

Summary
RAS pathway mutations, which are present in 30% of patients with chronic myelomonocytic leukemia (CMML) at diagnosis, confer a high risk of resistance to and progression after hypomethylating agent (HMA) therapy, the current standard of care for the disease. Here, using single-cell, multi-omics technologies, we seek to dissect the biological mechanisms underlying the initiation and progression of RAS pathway-mutated CMML. We identify that RAS pathway mutations induce transcriptional reprogramming of hematopoietic stem and progenitor cells (HSPCs) and downstream monocytic populations in response to cell-intrinsic and -extrinsic inflammatory signaling that also impair the functions of immune cells. HSPCs expand at disease progression after therapy with HMA or the BCL2 inhibitor venetoclax and rely on the NF-κB pathway effector MCL1 to maintain survival. Our study has implications for the development of therapies to improve the survival of patients with RAS pathway-mutated CMML.
Keywords: CMML, progression, RAS, MCL1, NF-kB, transcriptomics, multiomics
Graphical abstract
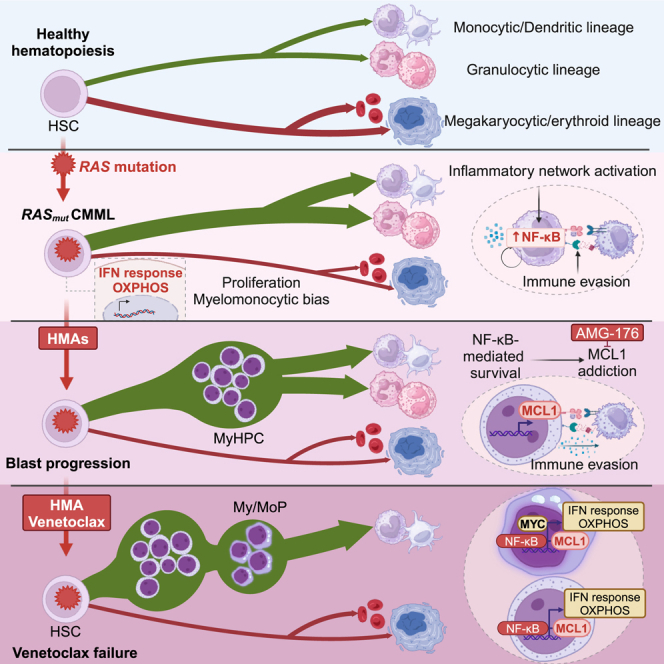
Highlights
-
•
RAS mutations induce inflammatory network activation and immune evasion in CMML
-
•
Expansion of NF-κB-dependent HSPCs drives blast progression (BP) after HMA therapy
-
•
RAS mutant CMML HSPCs rely on MCL1 for survival during BP
-
•
NF-κB pathway activation in RAS mutant HSPCs drives venetoclax resistance
In brief, Montalban-Bravo et al. show that RAS pathway mutations induce multi-step reprogramming of distinct cellular populations in CMML. Expansion of myeloid progenitors at blast progression following hypomethylating agent therapy is driven by NF-κB pathway activation and MCL1 dependency. These data support the potential of NF-κB-targeted therapies in CMML.
Introduction
Chronic myelomonocytic leukemia (CMML), a clonal disorder of mutant hematopoietic stem cells (HSCs),1 is characterized by myelodysplastic and myeloproliferative bone marrow (BM) features,2,3 and a high risk of progression to acute myeloid leukemia (AML).4,5,6 Hypomethylating agent (HMA) therapy, the current standard of care for most patients with CMML,7 can overcome CMML cells’ aberrant proliferation and achieve improved outcomes in some patients. However, most patients only have transient responses to HMA therapy, owing to these agents’ inability to effectively deplete HSCs and decrease tumor burden. CMML patients whose disease undergoes transformation to AML upon HMA therapy failure have dismal clinical outcomes.8,9
Despite advances in the genetic characterization of CMML, the development of alternative frontline treatments or more effective second-line therapies to improve the outcomes of CMML patients with high-risk biological features has been delayed because of an incomplete understanding of the ways in which different hematopoietic populations that persist throughout HMA therapy contribute to disease maintenance and progression.
Mutations in RAS pathway signaling genes (BRAF, CBL, KRAS, NF1, NRAS, and PTPN11) confer adverse biological features that increase the risk of disease progression and poor overall survival, particularly when they are concurrently present with loss-of-function mutations in the ASXL transcriptional regulator 1 gene, ASXL1.10
Herein, we used single-cell technology-based approaches to elucidate the biological and molecular landscape of RAS pathway-mutated CMML to guide the selection of future therapeutic interventions and achieve durable responses in CMML patients in whom blast progression (BP) occurs after failure to HMA therapy.
Results
Mutations in RAS pathway signaling genes predict a high risk of CMML BP after HMA therapy failure
We first evaluated whether specific mutations predict a high risk of CMML BP in a cohort of 108 CMML patients who received HMA therapy (Table S1). After a median follow-up of 19 months (95% confidence interval [CI], 15.8–23.9 months), 57 patients experienced HMA therapy failure; 36 patients had BP at the time of therapy failure. Mutations in RAS pathway genes were significantly associated with BP (odds ratio = 3.35; 95% CI, 1.46–7.70; p = 0.004) (Figure 1A) and shorter time to BP (hazard ratio = 2.21; 95% CI, 1.13–4.33; p = 0.021) (Figure 1B). Similarly, logistic regression analysis showed that RAS pathway mutations were associated with a higher risk of BP (p = 0.01158) (Table S2). To assess whether BP was associated with mutations that were not detected at diagnosis or with the clonal expansion of pre-existing mutations, we sequenced BM cells isolated from samples collected at the time of BP after HMA failure from 22 of the 36 patients and compared the cells’ genomic landscape with that of BM cells isolated at diagnosis (Figure 1C). Among 22 patients with BP, 14 (64%) had RAS pathway mutations at diagnosis, and 20 (91%) had RAS pathway mutations at BP. Nine patients (41%) acquired newly detectable RAS pathway mutations at BP (4 patients had no detectable RAS pathway mutations at diagnosis, and 5 patients acquired other RAS pathway mutations). Of the 14 patients with RAS pathway-mutated CMML at diagnosis, 10 had BP without RAS pathway mutation-induced clonal evolution (Figure 1D).
Figure 1.

Mutations in RAS pathway signaling genes predict a high risk of CMML BP after HMA therapy failure
(A) Bar chart showing the frequencies of detectable mutations and cytogenetic abnormalities among 108 CMML patients who received HMA therapy and whose disease progressed (green) or did not progress (blue). Asterisks indicate significantly different frequency changes (∗p < 0.05).
(B) Kaplan-Meier survival curves showing the cumulative incidence of BP after HMA therapy in previously untreated CMML patients with or without RAS pathway mutations. N, number; HR, hazard ratio; CI, confidence interval.
(C) Bar chart showing the overall frequencies of detectable mutations among 22 CMML patients whose disease progressed and in whom targeted sequencing was performed at the time of BP. Mutations at diagnosis and BP are indicated by pink and green, respectively. Paired samples were analyzed.
(D) Detected mutations and their variant allele frequencies (VAFs) in matched samples obtained at diagnosis and at the time of BP in the 22 CMML patients shown in (C). Columns represent the mutations and VAFs from sequential samples of individual CMML patients at diagnosis and BP. Patient identifiers are shown at the top of each column. Asterisks indicate the presence of multiple mutations in a particular gene. The numbers of RAS mutations are shown in red gradient; the VAFs of each mutation are shown in blue gradient.
These results were validated using single-cell DNA sequencing coupled with cell-surface immunophenotyping analysis of mononuclear cells (MNCs) isolated from sequential BM samples obtained at the time of diagnosis or BP from two representative RAS pathway-mutated CMML patients whose disease never responded to therapy (Figures S1A and S1B) or underwent clonal evolution after an initial response (Figures S2A and S2B). Taken together, these data demonstrate that patients with RAS pathway-mutated CMML have a high risk of BP at the time of HMA therapy failure. This observation has important clinical implications in light of our recent study showing that RAS pathway mutations also drive resistance to and/or BP following venetoclax-based second-line therapy.11 These data underscore the urgent need to dissect the biological mechanisms of RAS pathway mutation-induced therapy resistance, as such an understanding could lead to the development of future therapeutic approaches to prevent or overcome disease progression.
RAS pathway mutations activate cell-intrinsic and -extrinsic inflammatory networks
To dissect the molecular mechanisms underlying the progression of RAS pathway-mutated CMML, we first evaluated the molecular determinants of disease initiation. We performed single-cell RNA sequencing (scRNA-seq) analysis of lineage-negative (Lin–) CD34+ hematopoietic stem and progenitor cells (HSPCs) isolated from five untreated RAS pathway mutant CMML patients and two age-matched healthy donors (HDs) (Table S3). This analysis identified eight cellular clusters driven by the differentiation profile of the cells (Figure 2A), which we defined based on the differential expression of validated lineage-specific transcriptional factors (TFs) and cellular markers12,13 (Figure S3A; Table S4). Compared with HSPCs from HDs, Lin–CD34+ HSPCs from RAS pathway mutant CMML patients had a predominant myeloid differentiation route with higher frequencies of early myeloid hematopoietic progenitor cells (eMyHPCs) (clusters 1 and 3, characterized by the high expression of CD34, BTF3, and CEBPA but low expression of CD38) and more differentiated MyHPCs (dMyHPCs) (clusters 0, 4, and 6, marked by the expression of CEBPD and/or CEBPA, as well as that of MPO) at the expense of more primitive HSCs (cluster 2, marked by the high expression of MLLT3, MEG3, and CLEC9A), and erythroid/megakaryocyte (Ery/Mk) HPCs (clusters 5 and 7, marked by the expression of KLF1, GATA1, and GATA2) (Figure 2B). Differential expression analysis revealed that genes upregulated in RAS pathway mutant CMML HSCs compared with HD HSCs were mainly involved in oxidative phosphorylation, interferon (IFN) response, and apoptosis (Figures 2C and S3B). Similar results were observed in eMyHPCs and dMyHPCs (Figure 2C).
