

Abstract
Retinoblastoma is the most common pediatric ocular malignancy. It is triggered by a biallelic mutation in the RB1 gene or MYCN oncogene amplification. Retinoblastomas can be unilateral (60%–70%) or bilateral (30%–40%); bilateral tumors are always heritable and present at an earlier age as compared to unilateral ones (18–24 months vs. 36 months in India). High prevalence rates, delayed presentation, and inaccessibility to healthcare lead to worse outcomes in developing countries. The past few decades have seen a paradigm change in the treatment of retinoblastomas, shifting from enucleation and external beam radiotherapy to less aggressive modalities for eye salvage. Multimodality treatment is now the standard of care and includes intraarterial or intravenous chemotherapy along with focal consolidation therapies such as transpupillary thermotherapy, cryotherapy, and laser photocoagulation. Intravitreal and intracameral chemotherapy can help in controlling intraocular seeds. Advanced extraocular or metastatic tumors still have a poor prognosis. Genetic testing, counseling, and screening of at-risk family members must be incorporated as essential parts of management. A better understanding of the genetics and molecular basis of retinoblastoma has opened up the path for potential targeted therapy in the future. Novel recent advances such as liquid biopsy, prenatal diagnosis, prognostic biomarkers, tylectomy, and chemoplaque point to promising future directions.
Keywords: Genetics, intra-arterial chemotherapy, intravenous chemotherapy, liquid biopsy, recent advances, retinoblastoma, review, update
Retinoblastoma is the most common primary intraocular cancer in children, comprising 2.5%–4% of all pediatric malignancies.[1] It is an aggressive, debilitating, and lethal cancer; timely institution of a multimodality management approach can not only save the life but also save the eye in many patients. In low- and middle-income countries (LMIC), delayed presentation with advanced disease stage limits the use of conservative treatment options and results in considerable fatality.
This article provides a comprehensive updated overview of retinoblastoma with an emphasis on genetics and management, highlighting the recent advances. PubMed was searched using the keywords “retinoblastoma,” “genetics,” “grouping,” “treatment,” “chemoreduction,” “chemotherapy,” “focal therapy,” “radiotherapy,” “orbital retinoblastoma,” “liquid biopsy,” and “recent advances.”
History
Retinoblastoma was described as early as in the 16th century by Pawius, but it was Wardrop, in the early 1800s, who recognized it as originating from the eye and performed the first enucleation for it. The tumor was then known as “fungus hematodes,” and the term retinoblastoma was coined by Verhoeff in 1922.[2]
Epidemiology
The incidence of retinoblastoma is constant across all populations, ranging from one case per 15,000 to 20,000 live births.[3,4,5,6] There is no established geographic, ethnic, or sex predilection, but disease prevalence is higher in Asia and Africa due to the high birth rates in these regions.[3,4] Out of 7000–8000 new annual retinoblastoma cases, half are from Asia (excluding Japan), with India contributing as much as 1500–2000 cases.[5,6] Mortality rates are also staggeringly high in the LMIC, ranging from 20–40% in Latin America and Asia (excluding Japan) to 70% in Africa when compared to rates of 3%–5% in Japan, North America, and Europe.[3,6] This can be attributed to a plethora of socioeconomic factors such as inaccessible healthcare, high treatment costs, inadequate nutrition in pregnancy (predisposing to sporadic mutations), and low maternal education levels, eventually leading to delayed presentation and worse prognosis.[1,4,5] The median age of diagnosis of retinoblastoma in India is 29–33 months (unilateral: 36 months, bilateral: 18–24 months), which is comparable to rates reported from Africa and other Asian countries.[3,7,8,9,10,11] The mean age of diagnosis in Canada and the USA is relatively lower (unilateral: 24–27 months, bilateral: 12–15 months).[3,12]
Biology and genetics
Molecular basis
Retinoblastoma is incited by inactivation of the RB1 gene located on chromosome 13q14. Identified in 1986, RB1 was the first tumor suppressor gene to be discovered.[4] It produces pRB, a cell cycle regulatory protein that, in its hypo-phosphorylated state, inhibits the transition from the G1 to S phase of the cell cycle by blocking E2F transcription factors. The lack of functional pRB in RB1 mutation results in uncontrolled cell division.[4,13,14] Various E2F-independent pathways of pRB are also thought to play a role in retinoblastoma suppression, but these are not yet clearly understood.[13,14]
Cell of origin
Recent evidence suggests that retinoblastoma originates from cone photoreceptor precursor cells in the retina.[13] It has been postulated that the cone cell line is more susceptible to pRB deficiency due to a lack of compensatory mechanisms, leading to tumorigenesis.[15] The localization of tiny intraretinal RB in infants to the inner nuclear layer (INL) using OCT indicates that cone precursors lose their ability to migrate to the outer retina after RB1 mutation and remain in the INL.[13,16,17]
Gene mutations
There has been a recent overhaul to the traditional belief that the biallelic mutation of the RB1 gene (RB1-/-) triggers all retinoblastomas. RB1-/- alone can only cause tumor initiation but is inadequate for tumor progression, as evidenced by RB1-/- retinomas, the benign counterpart or premalignant precursor of retinoblastomas.[13,14,16] Further genetic or epigenetic changes (SYK expression) and mutations (M3 to Mn) are needed along with RB1-/- to drive retinoblastoma formation.[13,14] Analysis of somatic copy number alterations (SCNA) has identified additional genes such as CRB1, NEK7, SOX4, and NUP205 as potential driver genes.[18] The mutations in the RB1 gene usually arise de novo and can be point mutations, deletions, or promoter methylations.[13,16]
A small subset of retinoblastomas interestingly does not possess any RB1 mutations (RB1+/+). Some of them have high-level MYCN oncogene amplification (MYCNA). RB1+/+MYCNA tumors comprise 1.4% of unilateral retinoblastomas and have separate implications for the patient.[19] They are nonhereditary and have no risks for contralateral retinoblastoma, familial transmission, or other secondary cancers.[14] In yet others, both RB and MYCN genes are normal; these have been attributed possibly to 13q chromothripsis.[20,21]
Inheritance patterns and genetic subtypes
Knudson's groundbreaking two-hit hypothesis (1971) proposed that two consecutive mutational events (M1, M2) are required for retinoblastoma formation.[5,13,14] M1 (first hit) is the mutation of the first RB1 allele, while M2 (second hit) is the loss of the remaining normal allele with duplication of the mutated M1 allele. This biallelic mutation is known as loss of heterozygosity.[16] This theory forms the basis of the genetic subtypes of retinoblastoma.
