (OCR) of venetoclax-treated (400 nM for 18 h) or untreated total NK cells (n = 4, biological replicates).
(K) Spare respiratory capacity calculated from (J).
(L) ATP assay was conducted to evaluate total ATP production, mitochondrial oxidative phosphorylation-derived ATP (mito-ATP), and glycolysis-derived ATP (glyco-ATP) in total NK cells treated with 400 nM venetoclax for 18 h or left untreated. Statistical analysis was performed for mito-ATP between the control and venetoclax-treated groups (n = 9–10, biological replicates).
(M) The p-p65 expression level in total NK cells treated with or without 400 nM venetoclax for 18 h was detected by flow cytometry (n = 8, biological replicates). The results represent three independent experiments using NK cells from different donors.
(N and O) Confocal microscopy images (N) and quantification of p-p65 intranuclear signal intensity (O) in total NK cells treated with or without 400 nM venetoclax for 18 h. The results represent three independent experiments using NK cells from different donors.
(P) Quantitative real-time PCR data showing changes in BCL-xL mRNA expression in CB-NK cells after 400 nM venetoclax treatment for 18 h (n = 6, biological replicates). The results represent three independent experiments using NK cells from different donors.
Statistical significance was determined by unpaired Student’s t tests (D, H, K, L, O, and P), paired Student’s t tests (F, G, and M), hypergeometric tests (A and I), and the Mann-Whitney test (E). Data are presented as mean ± SD.
See also Figure S5.
Our scRNA-seq data suggested that the C3 subpopulation, marked by IL18RAP (CD218b), a subunit of the interleukin-18 receptor complex known to activate the NF-κB signaling pathway,28 was associated with the positive regulation of NF-κB signaling (Figures 4C and 4D). Venetoclax treatment potentially activated NF-κB signaling in the C3 subpopulation, as indicated by the upregulation of NF-κB target genes (IFNG, CCL5, PRDX1, XIAP, and VIM) (Figure 5E). NF-κB pathway activation was further confirmed by elevated levels of phosphorylated p65 (p-p65) and IKKα (p-IKKα), as well as increased nuclear localization of p-p65 (Figures 5F–5H). These findings suggest that venetoclax treatment activates the NF-κB pathway in the C3 NK cell subpopulation.
Expanding beyond the C3 subpopulation, we observed enrichment of the OXPHOS-related signaling pathway in all venetoclax-treated NK cell subpopulations (except C4), with the C3 subpopulation showing the highest enrichment (Figure S5H). GO analysis of all venetoclax-treated NK cells indicated enrichment of genes related to the oxidative stress response and mitochondrial-protein-containing complex (Figure 5I). Venetoclax treatment not only significantly upregulated genes related to OXPHOS, ATP-synthesis-coupled electron transport, and the respiratory ETC in total NK cells (Figures S5I–S5K) but also enhanced mitochondrial function, as indicated by increased spare respiratory capacity and mitochondrial ATP production (Figures 5J–5L). Analysis of the MMP and mitochondrial mass in total NK cells revealed higher levels in venetoclax-treated NK cells than in untreated NK cells (Figures S5L and S5M). Moreover, NF-κB signaling was activated in total NK cells after venetoclax treatment (Figures 5M–5O), and this change was accompanied by an upregulation of the NF-κB target gene IFNG (Figure 1D). Venetoclax treatment also upregulated the expression of BCL-xL (a known NF-κB target29) and MCL-1 in NK cells, contributing to their evasion of apoptosis (Figure 5P and S5N–S5O). However, it had no impact on the expression of BCL-2 (Figure S5P). Overall, these findings suggest that venetoclax treatment increases mitochondrial energy metabolism and activates NF-κB signaling in both the CD161lowCD218b+ NK cell subset and total NK cell population.
Venetoclax activates NF-κB signaling to boost mitochondrial metabolism and facilitate IS formation in NK cells
In the C3 NK cell subpopulation, we observed a correlation between NF-κB activation and the OXPHOS signature (Figure 6A). Inhibiting NF-κB in C3 NK cells reduced the oxygen consumption rate, mitochondrial ATP production, MMP, and mitochondrial mass, counteracting the enhancements induced by venetoclax treatment (Figures 6B–6G). Extending beyond the C3 subpopulation, scRNA-seq data indicated a similar correlation between NF-κB activation and the OXPHOS signature in the total NK cell population (Figure 6H). Immunofluorescence staining demonstrated that NF-κB inhibition reversed the increase in the MMP and mitochondrial mass induced by venetoclax treatment in total NK cells (Figure 6I). These findings suggest that venetoclax enhances mitochondrial metabolism through NF-κB activation.
Figure 6.

Venetoclax enhances mitochondrial metabolism via NF-κB activation in NK cells
(A) Correlation between the NF-κB activation signature and OXPHOS signature in venetoclax-treated or untreated C3 NK cells.
(B) OCR kinetics in C3 NK cells treated with 400 nM venetoclax, 25 μg/mL SN50 (a cell-permeable inhibitor of NF-κB translocation) or venetoclax plus SN50 or left untreated.
(C–F) Quantification of basal OCR (C), maximal OCR (D), spare respiratory capacity (E), and ATP production rate (F) derived from (B), n = 9, biological replicates.
(G) Confocal microscopy images (left) showing MitoTracker Green and MitoTracker Red CMXRos staining in C3 NK cells treated with 25 μg/mL SN50, 400 nM venetoclax, or venetoclax plus SN50 or left untreated. The graphs (right) show the corresponding quantification of the mean fluorescence intensity (one data point per cell). Results from two independent experiments as presented.
(H) Correlation between the NF-κB activation signature and OXPHOS signature in venetoclax-treated or untreated total NK cells.
(I) Confocal microscopy images (left) and statistical analysis (right) illustrating MitoTracker Green and MitoTracker Red CMXRos staining in total NK cells treated with 25 μg/mL SN50, 400 nM venetoclax, or venetoclax plus SN50 or left untreated. The results represent two independent experiments.
Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons test (C–G and I) and hypergeometric test (A and H). Data are presented as mean ± SD. For (B), data are presented as mean ± SEM.
NF-κB activation modulates the activity of RhoA, a small GTPase that controls actin dynamics (critical for IS formation).30 Venetoclax treatment enhanced cytoskeletal remodeling, lytic granule movement, MTOC polarization, and binding avidity (Figures 3 and S3). These effects of venetoclax were also replicated in C3 NK cells (Figures S6A–S6C), implying that venetoclax promotes IS formation in an NF-κB-dependent manner. To address this, we performed rescue experiments using total CB-NK cells. Inhibition of NF-κB markedly reduced F-actin expression, whereas venetoclax alone increased F-actin expression (Figure S6D). Blocking NF-κB also hampered the polarization of lytic granules toward IS induced by venetoclax (Figures 7A and 7B). Furthermore, mitochondrial polarization toward the IS, crucial for the killing process,31 was accelerated by venetoclax and blocked by NF-κB inhibitors (Figures 7C and 7D). Additionally, blocking mitochondrial energy metabolism impaired lytic granule polarization during venetoclax treatment (Figures 7E–7H). These multifaceted impacts on cellular dynamics underscore the interconnected roles of NF-κB, F-actin, and mitochondrial functions in shaping the response of NK cells to venetoclax.
Figure 7.

Venetoclax boosts mitochondrial metabolism via NF-κB to facilitate IS formation in NK cells
(A) Representative confocal images of cell conjugates after NK cell/KG-1a cell contact under the indicated conditions stained with F-actin (red) and perforin (green). NK cells (1 × 105 cells per well) treated with venetoclax (400 nM), SN50 (25 μg/mL), or venetoclax plus SN50 or left untreated were co-cultured with KG-1a cells at a 1:1 ratio for 1 h. White arrows indicate perforin.
(B) Granule-to-synapse distance quantified for 33–47 conjugates per group. The results represent three independent experiments. The distance from the perforin to the IS was determined as described in Figure S3G.
(C) MitoTracker Deep Red (cyan), F-actin (red), and DAPI (blue) staining in cell conjugates after NK cell/KG-1a cell contact under the indicated conditions. NK cells (1 × 105 cells per well) treated with venetoclax (400 nM), SN50 (25 μg/mL), or venetoclax plus SN50 or left untreated were co-cultured with KG-1a cells for 30 min at a ratio of 1:1. White arrows indicate mitochondria.
(D) Mitochondria-to-synapse distance quantified for 39–71 conjugates per group. The results represent three independent experiments. The distance from the mitochondria to the IS was determined as described in Figure S3G.
(E) F-actin (red) and perforin (green) staining in cell conjugates after NK cell/KG-1a cell contact under the indicated conditions. NK cells (1 × 105 cells per well) treated with venetoclax (400 nM), oligomycin A (200 nM), or venetoclax plus oligomycin A or left untreated were co-cultured with KG-1a cells for 1 h at a ratio of 1:1. White arrows indicate perforin.
(F) The granule-to-synapse distance quantified for 32–45 conjugates per group. The results represent three independent experiments. The distance from perforin to the IS was determined as described in Figure S3G.
