

Significance
We have evaluated the effect of cytotoxic T lymphocyte-associatedantigen 4 (CTLA-4) Ig on antitumor response induced by CTLA-4 and PD-1 blockade in murine tumor models. Our results illustrate the potential to use CTLA-4 Ig blockade of T cell costimulation to reduce the impact of irAEs without negatively affecting antitumor efficacy.
Keywords: irAEs, myocarditis, immune checkpoint therapies, anti-CTLA-4, anti-PD-1
Abstract
Immune checkpoint therapies (ICT) improve overall survival of patients with cancer but may cause immune-related adverse events (irAEs) such as myocarditis. Cytotoxic T lymphocyte-associated antigen 4 immunoglobulin fusion protein (CTLA-4 Ig), an inhibitor of T cell costimulation through CD28, reverses irAEs in animal models. However, concerns exist about potentially compromising antitumor response of ICT. In mouse tumor models, we administered CTLA-4 Ig 1) concomitantly with ICT or 2) after ICT completion. Concomitant treatment reduced antitumor efficacy, while post-ICT administration improved efficacy without affecting frequency and function of CD8 T cells. The improved response was independent of the ICT used, whether CTLA-4 or PD-1 blockade. The frequency of Tregs was significantly decreased with CTLA-4 Ig. The resulting increased CD8/Treg ratio potentially underlies the enhanced efficacy of ICT followed by CTLA-4 Ig. This paradoxical mechanism shows that a CTLA-4 Ig regimen shown to reduce irAE severity does not compromise antitumor efficacy.
Metastatic melanoma typically has poor prognosis, historically resulting in the survival of most patients for less than a year (1). Immune checkpoint therapies (ICTs) that block cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), like ipilimumab, alleviate the inhibitory effects of CTLA-4 on T cell activation (2, 3). Antibodies that target programmed cell death protein 1 (PD-1), such as nivolumab, disrupt the interaction between PD-1 and PD-L1, restoring the cytotoxic activity of T cells (4, 5). However, ICT has the potential to induce irAEs, including rash, diarrhea, and nausea (any grade, ~20%). In most cases, these irAEs are non-life-threatening and tolerable for patients with cancer. On the contrary, while myocarditis is a rare event (0.54% vs. 1.22% in patients treated with anti-PD-1 alone or combination of anti-CTLA-4 and anti-PD-1 respectively), around 50% of these cases are fatal (6–8). Immune-related myocarditis is characterized by the infiltration of immune cells in the heart, and α-myosin appears to be the target of T cells implicated in ICT-driven cardiomyopathy (9).
CTLA-4 Ig is a soluble fusion protein of the CTLA-4 molecule. Due to its stronger binding affinity toward CD80 and CD86 on antigen-presenting cells (APCs) than the T cell costimulatory receptor CD28, CTLA-4 Ig attenuates the activation of T cells (10–12). It has been approved by the FDA as abatacept for the treatment of rheumatoid arthritis (13). In a prior publication, we introduced transgenic ctla4+/− pdcd1−/− mice, which manifest a myocarditis phenotype characterized by immune cell infiltration in heart. Over half of these mice do not survive beyond 2 mo. Treatment with CTLA-4 Ig decreases mortality in these mice (14). However, concerns remain that CTLA-4 Ig, due to its dampening effect on costimulation, could blunt the antitumor response associated with ICT. This study aims to assess whether CTLA-4 Ig would compromise antitumor response induced by ICT and explore the underlying mechanism in murine tumor models.
Results
Post-ICT CTLA-4 Ig Treatment Increases Antitumor Response.
In a previous study, we introduced ctla4+/− pdcd1−/− mice, demonstrating the myocarditis phenotype. It highlighted the effectiveness of CTLA-4 Ig in preventing myocarditis and improving the survival rate of these mice (14). To investigate whether CTLA-4 Ig that prevents myocarditis would affect the antitumor efficacy of ICT, we have implanted murine melanoma B16F10 into ctla4+/− pdcd1−/− mice. However, all the mice remained tumor-free and eventually died as previously described (14).
We then investigated the impact of a CTLA-4 Ig regimen on the ICT antitumor response independently of the myocarditis phenotype. We administered either anti-CTLA-4 (clone 9H10) or anti-PD-1 to C57BL/6J mice bearing B16F10 tumors on day 7. CTLA-4 Ig treatment was initiated on day 10, 13, or 16 (Fig. 1A and SI Appendix, Fig. S1A). Notably, starting CTLA-4 Ig treatment on day 10 diminished the effectiveness of ICT-driven antitumor response. Treatment of CTLA-4 Ig that was started on day 13 had no discernible effect, while treatment on day 16 improved the antitumor response (Fig. 1A and SI Appendix, Fig. S1A). The improved response suggests that sustained costimulation is not essential for the antitumor response.
Fig. 1.