Heritable retinoblastomas comprise 30%–40% of all retinoblastomas.[5] In these tumors, M1 is a germline mutation affecting all cells in the body, predisposing the patient to not only retinoblastoma but also other secondary non-ocular cancers (osteosarcoma, leiomyosarcoma, melanoma), especially when the child is treated with external beam radiation (EBRT).[5,16] M2 is a somatic mutation occurring in multiple retinal cells, predisposing to bilateral (80%) and multifocal (67%) retinoblastomas with an earlier age of onset.[4,5,14] Less commonly, heritable cases can be unilateral (10%–15%), trilateral (5%), or unifocal (33%).[14,16] Furthermore, 75% of the germline mutations are sporadic and arise de novo, while only 25% are familial and inherited.[1,5,12] De novo germline mutations can be either pre-conceptional or post-conceptional.[16] Pre-conceptional mutations occur during spermatogenesis in the paternal allele and are present in all cells; they have been associated with advanced paternal age.[1,4,16] Post-conceptional mutations are present in only some cells (mosaicism).[16] Heritable tumors have autosomal dominant inheritance, that is, any offspring of a parent with a germline RB1 mutation will have a 50% risk of inheriting the mutation. Penetrance is 90%; 10% are unaffected carriers who do not develop tumors.[14]
Non-heritable retinoblastomas comprise 60%–70% of all retinoblastomas.[5] In most of these tumors, both M1 and M2 are somatic and occur in a single retinal cell, leading to unilateral and unifocal tumors with a later age of onset.[4,5,14] In others, there is somatic amplification of the MYCN oncogene.[16]
13q deletion syndrome is caused by genomic deletion or translocation and is characterized by dysmorphic features, motor impairment, and neurodevelopmental delay.[5,14] The severity of phenotypic presentation depends on the loci involved relative to the 13q32 region.[4]
Clinical features and diagnosis
Age of presentation and laterality
Retinoblastoma is a tumor in young children; two-thirds are diagnosed by 2 years and 95% by 5 years.[1,5] Earlier age of presentation is associated with a higher probability of bilateral involvement.[1] Bilateral cases are heritable (100%) and usually present in the first year of life, whereas unilateral cases are non-heritable (85%–90%) and often present later in the second to third year.[1,5]
Ocular features and examination
Leucocoria or white pupillary reflex, usually noticed incidentally by parents on flash photography, is the most common presentation (60%–80%). Strabismus is the next common presentation and indicates foveal involvement by the tumor.[4,5] In LMIC, patients often present in advanced stages with proptosis, buphthalmos, painful red eye (secondary glaucoma), and decreased vision.
Diagnosis of retinoblastoma is clinical, and biopsy is contraindicated due to the risk of tumor dissemination.[13] Ocular examination under general anesthesia is critical for diagnosis and staging.[22] Anterior segment examination can reveal hyphema or pseudohypopyon (tumor material/exudate) or anterior chamber seeding (ACS) in retinoblastomas that extend anteriorly.[5] Rubeosis iridis can be present in advanced disease. Intraocular pressure (IOP) and corneal diameter should be measured in all patients. Corneal edema and raised IOP are seen in secondary glaucoma.[4] Dilated fundus examination with scleral indentation is necessary. The classic retinoblastoma appears as a discrete yellow-white retinal mass with dilated vessels; there may be associated vitreous hemorrhage (VH), subretinal fluid/seeds, and vitreous seeds.[4,5,22] Seeds can be morphologically classified as dust, clouds, or pearls.[14] Laterality, number of tumor foci, size, and location (relative to optic disc and macula) should be noted with accurate fundus drawings.[1,22] In newly diagnosed unilateral cases, it is important to carefully examine the normal contralateral eye at all visits as 1.5%–3% of these cases can reportedly develop a new retinoblastoma lesion in the fellow eye more than a month after diagnosis (metachronous retinoblastoma).[4]
Orbital involvement is common in LMIC and can present with gross fungating mass or a picture similar to orbital cellulitis (sterile inflammation from extensive necrosis of large tumor mass).[5] It is worth noting that although orbital cellulitis is associated with high-risk pathologic features (HRF), it does not automatically imply the presence of extraocular extension; the tumor may still be confined within the eye.[23,24] Orbital retinoblastomas have been classified into primary (detected at presentation), secondary (in socket post-enucleation), accidental (iatrogenic surgical perforation), overt (previously unrecognized episcleral nodule or optic nerve involvement noted during enucleation), and microscopic (on histopathology post-enucleation).[25]
Growth patterns
All retinoblastomas are of intraretinal origin but can progress differently with growth. Exophytic tumors grow away from the retina, causing subretinal seeding and retinal detachment (RD). Endophytic tumors grow into the vitreous, causing vitreous seeding. Others can exhibit a combined pattern.