(G) F-actin (red) and perforin (green) staining in cell conjugates after NK cell/KG-1a cell contact under the indicated conditions. NK cells (1 × 105 cells per well) treated with venetoclax (400 nM), rotenone (200 nM), or venetoclax plus rotenone or left untreated were co-cultured with KG-1a cells for 1 h at a ratio of 1:1. White arrows indicate perforin.
(H) The granule-to-synapse distance quantified for 24–63 conjugates per group. The results represent three independent experiments. The distance from perforin to the IS was determined as described in Figure S3G.
(I) The immune-synapse-binding avidity of NK cells, treated with venetoclax (400 nM) or venetoclax plus SN50 (25 μg/mL) or left untreated, to KG-1a cells was assessed via acoustic force microfluidic microscopy.
(J) Cell-binding avidity from (J) at 1,000 pN, n = 3.
(K and L) Representative flow cytometry plots and quantification of the specific killing ability of CB-NK cells (5 × 104 cells per well) treated with venetoclax (400 nM), oligomycin A (200 nM), rotenone (200 nM), venetoclax plus oligomycin A, or venetoclax plus rotenone or left untreated against KG-1a cells at a 2.5:1 ratio for 4 h (n = 11, biological replicates). The results represent six independent experiments. Representative flow plots showing NK cells derived from the same donor.
(M) Flow cytometry analysis of the percentage of Annexin V+ KG-1a cells co-cultured for 4 h at a 2.5:1 ratio with total NK cells (5 × 104 cells per well) pretreated with venetoclax (400 nM), SN50 (25 μg/mL), or venetoclax plus SN50 or left untreated (n = 13, biological replicates). The results represent six independent experiments.
Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons test (B, D, F, H, and J–M). Data are presented as mean ± SD.
See also Figure S6.
Expanding from cellular dynamics to NK cell interaction with target cells, we found that NF-κB inhibition significantly reduced the binding avidity between NK cells and target cells, as measured by z-Movi (Figures 7I and 7J). NF-κB inhibition also counteracted enhanced cytotoxicity and IFN-γ secretion induced by venetoclax in C3 NK cells (Figures S6E and S6F). Moreover, employing multiple inhibitors that block both mitochondrial-dependent energy metabolism and NF-κB signaling significantly reduced cytotoxicity and IFN-γ secretion in total NK cells treated with venetoclax (Figures 7K–7M, S6G, and S6H). Taken together, these findings indicate that venetoclax boosts mitochondrial metabolism via the NF-κB pathway, facilitating IS formation and enhancing NK cell cytotoxicity.
Discussion
Targeted anticancer drugs have shown immunomodulatory effects and have influenced therapeutic outcomes in both preclinical and clinical studies.32,33,34 Venetoclax, a BCL-2 inhibitor used for leukemia treatment, primarily induces apoptosis by inhibiting BCL-2 overexpression in leukemic cells.11 Recent studies have suggested a potential role for venetoclax in modulating T cell function.9,35 Additionally, venetoclax boosts mitochondrial apoptosis priming in a variety of cancer cells, making them more susceptible to NK cell-mediated cytotoxicity. This is further evidenced by the synergistic effect of venetoclax and preactivated NK cells in reducing tumor burden in humanized mouse models.18 Moreover, venetoclax has been shown to upregulate NKG2D ligand expression in AML cells, heightening their sensitivity to NK cell-mediated killing.36 Extending beyond cancer immunotherapy, recent studies have revealed a significant role of venetoclax in combating viral infections, such as HIV.37,38 It has been shown that venetoclax can both suppress HIV-infected CD4+ T cells and boost their elimination by NK cells.38 Despite these insights, the intricate relationship between venetoclax and its direct effects on NK cells remains largely unexplored, prompting us to unravel this intricate interplay.
In this study, we found that venetoclax directly activated human NK cells, augmenting their cytotoxicity against leukemia both in vitro and in vivo. Mechanistically, venetoclax enhanced NK cell avidity and IS formation in leukemic cells, driven by the activation of NF-κB and the enhancement of mitochondrial metabolism. Our study contributes a perspective by highlighting the potential of venetoclax as a multifaceted immunometabolic modulator to augment adoptive NK cell-based immunotherapy. These findings present a promising strategy for enhancing NK cell immunotherapy and provide valuable insights into the development of more effective cancer treatments.
IS formation driven by F-actin-dependent plasma membrane remodeling is crucial for NK cell cytotoxicity.22,39,40 In the present study, venetoclax treatment was shown to increase F-actin expression and lytic granule polarization to the IS, highlighting its role in modulating NK cell cytoskeletal dynamics and enhancing cytotoxicity. Cellular avidity encompasses various aspects of the cellular synapse and plays a critical role in the immune response.41,42 Recent studies have revealed a close association between the avidity and cytotoxicity of immune cells, particularly in the context of in vivo tumor elimination.42,43 Using acoustic force microscopy, we measured NK cell-binding avidity in AML cells at the single-cell level and revealed that venetoclax significantly increased this binding avidity. This finding provides insight into how venetoclax enhances the interaction of NK cells with target cells, opening avenues for exploring its therapeutic potential in various cancers and its effect on different NK cell subsets.
Using scRNA-seq and functional assays, we identified a sensitive NK cell subpopulation, denoted as C3, characterized by low CD161 and high CD218b expression, which showed the most significant response to venetoclax. The CD161lowCD218b+ NK subset exhibited enhanced cytotoxicity, degranulation, and IFN-γ secretion upon venetoclax treatment, indicating its potential role in NK cell-mediated immunotherapy. Considering that CD161 acts as an immune checkpoint inhibitor of NK cells,44 it would be interesting to explore the potential synergistic effect of combining venetoclax with CD161-blocking antibodies to further enhance NK cell cytotoxicity.
Venetoclax upregulated OXPHOS and ATP synthesis in both the C3 subpopulation and overall NK cell population, aiding NK cell activation and lytic granule polarization to the IS. Notably, efficient ATP production through OXPHOS is essential for optimal NK cell activity.45,46 Our study further revealed that venetoclax not only boosts mitochondrial metabolism but also promotes mitochondrial translocation toward the IS via the NF-κB signaling pathway. This orchestrated response addresses the heightened energy requirements for improved NK cell cytotoxicity. Recently, a study showed that venetoclax monotherapy had no significant effect on the survival of T cells and NK cells in different cancer types, suggesting the feasibility of combined T cell and NK cell therapy with venetoclax.47 Additionally, our study showed that venetoclax activated NF-κB signaling, which was accompanied by an increase in BCL-xL expression in NK cells. This observation aligns with the findings of a previous study showing a survival advantage afforded by increased BCL-xL expression following BCL-2 inhibition.48 Elucidating the intricate interplay among OXPHOS, NF-κB activation, and apoptosis evasion will pave the way for therapeutic interventions and advance our understanding of the broader immunological landscape impacted by venetoclax in the context of cancer immunotherapy.
The excessive activation of NF-κB and abnormal elevation of energy metabolism are hallmarks of cancer cells, making them promising targets in cancer therapy.49,50,51 Nevertheless, although our overall findings strongly suggest that venetoclax boosts NK cell cytotoxicity by promoting mitochondrial metabolism and IS formation via NF-κB, caution should be exercised in clinical trials involving combined use of venetoclax and NF-κB/metabolic inhibitors for NK cell-based immunotherapy. This combination might diminish the effectiveness of venetoclax in augmenting NK cell function. Given that targeting overexpressed NF-κB or metabolic pathways in cancer is a promising strategy, combining venetoclax with NF-κB/metabolic inhibitors could potentially compromise the efficacy of NK cell-based cancer immunotherapy. Another interesting finding in this report is that venetoclax, known as a BCL-2 inhibitor, significantly enhanced NK cell-mediated killing in a manner likely independent of BCL-2 inhibition. This enhancement was not observed with other BCL-2 inhibitors or with BCL-2 knockdown, consistent with findings in other cell types.52 Further research into the involvement of BCL-2 and alternative targets of venetoclax will deepen our understanding of the effect of venetoclax on NK cell function and help clarify its mode of action.
In summary, our study proposes a potentially feasible approach for augmenting NK cell-based immunotherapies. By identifying the potential of venetoclax to enhance NK cell function, including boosting mitochondrial metabolism, IS formation, and cytotoxicity, we uncovered a promising strategy to improve cancer treatment outcomes. Given the FDA approval of venetoclax, its existing endorsement can expedite the translation of our research into clinical practice, leading to more effective and targeted cancer therapies. Further investigations and clinical trials are crucial to validate and expand upon our findings, ultimately advancing the field of cancer immunotherapy and improving patient outcomes.