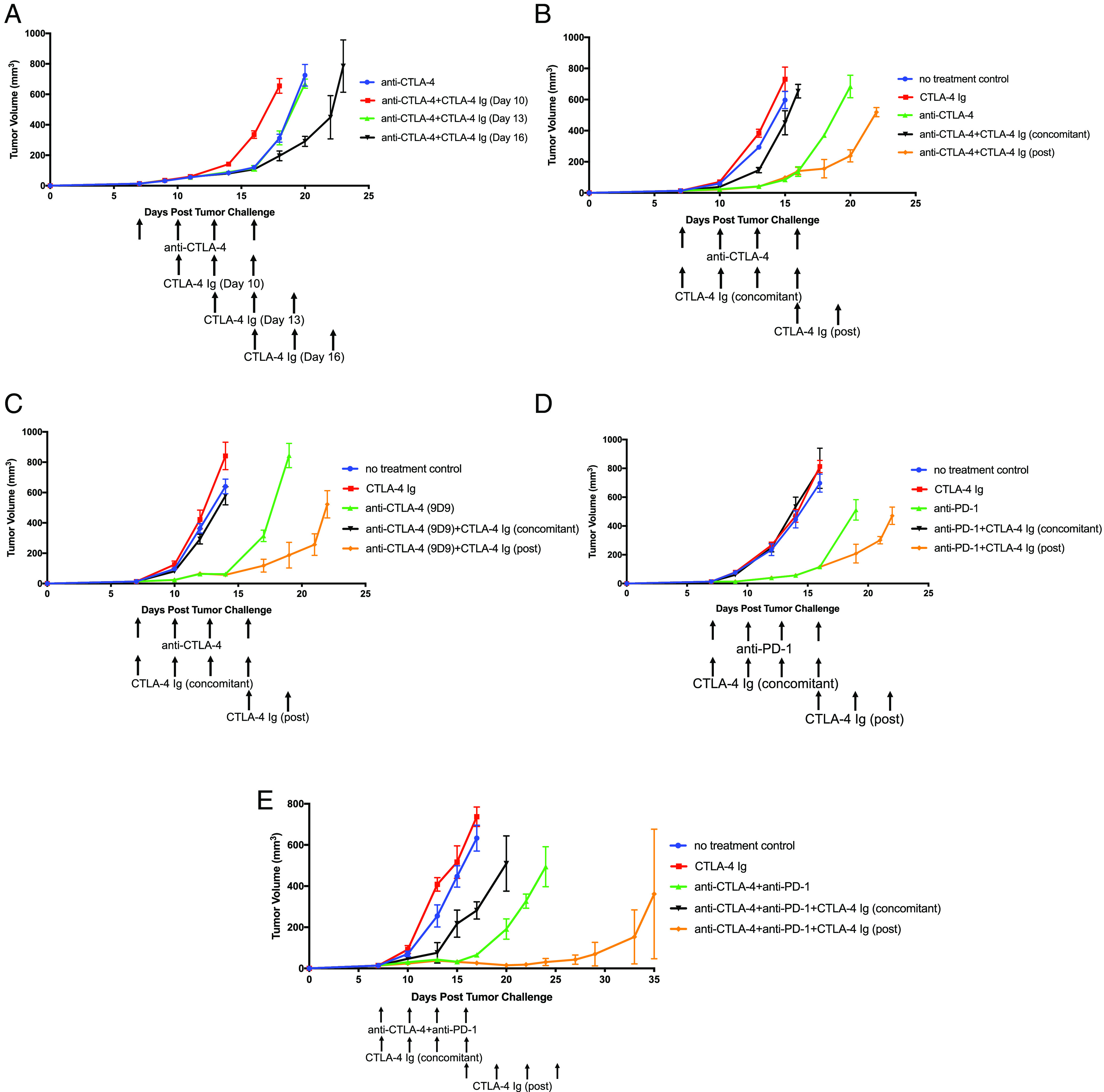
Post-ICT CTLA-4 Ig treatment increases antitumor efficacy. (A) Tumor growth curves of C57BL/6J mice harboring B16F10 tumors first treated with anti-CTLA-4 (clone 9H10) on days 7, 10, 13, and 16. Mice were then treated with CTLA-4 Ig which began on day 10, 13, or 16. Tumor growth curves of B16F10 tumors in C57BL/6J mice treated with (B) anti-CTLA-4 (clone 9H10), (C) anti-CTLA-4 (clone 9D9), (D) anti-PD-1, or (E) anti-CTLA-4 (clone 9H10) and anti-PD-1 in combination CTLA-4 Ig (concomitant). Mice were further treated with CTLA-4 Ig (post) on day 16. All experiments began with five mice per group. Representative experiment from duplicated experiments is shown.
We then aimed to determine whether these findings indicate a generalizable impact of CTLA-4 Ig on the antitumor response of ICT. To achieve this, we conducted similar experiments using the relatively high immunogenic murine colorectal MC38 to contrast with the poorly immunogenic B16F10. Additionally, various isotypes or ICT treatments, such as anti-CTLA-4 (clones 9H10 or 9D9), anti-PD-1, or a combination of anti-CTLA-4 and anti-PD-1, were utilized with CTLA-4 Ig, allowing us to differentiate the effects of CTLA-4 Ig from each ICT treatment. We administered CTLA-4 Ig to mice bearing B16F10 or MC38 tumors under two conditions: 1) concomitantly with ICT, beginning on day 7 (concomitant), or 2) after the full ICT regimen, starting on day 16 (post). Concomitant administration of CTLA-4 Ig with ICT significantly diminished antitumor efficacy, however, the antitumor efficacy improved when mice received CTLA-4 Ig (post) after ICT concluded (Fig. 1 B–E and SI Appendix, Fig. S1 B and C). These consistent observations across the two tumor models and ICT regimens imply that sustained costimulation for antitumor response is not necessary once the course of ICT treatment is completed. The improved antitumor response by CTLA-4 Ig (post) is irrespective of tumor type, specific ICT, or the isotype of the anti-CTLA-4 antibody. Moreover, it’s not simply a result of depleting anti-CTLA-4 with CTLA-4 Ig, which would impact treatment potency.
Because of the counterintuitive improvement of the antitumor response that results from inhibition of costimulation after ICT, we tested the effect of prolonged ICT on the antitumor response. When we extended anti-CTLA-4 treatment from ending on day 16 to ending on day 19, the antitumor response was unchanged (SI Appendix, Fig. S1D), demonstrating that T cells appeared to achieve peak activation after the initial 4 doses of anti-CTLA-4 ending on day 16.
In summary, treatment with CTLA-4 Ig improves the antitumor response after the completion of ICT. However, it diminishes treatment efficacy when administered concomitantly with ICT.