Atypical patterns include a rare diffuse infiltrating type that lacks a discrete mass and grows slowly with flat retinal infiltration. It has a male preponderance with a later age of presentation and is predictive of the presence of pathological HRF.[5,24,26] A diffuse anterior variant has also been reported that usually mimics uveitis with clinical signs of anterior chamber reaction, keratic precipitates, pseudohypopyon, and raised IOP; fundus examination may be normal due to the peripheral location of the tumor.[27] Other patterns include cavitary retinoblastoma with intratumoral cavities or pseudocysts.[28] This low-grade variant does not regress completely after chemotherapy or radiation, maintaining stable dimensions, but has a low risk of recurrence.[29]
Differential diagnoses
The common differentials for retinoblastoma include other causes of leukocoria such as Coats disease, persistent fetal vasculature (PFV), ocular toxocariasis, familial exudative vitreoretinopathy, congenital cataract, coloboma, stage V retinopathy of prematurity, endogenous endophthalmitis, organized VH, and RD. Some tumors such as astrocytic hamartoma, juvenile xanthogranuloma, and medulloepithelioma can also simulate retinoblastoma.[4,5]
Systemic features
Patients with 13q deletion have microcephaly, motor impairment, intellectual disability, and facial dysmorphic features such as thick anteverted ear lobes, telecanthus, hypertelorism, depressed nasal bridge, high broad forehead, and prominent philtrum.[1,4]
Primitive neuroectodermal tumors (PNET) are found in the brain in less than 10% of bilateral cases. These are termed “trilateral” retinoblastoma and usually involve the pineal region (pinealoblastoma) in 75% and the suprasellar/parasellar region in 25%. “Quadrilateral” cases are rare and involve both pineal and suprasellar areas. Infrequently, pineal cysts may be found in bilateral cases and are thought to represent forme-fruste trilateral tumors.[1]
Metastasis
Metastasis has been reported to occur in 5.8%–11% of retinoblastomas and can be direct via the optic nerve, hematogenous, or lymphatic spread via extrascleral extension (ESE).[30,31] The common sites of metastasis are the CNS, bones (painless palpable masses on craniofacial or long bones), bone marrow, lymph nodes (preauricular, submandibular, cervical), and liver.[5,32]
Pathology
Retinoblastomas are composed of small round blue hyperchromatic cells (neuroblasts) with a high nuclear to cytoplasmic ratio and high mitotic count, arranged in sheets, nests, and trabeculae. There are areas of necrosis (due to the tumor outgrowing its blood supply) and calcification. Flexner–Wintersteiner rosettes (ring of cells surrounding a central lumen), found in 70% of tumors, are characteristic of retinoblastoma. Fleurettes (foci of actual photoreceptor differentiation), seen in 15%–20% of tumors, indicate well-differentiated retinoblastomas. Homer–Wright rosettes (tangle of neural filaments at the center) are non-specific and can also be found in neuroblastoma or medulloblastoma. These should be differentiated from pseudorosettes, which are perivascular cuffs of tumor cells.[33] Tumor differentiation can range from well-differentiated (fleurettes or neuronal differentiation) to moderately differentiated (many rosettes) or poorly differentiated (no fleurettes or rosettes).[34]
Investigations
Ocular imaging
Wide-angle handheld fundus camera imaging is indispensable for photographic documentation and serial monitoring of retinoblastomas. RetCam, a contact lens imaging system with a field of view up to 130°, is commonly used.[35] RetCam has also been used for fluorescein angiography to characterize vascular features such as subclinical iris neovascularization and intrinsic tumor vessels.[36] In infants with a positive family history of retinoblastomas, portable handheld OCT can be used to detect tiny intraretinal tumors that are clinically invisible.[13,17,37] OCT can also prove vital during follow-up to evaluate foveal status, post-treatment regression patterns, and scarring and to detect new posterior pole lesions.[38]
Ultrasound B-scan is used to measure (largest basal dimension (LBD) and height) the intraocular tumor (heterogenous, hyperechoic mass arising from the retina) and demonstrate the pathognomonic finding of calcification (high amplitude echoes with acoustic shadowing). It can also detect vitreous seeds, RD, or choroidal thickening and differentiate it from other causes of leukocoria such as PFV.[13,22,39] UBM has been used to accurately define the anterior extent of tumors, detect uveal involvement, and identify angle closure.[34,40] MRI with high-resolution protocol is an extremely valuable modality to delineate extraocular involvement in advanced orbital disease and detect cerebrospinal fluid seeding, intracranial spread, or PNETs.[22,41] It can accurately detect HRF (such as postlaminar ONI) and massive choroidal invasion but is less sensitive to prelaminar ONI, ciliary body, or scleral invasion.[42] In patients with optic nerve enhancement, in the absence of nerve thickening, MRI cannot accurately predict ONI; treatment decisions such as institution of neoadjuvant chemotherapy should not be taken in these cases only based on MRI.[43,44] A recent report suggests that some enhancement patterns such as solid patterns on MRI correlate well with ONI as compared to tram-track or punctate patterns.[45] MRI can also help to distinguish retinoblastoma from other mimickers such as PFV and Coats disease.[46] Retinoblastomas appear hyperintense on T1-weighted images and hypointense on T2-weighted images and show contrast enhancement.[34] It is advisable to limit the use of CT scans due to the risk of secondary cancers from radiation in heritable retinoblastomas. Contrast-enhanced CT can be done in case of unavailability of MRI to detect extraocular extension.[13,22,34]
Systemic screening
Metastatic workup is not done routinely but is reserved for cases with extraocular spread or at least microscopic residual disease (MRD) post-enucleation.[5,47] Bone marrow and cerebrospinal fluid analysis have been recommended in all stage III and IV retinoblastomas.[47] Complete blood count, liver function tests, bone scintigraphy, bone marrow aspiration/biopsy, lumbar puncture, and full-body imaging are required in metastatic cases.[1,5]
Genetic testing
Bilateral cases can almost always be assumed to have a germline RB1 mutation; however, unilateral cases should not be presumed somatic, and 15% of them can possess germline mutation. Therefore genetic testing is ideally indicated in all children diagnosed with retinoblastoma with an unknown RB1 status. Detection of the RB1 mutation can help to identify at-risk children who may develop new tumors in the ipsilateral or contralateral eye, PNETs, or secondary cancers.[14] It is also essential for early tumor detection in predisposed persons, deciding follow-up schedules, genetic counseling, family planning, and reducing unnecessary testing and family screening.
