

Abstract
The toxic side effects of chemotherapy have long limited its efficacy, prompting expensive and long-drawn efforts to develop more targeted cancer therapeutics. An alternative approach to mitigate off-target toxicity is to develop a device that can sequester chemotherapeutic agents from the veins that drain the target organ before they enter systemic circulation. This effectively localizes the chemotherapy to the target organ, minimizing any hazardous side effects. 3D printing is ideal for fabricating these devices, as the geometric control afforded allows us to precisely dictate its hemodynamic performance in vivo. However, the existing materials compatible with 3D printing do not have drug-binding capabilities. Here, we report the stable coating of genomic DNA on a 3D-printed structure for the capture of doxorubicin. Genomic DNA is an effective chemotherapeutic-agent capture material due to the intrinsic DNA-targeting mechanism of action of these drugs. Stable DNA coatings were achieved through a combination of electrostatic interactions and ultraviolet C (UVC, 254 nm) cross-linking. These UVC cross-linked DNA coatings were extremely stable—leaching on average 100 pg of genomic DNA per mm2 of 3D-printed structure over a period of 30 min. In vitro studies of these materials in phosphate buffered saline and human serum demonstrated that they were able to capture, on average, 72 and 60 ng of doxorubicin per mm2 of structure, respectively. The stability and efficacy of these genomic DNA-coated 3D-printed materials represent a significant step forward towards the translation of these devices to clinical applications for the potential improvement of chemotherapy treatment.
Keywords: chemotherapy, genomic DNA, surface coating, 3D printing, stimuli-responsive, drug capture
Graphical Abstract
1. INTRODUCTION
Cancer is a global public health challenge that affects people in every world region, regardless of demographic and socioeconomic background. It is estimated that by 2040, cancer-related deaths will grow to approximately 16 million each year, placing a significant burden on healthcare systems around the world.1 Chemotherapy is one of the most powerful tools that we have to treat cancer today but is limited by its systemic toxicity: chemotherapeutic agents that are not taken up by cancer cells can kill healthy cells instead, which causes debilitating side effects as well as irreversible organ damage2, affecting patient survival.3 Many common chemotherapy drugs have greater efficacy at higher dosages,4 which requires the clinicians to often make the choice between maximizing tumor suppression and avoiding irreversible off-target toxicity. This limits the anticancer efficacy of these existing drugs. Over the past decades, enormous efforts have been made to develop new therapeutic agents that can target cancer cells more precisely while simultaneously minimizing the damage to healthy cells.5 These drug development cycles are extremely lengthy, lasting many years, and are prohibitively expensive, which translates into increasingly costlier drugs.6,7 The associated financial burden on the patients is significant and often causes nonadherence to treatment schedules as well as a reduction in overall quality of life.8 It is thus imperative to go beyond the small-molecule approach and develop new technologies and devices to reduce and/or eliminate off-target toxicity of well-established traditional drugs.
In 2014, Patel and co-workers proposed chemotherapy filtration devices (“ChemoFilters”) that could sequester excess chemotherapeutic agents from the bloodstream prior to them entering systemic circulation. Conceptually, these devices would be deployed in the vein draining the organ undergoing localized intra-arterial chemotherapy, forming a closed system where the drug would be prevented from leaving the organ and causing systemic toxicity. The main use of this ChemoFilter was to treat hepatocellular carcinoma (HCC) in the liver, as depicted schematically in Figure 1a. The liver was a model organ to deploy this device, as procedures like transarterial chemoembolization (TACE) already allowed the localized delivery of a high-dose of chemotherapeutics directly to the tumor, and the hepatic veins and inferior vena cava (IVC) provided direct downstream locations to deploy the ChemoFilter. The first generation of devices featured a simple Nitinol frame attached to a sulfonated ion-exchange membrane that could bind via electrostatic interactions to doxorubicin, a commonly used chemotherapy drug. In a simulated TACE treatment of HCC in swine, the deployment of these devices in the IVC resulted in an 85% reduction in the concentration of doxorubicin in the blood postfilter, demonstrating in vivo proof-of-concept of chemotherapy filtration.9
Figure 1.

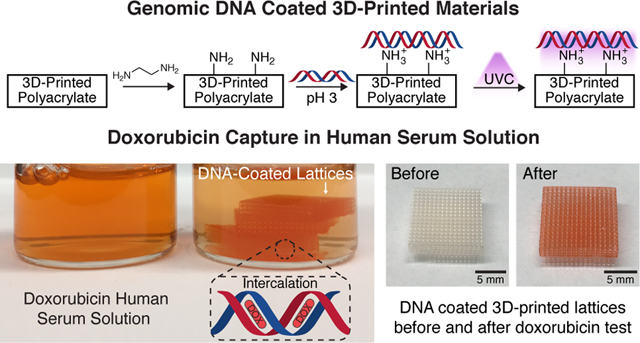
ChemoFilter concept and material design. (a) Schematic of clinical use of ChemoFilter in the treatment of HCC. Excess chemotherapeutic agents not absorbed by the tumor are captured by the ChemoFilter before they enter systemic circulation. (b) Doxorubicin intercalates into DNA and causes cell death. (c) 3D cubic lattice fabricated via digital light processing printing (left). A scanning electron microscope (SEM) image (top view) of the same cubic lattice (right). (d) Two synthetic approaches to coating 3D-printed polyacrylate materials with DNA. Top row: Coating of DNA followed by UVC cross-linking. Bottom row: Installation of surface amine groups followed by electrostatic-interaction assisted DNA coating and then subsequent UVC cross-linking.
Successive generations of ChemoFilter devices in subsequent years have explored different drug-binding materials based on ion exchange,10–12 Schiff base formation,13 or DNA interaction.14,15 DNA-based materials exploit the intrinsic DNA-targeting mechanism of action of many common chemotherapy drugs to capture them. The idea is to introduce external DNA into the body for them to be deliberately attacked by the chemotherapy drugs instead of healthy cell DNA. The specific design of the ChemoFilter structure is critical to its in vivo performance: it has to maximize surface area and interaction time for drug binding while minimizing hemodynamic disturbances, such as flow obstruction and induction of thrombosis. As such, there have been significant ongoing efforts to model the hemodynamic performances of these devices in vivo to determine their optimal geometry.16 However, the limited form factors of the drug-binding materials available today, such as membranes and micro/nanoparticles, render true optimization impractical—membranes are two-dimensional in nature and need to be attached to a support frame if they are to be used in more complex three-dimensional (3D) configurations,9,11 which places an inherent limit on the designs that can be achieved using them.
Similarly, particles need to be immobilized or encapsulated using another material to prevent them from circulating freely outside of their intended location within the body,10,15 restricting the ways that they can be implemented in a device.
Additive manufacturing (AM) offers a potential solution to this problem because it enables the fabrication of materials with arbitrarily complex geometries in a single processing step.17 Computational fluid dynamics models of ChemoFilter devices with different geometries have also suggested that 3D-printed architectures are ideal for this application because they can be easily optimized to have the desired hemodynamic performances.18,19 However, a substantial challenge with AM is that the existing materials compatible with it do not have drug-binding capabilities. Inspired by the coatings of sulfonated block copolymers on 3D-printed devices introduced by Oh et al.,12 we investigated two methodologies to coat genomic DNA onto commercially available 3D-printed polyacrylates for use as an efficient and stable drug capture material. In vitro studies confirm that these materials are able to sequester ∼72 and ∼60 ng of doxorubicin per mm2 of 3D-printed material (ng mm−2) from phosphate buffered saline (PBS) and human serum (HS) respectively and exhibit marginal DNA loss of ∼0.1 ng of DNA per mm2 of 3D-printed material (ng mm−2) over time.