Figure 2.

RAS pathway-mutated CMML cells activate cell-intrinsic and -extrinsic inflammatory networks
(A) UMAP of scRNA-seq data for pooled single Lin–CD34+ cells isolated from BM samples of two HDs (n = 895) and five RAS pathway mutant CMML patients (n = 3,161). Each dot represents one cell. Different colors represent the cluster cell-type identity (left) or sample origin (right). HSC, hematopoietic stem cells; eMyHPC, early myeloid progenitor cells; dMyHPC, differentiated myeloid progenitors; Ery/MkHPC, erythroid/megakaryocyte hematopoietic progenitor cells. Dashed lines indicate single clusters in each cell-type population.
(B) Distribution of HD (top) and RAS pathway mutant CMML (bottom) Lin–CD34+ cell types among the clusters shown in (A).
(C) Pathway enrichment analysis of the genes that were significantly upregulated in HSCs (left), eMyHPCs (middle), and dMyHPCs (right) from the five RAS pathway mutant CMML samples shown in (A) compared with those from HD samples (adjusted p ≤ 0.05). The top 10 hallmark gene sets are shown.
(D) UMAP of scRNA-seq data for pooled single MNCs isolated from BM samples of three HDs (n = 12,836), three RAS pathway wild-type (RASwt) (n = 16,038), and five RAS pathway mutant (RASmut) (n = 12,234) CMML patients. Each dot represents one cell. Different colors represent the cluster cell-type identity (left) or sample origin (right). HSC, hematopoietic stem cells; MKP, megakaryocyte precursors; Ery, erythroid precursors; MyHPC, myeloid hematopoietic progenitor cells; Mono, monocytes; CD16 Mono, non-classical CD16+ monocytes; cDC, classical dendritic cells; pDC, plasmacytoid dendritic cells; B cell, B lymphocytes; PC, plasma cells; nCD4T, naive CD4+ T cells; mCD4/CD8, memory CD4+ and CD8+ T cells; eCD8T, effector CD8+ T cells; NKC, natural killer cells. Dashed lines indicate single clusters in each cell-type population.
(E) Distribution of HD (top), and RAS pathway wild-type (RASwt) (middle) or mutant (RASmut) CMML (bottom) BM MNC populations among the clusters shown in (D).
(F) Pathway enrichment analysis of the genes that were significantly upregulated in RAS pathway wild-type (top left) or mutant (top right) CMML monocyte clusters compared with those in HD and RAS pathway mutant monocyte clusters compared with those in RAS pathway wild-type monocyte clusters (bottom left) (adjusted p ≤ 0.05). The top 10 hallmark gene sets are shown.
To evaluate the contribution of downstream myelo/monocytic (My/Mo) populations to disease maintenance, we performed scRNA-seq analysis of BM MNCs isolated from three HDs, and five untreated RAS pathway mutant CMML samples. To dissect the specific role of RAS pathway mutations in disease initiation, we also included BM MNCs from three untreated RAS pathway wild-type CMML samples. This analysis identified 18 cellular clusters inclusive of all major BM cell types that we defined based on the expression of lineage-specific TFs and cellular markers and using the single-cell transcriptome to protein prediction with deep neural network pipeline14,15 (Figures 2D and S3C; Table S5). Consistent with the predominant myeloid differentiation bias of CMML HSPCs, differential analysis of BM cell lineage composition revealed that the monocyte population (clusters 0 and 4) increased in BM CMML samples compared with that in BM HD samples, regardless of the presence of RAS pathway mutations (Figure 2E).
However, although CMML monocytes from RAS pathway wild-type CMML underwent transcriptional reprogramming compared with those from HDs, RAS pathway mutant monocytes had significantly enhanced upregulation of IFN and NF-κB signaling-mediated inflammatory responses compared with RAS pathway wild-type monocytes (Figures 2F and S4A).
Inflammatory networks modulate the immune microenvironment and contribute to immune escape.16 To assess whether CMML monocytes directly suppress the immune response and whether RAS pathway mutations modulate such interactions, we dissected the intercellular crosstalk and communication networks between CMML cells and all other BM cells. We inferred cell-to-cell communications from the combined expression of multi-subunit ligand-receptor complexes using CellPhoneDB, a repository of ligands and receptors and their interactions.17 After generating a homeostatic interactome of BM MNCs from HDs, we analyzed the cellular communication networks that were upregulated in RAS pathway wild-type and mutant CMML BM samples (Figure S4B; Table S6). Compared with HD MNCs, RAS pathway mutant CMML MNCs had significantly more ligand-receptor interactions involving monocytes, classical dendritic cells (cDCs), plasmacytoid DCs (pDCs), MyHPCs, effector CD8+ T (eCD8T) cells, and natural killer (NK) cells (Figure S4B). Monocytes, cDCs, and immune populations from patients with RAS pathway mutant CMML gained significantly more ligand-to-receptor interactions compared with those without RAS pathway mutations (Figure S4B). Specifically, expression levels of chemokine genes (CCL3 and CCL3L1) and cytokine genes (IL1B, TNFSF10, MIF, and HGF) involved in inflammatory signaling and NF-κB-mediated cell survival were significantly increased in CMML monocytes, cDCs, pDCs, and MyHPCs, and enriched in patients with RAS pathway mutations (Figure S4C). Monocytes and cDCs from patients with RAS pathway mutations expressed higher levels of the receptors of these ligands (CCR1, CCR5, CD74, and TNFRSF10B), which suggests that an aberrant feedback loop among different cell types preferentially contributes to CMML maintenance in RAS pathway mutant CMML. Together, these data are consistent with previous findings showing that, in other cancers, NF-κB signaling activation is essential for RAS pathway mutation-induced tumorigenesis.18,19,20,21
CMML monocytes, pDCs, and cDCs also gained cell-to-cell interactions with NK and eCD8T cells. Interactions involving the HLA-E-KLRC1/2, CDH1-KLRG1, LGALS9-HAVCR2, and TGFB1-TGBR1/3 ligand-receptor pairs (known to inhibit the immune cell functions22,23,24,25,26,27,28,29) were the most common (Figure S4C; Table S6). To evaluate whether CMML BM monocytes and immune cells spatially co-localized, we performed multiplex immunofluorescence analysis of BM biopsy sections obtained from CMML patients (n = 4) at the time of diagnosis. This analysis revealed that BM monocytes (CD14+CD68− cells) resided within a median of 19.73 μm (95% CI, 12.75–32.25 μm) from CD8+ T cells and 22.62 μm (95% CI, 15.73–32.17 μm) from NK cells (CD3−CD56+ cells; Figures S5A and S5B), which suggests that these cell populations interact with each other. Accordingly, both CMML NK cells (cluster 1) and eCD8T cells (cluster 3) had increased expression levels of immune checkpoint genes associated with these cells’ exhaustion (e.g., KLRG1, KLRC1, TIGIT, LAG3, CD244, B3GAT1, and CD160) compared with those from HDs (Figure S5C).30,31,32 To further characterize the functional state of CD8+ T and NK cells in CMML, we evaluated the expression of activation markers on these cells after antigen exposure. After co-culture with K562 AML cells, the frequencies of IFN-γ+ CD8+ T cells and activated CD16+ NK cells were significantly lower in RAS pathway mutant CMML but not in RAS pathway wild-type CMML, compared with those in HDs (Figure S5D). In addition, IFN-γ+ CD8+ T cells and NK cells from patients with RAS pathway mutant CMML, but not those from RAS pathway wild-type CMML, had significantly lower IFN-γ and perforin expression levels, respectively (Figure S5D).
Taken together, these data suggest that CMML HSPCs and downstream My/Mo cells undergo significant transcriptional rewiring and that RAS pathway mutations enhance the activation of cell-intrinsic and -extrinsic inflammatory networks in CMML monocyte populations to maintain cell proliferation and suppress the immune microenvironment, thus enabling immune escape and clonal expansion.
RAS pathway-mutated HSCs upregulate NF-κB transcriptional programs and drive CMML BP after HMA therapy failure
To evaluate the cellular and molecular dynamics of CMML progression, we performed scRNA-seq analysis of Lin–CD34+ HSPCs isolated from BM samples sequentially obtained from five RAS pathway mutant CMML patients at diagnosis and BP (Figures 3A and S6A; Table S7). HSPCs isolated from BM samples obtained at BP maintained aberrant differentiation toward the My/Mo lineage (Figure 3B) and had upregulated genes belonging to the NF-κB signaling pathway (Figure 3C). Importantly, MCL1, an anti-apoptotic member of the BCL2 family and a downstream effector of the NF-κB pathway, was significantly upregulated in HSCs (cluster 6) and eMyHPCs (clusters 0 and 1) at BP compared with those at diagnosis (Figures S6B and S6C; Table S8).
Figure 3.

RAS pathway-mutated HSCs undergo epigenetic reprogramming and drive CMML BP after HMA therapy failure
(A) UMAP of scRNA-seq data for pooled single Lin–CD34+ cells isolated from BM samples of five RAS pathway mutant CMML patients at diagnosis (n = 1,840) and at BP after HMA therapy failure (n = 1,711). Each dot represents one cell. Different colors represent the cluster cell-type identity (left) or sample origin (right). HSC, hematopoietic stem cells; eMyHPC, early myeloid hematopoietic progenitor cells; dMyHPC, differentiated myeloid hematopoietic progenitor cells; Ery/MkHPC, erythroid/megakaryocyte hematopoietic progenitor cells. Dashed lines indicate single clusters in each cell-type population.
(B) Distribution of Lin–CD34+ cell types at diagnosis (top) and BP (bottom) among the clusters shown in (A).
(C) Pathway enrichment analysis of the genes that were significantly upregulated in HSCs (left) and dMyHPCs (right) at the time of BP after HMA therapy failure compared with those at diagnosis (adjusted p ≤ 0.05). The top 10 hallmark gene sets are shown.