With available modern techniques, almost 95% of RB1 mutations can be detected.[14] Next-generation sequencing can identify up to 75% of all RB1 mutations.[5,16] For the remaining, other methods such as multiplex ligation-dependent probe amplification (MLPA), quantitative multiplex polymerase chain reaction (QM-PCR), or array comparative genomic hybridization (aCGH) are used. Karyotyping or fluorescent in-situ hybridization (FISH) can be used to detect large deletions or chromosomal translocations as in 13q deletion syndrome.[16] Splicing mutations cannot be detected by DNA sequencing either and may need reverse transcriptase-PCR (RT-PCR) or RNA sequence studies. When screening for known mutations, Sanger dideoxy sequencing or allele-specific-PCR (AS-PCR) can be more efficient.[14,16]
These tests require good-quality DNA samples, derived from peripheral blood, saliva, or tumor tissue (fresh/frozen tissue is superior to formalin-fixed/paraffin-embedded). In 97% of bilateral cases, the mutation can be detected solely from leucocyte DNA, while in 3%, tumor DNA is required. In unilateral cases, both are tested: tumor tissue first, followed by blood. If the mutation detected in tumor DNA is also present in blood, it indicates heritable retinoblastoma. If the mutation is absent in blood, it is most likely sporadic, but there can still be a very low risk (<1%) of heritable form due to mosaicism.[5,14,16] If a tumor tissue sample is not available and no mutation is detected in blood, the risk of an unidentified germline mutation is 1%–1.5%. In these patients, the contralateral eye should be screened at follow-up visits up to the age of 5 years and their offspring should also be under surveillance.[14]
Liquid biopsy
Recent cutting-edge research reports that the aqueous humor (AH) of eyes with retinoblastoma contains measurable levels of cell-free DNA (cfDNA) that have presumably diffused to the anterior chamber from necrotic or lytic tumor cells in the posterior segment. Only a small amount of AH (0.1 mL) is sufficient to obtain DNA for genetic analysis. Copy number variation profiling was used to confirm that cfDNA in the AH was indeed derived from the tumor. As biopsy is contraindicated in retinoblastomas, tumor-specific DNA for genetic analysis was only available from enucleated eyes to date. Liquid biopsy in the form of AH sampling overcomes this by providing a minimally invasive and safe method of procuring tumor-specific DNA in eye salvage cases. It is a means of “surrogate tumor biopsy,” which has the potential to revolutionize genomic profiling of retinoblastomas.[48]
Grouping and staging
The Reese–Ellsworth system (1963) classified retinoblastomas based on their predicted response to EBRT[49] [Table 1]. Its relevance is considerably less today as EBRT has been largely replaced with less aggressive management strategies.
Table 1.
Reese–Ellsworth grouping system
| Group: Chances of globe salvage | Characteristics |
|---|---|
| I: Very favorable | a) Solitary tumor, <4 DD, at/behind the equator |
| b) Multiple tumors, none >4 DD, all at/behind the equator | |
| II: Favourable | a) Solitary tumor, <4–10 DD, at/behind the equator |
| b) Multiple tumors, none >4–10 DD, all at/behind the equator | |
| III: Doubtful | a) Any lesion anterior to the equator |
| b) Solitary tumor, >10 DD, behind the equator | |
| IV: Unfavourable | a) Multiple tumors, some >10 DD |
| b) Any lesion anterior to the ora | |
| V: Very unfavourable | a) Massive tumors involving over half the retina |
| b) Vitreous seeding |
DD: disc diameter
The International Intraocular Retinoblastoma Classification (IIRC; Children Hospital of Los Angeles, 2005) and the International Classification of Retinoblastoma (ICRB; Philadelphia, 2006) are two versions of a grouping system that classifies tumors based on their predicted response to systemic chemotherapy[50,51] [Table 2 and Fig. 1]. The two are largely similar but have subtle inconsistencies in the group definitions. The differences in the descriptions of groups B–D are small enough to have no real-world clinical implications. However, it was found that the discrepancies in group E definitions can hypothetically lead to a tumor being categorized as very high risk (E) or low risk (B) depending on the version of classification used. For example, a tumor that occupies >50% of the globe without any destructive features is group E as per ICRB; as per IIRC, it can be group C–D depending on the degree of seeding or group B in the absence of seeds.[52] The lack of uniformity precludes consistent worldwide application in clinical and research scenarios.
Table 2.
International classifications of retinoblastoma
| Groups (risk of treatment failure with IVC) | Features | |
|---|---|---|
|
| ||
| IIRC | ICRB | |
| A (very low) | • Intraretinal tumors ≤3 mm • Located ≥1.5 mm from the optic disc and ≥3 mm from foveola |
Tumors ≤3 mm (LBD and thickness) |
| B (low) | • All intraretinal tumors not in group A • SRF within ≤5 mm of the tumor • No seeds |
• Tumors >3 mm • Tumors within ≤1.5 mm of the optic disc and ≤3 mm of the fovea • Clear SRF ≤3 mm of the tumor |
| C (moderate) | • Focal fine vitreous/subretinal seeds in discrete retinal tumors • SRF extending to ≤1 quadrant |
Subretinal seeds (C1), vitreous seeds (C2) or both (C3); all ≤3 mm from tumor |
| D (high) | • Diffuse vitreous/subretinal seeds and/or • Massive intraocular endophytic/exophytic tumor • SRF extending to >1 quadrant |
Subretinal seeds (D1), vitreous seeds (D2) or both (D3); all >3 mm from tumor |
| E (very high) | Anatomically or functionally destroyed eyes with ≥1 of the following: • Intractable NVG • Massive intraocular hemorrhage • Aseptic orbital cellulitis • Tumor anterior to anterior hyaloid • Tumor touching the lens • Diffuse infiltrating retinoblastoma • Phthisis or pre-phthisis |
• Massive tumor occupying >50% of globe • NVG • Media opacity due to hemorrhage (anterior chamber/vitreous/subretinal) • Invasion of post-laminar optic nerve, choroid (>2 mm), sclera, orbit, anterior chamber |
SRF: Subretinal, NVG: Neovascular glaucoma
Figure 1.
