

ABSTRACT
Immune checkpoint (IC) blockade and adoptive transfer of tumor-specific T-cells (ACT) are two major strategies to treat metastatic melanoma. Their combination can potentiate T-cell activation in the suppressive tumor microenvironment, but the autoimmune adverse effects associated with systemic injection of IC blockers persist with this strategy. ACT of tumor-reactive T-cells defective for IC expression would overcome this issue. For this purpose, PD-1 and TIGIT appear to be relevant candidates, because their co-expression on highly tumor-reactive lymphocytes limits their therapeutic efficacy within the tumor microenvironme,nt. Our study compares the consequences of PDCD1 or TIGIT genetic deletion on anti-tumor properties and T-cell fitness of melanoma-specific T lymphocytes. Transcriptomic analyses revealed down-regulation of cell cycle-related genes in PD-1KO T-cells, consistent with biological observations, whereas proliferative pathways were preserved in TIGITKO T-cells. Functional analyses showed that PD-1KO and TIGITKO T-cells displayed superior antitumor reactivity than their wild-type counterpart in vitro and in a preclinical melanoma model using immunodeficient mice. Interestingly, it appears that TIGITKO T-cells were more effective at inhibiting tumor cell proliferation in vivo, and persist longer within tumors than PD-1KO T-cells, consistent with the absence of impact of TIGIT deletion on T-cell fitness. Taken together, these results suggest that TIGIT deletion, over PD-1 deletion, in melanoma-specific T-cells is a compelling option for future immunotherapeutic strategies.
KEYWORDS: Adoptive cell transfer, CD8+ T cell clones, gene editing, immunotherapy, melanoma, PD-1, TIGIT
Introduction
Therapeutic strategies blocking the interaction between coinhibitory receptors on tumor-infiltrating T-cells and their ligands expressed on tumor cells have proven successful in multiple types of cancers1,2. However, immune checkpoint inhibitor (ICi) therapy typically yields an overall response rate of 20% to 40% in patients with solid tumors,3 and is frequently associated with immune-related adverse events (irAEs), due to the systemic injection of ICi. In order to limit these irAEs, strategies have emerged to directly inhibit IC signaling within the tumor microenvironment (TME). These include the use of genomic editing tools to invalidate IC expression in therapeutic T-cells. This approach is currently evaluated for CAR-T-cell therapy, with the aim to develop therapeutic T-cells with robust resistance to PD-L1 mediated inhibition within the TME.4 In solid tumors, CRISPR-Cas9-based deletion of the PDCD1 gene (coding PD-1) has been recently evaluated in preclinical models involving adoptive cell transfer (ACT) of TCR-engineered T-cells and of tumor-specific cytotoxic T-cells.5,6 This approach has been tested in a phase I clinical trial, on 3 patients with advanced cancers infused with TCR-engineered T-cells specific for NY-ESO-1 and inactivated for endogenous TCR and PD-1 expression.7 This first in human study documented the feasibility and safety of this new therapeutic strategy.
Nonetheless, few studies documented the possible unexpected consequences of such gene deletion on T-cell fitness – which refers to the ability of T-cells to expand, persist and exert effector functions after infusion – a crucial limiting factor for adoptive cell transfer efficacy.8,9 Notably, it has been reported that PDCD1 knock-out had a detrimental effect on the proliferation and persistence of T-cells. Several studies on CAR-T-cells targeting either EGFRvII,10 mesothelin11 or CD1912 reported that disruption of PDCD1 gene led to a slight but significant inhibition of CAR-T-cell expansion. Interestingly, a recent study also documented that conditional deletion of PD-1 in tumor-infiltrating Treg cells impaired their proliferative and suppressive abilities.13 These observations resonate with our results on melanoma-specific PD-1KO effector T-cell clones, that showed enhanced anti-tumor reactivity but decreased proliferation potential.14 It is thus critical to assess the impact of IC genomic deletion on the ability of CD8+ T-lymphocytes to expand, persist, and exert effector functions prior to considering the development of this therapeutic strategy. Whether this feature is specific to PD-1 deletion or could be observed when targeting another IC remains to be explored. Among IC, TIGIT has emerged as another promising target for immunotherapy, co-expressed with PD-1 on highly reactive effector T-cells,15–17 and on memory resident T-cells.18
In this study, we sought to compare the impact of TIGIT or PD-1 deletion in melanoma-specific CD8+ T-lymphocytes, in terms of T-cell fitness, and anti-tumor potential in a pre-clinical model.
Materials and methods
Cell lines and cell culture
Melanoma-specific CD8+ T lymphocytes were cultured in RPMI1640 medium supplemented with 8% human serum, 2 mM L-glutamine, 100 U/mL penicillin, 0.1 mg/mL streptomycin (Gibco) and 150 U/mL human recombinant IL-2 (Proleukin, Novartis Pharma). Polyclonal Melan-A-specific CD8+ T-cells were derived from patient’s PBMC, upon peptide stimulation and sorting with HLA-peptide coated beads.19 T-cell clones were derived from this polyclonal population by limiting dilution, and amplified on feeder cells as previously described.14
The human TAP deficient T2-cell line was purchased from the ATCC (CRL-1992). The melanoma cell line M113, registered in the Biocollection PC-U892-NL (CHU Nantes, France), was established from metastatic tumor fragments in the Unit of Cell Therapy of Nantes. This cell line was stably transfected with human PD-L1 gene (NM_014143.2, Sino Biological, HG10084-UT).14 From the T2 cell line, expressing or not PD-L1,20 we established two additional cell lines, stably transfected with the main TIGIT ligand, CD155 (NM_006505.3, Sino Biological, HG10109-UT). The expression of PD-L1 and CD155 on the cell lines was validated by flow cytometry, with anti-PD-L1 (Clone MIH1, BD Biosciences) and anti-CD155 (Clone SKII.4, BioLegend) monoclonal antibodies. The expression of HLA-A2 and Melan-A on the M113 cell line was also validated by flow cytometry, with anti-HLA-A2 (Clone BB7.2, BD Biosciences) and anti-MelanA (Clone A103, Santa Cruz Biotechnology) monoclonal antibodies.
PDCD1 and TIGIT editing in Melan-A-specific CD8+ T lymphocytes
PDCD1KO Melan-A specific T-cell clones were previously obtained from a polyclonal T-cell population derived from a melanoma patient.14,21 TIGIT editing was performed on this same starting polyclonal T-cell population, through electroporation of ribonucleic complexes with TIGIT sgRNA (0.45 µM final, IDT) (Table S1) and Cas9 protein (0.3 µM final) (IDT) into 106 CD3-activated T-lymphocytes, in 100 µL of serum-free medium (Nepa21 apparatus, Nepagene). After cloning by limiting dilution and amplification, screening and selection of TIGITKO T-cell clones were performed by flow cytometry, with anti-TIGIT antibody (Clone A15153G, BioLegend), after CD3 activation (OKT3 clone, CRL-8001, ATCC). The expression of PD-1, was systematically assessed on TIGITKO T-cell clones, upon co-labeling with anti – PD-1 (Clone EH12, BD Biosciences) antibody.
