

Abstract
Streptococcus pneumoniae (Sp), a leading cause of community-acquired pneumonia, can spread from the lung into the bloodstream to cause septicemia and meningitis, with a concomitant three-fold increase in mortality. Limitations in vaccine efficacy and a rise in antimicrobial resistance have spurred searches for host-directed therapies that target pathogenic immune processes. Polymorphonuclear leukocytes (PMNs) are essential for infection control but can also promote tissue damage and pathogen spread. The major Sp virulence factor, pneumolysin (PLY), triggers acute inflammation by stimulating the 12-lipoxygenase (12-LOX) eicosanoid synthesis pathway in epithelial cells. This pathway is required for systemic spread in a mouse pneumonia model and produces a number of bioactive lipids, including hepoxilin A3 (HXA3), a hydroxy epoxide PMN chemoattractant that has been hypothesized to facilitate breach of mucosal barriers. To understand how 12-LOX-dependent inflammation promotes dissemination during Sp lung infection and dissemination, we utilized bronchial stem cell-derived air-liquid interface (ALI) cultures that lack this enzyme to show that HXA3 methyl ester (HXA3-ME) is sufficient to promote basolateral-to-apical PMN transmigration, monolayer disruption, and concomitant Sp barrier breach. In contrast, PMN transmigration in response to the non-eicosanoid chemoattractant fMLP did not lead to epithelial disruption or bacterial translocation. Correspondingly, HXA3-ME but not fMLP increased release of neutrophil elastase (NE) from Sp-infected PMNs. Pharmacologic blockade of NE secretion or activity diminished epithelial barrier disruption and bacteremia after pulmonary challenge of mice. Thus, HXA3 promotes barrier disrupting PMN transmigration and NE release, pathological events that can be targeted to curtail systemic disease following pneumococcal pneumonia.
Keywords: Streptococcus pneumoniae, airway mucosal barrier, neutrophil transmigration, neutrophil elastase, 12-lipoxygenase
Introduction:
Streptococcus pneumoniae (Sp; also known as the pneumococcus) is a Gram-positive bacterium that asymptomatically colonizes the nasopharynx of 5–10% of healthy adults, but can spread to the lower respiratory tract and is the most frequent cause of community-acquired pneumonia (1). Subsequent bacterial translocation from the airway into the bloodstream can lead to invasive disease, such as septicemia and meningitis, events associated with a three-fold increase in mortality (2). Invasive pneumococcal infections result in approximately 14 million cases and one million deaths annually worldwide (3). Vaccination and antimicrobials are first-line strategies in combating pneumococcal diseases. However, the rapid rise of antibiotic resistance and the limited antigenic breadth of effective vaccines have fueled interest in treatment strategies that focus on diminishing tissue-destructive host immune responses (4–7).
Pneumococcal infection of lung mucosa drives robust recruitment of polymorphonuclear leukocytes (PMNs, or neutrophils), leading to the acute inflammation that is a hallmark of this infection (1). PMNs confront invading Sp with multiple antibacterial mechanisms, including release of reactive oxygen species (ROS) (8), neutrophil extracellular traps (NET) (9), and/or proteases such as cathepsin G (CG) and neutrophil elastase (NE) (10). Indeed, neutropenic individuals or neutrophil-depleted mice are highly susceptible to systemic Sp infection (11, 12). Nevertheless, sustained pulmonary accumulation of PMNs increases airway permeability with a concomitant risk of disseminated infection (13, 14). Protease inhibitors that diminish PMN infiltration also reduce bacteremia and lethality after Sp pulmonary challenge of mice (15, 16). Finally, mice that retain high numbers of pulmonary PMNs suffer higher levels of bacteremia and mortality (17–20), and depletion of PMNs 18 hours post-infection (h.p.i.) mitigates disease and pathogen spread (21).
Chemotactic cues not only recruit PMNs but also influence their tissue-destructive character (22–24). Hence, in addition to their recruitment, PMN-directed pathologies may result from enhanced tissue-damaging PMN activities (24). The major Sp virulence factor pneumolysin (PLY), a cytolysin that drives tissue damage and promotes early bacteremia (25–27), stimulates the 12-lipoxygenase (12-LOX) pathway in epithelial cells and results in the synthesis and apical secretion of eicosanoid PMN chemoattractants (17, 28, 29). Among 12-LOX-generated bioactive lipid mediators (30), the hydroxy epoxide hepoxilin A3 (HXA3) is a potent chemoattractant (31) that orchestrates mucosal inflammation during both intestinal (32, 33) and pulmonary infections (34). Like other chemoattractants (23, 35), HXA3 has both chemotactic and non-chemotactic effects on PMNs (36), triggering intracellular calcium release (36), promoting PMN survival (37), inducing NET formation (38), and stimulating the release of additional arachidonic acid metabolites (39). Notably, genetic ablation or chemical inhibition of 12-LOX drastically reduces PMN infiltration, bacteremia, and mortality following Sp lung challenge of mice (17, 29), suggesting that barrier disruption and systemic Sp disease could be mitigated by modulation of PMN effector functions that are enhanced by one or more products of the 12-LOX pathway.
The tissue-destructive functions of PMNs are dramatically altered upon exposure to bacterial factors (24, 40, 41), but the effect of HXA3 on PMNs in the context of Sp infection has not been examined. In addition, 12-LOX promotes the production of numerous bioactive lipids (30), and although HXA3 has been hypothesized to be the essential driver in PLY-promoted Sp dissemination from the lung, this eicosanoid has not been directly implicated in the Sp- or PLY-driven PMN chemotaxis. These limitations are in part a reflection of the instability of HXA3 in aqueous environments (32), as well as the lack of an easily manipulated in vitro experimental model that faithfully reflects Sp-mediated inflammation and bacterial translocation across an epithelial barrier. Indeed, the respiratory epithelial culture models previously applied to Sp infection are typically based on immortalized cell lines that lack the cellular diversity and bona fide barrier function integral to airway epithelium (42). Here, we characterized the role of PLY in promoting PMN transmigration and epithelial compromise using air-liquid interface (ALI) monolayers derived from bronchial stem cells that recapitulate key features of the airway epithelium. Moreover, ALI monolayers genetically ablated for 12-LOX deficient permitted the demonstration that HXA3 methyl ester (HXA3-ME), a stable and active version of HXA3, is sufficient to promote PMN transmigration and Sp barrier breach. Corresponding studies of the signaling capacities of HXA3-ME on PMN in the context of Sp infection showed that HXA3 is not only a central driver of PMN transmigration across infected epithelium but also enhances the tissue-damaging proteolytic activity of PMNs. Targeting this HXA3-promoted activity mitigated systemic disease following Sp pulmonary challenge of mice, illustrating its therapeutic potential as a host-directed therapy for Sp infection.
Results:
The 12-LOX pathway, stimulated by PLY-producing Sp, promotes PMN infiltration, lung permeability, and bacteremia following Sp lung infection in mice.
Activation of the airway epithelial cell 12-LOX pathway is triggered by Sp pneumolysin (29). We intratracheally (i.t.) inoculated BALB/c mice with 1x107 CFU of WT Sp TIGR4 or the isogenic PLY-deficient mutant Sp TIGR4 Δply. At 18 hours post infection (h.p.i.), the two strains reached similar lung burdens (Figure 1a, “WT” vs. “Δply”), consistent with previous reports (43, 44). Both strains also induced pulmonary inflammation, but consistent with the ability of PLY to stimulate the 12-LOX pathway and increase inflammation (17, 26), PMN pulmonary infiltration was 1.5-fold higher in mice infected with WT Sp compared to Sp Δply (p < 0.01; Figure 1b, “WT” vs. “Δply”). To assess damage to the lung barrier, at 18 h.p.i. we delivered 1 mg of 70 kDa FITC-dextran intravenously into infected mice and, after 30 minutes, measured the fluorescence signal in lung homogenates relative to that of serum. Infection by WT Sp increased lung permeability more than two-fold relative to uninfected mice (p < 0.01), whereas infection with Sp Δply had no effect (Figure 1c, “WT” vs. “Δply”). Mirroring the increased lung permeability to FITC-dextran, WT Sp infection resulted in a ten-fold higher level of bacteremia compared to Sp Δply infection (p < 0.05; Figure 1d, “WT” vs. “Δply”).
Figure 1. The 12-LOX pathway, stimulated by PLY-producing Sp, promotes PMN infiltration, lung permeability and bacteremia following Sp lung infection in mice.

BALB/c mice were infected i.t. with 1 × 107 CFU wild type (WT) or PLY-deficient mutant (Δply) TIGR4 Sp for 18 h, with or without i.p injection of 8 mg/kg of the 12-LOX inhibitor CDC. (a) Bacterial lung burden determined by measuring CFU in lung homogenates. (b) PMN infiltration determined by flow cytometric enumeration of Ly6G+. (c) Lung permeability quantitated by measuring the concentration of 70 kDa FITC-dextran in the lung relative to serum after i.v. administration. (d) Bacteremia measured by enumerating CFU in serum. Each panel is representative of three independent experiments, or pooled data from three independent experiments. Error bars represent mean ± SEM. Statistical analysis was performed using ordinary one-way ANOVA: *p-value < 0.05, **p-value < 0.01, ***p-value < 0.001, ****p-value < 0.0001.
We previously found that inhibition of 12-LOX activity by i.p injection of cinnamyl-3,4-dihydroxy-α-cyanocinnamate (CDC) did not affect Sp lung burden but curtailed PMN lung infiltration in C57BL/6 (B6) mice (17). Here, after infection of BALB/c mice with WT Sp, CDC treatment similarly diminished lung PMN infiltration (p < 0.0001) without altering lung burden (Figure 1a–b, “WT + CDC”). Lung barrier disruption and Sp dissemination also depended on 12-LOX activity because CDC treatment of Sp-infected mice resulted in lower FITC-dextran leakage (p < 0.01; Figure 1c) and bacteremia at 18 h.p.i. (p < 0.01; Figure 1d). Therefore, 12-LOX activation by PLY promoted PMN infiltration to the lungs, an event that correlated with increased lung permeability and Sp spread to the bloodstream.
The 12-LOX pathway promotes PMN transmigration and epithelial barrier breach upon apical infection of ALI monolayers by PLY-producing Sp.
Recent advances in airway stem cell biology have allowed for the generation of genetically tractable in vitro stem cell-derived epithelial cultures with organized architecture and functional attributes of the airway mucosa, including beating cilia, apical mucus production, and a robust junctional barrier (45). To identify key steps underlying the promotion of bacteremia by PLY and 12-LOX activation, we modeled interactions between Sp and PMNs at the airway epithelium by culturing human airway basal stem cells (BSCs) on 3 μm pore size Transwell filters. After growth to confluency, media was removed from the apical side of the monolayers, a step that triggers the differentiation of the stem cells to form a monolayer containing the diverse airway epithelial cell types (42), including ciliated cells, mucus-producing goblet cells, and secretory club cells, found in bona fide airway epithelium. We then added 1×106 PMNs isolated from human peripheral blood to the basolateral surface of these air-liquid interface (ALI) cultures and assessed their movement to the apical side upon Sp infection.
