

Abstract
The current study analyzed the intersecting biophysical, biochemical, and functional properties of extracellular particles (EPs) with the human immunodeficiency virus type‐1 (HIV‐1) beyond the currently accepted size range for HIV‐1. We isolated five fractions (Frac‐A through Frac‐E) from HIV‐infected cells by sequential differential ultracentrifugation (DUC). All fractions showed a heterogeneous size distribution with median particle sizes greater than 100 nm for Frac‐A through Frac‐D but not for Frac‐E, which contained small EPs with an average size well below 50 nm. Synchronized and released cultures contained large infectious EPs in Frac‐A, with markers of amphisomes and viral components. Additionally, Frac‐E uniquely contained EPs positive for CD63, HSP70, and HIV‐1 proteins. Despite its small average size, Frac‐E contained membrane‐protected viral integrase, detectable only after SDS treatment, indicating that it is enclosed in vesicles. Single particle analysis with dSTORM further supported these findings as CD63, HIV‐1 integrase, and the viral surface envelope (Env) glycoprotein (gp) colocalized on the same Frac‐E particles. Surprisingly, Frac‐E EPs were infectious, and infectivity was significantly reduced by immunodepleting Frac‐E with anti‐CD63, indicating the presence of this protein on the surface of infectious small EPs in Frac‐E. To our knowledge, this is the first time that extracellular vesicle (EV) isolation methods have identified infectious small HIV‐1 particles (smHIV‐1) that are under 50 nm. Collectively, our data indicate that the crossroads between EPs and HIV‐1 potentially extend beyond the currently accepted biophysical properties of HIV‐1, which may have further implications for viral pathogenesis.
Keywords: amphisomes, cART, exomeres, exosomes, extracellular particles, extracellular vesicles, HIV‐1, NRTIs, small HIV, smHIV‐1
1. INTRODUCTION
Human immunodeficiency virus type 1 (HIV‐1) is a causative agent of acquired immunodeficiency syndrome (AIDS), and since its discovery in 1981, it has caused approximately 40.1 million deaths worldwide (UNAIDS, 2023). By 2021, it was estimated that about 1.5 million people (about the population of West Virginia) contracted the virus and 1.1 million succumbed to the disease, while 40 million people live with HIV disease (UNAIDS, 2023). The introduction of combination antiretroviral therapy (cART) has significantly improved the quality of life in HIV‐1 patients and has reduced the incidence of opportunistic infections. Approximately 10 million HIV‐1 patients have received cART (Archin et al., 2014; Gueler et al., 2017). Modern cART regimens effectively target various stages of the HIV‐1 cycle, including viral entry, reverse transcription, integration, protease cleavage of viral polyproteins, and virion maturation (Archin et al., 2014). However, life‐long adherence to a cART regimen is required to keep the virus at bay, and cessation of the treatment results in viral rebound and subsequent CD4+ T‐cell depletion (Davey et al., 1999).
Extracellular vesicles (EVs) and particles are secreted by most cell types and play a key role in intercellular communication (Pelissier Vatter et al., 2021). Classically, EVs are surrounded by a lipid bilayer with a heterogeneous size distribution from 30 nm to several microns with a diverse biochemical composition (Welsh et al., 2024; Zijlstra & Di Vizio, 2018). Nonetheless, various biological fluids also contain non‐vesicular particles (NVEPs) including exomeres and supermeres, which carry functional cargo to cells without a defined lipid bilayer (Anand et al., 2021; Q. Zhang, Jeppesen, Higginbotham, Graves‐Deal, et al., 2021; Q. Zhang et al., 2019). Additionally, the complexity of different biological fluids is further increased by the presence of viruses alongside EVs and NVEPs, which requires further consideration. Therefore, the collective term extracellular particles (EPs) aptly describes the varied entities (EVs, NVEPs, and viruses) present alongside diverse biofluid mixtures, as classified in MISEV 2023 (Welsh et al., 2024).
Generally, EPs are present in various biological fluids and have been shown to maintain homeostasis of the innate and adaptive immune response (Pelissier Vatter et al., 2021; Robbins & Morelli, 2014). Egress of EPs from the donor cells is a well‐regulated process (Colombo et al., 2014; van Niel et al., 2018) that is also hijacked by different viruses to facilitate their exit (Margolis & Sadovsky, 2019; Nolte‐’t Hoen et al., 2016). Intercellular communications are disrupted during viral infections with EPs containing viral cargo, such as viral proteins and nucleic acids (Fleming et al., 2014; Ipinmoroti & Matthews, 2020; Nolte‐’t Hoen et al., 2016). Viruses, as obligate intracellular parasites, rewire cellular machinery (Summers, 2009) to produce toxic molecular byproducts to the host cells and are packaged and exocytosed from the infected cells in EPs (Yang et al., 2021). EPs have been implicated in the pathophysiology of various viral infections, such as coronavirus, human T‐cell leukaemia virus type 1 (HTLV‐1), hepatitis C virus (HCV), human adenovirus, and HIV‐1 (Giannessi et al., 2020; Ipinmoroti & Matthews, 2020; Martins & Alves, 2020; Schwab et al., 2015). For example, HIV‐1 can promote viral dissemination by incorporating Env glycoprotein (gp) (Arakelyan et al., 2017) or short viral transcripts into EPs (Narayanan et al., 2013) that can then enter and regulate recipient cells, making them more susceptible to viral infections (Narayanan et al., 2013; Sampey et al., 2016).
Furthermore, previous observations have shown that HIV‐1 can exit via alternative egress routes from monocytic cells. These HIV‐1 alternative egress routes were linked to the cell physiology state, indicating that the virus can either bud off at the plasma membrane from activated cells (Suyama et al., 2009) or bud inside into large multivesicular bodies (MVBs) from which it can exit through the exocytic pathway, similar to EVs (Deneka et al., 2007). These findings raised new questions regarding currently accepted egress routes of HIV‐1 (Pérez et al., 2019; Soares, 2014). Therefore, the “Trojan exosome hypothesis” (Gould et al., 2003) merged multiple aspects of the exocytic pathways and provided an alternative approach to HIV‐1 exit, which argues that the virus has a unique lipid and host cell protein composition. The striking resemblance between HIV‐1 and EVs, arising from their shared convergent evolutionary past (Wurdinger et al., 2012), presents complex scientific questions that require further elucidation (Margolis & Sadovsky, 2019).
The heterogeneous nature of EPs and viruses presents many technical challenges addressed by different separation and analysis techniques (Liangsupree et al., 2021; McNamara & Dittmer, 2020). We and others have successfully separated EPs from HIV‐1 by fractionation (DeMarino et al., 2019; Dettenhofer & Yu, 1999). However, EP markers were still present in the denser viral fraction, indicating that separations between host EPs and virus are practically very difficult (Nolte‐’t Hoen et al., 2016). Efforts are being made to guide the field toward better separation and molecular and functional characterization techniques of the EP fractions (Hill et al., 2013; Mateescu et al., 2017; Van Deun et al., 2017; Welsh et al., 2024; Witwer et al., 2013). With the advent of asymmetric‐flow field flow fractionation (AF4), the isolation of small nanoparticles under 50 nm, termed exomeres (H. Zhang & Lyden, 2019; H. Zhang et al., 2018), was possible, opening new frontiers into the field of EPs (Latifkar et al., 2019). Exomeres can also be isolated by ultracentrifugation (UC) methods (Q. Zhang et al., 2019) and have been shown to carry functional cargoes, such as RNA that could be transferred to recipient cells (Q. Zhang et al., 2019) or host receptor proteins that can bind to viral particles (Q. Zhang, Jeppesen, Higginbotham, Franklin, et al., 2021). Recently, further purification of supernatant from exomeres has led to the identification of new particles—supermeres, which are small amembranous nanoparticles enriched in RNA and distinct protein biomarkers (Q. Zhang, Jeppesen, Higginbotham, Graves‐Deal, et al., 2021).
However, newer technologies have not been used to study viral particles and EPs in the context of HIV‐1 infection, raising the question of whether infectious particles exist below the accepted viral size range for HIV‐1. In the current work, we wish to address the knowledge gap that covers the possibility of HIV‐1 under the currently accepted size range. To this extent, we expanded a previously accepted and extensively used sequential differential ultracentrifugation (DUC) method (Théry et al., 2006) by employing higher g force with longer spin times to recover smaller EPs (<100 nm).
Not surprisingly, with our methods, we isolate EPs larger and smaller than the current size of HIV‐1 that carry viral protein and nucleic acid cargoes. These included infectious large (2K), medium (100K), and very small (167K/18 h) viral particles that we collectively refer to as EPs. Furthermore, we designed a modified virus recovery assay (VRA) from a previously described method (Laird et al., 2013), which indicates that all the collected EPs were infectious, including the novel small‐sized EPs under 100 nm. Additionally, we interfered with the HIV‐1 infectious cycle by either blocking the virus entry with HIV‐1 envelope (Env)‐specific monoclonal antibodies (mAbs) or by using nucleoside reverse transcriptase inhibitors (NRTIs) during the culture of the EPs producer cells. The interference experiments not only indicated that the small fraction was sensitive to all the Env‐specific mAbs, revealing the presence of a fully functional Env gp120 but also that small EPs recovered from NRTIs‐treated producer cells were still infectious when compared to the other fractions, indicating the uniqueness of the EPs in Frac‐E. Immunodepletion of small EPs with a mAb that targets CD63 significantly reduced the infectivity, suggesting that these small infectious HIV‐1 virions (smHIV‐1) are potentially released from the infected cells by an alternative route to the currently accepted exit model for T‐cells. Thus, in our present work, we show the existence of unique, infectious particles smaller than the currently accepted size for HIV‐1. Lastly, this methodology could be potentially employed for other viruses or pathogens originating from eukaryotic or prokaryotic cells uncovering the prospect for alternative biogenetic pathways for such infectious agents.
2. MATERIALS AND METHODS
2.1. Cell lines and cell maintenance
The HIV‐1 lymphadenopathy‐associated virus (LAV) infected T‐cell lines J1.1LAV (ARP, cat#1340) (Folks et al., 1987; Perez et al., 1991), ACH‐2(LAV) (ARP, cat#349) (Folks et al., 1989), and the promonocytic U1(LAV) (Folks et al., 1988) (ARP, cat#165), cell lines were obtained through the NIH AIDS Reagent Program. The MOLT‐4 T‐cell line was obtained from ATCC (crl‐1582™). The HIV‐1 Rev‐dependent A3R5.7 GFP/Luc was purchased from Cube BioSystems (cat#CUBRC0012). The A3R5.7 GFP/Luc cell line is an A301‐derived T‐cell line expressing CD4, CXCR4, CCR5 a4b7, and a GFP/Luc reporter gene under the control of Rev responsive element (RRE) promoter (McLinden et al., 2013; Wu et al., 2007).
All the cell lines were cultured in RPMI‐1640 with 2 mM L‐glutamine (Quality Biological, cat#112‐025‐131) supplemented with 10% heat‐inactivated fetal bovine serum (FBS) (Gemini Bio‐Products, cat#100‐106), 100 U/mL penicillin and 100 µg/mL streptomycin (Quality Biological, cat#120‐095‐721). Additionally, the A3R5.7 GFP/Luc RPMI 10% FBS base media was supplemented with 1 µg/mL puromycin (Fisher Scientific, cat#A1113880) and 1 µg/mL G418 (Sigma‐Aldrich, cat#A1720). The cell lines were regularly passaged at 3–4‐day intervals and maintained under standard cell culture conditions in an incubator at 37°C with a relative humidity of 90% and 5% CO2. Cell counts for cell maintenance and subsequent isolation steps were executed with Muse® cell analyzer (Luminex Corporation) according to the manufacturer's instructions using the Muse® count a viability reagent (Luminex, cat#MCH10103) and by using KOVA® Glasstic Slide with Counting Grids hemocytometer (KOVA®, cat# 87144) with trypan blue (1:1) for a total count of cells/mL.
2.2. Small‐scale isolation of EPs
2.2.1. Cell viability assay
Donor J1.1LAV and U1LAV cells (5 × 105 cells/mL) were cultured in technical triplicates in 100 µL of fresh EV‐depleted (FBS, ultracentrifuged at 100,000 × g speed for 90 min to deplete EVs) RPMI media (EVDM) in a 96‐well assay plate (Corning®, cat#3610). Cell cultures were separated into four parts, referring to the different time points: cells in complete RPMI with 10% FBS before starvation in 1% FBS (‐72 h), after 72 h of starvation in 1% FBS (0 h), 6 h post‐release in 20% FBS media (6 h) and 24 h post‐release (24 h). CellTiter‐Glo® reagent (Promega, cat#G7572) was used at a 1:1 ratio to quantify cell viability on a GloMax® Explorer reader (Promega). EPs‐depleted RPMI media alone was used for background measurements and subsequent normalization.
2.3. Isolation of EPs by DUC
HIV‐1‐infected T‐cells (J1.1LAV) at a cell density of approximately 5 × 107 cells/mL were synchronized in the G0 phase of the cell cycle via serum starvation for 72 h, washed and released into serum‐rich media with a mixture of phytohaemagglutinin and interleukin 2 (PHA/IL‐2) to induce viral gene expression. The samples were collected at 6 and 24 h post‐release. Cells were separated from the supernatant by low‐speed centrifugation (500 × g for 10 min), and supernatants were transferred into 26.3 mL polycarbonate bottles (cat. # 355654; Beckman Coulter Inc.). The bottles were placed in a 70Ti (Beckman) rotor and spun at 2000 × gavg (4,400 rpm, Frac‐A) for 45 min. The resulting supernatant was added to a new set of bottles and weighed for further UC. At the same time, the pellets were resuspended with PBS, spun down, and consolidated into a single 1.5 mL tube yielding Frac‐A (150–200 µL per sample). Next, the supernatant was spun at 10,000 × gavg (9900 rpm, Frac‐B) for 45 min. The resulting supernatant was added to eight UC tubes (each with 22.5 mL of supernatant) for further UC. At the same time, the pellet was resuspended with PBS (as described above) and consolidated into a single 1.5 mL tube, yielding the Frac‐B EPs. Next, the supernatant was spun at 100,000 × g for 90 min (Frac‐C). Then, the supernatant was centrifuged at 167,000 × g for 4 h (Frac‐D) and then at 167,000 × g for 16 h (Frac‐E) respectively. All isolated EPs were frozen at −80°C for later downstream assays.
2.4. EPs isolation using size exclusion chromatography: Izon qEV columns
The qEV 70 nm exclusion columns were used according to the manufacturer's protocol. Briefly, qEV single/70 nm columns were washed with 1 mL PBS before loading 150 µL of J1.1LAV, and U1LAV large EPs (Frac‐A) were resuspended in PBS on top of each column. A 1 mL void volume was then collected before collecting 40 200 µL fractions. The fractions were sequentially pooled into five (1 mL) fractions per pool, then concentrated with Nanotrap® particles (described below), followed by NTA, WB, qPCR, or functional assays.
2.5. Primary cell culture and large EPs isolation
Healthy peripheral blood mononuclear cells (PBMC) (Precision For Medicine, Frederick, MD) were thawed and rested in complete RPMI overnight according to the manufacturer's guidelines. Phorbol 12‐Myristate 13‐Acetate (PMA; cat: 16561‐29‐8, Cayman Chemical) and PHA were added the next day, and treatment continued every other day for up to ten days. On day 10, T‐cell cells in suspension were separated and infected with HIV‐1 89.6 (MOI: 10) with the addition of Infectin™ and cultured for another 4 days. Cells were then removed by 500 × g spin for 10 min, and supernatants were centrifuged at 2000 × g for 45 min, washed in PBS, and fractionated by qEV single/70 nm column.
2.6. Infectivity assays
Large EPs (Frac‐A, 2K) from J1.1LAV (HIV‐1 infected T‐cells) and U1LAV (HIV‐1‐infected myeloid cells) from supernatants collected at 6 and 24 h time points were used to treat uninfected T‐cells (CEM, Jurkat) and uninfected myeloid cells (U937). A total of 106 uninfected cells were resuspended in 100 µL of resuspended EPs sample and incubated for 8 h Cells were then washed with PBS and placed in 1 mL of fresh complete RPMI 1640 media and incubated for 48 h Viral replication was assessed by WB analysis for HIV‐1 proteins gp120, Nef, and p24.
To assess the infectivity of fractionated particles, we next incubated fractions 1–5 with either uninfected Jurkat T‐cells or U937 myeloid cells for 8 h along with the addition of Infectin™ (to increase the efficiency of viral entry). Cells were then washed, plated with fresh complete media, and incubated for 48 h at 37°C. Next, the cells were pelleted, washed, lysed, and probed for viral proteins using WB for the presence of HIV‐1 proteins gp120, Nef, and p24.
Next, we studied the release of large EPs from HIV‐1‐infected primary cells. PBMC from three donors were treated with PHA and PMA and allowed to grow for 10 days in culture to induce T‐cell growth and macrophage differentiation. Primary T‐cells were then separated and infected with HIV‐1 89.6 (MOI: 10) with the addition of Infectin™ and cultured for another 4 days. Cells were then removed, and supernatants were centrifuged at 2,000 × g for 45 min to collect large EPs. Upon fractionation by sizing columns, large EPs fractions were used to infect naïve Jurkat cells. The recipient cells were separated in half, collected 3‐ and 10 days post‐infection, respectively, and processed for downstream qPCR to assess viral replication.
2.7. EPs and virion capture using Nanotrap® particles
We utilized Nanotrap® particles (Ceres Nanosciences, Inc.) for EPs and virion isolations. Equal amounts of each Nanotrap® particle (NT80, NT82, NT86) and 1XPBS without Calcium and Magnesium were mixed and resuspended to make a 30% slurry. To capture EPs and/or virions, we added 40 µL of the slurry to 500 µL of supernatant, and the samples were rotated overnight at 4°C. The particles were separated and washed with PBS, and the pellets were processed for downstream assays.
2.8. Preparation of whole cell extracts
Cells were centrifuged at 300 × g for 10 min and washed with PBS. The pellet was resuspended in lysis buffer [50 mM Tris‐HCl (pH 7.5), 120 mM NaCl, 5 mM EDTA, 0.5% Nonidet NP‐40, 50 mM NaF, 0.2 mM Na3VO4, 1 mM DTT, and one complete protease inhibitor cocktail tablet/50 mL (Roche Applied Science, Mannheim, Germany)]. The mixture was incubated on ice for 30 min with vortexing every 5 min. Cell debris was removed via centrifugation at 12,000 × g at 4 °C for 10 min. Protein concentration was measured by Bradford assay according to the manufacturer's guideline (Bio‐Rad).
2.9. Large‐scale isolation of EPs
2.9.1. Cell culture with EPs‐depleted medium
Cell lines and uninfected primary PBMC used for isolating large‐scale EPs (150 mL plus) were cultured in an EPs‐depleted base medium (EPDM). The EPDM was prepared as previously described by Théry et al. (2006) with the following modifications. For cell lines, a stock 20% FBS RPMI base medium was ultracentrifuged with Optima XL ultracentrifuge (Beckman Coulter; Indianapolis, IN) for 18 h at 180,000 × gavg using Type 45 Ti rotor (39,311 rpm k‐Factor 174). Subsequently, the medium was carefully removed so as not to disturb the pellet on the side of the tube, and 5 mL of medium was left behind at the bottom of the tube. Next, the 20% FBS RPMI was diluted with serum‐free RPMI at a dilution factor 1:1 to obtain the EPDM. Similar to above, the PBMC (obtained from the Walter Reed Army Institute of Research protocol#1386 RV‐229B), were grown in EP‐depleted CTS™ OpTimizer™ T‐cell expansion serum‐free medium (ThermoFisher scientific cat# A1048501; EPDM‐CTS™) and used for HIV‐1 infection. Post depletion, the medium was further supplemented with 100 U/mL penicillin and 100 µg/mL streptomycin (Quality Biological, cat#120‐095‐721) and 2 mM L‐Glutamine (Quality Biological, cat#118‐084‐721), and passed through a 0.22 µm filter (stericup vacuum device, Millipore Sigma, cat#S2GPU05RE).
2.10. Total EV and particle isolation by UC
J1.1LAV cell line was seeded at a concentration of 5 × 105 cells/mL in a T225 flask (Thermo Fisher Scientific, cat#159933) and maintained under standard cell culture conditions for 4 days. Following the 4‐day culture, the cells were separated from the cell culture conditioned medium (CCM) by a 300 g force spin with a Sorvall Legend RT (model#75004377) benchtop centrifuge to obtain the CCM. Subsequently, the CCM was centrifuged for 18 h with a Type 45 Ti rotor in 167,000 × gavg (37,900 rpm, k‐factor 187) to obtain J1.1LAV total EPs pellets. The obtained pellets were resuspended in 60 mL particle‐free phosphate buffer saline (pfPBS) for an additional wash step and repeated at the same speed and time. The particle pfPBS was obtained by filtering the phosphate buffer saline through a 20 nm Whatman® Anotop® syringe filter. Total EPs pellets or Fraction‐0 (Frac‐0) were resuspended in 150–200 µL pfPBS and stored at −80°C, as suggested by other groups (Folks et al., 1989; Perez et al., 1991), for further downstream analysis.