Sequencing of the edited PDCD1 and TIGIT genes and of TCR genes
The genomic DNA from WT, PD-1KO and TIGITKO T-cell clones was purified using the QIAamp DNA Mini Kit (QIAGEN) from 2 × 106 T-cells. The DNA fragment spanning TIGIT-and PDCD1 editing target sites were amplified by PCR using primer pairs indicated in Table S1. Amplified fragments were sequenced using gene specific primers (Table S1) by Eurofins, after a step of TOPO® TA cloning for T-cell clones exhibiting bi-allelic editing events, according to manufacturer’s instructions (Thermo Scientific).
For TCR sequencing, the total RNA from 2 × 106 T lymphocytes was extracted from antigen-specific T-cell clones using RNeasy Mini Kit (QIAGEN). Potential genomic DNA contamination was eliminated through the RNase-Free DNAse Set (QIAGEN). RNA quantification was determined using a Nanodrop ND-1000 spectrophotometer (Thermo Scientific) and quality was assessed using the Agilent RNA 6000 Nano kit (Agilent Technologies) with a 2100 Bioanalyzer instrument (Agilent Technologies). Reverse transcriptions, PCR amplifications and sequencing were performed as described and we have followed throughout the manuscript the IMGT TCR nomenclature.22–24
Western blot analysis
Whole cell lysates were prepared as previously described from CD3-activated T-cell clones.14 The primary antibodies targeting PD-1, TIGIT (Clones D4W2J and E5Y1W, Cell Signaling Technology) and Actin (Clone ACTN05 (C4), Invitrogen) were incubated on the membranes overnight at 4°C in TBS-T 5% milk. After washing in TBS-T, the membrane was incubated for 1 hr at room temperature with secondary antibodies (HRP-conjugated goat anti-mouse or goat anti-rabbit polyclonal antibodies, Jackson Immunology Research). The revelation was achieved using ClarityTM, Western ECL Substrate (Bio-Rad), and results were visualized via an image acquisition station (Azure 300, Azure Biosystems).
T-cell clone proliferation
Amplification of T-cell clones was monitored by calculating the number of T-cell divisions after 14 days on feeder cells as previously described.19 Proliferation abilities of T-cell clones were assessed upon CFSE (CellTrace™ Invitrogen) labeling, followed by activation with a range of anti-CD3 mAb OKT3 (0–800 ng/µL) for 5 days. Proliferation was analyzed by flow cytometry using the FlowJo software (BD Biosciences) proliferation modeling tool.
Ultraplex RNA-seq library construction
QIAseq UPXome libraries (QIAGEN) were prepared with 10 ng of total human RNA from OKT3 activated T-cell clones (biological triplicates) according to the manufacturer’s handbook. The libraries are made using random hexamers with QIAseq FastSelect – rRNA HMR reagent to block ribosomal RNA. After cDNA synthesis each sample type was pooled and the library was amplified with primers that added unique dual library indices. Libraries were quality controlled for size using Agilent TapeStation High Sensitivity D5000 screentapes and the amount of library was quantitated using ThermoFisher’s Qubit Fluorimeter.
Illumina NGS sequencing and data analysis using RNA-seq Analysis Portal (RAP)
RNA-seq libraries were run on an Illumina NextSeq 500 using 2 × 74 paired end reads with dual 10 base pair unique indexing. The resulting FASTQ files were analyzed on QIAGEN’s GeneGlobe Data Analysis Center using the RNA-seq Analysis Portal (https://www.qiagen.com/us/shop/genes-and-pathways/data-analysis-center-overview-page). RAP is a cloud-based platform that provides primary read alignment and demultiplexed UMI values using CLC Genomics which are then used for differential gene expression and pathway analysis using Ingenuity® Pathway Analysis.
qPCR on differentially expressed genes
To complete the RT2 Profiler PCR Array (QIAGEN) procedure, 100ng of RNA samples were first converted to first-strand cDNA using RT2 First Strand Kit according to the manufacturer’s instructions. Then, the cDNA templates were mixed with the ViiA7 Real-Time PCR System (Thermo Scientific) and ready-to-use RT2 qPCR Master Mixes and then aliquoted into each well of the same plate containing predispensed gene-specific primer sets (QIAGEN). After qPCR, relative expression was determined using ΔΔCt method.
Functional avidity of WT, PD-1KO and TIGITKO T-cell clones
Functional avidity of WT and IC-edited Melan-A-specific T-cell clones was evaluated after co-culture with TAP-deficient T2 cells loaded with a range of Melan-AA27L (ELAGIGILTV) peptide at the effector/target ratio 1/2. CD107a mobilization was measured after 3 hrs of co-culture at 37°C in the presence of mAb specific for CD107a (Clone H4A3, BioLegend). T lymphocytes were then stained with anti-CD8 mAb (Clone RPA-T8, BioLegend) and analyzed by flow cytometry.
IFN-γ production of WT, PD-1KO and TIGITKO T-cell clones
After 12 h of stimulation at 37°C with T2 cells expressing or not PD-L1 and CD155, loaded with 10 µM of the Melan-AA27L peptide, the production of IFN-γ was determined in supernatants by IFN-γ-specific ELISA (Invitrogen) according to the manufacturer’s recommendations. The percentage of inhibition of IFN-γ secretion was calculated as follows: % inhibition= (1- ([IFN-γ] upon activation with IC-expressing T2 cells/[IFN-γ] upon activation with wild type T2 cells)) x 100.
Mouse xenograft model
Six to eight-week-old female NSG mice (Charles River laboratory) with unrestricted access to food and water were kept under specific pathogen-free conditions in the UTE animal facility (SFR Santé, Nantes, license number: D44278). The animal experiments were performed in accordance with the recommendations established by the Ethic Committee for Animal Experiments of Nantes University (APAFIS#35425–2022021413339813v2) and the FELASA (Federation of Laboratory Animal Science Associations). Subcutaneous xenograft tumors were established by injections of 106 PD-L1+/CD155+ human melanoma into the flank of NSG mice. At day seven, tumor-bearing mice were randomly allocated into four groups with homogeneous tumor volumes (Fig. S6A) which received intravenously either: DPBS (n = 9), 5 × 106 WT4 (n = 8), 5 × 106 PD-1KOP6 (n = 9) and 5 × 106 TIGITKOT1 (n = 9) T-cell clones in DPBS. Intravenous injections were repeated twice at days 14 and 21 after engraftment. Tumor burdens were measured by an electronic caliper, and the tumor volume was calculated based on the following formula: volume = Length x Width x Height x π/6.25 Mice were sacrificed taking into account the appearance of necrosis in tumors, weight loss (20% of initial weight) and tumor size (>1000 mm3), in accordance with national and international policies. Tumors were removed at the time of sacrifice, at the end of tumor growth monitoring, with the exception of one tumor per group, removed 30 h after the last T-cell injection.