Two hours of apical infection with 1×107 Sp/Transwell induced robust PLY-dependent PMN transmigration across human ALI monolayers, with WT Sp triggering two-fold greater migration compared to Sp Δply (p < 0.0001; Figure 2a, “Human ALI”). WT Sp infection of monolayers pretreated with CDC failed to trigger PMN transmigration (Figure 1a, “WT+CDC”), suggesting that PMN transmigration across Sp-infected ALI monolayers was dependent on eicosanoid lipid mediators produced by 12-LOX, recapitulating our findings during pulmonary Sp challenge in mice.
Figure 2. The 12-LOX pathway promotes PMN transmigration and epithelial barrier breach upon apical infection of ALI monolayers by PLY-producing Sp.

Human BSC-derived ALI monolayers (left column) or WT B6 and 12-LOX-deficient Alox15−/− mouse BSC-derived ALI monolayers (right column) were apically infected with 1 × 107 WT or Δply Sp in the presence of basolateral PMNs. (a) After 2 hours of PMN migration, the degree of transmigration as determined by MPO activity in the apical chamber. (b) PMN infiltration and monolayer integrity assessed by fluorescence confocal microscopy after staining nuclei with DAPI and F-actin with fluorescent phalloidin. For clarity, images shown are of extended projections (all z-sections collapsed into 1 plane). Arrows indicate examples of PMN nuclei. Scale bar = 40 μm for all images. Quantitation of epithelial retention is shown in the graph below the images, performed by enumerating epithelial cell nuclei relative to uninfected ALI in five images per experiment. (c) Epithelial permeability measured by HRP flux relative to monolayers infected with WT Sp. (d) Sp translocation quantitated by measuring basolateral CFU. Each panel is representative of three independent experiments, or pooled data from three independent experiments. Error bars represent mean ± SEM. Statistical analysis was performed using ordinary one-way ANOVA: *p-value < 0.05, **p-value < 0.01, ***p-value < 0.001, ****p-value < 0.0001.
Given the correlation between PMN infiltration and barrier disruption in vivo, we visualized monolayers by fluorescence confocal microscopy. PMNs were distinguished from ALI cells by staining cell nuclei with DAPI and visualizing their F-actin with fluorescent phalloidin. Upon infection with WT Sp, PMNs, identified by their multi-lobed nuclei, were found to infiltrate the epithelial monolayers in great numbers. Infection with Sp Δply resulted in reduced but detectable PMN infiltration (Figure 2b, “Human ALI”, yellow arrows). On the other hand, epithelial cells were lost from the Transwell filters post-PMN transmigration. To quantitate epithelial cell loss, we optimized a CellProfiler pipeline to distinguish epithelial cells from PMNs based on the size and shape of their nuclei (see Methods). Quantitation of each cell type indicated that infection with WT Sp and concomitant PMN migration triggered a 64% loss in epithelial cells from the monolayer (Figure 2b, “WT Sp”). This loss was entirely dependent on the presence of PMNs (Figures S1a-b). It was also partially dependent on PLY, because infection with Sp Δply resulted in a 41% (and significantly lower) loss of epithelial cells (p < 0.01; Figure 2b, “Sp Δply”).
To quantitate epithelial barrier function, we measured leakage of the basally loaded tracer protein HRP into the apical chamber. A 17-fold increase in HRP flux was observed after PMN transmigration induced by apical infection of ALI monolayers by WT Sp (p < 0.0001; Figure 2c, “Human ALI”). This level of leakage was 1.5-fold higher compared to monolayers that had been pretreated with CDC or monolayers that were infected with Sp Δply (p < 0.01; Figure 2c, “Human ALI”). The diminished HRP leakage observed in the latter conditions correlated with a 25- or 9-fold decrease in cross-monolayer bacterial movement (p < 0.0001; Figure 2d, “Human ALI”). As predicted, disruption to barrier integrity depended entirely on the presence of PMNs (Fig S1c-d).
We then tested the effect of genetic ablation of 12-LOX by generating ALI monolayers from WT or 12-LOX-deficient Alox15−/− mice (Figure 2a, “Mouse ALI”). Infection of ALI monolayers from B6 mice with WT Sp induced PMN transmigration 7-fold higher than basal (uninfected) levels (p < 0.0001) and 2-fold higher (p < 0.0001) than that induced by Sp Δply (Figure 2a, “B6”). In contrast, Alox15−/− ALI monolayers failed to trigger significant PMN transmigration during infection by either WT or PLY-deficient Sp (Figure 2a, “Alox15−/−”).
Confocal microscopy analysis of monolayers after PMN migration revealed that WT infection was associated with a 68% loss of B6 ALI monolayer compared to a 12% loss of Alox15−/− ALI monolayers (p < 0.0001; Figure 2b, “Mouse ALI”). Correspondingly, a 30-fold increase in HRP flux was detected across monolayers infected with WT Sp compared to uninfected monolayers (p < 0.0001; Figure 2c, “Mouse ALI”). This increase in HRP flux was promoted by both PLY and 12-LOX, because (a) Δply Sp infection of WT B6 monolayers resulted in 2-fold lower flux (p < 0.05); and (b) WT Sp infection of Alox15−/− ALI monolayers resulted in 4-fold lower flux (p < 0.0001; Figure 2c, “Mouse ALI”).
The PLY- and 12-LOX-dependent barrier disruption correlated with enhanced Sp translocation across ALI monolayers, as WT Sp translocation across B6 monolayers was 3-fold higher than that of Sp Δply (p < 0.05) and 10-fold higher than that of WT Sp across Alox15−/− monolayers (p < 0.001; Figure 2d, “Mouse ALI”). Notably, although PLY has diverse effects on mammalian cells (46, 47), upon infection of 12-LOX-deficient ALI, the presence or absence of PLY had no effect on barrier disruption and bacterial translocation. Thus, not only is 12-LOX-dependent PMN transmigration required for barrier breach during Sp infection of ALI monolayers, but the critical role of PLY in this process is the induction of the 12-LOX pathway.
A soluble factor produced by ALI monolayers via the 12-LOX pathway upon apical Sp infection promotes both PMN migration and barrier disruption.
Infection of WT but not 12-LOX-deficient ALI monolayers by Sp triggered PMN migration and barrier breach (Fig. 2; “Mouse ALI”; “Alox15−/−”). To detect putative soluble factor(s) produced by infected epithelium via the 12-LOX pathway, we first collected apical supernatants from B6 ALI monolayers that had been infected with WT Sp (herein referred to as “WT supernatant”), or as controls, infected with Δply Sp (“Δply supernatant”) or left uninfected (“uninfected supernatant”). (We did not include these supernatants of Alox15−/− ALI cultures because these monolayers did not support PMN migration under any conditions; Fig. 2a). Detecting factors that are capable of drawing PMNs across Sp-infected ALI monolayers and facilitating bacterial translocation is confounded by the further production of 12-LOX-derived products by infected cells. Hence, we added these supernatants to Alox15−/− (not WT B6) ALI monolayers that had been apically infected with WT Sp. The addition of WT supernatant triggered PMN transmigration across infected Alox15−/− ALI monolayers at a 25- and 2-fold higher level than that triggered by uninfected supernatant and Δply supernatant, respectively (Figure 3a).
Figure 3. A soluble factor produced by ALI monolayers via the 12-LOX pathway upon apical Sp infection promotes both PMN migration and barrier disruption.
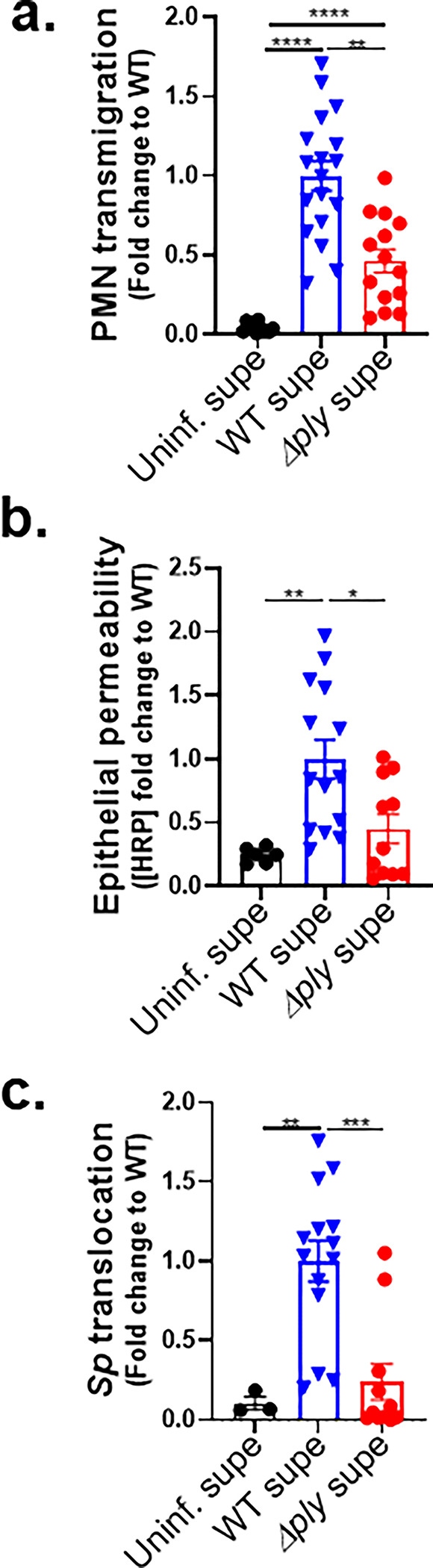
Alox15−/− mouse BSC-derived ALI monolayers were apically infected with 1 × 107 WT Sp and transferred into apical chambers containing supernatant generated from WT Sp infection (WT supe) or Δply infection (Δply supe) of B6 mouse BSC-derived ALI monolayers. (a) After two hours of PMN migration, the degree of transmigration as determined by MPO activity in the apical chamber. (b) Epithelial permeability measured by HRP flux relative to monolayers infected with WT Sp. (c) Sp translocation quantitated by measuring basolateral CFU. Each panel is representative of three independent experiments, or pooled data from three independent experiments. Error bars represent mean ± SEM. Statistical analysis was performed using ordinary one-way ANOVA: *p-value < 0.05, **p-value < 0.01, ***p-value < 0.001.
To determine if PMN migration in response to a 12-LOX-dependent soluble factor (or factors) disrupted the infected monolayer, we measured cross-epithelial horseradish peroxidase (HRP) leakage. WT supernatant induced 4- and 2-fold more leakage than uninfected supernatant and Δply supernatant, respectively (Figure 3b). In turn, HRP leakage correlated with bacterial movement because WT supernatant was associated with 10- and 5-fold higher Sp translocation than uninfected and Δply supernatant, respectively (Figure 3c). That supernatant of epithelium infected with WT Sp was sufficient to rescue PMN migration across Alox15−/− ALI monolayers, as well as concomitant barrier disruption and Sp translocation, affirmed the presence of a soluble mediator (or mediators) in the epithelial apical supernatant that acts as a PMN chemoattractant and drives barrier breach during Sp infection.
Upon Sp infection of ALI monolayers, PMN transmigration induced by HXA3 but not fMLP promotes barrier breach.