(D) UMAP of scATAC-seq data for pooled Lin–CD34+ cells isolated from BM samples obtained from three RAS pathway mutant CMML patients at diagnosis (n = 5,066) and at BP after HMA therapy failure (n = 8,603). Each dot represents one cell. Different colors represent the cluster identity (left) or sample of origin (right). HSC, hematopoietic stem cells; MyHPC, myeloid progenitor cells; Ery/MkHPC, erythroid/megakaryocyte hematopoietic progenitor cells.
(E) Pathway enrichment analysis of genes whose promoters were enriched in open chromatin regions in HSCs (clusters 0 and 4, shown in D) at the time of BP as compared with those at diagnosis (p ≤ 10−4). The top 10 hallmark gene sets are shown.
(F) Pathway enrichment analysis of genes whose distal elements were enriched in open chromatin regions in HSCs (clusters 0 and 4, shown in D) at the time of BP as compared with those at diagnosis (adjusted p ≤ 0.05). The top 10 hallmark gene sets are shown.
To evaluate whether HSPCs’ transcriptional changes at BP resulted from epigenetic reprogramming in the more primitive HSCs, we performed single-cell assays for transposase-accessible chromatin with high-throughput sequencing (scATAC-seq) to profile the chromatin accessibility landscape in Lin–CD34+ HSPCs isolated from BM samples sequentially obtained from three RAS pathway mutant CMML patients at diagnosis or BP. Our analysis identified five clusters with distinct TF binding motif enrichment in the open chromatin regions (Figures 3D and S6D; Table S9). MyHPCs (clusters 1 and 2) were characterized by open chromatin regions in the binding motifs of the myeloid TFs SPI1B and CEBPA and TFs belonging to the FOS and JUN families. HSCs (clusters 0 and 4) had the highest activities of TFs involved in stemness maintenance, such as HLF and TFs belonging to the nuclear retinoid receptor and EGR families. Ery/MkHPCs (cluster 3) were characterized by open chromatin regions in binding motifs for GATA TFs.
Consistent with our transcriptomic data, HSCs at BP had increased open chromatin peaks at the promoters of genes involved in NF-κB pathway activation and inflammatory response pathways (Figure 3E), including MCL1 (Figure S6E; Table S10). HSCs also showed increased open chromatin peaks of genes involved in NF-κB pathway activation and inflammatory response pathways at the genes’ distal elements, which define cell identity and differentiation trajectories more precisely than promoter regions do33 (Figures 3F and S6F).
Taken together, these data suggest that RAS pathway-mutated CMML HSCs exacerbate the activation of inflammatory and NF-κB pathway transcriptional programs and promote transcriptional upregulation of NF-κB signaling-mediated anti-apoptotic pathways to maintain survival at BP after HMA failure.
RAS pathway-mutated CMML cells rely on MCL1 overexpression to maintain their survival at BP
To elucidate whether MyHPCs’ transcriptional and epigenetic reprogramming drives BP, we performed scRNA-seq analysis of MNCs isolated from sequential RAS pathway-mutated CMML BM samples obtained from six CMML patients at diagnosis and BP (Figures 4A and S7A; Table S11). MNCs at BP had a significantly higher frequency of CD34+ MyHPCs (22.5% vs. 2.8%, respectively; clusters 7, 8, and 14) compared with those at diagnosis (Figures 4B and S7B). These results were confirmed by flow cytometry analysis of Lin–CD34+ cells in 70% of the patients (Figure S7C), which suggests that BP after HMA therapy is mostly driven by the expansion of the HSPC compartment. Consistent with our results in the Lin–CD34+ compartment, differential expression analysis confirmed that MyHPCs at BP had upregulated genes involved in TNF-α-mediated NF-κB activation (Figure S7D), including MCL1 (Figures S7E and S7F; Table S12). The upregulation of these genes was maintained in downstream My/Mo progenitors (My/MoPs; clusters 5 and 17) and was significantly increased in the monocytic populations (clusters 2, 3, 11, 15, and 20) (Figures 4C and S7G; Table S12). Consistent with these transcriptomic results, CD34+ BM cells from patients with RAS pathway mutant CMML at BP (n = 2) had higher MCL1 protein expression than did cells from patients at diagnosis (n = 3; Figure S7H).
Figure 4.

RAS pathway-mutated CMML cells rely on MCL1 overexpression to maintain their survival at BP
(A) UMAP of scRNA-seq data for pooled single MNCs isolated from BM samples of six RAS pathway mutant CMML patients at diagnosis (n = 16,372) and at BP after HMA therapy failure (n = 19,541). Each dot represents one cell. Different colors represent the cluster cell-type identity (left) or sample of origin (right). MyHPC, myeloid hematopoietic progenitor cells; My/MoP, myelo/monocytic progenitors; Mono, monocytes; cDC, classical dendritic cells; pDC, plasmacytoid dendritic cells; MKP, megakaryocyte precursors; Ery-E, early erythroid precursors; Ery-L, late erythroid precursors; B cell, B lymphocytes; PC, plasma cells; nCD4T, naive CD4+ T cells; mCD4T, memory CD4+ T cells; eCD8T, effector CD8+ T cells; NKC, natural killer cells. Dashed lines indicate single clusters in each cell-type population.
(B) Distribution of MNC populations at diagnosis (top) and progression (bottom) among the clusters shown in (A).
(C) Pathway enrichment analysis of the genes that were significantly upregulated in the monocytic populations (clusters 2, 3, 11, 15, and 20) shown in (A) at the time of BP after HMA therapy failure compared with those at diagnosis (adjusted p ≤ 0.05). The top 10 hallmark gene sets are shown.
(D) Numbers of live Lin–CD34+CD38– HSCs and Lin–CD34+CD38+ MyHPCs from CMML patients with BP after treatment with vehicle or AMG-176 (n = 4, 20 nM) for 48 h. Lines represent means ± SD. Statistical significance was calculated using a two-tailed Student’s t test (∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
(E) UMAP of scRNA-seq data for pooled single MNCs isolated from BM samples obtained from a representative CMML patient at the time of BP after HMA therapy failure (n = 6,209) and subsequent failure to venetoclax-based therapy (n = 6,795). Each dot represents one cell. Different colors represent the cluster cell-type identity (left) or the sample of origin (right). HSC, hematopoietic stem cells; MyHPC, myeloid hematopoietic progenitor cells; My/MoP, myelo/monocytic progenitors; Mono, monocytes; Ery/MkHPC, erythroid/megakaryocytic hematopoietic progenitor cells; Ery-E, early erythroid precursors; Ery-L, late erythroid precursors; Pre-E, pre-erythrocytes; mCD8T, memory CD8+ T cells; eCD8T, effector CD8+ T cells; NKC, natural killer cells.
(F) Pathway enrichment analysis of the genes that were significantly upregulated in MyHPCs at the time of venetoclax failure compared with those at the time of BP after HMA therapy failure (adjusted p ≤ 0.05). The top 10 hallmark gene sets are shown.
(G) Distribution of myeloid cell types among the myeloid compartments at BP after HMA therapy failure (left) and venetoclax-based therapy failure (right).
MNCs at BP exacerbated the cellular communication networks between cDCs, MyHPCs, My/MoPs, pDCs, monocytes, and eCD8T cells, compared with MNCs at baseline. Exacerbation of the cellular communication networks mainly occurred through immune-suppressive interactions between the LGALS9-HAVCR2 ligand-receptor pair, as well as increased CCL3/CCR1 and HGF/CD44 interactions between monocytes, MyMoPs and MyHPCs (Figures S8A and S8B; Table S13).
To determine whether MCL1 upregulation was a hallmark of BP in RAS pathway mutant CMML or a general mechanism of treatment resistance and progression in CMML, we performed scRNA-seq analysis of BM MNCs isolated from RAS pathway wild-type CMML samples at diagnosis (n = 3) and BP after HMA failure (n = 3) (Figures S9A and S9B; Table S14). RAS pathway wild-type BM MNCs at progression had a higher frequency of MyHPCs compared with those at diagnosis (8.1% vs. 3.9%, respectively; cluster 7; Figure S9C). MyHPCs at BP upregulated genes involved in TNF-α-mediated NF-κB activation but not MCL1 (Figures S9D and S9E; Table S15). Similar data were also observed in downstream My/MoPs and monocytes (Figure S9D; Table S15). Consistent with these findings, CD34+ BM cells from patients with RAS pathway wild-type CMML at diagnosis (n = 3) and BP (n = 3) had similar MCL1 protein expression levels (Figure S7H).
Together, these data suggest that only RAS pathway-mutated CMML MyHPCs and monocytes rely on MCL1-driven anti-apoptotic pathways to maintain survival and expand after therapy failure. To test this hypothesis, we treated Lin–CD34+ HSPCs isolated from the BM of patients with RAS pathway-mutated CMML with the MCL1 inhibitor AMG-17634 (at a dose that did not deplete Lin–CD34+CD38– or Lin–CD34+CD38+ HSPCs isolated from the BM of HDs in co-culture system with mesenchymal stromal cells; Figure S10A). AMG-176 significantly decreased the numbers of Lin–CD34+CD38– and Lin–CD34+CD38+ HSPCs isolated from BM samples obtained from patients with RAS pathway-mutated CMML at BP (Figure 4D). AMG-176 did not significantly affect the survival of HSPCs isolated from BM samples obtained from patients at diagnosis (Figure S10B), which confirms that CMML HSPCs maintain an intact apoptotic program at disease initiation. Consistent with our scRNA-seq analysis showing that MCL1 was not upregulated in RAS pathway wild-type HSPCs, the treatment with AMG-176 did not deplete RAS pathway wild-type HSPCs at the time of BP after HMA failure (Figure S10C).
Importantly, BCL2 was not overexpressed in either RAS pathway mutant CMML HSPCs or downstream My/Mo populations at BP after HMA therapy failure (Figure S10D). BCL2 expression was significantly downregulated at progression in MyHPCs and My/MoPs from CMML patients without detectable RAS pathway mutations at the time of BP (Figure S10E). These findings are consistent with our previous clinical observation that CMML patients in whom HMA therapy has failed do not benefit from second-line therapy with venetoclax.11 Indeed, scRNA-seq analysis of MNCs isolated from sequential BM samples obtained from one representative RAS pathway mutant CMML patient whose disease progressed after HMA therapy failure and did not respond to venetoclax therapy (Figures 4E and S10F; Table S16) revealed that MyHPCs further exacerbate the expression of genes involved in TNF-α-mediated NF-κB pathway activation (Figure 4F), including MCL1 (Figure S10G). Venetoclax failure was associated with a significant expansion of downstream My/MoPs in the myeloid compartment (Figure 4G) and these cells’ high expression of MCL1 (Figure S10G).