CTLA-4 Ig (Post) Blocks Costimulation Pathway.
The observation that CTLA-4 Ig (concomitant) reduces antitumor efficacy of ICT aligns with our comprehension on the mechanism of CTLA-4 Ig, involving the inhibition of T cell activation (12, 13). To investigate whether the improved antitumor response resulting from CTLA-4 Ig (post) has an inhibitory effect on the costimulation pathway, mice that received CTLA-4 Ig (post) were also subject to blockade of CD80 and CD86 or blockade of CD28. Our results demonstrated the antitumor response induced by CTLA-4 Ig (post) was similar to that induced when CD80 (post) and CD86 (post), or CD28 (post), were blocked after ICT (Fig. 2 A and B). Subsequently, we assessed the frequency of costimulatory molecules on CD45+ CD3- CD11c+ APCs in tumors on day 24. The frequency of CD80+ and CD86+ APCs remained similar from mice treated with only anti-CTLA-4 and those treated with a combination of anti-CTLA-4 and CTLA-4 Ig (post) (Fig. 2 C, D, and H). We also investigated whether CTLA-4 Ig (post) impacted antigen presentation and found no change in the frequency of H-2Db+ and MHCII+ APCs (Fig. 2 E, F, and H). Previous studies have demonstrated that activated T cells produce IFN-γ, subsequently increasing the frequency of PD-L1-expressing cells (15, 16). Since CTLA-4 Ig (post) can curtail T cell activation and IFN-γ production, we examined the frequency of PD-L1+ cells. However, the frequency of PD-L1+ APCs remained unaffected by CTLA-4 Ig (post) (Fig. 2 G and H). In summary, our findings indicate that the improved antitumor response induced by CTLA-4 Ig (post) is likely due to the blockade of costimulation, maintaining the same target pathway without any apparent influence on APCs.
Fig. 2.
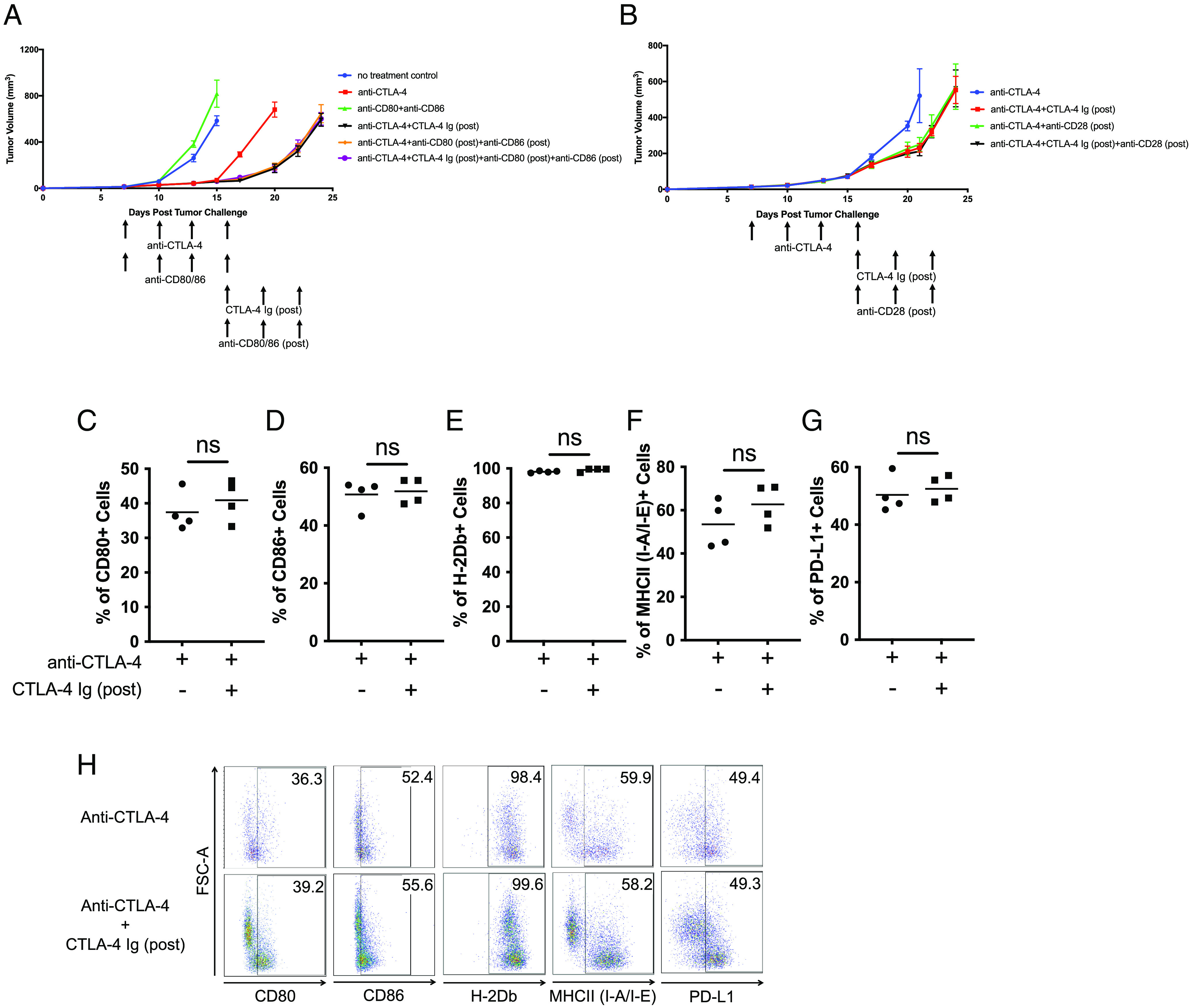
CTLA-4 Ig (post) blocks costimulation through CD80/CD86/CD28 pathway. Tumor growth curves of C57BL/6J mice harboring B16F10 tumors initially treated with anti-CTLA-4 (clone 9H10). Once the initial treatments were completed, mice were further treated with CTLA-4 Ig (post) in combination with (A) anti-CD80 (post) and anti-CD86 (post) or (B) anti-CD28 (post). Dot-plot representation of frequency of CD45+ CD3− CD11c+ APCs that express (C) CD80+, (D) CD86+, (E) MHCI (H-2Db)+, (F) MHCII (I-A/I-E)+, and (G) PD-L1+ in tumor on day 24. (H) Representative FACS plots demonstrating percentages of CD45+ CD3− CD11c+ APCs that express CD80, CD86, MHCI (H-2Db), MHCII (I-A/I-E), and PD-L1. All experiments began with five mice per group. Representative experiment from duplicated experiments is shown.