Immunohistochemistry (IHC) and immunofluorescence (IF)
Tumors were collected, formalin-fixed and paraffin-embedded. IHC was performed on 3 µm paraffin sections of each tumor, using anti-PD-L1, anti-CD155 (clones E1L3N and D8A5G, Cell Signaling technology), and anti Ki67 (clone MIB1, Agilent) primary antibodies, followed by the Peroxidase/DAB Envision detection system (Agilent) on an automated platform (Dako Autostainer). The sections were counterstained with Mayer’s hematoxylin and mounted with xylene-based media. Images for analysis were acquired as whole slide images with a Nanozoomer 2.0 Hamamatsu slide scanner. Ki67+ cells were quantified on whole tumor-tissue sections, with the open source software Qupath, using the positive detection workflow.26
T-cell infiltration was quantified by immunofluorescence staining with anti-hCD3 specific antibody. After blocking with Animal Free Blocker (SP-5030-250 Vector Labs) for 30 min, sections were incubated at room temperature for 60 min with primary rabbit polyclonal antibody against human CD3 (Dako). Sections were then incubated with a secondary antibody (Fab’2 donkey anti-rabbit IgG AF568, Abcam) for 30 min at room temperature. Sections were slightly counterstained 3 min with the Invitrogen NucBlue Live ReadyProbes (Hoechst 33,342), and mounted with Prolong Gold Antifade Reagent (Life Technologies). Image acquisition was performed using the Zeiss Cell Discoverer 7 microscopy workstation, where samples were acquired as whole sections. Analysis of the T-cell infiltrated areas has been achieved using QuPath open-source software,26 and cell segmentation was carried out using the Stardist module.27 The frequency of T-cell infiltration was calculated on the basis of CD3 count divided by the total number of cells.
Results
Production and characterization of TIGITKO Melan-A-specific CD8+ T-cell clones
We knocked-out TIGIT gene in a Melan-A specific CD8+ T-cell population highly expressing PD-1 and TIGIT, previously used to derive PD-1KO T-cell clones14 (Table S1). Using flow cytometry, we selected 5 TIGITKO T-cell clones negative for TIGIT expression (Figure 1(a), middle panel). Recombination events occurred on the two TIGIT alleles (Table S2), with deletion ranging from 1 to 130nt, resulting in a frameshift leading to a premature stop-codon or the deletion of the start codon. Furthermore, we also checked the integrity of PDCD1 gene in TIGITKO T-cells and documented that PDCD1 alleles remained unchanged in TIGITKO T-cell clones (Table S2). Nonetheless, the comparison of PD-1 expression on WT and TIGITKO T-cell clones revealed that PD-1 expression was decreased on all TIGITKO clones, both in terms of percentage of expression and median fluorescence (Figure 1(b)). We further confirmed by Western blot analysis the total absence of TIGIT in TIGITKO T-cell clones, both membrane-bound and cytoplasmic. This analysis also revealed a dramatic decreased expression of PD-1, that was surprisingly almost no longer detectable in 4/5 TIGITKO T-cell clones (Figure 1(c)). As PD-1 membrane expression was still visible, albeit decreased, by flow cytometry, the latter result can probably be explained by the lower sensitivity of western blot compared with flow cytometry.
Figure 1.
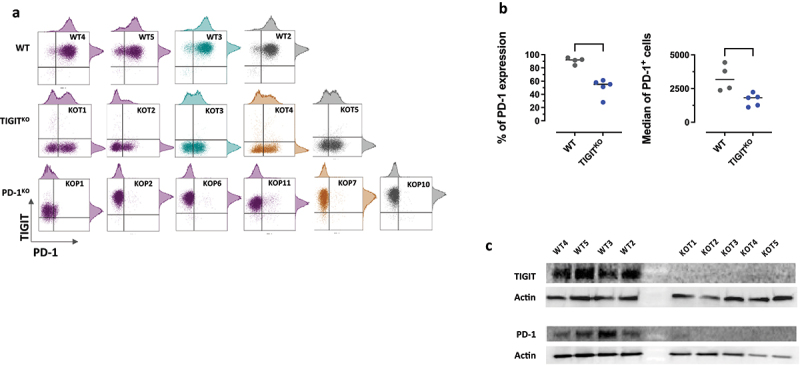
Phenotypic characterization of WT and gene-edited T-cell clones. (a) PD-1 and TIGIT expression was measured by flow cytometry after CD3 activation. Colored groups (purple, blue and yellow) identify T-cell clones sharing the same TCR. Grey T-cell clones express unique TCR. (b) Comparison of the % of PD-1 expression (left) and median fluorescence intensity (right) between WT and TIGITKO T-cell clones (Unpaired T-test, ***untailed p-value <0.001, * <0.05). (c) Western-blot analysis of TIGIT and PD-1 expression on CD3-activated WT and TIGITKO T-cell clones. Actin was used as a loading control.
Maintenance of high levels of the activator receptor CD226, subject to synergistic inhibition by PD-1 and TIGIT and essential for optimal T-cell activation in ACT,28–30 was also validated by flow cytometry on all T-cell clones (Figure S1).
TCR sequencing revealed that 2/5 TIGITKO T-cell clones shared the same TCR as 2 WT and 4 PD-1KO T-cell clones previously described.14 For further experiments, we focused on WT, PD-1KO and TIGITKO T-cell clones expressing the same TCR (underlined in Table S2 and in purple in Figure 1(a)) to overcome the potential differences in T-cell activation linked to the expression of TCR with different affinities.
PD-1KO T-cells exhibit a lower proliferation ability than WT and TIGITKO T-cells
We first evaluated the proliferation abilities of WT (WT4 and WT5), PD-1KO (KOP1, KOP2, KOP6 and KOP11) and TIGITKO T-cell clones (KOT1 and KOT2), sharing the same TCR, by counting the number of divisions that occurred after 14 days of T-cell amplification on irradiated feeder cells. The number of cell divisions made by WT and TIGITKO T-cell clones was similar (about 7 divisions), while the number of divisions of PD-1KO clones was reduced by about half (Figure 2(a)). These results were further confirmed upon CFSE labeling of T-cell clones, followed by a stimulation with a range of anti-CD3 antibody during 5 days (Figure 2(b)). At maximum CD3 stimulation, the frequency of proliferative PD-1KO T-cells (around 28%) was significantly lower than that of WT and TIGITKO T-lymphocytes (p < 0.0001), reaching around 45% (Figure 2(c)). At this point, around 60% of PD-1KO T-cells remained undivided, whereas WT and TIGITKO T-cell clones were able to complete between 1 and 5 divisions (Figure S2). RNA sequencing analysis was further undertaken to compare the impact of genetic deletion of the PDCD1 and TIGIT genes, particularly on the expression of cell cycle-related genes.