2.11. Preparation of HIV‐1 infected PBMC for EPs isolation
To generate EPs obtained from HIV‐1 infected primary PBMC, we utilized PBMC sourced from an HIV‐1 negative donor, collected via leukapheresis within the framework of the Walter Reed Army Institute of Research IRB approved clinical study RV229C (WRAIR#1386) protocol. We infected the PBMC with HIV‐1 BaL and LAV, similar to what we previously have described (Brown et al., 2005), with the following modifications. Cells were thawed and resuspended at two million cells/mL in EPDM‐CTS™ supplemented with 100 U/mL penicillin and 100 µg/mL streptomycin (Quality Biological, cat#120‐095‐721) and 2 mM L‐Glutamine (Quality Biological, cat#118‐084‐721), 10 units/mL interleukin‐2 (IL‐2) (SigmaMilipore cat#10799068001) and 1 µg/mL phytohemagglutinin (SigmaMilipore cat# 232‐718‐7) for 16 to 18 h at 37°C under standard conditions. After the incubation, PBMC were washed with fresh EPDM‐CTS™ supplemented with IL‐2 medium and cultured for 3–4 days prior to infections. Next, cells were centrifuged at 300 × g force spin for 10 min, and infected 25–450 million PBMC pellets with 1–3 mL BaL stock virus and 25 million PBMC with 200 µL Frac‐0 J1.1LAV for 1 h at 37°C under standard conditions. After 1 h of incubation, the cell pellets were resuspended in 10 to 50 mL PBMC culture medium and incubated overnight at 37°C under standard conditions. The next day, the infected cells were washed three times and resuspended at a final cell concertation of 1.5–2 million cells/mL in EPDM‐CTS™ supplemented with IL‐2 and incubated for 12–13 days at 37°C under standard conditions. The supernatants were collected at days 12–13 post‐infection, and EPs were either isolated by DUC or SDF as described in the respective sections.
2.12. Fractionation of CCM by DUC
For the fractionation DUC, we used day 4 cell CCM of J1.1LAV cell lines, ACH‐2LAV, and day 12 of CCM from PBMC infected with BaL. The J1.1LAV and ACH‐2 were seeded in 180 mL to 360 mL EPDM at a cell density of 5 × 105 cells/mL, while BaL‐infected PBMC were calibrated at a cell density of 2 × 106 cells/mL in 245 mL EPDM‐CTS™, in T‐225 flasks (Thermo Fisher Scientific, cat#159933). For the DUC of J1.1LAV NRTIs, the EPDM was supplemented with 10 µM Tenofovir disoproxil fumarate (ARP, cat#10198) and 10 µM Emtricitabine (ARP, cat#10071), which were added to the cell culture at day 0, and day 2. Preceding the DUC, the Muse® cell analyzer recorded final cell counts as described above. First, the cells were pelleted at 300 × g force spin for 10 min to obtain CCM. For the subsequent DUC fractionation steps to obtain EPs subpopulation, we combined the previously described methods by Théry et al. (2006) and Q. Zhang et al. (2019). Each step in the DUC was executed twice at 4°C to include an additional 60 mL pfPBS wash step of the fractions, as depicted in Figure 1. The initial step in DUC, the CCM was spun at 2000 g force with the Sorvall Legend RT (model#75004377) benchtop centrifuge for 20 min to obtain Frac‐A. Next, each DUC of the CCM was executed with a fixed angel Type 45 Ti rotor (Beckman Coulter, Indianapolis, IN) using the Optima XL ultracentrifuge (Beckman Coulter). The DUC steps parameters were as follows: Frac‐B 10,000 × gavg force spin for 40 min (9,300 rpm, k: factor 3,100); Frac‐C 100,000 × gavg force spin for 90 min (29,300 rpm k: factor 313); Frac‐D 167,000 × gavg force spin for 3 h (37,900 rpm k‐factor 187); Frac‐E 167,000 × gavg force spin for 18 h (37,900 rpm, k: factor 187). After the pfPBS wash step, the pellets were resuspended in 150–200 µL pfPBS, and the obtained fractions were stored at −80°C for downstream characterizations.
FIGURE 1.
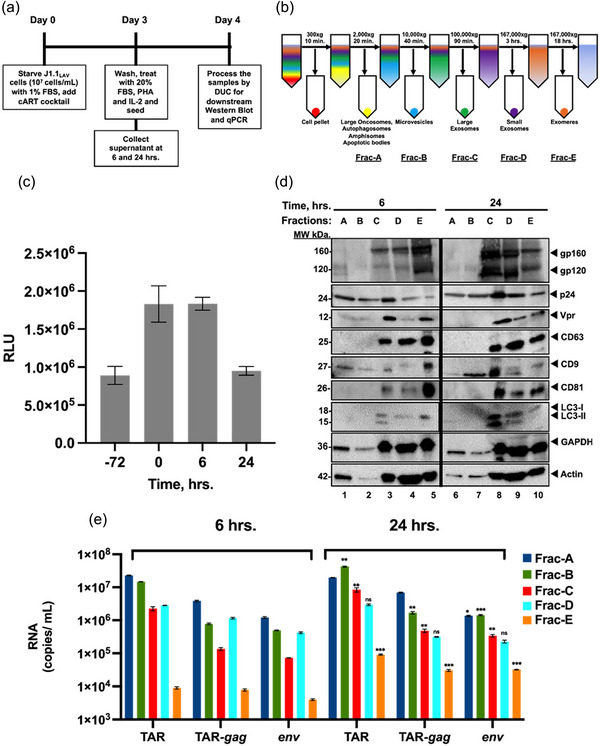
Isolation and characterization of early versus late extracellular vesicles from HIV‐1‐infected T‐cells. (a) J1.1 cells (5 × 107 cells/mL) were serum‐starved in 1% FBS media to induce cell cycle synchronization (G0 stage of the cell cycle) and incubated with cART cocktail. Next, the cells were released in complete media containing 20% EPs‐depleted serum and PHA/IL‐2 (1 µg/mL and 25 units/mL, respectively) to increase HIV‐1 transcription and reverse latency; (b) Schematics of the DUC steps used to isolate various EPs populations including 2K, 10K, 100K, 167K/4 h, 167K/16 h (as Fracs A‐E); (c) Сell viability analysis of HIV‐1 infected T‐cells before serum starvation (‐72 h), before release (0 h), 6 h and 24 h post‐release; (d) EPs pellets of the fractions A‐E for 6 (lanes 1−5) and 24 h (lanes 6−10) were first processed on a G‐50 Sephadex spin column to remove ∼50% serum albumin (packed bed volume of 0.5 mL and spin of 2,000 × g for 2 min) and then analyzed by Western blotting for exosomal markers CD9, CD81 and CD63, autophagy markers (LC3‐I, and LC3‐II), HIV‐1 viral proteins (gp120, Vpr and p24), actin and GAPDH as a control; (e) total RNA from the pellets was extracted and analyzed by RT‐qPCR (technical triplicates) for HIV‐1 transcripts (TAR, TAR‐gag and env), a two‐tailed Student's t‐test was used to assess significance: **P < 0.01, ***P < 0.001.
2.13. Alternative small EPs isolation by serial differential filtration from CCM
For the alternative isolation of small EPs, we serially differentially filtered (SDF) CCM, as depicted in (Figure S11 a), similar to what Heinemann and Vykoukal described previously (Heinemann & Vykoukal, 2017). Consequently, we used SDF 150 mL of day 4 J1.1LAV CCM or 10 mL small volume CCM collected at day 12 from PBMC infected with BaL or LAV Frac‐0 virus stocks. Thus, for a larger volume of 150 mL J1.1LAV CCM, first, we filtered through 450 nm followed by 220 nm cup filters (Millipore, Stericup® cat# S2HVU05RE, and cat# S2GPU05RE). After these two initial filtration steps, we moved the filtrate into a third and, respectively, fourth vacuum cup filtration system (Sterlitech, cat#200320‐30) equipped with 80 nm and 50 nm polycarbonate track etched membranes filters (Sterlitech, cat#PCT00847100, and cat#PCT00547100). The 10 mL CCM derived from the PBMC infected with HIV‐1 BaL and LAV were passed sequentially through 450 nm and, respectively, 220 nm Steriflip® filter devices (Millipore, cat# SE1M003M00 and SE1M179M6) after which we passed the CCM through two KS 13 syringe filter devices (Sterlitech, cat#17301000) equipped with an 80 nm and a 50 nm polycarbonate track etched membranes (Sterlitech, cat# PCT00813100, and PCT00513100). Next, we concentrated the filtrates with a cold sterilized 100 kDa Amicon® (Sigma cat#UFC9100, and cat#UFC8100) centrifugal concentration device according to the manufacturer's specifications, from 150 mL to 650 µL respectively and from 10 mL to 80 µL. Finally, the obtained 650 µL and 80 µL retentate was resuspended two times in 15 mL and in 4 mL pfPBS and reconcentrated to 650 µL and 80 µL, respectively, after which it was sub‐aliquoted and stored at −80°C for downstream analysis.
2.14. Nanoparticle tracking analysis
Nanoparticle tracking analysis (NTA) was performed with a multispectral instrument, ViewSizer 3000 (HORIBA Instruments Incorporated, Piscataway, NJ), equipped with a blue (450 nm), green (520 nm), and a red (635 nm) laser, which can detect individual particles in the size range of 10–2000 nm. Since this system is cuvette‐based, it allows for better decontamination and cleaning procedures of the cuvette between each sample. Before NTA analysis, all the fractions were diluted 1:500 to 1:50000 in pfPBS to a final volume of 2 mL, bringing the particle concentration in the range of 5 × 106 to 2 × 108 particles per mL, as recommended by the manufacturer. For the acquisition of EPs with the ViewSizer 3000 instrument, we followed the methodology previously described by Comfort et al. (2021), with the following instrument settings: frame rate 30 fps; exposure 15 ms; laser power blue 210 mW, green 12 mW, red 8 mW; frames per video 300 frames; and gain 30 dB.; temperature control active set to 22°C. We recorded 50 videos for each sample. Between each frame, the instrument was set to 5 s of stirring at 350 rpm and 3 s of wait time before each video to ensure that entirely independent sets of particles were captured. The acquired frames were processed as previously described by Comfort et al. (2021) with the proprietary ViewSizer 3000 advanced software set to the following settings: disabled autodetection override, feature diameter set to 30, detection threshold set to the default polydisperse sample, and detection threshold manual set to 0.8. After the ViewSizer 3000 software processed all the frames, the following parameters that define the particle size distribution (PSD) were obtained: particle concentration (particles/mL); modal size [nm]; the standard deviation of the size [nm]; and D10 [nm] D50 [nm] D90 [nm]. The D10 is where 10% and less the 10% of the sample is contained, D50 is where 50% and less than 50% of the sample is contained, also called the median, and D90 is where 90% and less the 90% of the sample is contained. The constant bin table with 1 nm bin width, calculated by the ViewSizer 3000 software, was used to graph the NTA PSD data. Additionally, we tested the instrument's capabilities to detect standard silica nanospheres (NanoXact™) with particles in size range of 20 nm to 500 nm, which are commercially available from nanoComposix (20 nm cat#SISN20, 50 nm cat#SISN50, 100 nm cat#SISN100, 200 nm cat#SISN200, 500 nm, cat#SISN500). The graphed PSD results in the supplemental (Figure S1a‐e) and the tabulated PSD in supplementary Table S1 collectively indicated that the instrument can measure particles with size ranges from 20 nm to 500 nm.
2.15. Transmission electron microscopy
In brief, the transmission electron microscopy (TEM) was carried out as previously described (Jung & Mun, 2018; Théry et al., 2006) with minor modifications. Formvar grids (Electron Microscopy Sciences, cat#FCF400‐CU) were positively charged with PELCO easiGlow™ (Ted Pella, Inc. cat#9100S) set for 25 s at 15 mAps. Next, the fractions containing the EPs were adsorbed for 1 min onto the formvar grids followed by a 5 min 2% glutaraldehyde fixation (Electron Microscopy Sciences, cat#16500) and then washed three with ultrapure water, after which the samples were negatively stained with 2% uranyl acetate (UA) (Electron Microscopy Sciences, cat#22400‐2) for 1 min after which the excess UA was wicked away, and the grids airdried. Images were acquired with JEOL JEM‐1400 series transmission electron microscope equipped with an AMT‐XR611M digital camera system.
2.16. EPs biochemical characterization
Biochemical characterization included the following assays: total protein; total lipid; HIV‐1 capsid p24 antigen capture ELISA; quantification of HIV‐1 TAR, TAR‐gag, env, RNA by RT‐qPCR; WB for EPs and HIV‐1 protein markers; Dot blotting for SDS membrane protection assay.
2.17. Total protein assay
Protein concentration was determined for all EPs fractions using the Pierce enhanced microbicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, cat#23235) according to the manufacturer's instructions with the following modification. EPs fractions were lysed in 2% SDS at a dilution factor of 1:10 for 15 min at room temperature (RT) and then further serial diluted five‐fold. Next, 150 µL of standards and samples were transferred to a transparent flat‐bottom 96‐well plate (Corning®, cat#3595) and mixed with 150 µL of working reagent. The plates were placed in an incubator at 60°C for 30 min then cooled to RT, and the absorbance of the mixture was measured with a VersaMax™ plate reader (Molecular Devices San Jose, CA) at a 562 nm wavelength. A four‐parameter logistic curve fit was used to extrapolate the concentrations of the samples with SoftMax Pro 7.1 software (Molecular Devices San Jose, CA).
2.18. Total lipid assay
The total lipid content of each fraction was determined with an improved, high‐sensitivity 96‐well plate format, as previously described by Visnovitz et al. (2019). In brief, a lipid standard was prepared from 1.2‐Dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC) (Sigma, cat#P6354), and used to construct the standard curve (6.25–400 µg/mL) that was stored at 4°C for up to a month. To measure the total lipid concentration of the EPs fractions, 40 µL of sample and standard was mixed with 200 µL of 96% sulfuric acid (Sigma, cat#258105) in 1.5 mL Eppendorf tubes (Eppendorf, cat#0030 120‐086) and incubated in heat blocks with the tube caps open at 90°C for 20 min, in a chemical fume hood. The tubes were then recapped and cooled to RT for 5 min at +4°C. Next, we added 120 µL of phospho‐vanillin reagent (Sigma, cat#V1104) prepared at 1 mg/mL in 17% phosphoric acid (Sigma, cat#3452445) to each tube followed by brief vortexing; 280 µL of each standard and sample were transferred to a clear 96‐well flat‐bottom microplate (Corning®, cat#3591) and further incubated for 1 h at 37°C. The 540 nm absorbance was then measured with a VersaMax™ plate reader (Molecular Devices San Jose, CA), and the concentration of the total lipid content was extrapolated from the standard curve with a four‐parameter logistic curve fit using the SoftMax Pro 7.1 software (Molecular Devices, San Jose, CA).
2.19. HIV‐1 p24 antigen capture ELISA
The HIV‐1 p24 concentration in each fraction was determined by a commercially available p24 capture ELISA kit (Advanced Bioscience Laboratories, cat#5421). In brief, samples were treated with the kit‐provided disruption buffer and then serially diluted to be within the detection range of 3.1–100 pg/mL of the p24 kit. The p24 concentration was then determined per the manufacturer's instructions. A four‐parameter logistic curve fit was used to extrapolate the p24 concentrations of fractions, and the absolute values were calculated using SoftMax Pro 7.1 software (Molecular Devices, San Jose, CA).
2.20. RNA isolation, reverse transcription, and quantitative real‐time PCR
Reverse transcription‐quantitative real‐time PCR (RT‐qPCR) analysis of HIV‐1 RNA was executed on all EPs fractions, similar to the method described by Barclay et al. (2017). In brief, RNA was isolated by TRIzol (Thermo Fisher Scientific, cat#15596026)/chloroform method as per the manufacturer's recommendation. The total RNA was used to generate cDNA utilizing the GoScript Reverse Transcription System (Promega, cat#5003) and specific HIV‐1 reverse primers for RT‐qPCR as follows: HIV‐1 trans‐activation response (TAR) reverse (5′‐ CAA CAG ACG GGC ACA CAC TAC‐3′; 99–119); Gag reverse (5′‐GCT GGT AGG GCT ATA cat TCT TAC‐3′; 1155–1178) and Envelope (env) reverse (5′‐TGG GAT AAG GGT CTG AAA CG‐3′;7917‐7936). Next, qPCR was performed with 2 µL of cDNA using iQ supermix (Bio‐Rad, cat#1708860) using the following pairs of primers/probes specific for target TAR sequences: TAR ‐Forward (5′‐GGT CTC TCT GGT TAG ACC AGA TCT G‐3′, Tm = 60°C); TAR‐Reverse: (5′‐CAA CAG ACG GGC ACA CAC TAC‐3′, Tm = 58°C) and TAR Prob (5′−56‐FAM‐AGC CTC AAT AAA GCT TGC CTT GAG TGC TTC‐36‐TAMSp‐3′). Serially diluted DNA from HIV‐1 8E5 cells (NIH AIDS Reagent Program) was used as a standard with the following PCR conditions: 50°C for 2 min (1 cycle), 95°C for 3 min (1 cycle), 95°C for 15 s, and 60°C for 40 s (41 cycles). Quantification of RNA (copies/mL) was determined from each sample's average cycle threshold (Ct) value relative to the standard curve. All reactions were carried out in triplicate on the Bio‐Rad CFX96 Real‐Time PCR Detection System.
2.21. Western blot analysis
For Western blot (WB) analysis, 15 µL fractions were mixed with an equal volume of 2X Laemmli buffer and heated to 90°C for 3 mins. A total of 30 µL of each sample was loaded on a 4−20% Tris/glycine 1.0 mm gel (Invitrogen, Carlsbad, CA) and separated at 180 V for 1 h. The electrophoretically separated proteins were transferred from the gel onto the PVDF membrane (Immobilon®‐P, Millipore, Burlington, MA) at 50 mA overnight. The PVDF membrane was blocked in 5% milk in PBS‐T (PBS with 0.1% Tween‐20) for 30 min before an overnight incubation at 4°C in PBS‐T with the primary antibody against the protein of interest. The following commercially available primary antibodies were used: α‐Lamp1 (Santa Cruz Biotechnology, cat#sc‐20011, 1:200), α‐Rab5 (Santa Cruz Biotechnology, cat#sc‐46692, 1:200), α‐Rab7 (Santa Cruz Biotechnology, cat#sc‐376362, 1:200), α‐MAP LC3 α/β (Santa Cruz Biotechnology, cat#sc‐398822, 1:200), α‐CD45 (Santa Cruz Biotechnology, cat#sc‐53666, 1:200); α‐ICAM‐1 (Santa Cruz Biotechnology, cat#sc‐8439, 1:200); α‐CD63 (Systems Biosciences, cat#EXOAB‐CD63A‐1, 1:1000); α‐CD9 (Systems Biosciences, cat#EXOAB‐CD9A‐1, 1:1000); α‐CD81 (Systems Biosciences, cat#EXOAB‐CD81A‐1, 1:1000); α‐Flotillin‐1 (Santa Cruz Biotechnology, cat#sc‐74566, 1:200); α‐TSG101 (Santa Cruz Biotechnology, cat#sc‐22774, 1:200); α‐Alix (Santa Cruz Biotechnology, cat#sc‐49268, 1:200); α‐Hsp70 (Santa Cruz Biotechnology, cat#sc‐1060‐R, 1:200); α‐GAPDH (Santa Cruz Biotechnology, cat#sc‐47724, 1:200); α‐CD44 (Abcam, cat# ab157107, 1:1000); α‐Actin (Abcam, cat#ab‐49900, 1:5000). Additionally, the following antibodies available from the NIH HIV‐1 AIDS Reagent programme were also used for WB analysis: α‐gp120 (ARP, cat#522; 1:1000); α‐Nef (ARP, cat#3689; 1:1000); α‐p24 (ARP, cat#4121, 1:1000); and α‐integrase (IN) (ARP, cat#7374, 1:500). The next day, the membranes were washed five times with PBS‐T wash buffer and further incubated with the appropriate HRP‐conjugated secondary antibodies as follows: mouse anti‐goat IgG‐HRP (Santa Cruz Biotechnology, cat#sc‐2354, 1:5000), goat anti‐mouse poly HRP (Invitrogen, cat#32230, 1:5000), and goat anti‐rabbit HRP (System Biosciences, 1:5000), for 2 h at 4°C. The HRP luminescence was activated with a Clarity Western enhanced chemiluminescence (ECL) Substrate (Bio‐Rad, Hercules, CA) and imaged using a ChemiDoc Touch Imaging System (Bio‐Rad, Hercules, CA).
2.22. SDS membrane protection assay and dot blotting
For the SDS membrane protection assay and dot blot analysis, 15 µL EPs samples were either first fixed with 15 µL 4% paraformaldehyde for 5 min or mixed with 15 µL 4% SDS and incubated for 30 min at RT. After that, 2.5 µL of EPs samples were dotted on 0.45 µm nitrocellulose membranes and airdried. Next, individual membranes were blocked as described for WB and probed with primary antibodies for 2.5 h at RT. The following primary antibodies were used: α‐ß‐Actin (Abcam, cat#8227, 1:1500); α‐HIV‐1 p24 (ARP, cat#3537, 1:500); and α‐HIV‐1 integrase 2C11 (ARP, cat#7374, 1:500). Appropriate secondary antibodies were then added, blots were washed and then developed as described above for WB.