CTLA-4 Ig (Post) Does Not Influence Frequency and Function of CD8 T Cells.
We investigated whether the improved antitumor response induced by CTLA-4 Ig (post) is a T cell–mediated response. The antitumor response was significantly reduced when either CD4 or CD8 T cells were depleted during CTLA-4 Ig (post) treatment (Fig. 3 A and B and SI Appendix, Fig. S2 A–F). These findings suggest that the improved antitumor response resulting from CTLA-4 Ig (post) is a T cell–mediated response which relies on CD4 and CD8 T cells.
Fig. 3.
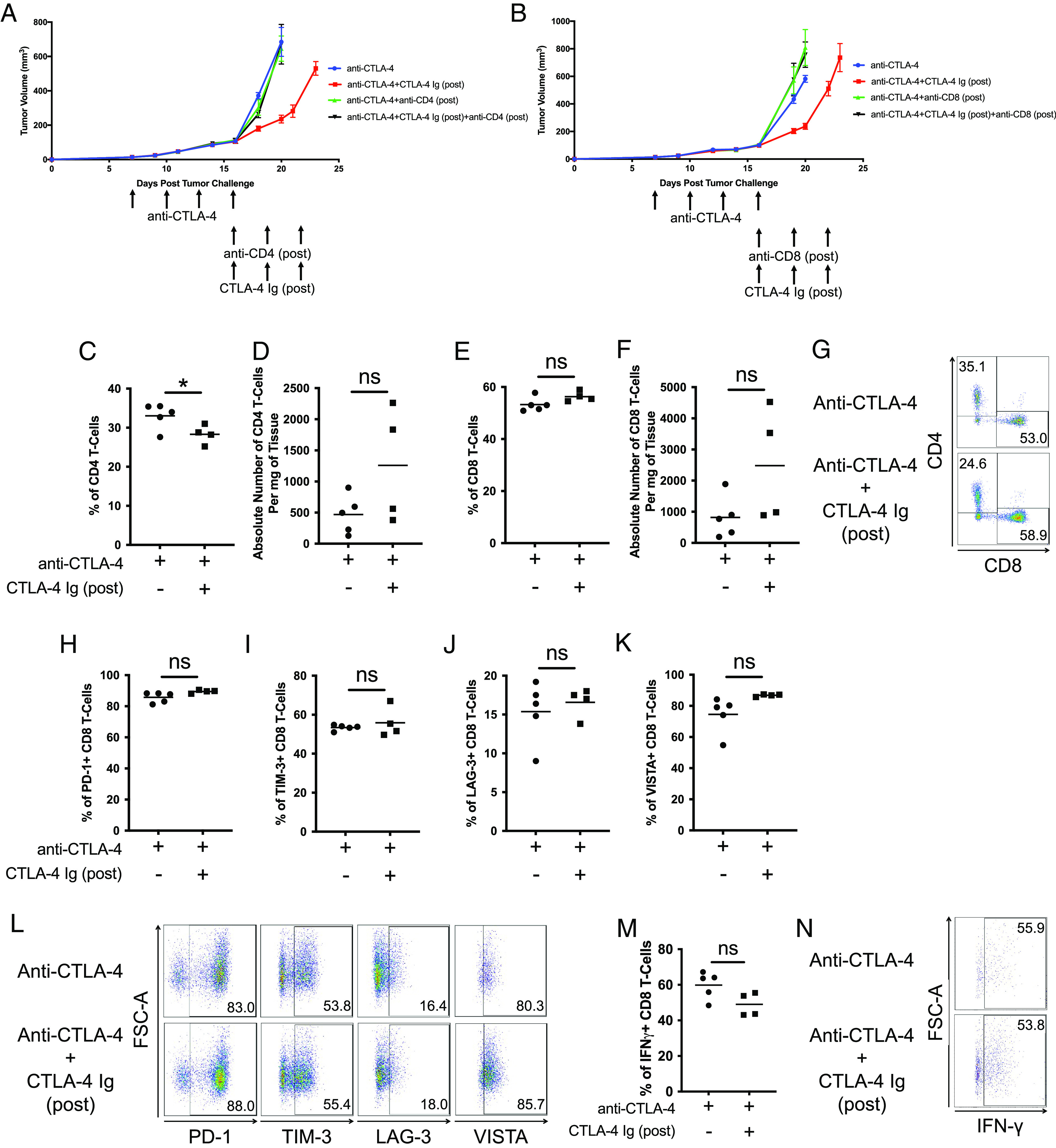
CTLA-4 Ig (post) has no effect on the CD8 T cells. Tumor growth curves of C57BL/6J mice harboring B16F10 tumors were initially treated with anti-CTLA-4 (clone 9H10). Once the initial treatments were completed, mice were further treated with CTLA-4 Ig (post) in combination with (A) anti-CD4 (post) or (B) anti-CD8 (post). Dot-plot representation of (C) frequency and (D) absolute number of CD4 T cells in B16F10 tumor on day 24. Dot-plot representation of (E) frequency and (F) absolute number of CD8 T cells in B16F10 tumor on day 24. (G) Representative FACS plots demonstrating percentages of CD4 and CD8 T cells. Dot-plot representation of frequency of (H) PD-1+, (I) TIM-3+, (J) LAG-3+, and (K) VISTA+ CD8 T cells. (L) Representative FACS plots demonstrating percentages of PD-1+, TIM-3+, LAG-3+, and VISTA+ CD8 T cells. Dot-plot representation of (M) frequency of IFN-γ+ CD8 T cells. (N) Representative FACS plots demonstrating percentages of IFN-γ+ CD8 T cells. All experiments began with five mice per group. Representative experiment from duplicated experiments is shown.