Figure 2.
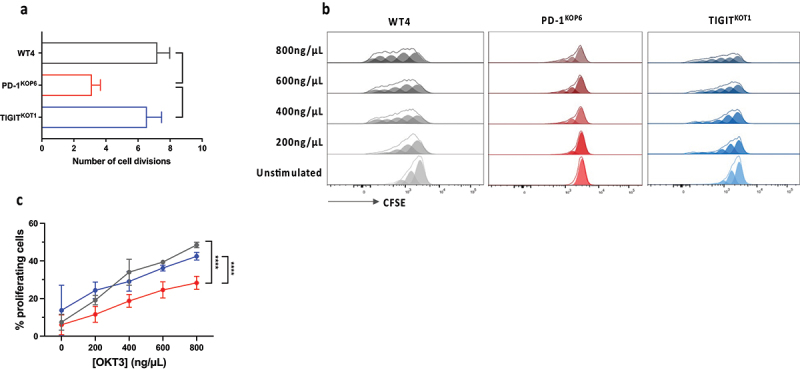
PD-1KO T-cells exhibit a lower proliferation ability than WT and TIGITKO T-cells. (a) Number of divisions of WT4, PD-1KOP6, TIGITKOT1 T-cell clones, after 14 days of amplification on feeder cells (n = 2). Data are represented as mean ± SD. Number of divisions was calculated according to this formula: n = log2(N/N0), where n= number of divisions, N = final number of T-cells and N0=initial number of seeded T-cells (*p < 0.05, One-way ANOVA and Tukey’s multiple comparison test). (b) WT4, PD-1KOP6, TIGITKOT1 T-cell clones were labeled with CFSE (85 nM) and cultivated with a range of anti-CD3 mAb OKT3 (0-800ng/µL). Proliferation was analyzed after 5 days by flow cytometry. (c) Percentage of proliferating T-cells (n = 2), following CD3 activation, represented as mean ± SEM (****p < 0.0001, Two-way ANOVA and Tukey’s multiple comparison test).
PDCD1 knock out has a detrimental impact on gene expression profile of activated T-cells in contrast to TIGIT knock-out
Heatmaps in Figure 3 illustrate differentially expressed genes (DEG) between WT and TIGITKO T-cell clones and WT and PD-1, sharing the same TCR (indicated in purple in Figure 1(a)). Relatively few DEG were found in TIGITKO T-cell clones (8 under-expressed, indicated in blue and 22 over-expressed, indicated in red). In contrast, PD-1 ablation has a more important impact on the number of DEG, with 171 under-expressed and 185 over-expressed, and we found that three pathways, all related to cell cycle and proliferation were significantly inhibited in PD-1KO T-cell clones (Table S3). RNAseq analyses were also conducted on all the T-cell clones, regardless of the TCR expressed (4 WT, 6 PD-1KO and 5 TIGITKO T-cell clones, with biological triplicates) and we confirmed the downmodulation of genes involved in cell cycle in all PD-1KO T-cell clones, supporting the robustness of our results (Figure S3B).
Figure 3.
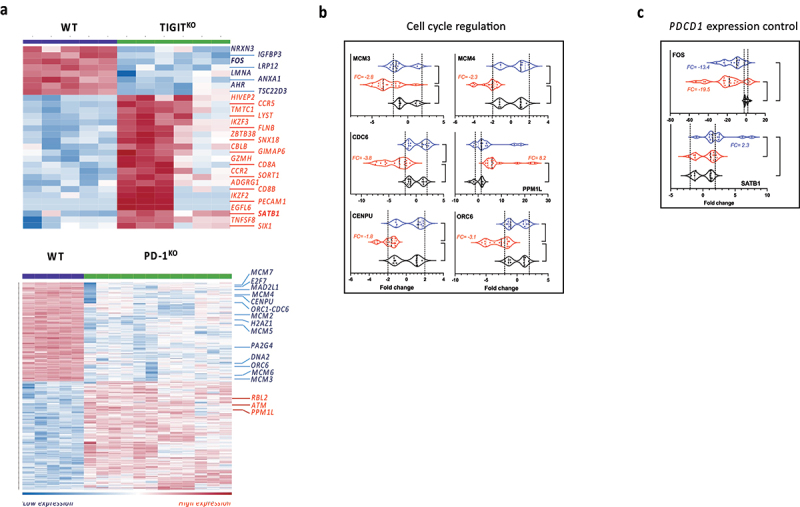
PDCD1 knock out has a detrimental impact on gene expression profile of activated T-cells in contrast to TIGIT knock-out. (a) Heatmap reporting scale expression of up- and down-regulated genes between WT and TIGITKO (upper panel) and WT and PD-1KO T-cell clones (lower panel). RNAseq was performed on CD3-activated T-cell clones sharing the same TCR in biological triplicates (Maximum FDR p-value = 0.001 and minimum absolute fold-change = 2). (b) Expression of cell cycle regulation genes, and (c) expression of genes involved in PDCD1 modulation, measured by RT-qPCR in WT (black, n = 4), PD-1KO (red, n = 6) and TIGITKO (blue, n = 5) activated T-cell clones. qPCR on each gene was performed on all CD3-activated T-cell clones, whatever TCR expressed, in biological triplicates (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, One-way ANOVA and Tukey’s multiple comparison test).
In order to further validate these results, we used qPCR to measure the expression of 6 selected genes (MCM3, MCM4, CDC6, PPM1L, CENPU and ORC6) related to cell cycle (underlined in Table S3), on all these 15 T-cell clones (regardless of the TCR expressed (Figure 3(b)). Results confirmed the down-regulation of MCM3, MCM4, CDC6, CENPU and ORC6, and the overexpression of PPM1L, involved in apoptosis signaling regulation,28 in all 6 PD-1KO T-cell clones, while remaining unchanged in the 5 TIGITKO T-cell clones.
Conversely, very few genes were modulated following TIGIT ablation (Figure 3(a), upper panel). Considering that the most striking feature of TIGITKO T-cell clones was their decreased PD-1 expression, we focused on 12 genes known to control PDCD1 gene expression29 (Table S4). Among these genes, we found that the expression of FOS, that directly activates PDCD1 promotor,30 was dramatically reduced in TIGITKO T-cell clones (Fold change= −115.49 and FDR p-value = 1.05e-4) (Figure 3(a)). Additionally, we also found that SATB1, that down-regulates PDCD1 expression through epigenetic mechanisms31 was significantly overexpressed in TIGITKO T-cell clones (Table S4 and Figure 3(a)). RNAseq and qPCR conducted on all T-cell clones confirmed the down modulation of FOS and the overexpression of SATB1 in all TIGITKO T-cell clones (Figure S3A and Figure 3(b)).