The 12-LOX pathway generates a number of bioactive lipids, but based on mucosal infection by several bacterial pathogens (32, 34, 48), hepoxilin A3 (HXA3) is a prime candidate for the 12-LOX-dependent chemoattractant secreted into the apical supernatant by infected B6 ALI monolayers. To test whether HXA3 is sufficient to trigger PMN transmigration, barrier disruption, and bacterial translocation in vitro, we added HXA3 methyl ester (HXA3-ME), a stable synthetic form of HXA3, to the apical chamber of Alox15−/− ALI monolayers infected with WT Sp, and monitored transmigration of basolateral PMNs. As controls, the well-characterized non-eicosanoid PMN chemoattractant N-formyl-L-methionyl-L-leucyl-phenylalanine (fMLP) induced PMN transmigration, whereas the HBSS buffer control did not (Figure 4a, “HBSS”, “fMLP”). We found that the apical addition of HXA3-ME induced PMN transmigration equivalent to that triggered by fMLP (Figure 4a, “HXA3”), Indicating that HXA3 is sufficient to induce PMN migration across Sp-infected ALI monolayers.
Figure 4. Upon Sp infection of ALI monolayers, PMN transmigration induced by HXA3 but not fMLP promotes barrier breach.
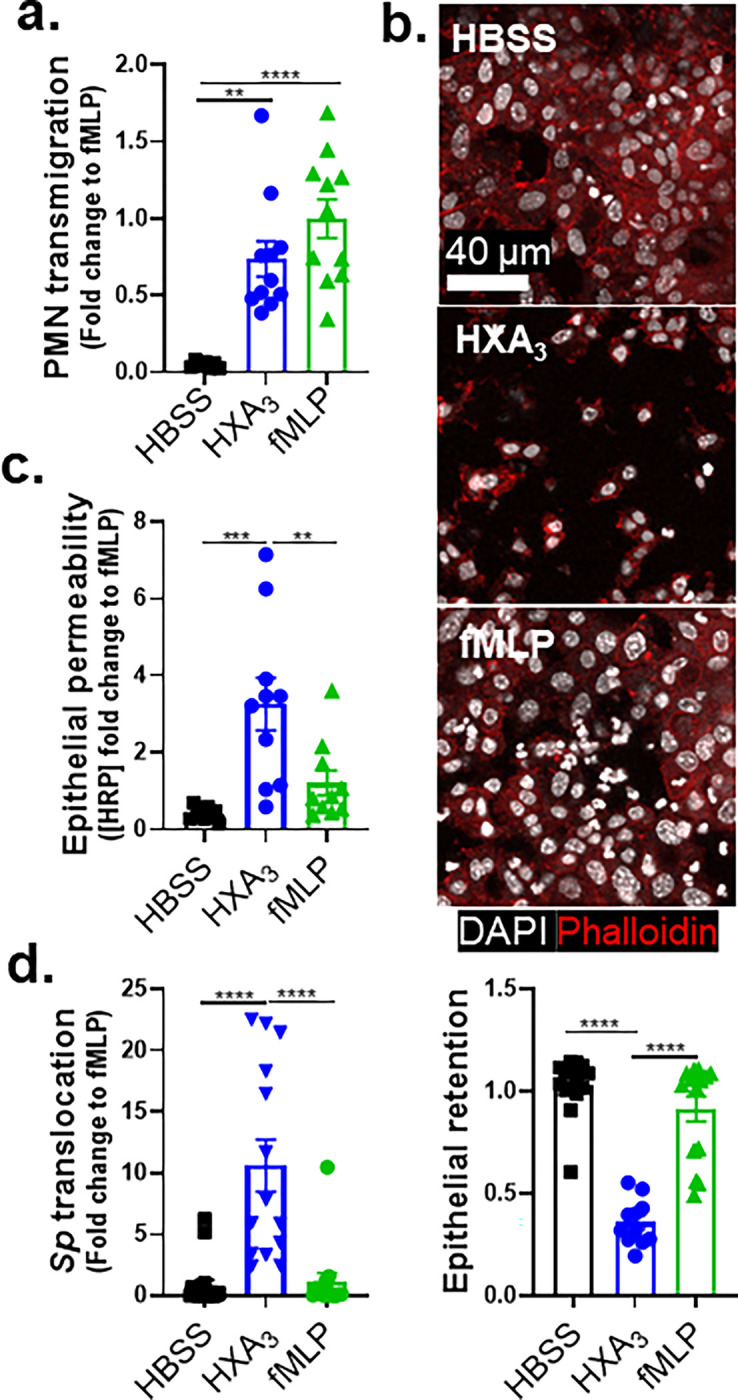
Alox15−/− mouse BSC-derived ALI monolayers were apically infected with 1 × 107 WT Sp and transferred into apical chambers containing 10 nM HXA3 methyl ester (“HXA3”), or 10 μM fMLP, in the presence of basolateral PMNs. (a) After two hours of PMN migration, the degree of transmigration as determined by MPO activity in the apical chamber. (b) Monolayer integrity assessed by fluorescence confocal microscopy after staining nuclei with DAPI and F-actin with fluorescent phalloidin. For clarity, images shown are of extended projections (all z-sections collapsed into 1 plane). Scale bar = 40 μm for all images. Shown below the images is epithelial retention quantitated by enumerating epithelial cell nuclei relative to uninfected monolayers in five images per experiment. (c) Epithelial permeability measured by HRP flux relative to monolayers infected with WT Sp. (d) Sp translocation quantitated by measuring basolateral CFU. Each panel is representative of three independent experiments, or pooled data from three independent experiments. Error bars represent mean ± SEM. Statistical analysis was performed using ordinary one-way ANOVA: *p-value < 0.05, **p-value < 0.01, ***p-value < 0.001, ****p-value < 0.0001.
In addition to inducing PMN migration, chemoattractants can alter other PMN functional responses (24), and HXA3 influences a variety of PMN behaviors (36), such as intracellular calcium release (36), apoptosis inhibition (37), and NETosis (38). Indeed, despite similar levels of PMN transmigration in response to HXA3 and fMLP, PMN transmigration induced by fMLP was associated with retention of the epithelial monolayer integrity (Figure 4b), minimal HRP flux (Figure 4c), and the absence of Sp transepithelial movement (Figure 4d, “fMLP”), whereas that mediated by HXA3-ME induced loss of 64% of the monolayer, a 4-fold increase in HRP leakage, and a 10-fold increase in Sp translocation (Figure 4b–d, “HXA3”). These data indicate that HXA3 induces a mode of PMN transmigration capable of promoting barrier disruption and bacterial translocation.
HXA3-stimulated PMNs generate robust NE in response to Sp infection.
Previous studies show that PMNs respond to purified HXA3 by resisting apoptosis (37) and generating NETs (38), events that may reinforce PMN inflammatory potential. However, the potentially tissue-damaging state of PMNs is greatly influenced by exposure to microbial pathogens (24). Hence, to identify key features ofPMNs that may lead to monolayer disruption and Sp translocation, we compared the effect of HXA3 and fMLP on various PMN responses in the context ofSp infection. To begin this analysis, we first characterized PMN activities in response to Sp in the absence of chemoattractant. After 30 min of infection with Sp, 94% of PMNs remained viable, i.e., membrane impermeable to propidium iodide (PI; Figure S2a), and PMNs killed 70% of opsonized Sp (Figure S2b). Infection with Sp triggered >7-fold increases in NETosis, PMN apoptosis, and ROS production (Figures S2c, e-f), and >2-fold increases in MMP and NE release (Figure S2d, g). (The relative log-fold changes in various activities of infected PMN parameters induced by Sp infection are provided in a radar plot; Figure S2h).
We next profiled the effect of HXA3 and fMLP on Sp-induced responses of infected PMNs. fMLP treatment resulted in a slight increase in membrane-permeant PMNs compared to HBSS or HXA3 treatment (10% versus 6%; Figure 5a). Nevertheless, fMLP- and HXA3-treated PMNs were equally proficient as HBSS-treated PMNs at opsonophagocytic killing (Figure 5b). The presence or absence of fMLP or HXA3 also did not affect NETosis or MMP secretion by infected PMNs (Figure 5c, d). fMLP treatment resulted in slightly higher levels of apoptosis, reflected by surface levels of Annexin V compared to untreated or HXA3-treated PMNs (Figure 5e), a finding consistent with the observation that HXA3 diminishes PMN apoptosis (37). Finally, HXA3 resulted in slightly higher ROS production than fMLP (10% versus 8%, Figure 5f).
Figure 5. HXA3 enhances NE secretion by Sp-infected PMNs.

1 × 106 PMNs were infected with 1 × 107 Sp after treatment with control HBSS, 10 μM fMLP, or 10 nM HXA3 methyl ester (“HXA3”), and evaluated for functional performance via (a) PMN membrane permeability determined by propidium iodide staining (PI+), (b) opsonophagocytic killing quantitated by plating for CFU, (c) NETosis determined by Sytox and anti-MPO staining (Sytox+ MPO+), (d) released MMP activity by substrate conversion and expressed relative to uninfected PMNs, (e) apoptosis determined by lack of straining by propidium iodide and positive staining of Annexin V (PI− Annexin V+), (f) ROS production by intracellular oxidation of substrate (DCF+), and (g) released NE activity by substrate conversion and expressed relative to uninfected PMNs. (h) Sp-infected PMNs were treated with HXA3 methyl ester in the presence or absence of 50 μM Nexinhib20 (Nex.) and relative NE activity in supernatant quantitated by substrate conversion as in panel g. (i) Radar plot summary of log fold change in PMN activities in (a-g). Each panel shown is representative of three independent experiments. Error bars represent mean ± SEM. Statistical analysis was performed using ordinary one-way ANOVA: *p-value < 0.05, **p-value < 0.01, ***p-value < 0.001, ****p-value < 0.0001.
The greatest chemoattractant-dependent difference detected in Sp-infected PMNs was NE activity. fMLP stimulation appeared to diminish NE activity compared to PBS, although this difference did not reach statistical significance (Figure 5g). In contrast, HXA3 resulted in an almost 2-fold increase relative to the control (P<0.001). NE has been implicated in severe lung injury during Sp infection (49, 50) and is delivered by PMNs largely through the release of exosomes and primary granules (51). The increase in HXA3-triggered NE activity was eliminated by Nexinhib20, which blocks NE release by both exosomes and primary granules (52) (Figure 5h, “Nex”). The relative changes in various activities of infected PMN parameters induced by fMLP or HXA3 are summarized in Figure 5i.
PLY-producing Sp promotes release of NE and primary granules in a 12-LOX-dependent manner during experimental lung infection.
Given the enhanced NE release by HXA3-stimulated, Sp-infected PMNs in vitro, we assessed in vivo degranulation of primary granules, a major mechanism of NE release (51) of PMNs. 18 hours after i.t. infection of BALB/c mice with Sp, we measured the relative level of the primary granule marker CD63 on the surface of pulmonary PMNs. CD63 surface expression was 3.5-fold higher on PMNs from lungs of mice infected with WT Sp compared to uninfected mice (Figure 6b, “Uninf.” vs. “WT”). This elevated level was reduced by 25% during infection with Sp Δply (P<0.05; Figure 6b, “Δply”), suggesting that PLY-induced HXA3 production contributed significantly to degranulation. Consistent with this, inhibition of 12-LOX with CDC after infection with WT Sp infection resulted in a similar decrease in PMN CD63 surface expression (Figure 6a, “CDC”).