2.23. Cell outgrowth assay
We designed a cell outgrowth assay to check for the possibility of contaminating cells during our large‐scale isolation of EPs. To this extent, the fractions were stored at +4°C or −80°C overnight. Following the overnight incubations, triplicate volumes of 25 µL each fraction (Frac‐A trough Frac‐E) were added to 225 µL of EPDM in a 96‐well flat‐bottom tissue culture plates (Corning®, cat#3585) and incubated under standard conditions 37°C, 90% relative humidity, and 5% CO2, for an additional 10 days. Additionally, we plated 3000 J1.1LAV cells/well as control in 250 µL EPDM. After the 10‐day incubation period, the volumes in the 96 wells were carefully adjusted to 100 µL, and 100 µL of CellTiter‐Glo® (Promega, cat#G9241) was added per well and resuspended. The total volumes of each well were transferred to 96‐well black well tissue culture plates (PerkinElmer, cat#6005270), and the resulting luminescence was recorded according to the manufacturer's instruction with the EnVision® multimode plate reader (PerkinElmer), where we recorded the relative light units (RLU). The recorded RLU was tabulated and the results were graphed.
2.24. MOLT‐4 VRA
VRA was executed by modifying the virus outgrowth assay previously described by Laird et al. (2013). In brief, 2 × 105 MOLT‐4 target cells/well were pelleted in 96‐well V‐bottom plates (Corning®, cat#3894), and equal amounts (20–25 µL) of fractions were added to individual wells. The VRA mixture was incubated for 1 h at 37°C, 90% relative humidity, and 5% CO2, after which 130 µL of EPDM was added to each well and incubated overnight. The next day, four wash steps were executed with EPDM to ensure that the initial fraction input was washed away. After the four wash steps, the cell suspension was transferred to 48‐well plates (Corning®, cat#3548), and the final volume was adjusted to 1 mL/well and incubated for an additional 4 days under standard conditions. If the cell cultures were carried beyond 4 days to accommodate the increased cell growth, the well contents were transferred to a 24‐well plate (Corning®, cat#3527), and the final volumes were adjusted to 2 mL media. On days 4, 7, and 14, 50 µL CCM/well was carefully collected from the VRA plates to either measure the amount of HIV‐1 p24 by capture ELISA or 50 µL CCM per well was transferred onto A3R5.7 GFP/Luc reporter cells, as described below, to confirm the presence of replication capable HIV‐1 virions.
2.25. MOLT‐4 VRA A3R5.7 GFP/Luc HIV‐1 reporter assay
A reporter assay was performed to confirm the presence of the infectious HIV‐1 virions in the supernatant of the MOLT‐4 VRA. Briefly, A3R5.7 GFP/Luc HIV‐1 reporter cells were pre‐mixed with 12.5 µg/mL DEAE‐Dextran hydrochloride (Sigma, cat#9985) and 1 × 105 cells per well in 50 µL were seeded in 96‐well black tissue culture plates (PerkinElmer, cat#6005270). Next, 50 µL of MOLT‐4 VRA supernatant was transferred onto the cells in triplicate and cultured under standard conditions overnight. An additional 100 µL of A3R5.7 GFP/Luc culture media (described in section 2.1) was added to each well and cultured for another 3 days. After this step, 100 µL of cell culture supernatant was removed from every well, and 100 µL/well of reconstituted Bright‐Glow™ (Promega, cat#E2610) was added and incubated for 5 min at RT to develop luminescence output, as expressed by RLU. The RLU was measured with the EnVision® multimode plate reader (PerkinElmer).
2.26. MOLT‐4 VRA cytopathic effects
Cytopathic effects (CPE) of MOLT‐4 VRA target cells as assessed by syncytial cell formation were captured by phase‐contrast microscopy with a Zeiss Axiovert 40 CFL inverted microscope equipped with a 12 volt, 35‐watt halogen lamp for phase‐contrast images. For the 10X images, we used a 10/0.25 A‐Plan Zeiss objective. The images were obtained with an AxioCam MRm CCD monochrome microscope camera and processed with ZEN 2012 (blue edition Carl Zeiss Microscopy GmbH, 2011). The syncytial cells were observed under phase contrast and enumerated with a cell counting device.
2.27. MOLT‐4 VRA antibody blocking
Antibody blocking experiments were performed with HIV‐1 non‐neutralizing antibodies (non‐Nab) as negative controls or with HIV‐1 broadly neutralizing antibodies (bNAbs). The following reagents were obtained commercially or through NIH HIV‐1 regent programme: non‐Nab COV2‐2196 (AstraZeneca, cat#AZD8955); non‐Nab against HIV‐1 gp120 CD4 binding site, 48d (IgG1) (ARP, cat#1756); bNAb against HIV‐1 gp41, 10E8 (IgG1) (ARP, cat#12294); bNAb against HIV‐1 gp120 CD4 binding site (CD4bs), VRC01 (IgG1) (ARP, cat#12033); bNAb against HIV‐1 gp120 variable loop1/loop2 (V1V2), PG16 (IgG1) (ARP, cat#12150). Experiments were performed in duplicate, where 25 µL of EPs fractions and 25 µL of mAbs at a final concentration of 6.25 µg/mL were mixed in 96‐well V‐bottom plates and incubated for 1 h at 37°C. The mixture was then added to VRA MOLT‐4 target cells and cultured as described above for the MOLT‐4 VRA. The amount of blocking was expressed as a percent reduction of p24 at day 4 by using the p24 pg/mL of PBS‐treated wells, compared to the antibody treated wells.
2.28. Direct stochastic optical reconstruction microscopy
Direct stochastic optical reconstruction microscopy (dSTORM) was performed on EPs derived from J1.1LAV cells. Before dSTORM analysis, we conjugated Atto 488 (Abcam, cat#269896) to either bNAb, VRC01, or PG16, and ALEXA® 647 (Abcam, cat#269823) to the α‐HIV‐1 integrase 2C11 mAb (as mentioned above) according to manufacturer's instruction. Additionally, human IgG1 isotype control (BioLegend, cat#401402) was conjugated to ALEXA® 647 as a control for agp120 staining, and mouse IgG1K isotype control (BioLegend, cat# 401402) to Atto 488 as a control for α‐HIV‐1 integrase.
EPs were immunolabeled and imaged using the ONI EVs Profiler kit (#EV‐MAN‐1.0, Oxford Nanoimaging (ONI), UK) as described previously (Santos et al., 2021). Briefly, 1.0 × 107 EPs were immobilized on microfluidic chips coated with PEG‐Biotin, fixed with F1 solution for 10 min, and permeabilized with P1 for 10 min. EPs were then incubated for 50 min with fluorescently labelled mAbs diluted in P1 buffer: CD9‐Atto488 or CD9‐Cy3B (used with the HIV‐1 specific mAb combinations), CD63‐Cy3B, CD81‐AlexaFluor®647, VRC‐01 Atto488, PG16 Atto488, and 2C11 AL647. Finally, samples were again fixed with F1 for 10 min, and freshly prepared STORM‐imaging buffer was added prior to image acquisition. All solutions and antibodies against CD9, CD63, and CD81 were provided in the kit. Using the Nanoimager S Mark II (ONI) high‐resolution microscope with 100X oil‐immersion objective, labelled proteins were imaged sequentially at 25, 35, and 50% power for the 640, 561, and 488 nm lasers, respectively, at 1,000 frames per channel with the angle of illumination set to 52.5°. Before the start of the imaging session, channel mapping was calibrated using 0.1 µm TetraSpeck™ beads (Thermo Fisher Scientific, cat#T7279). Data were processed using NimOS software (version 1.19, ONI). Subpopulation analyses of EPs expressing one, two, or three markers were analyzed using ONI's online platform, CODI (https://alto.codi.bio/,version v0.34.2). Herein, we performed a density‐based clustering analysis with drift correction and filtering to evaluate each vesicle.
2.29. EPs immunoprecipitation and immunodepletion
To assess the presence of amphisome markers on large EPs, we immunoprecipitated Frac‐A. To this extent, the Frac‐A of J1.1LAV and U1LAV cells were incubated with antibodies against Lamp1, Rab5, Rab7, and LC3‐I/II. Briefly, HIV‐1‐infected J1.1LAV and U1LAV cells with a cell density of approximately 5 × 107 were maintained for 5 days. The conditioned media was centrifuged at 2,000 × g for 45 min and EPs pellets were resuspended in PBS. Next, Frac‐A material was equally separated into five parts and treated with antibodies against amphisome markers—α‐Lamp1, α‐Rab5, α‐Rab7, α‐ MAP LC3 α/β and α‐IgG (Santa Cruz Biotechnology, cat#sc‐5275) as isotype control. Next, Protein A/G magnetic beads (Thermofisher ChIP grade Protein A/G Magnetic Beads, cat# 26162) were used to pull down antibody‐EPs complexes. The obtained pull down material was further processed for downstream WB and qPCR against HIV‐1 proteins and RNAs, respectively.
For the immunodepletion 25 µL Frac‐E EPs were mixed with 4 µg anti‐CD63 (Santa Cruz Biotechnology, cat#sc‐5275) or with 4 µg IgG1K mouse isotype control (BioLegend, cat#401402) for 30 min at +4°C with gentle rocking. After the antibody and Frac‐E incubation, 50 µL of prewashed protein‐G‐coated magnetic beads (Thermo Fisher, cat#1003D) were incubated with the immunoprecipitated complex for 30 min at RT. The resulting CD63 or IgG1K immunoprecipitated complex bound to the protein‐G‐beads were separated from the rest of the input Frac‐E under a strong magnetic field. The unbound Frac‐E was removed and transferred onto MOLT‐4 VRA target cells to check for the depletion of CD63+ HIV‐1. Concurrently, the CD63 or IgG1K bead immunoprecipitated complex was washed two times with PBS and treated with 30 µL 1X Laemmli buffer for 30 min at RT, after which the magnetic beads were retained under a strong magnetic field to separate them from the resulting lysates. The resulting lysates were electrophoretically separated and blotted as described above in the WB analysis section, where we probed for p24 with rabbit anti‐HIV‐1 p24 (ARP, cat#4250).
2.30. Image analysis
TEM and phase‐contrast microscopy micrographs were sized and annotated with NIH ImageJ software version 1.52k, according to the ImageJ user guide (Schneider et al., 2012).
2.31. EV‐TRACK
For the large‐scale isolations, we have submitted all relevant data of our experiments to the EV‐TRACK knowledgebase (EV‐TRACK ID: EV230011).
2.32. Statistical analysis
For statistical comparisons, numerical data were tabulated in Microsoft Excel Version 16.55. Statistical significance and data graphing were performed using GraphPad Prism 9.4 software (GraphPad Software Inc.). Error bars for all the graphs represent the mean with standard deviation. Normal distributions of the data were determined before analysis using the Shapiro‐Wilk test. Student's t‐test, Mann‐Whitney U test, or two‐way ANOVA test with Tukey's post‐hoc analysis was used according to the distribution of the data with *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 representing significance levels.
3. RESULTS
3.1. Large EPs are enriched in viral and cellular proteins
We have previously observed the presence of various viral and host proteins in EPs from HIV‐1‐infected cells and were able to assess infectivity of early (6 h) and late (24 h) total EPs released from the cells (Barclay et al., 2017; DeMarino et al., 2018; Kim et al., 2021; Narayanan et al., 2013; Sampey et al., 2016). Importantly, we found that many viral byproducts (i.e., p24, env, TAR RNA) were present in early release EPs (6 h post‐release) but were not infectious. Therefore, we decided to separate EPs based on their sedimentation rate using UC. HIV‐1‐infected T‐cells were synchronized in the G0 phase of the cell cycle via serum starvation for 72 h, washed, and released into serum‐rich media with PHA/IL‐2 to induce viral gene expression (Figure 1a). Here, the rationale was to see whether synchronized cells were also able to release EPs and viruses at different time points. To better understand the mechanisms of EPs release, cells and supernatants were collected at 6 and 24 h post‐release, and supernatants were subjected to DUC at speeds of 2,000 × g for 45 min (Frac‐A); 10,000 × g for 45 min (Frac‐B); 100,000 × g for 1.5 h (Frac‐C); 167,000 × g for 4 h (Frac‐D) and 16 h (Frac‐E), respectively (Figure 1b), and used for downstream assays. Cell viability was measured before starvation (72 h) and at 0 h, 6 h, and 24 h post block and release. Data in Figure 1c show that cells grew after release (6 h) and declined in the 24 h sample, possibly due to cell cycle block or viral activation (i.e., Vpr related). We then utilized the EPs fractions for WB against viral and host cellular proteins required for biogenesis, including tetraspanin CD63 and autophagy marker LC3 I/II. Data in Figure 1d shows that EPs had high gp160/gp120 and Vpr expression in Fracs C‐D. Viral p24 protein was also present in fractions A‐E, but more so in the 24 h samples. CD9 a commonly found tetraspanin protein in EPs from virally infected cells was also present in all fractions; however, more in the 24 h samples. Two other tetraspanin proteins CD63 and CD81 were also present in Fracs C‐E. Autophagy marker LC3‐I/II was primarily found in fractions D and E. Overall, the highest amount of many of the proteins tested (viral and cellular) was found in the 24 h samples. Collectively, these data indicate a time‐dependent accumulation of exosomal, autophagy, and viral protein markers when cells are released from the cell cycle block.
3.2. Presence of viral RNAs in differentially centrifuged EPs fractions over time
Previously, we have shown time‐dependent accumulation of multiple HIV‐1 RNAs in both intracellular and extracellular environments (Kim et al., 2021). Here, we processed the isolated EPs fractions for the presence of short (TAR), long non‐coding (TAR‐gag), and genomic HIV‐1 RNAs (env). Each EPs fraction was processed for RNA isolation and used in RT‐qPCR reactions utilizing our established primers for HIV‐1 RNAs. Data in Figure 1e shows the presence of all three transcripts in each fraction at 6 and 24 h post‐release and a significant increase of all three transcripts at 24 h. Collectively, our data indicate that similar to viral and host protein increase in the extracellular environment, there was a time‐dependent accumulation of all three species of extracellular RNAs in various particles isolated with different sedimentation rates.
3.3. Large EPs contain amphisome‐specific proteins
We have previously shown that samples obtained from the 24 h blocked and released cells contained a mixture of EPs and infectious virions (Kim et al., 2021; Schneider et al., 2012). However, we could not characterize EPs from the 24 h samples due to insufficient material for downstream assays. Data from Figure 1 indicated that p24 was present in multiple fractions, including the large Frac‐A samples. Therefore, we decided to test whether Frac‐A from J1.1LAV cells could potentially contain intact virions and initiate viral replication in naïve recipient cells. Samples from blocked and released cells from both 6 h and 24 h time points were processed for the presence of viral RNA. Data in Figure 2a show presence of all three TAR, TAR‐gag, and env RNAs, and as expected, increased levels from the 24 h samples. Next, we added both 6 and 24 h samples to uninfected T‐cells (CEM) or myeloid (U937) cells and scored for the presence of viral proteins as a sign of active replication.
FIGURE 2.
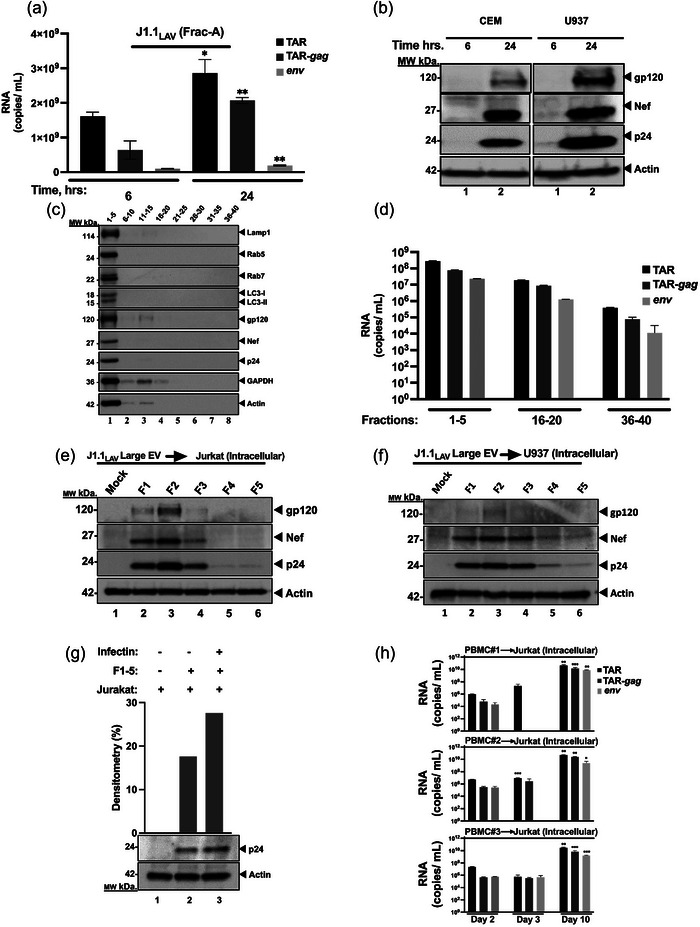
J1.1LAV large extracellular vesicles (Frac‐A) isolation and characterization. (a) Total RNA from Frac‐A pellets collected at 6 and 24 h was extracted and analyzed by RT‐qPCR for HIV‐1 transcripts (TAR, TAR‐gag and env); (b) Frac‐A samples collected post‐release from J1.1 cells at 6 and 24 h were used to treat naïve CEM and U937 cells. A total of 106 naïve cells were resuspended in 100 µL supernatant and 200 µL of media and incubated for 8 h, then the supernatants were removed, and cells were placed in 1 mL fresh media and incubated for 48 h The cells were then collected and processed for Western blot and probed for HIV‐1 proteins; (c) Large J1.1 EPs (Frac‐A) were loaded onto Izon qEV 70 nm single columns. Forty fractions were collected and pooled in sets of 5. Pooled fractions were then nanotrapped and Western‐blotted for Lamp1, Rab5, Rab7, LC3, gp120, Nef, p24, GAPDH, and actin using Western blot; (d) HIV‐1 RNA content of pooled SEC fractions derived from large EVs population produced by J1.1 cells. Next, the fractions 1‐5 were used to treat naïve Jurkat (e), and U937 cells (f). A total of 106 naïve cells were resuspended in 100 µL supernatant, and 200 µL of media and 50 µL of Infectin™ and incubated for 8 h, then the supernatants were removed, and cells were placed in 1 mL fresh media and incubated for 2 days. The cells were then harvested and pelleted for Western blot analysis. Separately the pooled fractions 1‐5 were tested on infectivity with the use of Infectin™ as a control (g). Densitometry percent count of HIV‐1 p24 expression was determined relative to actin. (h) Next, infectivity of large EPs from HIV‐1‐infected primary cells was assessed. PBMCs from three donors were treated with PHA and IL‐2 and allowed to grow for 5 days in culture. The PBMCs were then infected with HIV‐1 89.6 (MOI: 10) with Infectin™ and cultured for another 4 days. Cells were then removed, and supernatants were centrifuged at 2,000 × g for 45 min to collect large EPs, that were fractionated by sizing columns and used to infect recipient cells. The recipient cells were separated in half and collected in 3‐ and 10 days post‐infection, respectively, and F#2 represents the starting material from fraction #2 used for infection of Jurkat cells; total RNA from fractionated EPs and recipient cells was isolated. Using 3′‐end primers specific to TAR, TAR‐gag and env regions, cDNA was produced. RNA levels were assessed by RT‐qPCR with TAR‐specific primers. Student's t‐tests compared HIV‐1 RNA copy numbers from the fractions used for infection and from the recipient cells collected at 3‐ and 10‐days post‐infection. *P < 0.05; **P < 0.01, ***P < 0.001. Error bars, SD.
Data in Figure 2b indicate that the 24 h Frac‐A samples (large EPs) contained replicating virions and infected CEM and U937 recipient cells (lane 2). This was evident by the presence of new gp120, Nef, and p24 in the infected cells. We next asked whether the large Frac‐A EPs could further be purified using size exclusion chromatography. Data in Figure 2c show a series of WB for the presence of amphisome and viral markers (Chen et al., 2018; Sanchez‐Wandelmer & Reggiori, 2013). The large Frac‐A EPs were separated on Izon qEV 70, pooled together in sets of five and enriched for EPs by NT80/82/86 Nanotrap® particles and processed for downstream WB. All markers of amphisomes (large EPs released from the cells) were present in fractions 1–5, including Lamp1, Rab5, Rab7, and LC3‐I/II markers. The same fractions also contained viral proteins, including gp120, Nef, and p24. We then proceeded to quantify the potential presence of viral RNAs in these samples. Data in Figure 2d indicates that all three RNAs were most abundant in the fraction 1–5 samples. However, unlike the data for the protein samples, fractions 16–40 all contained viral RNAs, albeit at lower levels in the late fractions (i.e., fractions 36–40). These RNAs are potentially either encapsulated in vesicles or free in solution that co‐purified with Frac‐A. Collectively, these data indicate that large Frac‐A EPs can be separated using a combination of DUC and size exclusion columns to obtain amphisomes containing infectious virions.