Limitations of the study
Our study revealed that venetoclax enhances mitochondrial metabolism through NF-κB signaling, thereby promoting IS formation and ultimately enhancing the cytotoxicity of NK cells. However, the precise molecular mechanisms through which NF-κB enhances mitochondrial metabolism remain to be elucidated. Further studies are required to fully understand how NF-κB regulates mitochondrial metabolism in venetoclax-treated NK cells. Additionally, our findings demonstrate that venetoclax can improve the efficiency of NK cell-mediated clearance of AML cells in AML patient-derived xenograft models. Nonetheless, the antitumor efficacy of this combination therapy needs to be confirmed in clinical trials.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD45-FITC (HI30) | Biolegend | Cat# 304038; RRID: AB_2562050 |
| Anti-human CD45-BV510 (HI30) | Biolegend | Cat# 304036; RRID: AB_2561940 |
| Anti-human CD45-APC/Cy7 (HI30) | Biolegend | Cat# 304014; RRID: AB_314402 |
| Anti-human CD3-PE/Cy7 (HIT3a) | Biolegend | Cat# 300316; RRID: AB_314052 |
| Anti-human CD3-PerCP/Cy5.5 (OKT3) | Biolegend | Cat# 317336; RRID: AB_2561628 |
| Anti-human CD3-APC/Cy7 (SK7) | Biolegend | Cat# 344818; RRID: AB_10645474 |
| Anti-human CD3-FITC (UCHT1) | Biolegend | Cat# 300406; RRID: AB_314060 |
| Anti-human CD56-APC (5.1H11) | Biolegend | Cat# 362504; RRID: AB_2563913 |
| Anti-human CD56-APC/Cy7 (HCD56) | Biolegend | Cat# 318332; RRID: AB_10896424 |
| Anti-human CD56-BV421 (5.1H11) | Biolegend | Cat# 362552; RRID: AB_2566061 |
| Anti-human CD56-PE/Cy7 (5.1H11) | Biolegend | Cat# 362510; RRID: AB_2563927 |
| Anti-human CD56-BV650 (5.1H11) | Biolegend | Cat# 362532; RRID: AB_2565602 |
| Anti-human CD56-Alexa Fluor 488 (HCD56) | Biolegend | Cat# 318312; RRID: AB_604102 |
| Anti-human CD33-APC (WM53) | Biolegend | Cat# 303408; RRID: AB_314352 |
| Anti-human IFN-γ-FITC (B27) | Biolegend | Cat# 506504; RRID: AB_315437 |
| Anti-human IFN-γ-PerCP/Cy5.5 (B27) | Biolegend | Cat# 506528; RRID: AB_2566187 |
| Anti-human CD161-BV785 (HP-3G10) | Biolegend | Cat# 339930; RRID: AB_2563968 |
| Anti-human NgR2-APC/Cy7 (P14) | Novus Biologicals | Cat# NBP2-52676APCCY7; RRID: N/A |
| Anti-human CD39-APC (A1) | Biolegend | Cat# 328210; RRID: AB_1953234 |
| Anti-human IL-18 R beta-PE (132029) | R&D Systems | Cat# FAB118P; RRID: AB_2280188 |
| Anti-human-Bcl-2- PE (A1) | Biolegend | Cat# 658707; RRID: AB_2563281 |
| Anti-human Phospho-IKKα/β (Ser176/180)-Alexa Fluor488 (16A6) | Cell Signaling Technology | Cat# 92255; RRID: N/A |
| Anti-human Phospho-p65 (Ser536) (93H1) | Cell Signaling Technology | Cat# 3033; RRID: AB_331284 |
| Anti-human Bcl-xL-PE (54H6) | Cell Signaling Technology | Cat# 13835; RRID: AB_2798326 |
| Anti-human MCL-1-Alexa Flour 647 (D2W9E) | Cell Signaling Technology | Cat# 78471; RRID: AB_331284 |
| Donkey anti-rabbit IgG-Alexa Fluor488 (Poly4064) | Biolegend | Cat# 406416; RRID: AB_2563203 |
| Cy3 AffiniPure Donkey Anti- Rabbit IgG (H + L) | Jackson Immunoresearch | Cat# 715-165-150; RRID: AB_2340813 |
| Alexa Fluor 647 Donkey Anti-Rabbit IgG (H + L) | Jackson Immunoresearch | Cat# 711-605-152; RRID: AB_2492288 |
| Anti-mouse IgG (H + L), F(ab')2 Fragment-Alexa Fluor 488 | Cell Signaling Technology | Cat# 4408; RRID: AB_10694704 |
| Anti-human-γ-Tubulin | Sigma-Aldrich | Cat# T3559; RRID: AB_477575 |
| Actin-Tracker Red-555 | Beyotime | Cat# C2203S; RRID:N/A |
| APC Annexin V | Biolegend | Cat# 640941; RRID: AB_2616657 |
| FITC Annexin V | Biolegend | Cat# 640945; RRID: AB_2629519 |
| Anti-human Perforin-FITC (B-D48) | Biolegend | Cat# 353310; RRID: AB_2571967 |
| Anti-human Perforin-BV421 (dG9) | Biolegend | Cat# 308122; RRID: AB_2566204 |
| Anti-human CD107a-FITC (H4A3) | Biolegend | Cat# 328606; RRID: AB_1186036 |
| Anti-human CD107a-PE (H4A3) | BD Pharmingen | Cat# 555801; RRID: AB_396135 |
| Anti-human/mouse Granzyme B- Alexa Fluor 700 (QA16A02) | Biolegend | Cat# 372222; RRID: AB_2728389 |
| PerCP/Cy5.5 Mouse IgG1, κ Isotype Ctrl (MOPC-21) | Biolegend | Cat# 400150; RRID: AB_893664 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074; RRID: AB_2099233 |
| Anti-human α-Tubulin (acetyl K40) | Abcam | Cat# ab179484; RRID: AB_2890906 |
| Anti-human BCL-2 (D55G8) | Cell Signaling Technology | Cat# 4223; RRID: AB_1903909 |
| Biological samples | ||
| Fresh human cord blood mononuclear cells | The First Affiliated Hospital of USTC | N/A |
| Fresh AML bone marrow mononuclear cells | The First Affiliated Hospital of USTC | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| eBioscience™ Permeabilization Buffer (10×) | Thermo Fisher Scientific | Cat# 00-8333-56 |
| eBioscience™ Fixation/Permeabilization Diluent | Thermo Fisher Scientific | Cat# 00-5223-56 |
| eBioscience™ Fixation/Permeabilization Concentrate | Thermo Fisher Scientific | Cat# 00-5123-43 |
| CellTrace Far Red Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34564 |
| CellTrace Violet Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34557 |
| Mito-Tracker Red CMXRos | Beyotime | Cat# C1035 |
| Mito-Tracker Green | Beyotime | Cat# C1048 |
| MitoTracker Deep Red | Thermo Fisher Scientific | Cat# M22426 |
| CFSE | MedChemExpress | Cat# HY-D0056 |
| Annexin-V-Binding Buffer | Biolegend | Cat# 422201 |
| 7-AAD Viability Staining Solution | Biolegend | Cat# 420404 |
| Venetoclax | MedChemExpress | Cat# HY-15531 |
| A-1331852 | MedChemExpress | Cat# HY-19741 |
| Maritoclax | MedChemExpress | Cat# HY-15613 |
| Lisaftoclax | MedChemExpress | Cat# HY-129179 |
| 5-Azacytidine | MedChemExpress | Cat# HY-10586 |
| Decitabine | MedChemExpress | Cat# HY- A0004 |
| BAY11-7082 | MedChemExpress | Cat# HY-13453 |
| Pyrrolidinedithiocarbamate | MedChemExpress | Cat# HY-18738 |
| SN50 | MedChemExpress | Cat# HY-P0151 |
| Rotenone | MedChemExpress | Cat# HY-B1756 |
| Oligomycin A | MedChemExpress | Cat# HY-16589 |
| Concanavalin A | Sigma-Aldrich | Cat# C5275 |
| Poly-L-Lysine | Sigma-Aldrich | Cat# P4707 |
| Ficoll cell separation solution | TBDscience | Cat# LTS1077 |
| Recombinant human IL-12 | PeproTech | Cat# 210-12 |
| Recombinant human IL-15 | PeproTech | Cat# 210-15 |
| DAPI Staining Solution | Biosharp | Cat# BL105A |
| Penicillin Streptomycin Amphotericin | Biosharp | Cat# BL142A |
| IMDM culture medium | Biosharp | Cat# BL312A |
| DMEM culture medium | Viva Cell Biosciences | Cat# C3113-0500 |
| RPMI1640 | Biosharp | Cat# BL303A |
| Fetal Bovine Serum | Sigma-Aldrich | Cat# F7524 |
| QuickBlock™ Blocking Buffer for Immunol Staining | Beyotime | Cat# P0260 |
| D-Luciferin | Gold-Biotechnology | Cat# 115144-35-9 |
| MethoCult™ Classic | STEMCELL Technologies | Cat# H4434 |
| Wright-Giemsa stain | Biosharp | Cat# BL800A |
| ProLong™ Gold Antifade Mounting Medium | Thermo Fisher Scientific | Cat# P10144 |
| Cell Counting Kit-8 | MedChemExpress | Cat# HY-K0301 |
| 5×protein loading buffer | Servicebio | Cat# G2013 |
| PageRuler Prestained Protein Ladder | Thermo Fisher Scientific | Cat# 26616 |
| 20×TBST buffer | Solarbio | Cat# T1082 |
| Rapid Transfer Buffer (20×) | NCM Biotech | Cat# WB4600 |
| Universal Antibody Diluent | NCM Biotech | Cat# WB500D |
| ECL Western Blotting Substrate | NCM Biotech | Cat# P2200 |
| NP40 | Beyotime | Cat# P0013F |
| Polyethylenimine (PEI) | Polysciences | Cat# 23966 |
| Polybrene | Sigma-Aldrich | Cat# H9268 |
| MOPS buffer | Epizyme | Cat# PS120S |