Figure 6. PLY-producing Sp promotes release of NE and primary granules in a 12-LOX-dependent manner during experimental lung infection.
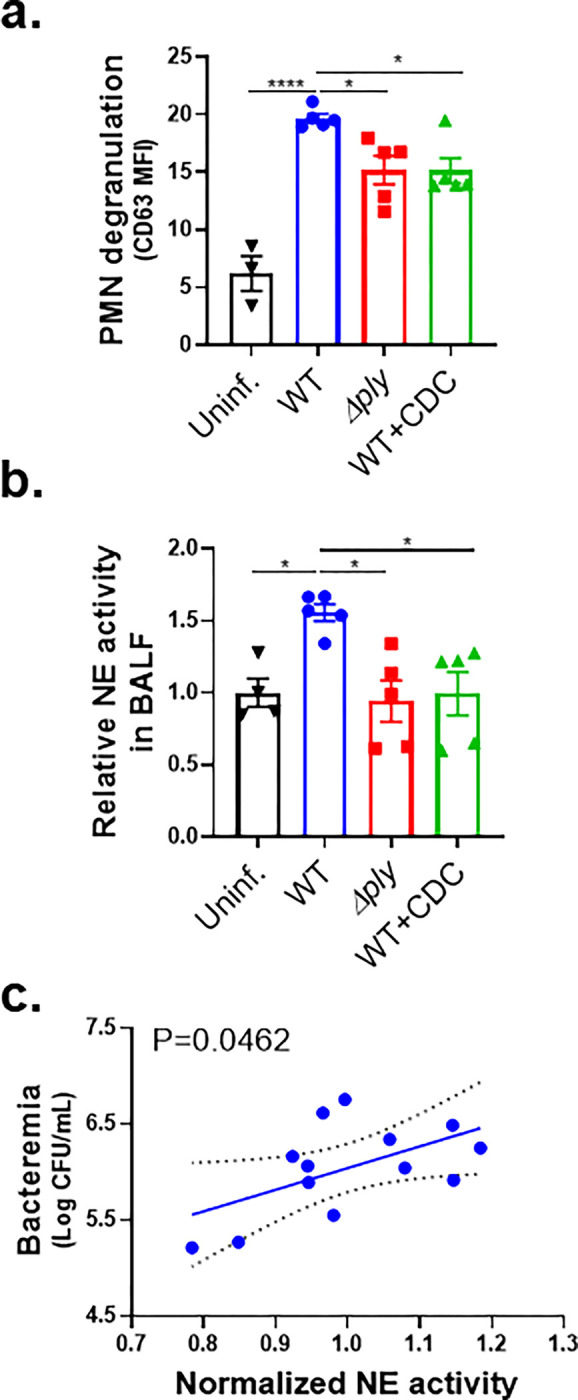
BALB/c mice were infected i.t. with 1 × 107 CFU WT or Δply Sp for 18 h, with or without i.p injection of 8 mg/kg of the 12-LOX inhibitor CDC. (a) NE activity in cell-free BALF determined by substrate conversion, expressed relative to the NE activity in cell-free BALF from uninfected mice. (b) FACS analysis of degranulation determined by CD63 expression on Ly6G+ lung infiltrating PMNs. (c) Correlation between normalized NE activity in (a) and bacteremia determined by enumerating CFU in serum. Each panel shown is representative of three independent experiments, or pooled data from three independent experiments. Error bars represent mean ± SEM. Statistical analysis was performed using ordinary one-way ANOVA: *p-value < 0.05, ***p-value < 0.001, ****p-value < 0.0001.
To determine if PMN degranulation corresponded to increased pulmonary NE levels bronchial alveolar lavage fluid (BALF) of BALB/c mice at 18 h.p.i. was centrifuged to remove PMNs and other cells, and then assessed for NE activity. Activity was 50% higher in WT Sp-infected mice compared to uninfected mice or mice infected with Sp Δply (Figure 6b, “Δply”), a finding consistent with previous reports (7). CDC treatment of infected mice, which dramatically decreases PMN lung infiltration (29), reduced BALF NE to levels indistinguishable from that of uninfected mice (Figure 6b, “CDC”). In mice infected with WT Sp, BALF NE activity significantly correlated with bacteremia (Figure 6c). These findings suggest that PLY-triggered HXA3 promotes lung-infiltrating PMNs to release NE during pulmonary Sp challenge, thus enhancing bacteremia.
Inhibition of NE release mitigates disruption of the lung epithelial barrier and bacteremia following Sp lung infection.
NE degrades epithelial cell junctions and extracellular matrices in vitro (53, 54) and has been implicated in the pathogenesis of several human disorders that involve inflammatory damage (51). To determine if inhibition of PMN degranulation or NE activity protects lung barrier function during Sp infection in mice, we delivered the degranulation inhibitor Nexinhib20 or the NE inhibitor Sivelestat (16) by intraperitoneal (i.p.) injection to BALB/c mice (see Methods), followed by Sp lung challenge. Neither inhibitor altered bacterial lung burden or PMN infiltration at 18 h.p.i (Figure 7a–b). Nexinhib20 significantly diminished PMN degranulation, measured by surface CD63, compared to vehicle-treated mice (Figure 7c); Sivelestat did not achieve significant effect. Notably, both Nexinhib20 and Sivelestat prevented an increase in the NE activity of BALF (Figure 7d).
Figure 7. Inhibition of NE release mitigates disruption of the lung epithelial barrier and bacteremia following Sp lung infection.

BALB/c mice were infected i.t. with 1 × 107 CFU WT Sp for 18 h, with or without i.p injection of 30 mg/kg Nexinhib20 (Nex) or 30 mg/kg Sivelestat (Siv) one hour prior to infection. (a) Bacterial lung burden determined by measuring CFU in lung homogenates; (b) PMN infiltration determined by flow cytometric enumeration of Ly6G+; (c) Degranulation determined by CD63 expression on Ly6G+ lung infiltrating PMNs by FACS; (d) Relative NE activity in BALF determined by substrate conversion; (e) Lung permeability determined by measuring the concentration of 70 kD FITC-dextran in lung relative to serum after i.v. administration; and (f) Bacteremia determined by enumerating CFU in serum. Each panel is representative of three independent experiments, or pooled data from three independent experiments. Error bars represent mean ± SEM. Statistical analysis was performed using ordinary one-way ANOVA: *p-value < 0.05, **p-value < 0.01, ****p-value < 0.0001.
The decrease in PMN degranulation associated with Nexinhib20 significantly protected the lung epithelial barrier, reducing epithelial barrier permeability to intravenous 70 kDa FITC dextran by 55% (Figure 7e, “Nex.”); Sivelestat treatment exhibited a similar trend, reducing permeability by 25% (Figure 7e, “Siv.”). Importantly, both inhibitors diminished bacteremia significantly by >10-fold (Figure 7f). These data suggest that NE release by HXA3-activated lung infiltrating PMNs contributes to barrier disruption.
Discussion
Lung infections by viral and bacterial pathogens, especially multi-drug resistant bacteria, remain a major cause of death and require searches for therapies that target infection-associated pathogenic host processes (4, 7). Pulmonary infiltration by PMNs can drive lung damage and concomitant transepithelial movement of pathogens, including Sp (55–57), leading to life-threatening systemic infection. Indeed, transepithelial migration of PMNs in response to activation of the 12-LOX pathway disrupts cultured epithelial monolayers (29) and promotes lethal bacteremia in a mouse Sp lung challenge model (17). However, PMNs are also key immune cells critical for early defense against Sp infection (58). Hence, efficacious host-directed therapies to combat Sp spread must selectively target PMN effector mechanisms that promote barrier disruption while leaving intact activities required for pathogen control. Identification of the critical pathologic activities of PMNs during Sp infection of the lung requires model systems that faithfully reflect key features of PMN-Sp interactions at the respiratory mucosa.
The bronchial BSC-derived ALI epithelial model recapitulates important aspects of the architecture of bona fide airway epithelium, including the diversity of cell types and the formation of mature apical junction complexes that facilitate a functional mucosal barrier (45, 59). Here, we show that Sp infection of human and murine BSC-derived ALI monolayers mirror essential features of epithelial barrier breach following pulmonary Sp challenge in mice (17, 60), including the requirement for PMN transmigration that is entirely dependent on 12-LOX and partially dependent on PLY (29). PLY does not trigger detectable PMN transmigration or concomitant bacterial translocation after genetic ablation of 12-LOX pathway, suggesting that PLY-triggered pro-inflammatory and barrier disrupting activity in the lung is entirely due its ability to stimulate this pathway. That a PLY-deficient Sp was still capable of triggering 12-LOX-dependent PMN migration across ALI monolayers, albeit at lower than wild-type levels (Figure 2), is consistent with previous work indicating that Sp is also capable of stimulating PMN transmigration via PLY-independent means (29).
Chemotactic cues can have remarkably diverse effects on PMNs, including altering effector functions, antimicrobial activity, and inflammatory potential (23, 61). For example, in models of sterile lung injury, infiltrating PMNs are apoptotic and produce tissue-repair molecules such as TGF-β, VEGF, and resolvins (62–64). Conversely, in cystic fibrosis (CF) models, PMNs undergo transcriptional changes that reduce bactericidal activity and enhance tissue-damaging degranulation (40, 65). Similarly, PMNs that migrate into COVID-19-infected airways display a hyperinflammatory phenotype that drives lung pathology (41). Here, we show that chemotactic cues ultimately lead to divergent infection outcomes in Sp infection of ALI monolayers. Based on analogy to mucosal infection by other pathogens (32–34), HXA3 was previously deemed likely to be the 12-LOX-dependent PMN chemoattractant driving acute inflammation during Sp infection (17, 18). Here, the experimental utility of 12-LOX-deficient ALI monolayers permitted the definitive identification of HXA3 as indeed being sufficient to induce PMN transmigration and mucosal barrier disruption triggered by Sp infection. In turn, this finding was essential to permit a direct comparison of Sp-driven chemotaxis with that triggered by a well-studied control chemoattractant, fMLP (23, 66), revealing that HXA3-promoted specific pro-inflammatory conditioning of PMNs is critical for epithelial monolayer destruction.
The identification of HXA3 as sufficient for PMN-mediated mucosal barrier breach during infection by Sp prompted an exploration of pathologically important HXA3 responses. Changes in PMN physiology upon stimulation by purified HXA3 include increased calcium flux, NETosis, and anti-apoptotic programs (32, 37, 38), but here we investigated HXA3 response in the context of Sp infection. By far the largest difference upon ex vivo treatment of Sp-infected PMNs with HXA3 compared to fMLP was a 4-fold higher level of NE activity (Figure 5). HXA3 alone does not enhance PMN NE activity (Figure 5), indicating that this response requires co-stimulation by both bacteria and chemoattractant and emphasizing the importance of including microbial agents in studies of PMN responses to infection-triggered chemotactic agents. Moreover, pulmonary PMNs from mice challenged i.t. with Sp exhibited PLY- and 12-LOX-promoted elevation of degranulation, a means to release NE, as well as elevated NE levels in BALF, indicating that HXA3 triggered NE release during mouse lung infection as well (Figure 6).