3.4. Presence of viral infectivity from large EPs
Size exclusion chromatography shown in Figure 2c demonstrates the hallmarks of both amphisome and viral markers. However, these fractions were mixed for material recovery needed for WB analysis. We, therefor, re‐ran the sizing column using fresh Frac‐A material and focused only on fractions 1–5 for infectivity. Each fraction was diluted and measured using NTA to determine concentration, peak, mean, and median sizes. Data in Figure S2a show that fraction 2 had the highest concentration (up to 1.1 × 1011 particles/mL), and the concentration gradually dropped in the next fractions. To further characterize particles, the mean (Figure S2b), median (Figure S2c), and peak diameter sizes (Figure S2d) were also assessed. As expected, the median diameter of the fractionated particles was in the range of 220–260 nm. Additionally, NTA of fractions 1–5 from HIV‐1‐infected monocytes (U1LAV) showed similar PSDs and concentration, with the latter being highest in fraction 2 (Figure S3a‐d).
The results in Figure 2e,f show that both Jurkat and U937 cells treated with qEV fraction 2 contained the highest rate of viral replication, followed by fractions 1, 3, and 4, respectively. Importantly, when we assessed the role of cofilin activation in infection by these large EPs, the results showed that samples treated with Infectin™ (S3 peptide that directly inhibits cofilin phosphorylation by LIM‐kina se1) (Yoder et al., 2008) showed increased levels of p24 expression in recipient cells (Figure 2g). Collectively, these data indicate that the large EPs, using simple centrifugation followed by use of sizing columns, can separate particles that contain infectious material.
3.5. Infectivity of large EPs collected from HIV‐1‐infected primary T‐cells
PBMC were infected with HIV‐1, and the large EPs were first centrifuged and then purified using size exclusion chromatography (Izon qEV 70). Upon addition to naïve Jurkat T‐cells, viral growth was primarily detected in samples from day 10 and minimally detected from day 3 samples (Figure 2h). Analysis of intracellular RNAs from these infected Jurkat cells showed only in day 10 samples increased TAR, TAR‐gag, and env RNA, indicating new viral progeny formation over background input material (F#2). Collectively, these data suggest that infection of primary cells with HIV‐1 also produced large EPs that can be infectious as detected in naïve recipient cells.
3.6. Presence of amphisome markers on large EPs
To further test the presence of amphisome markers on large EPs containing viral proteins (Figure 2c), we immunoprecipitated Frac‐A material with antibodies specific to previously described amphisome markers, including Lamp1, Rab5, Rab7, and LC3‐I/II (Schneider et al., 2012). HIV‐1‐infected J1.1LAV and U1LAV cells with a cell density of approximately 5 × 107 were maintained for 5 days, conditioned media was centrifuged at 2,000 × g for 45 min, and EPs pellet was resuspended in PBS. Next, Frac‐A material was loaded onto Izon qEV 70 nm single columns and fractions 1–5 were used for immunoprecipitations. These fractions were equally separated into five parts and treated with antibodies against amphisome markers—α‐Lamp1, α‐Rab5, α‐Rab7, α‐LC3‐I/II and α‐IgG as isotype control (Figures 3a and S3e). Next, Protein A/G magnetic beads were used to pull down possible antibody‐EPs complexes, and pulled‐down beads were processed for downstream WB and qPCR against HIV‐1 proteins and RNAs. WB analysis showed that the populations of large EPs pulled down by α‐Rab5 showed the strongest presence of gp160/120 protein (Figures 3b; lanes 3 and S3f; lanes 1, 2, 4 for U1 cells). The sample pulled down by α‐Rab5 antibody showed increased co‐precipitated of Nef and p24 proteins (Figure 3b; lanes 3 and 4 and Figure S3f; lane 1). Interestingly, the sample precipitated by α‐Rab7 antibody showed the lowest signal of gp160/120 among all IPs. Next, we isolated RNA from these IPs and looked for the presence of HIV‐1 RNA species (Figures 3c and S3g). Data indicated that large J1.1LAV EPs pulled down by α‐Lamp1 and α‐LC3‐I/II antibodies showed increased copy numbers of short non‐coding (TAR), however, other longer forms of the HIV RNA (TAR‐gag and genomic env) were present only in the α‐Lamp1 precipitates (Figure 3c). Collectively, these data suggest that large EPs, expressing amphisome markers on their surface (i.e., Rab5 and/or Lamp1) have an increased level of viral proteins and RNAs.
FIGURE 3.
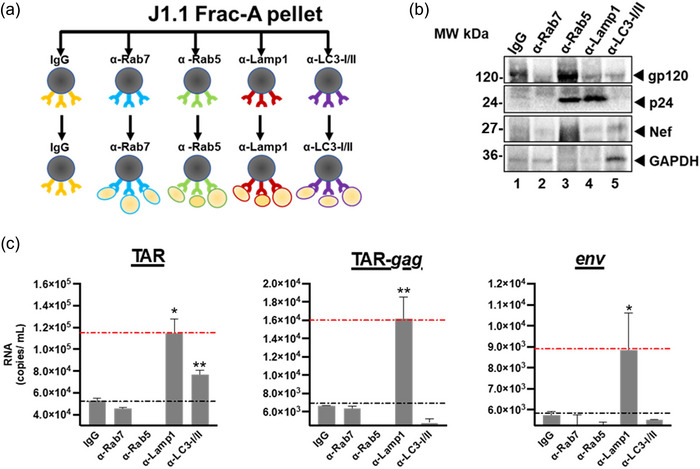
Immunoprecipitation from J1.1LAV Frac‐A. (a) Diagram of the immunoprecipitation performed with a‐Rab7, a‐Rab5, a‐Lamp1, and a‐LC3 antibodies using fractions 1‐5 from qEV 70 size exclusion chromatography; (b) IP‐ed samples were pulled‐down washed and probed for the presence of viral proteins gp120/160, p24, Nef, and GAPDH by WB and (c) viral RNAs such as TAR, TAR‐gag and env by qRT‐PCR where IgG served as control. Data represents mean ± standard deviation (SD) of three technical replicate measurements. Statistical significance was calculated with a two‐tailed unpaired Student's t‐test *P < 0.05, **P < 0.01 significance level.
3.7. Functional and biophysical analysis of J1.1LAV EPs indicate the presence of infectious HIV‐1 and various sizes of EPs
Since our experiments demonstrated the presence of infectious virus in Frac‐A EPs recovered from J1.1LAV cell cultures supernatants, we looked at the maximal infectivity as defined by p24 and induction of luciferase signal in A3R5.7 HIV‐1 reporter cells. These experiments were also critical in understanding EPs release in unsynchronized cells, as most HIV research has been performed using unsynchronized cell lines or primary cells. We therefore infected MOLT‐4 VRA target cells with concentrated J1.1LAV total EPs (collected and processed on day 4, as described in the large isolation methods section). We monitored the MOLT‐4 supernatants for HIV‐1 p24 and the presence of functional virus with A3R5.7 HIV‐1 reporter cells. The MOLT‐4 VRA results indicated that the J1.1LAV total EPs are infectious and that over time, the p24 from day 4 to 14 increased while the infectivity decreased (Figure 4a‐b), demonstrating that the optimal timepoint to collect infectious material from J1.1LAV cell culture medium (CCM) is on day 4 post‐plating. We next sequentially centrifuged the supernatant of day 4 J1.1LAV, as depicted in (Figure 1b) and previously described by others (Holcar et al., 2021; Momen‐Heravi et al., 2013; Théry et al., 2006; Zhou et al., 2020).
FIGURE 4.
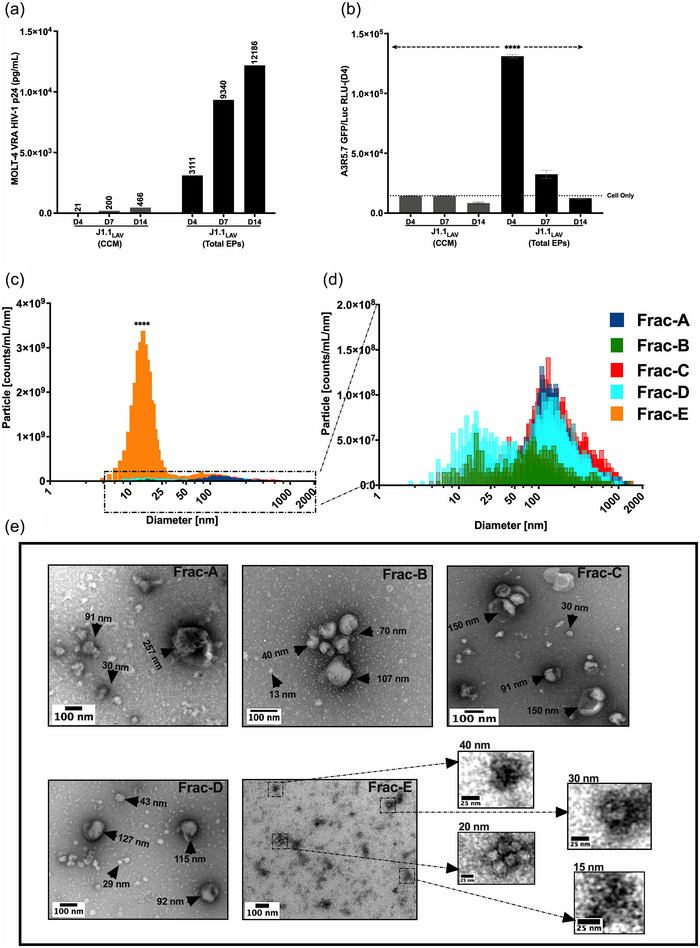
MOLT‐4 VRA of J1.1LAV cell culture conditioned media (CCM) and J1.1LAV ultracentrifuged (167,000 g × 18 H) total particles pellets (Total EPs 100 × concentrated CCM) and biophysical characterization of Fraction A through E. (a) J1.1 LAV (CCM) and J1.1LAV (Total Particles) MOLT‐4 VRA supernatant HIV‐1 p24 (pg/mL) assessment at day 4, 7 and 14; (b) Day 4, 7, and 14 MOLT‐4 VRA supernatant transferred onto A3R5.7 GFP/Luc HIV‐1 indicator cells, induction of firefly luciferase (Luc) expression measured as relative light units (RLU); (c) Graphical illustration of the particle size distribution (PSD) for J1.1 fractions, with Frac‐E in comparison to all the other fractions; (d) magnification of boxed in section from panel (a) for Frac‐A through Frac‐D; (e) TEM representative micrographs of Frac‐A through Frac‐E, black arrowheads and inserts indicate sizing of EPs contained in the fractions. The PSD graphs were generated with the data extracted from the constant bin tables with 1 nm bin sizes with an integration range set from 0 nm to 2,000 nm. Scale bars on the electron micrograph images represent 100 nm in the small inserts 25 nm. Data represents mean ± standard deviation (SD) of three technical replicate measurements. Statistical significance was calculated with One‐way ANOVA with Tukey's post‐hoc analysis multiple comparisons test with ****P < 0.0001 significance level.
We defined the size distributions of the recovered fractions, as previously reported by others (Phillips et al., 2021; Willms et al., 2018). The NTA analysis for fractions A‐D indicated an overlapping heterogeneous PSD (PSD) (Figure 4c, d, and Table 1). The modal sizes for fractions A‐D were skewed to the left compared to the D50 (or median size diameter) and mean (Figure 4d and Table 1), indicating that the mode would represent a better reflection of individual particle sizes. Interestingly, we recorded bimodal size distribution for Frac‐B and Frac‐D with two peaks around 20 nm and 116 nm (Figure 4d and Table 1). The modal sizes for Frac‐A, Frac‐C, and Frac‐D were between 110 nm and 132 nm (Table 1), which overlapped with the previously described HIV‐1 particle size (100–130 nm) (Freed, 2015; Fuller et al., 1997; McNamara & Dittmer, 2020; Wilk et al., 2001). Conversely, the D50 and modal size diameters of the particles contained in Frac‐E (Figure 4c) were significantly smaller, ranging from 15 nm to 20 nm, indicating that this fraction contained predominantly small particles. NTA indicated that the total particle concentration (particles/mL) was the highest for Frac‐E (Table 1), denoting the abundance of these particles in the J1.1LAV culture media.
TABLE 1.
ViewSizer 3000 NTA of each J1.1LAV EPs fractions particle size distribution (PSD) with particle size means, mode, standard deviation (SD) and particle concentration (particles/mL).
| J1.1LAV EPs PSD | |||||||
|---|---|---|---|---|---|---|---|
| Fractions | D10 | D50 | D90 (nm) | Mean | Mode | SD | Total concentration |
| (nm) | (nm) | (nm) | (nm) | (nm) | Particles/mL | ||
| A | 85 | 171 | 485 | 234 | 110 | 182 | 1.55E+10 |
| B | 29 | 111 | 326 | 152 | 17 | 139 | 4.06E+09 |
| C | 86 | 210 | 560 | 273 | 132 | 196 | 2.20E+10 |
| D | 44 | 177 | 440 | 219 | 116 | 165 | 1.70E+10 |
| E | 10 | 18 | 159 | 60 | 15 | 78 | 5.30E+10 |
Note: PSD is defined by D10, D50, and D90, where D10 is the point in the size distribution where 10% of the sample is contained, D50 is the point where 50% of the sample is included (median), and D90 is the point where 90% of the sample is contained. Data was generated from 50 videos (300 frames/video) processed with “Constant Bins Table” with 1 nm bin sizes set to integration range between 0 and 2000 nm.
The size heterogeneity measured with NTA was confirmed by TEM. In fractions A‐D, various particle shapes and sizes were observed, with some having cup‐shaped morphologies similar to what others previously described (Zhao et al., 2021) as exosomes (Figure 4e). We observed the presence of small and large individual particles that aggregated into larger particles, which the NTA would detect as a single large particle. The presence of large‐size aggregated particles detected by NTA in fractions A‐D would be included in the D90 size range (Table 1). Remarkably, for Frac‐E, we did not observe individual particles greater than 70 nm by TEM (Figure 4e), however, small particles were predominantly observed with electron‐dense foci in the size range of 15–40 nm. These particles occasionally aggregated into larger 60–70 nm particles (Figure 4e). Thus, using the enhanced ViewSizer 3000 NTA instrument in conjunction with TEM, a heterogeneous mixture of EPs with various sizes and morphologies was observed. Interestingly, we also observed that Frac‐E predominately contains EPs between 13 nm and 60 nm, indicating that with our modified DUC approach, we successfully isolated EPs in small‐size range previously defined by others as exomeres (H. Zhang et al., 2018).
3.8. Biochemical characterization of EPs
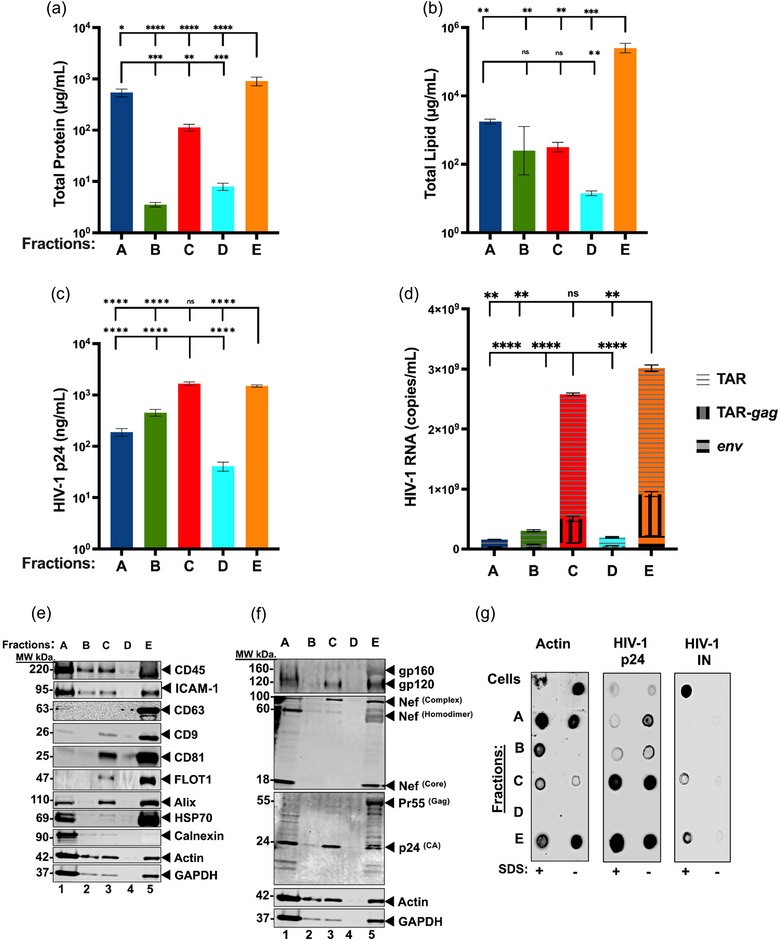
Following the sizing of the fractions, we sought to define the biochemical composition of the EPs. The total proteins, lipids, viral core proteins, and viral RNAs were measured. All fractions were isolated from the same conditioned media. Thus, the input quantities in all biochemical assays were normalized based on volume to avoid any bias from particle concentrations, as determined by NTA.
The results for the total protein and lipid content quantifications for each fraction indicated that Frac‐E contained significantly higher concentrations of total proteins and lipids. In contrast, protein and lipid content for other fractions were variable (Figure 5a,b). We calculated the particle‐to‐protein ratios, as previously described (Webber & Clayton, 2013), to indicate EV purity. The particle‐to‐protein ratios for Frac‐A and Frac‐E were less than 108 particles/µg protein (Table S2), indicating a higher amount of protein co‐isolated in the respective fractions. While Frac‐B, Frac‐C, and Frac‐D had ratios greater than 108 particles/µg protein (Table S2), implying these fractions had greater purity. Collectively, these results suggest that Frac‐A has larger aggerates of cell‐derived proteins. At the same time, Frac‐E is enriched in amembranous protein‐rich smaller EPs as previously described (Jeppesen et al., 2023; Tosar et al., 2022; Q. Zhang et al., 2019), thus implying the ratio does not apply to these respective fractions.
FIGURE 5.
Biochemical content analysis of J1.1LAV EPs. (a) Micro BCA total protein content (µg/mL); (b) Total lipid assay (µg/mL); (c) HIV‐1 p24‐antigen capture ELISA (pg/mL); (d) Cumulative RT‐qPCR for HIV‐1 RNA TAR TAR‐gag (copies/mL); (e) J1.1LAV EPs fractions western blotting (WB) probed for T‐cell marker (CD45), cell adhesion (ICAM‐1), extracellular vesicle tetraspanins markers (CD63, CD9, and CD81), extracellular cargo marker (TSG101, Alix, HSP70, Actin); (f) J1.1LAV EPs fractions WB for HIV‐1 protein content (gp120, Nef, and p24); (g) J1.1LAV EPs SDS membrane protection assay dot blotting of treated or not treated fractions probed for the presence Actin and HIV‐1 markers p24, Integrase (IN). For each measurement technical triplicates were executed. Data represents mean ± standard deviation (SD) of three technical replicate measurements. Statistical significance was calculated with One‐way ANOVA with Tukey's post‐hoc analysis multiple comparisons test with *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 significance level.
Since our previous work indicated that EPs recovered from various biological fluids (CCM, cerebral spinal fluid, and plasma) carry HIV‐1 proteins and RNA transcripts (TAR, TAR‐gag, env) (Narayanan et al., 2013; Sampey et al., 2016), we set out to characterize all the J1.1LAV T‐cell derived EPs fractions (Frac‐A trough Frac‐E) for their absolute content of viral HIV‐1 p24 core protein and RNA transcripts. The capture ELISA indicated significantly higher p24 core protein concentration in Frac‐C (Figure 5c) than in Frac‐A, Frac‐B, and Frac‐D. The levels of three HIV‐1 transcripts (TAR, TAR‐gag, env) were measured in all fractions using RT‐qPCR. Similar to the trends shown by the concentrations of HIV‐1 p24 core protein, Frac‐C and Frac‐E contained significantly higher levels of viral transcripts relative to the other fractions but equivalent to each other (Figure 5d). The higher levels of total HIV‐1 RNA transcripts measured in Frac‐C, compared to Frac‐A and Frac‐B aligned with our previously published findings (Barclay et al., 2017; Narayanan et al., 2013). Interestingly, we found that the smaller‐sized Frac‐E also contained significantly higher amounts of HIV‐1 RNA transcripts. In contrast, the intermediate Frac‐D had lower amounts of HIV‐1 viral transcripts (Figure 5d). These data suggest that Frac‐D contains small residual quantities of Frac‐C EPs or denser Frac‐E EPs. Thus, Frac‐D may be a transitional fraction between Frac‐C and Frac‐E and may be a necessary step to separate any residual larger and denser particles from the smaller and less dense particles.