| 4–12% gradient gel | Epizyme | Cat# LK308 |
| TRIzol® Reagent | Invitrogen | Cat# 15596018 |
| Critical commercial assays | ||
| NK Cell Isolation Kit human | Miltenyi Biotec | Cat# 130-092-657 |
| Dead Cell Removal Kit | Miltenyi Biotec | Cat# 130-090-101 |
| XF Cell Mito Stress Test Kit | Agilent | Cat# 103015-100 |
| HiScript III RT SuperMix for qPCR (+gDNA wiper) | Vazyme | Cat# R323-01 |
| ChamQ Universal SYBR qPCR Master Mix | Vazyme | Cat# Q711-02 |
| Deposited data | ||
| Single cell RNA-seq data | This paper | GEO: GSE237923 |
| Bulk RNA-seq data | This paper | GEO: GSE237922 |
| Original code | This paper | https://doi.org/10.5281/zenodo.10976537 |
| Experimental models: Cell lines | ||
| KG-1a | Laboratory of Changcheng Zheng | N/A |
| THP-1 | Laboratory of Changcheng Zheng | N/A |
| U937 | Laboratory of Changcheng Zheng | N/A |
| HEK293T | Laboratory of Changcheng Zheng | N/A |
| HL60-Luc | Laboratory of Haiming Wei | N/A |
| Experimental models: Organisms/strains | ||
| Mice: NOD-PrkdcscidIl2rgem1/Smoc | Shanghai Model Organisms Center | Cat# NM-NSG-001 |
| Oligonucleotides | ||
| BCL2L1 primer (human)-(qPCR) | This paper | F: GCAGCCGAGAGCCGAAAGG R: GAAGAGTGAGCCCAGCAGAACC |
| ENTPD1 primer (human)-(qPCR) | This paper | F: AGCCAGCAGAAGGAGTCTAACG R: CGCATCCAGCACAATCCCATAC |
| EPHB1 primer (human)-(qPCR) | This paper | F: GCGATGGCCCTGGATTATCTAC R: GCAGTAGCCGTTCTGGTGTC |
| GPC4 primer (human)-(qPCR) | This paper | F: AGTAGTGGAGAAGGAAGTGGAAG TG R: GCAGAAGACAGTGAGGAGGTAGG |
| ATP1B2 primer (human)-(qPCR) | This paper | F: GGTGGCTGTGAAGTTCCTGAATG R: CGATGTTGGCGGCGTTGATG |
| DKK3 primer (human)-(qPCR) | This paper | F: AAGGCAGAAGGAGCCACGAGTGC R: GGCCATTTTGGTGCAGTGACCCCA |
| RTN4RL2 primer (human)-(qPCR) | This paper | F: CCAGCACTCAGCGACTCTTCC R: ATGGTGGAGAGGTTGTTGGAGAAG |
| GAPDH primer (human)-(qPCR) | This paper | F: CCAGCAAGAGCACAAGAGGAA R: CAAGGGGTCTACATGGCAACT |
| BCL-2 primer (human)-(shRNA) | This paper | F: CCGGCCGGGAGATAGTGATGAAG TACTCGAGTACTTCATCACTATCTCC CGGTTTTTG R: AATTCAAAAACCGGGAGATAGTGA TGAAGTACTCGAGTACTTCATCACTA TCTCCCGG |
| Recombinant DNA | ||
| pLKO human shRNA1 BCL-2 | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism 8 | GraphPad | https://www.graphpad.com |
| Microsoft Excel | Microsoft Excel | https://www.microsoft.com |
| FlowJo software v10 | Flowjo, LLC | https://www.flowjo.com/solutions/flowjo |
| ImageJ | Fiji Image | https://imagej.nih.gov/ij/ |
| Cell Ranger (v3.1.0) | N/A | https://www.10xgenomics.com/cn/support/software/cell-ranger/latest |
| R (v4.1.2) | N/A | https://www.r-project.org/ |
| Python (v3.7.0) | N/A | N/A |
| LASX | Leica | N/A |
| MATLAB (v2018a) | N/A | https://www.mathworks.com |
| Seurat (v4.1.1) | N/A | https://satijalab.org/seurat/articles/install.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Fang Ni (fangni@ustc.edu.cn).
Materials availability
This study did not generate additional unique reagents.
Data and code availability
Bulk RNA-seq and single-cell RNA-seq data were deposited in GEO and are publicly available as of the date of publication. The accession numbers are listed in the key resources table. All original codes have been deposited at Zenodo (https://doi.org/10.5281/zenodo.10976537) and are publicly available on the date of publication. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Mice and housing conditions
Female 6-8-week-old M-NSG mice were purchased from Shanghai Model Organisms Center, Inc. All mice were housed under specific pathogen-free conditions and provided free access to food and water, in accordance with standard guidelines for animal care and husbandry. All experiments conducted on mice were performed in accordance with the National Guidelines for Animal Usage in Research (China) and approved by the Ethics Committee of the University of Science and Technology of China (USTC, Reference No. USTCACUC24120123020).
Cell lines
Human myeloid leukemia cell lines (KG-1a, THP-1, and U937) and HEK293T cells were generously gifted by Professor Changcheng Zheng (The First Affiliated Hospital of USTC). HL60-Luc cells were kindly provided by Professor Haiming Wei (USTC). THP-1, U937, HL60-Luc, KG-1a, and HEK293T cells were cultured in RPMI 1640 (Biosharp, Cat# BL303A), IMDM (Biosharp, Cat# BL312A) or DMEM (Viva Cell Biosciences, Cat# C3113-0500) supplemented with 10% FBS (Sigma, Cat# F7524) and 1× penicillin-streptomycin-amphotericin (Biosharp, Cat# BL142A). The cell lines were verified using short tandem repeat DNA fingerprinting. All cell lines tested negative for mycoplasma.
Human samples
Primary human AML (obtained from the bone marrow of AML patients) and cord blood (CB) samples were collected from the Department of Hematology and the Department of Obstetrics and Gynecology of the First Affiliated Hospital of USTC, respectively. The Review Committee of the Ethics Institution of the First Affiliated Hospital of USTC approved the collection and use of human tissues for this study (2022-KY-089; 2022-KY-022). The newly diagnosed human primary AML specimens used for the PDX model and NK cell cytotoxicity assays were obtained from patients who had not undergone any chemoradiotherapy at the time of specimen collection. Table S1 provides information on the patients with AML.
Method details
Human NK cell isolation and culture
Human NK cells were isolated from umbilical cord blood obtained from healthy donors or bone marrow obtained from patients with AML at the First Affiliated Hospital of the USTC. CB-NK cells were enriched by negative selection using a human NK Cell Isolation Kit (Miltenyi Biotec, Cat# 130-092-657) according to the manufacturer’s instructions. AML-NK cells were purified by flow sorting (FACSAria III, BD Biosciences) based on the DAPI−CD45highCD3−CD56+ signature. The isolated NK cells (purity >95%) were cultured in IMDM medium supplemented with 1× penicillin-streptomycin-amphotericin and 10% certified heat-inactivated FBS (IMDM complete medium). NK cells were activated using IL-12 (10 ng/mL, PeproTech, Cat# 210-12) and IL-15 (50 ng/mL, PeproTech, Cat#210-15). “Unstimulated” represents a resting state without cytokine activation. For venetoclax (MCE, Cat# HY-15531) treatment, 200–1000 nM venetoclax was added to NK cell culture 18–48 h before use. For pretreatment with the NF-κB inhibitors SN50 (25 μg/mL, MCE, Cat # HY-P0151), BAY 11–7082 (1 μM, MCE, Cat# HY-13453), and PDTC (5 μM, MCE, Cat# HY-18738), CB-NK cells were centrifuged and then resuspended in IMDM complete medium plus NF-κB inhibitors for 4 h, followed by 400 nM venetoclax for 18 h. For pretreatment with metabolism inhibitors, CB-NK cells were resuspended in IMDM complete medium plus Oligomycin A (200 nM, MCE, Cat# HY-16589) or rotenone (200 nM, MCE, Cat# HY-B1756) combined with venetoclax (400 nM) for 18 h. All cells were cultured in a humidified incubator (37°C, 5% CO2). For pretreatment with the BCL-xL inhibitor (A-1331852, MCE, Cat# HY-19741), MCL-1 inhibitor (Maritoclax, MCE, Cat# HY-15613). CB-NK cells were resuspended in IMDM complete medium and treated with the corresponding inhibitors for 18 h. Prior to co-culture experiments with AML cells, NK cells were washed twice with PBS and counted before each experiment to ensure the removal of residual venetoclax, thereby eliminating the possibility of venetoclax-induced apoptosis in AML cells.