2. RESULTS AND DISCUSSION
2.1. Material Design and Fabrication.
Doxorubicin, one of the most commonly used drugs to treat HCC, causes cytotoxicity by intercalating between the cell DNA base pairs and preventing its replication20,21 (Figure 1b). Surface functionalization of 3D-printed materials with DNA would allow us to exploit doxorubicin’s natural mechanism of action of binding to DNA against itself to sequester it from solution. The use of surface functionalization also allows us to utilize commercially available materials with a wide range of mechanical properties that have already been optimized for AM (Autodesk’s PR48, Formlabs’s Elastic Resin, and etc.) without the need for a lengthy customized photoresin development and optimization process—simplifying the fabrication process. Methods for functionalizing surfaces with DNA are well-established in the literature; they rely on specially designed synthetic DNA sequences whose functional groups can bind to a complementary moiety on the substrate.22–25 The main drawback to using synthetic DNA is its relatively high cost, which renders the fabrication of DNA-ChemoFilters prohibitively expensive and impractical.
An alternative approach is to functionalize the surface with genomic DNA, which is inexpensive and easily obtainable but does not possess reactive end-groups or functional groups that can be used to anchor it to a surface.26 The surface functionalization of genomic DNA remained a challenge until Blumenfeld et al. developed a methodology that utilized surface-bound alkylating agents that could covalently bind to genomic DNA.15 However, alkylation of DNA has been shown to lead to depurination at the alkylation site, effectively severing the bond between the DNA and the substrate.27–29 This is critical as the long-term impact of free-floating genomic DNA in the bloodstream is unclear: some studies indicate that the presence of foreign DNA in the bloodstream is not unusual;30,31 others indicate that it could lead to autoimmune diseases such as lupus.32,33 The possibility of depurination prompted us to consider alternative chemistries for genomic DNA functionalization, as the stability of these surface-DNA adducts is critical for the safe in vivo operation of these devices. Of particular interest was the work by Yamada et al., who showed that ultraviolet C (UVC, 254 nm) irradiation of dried genomic DNA films resulted in the formation of insoluble films that were stable in water for over 24 h at room temperature.34
Inspired by this approach, we first used a digital light processing (DLP) printer (Autodesk Ember) to fabricate 3D cubic lattices out of PR48, an open source acrylate-based photoresin (composition and mechanical properties in the Supporting Information). DLP is an attractive fabrication process because it offers high resolution (∼50 μm) at relatively fast print speeds.35 We chose cubic lattices to exemplify a 3D geometry that is easily fabricated with DLP but is challenging to produce using traditional manufacturing processes such as injection molding and extrusion molding. Each lattice was about 12 mm wide, 2.5 mm tall, and contained 16 × 16 × 3 cubic unit cells, with an opening of ∼500 μm with ∼250 μm wide beams, shown in Figure 1c. The surface area of each lattice was ∼1200 mm2. The dimensions of these lattices were chosen using some general considerations as to how they would be used in vivo: The ChemoFilter is meant to be deployed in the IVC, which has a diameter between 10–20 mm.36 As such, the overall size of the device needs to be within this size range for it to fit well within there. The largest components in blood are white blood cells at approximately 17 μm in size. The openings of the lattice were thus made sufficiently large to ensure that cells could flow through with little difficulty.
We explored two different methods of coating with genomic DNA, each described schematically in Figure 1d. In the first approach, coatings were deposited by soaking untreated lattices in an acidic solution (pH < 4) of salmon sperm genomic DNA (∼2000 base pairs long), followed by drying under vacuum, in a process similar to DNA combing. Under these conditions, the hydrophobic core of DNA is exposed and can interact with hydrophobic substrates, anchoring itself at random points along the strands to the substrate surface. Drying of the DNA solution results in a receding air–water interface, extending and fixing the DNA as the interface passes over it.37,38 The coated lattices were then irradiated with UVC light at an intensity of 47 mW/cm2 for 60 min on each major face of the lattice, soaked in PBS to remove any un-cross-linked DNA, and then irradiated with UVC light again for 60 min on each major face to produce the “DNA-UVC” lattices. Applying this coating procedure using a neutral solution of DNA resulted in a lower percentage of successful coats, with DNA strands often precipitating in fibrous masses within the lattice after drying instead of coating the surface (Figure S2). This is consistent with a previous study that reported that at neutral pH, little DNA denaturation takes place, which limits the amount of DNA combing that can occur on the surface.39
The second approach utilizes electrostatic interactions to facilitate the DNA coating process. DNA possesses negatively charged phosphate groups along its backbone, which can bind to positively charged substrates.39–41 One of the most commonly used cationic moieties is protonated amines, which have been used extensively in the literature to reversibly bind to DNA.42,43 We functionalized the surfaces of the 3D-printed lattices with free amines by treating them with neat ethylenediamine for 1 h—unreacted surface acrylates react with the diamine via a Michael addition to leave free surface amines.44 The presence and number of surface amines were verified and quantified using a colorimetric method based on the azo dye Orange II45 and determined to be 18.5 ± 0.5 nmol mm−2 (Figure S3). The aminated lattices were then coated with DNA in the same way as the first approach—soaked in an acidic DNA solution, dried under vacuum, and then subjected to the same UVC treatment—to produce “eDNA-UVC” coated lattices, where “eDNA” is used to denote electrostatically facilitated DNA coatings. The acidic nature of the DNA solution used here had a secondary function of protonating the surface amines and enabling their electrostatic interactions with the DNA.
2.2. DNA Coating Characterization.
To evaluate the impact of amination on the coating process, we characterized the DNA coated aminated and untreated lattices pre-UVC treatment using energy dispersive X-ray spectroscopy (EDS). Figure 2 shows the electron microscope images from DNA- (left panels) and eDNA-coated (right panels) lattices and their corresponding phosphorus elemental maps.
Figure 2.

Energy dispersive X-ray spectroscopy (EDS) characterization of DNA- (left panels) and eDNA-coated (right panels) lattices. (a) SEM image of a DNA-coated lattice and its phosphorus elemental map that shows inhomogeneous distribution. (b) A zoomed-in SEM image of a horizontal beam, its phosphorus elemental map, and EDS spectrum (bottom). Outlined rectangles in red represent grooves on the beam, rectangles in orange correspond to the ridges on the beam, and rectangles in green mark nodes that contain a vertical beam. (c) SEM image of an eDNA-coated lattice and its elemental map that shows homogeneously distributed phosphorus. (d) A zoomed-in SEM image of a horizontal beam, as well as its EDS spectrum, that shows phosphorus present in the grooves, along the ridges, and on the node.
The presence of phosphorus is indicative of DNA as it is the only possible source of phosphorus at this point in the coating process (Figure S4). EDS analysis of the untreated DNA-coated lattices (Figure 2a,b) indicates that the DNA coating was inhomogeneous. Analysis of the different morphological features, highlighted by the red, orange, and green boxes in Figure 2b, indicates that DNA mostly accumulated in the grooves (red box) on the horizontal beams. These grooves were introduced by the 3D printing process and are inevitable due to the layer-by-layer nature of the fabrication. No DNA was detected on the vertical beams (green box) or on the ridges (orange box) of the horizontal beams. The amount of phosphorus in the highlighted red region was approximately 8.8 at%, similar to that of pure DNA films that were cast on a flat silicon wafer (Figure S5). The selective accumulation of DNA in the grooves of the beams is currently the subject of a future study and is possibly due to contact angle pinning during the evaporation process. As the liquid interface recedes from the outside in, its contact angle is pinned on the grooves of the beams, resulting in deposition of DNA only in these regions.