Next, we analyzed by WB fractions A‐E host, EPs, and HIV‐1 protein marker levels, as suggested in MISEV 2023 (Welsh et al., 2024). Detection of CD45 and ICAM‐1 in Fracs A, B, C, and E confirmed that the EPs originated from a lymphocytic cell line (Figure 5e). We also detected CD9 and CD81, EPs tetraspanin markers in fractions A, C, and E (Figure 5e). However, we observed that CD63 was present predominantly in Frac‐E and detectable in Frac‐A (Figure 5e). Additionally, ALIX and Flotilin‐1, EPs markers involved in vesicular and membrane trafficking (Meister & Tikkanen, 2014; H. Zhang et al., 2018) and viral release (Frankel & Audhya, 2018; Larios et al., 2020; Odorizzi, 2006), were detected in fractions A, C, and E (Figure 5e). We observed that Frac‐A, Frac‐C, and Frac‐E contained ALIX, indicating that EPs are potentially released via the endosomal sorting complex required for transport (ESCRT) pathway. Of note, FLOT1 was detected in Frac‐C and to a greater extent in Frac‐E (Figure 5e), indicating that Frac‐E contains more lipids, further supporting the total lipid concentrations observed in Figure 5b. The cytosolic markers actin, GAPDH, and HSP70, known to be incorporated into EPs (Kowal et al., 2016; van Niel et al., 2018; H. Zhang et al., 2018), were present in Frac‐A, Frac‐B, Frac‐C, Frac‐E, but surprisingly not detectable in Frac‐D. However, HSP70, involved in the loading of EPs with proteins (van Niel et al., 2018), was detected only in Frac‐A and Frac‐E (Figure 5e).
The fractions were also tested for viral protein content by WB and probed for the HIV‐1 markers including gp120; Nef; and Gag. All three proteins were detected in Frac‐A, Frac‐C, and Frac‐E but not in Frac‐B and Frac‐D (Figure 5f). Furthermore, when we probed the fractions for Nef, we observed three distinct molecular weight species distributed in the EPs fractions. The first form was a higher molecular weight complex with an approximate 100 kDa molecular weight (MW) contained in Frac‐A, Frac‐C, and Frac‐E, which previously was described as a Nef complexed with other proteins such as actin and dynamin‐2 (Fackler et al., 1997; Pizzato et al., 2007) (Figure 5f). The second form had a MW of 60 kDa previously described as Nef homodimer (Campbell et al., 2008) present only in Frac‐A and Frac‐E (Figure 5f). The third form of Nef with MW of 18 kDa, previously described as Nef core processed by HIV‐1 protease and described as Nef incorporated into virions (Bukovsky et al., 1997; Welker et al., 1998, 1996) was detected only in Frac‐A and Frac‐E (Figure 5f). Additionally, we probed for the HIV‐1 p24Gag core protein which is processed by the viral protease from a larger precursor protein (Freed, 2015). We detected the presence of a single band of p24Gag HIV‐1 capsid protein in Frac‐C. This result indicates that this fraction contains mature Gag protein p24 processed from the polyprotein (Freed, 1998) (Figure 5f). However, in Frac‐A and Frac‐E, we also detected the unprocessed precursor HIV‐1 Gag protein Pr55Gag (Freed, 2015), as well as the p24Gag (Figure 5f), indicating that these fractions likely contain immature and mature HIV‐1 particles. It also appears that the p24 Gag in Frac‐E exists uniquely as a doublet; the reason for this is currently unclear.
As a final step, we explored the possibility of identifying membrane‐enclosed viral proteins, including viral integrase, that have been shown to bind to TAR (Barclay et al., 2017; DeMarino et al., 2018; Narayanan et al., 2013; Sampey et al., 2016). Therefore, we performed dot blots on fixed or SDS‐treated EPs to expose the luminal cargo. In addition to integrase, we probed for actin and HIV‐1 p24 to determine whether a protective membrane‐enclosed these proteins. For Frac‐A and Frac‐E, we detected actin inside and outside the EPs (Figure 5g). These findings could indicate that the co‐isolated cells in Frac‐A are lysed by freeze‐thawing (Tansey, 2006) and possibly a source of free actin. However, actin in Frac‐E may suggest that this fraction is either sensitive to cryodamage (Gelibter et al., 2022; Kusuma et al., 2018; Yuan et al., 2021) or contains co‐sedimented non‐membrane‐enclosed actin (Q. Zhang, Jeppesen, Higginbotham, Graves‐Deal, et al., 2021; Q. Zhang et al., 2019) (Figure 5g). Additionally, we found that all the fractions, especially Frac‐C and Frac‐E, were positive for p24 regardless of SDS treatment, indicating that these fractions contain both membrane‐protected and exposed HIV‐1 p24 (Figure 5g). Finally, we observed that HIV‐1 integrase was detectable in Frac‐C and Frac‐E only after SDS treatment (Figure 5g). The data indicates that Frac‐C and Frac‐E contain membrane‐enclosed integrase and potentially HIV‐1 virions.
3.9. Functional analysis of all fractions using a VRA, viral entry inhibition, and viral production interference
As the biophysical and biochemical characterization of the EPs indicated the potential presence of HIV‐1 virions in the isolated EPs, including in the novel Frac‐E, we tested the fractions for infectivity. A VRA (Figure 6a) was designed where the MOLT‐4 T‐cell line was used as target cells, similar to Laird et al. previously described quantitative virus outgrowth assay (Laird et al., 2013). To avoid input bias based on the biophysical or biochemical characterizations, we elected to normalize the amount of each fraction added to the target cells based on volume. Post day 4 of EPs addition to MOLT‐4 VRA target cells, the supernatant was tested for viral p24 antigen by capture ELISA. Since we previously observed that EVs contain mRNA transcripts (i.e., HTLV‐1) capable of expressing viral proteins in the recipient cells in the absence of functional virions (Jaworski et al., 2014), we used the A3R5.7 GFP/Luc HIV‐1 reporter T‐cell line to score for the presence of functional virions. The day 4 MOLT‐4 VRA supernatants were transferred onto A3R5.7 GFP/Luc HIV‐1 reporter T‐cells (Figure 6a), and then luciferase expression was assessed in addition to the p24 capture ELISA. CPE were also enumerated at day 4 (Figure S5a), as it has been described previously that the J1.1LAV HIV‐1 strain is capable of inducing the formation of syncytial cells (Figure S5b) (Lifson et al., 1986).
FIGURE 6.
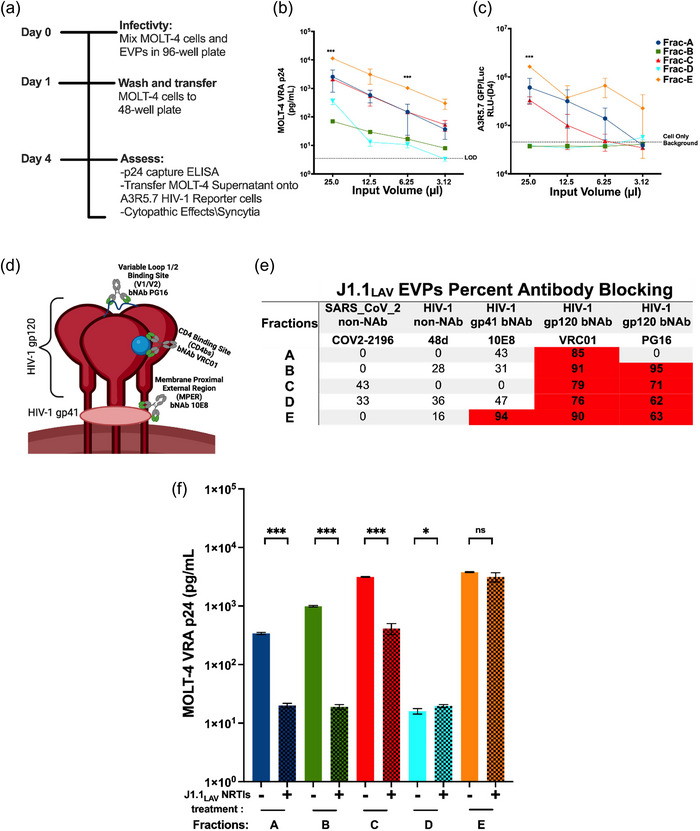
J1.1LAV EPs MOLT‐4 Virus recovery assay (VRA) with HIV‐1 monoclonal virus blocking and MOLT‐4 VRA assessment of viral production interference with NRTIs. (a) MOLT‐4 VRA assay schematics. (b) MOLT‐4 VRA supernatant HIV‐1 p24 pg/well levels measured on day 4 MOLT‐4 VRA initiation (LOD) Limit of Detection; (c) MOLT‐4 VRA supernatant transferred onto HIV‐1 A3R5.7 GFP/Luc viral indicator cells, where 4 days post‐transfer luciferase (Luc) activity was developed and measured as relative light units (RLU); (d) Model of HIV‐1 envelope glycoprotein (gp) with gp120 broadly neutralizing antibody (bNab) PG16 specific to the V1/V2 loop or VRC01 specific to the CD4bs, respectively, with gp41 specific bNab 10E8; (e) HIV‐1 bNAb blocking data, where equal volumes of EPs fractions were mixed with 12.5 µg/mL HIV‐1 bNAb to a final mAb concentration of 6.25 µg/mL or PBS (mock). The percent MOLT‐4 VRA infectivity reductions were calculated as a function of HIV‐1 p24 reduction compared to the PBS mock tread control wells. The data represents the percent average of two individual antibody blocking experiments where HIV‐1 non‐neutralizing antibody (non‐NAb); (f) NRTIs viral production interference, where the EPs fractions were recovered from naïve J1.1LAV or J1.1LAV NRTIs‐treated cells cultures and used in MOLT‐4 VRA and at day 4 p24 (pg/mL) was measurement with p24 capture ELISA, the input for the fractions was normalized within each pair based on the EPs counts. Data represents mean ± standard deviation (SD) of three technical replicate measurements. Statistical significance was calculated for panel (a and b) with One‐way ANOVA with Tukey's post‐hoc analysis multiple comparisons test and for panel (f) with two‐tailed unpaired Student's t‐test significance level *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 significance level.
The levels of p24 in the MOLT‐4 VRA indicated a dose‐dependent response for the various EPs inputs (25 µL; 12.5 µL; 6.25 µL; 3.12 µL), supporting the notion that these fractions indeed contain infectious virus (Figure 6b). Additionally, despite similar viral RNA content levels between Frac‐C and Frac‐E, Frac‐E was significantly more virulent than Frac‐C and the other fractions at the 25 µL and 12.5 µL inputs (Figure 6b). Furthermore, data from the A3R5.7 GFP/Luc HIV‐1 reporter cells showed trends similar to the MOLT‐4 p24 output (Figure 6c). The reporter cells demonstrated a significant amount of infectious virus in the MOLT‐4 VRA supernatants from Frac‐E (Figure 6c). Frac‐B and Frac‐D VRA supernatants did not induce reporter gene (Luc) expression in the reporter cells, indicating either a low virus titre or absence of infectious virions (Figure 6c). Collectively, the data obtained from the MOLT‐4 VRA assay indicate that Frac‐E, Frac‐C, and Frac‐A contained infectious virions.
Next, we asked whether we could block entry or interfere with virus production. First, inhibition of the infection of the MOLT‐4‐VRA target cells was assessed using three HIV‐1 broadly neutralizing monoclonal antibodies (bNAbs). Since the WB data (Figure 5f) indicated the presence of HIV‐1 Env in the EPs mixture and the MOLT‐4 VRA the presence of the infectious virus, we selected bNAbs known to bind Env and neutralize HIV‐1 infection (Figure 6d). The gp41 membrane proximal external region (MPER) of the Env involved in viral membrane and target cell membrane fusion was selected, as well as the gp120 part of the Env responsible for viral particle binding to target cell surface receptors (Wilen et al., 2012). The 10E8 bNAb that targets gp41 MPER, and two gp120 bNAbs that target either the CD4 binding site (CD4bs) (VRC01) or the apex of gp120 at the variable loop 1/2 region (PG16) (Chuang et al., 2019; Euler & Schuitemaker, 2012), were selected. Additionally, two non‐neutralizing mAbs (non‐NAb) were used as negative controls. A SARS‐CoV‐2 anti‐spike mAb COV2‐2196 (Kumar et al., 2021) and an HIV‐1 binding but not neutralizing mAb, 48d, which reacts with CD4‐induced gp120 (Thali et al., 1993; Zolla‐Pazner, 2004), served as the negative controls. For these experiments, we used a single input of mAbs adjusted to a final concentration of 6.25 µg/mL (after mixing with the EPs). At this single mAb concentration, the 10E8 bNAb showed <50% inhibition of fractions A‐D (Figure 6e), suggesting that this epitope was not as exposed in these fractions or there was antigen excess compared to the amount of mAb. However, 10E8 successfully blocked Frac‐E infectivity with 95% efficiency (Figure 6e), indicating adequate exposure of the gp41 MPER. The VRC01 CD4bs bNAb proficiently blocked virus entry into the MOLT‐4 cells, showing 74%–93% inhibition across the fractions (Figure 6e). In contrast, PG16 did not block Frac‐A infectivity, while it blocked the infectivity of all the other fractions (Figure 6e). The variability of the blocking efficiencies further supports the possibility of different viral subpopulations intermixed in the EPs. Interestingly, Frac‐E infectivity was blocked by all the bNAbs, indicating that these novel small‐size EPs contain smHIV‐1 virions with fully functional Env glycoproteins indispensable for virus particle binding and fusion (Lobritz et al., 2010; Sun et al., 2016).
We next asked whether we could restrict or interfere with the production of infectious EPs by culturing the J1.1LAV producer cells in a mixture of two NRTIs. Therefore, J1.1LAV cells were cultured for 4 days in the presence of the combination of two NRTIs, Tenofovir disoproxil fumarate (TDF) and emtricitabine (FTC) known to interfere with in vitro viral replication (Borroto‐Esoda et al., 2006). On day 4, the NRTIs‐treated cell culture had reduced cell counts, while the cell viability was similar to that of the untreated cells (Figure S6). The reduced cell count could be potentially attributed to the presence of TDF in culture, which was previously reported to induce cell cycle arrest (Brüning et al., 2012).
We then compared the infectivity of the EPs recovered from NRTI‐naïve or treated J1.1LAV cells using the MOLT‐4 VRA by adjusting the input volumes based on the total particle counts within each pair of EPs fractions. As expected, MOLT‐4 VRA reported a significant reduction in infectivity for Frac‐A, Frac‐B, and Frac‐C recovered from the NRTI‐treated cells. However, Frac‐E recovered from the NRTI‐treated cell culture had similar levels of infectivity to the Frac‐E recovered from untreated cells (Figure 6f). The MOLT‐4 VRA data and the reduced cell counts of the NRTI‐treated culture implied that the two drugs potentially interfere with the EPs biophysical and biochemical composition, similar to our previous observations (DeMarino et al., 2018). Consequently, we compared the biophysical and biochemical content of the EPs recovered from NRTIs‐naïve and NRTIs‐treated J1.1LAV cell culture.
The NTA PSD parameter D50 was slightly higher for fractions A‐D recovered from the NRTIs‐treated cell culture than those from untreated cells (Figure S7a‐b and Table S3). Additionally, the particle number/mL (Table S2‐3) was also somewhat increased for the fractions from treated cells. However, NTA for Frac‐E recovered from the NRTIs‐treated cells had similar D50 and modal size compared to the Frac‐E EPs recovered from the untreated cell supernatants, but with a decrease in total particle number/mL (Figure S7a; Tables S2‐3). The reduced total particle count for Frac‐E could be attributed to the NRTIs interfering with the cell cycle as described by others (Brüning et al., 2012), which could interfere with the total release number of Frac‐E EPs.
In addition to the sizing, we compared the biochemical content of EPs recovered from the NRTI‐naïve or treated cells. The total protein significantly decreased for Frac‐C and increased for Frac‐D, while the rest of the fractions remained unchanged between EPs recovered from treated and untreated cell cultures (Figure S8a). The total lipid comparison indicated that total lipid content increased in Frac‐A for the NRTIs‐ treated culture (Figure S8b). The viral p24 assessment indicated that for Frac‐A and Frac‐B, the levels of p24 decreased, while in Frac‐D, p24 levels increased for the NRTIs‐treated cell cultures. However, p24 levels in Frac‐C and Frac‐E EPs between treated and untreated cell cultures were similar (Figure S8c). The TAR RNA content of the EPs decreased in Frac‐A and Frac‐B and increased in Frac‐D for the NRTI‐treated cells, while no differences were observed for Frac‐C and Frac‐E (Figure S8d) irrespective of the treatment. For the longer transcript, TAR‐gag EPs recovered from the NRTIs‐treated cultures. There was a decrease in Frac‐A ‐D but no difference in Frac‐E EPs (Figure S8e). Finally, the env transcript recovered from Frac‐A, Frac‐B, Frac‐C, and Frac‐E of NRTIs‐treated cultures, compared with the untreated cultures, showed a significant decrease, while there was an increase in Frac‐D levels after treatment with NRTIs (Figure S8f). Although the viral transcript levels overall decreased in all the EPs recovered from the NRTIs‐treated cultures (compared with untreated culture EPs), the viral transcripts remained significantly elevated in Frac‐C and Frac‐E, suggesting preferential viral RNA packaging in those EPs fractions.
Additionally, we observed similar EPs and viral protein marker profiles by WB for EPs recovered from NRTIs‐treated cell cultures (Figure S8g and 7 h) compared to those recovered from the untreated cultures (Figure 5e,f). Furthermore, the membrane protection assay (Figure S8i) showed that membranes protected integrase in Frac‐C and Frac‐E. Finally, the particle counts‐to‐protein ratio indicated lower particle purity for Frac‐B recovered from NRTIs‐treated cell cultures (Table S2). Overall, the results indicated that treatment of the J1.1LAV with NRTIs interfered with the functionality, size, quantity, and quality of Fracs A‐E, further supporting our previous observations (DeMarino et al., 2018). Intriguingly, Frac‐E remained similarly infectious despite the antiretroviral treatment of the producer cells, further highlighting the distinctiveness of these particles (Figure 5f).
3.10. J1.1LAV Frac‐C and Frac‐E single‐EPs immunophenotyping
Since the biophysical, biochemical, and functional assays indicated differences between Frac‐C and Frac‐E, we next analyzed the phenotype of the respective fractions at single‐molecule‐sensitivity. To this end, we determined the molecular colocalization of tetraspanins and HIV‐1 markers on the J1.1LAV EPs by using direct stochastic optical reconstruction microscopy (dSTORM). In our approach for dSTORM phenotyping, we triple stained Frac‐C and Frac‐E with labelled mAbs for CD81, CD9, and CD63 tetraspanin markers or with labelled mAbs for CD9 or CD63 mixed with an additional labelled mAb against different HIV‐1 proteins.
The tetraspanin phenotyping (CD81+, CD9+, CD63+) indicated the presence of triple positive EPs in both Frac‐C and Frac‐E (Figure 7a). The percent of triple positive CD81+/CD9+/CD63+ EPs was two‐fold higher in Frac‐E than Frac‐C (Figure S9c‐d) and the double positive CD81+/CD63+ and CD9+/CD63+ were 10‐fold and five‐fold higher in Frac‐E than in Frac‐C, respectively (Figure S9c‐d). Conversely, the percent of double positive CD81+/CD9+ EPs was 25‐fold higher in Frac‐C than in Frac‐E (Figure S9c‐d). Additionally, the single positive EPs for CD81+ and CD9+ were five‐fold and 10‐fold higher in Frac‐C. Of note, the single positive CD63+ EPs were up to 10‐fold higher in Frac‐E (Figure S9c‐d). Overall, the cumulative tetraspanin fold differences indicated that CD63+ EPs were five‐fold higher in Frac‐E, CD9+ EPs were two‐fold higher in Frac‐C, and the CD81+ EPs present in both fractions were slightly higher in Frac‐E (Figure 7d). These data indicate that Frac‐C was enriched in CD9+ EPs. In contrast, Frac‐E was enriched with CD63+ EPs. Thus, we hypothesized that the HIV‐1 viral markers could also be differentially distributed in CD9+ and CD63+ Frac‐C and Frac‐E EPs.
FIGURE 7.