Subsequently, we investigated whether CTLA-4 Ig (post) increases the frequency of tumor-infiltrating lymphocytes (TILs). The group treated with CTLA-4 Ig (post) showed a lower intratumoral CD4 T cell frequency than the ICT alone group (Fig. 3 C, D, and G and SI Appendix, Fig. S3 A, B, and E). However, there were no discernible changes in the frequency of CD8 T cells (Fig. 3 E–G and SI Appendix, Fig. S3 C–E). Then, we investigated whether CTLA-4 Ig (post) could influence the differentiation state of CD8 T cells. As prior reports suggest terminally differentiated T cells are a distinct lineage that express inhibitory receptors following ICT, we evaluated the frequency of CD8 T cells expressing PD-1, TIM-3, LAG-3, and VISTA (17, 18). Approximately 90% of CD8 T cells in both groups express PD-1, and it was evident that CTLA-4 Ig (post) had no discernible effect on the frequency of CD8 T cells expressing these inhibitory receptors (Fig. 3 H–L and SI Appendix, Fig. S3 F–J). The expression of these inhibitory receptors implies that these CD8 TILs are fully activated, and CTLA-4 Ig (post) is incapable of reversing the status of these cells. To investigate whether these CD8 T cells remain functionally active and whether CTLA-4 Ig (post) could influence their activity, we stimulated these CD8 T cells ex vivo. However, we observed no significant change in the frequency of cytokine-producing cells (Fig. 3 M and N). The cytokine production suggests that CD8 T cells maintain their functional activity when subjected to CTLA-4 Ig (post) treatment.
Taken together, our findings align with existing literature in the field, which propose that a full course of ICT leads to terminal differentiation of T cells (17). This underscores the notion that once CD8 T cells have reached a state where their dependency on costimulation is no longer necessary, frequency and functionality of CD8 T cells are not affected by CTLA-4 Ig (post).
Antitumor Response Induced by CTLA-4 Ig (Post) Is Treg-Dependent.
The differentiation of T cells relies on costimulation (19, 20). However, the results in Fig. 3 suggest that the CD8 T cells are not affected in terms of frequency and cytokine production upon treatment with CTLA-4 Ig (post). This implies that costimulation is not necessary once CD8 T cells are fully activated subsequent to ICT treatments. In contrast, our findings in Fig. 3C and SI Appendix, Fig. S3A reveal a reduction in the overall frequency of intratumoral CD4 T cells upon CTLA-4 Ig (post) treatment. CTLA-4 Ig (post) led to a decreased frequency of Foxp3+ Tregs as a fraction of the overall CD4 T cell population (Fig. 4 A, B, and E and SI Appendix, Fig. S4 A, B, and E), along with a reduction in ICOS+ Tregs (Fig. 4 C–E and SI Appendix, Fig. S4 C–E). These observations suggest that, unlike CD8 T cells, Tregs require costimulation even after ICT treatments. Thus, the reduction in Tregs can improve the antitumor response. To test whether the antitumor response induced by CTLA-4 Ig (post) is dependent on Treg depletion, we conducted a study using Foxp3DTR mice bearing B16F10 tumors. Mice were treated with diphtheria toxin (DT) to deplete Tregs, in addition to receiving CTLA-4 Ig (post). When Tregs were depleted using DT, the antitumor responses induced with or without CTLA-4 Ig (post) treatment were similar (Fig. 4F and SI Appendix, Fig. S4 F–I). This suggests that the improved antitumor response by CTLA-4 Ig (post) depends on reducing the frequency of Tregs.
Fig. 4.