Flow cytometry
The antibodies used in this study are listed in the key resources table. NK cells were labeled with conjugated antibodies and analyzed by flow cytometry. IgG isotype controls were used for all the staining procedures. Surface staining was performed for 30 min at 4°C. For intracellular cytokine staining, NK cells were stimulated with 10 ng/mL IL-12 and 50 ng/mL IL-15 for 18 h. Following stimulation, the cells were stained for surface markers, fixed, and permeabilized for intracellular staining of IFN-γ, granzyme B, perforin, and p-IKKα. To evaluate NK cell degranulation, NK cells were incubated with target cells at a 2.5:1 ratio for 4 h with anti-CD107a antibody added at the start of the assay. For intracellular staining of p-p65, IL-12 and IL-15 pre-activated NK cells were fixed and permeabilized after surface marker staining and stained with anti-p-p65 primary antibodies, followed by an Alexa-Fluor-647-labeled secondary antibody. Flow cytometry was performed on a Beckman Coulter CytoFLEX flow cytometer, and the data were analyzed using FlowJo software. The cell gating strategies used in flow cytometry analysis and cell sorting were shown in Figure S7.
NK cell cytotoxicity assays
CFSE (5 μM, MCE, Cat# HY-D0056)-or CellTrace Violet Cell Proliferation Kit (5 μM, Thermo Fisher Scientific, Cat# C34557)-labeled target cells (KG-1a and THP-1 cell lines) were co-cultured with NK cells at various effector-to-target (E: T) ratios in 96-well plates. After a 4-h-co-culture, the cells were stained with Annexin V/7-AAD and analyzed by flow cytometry. In the primary AML cell experiments, primary AML cells were mixed with NK cells at a 2.5:1 ratio in 96-well plates. Primary AML cells were labeled using a CellTrace Violet Cell Proliferation Kit (5 μM, Cat# C34557, Thermo Fisher Scientific). After incubation, the cells were stained with anti-human antibodies (CD45, CD3, and CD33) at room temperature (RT) in the dark for 20 min and washed once with PBS. The cells were then stained with Annexin V/7-AAD and analyzed by flow cytometry. Specific killing (%) was calculated using the following formula: % specific killing = (% Annexin-V+ with NK cells - % Annexin-V+ without NK cells)/(100% - % Annexin-V+ without NK cells) ×100.
Colony formation assay
The clonogenic growth of AML cells exposed to NK cells was evaluated by co-culturing AML cell lines (KG-1a, THP-1, HL60-Luc, and U937) or primary AML cells from AML patients with venetoclax-treated or untreated NK cells for 4 h, at a ratio of 2.5:1. Subsequently, the cells were resuspended in MethoCult H4434 medium (StemCell Technologies, Cat# H4434) and plated in duplicate into 6-well plates. The plates were incubated for 14 days at 37°C and 5% CO2. Colonies were counted under a microscope, and a cluster of ≥30 cells was considered a colony. No CB-NK cells were added in the “Untreated” condition.
Lentiviral vector construction
The BCL-2-targeting shRNA (5′- CCGGGAGATAGTGATGAAGTA -3′) was cloned into the lentiviral vector (pLKO.1) containing an EGFP reporter gene. As a control, a pLKO.1 vector containing a scrambled shRNA sequence was prepared. Lentiviral particles were produced in 293T cells co-transfected with the constructed pLKO.1 vector, the packaging plasmid ps.pAX2, and the envelope plasmid pMD.2G. Transfection was performed using Polyethylenimine (PEI, Polyscience, Cat# 23966) according to the manufacturer’s instructions. Viral supernatants were harvested at 48 and 72 h post-transfection and stored at −80°C for future use.
Transduction of CB-NK cells
Purified CB-NK cells were cultured in preactivation medium (IMDM supplemented with 10% FBS, 10 ng/mL IL-12, and 10 ng/mL IL-15). After 24 h, these preactivated CB-NK cells were transduced with lentiviruses carrying either BCL-2-specific shRNA or a control sequence. This process involved centrifugation at 1700 rpm for 90 min at 37°C to facilitate viral entry. To enhance transduction efficiency, the medium was enriched with the previously mentioned cytokines and polybrene (8 μg/mL, Sigma-Aldrich, Cat# H9268). After 8 h, the cytokine-enriched medium was refreshed and EGFP+ NK cells were isolated 48 h after transduction by flow cytometric sorting (CytoFLEX SRT, Beckman).
Western blot
EGFP+ CB-NK cells were lysed with NP-40 lysis buffer (Beyotime, Cat# P0013F) containing a protease inhibitor cocktail (Sigma-Aldrich, Cat # 539138) on ice for 30 min. Lysates were then centrifuged at 12000 g for 15 min at 4°C to collect the supernatant. Proteins from the supernatant were separated on a 4–12% gradient SDS‒PAGE gel (EpiZyme, Cat# LK308) and transferred to PVDF membranes (Millipore, Cat# IPVH00010). The membranes were blocked with 5% fat-free milk for 30 min at room temperature (RT), followed by overnight incubation at 4°C with primary antibodies against BCL-2 (Cell Signaling Technology, Cat# 4223, 1:1000) and α-Tubulin (Abcam, Cat# ab179484, 1:1000). The membranes were then incubated with an HRP-conjugated anti-rabbit secondary antibody (Cell Signaling Technology, Cat# 7074,1:1000 dilution) for 1 h at RT. Protein bands were visualized using an enhanced chemiluminescence substrate (NCM Biotech, Cat# P2200).
Cell proliferation assay
The proliferation of CB-NK cells was determined using a Cell Counting Kit-8 (CCK-8, MCE, Cat# HY-K0301) according to the manufacturer’s manual. Briefly, 1 × 105 NK cells were seeded into each well of a 96-well plate. At 0, 2, 3, and 5 days post-culture, 100 μL of fresh IMDM (10% FBS, 10 ng/mL IL-12, and 10 ng/mL IL-15) combined with 10 μL of CCK-8 solution was added to each well. The plates were incubated for 2 h at 37°C in the dark. Subsequently, the absorbance was measured at 450 nm to determine cell proliferation.
Wright–Giemsa staining
Cell morphology was assessed using Wright–Giemsa staining, which stains the nucleus purple and the cytoplasm blue. In brief, cells were harvested, resuspended in PBS, and evenly spread onto a glass slide, which was subsequently stained with Wright–Giemsa staining solution following the manufacturer’s instructions. The stained glass slides were observed using a fluorescence inverted microscope system (Leica).
Xenograft mouse models
KG-1a cells (2×106), primary AML cells from the bone marrow of patients with AML (3×106), or HL60-Luc cells (5×106) were transferred to female 6-8-week-old M-NSG mice. Eight hours before the cancer cells were injected via the tail vein to establish a leukemia xenograft model, the mice were subjected to sublethal irradiation (2.5 Gy). After tumor formation, the mice were randomly assigned to three groups and treated with PBS, 5×106 NK cells or venetoclax-pretreated NK cells/mouse. AML burden was monitored by bioluminescence imaging using the IVIS Spectrum Imaging System (PerkinElmer) or detected by flow cytometry at the indicated time points. In all experiments, rhIL-15 (1 μg/mouse, PeproTech, Cat# 210-15) was administered intraperitoneally to mice every 3 days after NK infusion until euthanization.
Bioluminescence imaging
Mice received an intraperitoneal injection of D-luciferin potassium salt substrate (150 mg/kg, Gold Biotechnology, Cat#LUCK-1G). Images were acquired 12 min later using the IVIS Spectrum Imaging System (PerkinElmer), and the signal output was measured under anesthesia. The Living Image v 4.7.3 in vivo software package (PerkinElmer Inc.) was used to quantify signal intensity. The bioluminescent signal was expressed as radiance photons/sec/cm2/steradian (p/s/cm2/sr), and pseudocolor images were used to depict the signal intensities.
Mitochondrial membrane potential (MMP) and mitochondrial mass measurement
To evaluate the MMP in NK cells, 5×105 freshly isolated umbilical cord blood mononuclear cells (CBMCs) were treated with 100 nM MitoTracker Red CMXRos (Beyotime, Cat# C1035) at 37°C for 40 min in the dark. The CBMCs were then diluted in ice-cold PBS. Surface staining of NK cells was then performed for 20 min at RT, and the MitoTracker Red CMXRos fluorescence signal was promptly measured using a Beckman Coulter CytoFLEX instrument.
To evaluate the mitochondrial mass of NK cells, 5×105 freshly isolated CBMCs were treated with 100 nM MitoTracker Green (Beyotime, Cat# C1048) and incubated at 37°C for 40 min in the dark. After a single wash with ice-cold PBS, surface staining for NK cells was performed for 20 min at RT, and MitoTracker Green fluorescence was immediately measured using the Beckman Coulter CytoFLEX instrument.