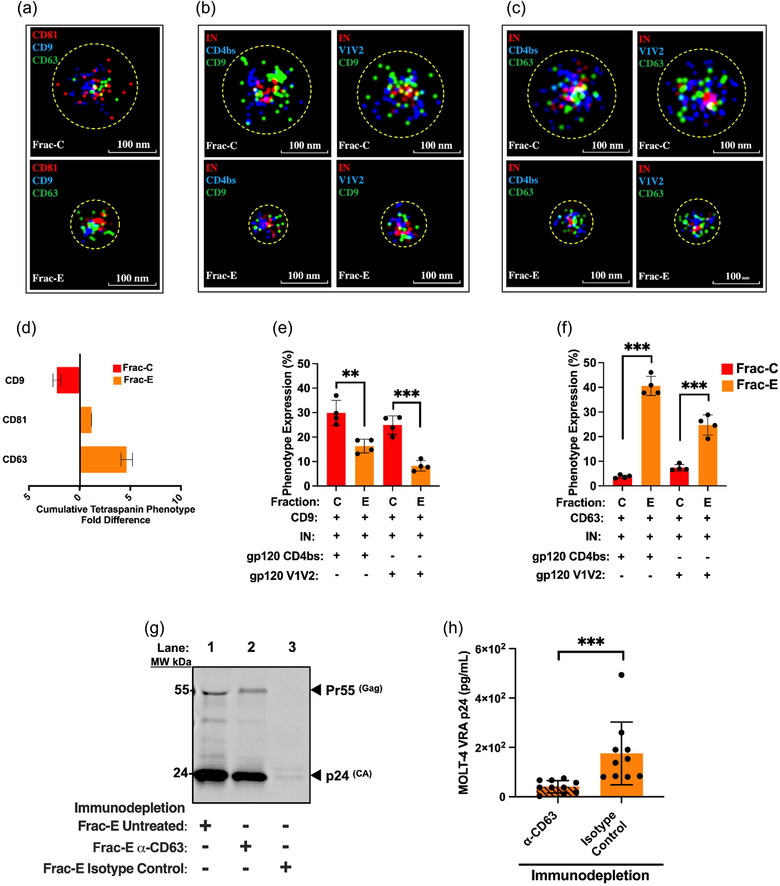
Analysis of J1.1LAV Frac‐C and Frac‐E with Frac‐E CD63 immunodepletion. (a) Representative dSTROM images for Frac‐C and Frac‐E EPs tetraspanin staining triple positive CD81+/CD9+/CD63+; (b) representative triple positive CD9+/IN+/CD4bs+ and CD9+/IN+/V1V2+ dSTROM images for Frac‐C and Frac‐E;(c) representative triple positive CD63+/IN+/CD4bs+ and CD63+/IN+/V1V2+ dSTROM images for Frac‐C and Frac‐E; (d) cumulative tetraspanin CD81+, CD9+, CD63+ percent phenotype fold difference between Frac‐C and Frac‐E; (e) CD9 triple positive with HIV‐1 IN+, HIV‐1 gp120 CD4bs+ or gp120 V1V2+ phenotype expression differences between Frac‐C and Frac‐E; (f) CD63 triple positive with HIV‐1 IN+, HIV‐1 gp120 CD4bs+ or gp120 V1V2+ phenotype expression differences between Frac‐C and Frac‐E; (g) Frac‐E CD63 or IgG isotype control immunodepleted complex retained on magnetic protein‐G capture beads resuspended in 1XSDS loading buffer under non‐reducing conditions and used for WB (as described in the methods sections), which was probed for p24 HIV‐1 core protein; (h) MOLT‐4 VRA of Frac‐E supernatants after CD63 and isotype control immunodepletion, results reflect three independent biological immunodepletion replicate experiments with three technical replicate measurements of HIV‐1 p24 by capture ELISA. dSTORM images were analyzed (n > 5000 EPs per quadruplicate experiment) at a magnification of 0.578 nm/pixels with the circles (yellow segmented line) representing the relative size of each EPs as measured in CODI using clustering analysis. Data represents mean ± standard deviation (SD) of either four technical replicate measurements (dSTORM) or five biological experiments with duplicate measurements (MOLT‐4 VRA Immunoprecipitation). Statistical significance for panels (e, f, and g) was calculated with unpaired two‐tailed Student's t‐test according to its distribution where the *P < 0.05; ***P < 0.001 significance.
To further validate the above hypothesis, we immunophenotyped Frac‐C and Frac‐E EPs with a cocktail of labelled mAbs against CD9 or CD63 in combination with labelled mAbs against HIV‐1 integrase (IN) (2C11), gp120 Env CD4 binding site (CD4bs, VRC01) or the gp120 Env V1V2 loop (PG16). We selected the HIV‐1 specific markers based either on our membrane protection assay findings (Figure 5g), which indicated HIV‐1 IN is luminal, or based on the reduction of infectivity with HIV‐1 gp120 Env‐specific bNAbs (VRC01 and PG16) (Figure 6e) that indicated the presence HIV‐1 gp120 Env on the outside of the infectious EPs. To correct for any background binding of the HIV‐1‐specific mAbs, we used isotype‐matched controls for the HIV‐1 markers to calculate the percent of EPs positivity for each mAb. The images of the permeabilized Frac‐C and Frac‐E EPs triple stained with either CD9 or CD63 and the HIV‐1 specific markers indicated that the viral IN is potentially centrally localized within the EPs (Figure 7b,c). Conversely, the CD9 or CD63 tetraspanins and the HIV‐1 Env markers were colocalized peripherally around the centrally situated IN (Figure 7b,c). This observation further implies a protective membrane around the HIV‐1 genome associated with IN, as previously described by others (Kessl et al., 2016). The isotype controls for IN and HIV‐1 Env mAbs showed negligible background staining (Figure S9a‐b), indicating a high specificity for the HIV‐1 mAb. The triple positive (CD9+/IN+/CD4bs+ or CD9+/IN+/V1V2+) EPs were significantly higher in Frac‐C than in Frac‐E (Figure 7e) and 2‐5‐fold higher, depending on the HIV‐1 mAb (Figure S9e‐h). In contrast, the percent of triple stained CD63+/IN+/CD4bs+ or CD63+/IN+/V1V2+ EPs were significantly higher in Frac‐E than in Frac‐C (Figure 7f), with 5‐ to 10‐fold higher in Frac‐E than in Frac‐C, depending on the HIV‐1 gp120 mAb used in the assay (Figure S9i‐l). The results shown in (Figure S9e‐l) further indicate the presence of a heterogeneous mixture of EPs that can be double or single positive for viral and EPs markers. Overall, the novel dSTORM single particle immunophenotyping of Frac‐C and Frac‐E EPs indicated that Frac‐C had a higher colocalization of HIV‐1 markers with CD9 (similar to studies from Dahmane et al., 2018) and Frac‐E had a higher colocalization of HIV‐1 markers with CD63 (similar to studies from Meerloo et al., 1993).
Therefore, we sought to determine whether the CD63+ EPs in Frac‐E are infectious. Thus, we immunodepleted Frac‐E EPs with α‐CD63 antibody and isotype control. The unbound Frac‐E was collected and used to infect MOLT‐4 VRA target cells while the bound Frac‐E to the magnetic protein‐G beads were treated with 1X loading buffer electrophoresed, blotted, and probed for HIV‐1 p24. The results indicated that α‐CD63 immunoprecipitation of Frac‐E captured HIV‐1 viral protein p24 onto the protein‐G beads (Figure 7g) and significantly reduced the infection of the unbound precipitate in the MOLT‐4 VRA assay when compared to the isotype control (Figure 7h). However, small residual infectivity remained after the immunoprecipitation of Frac‐E with α‐CD63 antibody, indicating that not all HIV‐1 particles in Frac‐E were CD63+, or not all CD63+ infectious particles were immunoprecipitated. This observation aligns with Frac‐E dSTORM data showing the absence of CD63− but the presence of IN+/CD4bs+ or IN+/V1V2 + EPs in Frac‐E (Figure S9g‐j).
3.11. Presence of HIV‐1 in small EPs isolated by DUC and through SDF from an alternative chronically infected T‐cell line and HIV‐1 infected primary PBMC
To further validate the existence of small infectious EPs in various cell culture supernatants of HIV‐1 infected cells, we conducted the isolation of Frac‐E by DUC from an alternative chronically infected T‐cell line, ACH‐2LAV, as well as from in vitro BaL‐infected PBMC. Sizing of ACH‐2LAV and infected BaL‐infected PBMC Frac‐C and Frac‐E EPs by NTA revealed the existence of small EPs subpopulations (Figure S10a‐e) in Frac‐E with similar size distribution to the J1.1LAV (Figure 4c‐d, and Table 1). Additionally, the MOLT‐4 VRA results indicated the presence of small infectious EPs in Frac‐E from both culture supernatants on day 4 for ACH‐2LAV and day 4, 7, and 14 for the BaL‐infected PBMC (Figure S10f‐g).
Subsequently, we aimed to isolate small EPs through an alternative method (Figure S11a) to validate the presence of infectious HIV‐1 below 50 nm. For this purpose, we implemented a series of serial differential filtration steps (SDF) as described in the methods section, nearly similar to what Heineman and Vykoukal have described (Heinemann & Vykoukal, 2017). We recovered small EPs by SDF from culture supernatants of J1.1LAV or infected PBMC with LAV or BaL. After the isolation, we sized and checked the infectivity of the small EPs by NTA and MOLT‐4 VRA. Not surprisingly, the sizing of the SDF EPs or filtered small EPs (fsEPs) (<50 nm) had a PSD of D50 under 50 nm, while the PSD for the EPs that passed only through the 450 nm filter had a D50 over 100 nm (Figure S11b‐e). Additionally, using TEM, we observed the presence of large and small particles that passed only through the 450 nm filter (Figure S11f), while for the fsEPs, we observed mainly small EPs with occasional aggregates (Figure S11e). The MOLT‐4 VRA indicated that the fsEPs derived from J1.1LAV were infectious at day 4 (Figure S11h), and similarly, at day 4, 7, and 14, the fsEPs recovered from LAV and BaL‐infected PBMC supernatants (Figure S11i‐J). These results collectively support the notion that the novel smHIV‐1 particles identified in Frac‐E through DUC from J1.1LAV can be successfully isolated using another infected T‐cell line and also from infected PBMCs using DUC and the alternative SDF method.
4. DISCUSSION
As the field of EPs expands beyond new boundaries toward large and smaller size particles, it has become more evident that EPs preparations are a mixture of various size vesicles and particles, capable of carrying proteins and nucleic acids that can induce physiological changes in the recipient cells (Anand et al., 2021; Clancy et al., 2021; Tosar et al., 2022; H. Zhang & Lyden, 2019; Q. Zhang et al., 2019). We and others have described EVs as close relatives of viruses with similar biophysical characteristics and capable of carrying viral proteins and nucleic acids (Hildreth, 2017; Martins & Alves, 2020; Narayanan et al., 2013; Nolte‐’t Hoen et al., 2016). Here, we demonstrate for the first time the presence of large and very smHIV‐1 virions beyond the currently accepted HIV‐1 size range of 120–140 nm (Freed, 2015; Fuller et al., 1997; McNamara & Dittmer, 2020; Wilk et al., 2001).
We have previously observed the presence of various viral and host proteins in EVs from HIV‐1‐infected cells, and we were able to assess their infectivity at early (6 h) and late (24 h) times post G0 release (Barclay et al., 2017; DeMarino et al., 2018; Kim et al., 2021; Narayanan et al., 2013; Sampey et al., 2016). Importantly, we found that many viral byproducts (i.e., p24, env, TAR RNA) were present in early‐release EVs (6 h post‐release), however, they were not infectious. These heterogeneous populations of EVs were then separated based on their sedimentation rates using UC. We started our analysis as an extension of the previous findings that infected cells synchronized at G0 phase of the cell cycle, followed by activation and release, produce non‐infectious EPs at 6 h, which contained classical exosomal markers, autophagy markers, and viral proteins and RNAs (DeMarino et al., 2022; Kim et al., 2021). Interestingly, when using UC of EPs into fractions (Fracs A‐E), we observed a unique large 2K sample (Frac‐A) collected at the 24‐h timepoint that contained infectious virions. The Frac‐A was further purified using size exclusion chromatography (Izon qEV 70), and the first few large fractions showed markers of amphisomes, including the presence of Lamp1, Rab5,7, and LC3‐I/II proteins. More importantly, virions in Frac‐A were blocked by the VRC01 (CD4 binding site) mAb but not by the PG16 (quaternary variable loop two binding site) mAb, suggesting that this form of the virus may not require a trimeric epitope formation to enter target cells. Therefore, the biogenesis of this virus may be different from that of the classical budding viruses in T‐cells, where both modes of entry (gp120 and receptor/co‐receptor) are required for the classically accepted HIV‐1 entry (i.e., present in Frac‐C). We also probed for the presence of Calnexin, an endoplasmic reticulum‐associated protein marker, as a potential indicative of cell contamination (Ramirez et al., 2018; Welsh et al., 2024), with the caveat that this marker was also described to make it to the plasma membrane (Okazaki et al., 2000; Wiest et al., 1997, 1995) from where it could be potentially incorporated into large EPs (Saludas et al., 2022). The WBs indicated that fractions A and B, especially when doing large‐scale preparations, were positive to varying degrees for Calnexin. Therefore, to double‐check for the possibility of cell contamination in the respective fractions, we stored freshly isolated EPs (Frac‐A through Frac‐E) at +4°C or ‐80°C overnight, after which we performed a cell outgrowth assay. We found that Frac‐A and Frac‐B, stored at +4°C, contained some intact cells (Figure S3) as indicated by the cell growth assay, however, when the samples were stored at ‐80°C prior to the cell outgrowth assay, there were no metabolically active or intact cells prior to our downstream assays (Tansey, 2006). Finally, experiments with EPs from patient material should shed light on the presence and significance of this form of virus and its mode of entry into cells.
Additionally, Frac‐B and Frac‐D‐treated MOLT‐4 VRA target cells revealed that these EPs carry HIV‐1 transcripts capable of inducing the expression of extracellular HIV‐1 p24 protein but do not necessarily warrant the production of enough virions needed to detect the induction of the reporter gene in the A3R5 GFP/Luc HIV‐1 reporter cells. However, the presence of syncytial cells in the MOLT‐4 VRA treated with the Frac‐B and Frac‐D implies that these EPs can still participate in the CPE by either forming viral biofilms via the ICAM‐1 that can induce cell aggregation and syncytial cell formation, as we have previously proposed (Pinto et al., 2021; Pinto et al., 2019). Alternatively, they might induce the expression of HIV‐1 gp120 on the surface of the cells with fusogenic properties (Lenardo et al., 2002). Furthermore, in contrast to the similarities in total viral RNA and p24 content between Frac‐C and Frac‐E, we observed that Frac‐E was significantly more infectious than all other fractions, possibly due to its higher concentration of total proteins and lipids, creating a more conducive environment for infection. Therefore, Frac‐E MOLT‐4 VRA assessment paired with NTA and TEM measurements indicated the existence of novel smHIV‐1 virions present in Frac‐E (Berman, 2019).
This unique form of the virus found in Frac‐E (<50 nm) has a size distribution similar to exomeres (H. Zhang & Lyden, 2019). Frac‐E contained CD63, CD81, and CD9, previously associated with small EVs (30‐60 nm) (Kowal et al., 2016; Q. Zhang et al., 2019). Intriguingly, HSP70 was enriched in Frac‐E despite being previously associated with slightly higher EPs sizes (Gandham et al., 2020; Lancaster & Febbraio, 2005; Welsh et al., 2024; van Niel et al., 2018; Q. Zhang et al., 2019). Furthermore, HSP70, a conserved major chaperone involved in protein folding, translocation, refolding, and degradation, is overexpressed in virally infected cells (Iyer et al., 2021) and was strongly detected in Frac‐E. HSP70 is sorted into intraluminal vesicles (Martins & Alves, 2020) and ultimately loaded into EPs (Gandham et al., 2020; Lancaster & Febbraio, 2005; Welsh et al., 2024; van Niel et al., 2018; Q. Zhang et al., 2019). Interestingly, HSP70 is overexpressed in HIV‐1 infected cells to overcome cell cycle arrest by the viral protein R (Vpr), as reported by Iordanskiy et al. (2004).
We expected a higher p24 concentration in Frac‐C since HIV‐1 virus preps are routinely isolated at an approximately 100,000 g force spin (McNamara & Dittmer, 2020; Webber & Clayton, 2013; Zhao et al., 2021). Furthermore, the 132 nm modal size of the particles in Frac‐C (Table 1) coincided with HIV‐1 size ranges (127‐140 nm), as previously determined by others (Freed, 2015; Phillips et al., 2021; McNamara & Dittmer, 2020; Willms et al., 2018). However, we did not expect to observe similar p24 levels in Frac‐C and Frac‐E (Figure 5c), despite Frac‐E predominantly containing smaller size EPs (Figure 4c; Table 1). In addition, more host cell proteins, including ALIX, were detected by WB in Frac‐A, Frac‐C, and Frac‐E, indicating that these EPs fractions contain proteins sorted by ESCRT machinery with an endosomal origin (Gandham et al., 2020; Pinto et al., 2021; Q. Zhang et al., 2019). Thus, our WB findings could suggest that these particles were recycled from the plasma membrane into endosomes and MVB and released to the outside through exocytosis (Lancaster & Febbraio, 2005). Furthermore, a combination of CD63, HSP70, and ALIX1 in Frac‐E detected by WB strongly suggests that most EPs in Frac‐E potentially have an endosomal origin, as previously defined by Iyer et al. (2021). Furthermore, the WB patterns for the viral proteins indicate that Frac‐C has mature HIV‐1 virions defined by a single band of p24Gag. However, Frac‐A and Frac‐E contain a heterogeneous mixture of EPs and HIV‐1 virions with various protein species indicated by various Gag and Nef molecular weights. Thus, WB findings for the viral cargoes further support that these fractions potentially contain a heterogeneous mixture of EPs and HIV‐1, with distinct differences noted among the fractions.
A caveat of our isolation approach is that Frac‐E has a significantly higher total protein concentration due to the long UC step (Salmond & Williams, 2021). Thus, the last step in our DUC approach to obtain Frac‐E might co‐isolate some soluble proteins of a non‐vesicular nature or exomeres and supermeres that have been described to have a higher protein content (Q. Zhang, Jeppesen, Higginbotham, Graves‐Deal, et al., 2021; Q. Zhang et al., 2019). Consequently, the elevated levels of proteins found in Frac‐E could explain why this fraction did not reach the purity level with an acceptable ratio of particles to total protein, as defined by Webber and Clayton (2013). Furthermore, we measured the highest total lipid levels in Frac‐E, suggesting the presence of membrane‐enclosed EPs.
We carried out a membrane protection assay to determine if viral markers in Frac‐E detected by WB are membrane‐enclosed. Since it was previously shown that integrase and viral TAR, TAR‐gag, and env RNA are binding partners (Kessl et al., 2016), we hypothesized that these two molecules would be membrane‐enclosed. The membrane protection assay, similar to that described by Osteikoetxea et al. (2015), indicated that integrase was enclosed by a protective membrane in Frac‐C and Frac‐E. However, actin and p24 were non‐luminal, indicating that these markers could be outside of the EPs due to the freeze‐thaw cycle and lysis (Tansey, 2006) inducing potential cryodamage due to storage (Gelibter et al., 2022; Kusuma et al., 2018; Yuan et al., 2021) or some that are on the surface of the EPs as previously described (Cvjetkovic et al., 2016). Consequently, the membrane protection assay indicated the presence of protected HIV‐1 integrase, previously described as binding the viral nucleic acid (Kessl et al., 2016), contained in Frac‐C and Frac‐E EPs.
In addition, we immunophenotyped Frac‐C and Frac‐E at single particle level using dSTORM technology for host and viral markers. We observed that Frac‐C and Frac‐E contained EPs triple positive for canonical EPs markers (CD81, CD9, CD63), as well as some of the EPs in the respective fractions were either double‐ or single positive for the respective markers, further emphasizing the heterogeneous nature of Frac‐C and Frac‐E EPs. Interestingly, the data revealed that Frac‐C was predominantly positive for CD9, whereas Frac‐E was predominantly positive for CD63, which also validated our WB findings. Thus, next, we sought to immunophenotype the virus contained in these two fractions with either CD9 or CD63 in combination with HIV‐1 integrase (protected by a membrane) and gp120 Env‐specific mAbs to the CD4bs and the V1V2 loop. The triple‐positive data indicated that the virus in Frac‐C contained significantly more CD9, while the virus in Frac‐E contained significantly more CD63. Furthermore, the presence of CD63 in the virus contained in Frac‐E could facilitate the viral fusion and integration described by Li et al. (2014) and increase its infectivity, which is in line with our immunodepletion data of Frac‐E with CD63 that indicated a significant reduction of viral infectivity. Also, the close‐up images of Frac‐C and Frac‐E viral EPs indicated that viral integrase may be centrally colocalized and surrounded by membrane‐bound tetraspanins and HIV‐1 gp120 Env trimer (V1V2 loop specific staining). Additionally, we observed that some of the EPs were either double or single positive for EPs and HIV‐1 markers, which indicates the presence of EPs that are sometimes mentioned as virus‐like particles (Nolte‐’t Hoen et al., 2016). Future knock‐out experiments of CD9 and CD63 in HIV‐1 latently infected cells will allow for better and more detailed analysis of the interacting partners for CD9 (i.e., ADAM10, MME, PTGFRN, ANXA6, etc.) and CD63 (i.e., TIMP1, ITGB1, SRC, AP4M1, etc.) with viral proteins and their role regarding the assembly of the novel smHIV‐1. Overall, the dSTORM findings strongly indicate the presence of phenotypically distinct smHIV‐1 particles released from the same J1.1LAV producer cell line in Frac‐E.
To further validate our results regarding the presence of smHIV‐1, we employed DUC and SDF to isolate smHIV‐1 from other sources, such as a chronically infected T‐cell line ACH‐2LAV and PBMC infected with the HIV‐1 BaL and LAV stock virus. Our findings demonstrate the presence of infectious smHIV‐1 in other cell culture supernatants obtained through additional methods, thereby providing further evidence for the presence smHIV‐1.
In conclusion, we have shown the existence of novel large and small‐size infectious EPs recovered from cell culture supernatants of HIV‐1 infected T‐cell lines and PBMC.