Disease manifestation in response to pathogens can be mitigated either by effective actions of the host immune system to clear the microbe or by control of infection-triggered immune responses that are detrimental to the host (67). NE, along with other serine proteases, contributes to Sp killing by PMNs ex vivo (10). However, we found that inhibition of NE activity during mouse lung infection by pretreatment with the NE inhibitor Sivelestat did not affect bacterial lung burden (Figure 7), nor did it alter PMN lung infiltration. Rather, inhibition of NE, which degrades extracellular matrix components (68) and alveolar epithelial cell junction proteins (69) that maintain epithelial integrity (70), decreased bacteremia by more than 90%. These findings indicate that, in the mouse model, the pathological activities of NE outweigh any beneficial role in direct pathogen killing (10, 12).
Nexinhib20 blocks formation of exosomes and degranulation of primary granules (52), the two means by which NE is released from PMNs. During mouse infection, this inhibitor diminished degranulation of lung PMNs as well as NE activity in BALF. Although Nexinhib20 diminishes surface localization of adhesion molecules and can limit PMN recruitment to sites of tissue damage (71), we found that this inhibitor did not alter PMN infiltration into the lung post-Sp challenge. Nexinhib20 has been shown to ameliorate PMN-directed tissue damage in models of myocardial ischemia-reperfusion (71) and pulmonary LPS-induced injury (72). Here we demonstrated the ability of the drug to mitigate injury during microbial infection. Treatment with Nexinhib20, like treatment with Sivelestat, did not alter pulmonary bacterial load (Figure 7), yet bacteremia was decreased >10-fold, corresponding to protection of pulmonary barrier function measured by leakage of 70 kDa dextran (Figure 7). Primary granules contain numerous proteases that may have diverse physiologically activities (73, 74), such as the activation or inactivation of cytokines and other biologically active host factors (75, 76), that may impact the course of Sp infection, so further characterization of the effects of Sivelestat and Nexinhib20 in vivo is required to garner a full understanding of how they diminish bacteremia.
NE has been implicated in the pathogenesis of several human disorders that involve inflammatory damage, including CF, chronic obstructive pulmonary disease, bronchopulmonary dysplasia, and acute respiratory distress syndrome (ARDS) (51). The pathogenic role of NE activity during Sp infection of the mouse lung revealed here is likely reflected in human infection because higher NE levels in BALF and serum of patients with bacterial pneumonia is associated with worse clinical outcomes (77–79). Sivelestat is clinically approved for the treatment of ARDS in Korea and Japan (80) and for COVID-19 induced ARDS in China (81). Although studies of efficacy in patients have yielded inconsistent results (51, 82–84), ongoing efforts to improve delivery, e.g., by nebulizer, have yielded favorable results in improving efficacy and limiting adverse effects (85). Similarly, intrapulmonary delivery of Nexinhib20-loaded nanoparticles in experimental animals increases drug availability and decreases LPS-induced acute lung injury (72). Future studies are required to determine the efficacy of NE inhibition in limiting Sp systemic disease.
Finally, HXA3 production is a conserved mucosal inflammatory response in a multitude of bacterial infections, and possibly in acute lung injury, asthma, and inflammatory bowel syndrome (22, 31, 32, 34, 86). Given the prominent role of PMNs in mediating tissue damage, targeted mitigation of HXA3-triggered changes in PMNs that promote damage but do not compromise host defense has potential efficacy for a broad range of disorders. The identification of such changes, such as excessive NE release, is an important step in developing such strategies.
Materials and Methods:
Bacterial strains and growth conditions
Mid-exponential growth phase aliquots of S. pneumoniae TIGR4 (serotype 4) were grown in Todd-Hewitt broth (BD Biosciences) supplemented with 0.5% yeast extract in 5% CO2 and Oxyrase (Oxyrase, Mansfield, OH), and frozen in growth media with 20% (v/v) glycerol. Bacterial titers in aliquots were confirmed by plating serial dilutions on Tryptic Soy Agar plates supplemented with 5% sheep blood (blood agar) (Northeast Laboratory Services, Winslow, ME). The TIGR4 PLY-deficient mutant (Δply) was a gift from Dr. Andrew Camilli (Tufts University School of Medicine, MA). For experiments, S. pneumoniae strains were grown in Todd-Hewitt broth, supplemented with 0.5% yeast extract and Oxyrase, in 5% CO2 at 37°C and used at mid-log to late log phase.
Murine infections
BALB/c mice, C57BL/6J mice, and Alox15 knockout (Alox15−/−) mice (B6.129S2-Alox15tm1Fun/J) were obtained from Jackson Laboratories. All animal experiments were performed in accordance with Tufts University Animal Care and Use Committee approved protocols. BALB/c mice were intratracheally challenged with 1×107 colony forming units (CFU) of S. pneumoniae in 50 μl phosphate-buffered saline (PBS) to induce pneumococcal pneumonia. Control mice received PBS. The role of 12-LOX on S. pneumoniae-induced inflammation in BALB/c mice was investigated by inhibiting this enzyme with cinnamyl-3,4-dihydroxy-α-cyanocinnamate (CDC) at 8 mg/kg, in 3% DMSO, 3% cremaphor EL (CrEL) in PBS as the vehicle. CDC was injected intraperitoneally (i.p.) twice daily, starting one day before infection. The role of NE on S. pneumoniae-induced inflammation was studied in BALB/c mice by treatment with Nexinhib20, which blocks release of primary granules (52) or Sivelestate, which inhibits this enzyme (16, 87). A single dose of Nexinhib20 at 30 mg/kg, in 3% DMSO, 3% CrEL in PBS, or Sivelestat at 30 mg/kg in PBS was injected i.p. 1 hour prior to infection. Mice were euthanized at 18 h.p.i.. Blood was obtained by cardiac puncture. Bronchoalveolar lavage fluid (BALF) was collected by washing the lungs twice with 1 ml PBS via a cannula, then whole lungs were removed and bacterial burden enumerated by plating lung homogenate on blood agar plates.
Assessing lung barrier function
For assessment of lung permeability, mice were intravenously injected with 70 kDa MW FITC-Dextran at 5 mg/kg 30 minutes prior to euthanasia. Whole lungs were isolated and homogenized in 1 ml PBS, which was then subjected to fluorescence quantitation using a Synergy H1 plate reader (BioTek). Readouts were normalized to fluorescence in the serum of the same animal, diluted 1:10 in PBS.
Measuring PMN infiltration and degranulation
For flow cytometric quantitation of lung PMNs, mice were euthanized at 18 h.p.i. and lung tissues were digested with 1 mg/ml Type II collagenase (Worthington) and 50 U/ml Dnase (Worthington) to obtain a single-cell suspension. Cells present in the suspension were stained on ice for 30 minutes with APC-conjugated anti-Ly-6G (clone 1A8) or PE-conjugated anti-CD63 (Biolegend) and then washed two times in FACS buffer (Biolegend). Cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences) and the fluorescence intensities of the stained cells were determined. Collected data were analyzed using FlowJo software (v10.7, BD) to determine the numbers of infiltrating (Ly6G+) PMNs, and their level of degranulation (mean fluorescence intensity of CD63).
Establishment of epithelial air-liquid interface monolayers
Human bronchial basal cells were isolated and expanded from lung tissue harvested from donors without lung disease through the New England Organ Bank under an IRB-approved protocol (MGH #2010P001354). In brief, using a previously published basal cell isolation protocol (45, 88), EpCAM+ epithelial basal cells were isolated from human trachea and mainstem bronchi tissue. Mouse airway basal cells were obtained from C57BL/6J (B6) or Alox15−/− mouse trachea.
Harvested basal cells were cultured in complete small airway epithelial growth media (SAGM) (Lonza, Cat. CC-3118), with propagation for up to 10 passages, using the dual SMAD inhibition protocol (45).To generate monolayers permissive to modeling PMN transmigration (59), Transwells with permeable (3 μm pore size) polycarbonate membrane inserts and a culture area of 0.33 cm2 (Corning product #3415) were collagen coated and seeded with 80 μl of the airway basal cells suspension (containing > 200,000 cells) in SAGM, resulting in a density of >6000 cells/mm2, and submerged in complete SAGM for airway basal cell recovery and expansion for 1–2 days to ensure monolayer confluence. The media in both chambers was then replaced with complete Pneumacult-ALI medium (StemCell Technology, Cat. 05001) for an additional day. To initiate air-liquid interface, ALI medium in the chamber contacting the cell apical surface was removed (designated as day 0). Media was changed every 1–2 days during differentiation.
ALI monolayers used in experiments were cultured for at least 21 days to allow for full maturation of both cilia and goblet cells, but no more than 34 days to avoid overgrowth or loss of epithelial barrier (42). Transepithelial electrical resistance was assessed using a voltmeter (EVOM2, Epithelial Voltohmmeter, World Precision Instruments, Inc.) prior to migration assays to ensure the establishment of a polarized epithelial barrier.
Infection of ALI monolayers
S. pneumoniae grown to log phase was washed and resuspended to 5×108 CFU/ml in Hanks' balanced salt solution (HBSS) supplemented with 1.2 mM Ca2+ and 0.5 mM Mg2+. 25 μl of bacterial suspension was added to the apical surface of the ALI monolayers (grown on the underside of the Transwells) by inverting the Transwells and incubating at 37°C with 5% CO2 for 2 hours to allow for attachment and infection of the ALI monolayers. After treatment, Transwells were placed in 24-well receiving plates containing HBSS with Ca2+ and Mg2+, and to allow for bacteria translocation, incubated for an additional 2 hours with or without the addition of 1×106 PMNs to the basolateral chamber. Detection of basally added horseradish peroxidase (HRP) in the apical chamber was used to assess ALI monolayer barrier integrity post-treatment. Buffer in the basolateral chambers was sampled and bacterial translocation across ALI monolayers was evaluated by plating serial dilutions on blood agar plates. Bacterial migration index was calculated as total CFUs in the basolateral chamber normalized to infection inoculum.
Production of cell supernatants containing HXA3
Epithelial cell supernatants were generated from B6 ALI monolayers infected with 1×107 WT or Δply S. pneumoniae for 1 hour at 37°C with 5% CO2, and then placed in 24-well receiving plates containing HBSS with Ca2+ and Mg2+ in the apical chamber for an additional 2 hours to allow for HXA3 generation. The apical chamber supernatants were then collected and transferred to new Transwells with ALI monolayers for PMN transmigration assays.
PMN transepithelial migration assays
Whole blood obtained from healthy human volunteers under an IRB-approved protocol (Tufts University protocol #10489) was used to isolate neutrophils using the Easysep direct human neutrophil isolation kit (Stemcell), and 1×106 PMNs were added to the basolateral chamber after two hours of apical infection of the ALI monolayers with S. pneumoniae. Purified HXA3 methyl ester (Cayman) at 10 nM and fMLP (Sigma) at 10 μM were supplemented apically as indicated. To test the effect of HXA3-containing cell supernatants, the apical media was replaced with cell supernatants prepared as described above.