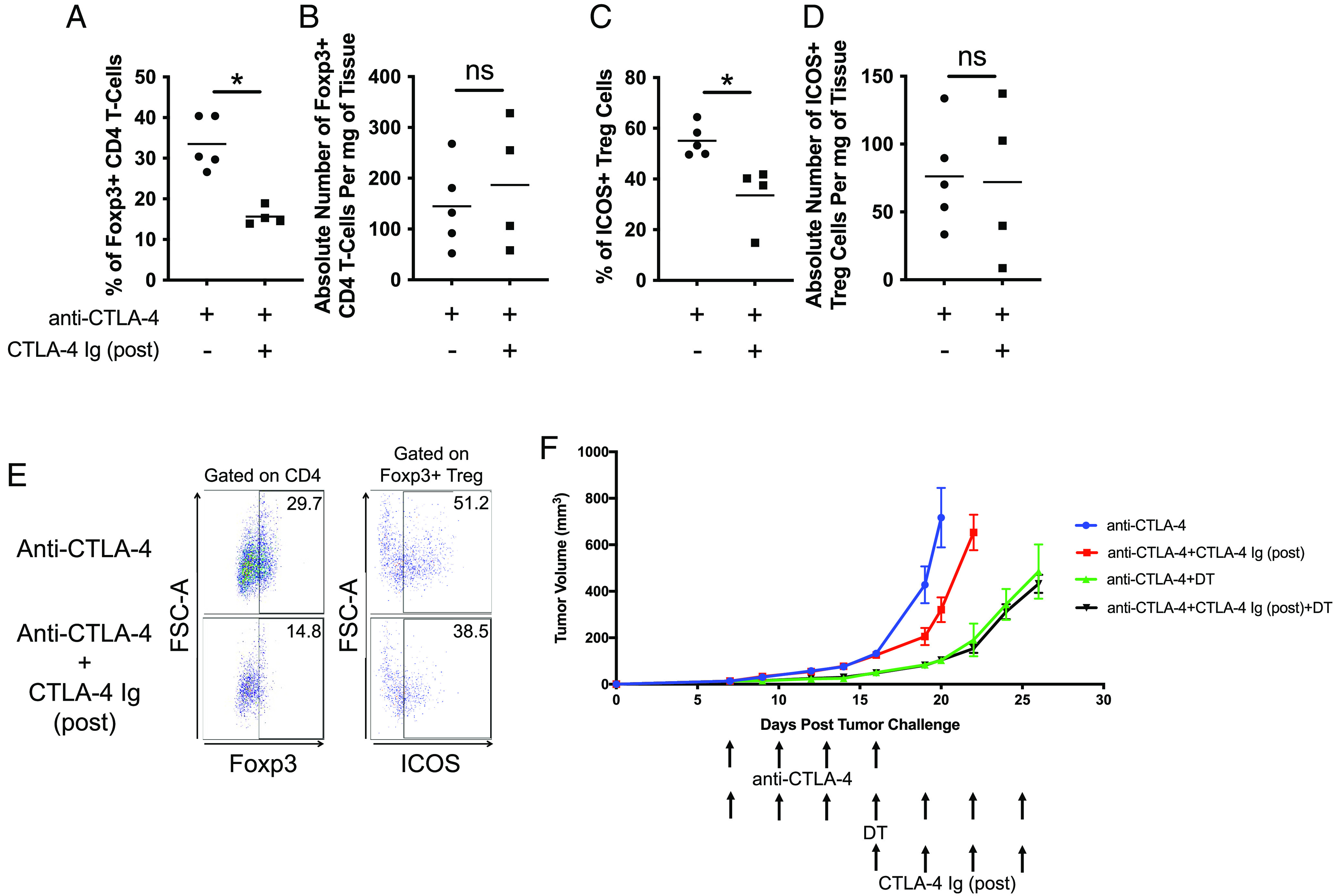
Antitumor response induced by CTLA-4 (post) is Treg-dependent. Mice were first treated with anti-CTLA-4 (clone 9H10) and then with CTLA-4 Ig (post). Dot-plot representation of (A) frequency and (B) absolute number of Foxp3+ CD4 T cells on day 24. Dot-plot representation of (C) frequency and (D) absolute number of ICOS+ Tregs on day 24. (E) Representative FACS plots demonstrating percentages of Foxp3+ CD4 and ICOS+ Tregs. (F) Tumor growth curves of B16F10 tumors in Foxp3DTR mice. Mice were first treated with anti-CTLA-4 (clone 9H10). Diphtheria toxin was administered to mice until the end of the experiment. Mice were then treated with CTLA-4 Ig (post) once the initial treatment of anti-CTLA-4 is finished. All experiments began with five mice per group. Representative experiment from duplicated experiments is shown.
Our research builds upon the findings outlined in our prior publication, which introduced transgenic ctla4+/− pdcd1−/− mice that manifest a myocarditis phenotype. It has been demonstrated that with CTLA-4 Ig, the severity of myocarditis can be reduced, leading to improved survival rates in mice (14). In this report, we investigate the effect of CTLA-4 Ig on the antitumor response induced by ICT in murine tumor models. We provide a mechanism through which CTLA-4 Ig improves the antitumor response when administered post-ICT by blocking Treg costimulation. Conversely, Tregs can potentially dampen the activity of reactive T cells that regulate myocarditis (21). It suggests a complex interplay between Tregs and the regulation of myocarditis. Additionally, while anti-CTLA-4 (clone 9H10) has the Fc chain necessary for antibody-dependent cellular cytotoxicity (ADCC) for Treg depletion, its effect is observable only within tumors and not in the spleen and lymph nodes (22). Even with anti-PD-1 antibodies lacking the Fc chain for Treg depletion, myocarditis can still be induced (23). These observations imply that myocarditis is unlikely to be solely the result of Treg depletion caused by therapeutic anti-CTLA-4 treatment. The relationship between Tregs and myocarditis regulation warrants further investigation. In conclusion, we propose that CTLA-4 Ig could hold potential for mitigating the severity of irAEs without compromising the antitumor response induced by ICT.
Discussion
CTLA-4 has a higher binding affinity and avidity for CD80 and CD86 than CD28 (10, 11, 24). The soluble fusion protein of human CTLA-4 Ig attenuates T cell activation by blocking CD28-mediated costimulation (12). We recently showed that CTLA-4 Ig has the potential to alleviate myocarditis and improves the survival of preclinical ctla4+/− pdcd1−/− mice. Supporting evidence from case studies further substantiates these outcomes (14). The primary objective of this report is to assess the effect of CTLA-4 Ig on the antitumor response induced by ICT. We have conducted experiments in wild-type C57BL/6J mice due to the inability of tumors to grow in the ctla4+/− pdcd1−/− mice (14). Our investigation focused on assessing T cell activation, terminally differentiated T cells, and costimulation for Tregs. We show that CTLA-4 Ig (concomitant) diminishes the efficacy of all ICT treatments, in line with our understanding that costimulation via CD28 on T cells is crucial for their activation (25, 26). Conversely, the administration of CTLA-4 Ig (post) after ICT treatment improves the antitumor response which suggests a generalizable impact of CTLA-4 Ig on ICT. CTLA-4 Ig (post) also does not affect the overall frequency and function of CD8 T cells (Fig. 3). This outcome is not surprising, given that CD28 engagement is no longer necessary for the survival or function of CD8 T cells once they have undergone initial costimulation. On the contrary, Tregs rely on costimulation for their survival even after ICT. Hence, the interruption of CD28 signaling by CTLA-4 Ig after activation leads to a decrease in the frequency of Tregs and improves antitumor response.