Avidity assessment
The microfluidic chips were coated with concanavalin A (5 mg/mL, Cat# C5275, Sigma-Aldrich) and dehydrated in a dry incubator. After chip rehydration with warm IMDM, KG-1a cells were seeded onto the chips at a density of 108 cells/ml. Continuous microscopic visualization was performed to ensure the absence of bubble formation and to ensure adequate cell density. After an initial incubation period of 2 h, the cells were provided with fresh medium and incubated for another hour. The effectors were stained using the CellTrace Far Red Cell Proliferation Kit (Thermo Fisher Scientific, Cat# C34564), according to the manufacturer’s instructions. Subsequently, the cells were seeded at a density of 106 cells/ml and incubated for 10 min before force was applied using a z-Movi cell avidity analyzer to detect effector detachment.
Conjugation assay
NK cell-to-target cell conjugation was evaluated, as previously described.53 Briefly, CBMCs were isolated from CB by Ficoll cell separation solution (TBDscience, Cat# LTS1077) density gradient centrifugation at RT. NK cells were enriched by negative selection using an NK Cell Isolation Kit according to the manufacturer’s instructions. After isolation, NK cells were labeled for 20 min using a CellTrace Violet Cell Proliferation Kit (5 μM, Thermo Fisher Scientific, Cat# C34557), according to the manufacturer’s instructions. KG-1a target cells were labeled for 20 min at 37°C with CFSE. After labeling, the cells were washed with PBS, mixed at a 1:4 E: T ratio, and centrifuged at 300 g for 1 s. The cells were co-cultured for 1 h at 37°C and the conjugates were quantified using flow cytometry.
RNA sequencing (RNA-seq) and data analysis
NK cells were enriched from CB by negative selection, using a human NK Cell Isolation Kit, to a purity of >95%. After venetoclax treatment, total cellular RNA was isolated using TRIzol (Invitrogen, Cat# 15596018) and the library was generated using the VAHTS Universal V8 RNA-seq Library Prep Kit. The library was sequenced in the single-read mode on an Illumina NovaSeq 6000 sequencing platform. Raw RNA-seq reads were mapped to the human GRCh38 assembly reference genome using the HISAT2 alignment tool (v2.0.1). Heatmaps were produced using unsupervised clustering, whereas differential gene expression analyses were conducted using edgeR (v3.28.1) with a log2-fold change (FC) of 1 and a p-value threshold of <0.05, indicating statistical significance. For Gene Ontology (GO) and pathway analysis of DEGs, the DEGs were uploaded to the Metascape website, and default parameters were used to perform GO and pathway analysis of the gene list. The list of the DEGs and enriched signaling pathways are provided in Tables S2 and S3.
Single-cell RNA-seq (scRNA-seq)
For scRNA-seq analysis of CB-NK cells, NK cells were isolated using the human NK Cell Isolation Kit to a purity of >95%. The scRNA-seq was performed on purified NK cells treated with or without venetoclax, derived from four distinct samples. To optimize sequencing efficiency and reduce batch effects, we pooled the four control samples into one group and the four venetoclax-treated samples into another prior to library preparation. Following 10× Genomics Single-Cell 3′ Reagent Kits (v3.1) guidelines, these pooled cells were counted and resuspended at a concentration of 1×106 cells/ml to target approximately 6,000 cells per library for sequencing. The construction of single-cell libraries strictly followed standard protocols provided by the manufacturer. Libraries were sequenced using the Illumina NovaSeq 6000 system.
scRNA-seq data analysis
Raw data (FASTQ files) for each sample were matched to the hg19 reference genome. UMI counts were quantified using the 10× Genomics Cell Ranger pipeline (v3.1.0)26 with the default settings. Quality control and dimensionality reduction analyses were conducted using R version 4.1.2 and the Seurat package version 4.1.2.54 Seurat objects for the venetoclax-treated and control groups were then created before filtering the samples using the following criteria: unique RNA feature counts of 500–3,000 and <10% mitochondrial genes. Genes that were expressed in <5 cells were excluded from analysis. The "IntegrateData" function was used to integrate the two datasets and then returned a new integrated Seurat object. The “RunPCA” function was used to compute the top 20 principal components (PCs) of the 2000 highly variably expressed genes. Cell clustering was performed using the “FindClusters” function with a resolution of 0.18. Cell clusters were visualized using uniform manifold approximation and projection (UMAP). To analyze the differential gene expression in our identified cell subpopulations, the “FindConservedMarkers” function of Seurat, which utilizes the Wilcoxon rank-sum test, was employed. In addition, differential gene expression analysis between the venetoclax-treated and untreated groups was performed using the “FindMarkers” function of Seurat, with multiple thresholds (p-value <0.001, |logFC| > 0.4 and min. pct >0.05).
For GO analysis of DEGs, the DEGs were uploaded to the Metascape website (http://metascape.org/gp/index.html) and GO analysis was performed using default settings. The gene sets were downloaded from MsigDB or published signatures55 and the signature score for each cell was calculated using the Seurat function "AddModuleScore”. For GSEA, normalized enrichment scores (NES) and p-values were calculated using the R package GSEA Base (v. 1.56.0). All scRNA-seq data were provided in Table S4. Marker genes of the five single-cell clusters, related to Figure 4, Table S5. DEGs in C3 subset cells (venetoclax vs. control), related to Figure 5, Table S6. List of signature/pathway genes used in this study, related to Figures 5, 6, S4, and S5, Table S7. DEGs in total NK cells (venetoclax vs. control), related to Figure 5.
Real-time quantitative PCR
Total RNA was isolated using the TRIzol Reagent (Invitrogen, Cat# 15596018). RNA was then reverse-transcribed using HiScript III RT SuperMix (Vazyme, Cat# R323-01) for RT-PCR. The reverse transcription products were subjected to real-time quantitative PCR using ChamQ Universal SYBR qPCR Master Mix (Vazyme, Cat# Q711-02). GAPDH mRNA level was used as an internal normalization control. The target gene primers are listed in the key resources table.
Oxygen consumption rate (OCR) and mitochondrial ATP measurement
Cells were seeded at a density of 2×105 cells per well on poly-L-lysine (Sigma-Aldrich, Cat# P4707)-coated XF96-well cell culture plates. OCR and real-time ATP rate assays were performed independently in a Seahorse XF Analyzer (Agilent Technologies), according to the manufacturer’s recommendations. Briefly, NK cells were incubated with Seahorse XF basal medium supplemented with 25 mM glucose, 2 mM glutamine, and 1 mM sodium pyruvate (adjusted to pH 7.4). Cells were incubated for 1 h in a non-CO2 incubator before OCR or ATP test compounds were serially injected. OCR was measured under basal conditions and after the addition of 2 μM Oligomycin A, 1.5 μM trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP), or 0.5 μM Antimycin & Rotenone, as indicated. The Seahorse XF Real-Time ATP Rate Assay kit (Agilent Technologies) was used to measure the total ATP and ATP fractions produced by mitochondrial OXPHOS or glycolysis. In the ATP Real-Time Rate Assay, the cells were treated with 2 μM Oligomycin A and 0.5 μM Antimycin & Rotenone (all drugs were purchased from Agilent Technologies).
Immunofluorescence and image acquisition
NK cells were fixed with 4% paraformaldehyde for 20 min and then blocked with QuickBlock Blocking Buffer for Immunol Staining (Beyotime, Cat# P0260). For p-p65 staining, NK cells were incubated with an anti-p-p65 primary antibody (1:800, Cell Signaling Technology, Cat# 3033) overnight at 4°C, followed by an Alexa Fluor 647-labeled secondary antibody (1:200) at RT for 1 h. For mitochondrial staining, live cells were incubated for 40 min at 37°C with MitoTracker Red CMXRos (100 nM, Beyotime, Cat# C1035) and MitoTracker Green (100 nM, Beyotime, Cat# C1048) in Hoechst 33342 staining solution (Beyotime, Cat# C1027) in the dark, and then fixed with 4% paraformaldehyde for 20 min. For F-actin staining, 4% paraformaldehyde-fixed cells were washed twice with PBS and then incubated with Alexa Fluor 555 phalloidin (1:200, Beyotime, Cat# C2203S) for 1 h at RT.