After two hours of transmigration, PMNs in the apical chamber were quantified by MPO activity assay, as described (29). Briefly, 50 μl of 10% Triton X-100 and 50 μl of 1 M citrate buffer were added to lyse transmigrated PMN, and 100 μl of lysed PMNs from each well was transferred to a 96-well plate. 100 μl of freshly prepared 2,2’-azinobis-3-ethylbenzotiazoline-6-sulfonic acid (ABTS) with hydrogen peroxide solution was added to each well and incubated in the dark at room temperature for 5–10 minutes. Absorbance at a wavelength of 405 nm was read on a microplate reader and measurement was converted to neutrophil number using a standard curve.
Fluorescence microscopy assessment of ALI monolayer integrity
At the end of the two hours of infection, followed by two hours of PMN transmigration across ALI monolayers, the degree of cell confluency of ALI monolayers on Transwell filters was assessed by fluorescence microscopy. To prepare samples for fluorescence microscopy, 4% paraformaldehyde fixed ALI monolayers were permeabilized with 0.1% Triton-X 100 in PBS with 3% BSA. ALI monolayers were then stained with DAPI (for nuclei) and Alexa Fluor 594 phalloidin (for F-actin), and visualized on excised filters with a Leica SP8 spectral confocal microscope (Leica). Epithelial cell retention on filters was quantitated by counting of DAPI-stained epithelial cell nuclei per field of view, carried out with CellProfiler pipeline optimized with size and roundness exclusion criteria for epithelial cell nuclei identification. Counts were normalized to uninfected controls.
Neutrophil elastase and metalloprotease activity
NE activity and MMP activity in soluble fraction of BALF from infected mice or PMN supernatants from 1×106 PMNs challenged with 1×107 CFU S. pneumoniae was determined using a PMN Elastase Fluorometric Activity Assay Kit (Abcam) and Fluorogenic MMP Substrate (Mca-PLAQAV-Dpa-RSSSR-NH2) (R&D Systems), respectively, following manufacturer's instructions. The area under the curve of kinetic substrate conversion curves over two hours was measured with a Synergy H1 plate reader (BioTek) and normalized to uninfected controls.
Opsonophagocytic (OPH) killing
The ability of neutrophils to kill pneumococci was assessed ex vivo as described previously (89), with modification. Briefly, 1×106 PMNs were incubated with 5 ×103 S. pneumoniae grown to mid-log phase and pre-opsonized with 10 μl rabbit complement (Pel-Freez) in 100 μl reactions in HBSS with Ca2+ and Mg2+. The reactions were incubated for 45 minutes at 37°C. Percent killing in comparison to incubations with no PMNs was determined by plating serial dilutions on blood agar plates.
Reactive oxygen species (ROS) production
Neutrophils were resuspended in HBSS with Ca2+ and Mg2+ containing 10 μM 2',7’-dichlorodihydrofluorescein diacetate (DCF) (Molecular Probes) to a final concentration of 1×107 cells/ml and gently agitated for 10 minutes at room temperature. 1×106 DCF-containing neutrophils were challenged with Sp at an MOI of 10 for 30 minutes at 37°C, then washed and resuspended in FACS buffer for analysis by a FACSCalibur flow cytometer (BD Biosciences). Collected data were analyzed using FlowJo software (v10.7, BD) to determine the numbers of ROS-producing DCF-positive cells.
Neutrophil extracellular trap formation (NETosis) and apoptosis by flow
1×106 neutrophils were challenged with Sp at an MOI of 10 for 30 minutes at 37°C, then washed and resuspended in FACS buffer. For NETosis analysis, cells were stained with a plasma membrane-impermeable DNA-binding dye, SYTOX™ AADvanced™ (Life Technologies, Carlsbad, CA), rabbit anti-myeloperoxidase (Abcam ab45977), and secondary goat anti-rabbit-Alexa Fluor 568 antibody (Invitrogen). For apoptosis analysis, cells were stained with FITC-conjugated Annexin V (BioLegend, San Diego, CA, USA), and propidium iodide (PI). Samples were read on a FACSCalibur flow cytometer (BD Biosciences), and collected data were analyzed using FlowJo software (v10.7, BD) to determine percent NETosis (MPO+ SYTOX+), and percent apoptosis (Annexin+ PI−).
Presentation of data and statistical analyses
Statistical and correlation analysis was carried out using GraphPad Prism (GraphPad Software, San Diego, CA). p values <0.05 were considered significant in all cases. For bacterial burdens, geometric mean ± geometric SD is shown; for all other graphs, the mean values ± SEM are shown. Due to intrinsic donor-to-donor variability of human PMN transmigration efficacy, experiments involving human donors were normalized within each experiment before pooling individual experiments. The conclusions drawn were those found to be reproducible and statistically significant across independent experiments.
Importance:
Streptococcus pneumoniae (Sp), a leading cause of pneumonia, can spread from the lung into the bloodstream to cause systemic disease. Limitations in vaccine efficacy and a rise in antimicrobial resistance have spurred searches for host-directed therapies that limit pathologic host immune responses to Sp. Excessive polymorphonuclear leukocyte (PMN) infiltration into Sp-infected airways promotes systemic disease. Using stem cell-derived respiratory cultures that reflect bona fide lung epithelium, we identified the eicosanoid hepoxilin A3 as a critical pulmonary PMN chemoattractant that is sufficient to drive PMN-mediated epithelial damage by inducing the release of neutrophil elastase. Inhibition of the release or activity of this protease in mice limited epithelial barrier disruption and bacterial dissemination, suggesting a new host-directed treatment for Sp lung infection.
Acknowledgements:
We thank Andrew Camilli for strains and Elsa Bou Ghanem, Stephania Libreros, Amanda Pulsifer, Rodney K. Tweten, Byran P. Hurley, and Beth A. McCormick for protocols and helpful discussions. This work was supported by NIH Awards AG071268 to JL and JM, and NIH Awards AI15081 and AI152499 to JMV.
Footnotes
Supplemental Figure 1. PMNs are required for epithelial cell detachment and barrier breach. Human BSC-derived ALI monolayers were apically infected with 1 × 107 WT or Δply Sp without basolateral PMNs. (a) After two hours, monolayer integrity was assessed by fluorescence confocal microscopy after staining nuclei with DAPI and F-actin with fluorescent phalloidin. For clarity, images shown are of extended projections (all z-sections collapsed into 1 plane). Scale bar = 40 μm for all images. (b) Epithelial retention was quantitated by enumerating epithelial cell nuclei relative to uninfected ALI. (c) Epithelial permeability was measured by HRP flux relative to monolayers infected with WT Sp. (d) Sp translocation was quantitated by measuring basolateral CFU. Each panel is a representative of three independent experiments. Error bars represent mean ± SEM.
Supplemental Figure 2. Sp infection alters PMN functional response profile. 1 × 106 PMNs were uninfected or infected with 1 × 107 Sp and evaluated for functional performance via (a) PMN membrane permeability determined by propidium iodide staining (PI+), (b) opsonophagocytic killing with or without the addition of complement opsonin, quantitated by plating for CFU, (c) NETosis determined by Sytox and anti-MPO staining (Sytox+ MPO+), (d) released MMP activity by substrate conversion and expressed relative to uninfected PMNs, (e) apoptosis determined by lack of straining by propidium iodide and positive staining of Annexin V (PI− Annexin V+), (f) ROS production by intracellular oxidation of substrate (DCF+), or (g) released NE activity by substrate conversion and expressed relative to uninfected PMNs (See Materials and Methods). (h) Radar plot summary of log fold change in PMN functional performance in (c-g). Each panel shown is representative of three independent experiments. Error bars represent mean ± SEM. Statistical analysis was performed using ordinary one-way ANOVA: **p-value < 0.01, ***p-value < 0.001, ****p-value < 0.0001.