In contrast, the eDNA-coated lattice had phosphorus distributed homogeneously throughout the structure (Figure 2c,d). The horizontal grooves (red box) showed a similar amount of phosphorus as the DNA-coated sample, at approximately 7.7 at%, with the ridges (orange box) and the vertical beams (green box) having 3.5 and 4.8 at% of phosphorus, respectively. The chemical composition revealed by EDS depends on the interaction volume, which implies that the lower amount of detected phosphorus corresponds to a thinner layer of DNA at these areas. These results indicate that both approaches successfully coated the 3D-printed substrates with DNA, with the amination approach showing markedly more uniform DNA coverage.
2.3. Stability of DNA Coatings.
To fully characterize the stability of the DNA coating, which is critical to the operation of the DNA-ChemoFilter, we invested significant efforts to systematically investigate the amount of DNA leaching into solution. Lattices were first soaked in PBS at 37 °C for 30 min while being vigorously shaken at 200 rpm. They were then removed from solution and blow-dried with air, and soaked again in a fresh solution of PBS under the same conditions. This procedure was repeated two more times, for a total of 4 × 30 min soaks. Ultraviolet–visible (UV–vis) spectroscopy was used to quantify the amount of DNA leached into solution by tracking the characteristic DNA absorbance peak at 260 nm. Figure 3a shows the results of the DNA leaching studies conducted on four different categories of lattices: DNA (Control), eDNA (Control), DNA-UVC, and eDNA-UVC-coated lattices. The control lattices were prepared using the exact same methodologies as their UV-treated counterparts except they were exposed to ambient light in lieu of the two UVC irradiation steps.
Figure 3.

DNA leaching studies conducted on four different categories of DNA-coated lattices. (a) Amount of DNA leached normalized by surface area over multiple PBS soaks, as determined by UV–vis spectroscopy. Data points and error bars correspond to the mean and error-propagated standard deviations of the set (n = 3). Plot on the right contains a zoomed-in section of the original plot, outlined by the green rectangle, which shows that all UVC-treated lattices show a considerable decrease in the amount of leached DNA (<1 ng mm−2). Each individual data point in the set is also shown. Phosphorus maps of the (b) DNA (Control), (c) eDNA (Control), (d) DNA-UVC, and (e) eDNA-UVC coated lattices. Little to no DNA was detected on the DNA (Control) and eDNA (Control) lattices. DNA can clearly be seen on the grooves of the horizontal beams for the DNA-UVC and eDNA-UVC lattices.
The results indicated that both types of control lattices leached a considerable amount of DNA over the four soaks, with the eDNA (Control) lattices leaching a total of 91.0 ± 10.6 ng mm−2 and the DNA (Control) lattices leaching a total of 76.4 ± 14.2 ng mm−2 (all values are mean ± error-propagated standard deviation and refer to the mass of DNA leached per mm2 of 3D-printed lattice; see Supporting Information for details on the relative leachings). The greater amount of leaching from the eDNA (Control) lattices is consistent with its more extensive DNA coverage (Figure 2c,d). The eDNA (Control) lattices leached DNA at a much faster rate than the DNA (Control) lattices, losing 85% of the total leached amount in the first soak, compared with 51% for the latter. This was likely a result of competitive binding of the phosphate anions present in PBS with the surface-functionalized amines,46 driving DNA desorption. By the fourth soak, the eDNA (Control) lattices leached 1.6 ± 0.5 ng mm−2, more than 4× lower than 5.3 ± 0.5 ng mm−2 for the DNA (Control) ones. EDS elemental mapping conducted on both types of control lattices after the leaching experiments (Figure 3b,c) indicates that this was due to the difference in the DNA coatings left on the surface. Phosphorus could only be found on the edges of some horizontal beams of the eDNA (Control) lattices, whereas it could still be clearly seen in some grooves of the horizontal beams in the DNA (Control) lattices.
The UVC-irradiated lattices leached considerably less DNA than their unirradiated controls (Figure 3a), with the eDNA-UVC lattices leaching a total of 5.1 ± 1.1 ng mm−2, approximately half of that of the DNA-UVC lattices, 11.7 ± 1.1 ng mm−2, over the four soaks. This implies that the UVC treatment was successful in producing insoluble cross-linked DNA coatings. It is possible that the additional electrostatic interactions helped enhance the stability of the cross-linked coatings. EDS elemental mapping of the irradiated lattices shows clear evidence of phosphorus remaining on the sample (Figure 3d,e), with the DNA-UVC phosphorus map post-leach resembling that of the as-coated lattices (Figure 2b). The eDNA-UVC phosphorus map showed that DNA was present on all the grooves of the horizontal beams and at the junctions between the horizontal and vertical beams (Figure 3e). There was no DNA on the ridges of the horizontal beams and on the lattice nodes, implying that the thinner coatings at those regions were likely not robust enough to prevent leaching into solution. Only the DNA on the grooves of the horizontal beams was left after the extensive soaking process. That notwithstanding, despite the larger amount of DNA present, the eDNA-UVC and DNA-UVC lattices only leached 0.1 ± 0.2 and 0.5 ± 0.1 ng mm−2 respectively at the end of the leaching experiments. This further highlights the stability of these UVC cross-linked DNA coatings. It is important to emphasize that the nonphysical values obtained from the leaching studies for the eDNA-UVC lattices were due to error propagation in the standard deviation (see Experimental Section/Methods).
The amount of DNA left on the surface of the lattices after the leaching experiments can be estimated by finding the difference between the total amounts of DNA leached from the Control lattices and that from the irradiated lattices. This includes the amount of DNA that was leached during the PBS soak between the UVC irradiations or ambient light exposures. Using this method, the amount of surface DNA on the eDNA-UVC and DNA-UVC lattices was determined to be 175.9 ± 32.8 and 116.8 ± 16.0 ng/mm2, respectively. These values underestimate the actual amount of DNA on the surface, since some DNA can still be detected on the Control lattices at the end of the leaching experiments (Figure 3b,c; see Supporting Information for a more detailed discussion). From the decreasing amount of DNA leached with each soak, we can assume that subsequent soaks in PBS would also result in approximately the same <1 ng amounts of DNA leached. This would represent a subsequent loss of ∼0.1 and ∼0.4% of the DNA present on the eDNA-UVC and DNA-UVC lattices, respectively. These results also indicate that the as-prepared coated materials should be used only after soaking in PBS for at least 30 min, such that the amount of DNA that would be leached into the body during operation is minimized. Although DNA degrades more rapidly in HS as compared to PBS, UVC-cross-linked DNA films have previously been shown to be resistant to nuclease34 and are thus expected to be similarly stable in HS.
As described earlier, the amount of DNA leached is an important metric of success, since the long-term impact of cell-free foreign DNA in the body is still relatively unknown. The evaluation and reduction of DNA leaching is thus crucial to the translation of these DNA-based ChemoFilters from benchtop to clinical application. While a finite amount of DNA was leached from our materials, it is worth noting that cell-free DNA is already present in our bodies, with cancer patients having anywhere between 0 and >1000 ng mL−1 of blood.47 In the future, this might provide us with some bounds as to what is acceptable in the human vascular system.