Functional properties of PD-1KO and TIGITKO T-cells are enhanced in response to ligand-expressing target cells
We generated from the HLA-A2 TAP-deficient T2 cell line, 3 T2 cell lines stably expressing PD-L1,20 CD155, or both ligands (Figure 4(a)). We first tested the relative avidity of 3 T-cell clones (WT4, PD-1KOP6 and TIGITKOT1 sharing the same TCR) on the wild-type T2 cell line (PD-L1neg/CD155neg) loaded with a range of Melan-AA27L peptide. As expected, these 3 T-cell clones exhibited a similar functional avidity on peptide-loaded T2 cells, with EC50 around 10−10M, measured through CD107a labeling (Figure 4(b)).
Figure 4.
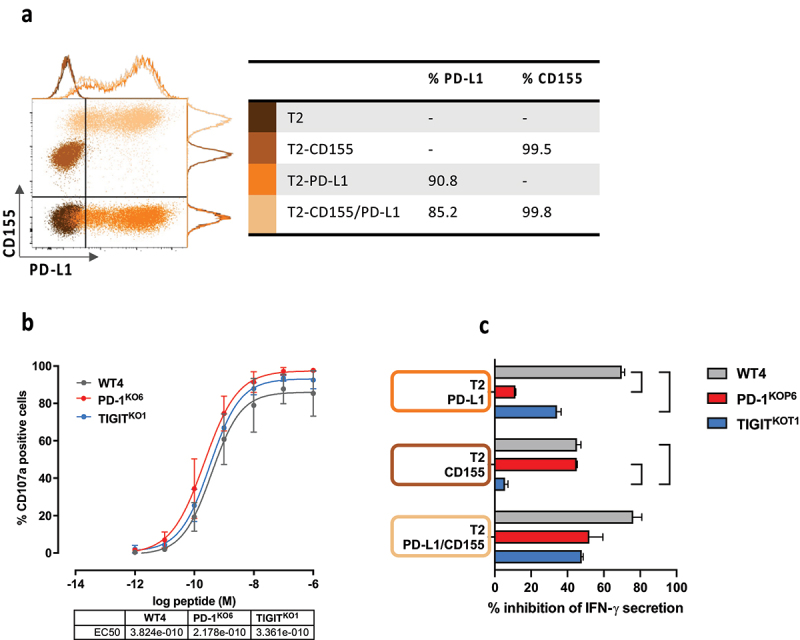
Functional properties of PD-1KO and TIGITKO T-cells are enhanced in response to ligand-expressing target cells.(a) Dot plot and histograms illustrating the expression of PD-L1 and CD155 on WT (dark brown), and CD155pos (light brown), PD-L1pos (orange) and PD-L1pos/CD155pos (yellow) T2 cells. Table illustrates the % of PD-L1 and CD155 expression on each type of T2 cells. (b) Functional avidities of WT4, PD-1KOP6 and TIGITKOT1 T-cell clones were evaluated by measuring CD107a degranulation in response to T2 cells loaded with a range of Melan-A peptide. Table illustrates the EC50 (M) of peptide concentration for each tested T-cell clone (n = 2, data are presented as mean ± SD). (c) WT and PD-L1 and/or CD155 expressing T2 cells were loaded with 100 nM of Melan-A peptide. After 24 h of co-culture with T-cell clones, IFN-γ production was measured by ELISA. % of inhibition was determined according to IFN-γ production in response to WT T2 cells (Data are represented as mean ± SD, *p < 0.05, Two-way ANOVA and Tukey’s multiple comparison test).
The relative inhibition of T-cell activation was further documented for ICKO T-cell clones, by measuring IFN-γ secretion, in response to the 4 types of T2 cell lines loaded with 10−9M of Melan-AA27L peptide. The percentage of inhibition was calculated relative to the amount of IFN-γ secreted after activation with wild-type T2 cell line loaded with peptide. As illustrated by Figure 4(c), WT4 T-cell clone (gray bars) was strongly inhibited by the expression of PD-L1 or CD155 on T2 cells, whereas PD-1KOP6 T-cell clone (red bars) was only inhibited by CD155 expression. As expected, the TIGITKOT1 T-cell clone (blue bars) was not inhibited by CD155 expression on T2 cells, and in accordance with its lower PD-1 expression, it was also less inhibited by PD-L1 expression on T2 cells than WT T-cell clone. Finally, the 3 T-cell clones were inhibited by the co-expression of CD155 and PD-L1 on T2 cells, even if this inhibition seemed less marked for the IC-edited T-cell clones (not significant).
TIGITKO T-cells significantly delayed the growth of melanoma tumors and persist longer within tumors, compared to WT and PD-1KO T-cells.
Anti-tumor efficacy of T-cell clones was assessed through their adoptive transfer in NSG mice, previously engrafted with the M113 melanoma cell line expressing HLA-A2, Melan-A antigen and both PD-L1 and CD155 (M113-CD155+/PD-L1+; Figure S4). The adoptive transfer of WT4 T-cell clone (gray line) was inefficient to delay the growth of melanoma tumors, compared to mice receiving DPBS injections (black line). This result confirmed our previous observations,14 showing that the overexpression of PD-L1 combined with the constitutive expression of CD155 on engrafted tumors, totally impaired the activation of PD-1+/TIGIT+ WT T-cells, while able to delay the growth of PD-L1neg melanoma tumors. In contrast, the adoptive transfer of PD-1KOP6 and TIGITKOT1 T-cell clones (respectively red and blue lines) significantly delayed the growth of melanoma tumors (2way ANOVA followed by Tukey’s comparison test, p < 0.0001) (Figure 5(a) and S5B). Interestingly, the adoptive transfer of TIGITKO T-cells resulted in a significantly better tumor growth control than the adoptive transfer of PD-1KO T-cells (p = 0.035).
Figure 5.