References:
- 1.Henriques-Normark B, Tuomanen EI. 2013. The pneumococcus: epidemiology, microbiology, and pathogenesis. Cold Spring Harb Perspect Med 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen H, Matsumoto H, Horita N, Hara Y, Kobayashi N, Kaneko T. 2021. Prognostic factors for mortality in invasive pneumococcal disease in adult: a system review and meta-analysis. Sci Rep 11:11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Backhaus E, Berg S, Andersson R, Ockborn G, Malmstrom P, Dahl M, Nasic S, Trollfors B. 2016. Epidemiology of invasive pneumococcal infections: manifestations, incidence and case fatality rate correlated to age, gender and risk factors. BMC Infect Dis 16:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zumla A, Rao M, Wallis RS, Kaufmann SH, Rustomjee R, Mwaba P, Vilaplana C, Yeboah-Manu D, Chakaya J, Ippolito G, Azhar E, Hoelscher M, Maeurer M, Host-Directed Therapies Network c. 2016. Host-directed therapies for infectious diseases: current status, recent progress, and future prospects. Lancet Infect Dis 16:e47–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cremers AJ, Mobegi FM, de Jonge MI, van Hijum SA, Meis JF, Hermans PW, Ferwerda G, Bentley SD, Zomer AL. 2015. The post-vaccine microevolution of invasive Streptococcus pneumoniae. Sci Rep 5:14952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farrell DJ, Klugman KP, Pichichero M. 2007. Increased antimicrobial resistance among nonvaccine serotypes of Streptococcus pneumoniae in the pediatric population after the introduction of 7-valent pneumococcal vaccine in the United States. Pediatr Infect Dis J 26:123–8. [DOI] [PubMed] [Google Scholar]
- 7.Sundaresh B, Xu S, Noonan B, Mansour MK, Leong JM, van Opijnen T. 2021. Host-informed therapies for the treatment of pneumococcal pneumonia. Trends Mol Med 27:971–989. [DOI] [PubMed] [Google Scholar]
- 8.Herring SE, Mao S, Bhalla M, Tchalla EYI, Kramer JM, Bou Ghanem EN. 2022. Mitochondrial ROS production by neutrophils is required for host antimicrobial function against Streptococcus pneumoniae and is controlled by A2B adenosine receptor signaling. PLoS Pathog 18:e1010700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mori Y, Yamaguchi M, Terao Y, Hamada S, Ooshima T, Kawabata S. 2012. alpha-Enolase of Streptococcus pneumoniae induces formation of neutrophil extracellular traps. J Biol Chem 287:10472–10481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Standish AJ, Weiser JN. 2009. Human neutrophils kill Streptococcus pneumoniae via serine proteases. J Immunol 183:2602–9. [DOI] [PubMed] [Google Scholar]
- 11.Evans SE, Ost DE. 2015. Pneumonia in the neutropenic cancer patient. Curr Opin Pulm Med 21:260–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hahn I, Klaus A, Janze AK, Steinwede K, Ding N, Bohling J, Brumshagen C, Serrano H, Gauthier F, Paton JC, Welte T, Maus UA. 2011. Cathepsin G and neutrophil elastase play critical and nonredundant roles in lung-protective immunity against Streptococcus pneumoniae in mice. Infect Immun 79:4893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lax S, Wilson MR, Takata M, Thickett DR. 2014. Using a non-invasive assessment of lung injury in a murine model of acute lung injury. BMJ Open Respir Res 1:e000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jose RJ, Williams AE, Mercer PF, Sulikowski MG, Brown JS, Chambers RC. 2015. Regulation of neutrophilic inflammation by proteinase-activated receptor 1 during bacterial pulmonary infection. J Immunol 194:6024–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamada K, Yanagihara K, Araki N, Harada Y, Morinaga Y, Izumikawa K, Kakeya H, Yamamoto Y, Hasegawa H, Kohno S, Kamihira S. 2011. In vivo efficacy of KRP-109, a novel elastase inhibitor, in a murine model of severe pneumococcal pneumonia. Pulm Pharmacol Ther 24:660–5. [DOI] [PubMed] [Google Scholar]
- 16.Yanagihara K, Fukuda Y, Seki M, Izumikawa K, Miyazaki Y, Hirakata Y, Tsukamoto K, Yamada Y, Kamhira S, Kohno S. 2007. Effects of specific neutrophil elastase inhibitor, sivelestat sodium hydrate, in murine model of severe pneumococcal pneumonia. Exp Lung Res 33:71–80. [DOI] [PubMed] [Google Scholar]
- 17.Bhowmick R, Maung N, Hurley BP, Ghanem EB, Gronert K, McCormick BA, Leong JM. 2013. Systemic disease during Streptococcus pneumoniae acute lung infection requires 12-lipoxygenase-dependent inflammation. J Immunol 191:5115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhowmick R, Clark S, Bonventre JV, Leong JM, McCormick BA. 2017. Cytosolic Phospholipase A2alpha Promotes Pulmonary Inflammation and Systemic Disease during Streptococcus pneumoniae Infection. Infect Immun 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marks M, Burns T, Abadi M, Seyoum B, Thornton J, Tuomanen E, Pirofski LA. 2007. Influence of neutropenia on the course of serotype 8 pneumococcal pneumonia in mice. Infect Immun 75:1586–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rubins JB, Charboneau D, Paton JC, Mitchell TJ, Andrew PW, Janoff EN. 1995. Dual function of pneumolysin in the early pathogenesis of murine pneumococcal pneumonia. J Clin Invest 95:142–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bou Ghanem EN, Clark S, Roggensack SE, McIver SR, Alcaide P, Haydon PG, Leong JM. 2015. Extracellular Adenosine Protects against Streptococcus pneumoniae Lung Infection by Regulating Pulmonary Neutrophil Recruitment. PLoS Pathog 11:e1005126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bruijnzeel PL, Uddin M, Koenderman L. 2015. Targeting neutrophilic inflammation in severe neutrophilic asthma: can we target the disease-relevant neutrophil phenotype? J Leukoc Biol 98:549–56. [DOI] [PubMed] [Google Scholar]
- 23.Metzemaekers M, Gouwy M, Proost P. 2020. Neutrophil chemoattractant receptors in health and disease: double-edged swords. Cell Mol Immunol 17:433–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giacalone VD, Margaroli C, Mall MA, Tirouvanziam R. 2020. Neutrophil Adaptations upon Recruitment to the Lung: New Concepts and Implications for Homeostasis and Disease. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jounblat R, Kadioglu A, Mitchell TJ, Andrew PW. 2003. Pneumococcal behavior and host responses during bronchopneumonia are affected differently by the cytolytic and complement-activating activities of pneumolysin. Infect Immun 71:1813–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Canvin JR, Marvin AP, Sivakumaran M, Paton JC, Boulnois GJ, Andrew PW, Mitchell TJ. 1995. The role of pneumolysin and autolysin in the pathology of pneumonia and septicemia in mice infected with a type 2 pneumococcus. J Infect Dis 172:119–23. [DOI] [PubMed] [Google Scholar]
- 27.Panagiotou S, Chaguza C, Yahya R, Audshasai T, Baltazar M, Ressel L, Khandaker S, Alsahag M, Mitchell TJ, Prudhomme M, Kadioglu A, Yang M. 2020. Hypervirulent pneumococcal serotype 1 harbours two pneumolysin variants with differential haemolytic activity. Sci Rep 10:17313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCormick BA. 2007. Bacterial-induced hepoxilin A3 secretion as a pro-inflammatory mediator. FEBS J 274:3513–3518. [DOI] [PubMed] [Google Scholar]
- 29.Adams W, Bhowmick R, Bou Ghanem EN, Wade K, Shchepetov M, Weiser JN, McCormick BA, Tweten RK, Leong JM. 2019. Pneumolysin Induces 12-Lipoxygenase-Dependent Neutrophil Migration during Streptococcus pneumoniae Infection. J Immunol doi: 10.4049/jimmunol.1800748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh NK, Rao GN. 2019. Emerging role of 12/15-Lipoxygenase (ALOX15) in human pathologies. Prog Lipid Res 73:28–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szabady RL, McCormick BA. 2013. Control of neutrophil inflammation at mucosal surfaces by secreted epithelial products. Front Immunol 4:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mrsny RJ, Gewirtz AT, Siccardi D, Savidge T, Hurley BP, Madara JL, McCormick BA. 2004. Identification of hepoxilin A3 in inflammatory events: a required role in neutrophil migration across intestinal epithelia. Proc Natl Acad Sci U S A 101:7421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mumy KL, Bien JD, Pazos MA, Gronert K, Hurley BP, McCormick BA. 2008. Distinct isoforms of phospholipase A2 mediate the ability of Salmonella enterica serotype typhimurium and Shigella flexneri to induce the transepithelial migration of neutrophils. Infect Immun 76:3614–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamang DL, Pirzai W, Priebe GP, Traficante DC, Pier GB, Falck JR, Morisseau C, Hammock BD, McCormick BA, Gronert K, Hurley BP. 2012. Hepoxilin A(3) facilitates neutrophilic breach of lipoxygenase-expressing airway epithelial barriers. J Immunol 189:4960–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Serhan CN, Chiang N, Dalli J, Levy BD. 2014. Lipid mediators in the resolution of inflammation. Cold Spring Harb Perspect Biol 7:a016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nigam S, Zafiriou MP, Deva R, Ciccoli R, Roux-Van der Merwe R. 2007. Structure, biochemistry and biology of hepoxilins: an update. FEBS J 274:3503–3512. [DOI] [PubMed] [Google Scholar]
- 37.Lin Z, Laneuville O, Pace-Asciak CR. 1991. Hepoxilin A3 induces heat shock protein (HSP72) expression in human neutrophils. Biochem Biophys Res Commun 179:52–6. [DOI] [PubMed] [Google Scholar]
- 38.Douda DN, Grasemann H, Pace-Asciak C, Palaniyar N. 2015. A lipid mediator hepoxilin A3 is a natural inducer of neutrophil extracellular traps in human neutrophils. Mediators Inflamm 2015:520871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pazos MA, Pirzai W, Yonker LM, Morisseau C, Gronert K, Hurley BP. 2015. Distinct cellular sources of hepoxilin A3 and leukotriene B4 are used to coordinate bacterial-induced neutrophil transepithelial migration. J Immunol 194:1304–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forrest OA, Ingersoll SA, Preininger MK, Laval J, Limoli DH, Brown MR, Lee FE, Bedi B, Sadikot RT, Goldberg JB, Tangpricha V, Gaggar A, Tirouvanziam R. 2018. Frontline Science: Pathological conditioning of human neutrophils recruited to the airway milieu in cystic fibrosis. J Leukoc Biol 104:665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eddins DJ, Yang J, Kosters A, Giacalone VD, Pechuan-Jorge X, Chandler JD, Eum J, Babcock BR, Dobosh BS, Hernandez MR, Abdulkhader F, Collins GL, Orlova DY, Ramonell RP, Sanz I, Moussion C, Eun-Hyung Lee F, Tirouvanziam RM, Ghosn EEB. 2023. Transcriptional reprogramming of infiltrating neutrophils drives lung pathology in severe COVID-19 despite low viral load. Blood Adv 7:778–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levardon H, Yonker LM, Hurley BP, Mou H. 2018. Expansion of Airway Basal Cells and Generation of Polarized Epithelium. Bio Protoc 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greene NG, Narciso AR, Filipe SR, Camilli A. 2015. Peptidoglycan Branched Stem Peptides Contribute to Streptococcus pneumoniae Virulence by Inhibiting Pneumolysin Release. PLoS Pathog 11:e1004996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacques LC, Panagiotou S, Baltazar M, Senghore M, Khandaker S, Xu R, Bricio-Moreno L, Yang M, Dowson CG, Everett DB, Neill DR, Kadioglu A. 2020. Increased pathogenicity of pneumococcal serotype 1 is driven by rapid autolysis and release of pneumolysin. Nat Commun 11:1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mou H, Vinarsky V, Tata PR, Brazauskas K, Choi SH, Crooke AK, Zhang B, Solomon GM, Turner B, Bihler H, Harrington J, Lapey A, Channick C, Keyes C, Freund A, Artandi S, Mense M, Rowe S, Engelhardt JF, Hsu YC, Rajagopal J. 2016. Dual SMAD Signaling Inhibition Enables Long-Term Expansion of Diverse Epithelial Basal Cells. Cell Stem Cell 19:217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pereira JM, Xu S, Leong JM, Sousa S. 2022. The Yin and Yang of Pneumolysin During Pneumococcal Infection. Front Immunol 13:878244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishimoto AT, Rosch JW, Tuomanen EI. 2020. Pneumolysin: Pathogenesis and Therapeutic Target. Front Microbiol 11:1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hurley BP, Siccardi D, Mrsny RJ, McCormick BA. 2004. Polymorphonuclear cell transmigration induced by Pseudomonas aeruginosa requires the eicosanoid hepoxilin A3. J Immunol 173:5712–20. [DOI] [PubMed] [Google Scholar]