2.4. In Vitro Evaluation of Drug Capture.
Figure 4 shows the doxorubicin binding capacity of the functionalized lattices tested in PBS and HS at 37 °C. Prior to these tests, the lattices were all subjected to 4 × 30 min PBS soaks at 37 °C, as described in the previous section to minimize the amount of DNA that would leach into solution during the drug-binding process. The mass of doxorubicin bound per square millimeter was used as the figure of merit to quantify and assess the binding efficiencies of our materials. Normalizing the amount of doxorubicin bound by the surface area allows us to estimate the surface area of the device that will be needed to capture a particular amount of doxorubicin and provide us with some design guidelines on the geometries that could be used. Figure 4a shows that the DNA-UVC lattices bound a total of 64.8 ± 10.3 and 48.1 ± 5.4 (mean ± error-propagated standard deviation) ng of doxorubicin mm−2 in 20 min from a 0.05 mg mL−1 solution of doxorubicin in PBS (blue points) and HS (orange points), respectively. This is almost double that of the DNA (Control) lattices, which bound 33.2 ± 6.5 and 23.1 ± 5.7 ng of doxorubicin mm−2 in PBS and HS, respectively. These results are consistent with the greater amount of DNA left on the DNA-UVC lattices compared to the DNA (Control) ones after the PBS soaks (Figure 3b,d). The approximately 30% reduction of doxorubicin binding in HS compared to PBS can be attributed to surface fouling from serum proteins,48 which will reduce the number of accessible DNA sites for doxorubicin binding.
Figure 4.

Doxorubicin capture in PBS and HS. The cumulative amount of doxorubicin bound over 20 min, as determined by fluorescence, normalized by surface area for the (a) DNA-UVC lattices and the associated controls in PBS (blue) and HS (orange) as well as (b) eDNA-UVC lattices and the associated controls in PBS (red) and HS (black). All points and error bars show the mean and error-propagated standard deviations of the set (n = 3). Each individual data point in the set is also shown. (c) DNA-UVC lattices and its controls and (d) eDNA-UVC lattices and its controls after the doxorubicin tests in PBS and HS. (e) Color of the doxorubicin HS solution at various time points during the doxorubicin binding experiments. (f) Comparison of color between the initial doxorubicin HS solution and the solution 20 min after the eDNA-UVC lattices have been introduced. The lattices also turned from white to red.
As an additional control, we also tested lattices that were UVC-treated but not coated in DNA. These UVC-only lattices exhibited very low binding capacities of 7.5 ± 6.1 ng mm−2 in PBS after 20 min, likely due to nonspecific physical adsorption. The extension of the error bar below 0 at the 10 min mark was due to error propagation in determining the standard deviation of the doxorubicin bound (see Experimental Section/Methods). Binding tests were not conducted in HS since the performance of the UVC-only materials in PBS was already poor. The extent of drug capture is easily visible: doxorubicin is a bright red drug, and the DNA-coated lattices are slightly off-white. Figure 4c clearly demonstrates the increasing extent of red staining: DNA-UVC > DNA (Control) > UVC only, which is consistent with the binding capacities observed. For the DNA-treated lattices, the underlying white substrate can still be seen among the red regions, which is due to the inhomogeneous DNA coatings observed without the amination treatment.
The aminated set of lattices showed a different trend: in PBS, the eDNA-UVC and eDNA (Control) lattices both bound a considerable amount of doxorubicin, at 72.4 ± 5.7 and 51.4 ± 4.9 ng of doxorubicin mm−2, respectively (Figure 4b, red points). This was despite the lack of DNA on the surface of the eDNA (Control) lattices (Figure 3c). To understand why the eDNA (Control) lattices were able to bind to doxorubicin, we conducted additional binding experiments using aminated lattices that were only exposed to UVC irradiation without any DNA (amine-UVC). The amine-UVC lattices were able to bind 46.6 ± 6.3 ng of doxorubicin mm−2 in PBS, indicating that the surface amines present on both the eDNA (Control) and amine-UVC lattices were responsible for doxorubicin binding. Visual observations of the three different treatment conditions, as shown in Figure 4d, indicated that the eDNA-UVC lattices turned red after the doxorubicin binding experiments, whereas the amine-UVC and eDNA (Control) coat lattices both turned purple instead. Doxorubicin is known to turn purple either under basic conditions49 or when complexed with transition metal cations.50 Since the doxorubicin tests were conducted in PBS, and the lattices had already been soaked in PBS for an extensive amount of time prior, it is reasonable to assume that the primary amines on the surface are completely protonated. It is thus unlikely that the observed purple color was due to pH changes. Furthermore, doxorubicin degrades rapidly in basic conditions, turning colorless over time.51 However, the purple colors of these lattices persist for months, indicating that the purple color likely did not arise from pH changes. As no transition metal cations were used anywhere in the study, the purple color observed also could not have been from complexation with a transition metal cation. All of these results suggest that the purple color is potentially due to a reaction between the doxorubicin and the protonated surface amines on the eDNA (Control) and amine-UVC lattices. The determination of the identity of the purple complex is outside the scope of this study and is a subject of future research.
In HS, the eDNA-UVC, eDNA (Control), and amine-UVC lattices bound 60.2 ± 6.1, 34.2 ± 5.0, and 27.0 ± 8.5 ng of doxorubicin mm−2, respectively (Figure 4b, black points). The ∼35–40% reduction in doxorubicin binding for the eDNA (Control) and amine-UVC lattices going from PBS to HS can be attributed to increased surface adsorption of serum proteins—most serum proteins are negatively charged at physiological pH, which will bind to the positively charged protonated surface amines via electrostatic interactions52—which would reduce the number of amine sites for doxorubicin binding. This was further corroborated by the reduced purple intensity of the eDNA (Control) and amine-UVC lattices after testing in HS as compared to in PBS (Figure 4d). Thus, while the eDNA (Control) and amine-UVC lattices were effective in binding to doxorubicin in PBS, they were unable to perform as well in HS. The results across both the DNA and eDNA sets of lattices indicated that the eDNA-UVC lattices were the most effective in capturing doxorubicin from both PBS and HS. Visual tracking of the doxorubicin human serum solution after the introduction of the lattices further confirmed this, with the red intensity of doxorubicin decreasing over time (Figure 4e) and a concurrent red staining of the off-white lattices (Figure 4f). The in vivo performance of these materials is currently the subject of a future study.