Taken together, our findings suggest that venetoclax therapy cannot overcome HMA failure-induced transcriptional reprogramming in My/MoPs and provide a rationale for targeting effectors of the NF-κB signaling pathway, such as MCL1, in patients with RAS pathway mutant CMML to improve the dismal outcome of CMML patients whose disease is resistant to available therapies.
Discussion
Whereas the dissection of the molecular landscape of CMML initiation and progression has significantly advanced our understanding of the pathogenesis of CMML,1,35,36,37,38 the development of more effective therapeutic approaches to improve patient survival has been delayed by our limited understanding of the ways in which genetic alterations affect distinct transcriptional states of My/Mo differentiation.
Mutations in RAS pathway genes, which are present in 30% of CMML patients,38 are enriched during disease progression in up to 90% of the cases and predict a higher risk of and a shorter time to relapse after HMA and venetoclax therapy.39 Currently, there are no other therapies that improve the survival duration of patients with RAS pathway-mutated CMML.
Using single-cell multi-omics technologies, we sought to dissect the biological mechanisms behind RAS pathway mutation-induced CMML evolution with the overall goal of identifying cellular vulnerabilities that could be therapeutically targeted to halt disease progression. We found that, at disease initiation, RAS pathway mutant CMML HSPCs significantly upregulated genes involved in the cell-intrinsic IFN signaling pathway such as IRF1, IRF7, IRF9, IFI44, IFI44L, IFIH1, IFIT3, or STAT2 that drive these cells’ differentiation toward the My/Mo lineage while maintaining an intact apoptotic program. Consistent with this observation and prior studies showing that KRAS or NRAS mutations directly activate intrinsic IFN-stimulated genes,40 IFN signaling activation in HSPCs was not associated with IFN receptor (IFNAR1, IFNAR2, IFNGR1, or IFNG2) or ligand (IFNG or IFNA) overexpression. In addition, this inflammatory reprogramming was exacerbated in downstream RAS pathway mutant monocyte populations, which expressed high levels of cytokines and cell surface receptors involved in NF-κB pathway activation and immune evasion. These results suggest that disease initiation and maintenance, as a result of RAS pathway mutations, rely on the activation of both cell-intrinsic and -extrinsic inflammatory networks in distinct cell populations and provide a rationale for using inhibitors of NF-κB-associated inflammatory signaling cascades as a frontline treatment for patients with RAS pathway-mutated CMML. These findings, which are consistent with previous studies showing the role of inflammatory cell populations in myeloid malignancies,41,42 have significant implications since several inflammation-targeting therapies that are currently in clinical development have shown great potential to treat patients with myeloid malignancies.43,44,45
Consistent with the long-standing observation that inhibition of apoptosis contributes to therapy resistance and cancer progression, we found that RAS pathway mutant CMML HSPCs isolated from BM samples at the time of BP depended on MCL1, an anti-apoptotic downstream effector of the NF-κB pathway, to maintain their survival and undergo clonal expansion. Consistent with this observation, we had demonstrated previously that TNF-α-mediated NF-κB pathway activation represents a cell-intrinsic adaptive mechanism to overcome cell death in response to therapeutic pressure.46 In addition, our findings align with prior data demonstrating that RAS mutations can directly induce NF-κB hyperactivation.21,47 Notably, targeting MCL1 activity with the small molecule AMG-176 only significantly depleted HSPCs from RAS mutant CMML but not those from RAS wild-type CMML, a finding that supports the selective use of MCL1 inhibitors to treat patients with RAS pathway mutant CMML in whom BP occurs at the time of HMA therapy failure. These results are consistent with previous findings showing that CMML monocytes rely on MCL1, but not BCL2, for survival,48 and that NRAS-mutant monocytic subclones that emerge at AML relapse depend on MCL1, not BCL2, for energy production.49 Consistent with this observation, our scRNA-seq analysis of BM MNCs from one representative patient with venetoclax-resistant disease confirmed that BCL2 inhibition cannot overcome the activation of NF-κB pathway-mediated inflammatory and survival mechanisms in HSPCs and downstream My/Mo populations.
In conclusion, this study highlights the importance of dissecting how specific genetic drivers affect the cell-of-origin in cancer to gain mechanistic insights into therapy failure and, thereby, develop selective therapeutic approaches to halt disease progression. Given that the RAS pathway mutation-induced reprogramming of CMML cells is a multi-step process that affects multiple biological signaling pathways (e.g., inflammation, apoptosis, and immune escape) in distinct BM cell types, our findings also suggest that only combination therapies that simultaneously target these pathways could effectively overcome disease progression and prolong the survival of patients whose disease is resistant to current therapeutic approaches.
Limitations of the study
In this study, we used 3′ RNA-seq by 10X Genomics, which evaluates RNA transcript expression levels for individual genes at the single-cell level but does not capture the entire RNA sequence, hence not allowing inference of the complete cDNA sequence and somatic mutation detection. Therefore, we were not able to correlate transcriptome to RAS pathway mutation status with single-cell resolution in all sequenced cases. Although we attempted to mitigate this intrinsic limitation to our sequencing technique by selecting samples with high RAS pathway mutant variant allele frequencies (VAFs), future studies will require the use of alternative single-cell sequencing technologies able to simultaneously capture genotype and transcriptome at the single-cell level to invariably characterize the specific features of RAS pathway mutant vs. wild-type cells. In an attempt to mitigate the impact of such a limitation in our identification of MCL1 upregulation as a preferential RAS mutant cell survival mechanism, we confirmed MCL1 upregulation at the protein level by evaluating RAS pathway mutant samples with high VAFs. In addition, although we were able to confirm the selective sensitivity of RAS pathway mutant Lin−CD34+ cells to MCL1 inhibition at the time of progression after HMA therapy failure, there are inherent limitations to the extent to which these studies can capture the in vivo effects of MCL1 inhibition and how this could affect distinct cell types and functionalities. Finally, although we validated our transcriptomic findings related to cell-cell communication networks between key BM populations with multiplex immunofluorescence and immunophenotypic immune cell characterization, future studies will require deeper investigation and validation of these interactions.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD2 | BD Biosciences | Cat#: 555326; RRID: AB_395733 |
| Anti-human CD3 | BD Biosciences | Cat#: 349201; RRID: AB_400405 |
| Anti-human CD4 | ThermoFisher | Cat#: MHCD0401; RRID:AB_10392546 |
| Anti-human CD7 | BioLegend | Cat#: 343104; RRID: AB_1659216 |
| Anti-human CD11b | ThermoFisher | Cat#: 11-0118-42; RRID: AB_1582242 |
| anti-human CD14 | BD Biosciences | Cat#: 347493; RRID: AB_400311 |
| Anti-human CD19 | BD Biosciences | Cat#: 340409; RRID: AB_400024 |
| Anti-human CD20 | BD Biosciences | Cat#: 555622; RRID: AB_395988 |
| Anti-human CD33 | ThermoFisher | Cat#: 11-0337-42; RRID: AB_1603221 |
| Anti-human CD56 | BD Biosciences | Cat#: 562794; RRID: AB_2737799 |
| Anti-human CD235 | BD Biosciences | Cat#: 559943; RRID: AB_397386 |
| Anti-human CD34 | BD Biosciences | Cat#: 562577; RRID: AB_2687922 |
| Anti-human CD38 | BioLegend | Cat#: 303534; RRID: AB_2561605 |
| Anti-human CD3 | BioLegend | Cat#: 317339; RRID: AB_2563407 |
| Anti-human CD4 | BioLegend | Cat#: 300502; RRID: AB_314070 |
| Anti-human CD8 | BioLegend | Cat#: 344710; RRID: AB_2044010 |
| Anti-human CD56 | BioLegend | Cat#: 362544; RRID: AB_2565922 |
| Anti-human CD16 | BioLegend | Cat#: 302028; RRID: AB_893262 |
| Anti-Human IFN-γ | BD Biosciences | Cat#: 554702; RRID: AB_398580 |
| Anti-human Perforin | BioLegend | Cat#: 308130; RRID: AB_2687190 |
| Zombie UV | BioLegend | Cat#: 423107 |
| Protein Transport Inhibitor Cocktail | eBioscience | Cat#: 00-4980-93 |
| Fc Receptor Binding Inhibitor Polyclonal Antibody | ThermoFisher | Cat#: 14-9161-73 |
| CD3e | Cell Signaling Technology | Cat#: 85061BF; RRID: AB_2721019 |
| CD4 | Abcam | Cat#: ab133616; RRID: AB_2750883 |
| CD8 | Thermo Scientific | Catalog#: MA5-13473; RRID: AB_11000353 |
| CD14 | Abcam | Cat#: ab183322; RRID: AB_2909463 |
| CD68, Clone PG-M1 | Dako | Cat#: M0876; RRID: AB_2074844 |
| CD56 | Dako | Cat#: M7304; RRID: AB_2750583 |
| DAPI | Akoya Biosciences | Cat#: FP1490; RRID: N/A |
| MCL1 | Cell Signaling Technology | Cat#: 4572; RRID:AB_2281980 |
| Vinculin | Sigma-Aldrich | Cat#: V9131; RRID:AB_477629 |
| Biological samples | ||
| Bone marrow aspirates from healthy donors | AllCells (Alameda, CA) and MD Anderson’s Department of Stem Cell Transplantation | N/A |
| Bone marrow aspirates from CMML patients | MD Anderson Bank | N/A |
| Human bone marrow-derived mesenchymal stem cells | Cells provided by Dr. M. Andreeff | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| AMG-176 | Med Chem Express | Cat#: HY-101565 |
| Critical commercial assays | ||
| Cytofix/Cytoperm kit | BD Biosciences | Cat#: 554714 |
| Deposited data | ||
| scRNA-seq data | This paper | GEO: GSE218390 |
| scATAC-seq data | This paper | GEO: GSE218390 |
| scDNA-seq data | This paper | GEO: GSE218390 |
| Experimental models: Cell lines | ||
| K562 cells | ATCC | ATCC CCL243 |
| Software and algorithms | ||
| SPSS 23.0 | SPSS, Inc | https://www.ibm.com/products/spss-statistics |
| R v4.1.2 | R Core Team | https://www.r-project.org |
| Seurat v4 R package ‘ | Hao et al.50 | https://github.com/satijalab/seurat |
| CellphoneDB | Vento-Tormo et al.51 | https://www.cellphonedb.org/ |
| GraphPad Prism version 10.0 | GraphPad software | https://www.graphpad.com/ |
| Spotfire | TIBCO | N/A |
| FlowJo | BD Biosciences | https://www.flowjo.com/ |
| GSEA | Metascape | https://metascape.org/ |
| Spotfire | TIBCO | https://www.spotfire.com/ |
| Other | ||
| Ficoll-Paque PLUS | ThermoFisher | Cat#:45-001-752 |
| CD34 Microbead Kit | Miltenyi Biotec | Cat#:130-046-702 |
| NK Cell Isolation Kit | Miltenyi Biotec | Cat#: 130-092-657 |
| Opal 9 Kit | Akoya Biosciences | Cat#: NEL797001KT |
| Qubit | Thermo Fisher Scientific | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Simona Colla (scolla@mdanderson.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
scRNA-seq, scATAC-seq, and scDNA-seq data are accessible at GEO under accession number GSE218390. No custom computer codes were generated in this study.