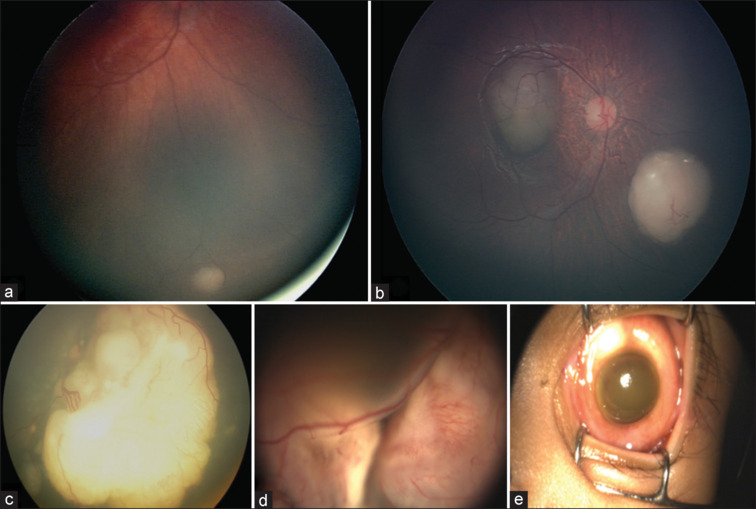
Grouping of retinoblastomas as per the International Classification: (a) Group A; (b) Group B; (c) Group C; (d) Group D; (e) Group E
The International Retinoblastoma Staging System (IRSS) (2006) takes histopathology and metastasis into account to classify advanced tumors based on their survival prognosis [Table 3]. Unlike other classification systems that are reserved for treatment-naïve intraocular retinoblastomas, IRSS can be used for extraocular and previously treated tumors.[53,54]
Table 3.
International staging system for retinoblastoma
| Stage | Features |
|---|---|
| 0 | Patients treated with salvage therapy |
| I | Enucleated eye with no microscopic residual disease |
| II | Enucleated eye with microscopic residual disease |
| III | Extraocular disease with regional spread a) Overt orbital disease b) Lymph node involvement (preauricular or cervical) |
| IV | Distant metastasis a) Hematogenous (without CNS spread) 1. Single lesion 2. Multiple lesions b) CNS (with/without other site) 1. Prechiasmatic lesion 2. CNS mass 3. Leptomeningeal and CSF disease |
The eighth edition of the American Joint Committee on Cancer (AJCC) staging (2017) for retinoblastoma includes the novel “heritability” (H) trait in addition to the classic “tumor, node, metastasis” (TNM) grades, making retinoblastomas the first malignancy where genetic predisposition has been taking into account in cancer staging [Table 4]. Furthermore, the AJCC pathological staging provides a logical progression with clear descriptions for each stage[34] [Table 5]. AJCC cT3c-e can predict the presence of HRF.[24] Survival in advanced intraocular tumors has also been correlated to cT3, with increasing metastatic mortality with increasing subcategories.[55] The AJCC grading is the first of its kind, incorporating both grouping (globe salvage prognosis) and staging (survival prognosis) with evidence-based definitions, and has been purported to soon become the gold standard.[54]
Table 4.
AJCC (8th edition) of retinoblastoma clinical TNMH classification
| cTNMH category | Features |
|---|---|
| cTX | Unknown evidence of intraocular tumor |
| cT0 | No evidence of an intraocular tumor |
| cT1 | Intraocular tumor, SRF ≤5 mm from any tumor a) Tumors ≤3 mm and >1.5 mm away from the optic disc and fovea b) Tumors >3 mm or within 1.5 mm of the optic disc or fovea |
| cT2 | Intraocular tumor with RD, vitreous seeds, or subretinal seeds a) SRF >5 mm from tumor b) Vitreous and/or subretinal seeds |
| cT3 | Advanced intraocular tumor a) Phthisis or pre-phthisis b) Invasion of the choroid, pars plana, ciliary body, zonules, lens, iris, or anterior chamber c) Raised IOP with NV and/or buphthalmos d) Hyphema and/or vitreous hemorrhage e) Aseptic orbital cellulitis |
| cT4 | Extraocular tumor involving the orbit, including the optic nerve a) Radiologic evidence of retrobulbar optic nerve involvement/thickening or orbital tissue involvement b) Clinically evident extraocular disease with proptosis and/or orbital mass |
| cNX | Regional lymph nodes cannot be assessed |
| cN0 | No regional lymph node involvement |
| cN1 | Evidence of involvement of preauricular, submandibular, and cervical lymph nodes |
| cM0 | No clinical features of intracranial or distant metastasis |
| cM1 | Distant metastasis without microscopic confirmation a) Distant site metastasis (bone marrow, liver) on radiologic or clinical tests b) CNS involvement on radiological imaging (excluding trilateral retinoblastoma) |
| pM1 | Distant metastasis with microscopic confirmation a) Pathologic evidence of distant site metastasis (bone marrow, liver, or others) b) Pathologic evidence of CNS parenchyma or CSF involvement |
| HX | Unknown or insufficient evidence of RB1 germline mutation |
| H0 | Normal RB1 alleles on blood test with high-sensitivity assays |
| H1 | Bilateral retinoblastoma, PNETs, family history of retinoblastoma, or molecular definition of a germline RB1 mutation |
Table 5.
AJCC (8th edition) of retinoblastoma pathological classification
| pT category | Definition |
|---|---|
| pTX | Unknown evidence of intraocular tumor |
| pT0 | No evidence of intraocular tumor |
| pT1 | Intraocular tumor without any local invasion, focal choroidal invasion, pre/intra-laminar ONI |
| pT2 | |
| pT3 | Intraocular tumor with local invasion |
| pT4 | a) Concomitant focal choroidal invasion and pre/intra-laminar ONI |
| b) Invasion of iris stroma and/or trabecular meshwork and/or Schlemm's canal | |
| Intraocular tumor with significant local invasion | |
| a) Massive choroidal infiltration (LBD >3 mm, or multiple foci of focal involvement totaling >3 mm, or full thickness choroid invasion) | |
| b) Retrolaminar ONI without involvement of transected end | |
| c) Partial thickness involvement of inner two-thirds of sclera | |
| d) Full thickness involvement of outer one-third of sclera and/or invasion into/around emissary channels | |
| Evidence of extraocular tumor: invasion of transected end of optic nerve or meningeal spaces around nerve, full thickness scleral invasion with involvement of episclera, adjacent fat, bone, extra-ocular muscle, conjunctiva or eyelids |
Treatment
The aims of retinoblastoma management are patient survival followed by globe salvage and lastly visual preservation. The choice of treatment modality depends on a plethora of clinical (age, laterality, staging, tumor dimensions, germline status, visual potential) and sociodemographic factors (financial constraints, cultural beliefs, family's psychosocial situation, distance from hospital, available institutional resources).