The coating strategy reported in this work is unique among other previously reported DNA-based materials in that it can be used to make devices with varying form factors. Previous generations of DNA-ChemoFilters utilized free-floating DNA or DNA-functionalized magnetic nanoparticles that required the use of porous mesh packets14 and magnetic rods15 respectively to fix the DNA to the device. These mesh packets and magnetic rods can only be deployed in certain configurations, which are not optimized for hemodynamic flow, rendering them unfeasible for in vivo operation since there is an increased risk of hemodynamic disturbances. In contrast, our coating strategy can be directly used on devices with geometries that have been optimized for hemodynamic flow. While the doxorubicin binding capabilities of the DNA-coated materials reported in this study are relatively lower compared to other DNA-ChemoFilters, this is due to the small amount of DNA used in these coatings (see Supporting Information). There are a few approaches that can be taken to increase the amount of DNA used: The intensity of UVC irradiation could be increased to enhance cross-linking. A higher concentration of the DNA coating solution could be used. Multiple coats of DNA could be used to increase the amount of DNA on the materials. The structure could also be adjusted to improve the coating coverage on them. There are some potential practical challenges associated with some of these approaches: As the geometry of the structure increases in complexity, it could be challenging for the DNA solution to completely infiltrate the structure and coat it completely. Heating the DNA solution to reduce its viscosity or the use of hydrophilic surfaces could help mitigate this and improve DNA penetration. Another potential challenge lies in irradiating the internal surfaces of these complex structures. The use of multiple high-powered UVC lamps or fiber optics could be used to address this issue and allow for complete irradiation of the entire surface. Once these have been investigated and optimized to ensure minimal DNA leaching and processing feasibility, the surface area can then be increased to further improve total binding capacity.
3. CONCLUSION
We have demonstrated a viable approach to fabricating stable genomic DNA-coated 3D-printed polymeric cubic lattices that are capable of capturing doxorubicin from human serum. The coatings were achieved via the simple immersion of aminated 3D cubic lattices into an acidic DNA solution, followed by vacuum drying and UVC irradiation. UVC irradiation was critical in creating stable cross-linked DNA coatings that were resistant to leaching while still capturing doxorubicin from solution. The eDNA-UVC lattices leached 0.1 ± 0.2 ng of DNA mm−2 over 30 min in PBS and captured 60.2 ± 6.1 ng of doxorubicin mm−2 of 3D-printed material in HS over 20 min. There is a lot of potential in utilizing the genomic DNA coating methodologies developed here on other substrates with different mechanical properties. For example, flexible substrates such as Nitinol or elastomers open up the possibility of deployable ChemoFilter devices, minimizing the invasiveness of the insertion procedure. While further development and optimization is needed for the clinical use of these devices, we believe that this work will provide a platform for DNA-based 3D-printed materials for chemotherapy applications that are inexpensive and simple to fabricate. The reduction of off-target toxicity in chemotherapy from a device approach has immense implications and has the potential to improve how we manage cancer. More generally, we hope that this concept of drug capture can be extended to tackle other problems in medicine that have to contend with off-target toxicities.
4. EXPERIMENTAL SECTION/METHODS
4.1. Materials.
Deoxyribonucleic acid sodium salt from salmon testes (DNA) (D1626, approximately 2000 base pairs long, Millipore Sigma), ethylenediamine (≥99%, Millipore Sigma), phosphate buffered saline 1× (PBS, Corning), hypo-opticlear human sera (HS, Sigma-Aldrich), methanol (Millipore Sigma), hydrochloric acid (37%, Millipore Sigma), doxorubicin hydrochloride salt (>99%, LC Laboratories), Orange II sodium salt (>85%, Sigma-Aldrich), sodium hydroxide concentrate (0.1 M, Millipore Sigma), and PR48 (Colorado Photopolymer Solutions) were used as received without further purification. Caution: Doxorubicin is highly toxic and needs to be handled with care.
4.2. Additive Manufacturing via Digital Light Processing Printing.
DLP was performed using a commercially available system (Autodesk Ember). CAD models of the cubic lattices (16 × 16 × 3 cubic unit cells, each with a characteristic opening dimension of approximately 500 μm and beam thickness of about 250 μm) were created with Solidworks and uploaded to the Autodesk Ember 3D printer for fabrication. The photoresin used was PR48. The print parameters are as follows: first layer — 5.0 s wait before exposure, 10.5 s exposure time, 3.0 rpm rotation speed; burn-in layers — 4 layers, 6.0 s wait before exposure, 7.0 s exposure time, 3 rpm rotation speed; model layer — 1.5 s wait before exposure, 5.5 s exposure time, 4 rpm rotation speed. After printing, the lattices were soaked in methanol for 5 h, blow-dried with compressed air, and then followed by a UV postcure (350–380 nm, 36W) (MelodySusie DR 301C) for 3 h. The lattices were then soaked in methanol for 2 × 360 min to leach out as many unreacted small molecules as possible. Finally, the lattices were dried under vacuum (National Appliance Company) at room temperature for 10 h to give the untreated lattices. Plates of 5 × 5 × 0.5 mm were also fabricated using a similar process.
4.3. Amination of PR48 Plates.
The untreated plates were soaked in a neat solution of ethylenediamine for varying lengths of time (15, 30, 60, 120, and 180 min) with gentle shaking at room temperature. The aminated plates were then washed with methanol three times and then soaked in methanol extensively to ensure that any ethylenediamine swollen in the sample is removed. This involved 4 × 30 min soaks in methanol, followed by another 2 × 360 min soaks in methanol. After each soak, the methanol was decanted, and fresh methanol was added. After all the soaks, the plates were dried under vacuum at room temperature for 10 h to give the aminated plates.
4.4. Orange II Assay.
The Orange II dye solution (15 mg/mL) was prepared using deionized water adjusted to pH 3 using hydrochloric acid. The aminated plates were then immersed in 5 mL of the Orange II solution for 30 min at room temperature with gentle shaking. The plates were then removed from solution and then washed 5 times with pH 3 water to remove excess unbound dye before drying under nitrogen. Each plate was then soaked in 10 mL of 0.1 M NaOH solution at 40 °C overnight with gentle shaking. The plate was then removed, and 100 μL of hydrochloric acid was added to the solution. The absorbance of the solution at 480 nm was then measured (Molecular Devices Flexstation 3) and correlated to the concentration of Orange II solution via the use of a calibration curve. By assuming that each Orange II molecule binds to a single surface amine, the surface density of accessible amines can then be determined.
4.5. Amination of PR48 Cubic Lattices.
The untreated lattices were soaked in a neat solution of ethylenediamine for 60 min with gentle shaking at room temperature. The aminated lattices were then washed with methanol three times and then soaked in methanol extensively to ensure that any ethylenediamine swollen in the sample is removed. This involved 4 × 30 min soaks in methanol, followed by another 2 × 360 min soaks in methanol. After each soak, the methanol was decanted and fresh methanol added. After all the soaks, the lattices were dried under vacuum at room temperature for 10 h to give the aminated lattices.
4.6. DNA Coating Procedures.
DNA (25 mg) was first dissolved in 10 mL of pH 3 water (deionized water adjusted to pH 3 with 37% hydrochloric acid) to give an acidic DNA solution of a concentration of 2.5 mg mL−1. Aminated or untreated lattices were then immersed in the acidic DNA solution for 60 min at room temperature, without any external agitation. Each lattice was immersed separately in 3 mL of the acidic DNA coating solution. The lattices were then carefully removed from the solution, placed on a PTFE mesh (0.045 × 0.025″ Opening Size, McMaster-Carr) on a glass Petri dish, and dried under vacuum at room temperature for 180 min. Use of the aminated PR48 lattices gave the eDNA-coated lattices. Use of the untreated PR48 lattices gave the DNA-coated lattices. Lattices were typically prepared in batches of three. Any postcoating procedures were conducted immediately after coating.
4.7. UVC Irradiation Procedures.
UVC irradiation was used to prepare the following lattices: DNA-UVC, eDNA-UVC, Amine-UVC, and UVC only. The UVC irradiation procedure is described in detail for the preparation of the DNA-UVC lattices but is identical for all the other lattices. They only differ in the type of lattices used.