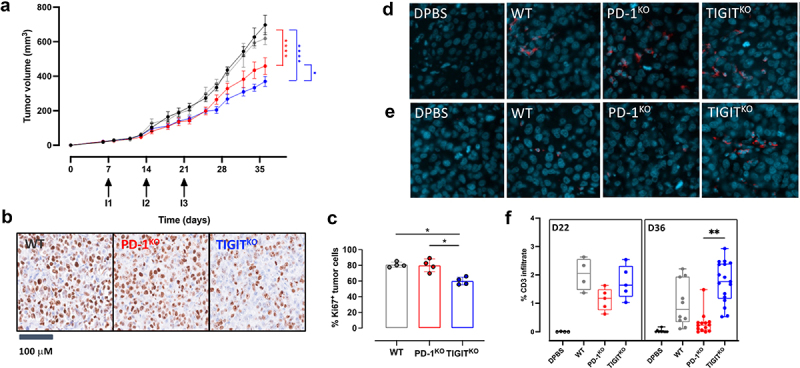
TIGITKO T-cells significantly delayed the growth of melanoma tumors and persist longer within tumors, compared to WT and PD-1KO T-cells. (a) Melanoma tumor (CD155+/PD-L1+) growth curves in NSG mice receiving 3 i.V. injections (Day 7, 14 and 21) of either DPBS (black line), 5 × 106 WT4 (gray line), PD-1KOP6 (red line) or TIGITKOT1 (blue line) melanoma specific T-cell clones (n = 9/group). Tumor volumes were measured from D0 to Day 36. Arrows represent T-cell injections and data are represented as mean ± SEM. (*p < 0.05 ****p < 0.0001, Two-way ANOVA and Tukey’s multiple comparison test). (b) Example of Ki67 staining of tumor cells from each treated group. Counterstaining was done with hematoxylin. (c) Percentages of Ki67+ tumor cells in each treated group (data are represented as mean ± SD; n = 4/group; *p = 0.055; One-way ANOVA and Kruskal-Wallis’ multiple comparison test). (d) T-cell infiltrate revealed by CD3 staining in tumors harvested at Day 22 (30 h after the third injection of T-cells) and (e) at day 36 (2 weeks after the third injection). (f) Percentage of CD3+ T-cells in tumors from each group, 30 hrs (D22, n = 1) and 2 weeks after the third injection of T-cells (D36, n = 3). Immunofluorescence staining was performed on 5 levels of sections/tumor, **p < 0.01; One-way ANOVA and Kruskal-Wallis’ multiple comparison test).
To go further, we investigated melanoma cell proliferation based on Ki67 labeling on 4 tumors from each group receiving T-lymphocytes, harvested either at day 22 (30 h after the third injection of T-cells) or at the day of sacrifice (Day 36, 2 weeks after the third injection). Overall, adoptive transfer of TIGITKO cell clones significantly reduced the fraction of proliferative tumor cells compared with WT (Adjusted p-value = 0.055) and PD-1KO T-cells (Adjusted p-value = 0.055), suggesting superior anti-tumor activity of TIGITKO T-cell clones over WT and PD-1KO T-cell clones (Figure 5(b-c)). In order to investigate whether this decreased proliferation of tumor cells was associated with a better persistence of TIGITKO T-cells, we documented by immunofluorescence the infiltration of infused T-cells in tumors harvested at D22 (30 h after the last injection) and at day 36 (2 weeks after the last injection). We documented a similar infiltration of transferred T-cells (WT or ICKO) 30 h after adoptive cell transfer (D22), clustered in different regions of the tumors with medians between 1.2 and 2% of T-cell infiltration (Figure 5(d), 5(f)). In contrast, 2 weeks after the third injection of T-cells (D36), we could still detect TIGITKO T-cells, with around 1.8% of T-cell infiltration, and to a lower extent WT-T-cells (0.8%), while PD-1KO have almost disappeared from tumors, with only 0.2% of T-cell infiltration detected (Figure 5(e–f)). Differences in infiltration between PD-1KO and TIGITKO T-cells were statistically significant (p < 0.01). In conclusion, the superior tumor growth control observed upon the adoptive transfer of TIGITKO T-cells over PD-1KO T-cells is associated with their enhanced persistence in tumors, consistent with their preserved proliferation abilities.
Discussion
In this study, we confirmed that PD-1 deletion, although beneficial for the anti-tumor function of melanoma-specific T-cell clones, was detrimental for their proliferation abilities. This suggests that this absence of PD-1 expression led to a phenotype similar to that of exhausted T-cells, consistent with a previous study demonstrating, in a mouse model, that the genetic absence of PD-1 led to the accumulation of more exhausted cytotoxic CD8+ T-cells.32 Nonetheless, these T-cell clones lack markers commonly found on exhausted T-cells (CD39, Tim-3) and their effector functions are preserved. Transcriptomic analyses highlighted the down-modulation of several cell cycle pathways in PD-1KO T-cell clones, a finding further confirmed by qPCR analyses. Genes specifically involved in chromosomal replication and DNA synthesis were significantly down-modulated, consistent with the lower proliferation of PD-1KO T-cells. Nevertheless, even if these observations are indisputably correlated, the link between PD-1 editing and decreased expression of these proteins remains to be explored.
The PD-1 signalosome has been recently deciphered and its interactors formally identified.33 In addition to main PD-1 interactors such as SHP-2 and SHP-1, other candidate interactors have been identified, such as MCM3 and BUB3 (a crucial component of the mitotic spindle assembly complex), detected in human T lymphocytes. Thus, a possible indirect functional interaction between PD-1 and such proteins could exist, and might contribute to the regulation of cell cycle. Indeed, the existence of a tonic PD-1 mediated signaling has been reported,34 contributing to impair T-cell functions, that could also have unexpected consequences on intrinsic T-cell properties such as proliferation potential.
In contrast to PD-1, the inhibitory mechanisms resulting from TIGIT binding are still unclear and under debate. Indeed, some studies have documented the recruitment of SHIP-1 to the intracellular portion of TIGIT, notably on NK cells,35,36 while others suggested that the ability of TIGIT to inhibit T-cell functions likely relied on the binding competition between TIGIT and CD226 for their shared CD155 ligand.37 However, a recent study showed that TILs found within renal and lung tumors rarely co-express TIGIT and CD226, suggesting that within certain tumors TIGIT exerts its co-inhibitory function independently of CD226.38 Thus, depending on the TME and the co-expression of TIGIT and CD226 by T lymphocytes, there would be two distinct mechanisms, one involving simple competition with CD226 for CD155 binding, and the other, in the absence of CD226, involving the intracellular domain of TIGIT and implying a negative signaling cascade. In our model, as all T lymphocytes co-express CD226, the inhibitory effect of TIGIT would be mainly caused by competition between the two molecules for CD155. This indirect mechanism of action may explain the lesser impact of deletion of this IC on T-cell fitness.
From a therapeutic standpoint the lower proliferation potential of PD-1KO T lymphocytes raises the question of the relevance of genomic deletion of this IC for cell therapy approaches. A less dramatic decrease in the proliferation of T-cells upon PDCD1 gene disruption has been already reported on CAR-T-cells10–12.It is quite logical that these observations are exacerbated in a clonal system, in which 100% of cells are negative for PD-1 expression, and this effect will undoubtedly be more moderate in polyclonal populations in which a proportion of PD-1+ lymphocytes remains present. Nevertheless, it has been clearly demonstrated that conditional deletion of PD-1 in Treg cell impaired their proliferative capacity,13 supporting its role in T-cell proliferation.