Using a modified DUC technique, we have demonstrated that HIV‐1 in the large Frac‐A EPs carries amphisome markers, while the small Frac‐E carries small endosomal CD63 markers. Additionally, by other alternative techniques such as size exclusion column chromatography, SDF, which were subsequently complemented with Iodixanol velocity gradient UC (data not shown), we provide evidence for the existence of multiple sizes of HIV‐1 released from infected T‐cells and PBMC. Additionally, our findings could have far‐reaching impacts on the role of various sizes of HIV‐1 and other viruses regarding their pathogenesis, influencing the fields of vaccine and drug development. Further investigation into the presence of these novel infectious particles recovered from various biological fluids, especially from HIV‐infected patient specimens, could produce insightful information regarding the antigenicity and the antiretroviral drug escape of HIV‐1.
AUTHOR CONTRIBUTIONS
Sebastian Molnar: Investigation (equal); methodology (equal); writing—original draft (equal); writing—review and editing (equal). Yuriy Kim: Investigation (equal); methodology (equal); writing—original draft (equal); writing—review and editing (equal). Lindsay Wieczorek: Conceptualization (supporting); investigation (supporting); methodology (supporting); writing—review and editing (supporting). Anastasia Williams: Investigation (supporting). Kajal Patil: Investigation (supporting). Pooja Khatkar: Investigation (supporting). Mark F. Santos: Investigation (supporting); methodology (supporting); writing—original draft (supporting). Gifty Mensah: Investigation (supporting). Aurelio Lorico: Conceptualization (supporting); investigation (supporting); methodology (supporting); project administration (supporting); supervision (supporting); writing—review and editing (supporting). Victoria Polonis: Conceptualization (equal); funding acquisition (equal); investigation (supporting); methodology (supporting); project administration (equal); resources (equal); supervision (equal); writing—original draft (supporting); writing—review and editing (supporting). Fatah Kashanchi: Conceptualization (equal); funding acquisition (equal); investigation (supporting); methodology (supporting); project administration (equal); resources (equal); supervision (equal); writing—original draft (supporting); writing—review and editing (supporting).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGEMENTS
We thank all the members of the Kashanchi lab for their assistance, as well as Mrs. Gwen Cox. This research was funded by National Institutes of Health (NIH) Grants (MH134389, AI043894, AI074410, AI078859, AI127351‐01 and NS099029) to F.K. and V.C. Also, by the cooperative agreement (W81XWH‐11‐2‐0174) between the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., and the U.S. Department of Defense (DoD). Additionally, we express our gratitude to Major Brittany Henschen and Sergeant Daniel Lozano from the Department of Pathology Walter Reed Army Institute of Research (WRAIR) for the help with TEM imaging and for Dr. Mangala Rao and Dr. Gary Matyas from the Laboratory of Adjuvant and Antigen Research WRAIR to facilitate access to specialized equipment. All authors declare no potential conflicts of interest, and the views expressed are those of the authors and should not be construed to represent the positions of the U.S. Army or the Department of Defense.
Molnar, S. M. , Kim, Y. , Wieczorek, L. , Williams, A. , Patil, K. A. , Khatkar, P. , Santos, M. F. , Mensah, G. , Lorico, A. , Polonis, V. R. , & Kashanchi, F. (2024). Extracellular Vesicle Isolation Methods Identify Distinct HIV‐1 Particles Released from Chronically Infected T‐cells. Journal of Extracellular Vesicles, 13, e12476. 10.1002/jev2.12476
Sebastian M. Molnar and Yuriy Kim authors contributed equally.
Victoria R. Polonis and Fatah Kashanchi senior authors contributed equally.
REFERENCES
- Anand, S. , Samuel, M. , & Mathivanan, S. (2021). Exomeres: A new member of extracellular vesicles family. Sub‐cellular biochemistry, 97, 89–97. [DOI] [PubMed] [Google Scholar]
- Arakelyan, A. , Fitzgerald, W. , Zicari, S. , Vanpouille, C. , & Margolis, L. (2017). Extracellular vesicles carry HIV env and facilitate HIV infection of human lymphoid tissue. Scientific reports, 7(1), 1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archin, N. M. , Sung, J. M. , Garrido, C. , Soriano‐Sarabia, N. , & Margolis, D. M. (2014). Eradicating HIV‐1 infection: Seeking to clear a persistent pathogen. Nature reviews. Microbiology, 12(11), 750–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay, R. A. , Schwab, A. , DeMarino, C. , Akpamagbo, Y. , Lepene, B. , Kassaye, S. , Iordanskiy, S. , & Kashanchi, F. (2017). Exosomes from uninfected cells activate transcription of latent HIV‐1. The Journal of biological chemistry, 292(28), 11682–11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, J. J. (2019). Changing how we think about infectious diseases. Taxonomic Guide to Infectious Diseases, 321–365. 10.1016/B978-0-12-817576-7.00008-0. PMCID: PMC7149514. [DOI] [Google Scholar]
- Borroto‐Esoda, K. , Vela, J. E. , Myrick, F. , Ray, A. S. , & Miller, M. D. (2006). In vitro evaluation of the anti‐HIV activity and metabolic interactions of tenofovir and emtricitabine. Antiviral therapy, 11(3), 377–384. [PubMed] [Google Scholar]
- Brown, B. K. , Darden, J. M. , Tovanabutra, S. , Oblander, T. , Frost, J. , Sanders‐Buell, E. , de Souza, M. S. , Birx, D. L. , McCutchan, F. E. , & Polonis, V. R. (2005). Biologic and genetic characterization of a panel of 60 human immunodeficiency virus type 1 isolates, representing clades A, B, C, D, CRF01_AE, and CRF02_AG, for the development and assessment of candidate vaccines. Journal of virology, 79(10), 6089–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüning, A. , Burger, P. , Gingelmaier, A. , & Mylonas, I. (2012). The HIV reverse transcriptase inhibitor tenofovir induces cell cycle arrest in human cancer cells. Investigational new drugs, 30(4), 1389–1395. [DOI] [PubMed] [Google Scholar]
- Bukovsky, A. A. , Dorfman, T. , Weimann, A. , & Göttlinger, H. G. (1997). Nef association with human immunodeficiency virus type 1 virions and cleavage by the viral protease. Journal of virology, 71(2), 1013–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, T. D. , Khan, M. , Huang, M. B. , Bond, V. C. , & Powell, M. D. (2008). HIV‐1 Nef protein is secreted into vesicles that can fuse with target cells and virions. Ethnicity & disease, 18(2), S2–19. [PMC free article] [PubMed] [Google Scholar]
- Chen, L. , Feng, Z. , Yue, H. , Bazdar, D. , Mbonye, U. , Zender, C. , Harding, C. V. , Bruggeman, L. , Karn, J. , Sieg, S. F. , Wang, B. , & Jin, G. (2018). Exosomes derived from HIV‐1‐infected cells promote growth and progression of cancer via HIV TAR RNA. Nature communications, 9(1), 4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang, G. Y. , Zhou, J. , Acharya, P. , Rawi, R. , Shen, C. H. , Sheng, Z. , Zhang, B. , Zhou, T. , Bailer, R. T. , Dandey, V. P. , Doria‐Rose, N. A. , Louder, M. K. , McKee, K. , Mascola, J. R. , Shapiro, L. , & Kwong, P. D. (2019). Structural survey of broadly neutralizing antibodies targeting the HIV‐1 env trimer delineates epitope categories and characteristics of recognition. Structure (London, England: 1993), 27(1), 196–206. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy, J. W. , Boomgarden, A. C. , & D'Souza‐Schorey, C. (2021). Profiling and promise of super metres. Nature cell biology, 23(12), 1217–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo, M. , Raposo, G. , & Théry, C. (2014). Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annual review of cell and developmental biology, 30, 255–289. [DOI] [PubMed] [Google Scholar]
- Comfort, N. , Cai, K. , Bloomquist, T. R. , Strait, M. D. , Ferrante, A. W., Jr , & Baccarelli, A. A. (2021). Nanoparticle tracking analysis for the quantification and size determination of extracellular vesicles. Journal of visualized experiments: JoVE, Mar 28(169). 10.3791/62447. PMID: 33843938; PMCID: PMC8243380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvjetkovic, A. , Jang, S. C. , Konečná, B. , Höög, J. L. , Sihlbom, C. , Lässer, C. , & Lötvall, J. (2016). Detailed analysis of protein topology of extracellular vesicles‐evidence of unconventional membrane protein orientation. Scientific reports, 6, 36338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmane, S. , Doucet, C. , Le Gall, A. , Chamontin, C. , Dosset, P. , Murcy, F. , Fernandez, L. , Pastene, D. S. , Rubinstein, E. , Mougel, M. , Nollmann, M. , & Milhiet, P.‐E. (2018). HIV‐1 specifically traps CD9 and CD81 tetraspanins within viral buds and induces their membrane depletion. Cold Spring Harbor Laboratory. [Google Scholar]
- Davey, R. T., Jr , Bhat, N. , Yoder, C. , Chun, T. W. , Metcalf, J. A. , Dewar, R. , Natarajan, V. , Lempicki, R. A. , Adelsberger, J. W. , Miller, K. D. , Kovacs, J. A. , Polis, M. A. , Walker, R. E. , Falloon, J. , Masur, H. , Gee, D. , Baseler, M. , Dimitrov, D. S. , Fauci, A. S. , & Lane, H. C. (1999). HIV‐1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proceedings of the National Academy of Sciences of the United States of America, 96(26), 15109–15114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMarino, C. , Barclay, R. A. , Pleet, M. L. , Pinto, D. O. , Branscome, H. , Paul, S. , Lepene, B. , El‐Hage, N. , & Kashanchi, F. (2019). Purification of high yield extracellular vesicle preparations away from virus. Journal of visualized experiments: JoVE, Sep 12(151). 10.3791/59876. PMID: 31566605. [DOI] [PubMed] [Google Scholar]
- DeMarino, C. , Cowen, M. , Khatkar, P. , Cotto, B. , Branscome, H. , Kim, Y. , Sharif, S. A. , Agbottah, E. T. , Zhou, W. , Costiniuk, C. T. , Jenabian, M. A. , Gelber, C. , Liotta, L. A. , Langford, D. , & Kashanchi, F. (2022). Cannabinoids reduce extracellular vesicle release from HIV‐1 infected myeloid cells and inhibit viral transcription. Cells, 11(4), 723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMarino, C. , Pleet, M. L. , Cowen, M. , Barclay, R. A. , Akpamagbo, Y. , Erickson, J. , Ndembi, N. , Charurat, M. , Jumare, J. , Bwala, S. , Alabi, P. , Hogan, M. , Gupta, A. , Noren Hooten, N. , Evans, M. K. , Lepene, B. , Zhou, W. , Caputi, M. , Romerio, F. , … Kashanchi, F. (2018). Antiretroviral drugs alter the content of extracellular vesicles from HIV‐1‐infected cells. Scientific reports, 8(1), 7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneka, M. , Pelchen‐Matthews, A. , Byland, R. , Ruiz‐Mateos, E. , & Marsh, M. (2007). In macrophages, HIV‐1 assembles into an intracellular plasma membrane domain containing the tetraspanins CD81, CD9, and CD53. The Journal of Cell Biology, 177(2), 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettenhofer, M. , & Yu, X. F. (1999). Highly purified human immunodeficiency virus type 1 reveals a virtual absence of Vif in virions. Journal of virology, 73(2), 1460–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euler, Z. , & Schuitemaker, H. (2012). Cross‐reactive broadly neutralizing antibodies: Timing is everything. Frontiers in immunology, 3, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fackler, O. T. , Kienzle, N. , Kremmer, E. , Boese, A. , Schramm, B. , Klimkait, T. , Kücherer, C. , & Mueller‐Lantzsch, N. (1997). Association of human immunodeficiency virus Nef protein with actin is myristoylation dependent and influences its subcellular localization. European journal of biochemistry, 247(3), 843–851. [DOI] [PubMed] [Google Scholar]
- Fleming, A. , Sampey, G. , Chung, M. C. , Bailey, C. , van Hoek, M. L. , Kashanchi, F. , & Hakami, R. M. (2014). The carrying pigeons of the cell: Exosomes and their role in infectious diseases caused by human pathogens. Pathogens and disease, 71(2), 109–120. [DOI] [PubMed] [Google Scholar]
- Folks, T. M. , Clouse, K. A. , Justement, J. , Rabson, A. , Duh, E. , Kehrl, J. H. , & Fauci, A. S. (1989). Tumor necrosis factor alpha induces expression of human immunodeficiency virus in a chronically infected T‐cell clone. Proceedings of the National Academy of Sciences of the United States of America, 86(7), 2365–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folks, T. M. , Justement, J. , Kinter, A. , Dinarello, C. A. , & Fauci, A. S. (1987). Cytokine‐induced expression of HIV‐1 in a chronically infected promonocyte cell line. Science (New York, N.Y.), 238(4828), 800–802. [DOI] [PubMed] [Google Scholar]
- Folks, T. M. , Justement, J. , Kinter, A. , Schnittman, S. , Orenstein, J. , Poli, G. , & Fauci, A. S. (1988). Characterization of a promonocyte clone chronically infected with HIV and inducible by 13‐phorbol‐12‐myristate acetate. Journal of immunology (Baltimore, Md.: 1950), 140(4), 1117–1122. [PubMed] [Google Scholar]
- Frankel, E. B. , & Audhya, A. (2018). ESCRT‐dependent cargo sorting at multivesicular endosomes. Seminars in cell & developmental biology, 74, 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed, E. O. (1998). HIV‐1 gag proteins: Diverse functions in the virus life cycle. Virology, 251(1), 1–15. [DOI] [PubMed] [Google Scholar]
- Freed, E. O. (2015). HIV‐1 assembly, release and maturation. Nature reviews. Microbiology, 13(8), 484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller, S. D. , Wilk, T. , Gowen, B. E. , Kräusslich, H. G. , & Vogt, V. M. (1997). Cryo‐electron microscopy reveals ordered domains in the immature HIV‐1 particle. Current biology: CB, 7(10), 729–738. [DOI] [PubMed] [Google Scholar]
- Gandham, S. , Su, X. , Wood, J. , Nocera, A. L. , Alli, S. C. , Milane, L. , Zimmerman, A. , Amiji, M. , & Ivanov, A. R. (2020). Technologies and standardization in research on extracellular vesicles. Trends in biotechnology, 38(10), 1066–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelibter, S. , Marostica, G. , Mandelli, A. , Siciliani, S. , Podini, P. , Finardi, A. , & Furlan, R. (2022). The impact of storage on extracellular vesicles: A systematic study. Journal of extracellular vesicles, 11(2), e12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannessi, F. , Aiello, A. , Franchi, F. , Percario, Z. A. , & Affabris, E. (2020). The role of extracellular vesicles as allies of HIV, HCV and SARS viruses. Viruses., 12(5), 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould, S. J. , Booth, A. M. , & Hildreth, J. E. (2003). The Trojan exosome hypothesis. Proceedings of the National Academy of Sciences of the United States of America, 100(19), 10592–10597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueler, A. , Moser, A. , Calmy, A. , Günthard, H. F. , Bernasconi, E. , Furrer, H. , Fux, C. A. , Battegay, M. , Cavassini, M. , Vernazza, P. , Zwahlen, M. , & Egger, M. , Swiss HIV Cohort Study, Swiss National Cohort . (2017). Life expectancy in HIV‐positive persons in Switzerland: Matched comparison with general population. AIDS (London, England), 31(3), 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann, M. L. , & Vykoukal, J. (2017). Sequential filtration: A gentle method for the isolation of functional extracellular vesicles. Methods in molecular biology (Clifton, N.J.), 1660, 33–41. [DOI] [PubMed] [Google Scholar]
- Heinemann, M. L. , & Vykoukal, J. (2017). Sequential filtration: A gentle method for the isolation of functional extracellular vesicles. In Kuo W. P., & Jia S., Eds., Extracellular vesicles: Methods and protocols. pp. 33–41. Springer New York. [DOI] [PubMed] [Google Scholar]
- Hildreth, J. E. K. (2017). HIV as trojan exosome: Immunological paradox explained? Frontiers in immunology, 8, 1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, A. F. , Pegtel, D. M. , Lambertz, U. , Leonardi, T. , O'Driscoll, L. , Pluchino, S. , Ter‐Ovanesyan, D. , & Nolte‐’t Hoen, E. N. (2013). ISEV position paper: Extracellular vesicle RNA analysis and bioinformatics. Journal of Extracellular Vesicles, 2(1), 22859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcar, M. , Kandušer, M. , & Lenassi, M. (2021). Blood nanoparticles—Influence on extracellular vesicle isolation and characterization. Frontiers in Pharmacology, 12, 773844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iordanskiy, S. , Zhao, Y. , Dubrovsky, L. , Iordanskaya, T. , Chen, M. , Liang, D. , & Bukrinsky, M. (2004). Heat shock protein 70 protects cells from cell cycle arrest and apoptosis induced by human immunodeficiency virus type 1 viral protein R. Journal of virology, 78(18), 9697–9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipinmoroti, A. O. , & Matthews, Q. L. (2020). Extracellular vesicles: Roles in human viral infections, immune‐diagnostic, and therapeutic applications. Pathogens (Basel, Switzerland), 9(12), 1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, K. , Chand, K. , Mitra, A. , Trivedi, J. , & Mitra, D. (2021). Diversity in heat shock protein families: Functional implications in virus infection with a comprehensive insight of their role in the HIV‐1 life cycle. Cell stress & chaperones, 26(5), 743–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski, E. , Narayanan, A. , Van Duyne, R. , Shabbeer‐Meyering, S. , Iordanskiy, S. , Saifuddin, M. , Das, R. , Afonso, P. V. , Sampey, G. C. , Chung, M. , Popratiloff, A. , Shrestha, B. , Sehgal, M. , Jain, P. , Vertes, A. , Mahieux, R. , & Kashanchi, F. (2014). Human T‐lymphotropic virus type 1‐infected cells secrete exosomes that contain Tax protein. The Journal of Biological Chemistry, 289(32), 22284–22305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen, D. K. , Zhang, Q. , Franklin, J. L. , & Coffey, R. J. (2023). Extracellular vesicles and nanoparticles: Emerging complexities. Trends in Cell Biology, 33(8), 667–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, M. K. , & Mun, J. Y. (2018). Sample preparation and imaging of exosomes by transmission electron microscopy. Journal of Visualized Experiments: JoVE, 00(131), 56482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl, J. J. , Kutluay, S. B. , Townsend, D. , Rebensburg, S. , Slaughter, A. , Larue, R. C. , Shkriabai, N. , Bakouche, N. , Fuchs, J. R. , Bieniasz, P. D. , & Kvaratskhelia, M. (2016). HIV‐1 integrase binds the viral RNA genome and is essential during virion morphogenesis. Cell, 166(5), 1257–1268. e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. , Mensah, G. A. , Al Sharif, S. , Pinto, D. O. , Branscome, H. , Yelamanchili, S. V. , Cowen, M. , Erickson, J. , Khatkar, P. , Mahieux, R. , & Kashanchi, F. (2021). Extracellular vesicles from infected cells are released prior to virion release. Cells, 10(4), 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal, J. , Arras, G. , Colombo, M. , Jouve, M. , Morath, J. P. , Primdal‐Bengtson, B. , Dingli, F. , Loew, D. , Tkach, M. , & Théry, C. (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proceedings of the National Academy of Sciences of the United States of America, 113(8), E968–E977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Chandele, A. , & Sharma, A. (2021). Current status of therapeutic monoclonal antibodies against SARS‐CoV‐2. PLoS pathogens, 17(9), e1009885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusuma, G. D. , Barabadi, M. , Tan, J. L. , Morton, D. A. V. , Frith, J. E. , & Lim, R. (2018). To protect and to preserve: Novel preservation strategies for extracellular vesicles. Frontiers in pharmacology, 9, 1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird, G. M. , Eisele, E. E. , Rabi, S. A. , Lai, J. , Chioma, S. , Blankson, J. N. , Siliciano, J. D. , & Siliciano, R. F. (2013). Rapid quantification of the latent reservoir for HIV‐1 using a viral outgrowth assay. PLoS pathogens, 9(5), e1003398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster, G. I. , & Febbraio, M. A. (2005). Exosome‐dependent trafficking of HSP70: A novel secretory pathway for cellular stress proteins. The Journal of Biological Chemistry, 280(24), 23349–23355. [DOI] [PubMed] [Google Scholar]