DNA-UVC.
Three DNA-coated lattices were placed on a PTFE mesh on a glass Petri dish. They were then irradiated with 254 nm UVC light (Analytik Jena UVGL-25, 8 mm from lattices) on the major face (16 × 16 unit cells) for 60 min, before being flipped onto the other major face, and irradiated for another 60 min. Following that, the lattices were added to 6 mL of PBS and gently shaken for 30 min at room temperature. The lattices were then briefly rinsed in deionized water and then blow-dried with compressed air. After drying, the lattices were placed on the PTFE mesh again and irradiated with 254 nm UVC light for 60 min on the major face, before being flipped onto the other major face, and irradiated for another 60 min. The total irradiation time was 240 min.
eDNA-UVC.
This is similar to the DNA-UVC irradiation procedure but using the eDNA-coated lattices instead.
Amine-UVC.
This is similar to the DNA-UVC irradiation procedure but using the aminated lattices instead.
UVC Only.
This is similar to the DNA-UVC irradiation procedure but using the untreated lattices instead.
4.8. DNA (Control) and eDNA (Control) Sample Preparation.
DNA (Control).
Three DNA-coated lattices were placed on a PTFE mesh on a glass Petri dish and exposed to ambient light for 60 min. After 60 min, it was flipped and exposed to ambient light for another 60 min. Following that, the lattices were added to 6 mL of PBS and gently shaken for 30 min at room temperature. The lattices were then briefly rinsed in deionized water and then blow-dried with compressed air. After drying, the lattices were placed on the PTFE mesh again and exposed to ambient light for 60 min before being flipped and exposed to ambient light for another 60 min.
eDNA (Control).
Similar to the DNA (Control) sample preparation but using the eDNA-coated lattices instead.
4.9. DNA Leaching Experiments.
The general procedure is outlined as follows: three lattices were soaked in 6 mL of PBS for 30 min at 37 °C and vigorously shaken at 200 rpm. The lattices were then removed and blow-dried with compressed air before being soaked in a fresh solution of PBS (6 mL, 37 °C, 200 rpm) for another 30 min. The process was repeated two more times, for a total of 4 × 30 min soaks. The PBS solutions were all stored for further measurements (DNA-PBS solutions). Following that, the lattices were briefly rinsed with deionized water and then blow-dried with compressed air. Each treatment condition was tested three times (n = 3). That is, in the case for the eDNA-UVC treatment, three separate sets of eDNA-UVC lattices were evaluated for their leaching parameters, with each set of lattices comprising three eDNA-UVC lattices.
4.10. DNA Leaching Quantification.
The DNA–PBS solutions obtained from the DNA leaching experiments were then measured using ultraviolet–visible (UV–vis) spectroscopy to quantify the amount of DNA in solution. UV–vis spectroscopy was performed on an Agilent Cary 60 from 200–400 nm. Baseline measurements using PBS were first established and then used to obtain the spectra from the DNA–PBS solutions. Representative absorption spectra can be found in the Supporting Information (Figure S6). The measured absorbance at 260 nm (Abs260) was then correlated to the concentration of DNA in PBS via the use of a calibration curve (Figure S7). The calibration curve was constructed by determining the Abs260 of PBS solutions with known concentrations of DNA. To ensure that the measurements accurately reflected just the concentration of DNA in solution, the spectra from the UVC-only lattices and the amine-UVC lattices were used as baselines also (Figure S8), i.e., the average Abs260 from the UVC-only lattices was converted to background “DNA” and then subtracted from the average DNA values from the DNA (Control) and DNA-UVC lattices. Similarly, the average Abs260 from the amine-UVC lattices was converted to background “DNA” and then subtracted from the average DNA values from the eDNA (Control) and eDNA-UVC lattices. The error-propagated standard deviation was determined from the standard deviations in the DNA leached from the treated lattices and its associated background “DNA” baseline.
4.11. Doxorubicin Binding Experiments.
The procedure is similar for both the PBS and HS experiments. A 1 mg mL−1 stock solution of doxorubicin in water was first prepared and vortexed to ensure complete dissolution. A 1 mL aliquot of this stock solution was then added to 19 mL of either PBS or HS to give the doxorubicin testing solution of a concentration of 0.05 mg mL−1. A 7.7 mL aliquot of solution was used as the testing volume. A 0.7 mL aliquot of solution was drawn as the initial 0 min time point. The remaining doxorubicin solution (7 mL) was then warmed up to 37 °C before three lattices were added in and then shaken at 200 rpm. Aliquots of 0.7 mL were then drawn at the 10 and 20 min mark. From each 0.7 mL aliquot, 3 × 0.1 mL were drawn and placed in a 96-well microplate. The concentrations of doxorubicin in each well were then measured by way of fluorescence on a microplate reader (Molecular Devices Flexstation 3). The measurement parameters were as follows: λex = 480 nm, λem = 550–590 nm, λcutoff = 530 nm, sensitivity = 100. Each treatment condition was tested three times (n = 3). That is, in the case for the eDNA-UVC treatment, three separate sets of eDNA-UVC lattices were evaluated for their doxorubicin binding capacities, with each set of lattices comprising three eDNA-UVC lattices.
4.12. Doxorubicin Bound Quantification.
The fluorescence intensity at 590 nm was correlated to the concentration of doxorubicin in PBS or HS via the use of a calibration curve (Figures S9 and S10). The calibration curve was constructed by determining the fluorescence intensity at 590 nm of PBS or HS solutions with known concentrations of doxorubicin. The amount of doxorubicin bound by the lattices was calculated by determining the average mass of doxorubicin in solution at each time point and subtracting it from that at t = 0 min for each set of lattices. The average amount of doxorubicin bound by each set of lattices was then averaged over the three sets to give the average mass of doxorubicin bound by the lattices. The error-propagated standard deviation at t = 10 and t = 20 min was determined from the standard deviations in the average amount of doxorubicin bound across the three sets and the error in determining the average amount of doxorubicin bound at each time point from each set of lattices.
4.13. Scanning Electron Microscopy and Energy Dispersive X-ray Spectroscopy.
The lattices were first coated in 10 nm of carbon (Leica EM ACE600) and then imaged in an SEM (Zeiss 1550VP FESEM) equipped with an Oxford X-Max SDD EDS system. A low voltage of 5 kV was used to improve the surface sensitivity of the elemental maps.
Supplementary Material
ACKNOWLEDGMENTS
This is a multidisciplinary effort sponsored by the National Cancer Institute (R01CA194533; principal investigator, Steven W. Hetts) involving investigators at multiple institutions. As such, all participants are part of the ChemoFilter Consortium and should be cited as collaborators. The Consortium’s members include the following individuals and institutions: Steven W. Hetts (University of California, San Francisco [hereafter, UCSF]), Mark W. Wilson (UCSF), Anand Patel (UCSF), Shuvo Roy (UCSF), Henry VanBrocklin (UCSF), Terilyn Moore (UCSF), Carol Stillson (UCSF), Aaron Losey (UCSF), Caroline Jordan (UCSF), Colin Yee (UCSF), Bridget Kilbride (UCSF), Jon Chan (UCSF), Nitash Balsara (University of California, Berkeley), Hee Jeung Oh (University of California, Berkeley), Julia R. Greer (Caltech), Daryl Yee (Caltech), Sankarganesh Krishnamoorthy (Caltech), Carl Blumenfeld (Caltech), Michael Schulz (Virginia Tech), Vitaliy Rayz (Purdue), and Nazanin Maani (Purdue). The authors would also like to acknowledge the following people at Caltech: Chi Ma for assistance with EDS analysis, Anthony Kwong and Prakriti Somani for assistance with carbon coatings, and Sammy Shaker for assistance with the experiments. In particular, the authors would also like to acknowledge Sankarganesh Krishnamoorthy and Jeong Hoon Ko for thoughtful discussions on the work.