-
•
The lead contact can provide any additional information required to reanalyze the data reported in this work paper upon request.
Experimental model and study participant details
Primary human samples
BM aspirates were obtained from patients with CMML who were seen in the Department of Leukemia at the University of Texas MD Anderson Cancer Center. Samples were obtained with the approval of the Institutional Review Board and in accordance with the Declaration of Helsinki. CMML diagnoses were assigned according to the World Health Organization criteria.3
RAS pathway mutations were identified by targeted amplicon-based next-generation sequencing (NSG).52 Genomic DNA was extracted from whole BM aspirate samples and was subject to targeted PCR-based sequencing using an NGS platform evaluating a total of 81 genes, as previously described.52 This analysis was performed within the MDACC CLIA-certified Molecular Diagnostics Laboratory after informed consent (additional details in supplemental information). For NGS-based analysis, the limit of detection for variant calling was 2%. Previously described somatic mutations registered at the Catalog of Somatic Mutations in Cancer (COSMIC: http://cancer.sanger.ac.uk/cosmic) were considered potential driver mutations.
All available samples carrying RAS pathway mutations were included in the study. Baseline BM aspirates were collected from patients before any treatment. Sequential BM samples were collected after HMA or venetoclax therapy failure. The clinical characteristics of the patients with RAS pathway mutated CMML are shown in Tables S1 and S3. BM samples from HDs were obtained from AllCells (Alameda, CA) and the Department of Stem Cell Transplantation at MD Anderson Cancer Center. Written informed consent was obtained from all donors.
MNCs were collected from each BM sample immediately after BM aspiration using the standard gradient separation approach with Ficoll-Paque PLUS (catalog number #45-001-752, Thermo Fisher Scientific). MNCs were cryopreserved and stored in liquid nitrogen until they were used. For cell sorting applications, MNCs were enriched in CD34+ cells using magnetic-activated cell sorting (MACS) with the CD34 Microbead Kit (catalog number #130-046-702, Miltenyi Biotec, Germany) and further purified by fluorescence-activated cell sorting (FACS) as described below.
Method Details
Clinical data analysis
A clinical dataset of 108 CMML patients treated with HMA therapy at the Department of Leukemia at the University of Texas MD Anderson Cancer Center was evaluated to identify predictors of therapy outcomes. HMA therapy failure was defined as a lack of response (based on IWG 2006 criteria) after at least 4 cycles of therapy or as relapse or progression after any number of cycles of therapy. Blast progression (BP) was defined as 1) the presence of >5% blasts in the BM at the time of primary HMA failure in patients with <5% blasts at baseline or an increase of at least 50% blasts in patients with 5–9% blasts at baseline; 2) BM blasts >20% or myeloid sarcoma regardless of primary or secondary failure; 3) BM blasts >5% at the time of secondary HMA failure (relapse or progression). Associations between gene mutations and BP were assessed using data from 108 patients whose samples were sequenced using the 81-gene panel; these analyses were performed at MD Anderson Cancer Center. Clinical datasets were analyzed using the SPSS 23.0 (SPSS, Inc., Chicago, IL, USA) and R (version 3.5.1) statistical software programs. Logistical regression analysis was performed using clinical, cytogenetic, and molecular characteristics in correlation with responses to HMA therapy. The dataset was randomly divided into a training set (30 patients with BP) and a testing set (5 patients with BP). A combination rule derived from selected features was trained using logistic regression in the training set and a fixed model in the testing set. Receiver operating characteristic (ROC) curves were generated using the “pROC” package in R (version 3.6.0). The 95% CIs for the areas under the ROC curves were estimated using the DeLong method.53 The chi-square or Fisher exact test was used to analyze differences between categorical variables. Survival curves were generated using the Kaplan-Meier method and compared using log rank tests. Responses to HMA- or venetoclax-based therapies were evaluated based on the International Working group 200354 and 200655 criteria for patients with secondary AML or CMML, respectively.
Flow cytometry analysis and fluorescence-activated cell sorting (FACS)
Quantitative flow cytometry and FACS analyses of Lin–CD34+ cells were performed using previously described staining protocols56,57 and antibodies against CD2, FITC, RPA-2.10, BD Biosciences, 555326; CD3, FITC, SK7, BD Biosciences, 349201; CD4, FITC, S3.5, Thermo Fisher, MHCD0401; CD7, FITC, 6B7, BioLegend, 343104; CD11b, FITC, ICRF44, Thermo Fisher, 11-0118-42; CD14, FITC, MφP9, BD Biosciences, 347493; CD19, FITC, SJ25C1, BD Biosciences, 340409; CD20, FITC, 2H7, BD Biosciences, 555622; CD33, FITC, P67.6, Thermo Fisher, 11-0337-42; CD56, FITC, B159, BD Biosciences, 562794; CD235a, FITC, HIR2, BD Biosciences, 559943; CD34, BV421, 581, BD Biosciences, 562577; CD38, APC, HIT2, BioLegend, 303534, as we described previously.58
Samples used for flow cytometry and FACS were acquired with a BD LSR Fortessa and a BD Influx Cell Sorter (BD Biosciences), respectively. The cell populations were analyzed using FlowJo software (https://www.flowjo.com). All experiments included single-stained controls and were performed at the South Campus Flow Cytometry & Cellular Imaging Facility at MD Anderson Cancer Center.
Multiplex imaging assay
BM core biopsies were used for multiplex immunofluorescence assessment. We optimized and validated a multiplex immunofluorescence panel using antibodies against CD3e, CD4, CD8, CD14, CD56, and CD68. Each antibody was assessed by multiplex immunofluorescence using the Opal 9 kit (catalog #NEL797001KT; Akoya Biosciences, Marlborough, MA), according to the following clones and dilutions: CD3e (clone D7A6E(AM), Cell Signaling Technology, 1:100), CD4 (clone EPR6855, Abcam, 1:200), CD8 (clone C8/144B, Thermo Scientific, 1:25), CD14 (clone SP192, Abcam, 1:100), CD56 (clone 123C3, Dako, 1:25), and CD68 (clone PG-M1, Dako, 1:50). The slides were imaged using the Vectra Polaris spectral imaging system (Akoya Biosciences, Marlborough, MA) using the fluorescence protocol at 10 nm λ from 420 nm to 720 nm. Both germinal center and interfollicular areas from lymph nodes with reactive lymphoid hyperplasia were used as positive controls. Each marker was analyzed at the single-cell level, and a supervised algorithm for phenotyping was built for each marker. Cell density for each marker and combinations of phenotypes were consolidated using Spotfire software (TIBCO Spotfire). The nearest neighbor analysis was performed using R version 4.2.1.
Western blot
BM CD34+ cells were enriched from BM MNCs using magnetic sorting with the CD34 Microbead Kit (Miltenyi Biotec). Cells were resuspended in Mammalian Cell & Tissue Extraction Kit buffer (BioVision Incorporated) and incubated on ice for 10 min. Lysates were then collected after centrifugation at 12,000 rpm at 4°C for 20 min. The amount of protein was quantified using the Qubit Protein Assay Kit and a Qubit Fluorometer (Thermo Fisher Scientific). SDS-PAGE and Western blotting were performed following standard protocols. Blotted membranes were incubated with primary monoclonal antibodies against human MCL1 (#4572S; 1:750 dilution; Cell Signaling Technology) and vinculin (hVIN-1; 1:2,000 dilution; Sigma-Aldrich). Membranes were developed using the SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific) in a KwikQuant Imager (Kindle Biosciences). Vinculin was used as a loading control, and lysates from the myeloma cell line JJN3 were used as positive controls.