For the lytic granule polarization assay, NK cells were mixed with KG-1a cells at a ratio of 1:1 and seeded on poly-L-lysine (Sigma-Aldrich, Cat# P4707)-coated coverslips. After fixation with 4% paraformaldehyde, the cells were blocked with QuickBlock Blocking Buffer for Immunol Staining for 30 min, followed by overnight incubation in blocking buffer containing anti-perforin (BioLegend, diluted 1:100) and anti-γ-tubulin antibodies (Sigma, diluted 1:200) at 4°C. Subsequently, the cells were incubated with Alexa Fluor-488-conjugated anti-mouse antibody (1:800), Alexa Fluor-647-conjugated anti-rabbit antibody (1:800), and Alexa Fluor 555 phalloidin (1:200) for 1 h at RT. DAPI (Biosharp, Cat# BL105A) was used for nuclear staining. For the mitochondrial polarization assay, NK cells previously labeled with MitoTracker Deep Red (100 nM, Thermo Fisher Scientific, Cat# M22426) were cultured with KG-1a cells for 30 min at a ratio of 1:1 and seeded on poly L-lysine-coated coverslips. After fixation with 4% paraformaldehyde, cells were blocked with QuickBlock Blocking Buffer for Immunol Staining for 30 min, followed by incubation in blocking buffer containing Alexa Fluor 555 phalloidin (1:200) for 1 h at RT. DAPI was used for nuclear staining. After washing, the cells were mounted on slides and imaged using a confocal laser-scanning microscope fitted with a 63× objective lens (Leica). Z-stacks (0.5 μm increments, 10 μm total) of Alexa 555, Alexa 647, or Alexa 488 were collected for each NK cell-target cell conjugate. Quantification of perforin or mitochondrial polarization was performed using projected images derived from z stack data. The NK cell area opposite the immune synapse was divided into five segments, with polarization indices of 0.2, 0.4, 0.6, 0.8, and 1.0 assigned by their respective distances from the synapse. The intensity index quantifies the ratio of perforin or mitochondria distribution relative to that of the immune synapse across these polarization indices. The code used to quantify the polarization distance between perforin and mitochondria was uploaded to GitHub.
Quantification and statistical analysis
GraphPad Prism 8 (GraphPad Software) was used for statistical analysis. Either the Student’s t-test or the Mann‒Whitney test was applied for comparisons between two groups. One-way ANOVA was used for the comparison of more than two groups. Survival analysis was conducted using the log rank (Mantel–Cox) test. Data were presented as mean ± SD, unless indicated in the figure legends.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2019YFA0801800 to F.N.), the Natural Science Foundation of China (32070916 and 82370159 to F.N.), the Anhui Natural Science Foundation for Distinguished Young Scholars (2108085J38 to F.N.), Research Funds of the Center for Advanced Interdisciplinary Science and Biomedicine of IHM (QYPY20220007to F.N.), and Fundamental Research Funds for the University of Science and Technology of China (WK9110000003 to C.Z.). We also thank the Joint Laboratory of Innovation in Life Sciences from the University of Science and Technology of China (USTC) and Changchun Zhuoyi Biological Co., Ltd.
Author contributions
F.N., H.W., and C.Z. conceived and conducted the project; F.N. and Y.W. designed the experiments; Y.W. performed most of the experiments and analyzed the data; B.H., T.L., and Y.W. analyzed the bulk and scRNA-seq data; Y.W. contributed to mouse models and cell culture; L.J., C.Z., X.S., N.Z., and Y.W. collected human samples and clinical information; H.W., M.W., X.L., and M.Z. provided comments and suggestions; Y.W. and F.N. wrote the manuscript; and F.N. supervised the project.
Declaration of interests
The authors declare no competing interests.
Published: May 21, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101580.
Contributor Information
Haiming Wei, Email: ustcwhm@ustc.edu.cn.
Changcheng Zheng, Email: zhengchch1123@ustc.edu.cn.
Fang Ni, Email: fangni@ustc.edu.cn.
Supplemental information
References
- 1.Labanieh L., Mackall C.L. CAR immune cells: design principles, resistance and the next generation. Nature. 2023;614:635–648. doi: 10.1038/s41586-023-05707-3. [DOI] [PubMed] [Google Scholar]
- 2.Maskalenko N.A., Zhigarev D., Campbell K.S. Harnessing natural killer cells for cancer immunotherapy: dispatching the first responders. Nat. Rev. Drug Discov. 2022;21:559–577. doi: 10.1038/s41573-022-00413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y., Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020;17:807–821. doi: 10.1038/s41423-020-0488-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimasaki N., Jain A., Campana D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020;19:200–218. doi: 10.1038/s41573-019-0052-1. [DOI] [PubMed] [Google Scholar]
- 5.Myers J.A., Miller J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021;18:85–100. doi: 10.1038/s41571-020-0426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu S., Galat V., Galat Y., Lee Y.K.A., Wainwright D., Wu J. NK cell-based cancer immunotherapy: from basic biology to clinical development. J. Hematol. Oncol. 2021;14:7. doi: 10.1186/s13045-020-01014-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamers-Kok N., Panella D., Georgoudaki A.M., Liu H., Özkazanc D., Kučerová L., Duru A.D., Spanholtz J., Raimo M. Natural killer cells in clinical development as non-engineered, engineered, and combination therapies. J. Hematol. Oncol. 2022;15:164. doi: 10.1186/s13045-022-01382-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laskowski T.J., Biederstädt A., Rezvani K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer. 2022;22:557–575. doi: 10.1038/s41568-022-00491-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee J.B., Khan D.H., Hurren R., Xu M., Na Y., Kang H., Mirali S., Wang X., Gronda M., Jitkova Y., et al. Venetoclax enhances T cell-mediated antileukemic activity by increasing ROS production. Blood. 2021;138:234–245. doi: 10.1182/blood.2020009081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leverson J.D., Sampath D., Souers A.J., Rosenberg S.H., Fairbrother W.J., Amiot M., Konopleva M., Letai A. Found in Translation: How Preclinical Research Is Guiding the Clinical Development of the BCL2-Selective Inhibitor Venetoclax. Cancer Discov. 2017;7:1376–1393. doi: 10.1158/2159-8290.CD-17-0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pollyea D.A., Stevens B.M., Jones C.L., Winters A., Pei S., Minhajuddin M., D'Alessandro A., Culp-Hill R., Riemondy K.A., Gillen A.E., et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018;24:1859–1866. doi: 10.1038/s41591-018-0233-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiNardo C.D., Pratz K.W., Letai A., Jonas B.A., Wei A.H., Thirman M., Arellano M., Frattini M.G., Kantarjian H., Popovic R., et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018;19:216–228. doi: 10.1016/s1470-2045(18)30010-x. [DOI] [PubMed] [Google Scholar]
- 13.Wei A.H., Montesinos P., Ivanov V., DiNardo C.D., Novak J., Laribi K., Kim I., Stevens D.A., Fiedler W., Pagoni M., et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo-controlled trial. Blood. 2020;135:2137–2145. doi: 10.1182/blood.2020004856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eichhorst B., Niemann C.U., Kater A.P., Fürstenau M., von Tresckow J., Zhang C., Robrecht S., Gregor M., Juliusson G., Thornton P., et al. First-Line Venetoclax Combinations in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2023;388:1739–1754. doi: 10.1056/NEJMoa2213093. [DOI] [PubMed] [Google Scholar]
- 15.Bhansali R.S., Pratz K.W., Lai C. Recent advances in targeted therapies in acute myeloid leukemia. J. Hematol. Oncol. 2023;16:29. doi: 10.1186/s13045-023-01424-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaweme N.M., Zhou F. Optimizing NK Cell-Based Immunotherapy in Myeloid Leukemia: Abrogating an Immunosuppressive Microenvironment. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.683381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu J., Niu T. Natural killer cell-based immunotherapy for acute myeloid leukemia. J. Hematol. Oncol. 2020;13:167. doi: 10.1186/s13045-020-00996-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan R., Ryan J., Pan D., Wucherpfennig K.W., Letai A. Augmenting NK cell-based immunotherapy by targeting mitochondrial apoptosis. Cell. 2022;185:1521–1538.e18. doi: 10.1016/j.cell.2022.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng X., Hou Z., Qian Y., Zhang Y., Cui Q., Wang X., Shen Y., Liu Z., Zhou Y., Fu B., et al. Tumors evade immune cytotoxicity by altering the surface topology of NK cells. Nat. Immunol. 2023;24:802–813. doi: 10.1038/s41590-023-01462-9. [DOI] [PubMed] [Google Scholar]