Systemic chemotherapy
Intravenous chemotherapy (IVC)
IVC is the monthly administration of 2–4 chemotherapy agents via a central or peripheral intravenous catheter for 3–12 cycles. Indications are bilateral retinoblastomas, germline tumors, positive family history, and HRF on pathologies such as choroidal invasion or ONI.[56,57] Apart from intraocular tumor control, IVC provides long-term protection against the development of pinealoblastomas, secondary cancers, and metastasis.[56,58,59] Visual outcomes are excellent as compared to focal modalities, which cause chorioretinal scarring.[56,57]
Chemoreduction is the use of IVC as a primary treatment to shrink the tumor. The most common regimen is the “triple-drug regimen” of vincristine, etoposide, and carboplatin (VEC), which has excellent salvage rates of 90%–100% for group A–C retinoblastomas[1,5] [Table 6]. Central tumors have a better outcome with chemotherapy as compared to peripheral ones.[1] Tumors with extensive seeding such as group D retinoblastomas usually have a poor response to IVC and require either EBRT or enucleation in 50% of cases.[1,5,51] Side effects of IVC are nausea, fever, pancytopenia, alopecia, and rarely, ototoxicity and renal toxicity.[57] Reports of infertility and secondary leukemia following IVC are controversial and have not been proven.[56,60] Granulocyte colony-stimulating factor (G-CSF) accelerates recovery from chemotherapy-induced myelosuppression but is usually not needed with standard VEC doses.[57] Less commonly used chemotherapy agents include cyclophosphamide, topotecan, and melphalan. Concerns of multidrug resistance with IVC have been addressed by the Toronto Protocol in which short infusions of chemotherapy are interspersed with short infusions of cyclosporin A.[3,61]
Table 6.
Dosage of chemotherapeutic drugs used in retinoblastoma treatment
| Drugs | Dosage |
|---|---|
| IVC | |
| Vincristine (Day 1) | SD: 1.5 mg/m2 (<3 years of age: 0.05 mg/kg) HD: 0.025 mg/kg |
| Etoposide (Day 1, 2) | SD: 150 mg/m2 (<3 years of age: 5 mg/kg) HD: 12 mg/kg |
| Carboplatin (Day 1) | SD: 560 mg/m2 (<3 years of age: 18.6 mg/kg) HD: 28 mg/kg |
| IAC | |
| Melphalan | 3–7.5 mg |
| Topotecan | 1–2 mg |
| IVitC | |
| Melphalan | 20–30 μg |
| Topotecan | 20–30 μg |
| ICamC | |
| Melphalan | 15–20 μg/0.05 mL |
| Topotecan | 7.5 μg/0.015 mL |
As compared to standard-dose chemoreduction, high-dose chemotherapy (HD-IVC) is used as an adjuvant or neoadjuvant modality. Adjuvant IVC (6–12 cycles) is used in patients with HRF or MRD post-enucleation; it significantly reduces the risk of metastasis from 24% to 4%.[62,63] Neoadjuvant IVC (3–6 cycles) is used to shrink large orbital tumors before surgery and EBRT.[25] Up to 12 cycles of systemic IVC have been used in patients with MRD, ESE, overt orbital tumors, and metastases.[5]
Intraarterial chemotherapy (IAC)
IAC is “chemosurgery” in which fluoroscopy-guided microcatheterization (through femoral and internal carotid artery) is used to deliver chemotherapeutic agents to the ophthalmic artery (OA) in a targeted manner. It was first introduced as “selective OA infusion” by Kaneko et al and the technique was later refined by Gobin et al.[64,65] The advent of IAC brought about a radical change in the management of retinoblastomas; it is now the standard of care for eye salvage in most high-income countries.[65,66] Super-selective delivery directly into the OA ensures that the eye receives 10 times the chemotherapeutic dose as compared to IVC, along with minimal systemic toxicity.[57] Indications are non-germline retinoblastoma, unilateral tumors, recurrent subretinal/vitreous seeds, and recurrent retinoblastoma after IVC or plaque brachytherapy.[56] IAC is contraindicated in patients with ESE, orbital cellulitis, neovascular glaucoma, PNETs, metastasis, or HRF on baseline MRI in group E tumors.[66] It is also avoided in patients below 4 months of age because of technical difficulties due to small caliber vessels; however, some cases of successful IAC have been reported in younger infants.[56,57,67] Moreover, its use is limited in LMIC because of the high cost, specialized training, and requirement of an interventional radiologist.[57] There is no standardized consensus yet on the ideal dose, number of agents, and cycles in IAC.[56,68,69] The common regimen for groups B and C is melphalan (5 mg) pushed slowly over 30 minutes in a pulsatile fashion every month for three cycles.[56,57,69] For D or E tumors with massive seeding, a higher dose or addition of topotecan may be needed[57] [Table 6]. Systemic complications include transient neutropenia, femoral artery occlusion, blue toe syndrome, and very rarely, carotid artery dissection, stroke, and death. Ocular complications are periocular side effects (edema, hyperemia, madarosis, blepharoptosis) and vascular events (retinal artery occlusion, ophthalmic artery spasm/obstruction, VH, choroidal ischemia).[57]
Primary IAC is used as the first-line treatment for non-germline, unilateral group B–E tumors.[57] Success rates are excellent, with globe salvage reaching 100% in groups B and C and 94% in D, while in group E, it is 36%.[56]
Secondary IAC is used as a second-line globe salvage therapy instead of enucleation in unilateral/bilateral tumors after failure of previous treatment (IVC/plaque/EBRT).[57,66] Globe salvage rates of 50%–72% have been reported.[70,71] Rates of complete regression were 94% for solid tumors, 95% for subretinal seeds, and 87% for vitreous seeds.[70]
Rescue IAC is used for retinoblastomas that recur after primary IAC failure; it provides tumor control in 75% and globe salvage in 67%.[72]
Minimal exposure IAC is when complete tumor control is attained after only 1–2 cycles of IAC and further cycles are withheld to avoid vascular toxicity and chemotoxicity.[68]