T cell and NK cell cytokine secretion assays
NK cells were isolated from BM MNCs obtained from HDs, and RAS pathway mutant or wild-type CMML patients by negative magnetic selection using the NK Cell Isolation Kit (Miltenyi Biotec). NK cells or BM MNCs were mixed with the human erythroleukemia cell line K562 at a target-to-effector ratio of 1:1. Cells were incubated for 4h at 37°C in 5% CO2 in the presence of a protein transport inhibitor cocktail (eBioscience 00-4980-93) for 4 h. After incubation, cells were harvested, washed with PBS, and stained with the viability dye Zombie UV. Cells were washed with PBS and resuspended in the presence of an Fc receptor-binding inhibitor antibody (ThermoFisher) for 20 min. NK cells and BM MNCs were stained with antibodies against CD3 (AF700, BioLegend). NK cells were further stained with antibodies against CD56 (PE-Dazle 594) and CD16 (PerCP-Cy5.5), whereas BM MNCs were stained with antibodies against CD4 (PE-Dazzle 594) and CD8 (PerCP-Cy5.5). Cells were then washed, fixed, and permeabilized using the Cytofix/Cytoperm kit (BD Biosciences) and intracellularly stained with antibodies against IFN-γ (APC, BD Biosciences) and perforin (BV711, BioLegend). Samples were acquired with a BD Fortessa (BD Biosciences), and cell populations were analyzed using FlowJo software (version 10.7.1, Ashland, OR).
scRNA-seq analysis and bioinformatic pipeline
ScRNA-seq analysis was performed as we described previously.58 Live Lin–CD34+ cells and live MNCs were isolated by FACS. Sample preparation and sequencing were performed at the Advanced Technology Genomics Core at MD Anderson Cancer Center. Sample concentration and cell suspension viability were evaluated using a Countess II FL Automated Cell Counter (Thermo Fisher Scientific). Samples were normalized for input onto the Chromium Single Cell A Chip Kit (10X Genomics), and single cells were lysed and barcoded for reverse transcription. Equal amounts of each uniquely indexed sample library were pooled together. Pooled libraries were sequenced using a NovaSeq6000 SP 100-cycle flow cell (Illumina). After sequencing, raw reads were aligned to the human genome (hg38), and the digital expression matrix was generated using cellranger count. Individual samples were merged to generate the digital expression matrix using cellranger aggr. The Seurat package in R was used to analyze the digital expression matrix. Cells with less than 100 genes and less than 500 unique molecular identifiers detected were not analyzed further. The Seurat function NormalizeData was used to normalize the raw counts. Variable genes were identified using the FindVariableGenes function. The Seurat ScaleData function was used to scale and center expression values in the dataset for dimensional reduction. Default parameters were used for the Seurat functions. When needed, samples were integrated using the Seurat functions FindIntegrationAnchors and IntegrateData. Principal component analysis and Uniform Manifold Approximation and Projection (UMAP) were used to reduce the dimensions of the data, and the first 2 dimensions were used in plots. To cluster the cells and determine the marker genes for each cluster, we used the FindClusters and FindAllMarkers functions, respectively. Differential expression analysis of the samples was performed using the FindMarkers function and the Wilcoxon rank-sum test. The Benjamini-Hochberg procedure was applied to adjust the false discovery rate. Functional enrichment analysis was performed using the Metascape software (https://metascape.org/gp/index.html#/main/step1).59 The human hallmark gene set was used. Analyses were performed using gene annotation available in 2020–2023.
CellphoneDB (v2.0.0)17 was used to analyze the ligand–receptor interactions. Briefly, each cell type was separated by disease classification, and a separate run was performed for each disease classification. The connectome web was plotted using the igraph package in R.
scATAC-seq analysis and bioinformatic pipeline
ScATAC-seq analysis was performed as we described previously.58 The scATAC-seq Low Cell Input Nuclei Isolation protocol (10X Genomics) was used to isolate nuclei from FACS-purified cells. Extracted nuclei were used for the consecutive steps of the scATAC-seq library preparation protocol following 10X Genomics guidelines. Equal molar concentrations of uniquely indexed samples were pooled together. Pooled libraries were sequenced using a NextSeq500 150-cycle flow cell (Illumina). Reads were aligned to human (hg38) genomes, and peaks were called using the cellranger-atac count pipeline. Individual samples were merged using the cellranger-atac pipeline to generate the peak-barcode matrix and TF-barcode matrix. To identify specific TF activity for each cell cluster, we used the R package Seurat to analyze the TF-barcode matrix. The raw counts were normalized by the sequencing depth for each cell and scaled for each TF using the NormalizeData and ScaleData functions. Principal component analysis and UMAP were applied to reduce the dimensions of the data, and the first 2 dimensions were plotted. The FindClusters function was used to cluster the cells. The FindAllMarkers function was used to determine the TF markers for each cluster. Differential analysis of TF activity in the samples was performed using the FindMarkers function and the Wilcoxon rank-sum test. Cluster identity was determined based on the activity of master regulators of lineage commitment, as we60 and others33,61 described previously. Cluster-specific peaks were determined using the FindAllMarkers function, and differentially accessible peaks between the samples were determined using the FindMarkers function. Each peak was associated with a specific gene based on its distance to that gene’s transcription start site (TSS). Peaks overlapping with a promoter region (−1,000 bp, +100 bp) of any TSS were annotated as peaks in promoters, whereas peaks not in promoter regions but within 200 kb of the closest TSS were annotated as peaks in the distal elements. Peaks not mapped in either the promoters or distal elements were annotated as peaks in intergenic regions.
scDNA and protein-seq analysis
Simultaneous analyses of DNA mutations and the cell-surface immunophenotype (scDNA and protein-seq) were performed as we described previously62 and according to the Mission Bio protocol using the custom-designed 37-gene myeloid panel kit and 48 oligo-conjugated antibodies against all major BM cell types (Biolegend). Briefly, cryopreserved BM MNCs were thawed, quantified, and then stained with the pool of the oligo-conjugated antibodies. Stained cells were washed and loaded onto the Tapestri machine for single-cell encapsulation, lysis, and barcoding. DNA libraries were extracted from the droplets followed by the purification using Ampure XP beads (Beckman Coulter). Then, the supernatant was incubated with biotinylated oligonucleotides (Integrated DNA Technologies) to capture the antibody tags, followed by purification using streptavidin beads (Thermo Fisher Scientific). Purified DNA and antibody-tagged libraries were indexed and then sequenced on the Illumina NovaSeq 6000 or NextSeq 500 systems with 150 bp paired-end multiplexed runs.
The resulting files containing DNA and protein data were visualized using the Mission Bio Mosaic library version 1.8. Only manually curated and whitelisted variants were used. Variants were filtered using the below setting: min_dp = 5, min_gq = 0, min_vaf = 21, max_vaf = 100, min_prct_cells = 0, min_mut_prct_cells = 0, and min_std = 0. Protein reads were normalized by centered log ratio, and subsequently underwent dimensionality reduction and clustering using Mosaic ‘run_pca’ (components = 15), ‘run_umap’ (attribute = 'pca', n_neighbors = 20, metric = 'cosine', min_dist = 0), and ‘cluster’ (attribute = 'umap', method = 'graph-community', k = 150). Default parameters were used unless otherwise specified, and randomness was controlled in all steps. Heatmaps were separately visualized in R using the ComplexHeatmap package (https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html).
Primary cell culture assays
FACS-purified Lin–CD34+ HSPCs were resuspended in cytokine-free sterile RPMI medium supplemented with 10% FBS, 1% penicillin-streptomycin, and 0.1% amphotericin B and plated in 48-well plates previously seeded with low-passage (p ≤ 4) healthy BM-derived human mesenchymal cells. Co-cultures were incubated at 37°C in a 5% CO2 atmosphere. After treatment with vehicle or AMG-176 (20 nM) for 48 h, cells were harvested and stained for quantitative flow cytometric analysis using the antibody panel described above and with AccuCheck Counting Beads (Thermo Fisher Scientific) added to each tube.
Quantification and statistical analysis
Statistical analysis of flow cytometry data was performed using Prism 8 software (https://www.graphpad.com). The figure legends include the statistical test(s) used in each experiment. Statistical significance was represented as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. In all analyses involving human samples, investigators were blinded to sample annotations and patient outcomes. For replicated experiments, the number of replicates is indicated in the figure legends. No statistical method was used to predetermine the sample size. No data were excluded from the analyses. The experiments were not randomized. Statistical analysis for blast progression and survival in the clinical cohort was performed as specified in the “clinical data analysis” section (method details).
Acknowledgments
This work was supported by philanthropic contributions to MD Anderson’s AML/MDS Moon Shot, the Edward P. Evans Foundation, and the National Institutes of Health through a Leukemia SPORE grant awarded to the MD Anderson Cancer Center (P50 CA100632). J.J.R.-S. is a recipient of the Odyssey Fellowship awarded by the MD Anderson Cancer Center. This work used MD Anderson’s South Campus Flow Cytometry & Cellular Imaging Facility and the Advanced Technology Genomics Core at the MD Anderson Cancer Center; the facilities are supported in part by the National Institutes of Health (National Cancer Institute) through MD Anderson’s Cancer Center Support Grant (P30 CA16672). The authors thank Joseph Munch for assistance with manuscript editing. The authors give thanks for the technical support of the following Translational Molecular Pathology-Immunoprofiling Laboratory members: Daniela E. Duenas, Mario L. Marques-Piubelli, Mei Jiang, Beatriz Sanchez-Espidion, Salome McAllen, and Jianling Zhou. The graphical abstract was created with Biorender.com.
Author contributions
G.M.-B. and S.C. designed the research. N.T., J.J.R.-S., I.G.-G., H.Y., V.A., and B.W. performed the experiments. F.M. analyzed scRNA and scATAC-seq data. S.L., R.K.-S., X.S., and J.Z. analyzed targeted DNA sequencing data. Y.J.K., T.T., and K.T. performed scDNA and protein-seq experiments. K.D. analyzed the flow cytometry data. G.M.-B. and J.J.R.-S. identified the clinical samples included in the study. G.M.-B. and F.D. analyzed the clinical data. G.A.-A. provided the HD samples. H.K. and G.G.-M. made critical intellectual contributions throughout the study. G.M.-B. and S.C. wrote the manuscript.
Declaration of interests
G.M.-B. declares research support from Rigel Pharmaceuticals, IFM Therapeutics, and Takeda Oncology. K.T. declares support from Symbio Pharmaceuticals, Novartis, Celgene/BMS, and GSK, and honoraria from Mission Bio and Illumina. H.K. declares research support from and an advisory role at Actinium and research support from AbbVie, Agio, Amgen, Ariad, Astex, Bristol Myers Squibb, Cyclacel, Daiichi-Sankyo, Immunogen, Jazz Pharma, Novartis, and Pfizer. G.G-M. declares research support from and an advisory role at Bristol Myers Squibb, Astex, and Helsinn, and research support from Amphivena, Novartis, AbbVie, H3 Biomedicine, Onconova, and Merck. S.C. declares research support from Amgen.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work, S.C. used Grammarly to correct grammar mistakes. After using this tool, S.C. reviewed and edited the content as needed. S.C. takes full responsibility for the content of the publication.