Funding
This work was supported by a grant from the National Cancer Institute (R01CA194533). The content is the sole responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.1c05209.
Additive manufacturing details, Orange II assay, DNA coatings characterization (optical and EDS), estimation of DNA of surface, additional DNA leaching quantification details (UV–vis), doxorubicin quantification details (PDF)
Special Issue Paper
This paper missed the Novel Stimuli-Responsive Materials for 3D Printing special issue.
Complete contact information is available at: https://pubs.acs.org/10.1021/acsami.1c05209
The authors declare the following competing financial interest(s): A patent has been filed on this work.
Contributor Information
Daryl W. Yee, Division of Engineering and Applied Science, California Institute of Technology, Pasadena, California 91125, United States
Steven W. Hetts, Department of Radiology and Biomedical Imaging, University of California – San Francisco, San Francisco, California 94107, United States
Julia R. Greer, Division of Engineering and Applied Science, California Institute of Technology, Pasadena, California 91125, United States
REFERENCES
- (1).American Cancer Society. Global Cancer Facts & Figures, 4th ed.; American Cancer Society, 2018. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/global-cancer-facts-and-figures/global-cancer-facts-and-figures-4th-edition.pdf (accessed 2021-04-16). [Google Scholar]
- (2).Yeh E; Bickford C. Cardiovascular Complications of Cancer Therapy: Incidence, Pathogenesis, Diagnosis, and Management. J. Am. Coll. Cardiol 2009, 53, 2231–2247. [DOI] [PubMed] [Google Scholar]
- (3).Cleeland C; Allen J; Roberts S; Brell J; Giralt S; Khakoo A; Kirch R; Kwitkowski V; Liao Z; Skillings J. Reducing the toxicity of cancer therapy: recognizing needs, taking action. Nat. Rev. Clin. Oncol 2012, 9, 471–478. [DOI] [PubMed] [Google Scholar]
- (4).Porrata L; Adjei A. The pharmacologic basis of high dose chemotherapy with haematopoietic stem cell support for solid tumours. Br. J. Cancer 2001, 85, 484–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kroschinsky F; Stölzel F; von Bonin S; Beutel G; Kochanek M; Kiehl M; Schellongowski P. New drugs, new toxicities: severe side effects of modern targeted and immunotherapy of cancer and their management. Crit. Care 2017, 21, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Siddiqui M; Rajkumar S. The high cost of cancer drugs and what we can do about it. Mayo Clin. Proc 2012, 87, 935–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).World Health Organization. Technical report: pricing of cancer medicines and its impacts: a comprehensive technical report for the World Health Assembly Resolution 70.12: operative paragraph 2.9 on pricing approaches and their impacts on availability and affordability of medicines for the prevention and treatment of cancer; World Health Organization, 2018. https://apps.who.int/iris/handle/10665/277190 (accessed 2021-04-16). [Google Scholar]
- (8).Yousuf Zafar S. Financial Toxicity of Cancer Care: It’s Time to Intervene. J. Natl. Cancer Inst 2016, 108, djv370. [DOI] [PubMed] [Google Scholar]
- (9).Patel A; Saeed M; Yee E; Yang J; Lam G; Losey A; Lillaney P; Thorne B; Chin A; Malik S; Wilson M; Chen X; Balsara N; Hetts S. Development and validation of endovascular chemotherapy filter device for removing high-dose doxorubicin: preclinical study. J. Med. Devices 2014, 8, 041008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yee C; McCoy D; Yu J; Losey A; Jordan C; Moore T; Stillson C; Oh H; Kilbride B; Roy S; Patel A; Wilson M; Hetts S. Endovascular Ion Exchange Chemofiltration Device Reduces Off-Target Doxorubicin Exposure in a Hepatic Intra-arterial Chemotherapy Model. Radiology: Imaging Cancer 2019, 1, e190009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chen X; Oh H; Yu J; Yang J; Petzetakis N; Patel A; Hetts S; Balsara N. Block Copolymer Membranes for Efficient Capture of a Chemotherapy Drug. ACS Macro Lett. 2016, 5, 936–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Oh H; Aboian M; Yi M; Maslyn J; Loo W; Jiang X; Parkinson D; Wilson M; Moore T; Yee C; Robbins G; Barth F; DeSimone J; Hetts S; Balsara N. 3D Printed Absorber for Capturing Chemotherapy Drugs before They Spread through the Body. ACS Cent. Sci 2019, 5, 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Krishnamoorthy S; Grubbs R. Aldehyde-Functionalized Magnetic Particles to Capture Off-Target Chemotherapeutic Agents. ACS Omega 2020, 5, 29121–29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Aboian M; Yu JF; Gautam A; Sze C; Yang J; Chan J; Lillaney P; Jordan C; Oh H; Wilson D; Patel A; Wilson M; Hetts S. In vitro clearance of doxorubicin with a DNA-based filtration device designed for intravascular use with intra-arterial chemotherapy. Biomed. Microdevices 2016, 18, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Blumenfeld C; Schulz M; Aboian M; Wilson M; Moore T; Hetts S; Grubbs R. Drug capture materials based on genomic DNA-functionalized magnetic nanoparticles. Nat. Commun 2018, 9, 2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Maani N. CFD MODELING IN DESIGN AND EVALUATION OF AN ENDOVASCULAR CHEMOFILTER DEVICE. Ph.D. Dissertation, Purdue University: West Lafayette, IN, 2019. https://hammer.purdue.edu/articles/thesis/CFD_MODELING_IN_DESIGN_AND_EVALUATION_OF_AN_ENDOVASCULAR_CHEMOFILTER_DEVICE/11303480/1 (accessed 2021-04-16). [Google Scholar]
- (17).Wong K; Hernandez A. A Review of Additive Manufacturing. ISRN Mech. Eng 2012, 2012, 208760. [Google Scholar]
- (18).Maani N; Hetts S; Rayz V. A two-scale approach for CFD modeling of endovascular Chemofilter device. Biomech. Model. Mechanobiol 2018, 17, 1811–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Maani N; Diorio T; Hetts S; Rayz V. Computational modeling of drug transport and mixing in the chemofilter device: enhancing the removal of chemotherapeutics from circulation. Biomech. Model. Mechanobiol 2020, 19, 1865–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Temperini C; Messori L; Orioli P; Bugno C; Animati F; Ughetto G. The crystal structure of the complex between a disaccharide anthracycline and the DNA hexamer d (CGATCG) reveals two different binding sites involving two DNA duplexes. Nucleic Acids Res. 2003, 31, 1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Gnapareddy B; Reddy Dugasani S; Ha T; Paulson B; Hwang T; Kim T; Hoon Kim J; Oh K; Park S. Chemical and Physical Characteristics of Doxorubicin Hydrochloride Drug-Doped Salmon DNA Thin Films. Sci. Rep 2015, 5, 12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Daniel S; Rao T; Rao K; Rani S; Naidu G; Lee H; Kawai T. A review of DNA functionalized/grafted carbon nanotubes and their characterization. Sens. Actuators, B 2007, 122, 672–682. [Google Scholar]