Tandem IAC is simultaneous bilateral IAC for advanced tumors. There is controversy surrounding it because of the risk of vascular complications in the better eye; moreover, it has no effect in preventing pinealoblastomas or metastasis.[57,66] Carboplatin can be used as an alternative drug to reduce the cumulative dose toxicity of melphalan in tandem IAC.[66]
Bridge IAC is combined sequential treatment in neonates, initiated with IVC, and followed by IAC once the child reaches 3 months of age and a weight of 6 kg.[73]
Periocular chemotherapy
Carboplatin or topotecan are injected in the sub-Tenon space as an adjunct to IVC in bilateral advanced or recurrent tumors with subretinal/vitreous seeds. It rapidly achieves a high intraocular concentration of the drug as compared to IVC but can cause complications such as eyelid edema, ecchymosis, orbital fat atrophy, fibrosis, ocular motility issues, and optic atrophy.[56]
Intrathecal chemotherapy
Intrathecal topotecan (0.4 mg) has been used to treat CNS metastasis in retinoblastoma, but the prognosis is guarded.[74]
Intraocular chemotherapy
Intravitreal chemotherapy (IVitC)
IVitC is indicated as a second line of treatment for recalcitrant or recurrent vitreous seeds, with success rates of 90%–100%.[56] Vitreous seeds are tumor cell aggregates that do not respond well to IVC as the vitreous is avascular. Due to a lack of blood supply, intravenous agents do not attain sufficient concentration within the vitreous to cause seed regression; IVitC overcomes this.[5] A typical regimen comprises six intravitreal injections of melphalan given weekly or biweekly.[56] Presence of seeds/tumor at the injection site, bullous RD, VH, and anterior segment invasion must be ruled out before injecting. Injection is given through the pars plana, 3–3.5 mm behind limbus, in a quadrant lacking and 2 clock hours away from vitreous seeds.[75] Adverse effects include retinal pigment epithelial mottling and focal cataract. The risk of needle-track tumor dissemination through the injection site has been curtailed by safety enhancement techniques such as prior paracentesis and post-procedure triple freeze-thaw cryotherapy at the site. Other drugs used include thiotepa (historical), carboplatin, methotrexate, and topotecan, of which the latter seems most promising.[56,75] Precision IVitC is a modified technique in which the drug is injected near a localized area of vitreous seeds under indirect ophthalmoscopy and not dispersed inside the whole eye.[76]
Intracameral chemotherapy (ICamC)
ICamC is the injection of melphalan (15–20 µg/0.05 mL) or topotecan (7.5 µg/0.015 mL) into the anterior chamber through a transcorneal approach to treat ACS.[57] Indications include both primary (seen at presentation, usually in diffuse infiltrating or anterior diffuse types) and secondary (appearing during the course of conservative treatment) ACS.[77] Until very recently, eyes with ACS were an absolute indication for enucleation and were also considered to pose a high risk of metastasis. IVC, periocular, or IVitC do not create high enough levels of chemotherapeutic agents in the aqueous to cause seed regression. However ICamC can cause complete ACS regression, and in eyes with an intact anterior hyaloid, globe salvage is also achievable in 71%.[78] Aqueous taps for cytopathology or cell culture can assess response to ICamC; injections are continued monthly till one additional injection after a negative tap.[57,78] Complications include iris heterochromia and cataract.[57]
Both IVitC and ICamC are ancillary modalities that are only used in conjunction with the treatment of the source of seeding.
Focal therapy
It is indicated for tumor consolidation after chemoreduction by IVC or IAC to treat small recurrent tumors or subretinal seeds and occasionally as primary treatment for group A or B tumors.[5]
Transpupillary thermotherapy (TTT)
Diode laser (infrared, 810 nm) by indirect ophthalmoscopy is used to induce sub-photocoagulation levels of hyperthermia (45°C–60°C) leading to cell apoptosis. Multiple spots (1.2 mm, 300–600 mW) are applied to the whole tumor till a grey-white uptake is noted. Monthly sessions are done with the endpoint of attaining a flat chorioretinal scar or complete calcification; a total of 2–6 sittings may be needed.[57,79] In a study, 92% of tumors with LBD <1.5 DD were successfully treated with TTT alone.[80] TTT has a synergistic effect with IVC as it enhances the effect of carboplatin.[79] Complications include focal cataract and iris atrophy.
Cryotherapy
Monthly sessions of triple freeze-thaw trans-scleral cryotherapy for small peripheral tumors can destroy cells by lowering the temperature to −90°C.[79] Cryotherapy has a synergistic effect by enhancing drug availability to the tumor when chemotherapy is administered within 48 hours of the thermal disruption.[57] Complications include large chorioretinal scar, transient serous RD, and rhegmatogenous RD.
Laser photocoagulation
It was previously used to coagulate vessels and restrict blood supply to small posterior tumors but is now avoided as it can restrict chemotherapeutic agents from reaching the tumor.[22]
Radiotherapy
Plaque brachytherapy
It is indicated as a secondary treatment for chemoresistant recurrent medium-sized (LBD ≤16 mm, height ≤9 mm) tumors after failed IVC/IAC and as a primary treatment for diffuse anterior retinoblastomas without a posterior component.[57] I125 or Ru106 episcleral plaques are used to deliver 40 Gy to the tumor apex over 2–4 days achieving tumor control in 79%.[79,81] Common complications are radiation papillopathy and retinopathy. Advantages over EBRT include shorter treatment duration, no orbital growth stunting, and no secondary cancers.