In conjunction with our previous publication (14), our current research affirms that CTLA-4 Ig is a potential therapeutic approach for effectively managing irAEs without compromising the antitumor response, as evidenced by our mouse models. As we extrapolate the applicability of our findings regarding CTLA-4 Ig to patients with cancer, several uncertainties persist: 1) The complex interplay between Tregs and myocarditis regulation. We have shown that CTLA-4 Ig (post) can reduce the frequency of Tregs; however, Tregs can dampen the activity of reactive T cells that regulate myocarditis (21). 2) There is a likelihood that the absence of costimulatory signals during activation may induce anergy in T cells, resulting in their failure to target α-myosin (9, 27). 3) Effect of CTLA-4 Ig on the Tregs in the periphery and lymphoid tissues. As CTLA-4 blockade has been shown to expand Tregs in the periphery (28–30), CTLA-4 Ig may have a similar effect on reducing Treg frequency in those tissues. 4) The optimal timing for initiating CTLA-4 Ig treatment in patients with cancer. It is necessary to study pharmacokinetics of ICT and CTLA-4 Ig and the functionality of activated T cells so that CTLA-4 Ig can be administered without compromising the antitumor response. While we cannot completely exclude these factors, thorough investigation in human subjects is crucial.
In clinical settings, irAEs have been observed to manifest early in treatment, but their frequency tends to increase later in treatment course or even after the completion of ICT (6, 31, 32). The management of irAEs typically involves systemic immunosuppressive approaches, such as steroids or antibodies like infliximab. Interestingly, a significant number of patients, especially those receiving immunosuppressants later in the course of ICT, still achieve complete clinical responses (31–34). This observation strongly suggests that CTLA-4 Ig treatment could be a viable option for addressing ICT-induced myocarditis within a clinical setting. The future holds promise for clinical studies that explore the administration of CTLA-4 Ig following ICT, providing more insights into its potential benefits and applicability.
Materials and Methods
Mice, Cell Lines, and Reagents.
First, 8- to 12-wk-old female C57BL/6J and Foxp3DTR mice were purchased from The Jackson Laboratory. All animal experiments were performed in accordance with The University of Texas MD anderson cancer center (MDACC) institutional animal care and use committee (IACUC) guidelines in a MDACC AAALAC accredited barrier facility vivarium. All experiments are performed at least twice. B16F10 and MC38 are murine cancer cell lines cultured in Dulbecco’s Modified Eagle Medium (HyClone, #SH30243.01) supplemented with 10% FBS (Gibco, #10082-147) and 1% penicillin and streptomycin (Caisson, #PSL01-100ML).
In Vivo Experiments.
C57BL/6J and Foxp3DTR mice (Jackson Laboratory) were housed and allowed to acclimate to the housing facility for 1 wk prior to experiments. Mice were then randomized in staged fashion before any treatment. For the tumor model experiments, tumor cells were implanted subcutaneously. Each treatment group is started with five mice with tumor diameter reached 3 to 4 mm on day 7. For checkpoint blockade antibodies treatments, four doses of antibodies were administered by intraperitoneal injection every 3 d. Anti-CTLA-4 antibodies (clone, 9H10, and 9D9) (BioXcell, #BE0313 and BE0164) and anti-PD-1 antibodies (clone, RMP1-14) (BioXcell, #BE0099) were used. In most experiments, anti-CTLA-4 (clone, 9H10) was used. Both 9H10 and 9D9 clone were administered using doses of 200 μg for initial injection and 100 μg for subsequent three injections. For anti-PD-1 treatments, clone RMP1-14 was administered using doses of 250 μg per injection. CTLA-4 Ig (BioXCell, #BE0099), anti-CD80 (clone, 1G10) (BioXCell, #BE0134), anti-CD86 (clone, GL-1) (BioXCell, #BE0025), and anti-CD28 (clone, 37.51) (BioXcell, #BE0015) were used. Then, 250 μg was administered per injection. Anti-CD4 (clone, GK1.5) (BioXCell, #BE0003-1) and anti-CD8 (clone, 2.43) (BioXcell, #BE0061) were used. Additionally, 500 μg was administered as initial injection followed by 250 μg as subsequent injections until mice were euthanized. Diphtheria toxin (DT) (Millipore, #D0564) were treated to mice at 10 μg/kg every 3 d until the end of experiment. Tumors in mice were measured three times per week. All mice were euthanized using CO2 and subsequent cervical dislocation when tumors reached 14 mm in maximum diameter or when the end of the study period was reached.
Flow Cytometry Analysis.
Tumors were harvested from mice. Dead cells were discriminated with live/dead viability dye (Thermo Fisher Scientific, #L34962). Antibodies for CD3 (Thermo Fisher Scientific, #45-0031-82), LAG-3 (Thermo Fisher Scientific, #11-231-82), Foxp3 (Thermo Fisher Scientific, #58-5773-82), TIM-3 (Thermo Fisher Scientific, #25-5870-82), CD11c (Thermo Fisher Scientific, #25-0114-82), MHCI (Thermo Fisher Scientific, #28-14-B), CD4 (BioLegend, #100559), CD8 (BioLegend, #100714), PD-1 (BioLegend, #135220), ICOS (BioLegend, #117406), CD86 (BioLegend, #105030), CD45.2 (BD Biosciences, #563685), VISTA (BD Biosciences, #742723), PD-L1 (BD Biosciences, #741014), CD80 (BD Biosciences, #562611), MHCII (BD Biosciences, #563415), and analyzed with an LSR-II flow cytometer (BD Biosciences), followed by Flow-Jo software (Tree-Star) analysis as previously described. Intracellular IFN-γ staining was done as previously described (35).