In contrast, TIGIT deletion has no impact on T-cell proliferation, and globally results in very little gene expression modulation. Among the few genes affected are FOS and SATB1, both involved in regulating PD-1 expression, which are modulated in TIGITKO T clones. This result is both intriguing and exciting because simple deletion of TIGIT would decrease PD-1 expression. This suggests that beyond their synergy in inhibiting T-cell activation, PD-1 and TIGIT expression could also be coordinated. This also suggests that although the complete deletion of PD-1 in CD8+ T lymphocytes has a highly detrimental impact on their proliferative capacity, maintaining a basal level of PD-1 expression appears to preserve proliferative functions.
Our results obtained in NSG mice revealed a better tumor growth control upon TIGITKO T-cells injection, even compared to PD-1KO T-cells. This better tumor control is associated with an increased persistence of TIGITKO T-cells within tumors (for up to two weeks), compared to PD-1KO T-cells, consistent with their respective proliferation abilities. Furthermore, tumor cell proliferation, assessed by Ki67 labeling, clearly showed that a lower fraction of tumor cells were able to proliferate upon adoptive transfer of TIGITKO T-cell clones, suggesting an enhanced control of tumor growth, consistent with their superior persistence within TME.
Altogether, our results strongly suggest that the ablation of TIGIT in melanoma-specific T lymphocytes could be a promising option for future therapeutic strategies in metastatic melanoma, as this potentiates their activity in the TME, while preserving their persistence. More generally, this study also underlines the crucial need to carefully document the mechanisms of action of ICs in order to anticipate consequences of their genetic ablation on cellular functions to predict whether deletion of a given IC will be beneficial in cancer immunotherapy.
Supplementary Material
Acknowledgments
We thank the core facilities from UMS BioCore : Cytometry and cell sorting “Cytocell”, cellular and tissular imaging “MicroPICell”, recombinant proteins “P2R” and animal facility “UTE”, for expert technical assistance.
Funding Statement
This work was supported by “Fondation ARC” [Project ARCPGA2021120004323_4875 to BM; ARC PJA3 to NL; fellowship n° 2021030003517 to LM] and the Ligue Nationale contre le Cancer [fellowship to GC and Team certification EL2022.LNCC/NaL to NL] This work was performed in the context of the “LabEx IGO” program [ANR-11-LABX-0016-01].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding authors on reasonable request (nathalie.labarriere@inserm.fr).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2024.2376782
References
- 1.Topalian SL, Drake CG, Pardoll DM.. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–10. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 3.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Razeghian E, Nasution MKM, Rahman HS, Gardanova ZR, Abdelbasset WK, Aravindhan S, Bokov DO, Suksatan W, Nakhaei P, Shariatzadeh S. et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. STEM Cell Res Ther. 2021;12(1):428. doi: 10.1186/s13287-021-02510-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ouchi Y, Patil A, Tamura Y, Nishimasu H, Negishi A, Paul SK, Takemura N, Satoh T, Kimura Y, Kurachi M. et al. Generation of tumor antigen-specific murine CD8+ T cells with enhanced anti-tumor activity via highly efficient CRISPR/Cas9 genome editing. Int Immunol. 2018;30(4):141–154. doi: 10.1093/intimm/dxy006. [DOI] [PubMed] [Google Scholar]
- 6.Chamberlain CA, Bennett EP, Kverneland AH, Svane IM, Donia M, Ö M. Highly efficient PD-1-targeted CRISPR-Cas9 for tumor-infiltrating lymphocyte-based adoptive T cell therapy. Mol Ther - Oncolytics. 2022;24:417–428. doi: 10.1016/j.omto.2022.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F. et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367(6481):eaba7365. doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krug A, Martinez-Turtos A, Verhoeyen E. Importance of T, NK, CAR T and CAR NK cell metabolic fitness for effective anti-cancer therapy: a continuous learning process allowing the optimization of T, NK and CAR-Based anti-cancer therapies. Cancers. 2021;14(1):183. doi: 10.3390/cancers14010183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehta PH, Fiorenza S, Koldej RM, Jaworowski A, Ritchie DS, Quinn KM. T cell fitness and autologous CAR T cell therapy in haematologic malignancy. Front Immunol. 2021;12:780442. doi: 10.3389/fimmu.2021.780442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakazawa T, Natsume A, Nishimura F, Morimoto T, Matsuda R, Nakamura M, Yamada S, Nakagawa I, Motoyama Y, Park Y-S. et al. Effect of CRISPR/Cas9-Mediated PD-1-Disrupted primary human third-generation CAR-T cells targeting EGFRvIII on in vitro human glioblastoma cell growth. Cells. 2020;9(4):998. doi: 10.3390/cells9040998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, Ma X, Wei F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. 2019;68(3):365–377. doi: 10.1007/s00262-018-2281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalinin RS, Ukrainskaya VM, Chumakov SP, Moysenovich AM, Tereshchuk VM, Volkov DV, Pershin DS, Maksimov EG, Zhang H, Maschan MA. et al. Engineered Removal of PD-1 from the Surface of CD19 CAR-T cells results in increased activation and diminished survival. Front Mol Biosci. 2021;8:745286. doi: 10.3389/fmolb.2021.745286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim MJ, Kim K, Park HJ, Kim G-R, Hong KH, Oh JH, Son J, Park DJ, Kim D, Choi J-M. et al. Deletion of PD-1 destabilizes the lineage identity and metabolic fitness of tumor-infiltrating regulatory T cells. Nat Immunol. 2023;24(1):148–161. doi: 10.1038/s41590-022-01373-1. [DOI] [PubMed] [Google Scholar]
- 14.Marotte L, Simon S, Vignard V, Dupre E, Gantier M, Cruard J, Alberge J-B, Hussong M, Deleine C, Heslan J-M. et al. Increased antitumor efficacy of PD-1-deficient melanoma-specific human lymphocytes. J Immunother Cancer. 2020;8(1):e000311. doi: 10.1136/jitc-2019-000311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME. et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon S, Vignard V, Varey E, Parrot T, Knol A-C, Khammari A, Gervois N, Lang F, Dreno B, Labarriere N. Emergence of high-avidity melan-A–Specific clonotypes as a reflection of Anti–PD-1 clinical efficacy. Cancer Res. 2017;77(24):7083–7093. doi: 10.1158/0008-5472.CAN-17-1856. [DOI] [PubMed] [Google Scholar]