Published: May 22, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101585.
Supplemental information
References
- 1.Itzykson R., Kosmider O., Renneville A., Morabito M., Preudhomme C., Berthon C., Adès L., Fenaux P., Platzbecker U., Gagey O., et al. Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121:2186–2198. doi: 10.1182/blood-2012-06-440347. [DOI] [PubMed] [Google Scholar]
- 2.Arber D.A., Orazi A., Hasserjian R., Thiele J., Borowitz M.J., Le Beau M.M., Bloomfield C.D., Cazzola M., Vardiman J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 3.Vardiman J.W., Thiele J., Arber D.A., Brunning R.D., Borowitz M.J., Porwit A., Harris N.L., Le Beau M.M., Hellström-Lindberg E., Tefferi A., Bloomfield C.D. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–951. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 4.Patnaik M.M., Padron E., LaBorde R.R., Lasho T.L., Finke C.M., Hanson C.A., Hodnefield J.M., Knudson R.A., Ketterling R.P., Al-kali A., et al. Mayo prognostic model for whom-defined chronic myelomonocytic leukemia: ASXL1 and spliceosome component mutations and outcomes. Leukemia. 2013;27:1504–1510. doi: 10.1038/leu.2013.88. [DOI] [PubMed] [Google Scholar]
- 5.Onida F., Kantarjian H.M., Smith T.L., Ball G., Keating M.J., Estey E.H., Glassman A.B., Albitar M., Kwari M.I., Beran M. Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood. 2002;99:840–849. doi: 10.1182/blood.v99.3.840. [DOI] [PubMed] [Google Scholar]
- 6.Patnaik M.M., Itzykson R., Lasho T.L., Kosmider O., Finke C.M., Hanson C.A., Knudson R.A., Ketterling R.P., Tefferi A., Solary E. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia. 2014;28:2206–2212. doi: 10.1038/leu.2014.125. [DOI] [PubMed] [Google Scholar]
- 7.Pleyer L., Leisch M., Kourakli A., Padron E., Maciejewski J.P., Xicoy Cirici B., Kaivers J., Ungerstedt J., Heibl S., Patiou P., et al. Outcomes of patients with chronic myelomonocytic leukaemia treated with non-curative therapies: a retrospective cohort study. Lancet. Haematol. 2021;8:e135–e148. doi: 10.1016/S2352-3026(20)30374-4. [DOI] [PubMed] [Google Scholar]
- 8.Alfonso A., Montalban-Bravo G., Takahashi K., Jabbour E.J., Kadia T., Ravandi F., Cortes J., Estrov Z., Borthakur G., Pemmaraju N., et al. Natural history of chronic myelomonocytic leukemia treated with hypomethylating agents. Am. J. Hematol. 2017;92:599–606. doi: 10.1002/ajh.24735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Montalban-Bravo G., Kanagal-Shamanna R., Li Z., Hammond D., Chien K., Rodriguez-Sevilla J.J., Sasaki K., Jabbour E., DiNardo C., Takahashi K., et al. Phenotypic subtypes of leukaemic transformation in chronic myelomonocytic leukaemia. Br. J. Haematol. 2023;203:581–592. doi: 10.1111/bjh.19060. [DOI] [PubMed] [Google Scholar]
- 10.Itzykson R., Kosmider O., Renneville A., Gelsi-Boyer V., Meggendorfer M., Morabito M., Berthon C., Adès L., Fenaux P., Beyne-Rauzy O., et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J. Clin. Oncol. 2013;31:2428–2436. doi: 10.1200/JCO.2012.47.3314. [DOI] [PubMed] [Google Scholar]
- 11.Montalban-Bravo G., Hammond D., DiNardo C.D., Konopleva M., Borthakur G., Short N.J., Ramos-Perez J., Guerra V., Kanagal-Shamanna R., Naqvi K., et al. Activity of venetoclax-based therapy in chronic myelomonocytic leukemia. Leukemia. 2021;35:1494–1499. doi: 10.1038/s41375-021-01240-2. [DOI] [PubMed] [Google Scholar]
- 12.Paul F., Arkin Y., Giladi A., Jaitin D.A., Kenigsberg E., Keren-Shaul H., Winter D., Lara-Astiaso D., Gury M., Weiner A., et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell. 2015;163:1663–1677. doi: 10.1016/j.cell.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 13.Giladi A., Paul F., Herzog Y., Lubling Y., Weiner A., Yofe I., Jaitin D., Cabezas-Wallscheid N., Dress R., Ginhoux F., et al. Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat. Cell Biol. 2018;20:836–846. doi: 10.1038/s41556-018-0121-4. [DOI] [PubMed] [Google Scholar]
- 14.Lian Q., Xin H., Ma J., Konnikova L., Chen W., Gu J., Chen K. Artificial-cell-type aware cell-type classification in CITE-seq. Bioinformatics. 2020;36:i542–i550. doi: 10.1093/bioinformatics/btaa467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Z., Ye C., Wang J., Zhang N.R. Surface protein imputation from single cell transcriptomes by deep neural networks. Nat. Commun. 2020;11:651. doi: 10.1038/s41467-020-14391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamarsheh S., Groß O., Brummer T., Zeiser R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020;11:5439. doi: 10.1038/s41467-020-19288-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 18.Chenette E.J. Cancer: A Ras and NF-kappaB pas de deux. Nat. Rev. Drug Discov. 2009;8:932. doi: 10.1038/nrd3060. [DOI] [PubMed] [Google Scholar]
- 19.Datta J., Bianchi A., De Castro Silva I., Deshpande N.U., Cao L.L., Mehra S., Singh S., Rafie C., Sun X., Chen X., et al. Distinct mechanisms of innate and adaptive immune regulation underlie poor oncologic outcomes associated with KRAS-TP53 co-alteration in pancreatic cancer. Oncogene. 2022;41:3640–3654. doi: 10.1038/s41388-022-02368-w. [DOI] [PubMed] [Google Scholar]
- 20.Daniluk J., Liu Y., Deng D., Chu J., Huang H., Gaiser S., Cruz-Monserrate Z., Wang H., Ji B., Logsdon C.D. An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J. Clin. Invest. 2012;122:1519–1528. doi: 10.1172/JCI59743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamarsheh S., Osswald L., Saller B.S., Unger S., De Feo D., Vinnakota J.M., Konantz M., Uhl F.M., Becker H., Lübbert M., et al. Oncogenic Kras(G12D) causes myeloproliferation via NLRP3 inflammasome activation. Nat. Commun. 2020;11:1659. doi: 10.1038/s41467-020-15497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morandi F., Airoldi I., Pistoia V. IL-27 driven upregulation of surface HLA-E expression on monocytes inhibits IFN-gamma release by autologous NK cells. J. Immunol. Res. 2014;2014 doi: 10.1155/2014/938561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pereira B.I., Devine O.P., Vukmanovic-Stejic M., Chambers E.S., Subramanian P., Patel N., Virasami A., Sebire N.J., Kinsler V., Valdovinos A., et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat. Commun. 2019;10:2387. doi: 10.1038/s41467-019-10335-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramsuran V., Naranbhai V., Horowitz A., Qi Y., Martin M.P., Yuki Y., Gao X., Walker-Sperling V., Del Prete G.Q., Schneider D.K., et al. Elevated HLA-A expression impairs HIV control through inhibition of NKG2A-expressing cells. Science. 2018;359:86–90. doi: 10.1126/science.aam8825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banh C., Fugère C., Brossay L. Immunoregulatory functions of KLRG1 cadherin interactions are dependent on forward and reverse signaling. Blood. 2009;114:5299–5306. doi: 10.1182/blood-2009-06-228353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lou C., Wu K., Shi J., Dai Z., Xu Q. N-cadherin protects oral cancer cells from NK cell killing in the circulation by inducing NK cell functional exhaustion via the KLRG1 receptor. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2022-005061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang R., Sun L., Li C.F., Wang Y.H., Yao J., Li H., Yan M., Chang W.C., Hsu J.M., Cha J.H., et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 2021;12:832. doi: 10.1038/s41467-021-21099-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slattery K., Woods E., Zaiatz-Bittencourt V., Marks S., Chew S., Conroy M., Goggin C., MacEochagain C., Kennedy J., Lucas S., et al. TGFbeta drives NK cell metabolic dysfunction in human metastatic breast cancer. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2020-002044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chakravarthy A., Khan L., Bensler N.P., Bose P., De Carvalho D.D. TGF-beta-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018;9:4692. doi: 10.1038/s41467-018-06654-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu L., Liu X., Wang X., Yan F., Wang P., Jiang Y., Du J., Yang Z. TIGIT(+) TIM-3(+) NK cells are correlated with NK cell exhaustion and disease progression in patients with hepatitis B virusrelated hepatocellular carcinoma. OncoImmunology. 2021;10 doi: 10.1080/2162402X.2021.1942673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Juno J.A., Stalker A.T., Waruk J.L., Oyugi J., Kimani M., Plummer F.A., Kimani J., Fowke K.R. Elevated expression of LAG-3, but not PD-1, is associated with impaired iNKT cytokine production during chronic HIV-1 infection and treatment. Retrovirology. 2015;12:17. doi: 10.1186/s12977-015-0142-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan I.S., Knutsdottir H., Ramakrishnan G., Padmanaban V., Warrier M., Ramirez J.C., Dunworth M., Zhang H., Jaffee E.M., Bader J.S., Ewald A.J. Cancer cells educate natural killer cells to a metastasis-promoting cell state. J. Cell Biol. 2020;219 doi: 10.1083/jcb.202001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corces M.R., Buenrostro J.D., Wu B., Greenside P.G., Chan S.M., Koenig J.L., Snyder M.P., Pritchard J.K., Kundaje A., Greenleaf W.J., et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016;48:1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caenepeel S., Brown S.P., Belmontes B., Moody G., Keegan K.S., Chui D., Whittington D.A., Huang X., Poppe L., Cheng A.C., et al. AMG 176, a Selective MCL1 Inhibitor, Is Effective in Hematologic Cancer Models Alone and in Combination with Established Therapies. Cancer Discov. 2018;8:1582–1597. doi: 10.1158/2159-8290.CD-18-0387. [DOI] [PubMed] [Google Scholar]