- (23).Escorihuela J; Bañuls M; Puchades R; Maquieira A. DNA microarrays on silicon surfaces through thiol-ene chemistry. Chem. Commun 2012, 48, 2116–2118. [DOI] [PubMed] [Google Scholar]
- (24).Heyries K; Blum L; Marquette C. Direct Poly-(dimethylsiloxane) Surface Functionalization with Vinyl Modified DNA. Chem. Mater 2008, 20, 1251–1253. [Google Scholar]
- (25).Joos B; Kuster H; Cone R. Covalent Attachment of Hybridizable Oligonucleotides to Glass Supports. Anal. Biochem 1997, 247, 96–101. [DOI] [PubMed] [Google Scholar]
- (26).Liu X; Diao H; Nishi N. Applied chemistry of natural DNA. Chem. Soc. Rev 2008, 37, 2745–2757. [DOI] [PubMed] [Google Scholar]
- (27).Gates K. An Overview of Chemical Processes That Damage Cellular DNA: Spontaneous Hydrolysis, Alkylation, and Reactions with Radicals. Chem. Res. Toxicol 2009, 22, 1747–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Osborne M; Wilman D; Lawley P. Alkylation of DNA by the Nitrogen Mustard Bis-(2-chloroethyl) methylamine. Chem. Res. Toxicol 1995, 8, 316–320. [DOI] [PubMed] [Google Scholar]
- (29).Kohn K; Spears C. Stabilization of nitrogen-mustard alkylations and inter-strand crosslinks in DNA by alkali. Biochim. Biophys. Acta, Nucleic Acids Protein Synth. 1967, 145, 734–741. [DOI] [PubMed] [Google Scholar]
- (30).Spisak S; Solymosi N; Ittzes P; Bodor A; Kondor D; Vattay G; Bartak B; Sipos F; Galamb O; Tulassay Z; Szállási Z; Rasmussen S; Sicheritz-Ponten T; Brunak S; Molnár B; Csabai I. Complete Genes May Pass from Food to Human Blood. PLoS One 2013, 8, e69805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Aucamp J; Bronkhorst A; Badenhorst C; Pretorius P. The diverse origins of circulating cell-free DNA in the human body: a critical re-evaluation of the literature. Biological Reviews 2018, 93, 1649–1683. [DOI] [PubMed] [Google Scholar]
- (32).Bai Y; Tong Y; Liu Y; Hu H. Self-dsDNA in the pathogenesis of systemic lupus erythematosus. Clin. Exp. Immunol 2018, 191, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Glenting J; Wessels S. Ensuring safety of DNA vaccines. Microb. Cell Fact 2005, 4, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yamada M; Kato K; Nomizu M; Sakairi N; Ohkawa K; Yamamoto H; Nishi N. Preparation and Characterization of DNA Films Induced by UV Irradiation. Chem. - Eur. J 2002, 8, 1407–1412. [DOI] [PubMed] [Google Scholar]
- (35).Yee D; Greer J. Three-dimensional chemical reactors: in-situ materials synthesis to advance vat photopolymerization. Polym. Int 2021, 70, 964. [Google Scholar]
- (36).Mookadam F; Warsame T; Yang H; Emani U; Appleton C; Raslan S. Effect of positional changes on inferior vena cava size. European Journal of Echocardiography 2011, 12, 322–325. [DOI] [PubMed] [Google Scholar]
- (37).Esmail Nazari Z; Gurevich L. Molecular Combing of DNA: Methods and Applications. J. Self-Assem. Mol. Electron 2013, 1, 125–148. [Google Scholar]
- (38).Bensimon A; Simon A; Chiffaudel A; Croquette V; Heslot F; Bensimon D. Alignment and sensitive detection of DNA by a moving interface. Science 1994, 265, 2096–2098. [DOI] [PubMed] [Google Scholar]
- (39).Allemand J; Bensimon D; Jullien L; Bensimon A; Croquette V. pH-dependent specific binding and combing of DNA. Biophys. J 1997, 73, 2064–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kim H; Bae I; Cho S; Boo J; Lee B; Heo J; Chung I; Hong B. Synthesis and characteristics of NH 2-functionalized polymer films to align and immobilize DNA molecules. Nanoscale Res. Lett 2012, 7, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Jewell C; Lynn D. Surface-mediated delivery of DNA: cationic polymers take charge. Curr. Opin. Colloid Interface Sci. 2008, 13, 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Nakagawa T; Tanaka T; Niwa D; Osaka T; Takeyama H; Matsunaga T. Fabrication of amino silane-coated microchip for DNA extraction from whole blood. J. Biotechnol 2005, 116, 105–111. [DOI] [PubMed] [Google Scholar]
- (43).Nakagawa T; Hashimoto R; Maruyama K; Tanaka T; Takeyama H; Matsunaga T. Capture and release of DNA using aminosilane-modified bacterial magnetic particles for automated detection system of single nucleotide polymorphisms. Biotechnol. Bioeng 2006, 94, 862–868. [DOI] [PubMed] [Google Scholar]
- (44).Farrer R; LaFratta C; Li L; Praino J; Naughton M; Saleh B; Teich M; Fourkas J. Selective Functionalization of 3-D Polymer Microstructures. J. Am. Chem. Soc 2006, 128, 1796–1797. [DOI] [PubMed] [Google Scholar]
- (45).Yee D; Schulz M; Grubbs R; Greer J. Functionalized 3D Architected Materials via Thiol-Michael Addition and Two-Photon Lithography. Adv. Mater 2017, 29, 1605293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Laucirica G; Marmisollé W; Azzaroni O. Dangerous liaisons: anion-induced protonation in phosphate–polyamine interactions and their implications for the charge states of biologically relevant surfaces. Phys. Chem. Chem. Phys 2017, 19, 8612–8620. [DOI] [PubMed] [Google Scholar]
- (47).Kustanovich A; Schwartz R; Peretz T; Grinshpun A. Life and death of circulating cell-free DNA. Cancer Biol. Ther 2019, 20, 1057–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Blaszykowski C; Sheikh S; Thompson M. Surface chemistry to minimize fouling from blood-based fluids. Chem. Soc. Rev 2012, 41, 5599–5612. [DOI] [PubMed] [Google Scholar]
- (49).Cheung B; Sun T; Leenhouts J; Cullis P. Loading of doxorubicin into liposomes by forming Mn2+-drug complexes. Biochim. Biophys. Acta, Biomembr 1998, 1414, 205–216. [DOI] [PubMed] [Google Scholar]
- (50).Abraham S; Edwards K; Karlsson G; MacIntosh S; Mayer LD; McKenzie C; Bally M. Formation of transition metal–doxorubicin complexes inside liposomes. Biochim. Biophys. Acta, Biomembr 2002, 1565, 41–54. [DOI] [PubMed] [Google Scholar]
- (51).Kaushik D; Bansal G. Four new degradation products of doxorubicin: An application of forced degradation study and hyphenated chromatographic techniques. J. Pharm. Anal 2015, 5, 285–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Abstiens K; Maslanka Figueroa S; Gregoritza M; Goepferich AM Interaction of functionalized nanoparticles with serum proteins and its impact on colloidal stability and cargo leaching. Soft Matter 2019, 15, 709–720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.