- Larios, J. , Mercier, V. , Roux, A. , & Gruenberg, J. (2020). ALIX‐ and ESCRT‐III‐dependent sorting of tetraspanins to exosomes. The Journal of Cell Biology, 219(3), e201904113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latifkar, A. , Hur, Y. H. , Sanchez, J. C. , Cerione, R. A. , & Antonyak, M. A. (2019). New insights into extracellular vesicle biogenesis and function. Journal of Cell Science, 132(13), jcs222406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenardo, M. J. , Angleman, S. B. , Bounkeua, V. , Dimas, J. , Duvall, M. G. , Graubard, M. B. , Hornung, F. , Selkirk, M. C. , Speirs, C. K. , Trageser, C. , Orenstein, J. O. , & Bolton, D. L. (2002). Cytopathic killing of peripheral blood CD4(+) T lymphocytes by human immunodeficiency virus type 1 appears necrotic rather than apoptotic and does not require env. Journal of virology, 76(10), 5082–5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G. , Endsley, M. A. , Somasunderam, A. , Gbota, S. L. , Mbaka, M. I. , Murray, J. L. , & Ferguson, M. R. (2014). The dual role of tetraspanin CD63 in HIV‐1 replication. Virology journal, 11, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liangsupree, T. , Multia, E. , & Riekkola, M. L. (2021). Modern isolation and separation techniques for extracellular vesicles. Journal of chromatography. A, 1636, 461773. [DOI] [PubMed] [Google Scholar]
- Lifson, J. D. , Feinberg, M. B. , Reyes, G. R. , Rabin, L. , Banapour, B. , Chakrabarti, S. , Moss, B. , Wong‐Staal, F. , Steimer, K. S. , & Engleman, E. G. (1986). Induction of CD4‐dependent cell fusion by the HTLV‐III/LAV envelope glycoprotein. Nature, 323(6090), 725–728. [DOI] [PubMed] [Google Scholar]
- Lobritz, M. A. , Ratcliff, A. N. , & Arts, E. J. (2010). HIV‐1 entry, inhibitors, and resistance. Viruses., 2(5), 1069–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis, L. , & Sadovsky, Y. (2019). The biology of extracellular vesicles: The known unknowns. PLoS Biology, 17(7), e3000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins, S. T. , & Alves, L. R. (2020). Extracellular vesicles in viral infections: Two sides of the same coin? Frontiers in cellular and infection microbiology, 10, 593170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateescu, B. , Kowal, E. J. , van Balkom, B. W. , Bartel, S. , Bhattacharyya, S. N. , Buzás, E. I. , Buck, A. H. , de Candia, P. , Chow, F. W. , Das, S. , Driedonks, T. A. , Fernández‐Messina, L. , Haderk, F. , Hill, A. F. , Jones, J. C. , Van Keuren‐Jensen, K. R. , Lai, C. P. , Lässer, C. , Liegro, I. D. , … Nolte‐’t Hoen, E. N. (2017). Obstacles and opportunities in the functional analysis of extracellular vesicle RNA—An ISEV position paper. Journal of extracellular vesicles, 6(1), 1286095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLinden, R. J. , Labranche, C. C. , Chenine, A. L. , Polonis, V. R. , Eller, M. A. , Wieczorek, L. , Ochsenbauer, C. , Kappes, J. C. , Perfetto, S. , Montefiori, D. C. , Michael, N. L. , & Kim, J. H. (2013). Detection of HIV‐1 neutralizing antibodies in a human CD4⁺/CXCR4⁺/CCR5⁺ T‐lymphoblastoid cell assay system. PLoS ONE, 8(11), e77756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara, R. P. , & Dittmer, D. P. (2020). Modern techniques for the isolation of extracellular vesicles and viruses. Journal of Neuroimmune Pharmacology: the official journal of the Society on NeuroImmune Pharmacology, 15(3), 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerloo, T. , Sheikh, M. A. , Bloem, A. C. , de Ronde, A. , Schutten, M. , van Els, C. A. , Roholl, P. J. , Joling, P. , Goudsmit, J. , & Schuurman, H. J. (1993). Host cell membrane proteins on human immunodeficiency virus type 1 after in vitro infection of H9 cells and blood mononuclear cells. An immuno‐electron microscopic study. The Journal of General Virology, 74(1), 129–135. [DOI] [PubMed] [Google Scholar]
- Meister, M. , & Tikkanen, R. (2014). Endocytic trafficking of membrane‐bound cargo: A flotillin point of view. Membranes, 4(3), 356–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momen‐Heravi, F. , Balaj, L. , Alian, S. , Mantel, P. Y. , Halleck, A. E. , Trachtenberg, A. J. , Soria, C. E. , Oquin, S. , Bonebreak, C. M. , Saracoglu, E. , Skog, J. , & Kuo, W. P. (2013). Current methods for the isolation of extracellular vesicles. Biological Chemistry, 394(10), 1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan, A. , Iordanskiy, S. , Das, R. , Van Duyne, R. , Santos, S. , Jaworski, E. , Guendel, I. , Sampey, G. , Dalby, E. , Iglesias‐Ussel, M. , Popratiloff, A. , Hakami, R. , Kehn‐Hall, K. , Young, M. , Subra, C. , Gilbert, C. , Bailey, C. , Romerio, F. , & Kashanchi, F. (2013). Exosomes derived from HIV‐1‐infected cells contain trans‐activation response element RNA. The Journal of Biological Chemistry, 288(27), 20014–20033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte‐’t Hoen, E. , Cremer, T. , Gallo, R. C. , & Margolis, L. B. (2016). Extracellular vesicles and viruses: Are they close relatives? Proceedings of the National Academy of Sciences of the United States of America, 113(33), 9155–9161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odorizzi, G. (2006). The multiple personalities of Alix. Journal of Cell Science, 119(15), 3025–3032. [DOI] [PubMed] [Google Scholar]
- Okazaki, Y. , Ohno, H. , Takase, K. , Ochiai, T. , & Saito, T. (2000). Cell surface expression of calnexin, a molecular chaperone in the endoplasmic reticulum. The Journal of Biological Chemistry, 275(46), 35751–35758. [DOI] [PubMed] [Google Scholar]
- Osteikoetxea, X. , Sódar, B. , Németh, A. , Szabó‐Taylor, K. , Pálóczi, K. , Vukman, K. V. , Tamási, V. , Balogh, A. , Kittel, Á. , Pállinger, É. , & Buzás, E. I. (2015). Differential detergent sensitivity of extracellular vesicle subpopulations. Organic & biomolecular chemistry, 13(38), 9775–9782. [DOI] [PubMed] [Google Scholar]
- Pelissier Vatter, F. A. , Cioffi, M. , Hanna, S. J. , Castarede, I. , Caielli, S. , Pascual, V. , Matei, I. , & Lyden, D. (2021). Extracellular vesicle‐ and particle‐mediated communication shapes innate and adaptive immune responses. The Journal of experimental medicine, 218(8), e20202579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez, P. S. , Romaniuk, M. A. , Duette, G. A. , Zhao, Z. , Huang, Y. , Martin‐Jaular, L. , Witwer, K. W. , Théry, C. , & Ostrowski, M. (2019). Extracellular vesicles and chronic inflammation during HIV infection. Journal of extracellular vesicles, 8(1), 1687275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, V. L. , Rowe, T. , Justement, J. S. , Butera, S. T. , June, C. H. , & Folks, T. M. (1991). An HIV‐1‐infected T cell clone defective in IL‐2 production and Ca2+ mobilization after CD3 stimulation. Journal of Immunology (Baltimore, Md.: 1950), 147(9), 3145–3148. [PubMed] [Google Scholar]
- Phillips, W. , Willms, E. , & Hill, A. F. (2021). Understanding extracellular vesicle and nanoparticle heterogeneity: Novel methods and considerations. Proteomics, 21(13‐14), e2000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto, D. O. , Al Sharif, S. , Mensah, G. , Cowen, M. , Khatkar, P. , Erickson, J. , Branscome, H. , Lattanze, T. , DeMarino, C. , Alem, F. , Magni, R. , Zhou, W. , Alais, S. , Dutartre, H. , El‐Hage, N. , Mahieux, R. , Liotta, L. A. , & Kashanchi, F. (2021). Extracellular vesicles from HTLV‐1 infected cells modulate target cells and viral spread. Retrovirology, 18(1), 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto, D. O. , DeMarino, C. , Pleet, M. L. , Cowen, M. , Branscome, H. , Al Sharif, S. , Jones, J. , Dutartre, H. , Lepene, B. , Liotta, L. A. , Mahieux, R. , & Kashanchi, F. (2019). HTLV‐1 extracellular vesicles promote cell‐to‐cell contact. Frontiers in microbiology, 10, 2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzato, M. , Helander, A. , Popova, E. , Calistri, A. , Zamborlini, A. , Palù, G. , & Göttlinger, H. G. (2007). Dynamin 2 is required for the enhancement of HIV‐1 infectivity by Nef. Proceedings of the National Academy of Sciences of the United States of America, 104(16), 6812–6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez, M. I. , Amorim, M. G. , Gadelha, C. , Milic, I. , Welsh, J. A. , Freitas, V. M. , Nawaz, M. , Akbar, N. , Couch, Y. , Makin, L. , Cooke, F. , Vettore, A. L. , Batista, P. X. , Freezor, R. , Pezuk, J. A. , Rosa‐Fernandes, L. , Carreira, A. C. O. , Devitt, A. , Jacobs, L. , … Dias‐Neto, E. (2018). Technical challenges of working with extracellular vesicles. Nanoscale, 10(3), 881–906. [DOI] [PubMed] [Google Scholar]
- Robbins, P. D. , & Morelli, A. E. (2014). Regulation of immune responses by extracellular vesicles. Nature reviews. Immunology, 14(3), 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmond, N. , & Williams, K. C. (2021). Isolation and characterization of extracellular vesicles for clinical applications in cancer—Time for standardization? Nanoscale advances, 3(7), 1830–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saludas, L. , Garbayo, E. , Ruiz‐Villalba, A. , Hernández, S. , Vader, P. , Prósper, F. , & Blanco‐Prieto, M. J. (2022). Isolation methods of large and small extracellular vesicles derived from cardiovascular progenitors: A comparative study. European journal of pharmaceutics and biopharmaceutics: official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik e.V, 170, 187–196. [DOI] [PubMed] [Google Scholar]
- Sampey, G. C. , Saifuddin, M. , Schwab, A. , Barclay, R. , Punya, S. , Chung, M. C. , Hakami, R. M. , Zadeh, M. A. , Lepene, B. , Klase, Z. A. , El‐Hage, N. , Young, M. , Iordanskiy, S. , & Kashanchi, F. (2016). Exosomes from HIV‐1‐infected cells stimulate production of pro‐inflammatory cytokines through trans‐activating response (TAR) RNA. The Journal of biological chemistry, 291(3), 1251–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Wandelmer, J. , & Reggiori, F. (2013). Amphisomes: Out of the autophagosome shadow? The EMBO journal, 32(24), 3116–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, M. F. , Rappa, G. , Karbanová, J. , Fontana, S. , Bella, M. A. D. , Pope, M. R. , Parrino, B. , Cascioferro, S. M. , Vistoli, G. , Diana, P. , Cirrincione, G. , Arena, G. O. , Woo, G. , Huang, K. , Huynh, T. , Moschetti, M. , Alessandro, R. , Corbeil, D. , & Lorico, A. (2021). Itraconazole inhibits nuclear delivery of extracellular vesicle cargo by disrupting the entry of late endosomes into the nucleoplasmic reticulum. Journal of extracellular vesicles, 10(10), e12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, C. A. , Rasband, W. S. , & Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nature methods, 9(7), 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab, A. , Meyering, S. S. , Lepene, B. , Iordanskiy, S. , van Hoek, M. L. , Hakami, R. M. , & Kashanchi, F. (2015). Extracellular vesicles from infected cells: Potential for direct pathogenesis. Frontiers in microbiology, 6, 1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares, H. (2014). HIV‐1 intersection with CD4 T cell vesicle exocytosis: Intercellular communication goes viral. Frontiers in immunology, 5, 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers, W. C. (2009). Virus infection. Encyclopedia of Microbiology, 546–552. 10.1016/B978-012373944-5.00323-0. PMCID: PMC7149757. [DOI] [Google Scholar]
- Sun, M. , Li, Y. , Zheng, H. , & Shao, Y. (2016). Recent progress toward engineering HIV‐1‐specific neutralizing monoclonal antibodies. Frontiers in immunology, 7, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama, M. , Daikoku, E. , Goto, T. , Sano, K. , & Morikawa, Y. (2009). Reactivation from latency displays HIV particle budding at plasma membrane, accompanying CD44 upregulation and recruitment. Retrovirology, 6, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey, W. P. (2006). Freeze‐thaw lysis for extraction of proteins from Mammalian cells. CSH protocols, 2006(7). 10.1101/pdb.prot4614 [DOI] [PubMed] [Google Scholar]
- Thali, M. , Moore, J. P. , Furman, C. , Charles, M. , Ho, D. D. , Robinson, J. , & Sodroski, J. (1993). Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120‐CD4 binding. Journal of virology, 67(7), 3978–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry, C. , Amigorena, S. , Raposo, G. , & Clayton, A. (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Current protocols in cell biology, 30(1), 3.22.1–3.22.29. [DOI] [PubMed] [Google Scholar]
- Tosar, J. P. , Cayota, A. , & Witwer, K. (2022). Exomeres and supermeres: Monolithic or diverse? Journal of extracellular biology, 1(6), e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UNAIDS . (2023). Global HIV & AIDS statistics — Fact Sheet .
- Van Deun, J. , Mestdagh, P. , Agostinis, P. , Akay, Ö. , Anand, S. , Anckaert, J. , Martinez, Z. A. , Baetens, T. , Beghein, E. , Bertier, L. , Berx, G. , Boere, J. , Boukouris, S. , Bremer, M. , Buschmann, D. , Byrd, J. B. , Casert, C. , Cheng, L. , … Hendrix, A. , EV‐TRACK Consortium . (2017). EV‐TRACK: Transparent reporting and centralizing knowledge in extracellular vesicle research. Nature methods, 14(3), 228–232. [DOI] [PubMed] [Google Scholar]
- van Niel, G. , D'Angelo, G. , & Raposo, G. (2018). Shedding light on the cell biology of extracellular vesicles. Nature reviews. Molecular cell biology, 19(4), 213–228. [DOI] [PubMed] [Google Scholar]
- Visnovitz, T. , Osteikoetxea, X. , Sódar, B. W. , Mihály, J. , Lőrincz, P. , Vukman, K. V. , Tóth, E. Á. , Koncz, A. , Székács, I. , Horváth, R. , Varga, Z. , & Buzás, E. I. (2019). An improved 96 well plate format lipid quantification assay for standardisation of experiments with extracellular vesicles. Journal of extracellular vesicles, 8(1), 1565263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber, J. , & Clayton, A. (2013). How pure are your vesicles? Journal of extracellular vesicles, 2(1), 19861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker, R. , Harris, M. , Cardel, B. , & Kräusslich, H. G. (1998). Virion incorporation of human immunodeficiency virus type 1 Nef is mediated by a bipartite membrane‐targeting signal: Analysis of its role in enhancement of viral infectivity. Journal of virology, 72(11), 8833–8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welker, R. , Kottler, H. , Kalbitzer, H. R. , & Kräusslich, H. G. (1996). Human immunodeficiency virus type 1 Nef protein is incorporated into virus particles and specifically cleaved by the viral proteinase. Virology, 219(1), 228–236. [DOI] [PubMed] [Google Scholar]
- Welsh, J. A. , Goberdhan, D. C. I. , O'Driscoll, L. , Buzas, E. I. , Blenkiron, C. , Bussolati, B. , Cai, H. , Di Vizio, D. , Driedonks, T. A. P. , Erdbrügger, U. , Falcon‐Perez, J. M. , Fu, Q. L. , Hill, A. F. , Lenassi, M. , Lim, S. K. , Mahoney, M. G. , Mohanty, S. , Möller, A. , Nieuwland, R. , … Witwer, K. W. (2024). Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. Journal of extracellular vesicles, 13(2), e12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiest, D. L. , Bhandoola, A. , Punt, J. , Kreibich, G. , McKean, D. , & Singer, A. (1997). Incomplete endoplasmic reticulum (ER) retention in immature thymocytes as revealed by surface expression of “ER‐resident” molecular chaperones. Proceedings of the National Academy of Sciences of the United States of America, 94(5), 1884–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiest, D. L. , Burgess, W. H. , McKean, D. , Kearse, K. P. , & Singer, A. (1995). The molecular chaperone calnexin is expressed on the surface of immature thymocytes in association with clonotype‐independent CD3 complexes. The EMBO journal, 14(14), 3425–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilen, C. B. , Tilton, J. C. , & Doms, R. W. (2012). HIV: Cell binding and entry. Cold Spring Harbor perspectives in medicine, 2(8), a006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk, T. , Gross, I. , Gowen, B. E. , Rutten, T. , de Haas, F. , Welker, R. , Kräusslich, H. G. , Boulanger, P. , & Fuller, S. D. (2001). Organization of immature human immunodeficiency virus type 1. Journal of virology, 75(2), 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willms, E. , Cabañas, C. , Mäger, I. , Wood, M. J. A. , & Vader, P. (2018). Extracellular vesicle heterogeneity: Subpopulations, isolation techniques, and diverse functions in cancer progression. Frontiers in immunology, 9, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witwer, K. W. , Buzás, E. I. , Bemis, L. T. , Bora, A. , Lässer, C. , Lötvall, J. , Nolte‐’t Hoen, E. N. , Piper, M. G. , Sivaraman, S. , Skog, J. , Théry, C. , Wauben, M. H. , & Hochberg, F. (2013). Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. Journal of extracellular vesicles, May 27(2). 10.3402/jev.v2i0.20360. PMID: 24009894; PMCID: PMC3760646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y. , Beddall, M. H. , & Marsh, J. W. (2007). Rev‐dependent lentiviral expression vector. Retrovirology, 4, 12–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurdinger, T. , Gatson, N. N. , Balaj, L. , Kaur, B. , Breakefield, X. O. , & Pegtel, D. M. (2012). Extracellular vesicles and their convergence with viral pathways. Advances in virology, 2012, 767694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L. , Li, J. , Li, S. , Dang, W. , Xin, S. , Long, S. , Zhang, W. , Cao, P. , & Lu, J. (2021). Extracellular vesicles regulated by viruses and antiviral strategies. Frontiers in cell and developmental biology, 9, 722020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder, A. , Yu, D. , Dong, L. , Iyer, S. R. , Xu, X. , Kelly, J. , Liu, J. , Wang, W. , Vorster, P. J. , Agulto, L. , Stephany, D. A. , Cooper, J. N. , Marsh, J. W. , & Wu, Y. (2008). HIV envelope‐CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell, 134(5), 782–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, F. , Li, Y. M. , & Wang, Z. (2021). Preserving extracellular vesicles for biomedical applications: Consideration of storage stability before and after isolation. Drug delivery, 28(1), 1501–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Freitas, D. , Kim, H. S. , Fabijanic, K. , Li, Z. , Chen, H. , Mark, M. T. , Molina, H. , Martin, A. B. , Bojmar, L. , Fang, J. , Rampersaud, S. , Hoshino, A. , Matei, I. , Kenific, C. M. , Nakajima, M. , Mutvei, A. P. , Sansone, P. , Buehring, W. , … Lyden, D. (2018). Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field‐flow fractionation. Nature cell biology, 20(3), 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , & Lyden, D. (2019). Asymmetric‐flow field‐flow fractionation technology for exomere and small extracellular vesicle separation and characterization. Nature protocols, 14(4), 1027–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Higginbotham, J. N. , Jeppesen, D. K. , Yang, Y. P. , Li, W. , McKinley, E. T. , Graves‐Deal, R. , Ping, J. , Britain, C. M. , Dorsett, K. A. , Hartman, C. L. , Ford, D. A. , Allen, R. M. , Vickers, K. C. , Liu, Q. , Franklin, J. L. , Bellis, S. L. , & Coffey, R. J. (2019). Transfer of functional cargo in exomeres. Cell reports, 27(3), 940–954. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Jeppesen, D. K. , Higginbotham, J. N. , Franklin, J. L. , Crowe, J. E., Jr , & Coffey, R. J. (2021). Angiotensin‐converting enzyme 2‐containing small extracellular vesicles and exomeres bind the severe acute respiratory syndrome coronavirus 2 spike protein. Gastroenterology, 160(3), 958–961. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Jeppesen, D. K. , Higginbotham, J. N. , Graves‐Deal, R. , Trinh, V. Q. , Ramirez, M. A. , Sohn, Y. , Neininger, A. C. , Taneja, N. , McKinley, E. T. , Niitsu, H. , Cao, Z. , Evans, R. , Glass, S. E. , Ray, K. C. , Fissell, W. H. , Hill, S. , Rose, K. L. , Huh, W. J. , … Coffey, R. J. (2021). Supermeres are functional extracellular nanoparticles replete with disease biomarkers and therapeutic targets. Nature cell biology, 23(12), 1240–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Liu, Y. , Liu, H. , & Tang, W. H. (2019). Exosomes: Biogenesis, biologic function and clinical potential. Cell & bioscience, 9, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Z. , Wijerathne, H. , Godwin, A. K. , & Soper, S. A. (2021). Isolation and analysis methods of extracellular vesicles (EVs). Extracellular vesicles and circulating nucleic acids, 2, 80–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y. , McNamara, R. P. , & Dittmer, D. P. (2020). Purification methods and the presence of RNA in virus particles and extracellular vesicles. Viruses., 12(9), 917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zijlstra, A. , & Di Vizio, D. (2018). Size matters in nanoscale communication. Nature Cell Biology, 20(3), 228–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolla‐Pazner, S. (2004). Identifying epitopes of HIV‐1 that induce protective antibodies. Nature reviews. Immunology, 4(3), 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information