- 35.Awada H., Nagata Y., Goyal A., Asad M.F., Patel B., Hirsch C.M., Kuzmanovic T., Guan Y., Przychodzen B.P., Aly M., et al. Invariant phenotype and molecular association of biallelic TET2 mutant myeloid neoplasia. Blood Adv. 2019;3:339–349. doi: 10.1182/bloodadvances.2018024216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carr R.M., Vorobyev D., Lasho T., Marks D.L., Tolosa E.J., Vedder A., Almada L.L., Yurcheko A., Padioleau I., Alver B., et al. RAS mutations drive proliferative chronic myelomonocytic leukemia via a KMT2A-PLK1 axis. Nat. Commun. 2021;12:2901. doi: 10.1038/s41467-021-23186-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y., He L., Selimoglu-Buet D., Jego C., Morabito M., Willekens C., Diop M.K., Gonin P., Lapierre V., Droin N., et al. Engraftment of chronic myelomonocytic leukemia cells in immunocompromised mice supports disease dependency on cytokines. Blood Adv. 2017;1:972–979. doi: 10.1182/bloodadvances.2017004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patel B.J., Przychodzen B., Thota S., Radivoyevitch T., Visconte V., Kuzmanovic T., Clemente M., Hirsch C., Morawski A., Souaid R., et al. Genomic determinants of chronic myelomonocytic leukemia. Leukemia. 2017;31:2815–2823. doi: 10.1038/leu.2017.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montalban-Bravo G., Hammond D., DiNardo C.D., Konopleva M., Borthakur G., Short N.J., Ramos-Perez J., Guerra V., Kanagal-Shamanna R., Naqvi K., et al. Activity of venetoclax-based therapy in chronic myelomonocytic leukemia. Leukemia. 2021;35:1494–1499. doi: 10.1038/s41375-021-01240-2. [DOI] [PubMed] [Google Scholar]
- 40.Reggiardo R.E., Maroli S.V., Halasz H., Ozen M., Hrabeta-Robinson E., Behera A., Peddu V., Carrillo D., LaMontagne E., Whitehead L., et al. Mutant KRAS regulates transposable element RNA and innate immunity via KRAB zinc-finger genes. Cell Rep. 2022;40 doi: 10.1016/j.celrep.2022.111104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferrall-Fairbanks M.C., Dhawan A., Johnson B., Newman H., Volpe V., Letson C., Ball M., Hunter A.M., Balasis M.E., Kruer T., et al. Progenitor Hierarchy of Chronic Myelomonocytic Leukemia Identifies Inflammatory Monocytic-Biased Trajectory Linked to Worse Outcomes. Blood Cancer Discov. 2022;3:536–553. doi: 10.1158/2643-3230.BCD-21-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen X., Eksioglu E.A., Zhou J., Zhang L., Djeu J., Fortenbery N., Epling-Burnette P., Van Bijnen S., Dolstra H., Cannon J., et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Invest. 2013;123:4595–4611. doi: 10.1172/JCI67580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia-Manero G., Winer E.S., DeAngelo D.J., Tarantolo S.R., Sallman D.A., Dugan J., Groepper S., Giagounidis A., Gotze K.S., Metzeler K., et al. Phase 1/2a study of the IRAK4 inhibitor CA-4948 as monotherapy or in combination with azacitidine or venetoclax in patients with relapsed/refractory (R/R) acute myeloid leukemia or lyelodysplastic syndrome. J. Clin. Oncol. 2022;40:7016. doi: 10.1200/JCO.2022.40.16_suppl.7016. [DOI] [Google Scholar]
- 44.Hunter A.M., Newman H., Dezern A.E., Steensma D.P., Niyongere S., Roboz G.J., Mo Q., Chan O., Gerds A., Sallman D.A., et al. Integrated Human and Murine Clinical Study Establishes Clinical Efficacy of Ruxolitinib in Chronic Myelomonocytic Leukemia. Clin. Cancer Res. 2021;27:6095–6105. doi: 10.1158/1078-0432.CCR-21-0935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walker A.R., Byrd J.C., Blachly J.S., Bhatnagar B., Mims A.S., Orwick S., Lin T.L., Crosswell H.E., Zhang D., Minden M.D., et al. Entospletinib in Combination with Induction Chemotherapy in Previously Untreated Acute Myeloid Leukemia: Response and Predictive Significance of HOXA9 and MEIS1 Expression. Clin. Cancer Res. 2020;26:5852–5859. doi: 10.1158/1078-0432.CCR-20-1064. [DOI] [PubMed] [Google Scholar]
- 46.Ganan-Gomez I., Yang H., Ma F., Montalban-Bravo G., Thongon N., Marchica V., Richard-Carpentier G., Chien K., Manyam G., Wang F., et al. Stem cell architecture drives myelodysplastic syndrome progression and predicts response to venetoclax-based therapy. Nat. Med. 2022;28:557–567. doi: 10.1038/s41591-022-01696-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tago K., Funakoshi-Tago M., Ohta S., Kawata H., Saitoh H., Horie H., Aoki-Ohmura C., Yamauchi J., Tanaka A., Matsugi J., Yanagisawa K. Oncogenic Ras mutant causes the hyperactivation of NF-kappaB via acceleration of its transcriptional activation. Mol. Oncol. 2019;13:2493–2510. doi: 10.1002/1878-0261.12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sevin M., Debeurme F., Laplane L., Badel S., Morabito M., Newman H.L., Torres-Martin M., Yang Q., Badaoui B., Wagner-Ballon O., et al. Cytokine-like protein 1-induced survival of monocytes suggests a combined strategy targeting MCL1 and MAPK in CMML. Blood. 2021;137:3390–3402. doi: 10.1182/blood.2020008729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pei S., Pollyea D.A., Gustafson A., Stevens B.M., Minhajuddin M., Fu R., Riemondy K.A., Gillen A.E., Sheridan R.M., Kim J., et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020;10:536–551. doi: 10.1158/2159-8290.CD-19-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vento-Tormo R., Efremova M., Botting R.A., Turco M.Y., Vento-Tormo M., Meyer K.B., Park J.-E., Stephenson E., Polański K., Goncalves A., et al. Single-cell reconstruction of the early maternal–fetal interface in humans. Nature. 2018;563:347–353. doi: 10.1038/s41586-018-0698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanagal-Shamanna R., Singh R.R., Routbort M.J., Patel K.P., Medeiros L.J., Luthra R. Principles of analytical validation of next-generation sequencing based mutational analysis for hematologic neoplasms in a CLIA-certified laboratory. Expert Rev. Mol. Diagn. 2016;16:461–472. doi: 10.1586/14737159.2016.1142374. [DOI] [PubMed] [Google Scholar]
- 53.DeLong E.R., DeLong D.M., Clarke-Pearson D.L. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–845. [PubMed] [Google Scholar]
- 54.Cheson B.D., Bennett J.M., Kopecky K.J., Büchner T., Willman C.L., Estey E.H., Schiffer C.A., Doehner H., Tallman M.S., Lister T.A., et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J. Clin. Oncol. 2003;21:4642–4649. doi: 10.1200/JCO.2003.04.036. [DOI] [PubMed] [Google Scholar]
- 55.Cheson B.D., Greenberg P.L., Bennett J.M., Lowenberg B., Wijermans P.W., Nimer S.D., Pinto A., Beran M., de Witte T.M., Stone R.M., et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108:419–425. doi: 10.1182/blood-2005-10-4149. [DOI] [PubMed] [Google Scholar]
- 56.Woll P.S., Kjällquist U., Chowdhury O., Doolittle H., Wedge D.C., Thongjuea S., Erlandsson R., Ngara M., Anderson K., Deng Q., et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell. 2014;25:794–808. doi: 10.1016/j.ccr.2014.03.036. [DOI] [PubMed] [Google Scholar]
- 57.Will B., Zhou L., Vogler T.O., Ben-Neriah S., Schinke C., Tamari R., Yu Y., Bhagat T.D., Bhattacharyya S., Barreyro L., et al. Stem and progenitor cells in myelodysplastic syndromes show aberrant stage-specific expansion and harbor genetic and epigenetic alterations. Blood. 2012;120:2076–2086. doi: 10.1182/blood-2011-12-399683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adema V., Ma F., Kanagal-Shamanna R., Thongon N., Montalban-Bravo G., Yang H., Peslak S.A., Wang F., Acha P., Sole F., et al. Targeting the EIF2AK1 signaling pathway rescues red blood cell production in SF3B1-mutant myelodysplastic syndromes with ringed sideroblasts. Blood Cancer Discov. 2022;3:554–567. doi: 10.1158/2643-3230.BCD-21-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thongon N., Ma F., Santoni A., Marchesini M., Fiorini E., Rose A., Adema V., Ganan-Gomez I., Groarke E.M., Gutierrez-Rodrigues F., et al. Hematopoiesis under telomere attrition at the single-cell resolution. Nat. Commun. 2021;12:6850. doi: 10.1038/s41467-021-27206-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buenrostro J.D., Corces M.R., Lareau C.A., Wu B., Schep A.N., Aryee M.J., Majeti R., Chang H.Y., Greenleaf W.J. Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell. 2018;173:1535–1548.e16. doi: 10.1016/j.cell.2018.03.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morita K., Wang F., Jahn K., Hu T., Tanaka T., Sasaki Y., Kuipers J., Loghavi S., Wang S.A., Yan Y., et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat. Commun. 2020;11:5327. doi: 10.1038/s41467-020-19119-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
scRNA-seq, scATAC-seq, and scDNA-seq data are accessible at GEO under accession number GSE218390. No custom computer codes were generated in this study.
-
•
The lead contact can provide any additional information required to reanalyze the data reported in this work paper upon request.