- 49.Domon H, Oda M, Maekawa T, Nagai K, Takeda W, Terao Y. 2016. Streptococcus pneumoniae disrupts pulmonary immune defence via elastase release following pneumolysin-dependent neutrophil lysis. Sci Rep 6:38013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Domon H, Terao Y. 2021. The Role of Neutrophils and Neutrophil Elastase in Pneumococcal Pneumonia. Front Cell Infect Microbiol 11:615959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeng W, Song Y, Wang R, He R, Wang T. 2023. Neutrophil elastase: From mechanisms to therapeutic potential. J Pharm Anal 13:355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson JL, Ramadass M, He J, Brown SJ, Zhang J, Abgaryan L, Biris N, Gavathiotis E, Rosen H, Catz SD. 2016. Identification of Neutrophil Exocytosis Inhibitors (Nexinhibs), Small Molecule Inhibitors of Neutrophil Exocytosis and Inflammation: DRUGGABILITY OF THE SMALL GTPase Rab27a. J Biol Chem 291:25965–25982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ginzberg HH, Cherapanov V, Dong Q, Cantin A, McCulloch CA, Shannon PT, Downey GP. 2001. Neutrophil-mediated epithelial injury during transmigration: role of elastase. Am J Physiol Gastrointest Liver Physiol 281:G705–17. [DOI] [PubMed] [Google Scholar]
- 54.Young RE, Voisin MB, Wang S, Dangerfield J, Nourshargh S. 2007. Role of neutrophil elastase in LTB4-induced neutrophil transmigration in vivo assessed with a specific inhibitor and neutrophil elastase deficient mice. Br J Pharmacol 151:628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clarke TB, Francella N, Huegel A, Weiser JN. 2011. Invasive bacterial pathogens exploit TLR-mediated downregulation of tight junction components to facilitate translocation across the epithelium. Cell Host Microbe 9:404–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weight CM, Venturini C, Pojar S, Jochems SP, Reine J, Nikolaou E, Solorzano C, Noursadeghi M, Brown JS, Ferreira DM, Heyderman RS. 2019. Microinvasion by Streptococcus pneumoniae induces epithelial innate immunity during colonisation at the human mucosal surface. Nat Commun 10:3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pechous RD. 2017. With Friends Like These: The Complex Role of Neutrophils in the Progression of Severe Pneumonia. Front Cell Infect Microbiol 7:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nemeth T, Sperandio M, Mocsai A. 2020. Neutrophils as emerging therapeutic targets. Nat Rev Drug Discov 19:253–275. [DOI] [PubMed] [Google Scholar]
- 59.Yonker LM, Mou H, Chu KK, Pazos MA, Leung H, Cui D, Ryu J, Hibbler RM, Eaton AD, Ford TN, Falck JR, Kinane TB, Tearney GJ, Rajagopal J, Hurley BP. 2017. Development of a Primary Human Co-Culture Model of Inflamed Airway Mucosa. Sci Rep 7:8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garcia-Suarez Mdel M, Florez N, Astudillo A, Vazquez F, Villaverde R, Fabrizio K, Pirofski LA, Mendez FJ. 2007. The role of pneumolysin in mediating lung damage in a lethal pneumococcal pneumonia murine model. Respir Res 8:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Palomino-Segura M, Sicilia J, Ballesteros I, Hidalgo A. 2023. Strategies of neutrophil diversification. Nat Immunol 24:575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peiseler M, Kubes P. 2019. More friend than foe: the emerging role of neutrophils in tissue repair. J Clin Invest 129:2629–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blazquez-Prieto J, Lopez-Alonso I, Huidobro C, Albaiceta GM. 2018. The Emerging Role of Neutrophils in Repair after Acute Lung Injury. Am J Respir Cell Mol Biol 59:289–294. [DOI] [PubMed] [Google Scholar]
- 64.Zemans RL, Briones N, Campbell M, McClendon J, Young SK, Suzuki T, Yang IV, De Langhe S, Reynolds SD, Mason RJ, Kahn M, Henson PM, Colgan SP, Downey GP. 2011. Neutrophil transmigration triggers repair of the lung epithelium via beta-catenin signaling. Proc Natl Acad Sci U S A 108:15990–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Margaroli C, Moncada-Giraldo D, Gulick DA, Dobosh B, Giacalone VD, Forrest OA, Sun F, Gu C, Gaggar A, Kissick H, Wu R, Gibson G, Tirouvanziam R. 2021. Transcriptional firing represses bactericidal activity in cystic fibrosis airway neutrophils. Cell Rep Med 2:100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gauthier JF, Fortin A, Bergeron Y, Dumas MC, Champagne ME, Bergeron MG. 2007. Differential contribution of bacterial N-formyl-methionyl-leucyl- phenylalanine and host-derived CXC chemokines to neutrophil infiltration into pulmonary alveoli during murine pneumococcal pneumonia. Infect Immun 75:5361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ayres JS, Schneider DS. 2012. Tolerance of infections. Annu Rev Immunol 30:271–94. [DOI] [PubMed] [Google Scholar]
- 68.Zhu Y, Huang Y, Ji Q, Fu S, Gu J, Tai N, Wang X. 2021. Interplay between Extracellular Matrix and Neutrophils in Diseases. J Immunol Res 2021:8243378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boxio R, Wartelle J, Nawrocki-Raby B, Lagrange B, Malleret L, Hirche T, Taggart C, Pacheco Y, Devouassoux G, Bentaher A. 2016. Neutrophil elastase cleaves epithelial cadherin in acutely injured lung epithelium. Respir Res 17:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Isono T, Hirayama S, Domon H, Maekawa T, Tamura H, Hiyoshi T, Sirisereephap K, Takenaka S, Noiri Y, Terao Y. 2023. Degradation of EGFR on lung epithelial cells by neutrophil elastase contributes to the aggravation of pneumococcal pneumonia. J Biol Chem 299:104760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu W, Cronin CG, Cao Z, Wang C, Ruan J, Pulikkot S, Hall A, Sun H, Groisman A, Chen Y, Vella AT, Hu L, Liang BT, Fan Z. 2022. Nexinhib20 Inhibits Neutrophil Adhesion and beta(2) Integrin Activation by Antagonizing Rac-1-Guanosine 5'-Triphosphate Interaction. J Immunol 209:1574–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mejias JC, Forrest OA, Margaroli C, Frey Rubio DA, Viera L, Li J, Xu X, Gaggar A, Tirouvanziam R, Roy K. 2019. Neutrophil-targeted, protease-activated pulmonary drug delivery blocks airway and systemic inflammation. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Raevens S, Van Campenhout S, Debacker PJ, Lefere S, Verhelst X, Geerts A, Van Vlierberghe H, Colle I, Devisscher L. 2020. Combination of sivelestat and N-acetylcysteine alleviates the inflammatory response and exceeds standard treatment for acetaminophen-induced liver injury. J Leukoc Biol 107:341–355. [DOI] [PubMed] [Google Scholar]
- 74.Yang T, Zhang J, Sun L, Zhu X, Li J, Wang J, Chen H, Bao R, Deng X, Hou J, Liu Y. 2012. Combined effects of a neutrophil elastase inhibitor (sivelestat sodium) and a free radical scavenger (edaravone) on lipopolysaccharide-induced acute lung injury in rats. Inflamm Res 61:563–9. [DOI] [PubMed] [Google Scholar]
- 75.Domon H, Nagai K, Maekawa T, Oda M, Yonezawa D, Takeda W, Hiyoshi T, Tamura H, Yamaguchi M, Kawabata S, Terao Y. 2018. Neutrophil Elastase Subverts the Immune Response by Cleaving Toll-Like Receptors and Cytokines in Pneumococcal Pneumonia. Front Immunol 9:732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Clancy DM, Sullivan GP, Moran HBT, Henry CM, Reeves EP, McElvaney NG, Lavelle EC, Martin SJ. 2018. Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep 22:2937–2950. [DOI] [PubMed] [Google Scholar]
- 77.Wilkinson TS, Conway Morris A, Kefala K, O'Kane CM, Moore NR, Booth NA, McAuley DF, Dhaliwal K, Walsh TS, Haslett C, Sallenave JM, Simpson AJ. 2012. Ventilator-associated pneumonia is characterized by excessive release of neutrophil proteases in the lung. Chest 142:1425–1432. [DOI] [PubMed] [Google Scholar]
- 78.Tagami T, Kushimoto S, Tosa R, Omura M, Yonezawa K, Akiyama G, Hirama H, Yokota H. 2011. Plasma neutrophil elastase correlates with pulmonary vascular permeability: a prospective observational study in patients with pneumonia. Respirology 16:953–8. [DOI] [PubMed] [Google Scholar]
- 79.Matsuse H, Yanagihara K, Mukae H, Tanaka K, Nakazato M, Kohno S. 2007. Association of plasma neutrophil elastase levels with other inflammatory mediators and clinical features in adult patients with moderate and severe pneumonia. Respir Med 101:1521–8. [DOI] [PubMed] [Google Scholar]
- 80.Aikawa N, Kawasaki Y. 2014. Clinical utility of the neutrophil elastase inhibitor sivelestat for the treatment of acute respiratory distress syndrome. Ther Clin Risk Manag 10:621–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sahebnasagh A, Saghafi F, Safdari M, Khataminia M, Sadremomtaz A, Talaei Z, Rezai Ghaleno H, Bagheri M, Habtemariam S, Avan R. 2020. Neutrophil elastase inhibitor (sivelestat) may be a promising therapeutic option for management of acute lung injury/acute respiratory distress syndrome or disseminated intravascular coagulation in COVID-19. J Clin Pharm Ther 45:1515–1519. [DOI] [PubMed] [Google Scholar]
- 82.Maki C, Inoue Y, Ishihara T, Hirano Y, Kondo Y, Sueyoshi K, Okamoto K, Tanaka H. 2020. Evaluation of appropriate indications for the use of sivelestat sodium in acute respiratory distress syndrome: a retrospective cohort study. Acute Med Surg 7:e471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tagami T, Tosa R, Omura M, Fukushima H, Kaneko T, Endo T, Rinka H, Murai A, Yamaguchi J, Yoshikawa K, Saito N, Uzu H, Kase Y, Takatori M, Izumino H, Nakamura T, Seo R, Kitazawa Y, Sugita M, Takahashi H, Kuroki Y, Irahara T, Kanemura T, Yokota H, Kushimoto S. 2014. Effect of a selective neutrophil elastase inhibitor on mortality and ventilator-free days in patients with increased extravascular lung water: a post hoc analysis of the PiCCO Pulmonary Edema Study. J Intensive Care 2:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zeiher BG, Artigas A, Vincent JL, Dmitrienko A, Jackson K, Thompson BT, Bernard G, Group SS. 2004. Neutrophil elastase inhibition in acute lung injury: results of the STRIVE study. Crit Care Med 32:1695–702. [DOI] [PubMed] [Google Scholar]
- 85.Barth P, Bruijnzeel P, Wach A, Sellier Kessler O, Hooftman L, Zimmermann J, Naue N, Huber B, Heimbeck I, Kappeler D, Timmer W, Chevalier E. 2020. Single dose escalation studies with inhaled POL6014, a potent novel selective reversible inhibitor of human neutrophil elastase, in healthy volunteers and subjects with cystic fibrosis. J Cyst Fibros 19:299–304. [DOI] [PubMed] [Google Scholar]
- 86.Rossaint J, Nadler JL, Ley K, Zarbock A. 2012. Eliminating or blocking 12/15-lipoxygenase reduces neutrophil recruitment in mouse models of acute lung injury. Crit Care 16:R166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Iwata K, Doi A, Ohji G, Oka H, Oba Y, Takimoto K, Igarashi W, Gremillion DH, Shimada T. 2010. Effect of neutrophil elastase inhibitor (sivelestat sodium) in the treatment of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): a systematic review and meta-analysis. Intern Med 49:2423–32. [DOI] [PubMed] [Google Scholar]
- 88.Gonzalez-Juarbe N, Bradley KM, Shenoy AT, Gilley RP, Reyes LF, Hinojosa CA, Restrepo MI, Dube PH, Bergman MA, Orihuela CJ. 2017. Pore-forming toxin-mediated ion dysregulation leads to death receptor-independent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ 24:917–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bou Ghanem EN, Clark S, Du X, Wu D, Camilli A, Leong JM, Meydani SN. 2015. The alpha-tocopherol form of vitamin E reverses age-associated susceptibility to streptococcus pneumoniae lung infection by modulating pulmonary neutrophil recruitment. J Immunol 194:1090–9. [DOI] [PMC free article] [PubMed] [Google Scholar]