- 21.Mukherjee M., Mace E.M., Carisey A.F., Ahmed N., Orange J.S. Quantitative Imaging Approaches to Study the CAR Immunological Synapse. Mol. Ther. 2017;25:1757–1768. doi: 10.1016/j.ymthe.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phatarpekar P.V., Overlee B.L., Leehan A., Wilton K.M., Ham H., Billadeau D.D. The septin cytoskeleton regulates natural killer cell lytic granule release. J. Cell Biol. 2020;219 doi: 10.1083/jcb.202002145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lagrue K., Carisey A., Morgan D.J., Chopra R., Davis D.M. Lenalidomide augments actin remodeling and lowers NK-cell activation thresholds. Blood. 2015;126:50–60. doi: 10.1182/blood-2015-01-625004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang C., Siebert J.R., Burns R., Gerbec Z.J., Bonacci B., Rymaszewski A., Rau M., Riese M.J., Rao S., Carlson K.S., et al. Heterogeneity of human bone marrow and blood natural killer cells defined by single-cell transcriptome. Nat. Commun. 2019;10:3931. doi: 10.1038/s41467-019-11947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Freud A.G., Mundy-Bosse B.L., Yu J., Caligiuri M.A. The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity. 2017;47:820–833. doi: 10.1016/j.immuni.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8 doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaplanski G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol. Rev. 2018;281:138–153. doi: 10.1111/imr.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen C., Edelstein L.C., Gélinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L) Mol. Cell Biol. 2000;20:2687–2695. doi: 10.1128/MCB.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J.G., Islam R., Cho J.Y., Jeong H., Cap K.C., Park Y., Hossain A.J., Park J.B. Regulation of RhoA GTPase and various transcription factors in the RhoA pathway. J. Cell. Physiol. 2018;233:6381–6392. doi: 10.1002/jcp.26487. [DOI] [PubMed] [Google Scholar]
- 31.Abarca-Rojano E., Muñiz-Hernández S., Moreno-Altamirano M.M.B., Mondragón-Flores R., Enriquez-Rincón F., Sánchez-García F.J. Re-organization of mitochondria at the NK cell immune synapse. Immunol. Lett. 2009;122:18–25. doi: 10.1016/j.imlet.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 32.Petroni G., Buqué A., Zitvogel L., Kroemer G., Galluzzi L. Immunomodulation by targeted anticancer agents. Cancer Cell. 2021;39:310–345. doi: 10.1016/j.ccell.2020.11.009. [DOI] [PubMed] [Google Scholar]
- 33.Lizotte P.H., Hong R.L., Luster T.A., Cavanaugh M.E., Taus L.J., Wang S., Dhaneshwar A., Mayman N., Yang A., Kulkarni M., et al. A High-Throughput Immune-Oncology Screen Identifies EGFR Inhibitors as Potent Enhancers of Antigen-Specific Cytotoxic T-lymphocyte Tumor Cell Killing. Cancer Immunol. Res. 2018;6:1511–1523. doi: 10.1158/2326-6066.CIR-18-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parameswaran R., Ramakrishnan P., Moreton S.A., Xia Z., Hou Y., Lee D.A., Gupta K., deLima M., Beck R.C., Wald D.N. Repression of GSK3 restores NK cell cytotoxicity in AML patients. Nat. Commun. 2016;7 doi: 10.1038/ncomms11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee Y.G., Guruprasad P., Ghilardi G., Pajarillo R., Sauter C.T., Patel R., Ballard H.J., Hong S.J., Chun I., Yang N., et al. Modulation of BCL-2 in Both T Cells and Tumor Cells to Enhance Chimeric Antigen Receptor T-cell Immunotherapy against Cancer. Cancer Discov. 2022;12:2372–2391. doi: 10.1158/2159-8290.CD-21-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu H.Y., Li K.X., Pan W.Y., Guo M.Q., Qiu D.Z., He Y.J., Li Y.H., Huang Y.X. Venetoclax enhances NK cell killing sensitivity of AML cells through the NKG2D/NKG2DL activation pathway. Int. Immunopharmacol. 2022;104 doi: 10.1016/j.intimp.2021.108497. [DOI] [PubMed] [Google Scholar]
- 37.Arandjelovic P., Kim Y., Cooney J.P., Preston S.P., Doerflinger M., McMahon J.H., Garner S.E., Zerbato J.M., Roche M., Tumpach C., et al. Venetoclax, alone and in combination with the BH3 mimetic S63845, depletes HIV-1 latently infected cells and delays rebound in humanized mice. Cell Rep. Med. 2023;4 doi: 10.1016/j.xcrm.2023.101178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chandrasekar A.P., Cummins N.W., Natesampillai S., Misra A., Alto A., Laird G., Badley A.D. The BCL-2 Inhibitor Venetoclax Augments Immune Effector Function Mediated by Fas Ligand, TRAIL, and Perforin/Granzyme B, Resulting in Reduced Plasma Viremia and Decreased HIV Reservoir Size during Acute HIV Infection in a Humanized Mouse Model. J. Virol. 2022;96 doi: 10.1128/jvi.01730-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rak G.D., Mace E.M., Banerjee P.P., Svitkina T., Orange J.S. Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse. PLoS Biol. 2011;9 doi: 10.1371/journal.pbio.1001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong D.C.P., Ding J.L. The mechanobiology of NK cells- 'Forcing NK to Sense' target cells. Biochim. Biophys. Acta. Rev. Cancer. 2023;1878 doi: 10.1016/j.bbcan.2023.188860. [DOI] [PubMed] [Google Scholar]
- 41.Greenman R., Pizem Y., Haus-Cohen M., Goor A., Horev G., Denkberg G., Sinik K., Elbaz Y., Bronner V., Levin A.G., et al. Shaping Functional Avidity of CAR T Cells: Affinity, Avidity, and Antigen Density That Regulate Response. Mol. Cancer Ther. 2021;20:872–884. doi: 10.1158/1535-7163.MCT-19-1109. [DOI] [PubMed] [Google Scholar]
- 42.Leick M.B., Silva H., Scarfo I., Larson R., Choi B.D., Bouffard A.A., Gallagher K., Schmidts A., Bailey S.R., Kann M.C., et al. Non-cleavable hinge enhances avidity and expansion of CAR-T cells for acute myeloid leukemia. Cancer Cell. 2022;40:494–508.e495. doi: 10.1016/j.ccell.2022.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chockley P.J., Ibanez-Vega J., Krenciute G., Talbot L.J., Gottschalk S. Synapse-tuned CARs enhance immune cell anti-tumor activity. Nat. Biotechnol. 2023;41:1434–1445. doi: 10.1038/s41587-022-01650-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kyrysyuk O., Wucherpfennig K.W. Designing Cancer Immunotherapies That Engage T Cells and NK Cells. Annu. Rev. Immunol. 2023;41:17–38. doi: 10.1146/annurev-immunol-101921-044122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi C., Finlay D.K. Optimising NK cell metabolism to increase the efficacy of cancer immunotherapy. Stem Cell Res. Ther. 2021;12:320. doi: 10.1186/s13287-021-02377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pearce E.L., Pearce E.J. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Teh C.E., Peng H., Luo M.X., Tan T., Trussart M., Howson L.J., Chua C.C., Muttiah C., Brown F., Ritchie M.E., et al. Venetoclax treatment in patients with cancer has limited impact on circulating T and NK cells. Blood Adv. 2023;7:2733–2745. doi: 10.1182/bloodadvances.2022008221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kohlhapp F.J., Haribhai D., Mathew R., Duggan R., Ellis P.A., Wang R., Lasater E.A., Shi Y., Dave N., Riehm J.J., et al. Venetoclax Increases Intratumoral Effector T Cells and Antitumor Efficacy in Combination with Immune Checkpoint Blockade. Cancer Discov. 2021;11:68–79. doi: 10.1158/2159-8290.CD-19-0759. [DOI] [PubMed] [Google Scholar]
- 49.Dimitrakopoulos F.I.D., Kottorou A.E., Kalofonou M., Kalofonos H.P. The Fire Within: NF-κB Involvement in Non-Small Cell Lung Cancer. Cancer Res. 2020;80:4025–4036. doi: 10.1158/0008-5472.Can-19-3578. [DOI] [PubMed] [Google Scholar]
- 50.Taniguchi K., Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat. Rev. Immunol. 2018;18:309–324. doi: 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- 51.Xiao Y., Yu T.J., Xu Y., Ding R., Wang Y.P., Jiang Y.Z., Shao Z.M. Emerging therapies in cancer metabolism. Cell Metab. 2023;35:1283–1303. doi: 10.1016/j.cmet.2023.07.006. [DOI] [PubMed] [Google Scholar]
- 52.Roca-Portoles A., Rodriguez-Blanco G., Sumpton D., Cloix C., Mullin M., Mackay G.M., O'Neill K., Lemgruber L., Luo X., Tait S.W.G. Venetoclax causes metabolic reprogramming independent of BCL-2 inhibition. Cell Death Dis. 2020;11:616. doi: 10.1038/s41419-020-02867-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Witkowski M., Tizian C., Ferreira-Gomes M., Niemeyer D., Jones T.C., Heinrich F., Frischbutter S., Angermair S., Hohnstein T., Mattiola I., et al. Untimely TGFβ responses in COVID-19 limit antiviral functions of NK cells. Nature. 2021;600:295–301. doi: 10.1038/s41586-021-04142-6. [DOI] [PubMed] [Google Scholar]
- 54.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo C., Wu M., Huang B., Zhao R., Jin L., Fu B., Wang P., Wang D., Zheng M., Fang J., et al. Single-cell transcriptomics reveal a unique memory-like NK cell subset that accumulates with ageing and correlates with disease severity in COVID-19. Genome Med. 2022;14:46. doi: 10.1186/s13073-022-01049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNA-seq and single-cell RNA-seq data were deposited in GEO and are publicly available as of the date of publication. The accession numbers are listed in the key resources table. All original codes have been deposited at Zenodo (https://doi.org/10.5281/zenodo.10976537) and are publicly available on the date of publication. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.