Statistical Analysis.
Data were analyzed with GraphPad Prism 8 software (GraphPad Software, La Jolla, CA). Two-tailed, unpaired t test was used to analyze data points obtained on the day when a particular group of mice were euthanized.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was funded by cancer prevention and research institute of Texas (CPRIT) to J.P.A. (R1203). J.P.A. is a CPRIT Distinguished Scholar in Cancer Research. J.P.A. is a director of Parker Institute for Cancer Immunotherapy.
Author contributions
S.M., D.A.Ç., and J.P.A. designed research; S.M. and H.L. performed research; S.M. analyzed data; and S.M., D.A.Ç., J.J.M., and J.P.A. wrote the paper.
Competing interests
J.P.A. reports consulting, advisory roles, and/or stocks/ownership for Achelois, Adaptive Biotechnologies, Akoya Biosciences, Apricity, Bectas, BioAtla, BioNTech, Candel Therapeutics, Dragonfly, Earli, Enable Medicine, Hummingbird, ImaginAb, Lava Therapeutics, Lytix, Marker, Osteologic, PBM Capital, Phenomic AI, Polaris Pharma, Time Bioventures, Trained Therapeutix, Two Bear Capital, and Venn Biosciences.
Footnotes
Reviewers: B.R.B., University of Minnesota Twin Cities Office of Academic Clinical Affairs; and A.J.K., Vir Biotechnology Inc.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Tsao H., Atkins M. B., Sober A. J., Management of cutaneous melanoma. N. Engl. J. Med. 351, 998–1012 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Hodi F. S., et al. , Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schadendorf D., et al. , Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol.: Off. J. Am. Soc. Clin. Oncol. 33, 1889–1894 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L., Han X., Anti-PD-1/PD-L1 therapy of human cancer: Past, present, and future. J. Clin. Invest. 125, 3384–3391 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tumeh P. C., et al. , PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson D. B., et al. , Fulminant myocarditis with combination immune checkpoint blockade. N. Engl. J. Med. 375, 1749–1755 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larkin J., et al. , Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 381, 1535–1546 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Wolchok J. D., et al. , Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 377, 1345–1356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Axelrod M. L., et al. , T cells specific for alpha-myosin drive immunotherapy-related myocarditis. Nature 611, 818–826 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker L. S., Sansom D. M., The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat. Rev. Immunol. 11, 852–863 (2011). [DOI] [PubMed] [Google Scholar]
- 11.van der Merwe P. A., Bodian D. L., Daenke S., Linsley P., Davis S. J., CD80 (B7–1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J. Exp. Med. 185, 393–403 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Najafian N., Sayegh M. H., CTLA4-Ig: A novel immunosuppressive agent. Expert Opin. Invest. Drugs 9, 2147–2157 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Bluestone J. A., St Clair E. W., Turka L. A., CTLA4Ig: Bridging the basic immunology with clinical application. Immunity 24, 233–238 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Wei S. C., et al. , A genetic mouse model recapitulates immune checkpoint inhibitor-associated myocarditis and supports a mechanism-based therapeutic intervention. Cancer Discov. 11, 614–625 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Curran M. A., Montalvo W., Yagita H., Allison J. P., PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. U.S.A. 107, 4275–4280 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zaretsky J. M., et al. , Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei S. C., et al. , Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 170, 1120–1133.e1117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei S. C., et al. , Negative co-stimulation constrains T cell differentiation by imposing boundaries on possible cell states. Immunity 50, 1084–1098.e1010 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyddane C., et al. , Cutting Edge: CD28 controls dominant regulatory T cell activity during active immunization. J. Immunol. 176, 3306–3310 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Salomon B., et al. , B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 12, 431–440 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Tschope C., et al. , Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 18, 169–193 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simpson T. R., et al. , Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 210, 1695–1710 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Won T., et al. , Cardiac myosin-specific autoimmune T cells contribute to immune-checkpoint-inhibitor-associated myocarditis. Cell Rep. 41, 111611 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwartz J. C., Zhang X., Fedorov A. A., Nathenson S. G., Almo S. C., Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature 410, 604–608 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Krummel M. F., Allison J. P., CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 182, 459–465 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walunas T. L., et al. , CTLA-4 can function as a negative regulator of T cell activation. Immunity 1, 405–413 (1994). [DOI] [PubMed] [Google Scholar]
- 27.Jenkins M. K., Schwartz R. H., Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J. Exp. Med. 165, 302–319 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peggs K. S., Quezada S. A., Chambers C. A., Korman A. J., Allison J. P., Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 206, 1717–1725 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quezada S. A., Peggs K. S., Curran M. A., Allison J. P., CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J. Clin. Invest. 116, 1935–1945 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selby M. J., et al. , Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 1, 32–42 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Riudavets M., et al. , Immune-related adverse events and corticosteroid use for cancer-related symptoms are associated with efficacy in patients with non-small cell lung cancer receiving anti-PD-(L)1 blockade agents. Front Oncol. 10, 1677 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salem J. E., et al. , Abatacept/ruxolitinib and screening for concomitant respiratory muscle failure to mitigate fatality of immune-checkpoint inhibitor myocarditis. Cancer Discov. 13, 1100–1115 (2023). [DOI] [PubMed] [Google Scholar]
- 33.Pan E. Y., Merl M. Y., Lin K., The impact of corticosteroid use during anti-PD1 treatment. J. Oncol. Pharm. Pract. 26, 814–822 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Salem J. E., et al. , Abatacept for severe immune checkpoint inhibitor-associated myocarditis. N. Engl. J. Med. 380, 2377–2379 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Mok S., et al. , Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 74, 153–161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.