- 17.Simon S, Voillet V, Vignard V, Wu Z, Dabrowski C, Jouand N, Beauvais T, Khammari A, Braudeau C, Josien R. et al. PD-1 and TIGIT coexpression identifies a circulating CD8 T cell subset predictive of response to anti-PD-1 therapy. J Immunother Cancer. 2020;8(2):e001631. doi: 10.1136/jitc-2020-001631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edwards J, Tasker A, Pires da Silva I, Quek C, Batten M, Ferguson A, Allen R, Allanson B, Saw RPM, Thompson JF. et al. Prevalence and cellular distribution of novel immune checkpoint targets across longitudinal specimens in treatment-naïve melanoma patients: Implications for Clinical Trials. Clin Cancer Res. 2019;25(11):3247–3258. doi: 10.1158/1078-0432.CCR-18-4011. [DOI] [PubMed] [Google Scholar]
- 19.Labarriere N, Fortun A, Bellec A, Khammari A, Dreno B, Saïagh S, Lang F. A Full GMP Process to select and amplify epitope-Specific T Lymphocytes for adoptive immunotherapy of metastatic melanoma. Clin Dev Immunol. 2013;2013:932318. doi: 10.1155/2013/932318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simon S, Vignard V, Florenceau L, Dreno B, Khammari A, Lang F, Labarriere N. PD-1 expression conditions T cell avidity within an antigen-specific repertoire. Oncoimmunology. 2015;5(1):e1104448. doi: 10.1080/2162402X.2015.1104448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dréno B, Khammari A, Fortun A, Vignard V, Saiagh S, Beauvais T, Jouand N, Bercegay S, Simon S, Lang F. et al. Phase I/II clinical trial of adoptive cell transfer of sorted specific T cells for metastatic melanoma patients. Cancer Immunol Immunother. 2021;70(10):3015–3030. doi: 10.1007/s00262-021-02961-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lefranc MP, Giudicelli V, Busin C, Malik A, Mougenot I, Déhais P, Chaume D. LIGM-DB/IMGT: an integrated database of ig and TcR, Part of the immunogenetics database a. Ann N Y Acad Sci. 1995;764(1):47–47. doi: 10.1111/j.1749-6632.1995.tb55805.x. [DOI] [PubMed] [Google Scholar]
- 23.Dietrich P-Y, Le Gal F-A, Dutoit V, Pittet MJ, Trautman L, Zippelius A, Cognet I, Widmer V, Walker PR, Michielin O. et al. Prevalent role of TCR alpha-chain in the selection of the preimmune repertoire specific for a human tumor-associated self-antigen. J Immunol. 2003;170(10):5103–5109. doi: 10.4049/jimmunol.170.10.5103. [DOI] [PubMed] [Google Scholar]
- 24.Davodeau F, Difilippantonio M, Roldan E, Malissen M, Casanova JL, Couedel C, Morcet JF, Merkenschlager M, Nussenzweig A, Bonneville M. et al. The tight interallelic positional coincidence that distinguishes T-cell receptor Jalpha usage does not result from homologous chromosomal pairing during ValphaJalpha rearrangement. Embo J. 2001;20(17):4717–4729. doi: 10.1093/emboj/20.17.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24(3):148–154. doi: 10.1007/BF00300234. [DOI] [PubMed] [Google Scholar]
- 26.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG. et al. QuPath: Open source software for digital pathology image analysis. Sci Rep. 2017;7(1):16878. doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pécot T, Cuitiño MC, Johnson RH, Timmers C, Leone G. Deep learning tools and modeling to estimate the temporal expression of cell cycle proteins from 2D still images. PLoS Comput Biol. 2022;18(3):e1009949. doi: 10.1371/journal.pcbi.1009949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saito S, Matsui H, Kawano M, Kumagai K, Tomishige N, Hanada K, Echigo S, Tamura S, Kobayashi T. Protein phosphatase 2Cepsilon is an endoplasmic reticulum integral membrane protein that dephosphorylates the ceramide transport protein CERT to enhance its association with organelle membranes. J Biol Chem. 2008;283(10):6584–6593. doi: 10.1074/jbc.M707691200. [DOI] [PubMed] [Google Scholar]
- 29.Bally APR, Austin JW, Boss JM. Genetic and Epigenetic Regulation of PD-1 Expression. J Immunol. 2016;196(6):2431–2437. doi: 10.4049/jimmunol.1502643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao G, Deng A, Liu H, Ge G, Liu X. Activator protein 1 suppresses antitumor T-cell function via the induction of programmed death 1. Proc Natl Acad Sci. 2012;109(38):15419–15424. doi: 10.1073/pnas.1206370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stephen TL, Payne KK, Chaurio RA, Allegrezza MJ, Zhu H, Perez-Sanz J, Perales-Puchalt A, Nguyen JM, Vara-Ailor AE, Eruslanov EB. et al. SATB1 expression governs epigenetic repression of PD-1 in Tumor-Reactive T Cells. Immunity. 2017;46(1):51–64. doi: 10.1016/j.immuni.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med. 2015;212(7):1125–1137. doi: 10.1084/jem.20142237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Celis-Gutierrez J, Blattmann P, Zhai Y, Jarmuzynski N, Ruminski K, Grégoire C, Ounoughene Y, Fiore F, Aebersold R, Roncagalli R. et al. Quantitative interactomics in primary T Cells provides a rationale for concomitant PD-1 and BTLA Coinhibitor blockade in cancer immunotherapy. Cell Rep. 2019;27(11):3315–3330.e7. doi: 10.1016/j.celrep.2019.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernandes RA, Su L, Nishiga Y, Ren J, Bhuiyan AM, Cheng N, Kuo CJ, Picton LK, Ohtsuki S, Majzner RG. et al. Immune receptor inhibition through enforced phosphatase recruitment. Nature. 2020;586(7831):779–784. doi: 10.1038/s41586-020-2851-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li M, X P, D Y, L S, H G, C J, Z H, H N, C X, Z L. et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-γ production of natural killer cells via β-arrestin 2-mediated negative signaling. The J Biol Chem. 2014;289(25):17647–17657. doi: 10.1074/jbc.M114.572420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu S, Zhang H, Li M, Hu D, Li C, Ge B, Jin B, Fan Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013;20(3):456–464. doi: 10.1038/cdd.2012.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banta KL, Xu X, Chitre AS, Au-Yeung A, Takahashi C, O’Gorman WE, Wu TD, Mittman S, Cubas R, Comps-Agrar L. et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8+ T cell responses. Immunity. 2022;55(3):512–526.e9. doi: 10.1016/j.immuni.2022.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Worboys JD, Vowell KN, Hare RK, Ambrose AR, Bertuzzi M, Conner MA, Patel FP, Zammit WH, Gali-Moya J, Hazime KS. et al. TIGIT can inhibit T cell activation via ligation-induced nanoclusters, independent of CD226 co-stimulation. Nat Commun. 2023;14(1):5016. doi: 10.1038/s41467-023-40755-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding authors on reasonable request (nathalie.labarriere@inserm.fr).