EBRT
Previously a preferred treatment modality, EBRT is now avoided due to the risk (53% at the age of 50 years) of developing secondary malignancies after radiation in germline tumors. Other complications include dry eye, cataract, contracted socket, radiation retinopathy, and papillopathy. EBRT is indicated in MRD post-enucleation.[57] It has also been used as a last salvage attempt in advanced orbital tumors with visual potential where chemotherapy is not feasible or has failed. The standard dose is 40–50 Gy given over 3–4 weeks.[25,82]
Proton beam therapy
It can minimize radiation to the orbital bone and reduce the risk of secondary cancers.[22,82]
Enucleation
Enucleation with primary orbital implant is indicated in unilateral group E retinoblastomas without visual potential, extraocular spread, and recalcitrant tumors that have failed prior salvage options.[57] In many LMIC countries, enucleation is still the standard of care for unilateral group D tumors. Care should be taken to avoid perforation, use minimal manipulation, obtain a long optic nerve stump (>10 mm), procure tissue for genetics, submit globe for histopathology to look for HRF, and ensure cosmetic rehabilitation.[83] HRFs include ACS, iris/ciliary body infiltration, massive choroidal infiltration, lamina cribrosa invasion, retrolaminar optic nerve invasion, and scleral infiltration. Detection of HRF indicates a high risk of metastasis, and these patients require 4–6 cycles of adjuvant IVC. In those with MRD in the form of involvement of nerve transection, full-thickness scleral extension, or ESE, EBRT is needed along with 12 cycles of adjuvant IVC.
A simplified treatment algorithm is presented in Fig. 1 to assist decision-making in the choice of treatment for intraocular (as per ICRB groups) and extraocular retinoblastomas; however, the treatment plan should be tailor-made for each patient.
Follow-up
After initiating treatment, patients are followed every 4 weeks (if chemoreduction: 3 weeks along with every IVC cycle) till complete tumor regression. At each visit, treatment response and adverse effects are noted, with adjustments (dose escalation, treatment addition, salvage replacement with enucleation) made accordingly. Regression patterns include type 0 (no visible remnant), type 1 (completely calcified remnant), type 2 (completely noncalcified remnant), type 3 (partially calcified remnant), and type 4 (atrophic chorioretinal flat scar).[84] After complete tumor regression, patients are monitored every 3 months for the first year, every 6 months for the next 3 years or till 7 years of age, and thereafter, annually (ideally lifelong).[57] In germline RB, MRI of the brain and orbits must be repeated every 6 months till 5 years of age; pediatric oncologist consultation and surveillance for secondary extraocular malignancies are also required.[16,57]
Ancillary management
Counseling
Families of patients (and patients themselves, when older) should be referred for genetic counseling after diagnosis of retinoblastoma to educate them about the inheritance, risk of malignancy in other offspring and relatives, family planning, and available genetic testing. Psychosocial care should also be offered to parents to provide moral support, grief counseling, and coping mechanisms.
Family screening
Family members with risk of RB1 mutation should be offered blood tests for genetic status. If the proband's parents have the mutation, then each of their offspring will have a 50% risk of developing retinoblastoma. Furthermore, 10% of parents who are negative for the mutation may possess mosaicism; their offspring have a 2%–3% risk of inheriting the mutation.[14] If the proband has mosaicism, then testing of parents or siblings is unnecessary as mosaicism is not inherited, but children of mosaic proband must be tested.[16] Children with known germline status but no tumor can be monitored monthly till 1 year of age and then every 3 months till the age of 5 years.[14]
Perinatal diagnosis
Pre-implantation diagnosis involves testing an embryo for RB1-/- after in-vitro fertilization.[14] Prenatal chorionic villus sampling (10–14 weeks of gestation) or amniocentesis (>15 weeks of gestation) can detect the presence of RB1-/- and help in parental decision-making.[14,16] Risk of miscarriage during these two procedures are 1% and 0.1-0.5% respectively.[16] If a mutation is detected, high-resolution in-utero fetal ultrasound can occasionally detect tumors, and preterm delivery can be considered to institute early treatment.[14] Postnatal peripheral or cord blood can be used after delivery for genetic testing.[14]
Recent advances
Currently, the presence of HRF in histopathology is used to predict the development of metastasis. Along with AH sampling, analysis of cfDNA levels in blood plasma (liquid biopsy) is a highly promising noninvasive modality that can be used for predicting metastatic risk through molecular profiling. In addition to prognostication, monitoring of cfDNA in blood post-enucleation can predict impending extraocular relapse.[85,86] Suggested potential biomarkers for metastasis prediction are microRNA, cone-rod homeobox (CRX) or Otx-like homeobox transcriptor, and CTX messenger RNA.[87]
Novel treatment modalities that are being considered include targeted therapy (pevonedistat, nutlin-3, SYK inhibitor), immunotherapy (anti-GD2 ganglioside antibodies/CART cells, immune checkpoint inhibitors), and oncolytic viruses (VCN01 oncolytic adenovirus).[85] Other possible compounds are Matrix metalloproteinases (MMP2, MMP9), pentoxifylline, inhibitors of survivin, antiapoptotic Bcl-2 family proteins, methyltransferase, and kinesin proteins.[28]
Tylectomy or surgical resection of retinoblastoma has been recently reported by a study to benefit survival (96%) in patients undergoing eye salvage; results are comparable to enucleation (95%) but superior to survival in eye salvage without tylectomy (90%).[88]
Chemoplaque is a novel drug delivery system in which an episcleral plaque filled with topotecan is used for sustained release of drug into the vitreous; the clinical trial (STEP-RB) is currently in phase I.[89]
Conclusion
A three-pronged approach is the goal to tackling this lethal pediatric cancer: awareness and early detection, deciding on the best treatment approach, and long-term monitoring. Eye salvage treatments have a high success rate if instituted at the proper time. Novel techniques such as liquid biopsy and targeted therapy may come to play a larger role in the coming years, and the future seems bright. However, LMICs like India, which have a massive retinoblastoma burden, still have a long way to go. The disparity in the mortality rates between LMICs and HMICs can only be addressed through community awareness programs in developing countries to prevent delayed diagnosis. Cancer care in India is making leaps and bounds, but ocular cancer sadly remains a neglected field and is not available in most cancer centers in the country. Dedicated ocular oncology specialist services with accessible resources, especially genetic testing and IAC, are the need of the hour.
Financial support and sponsorship:
Nil.
Conflicts of interest:
There are no conflicts of interest.
References
- 1.Rodriguez-Galindo C, Orbach DB, VanderVeen D. Retinoblastoma. Pediatr Clin North Am. 2015;62:201–23. doi: 10.1016/j.pcl.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 2.Alzahem T, Alsarhani W, Albahlal A, Safieh LA, Aldahmash S, Alzahem T, et al. History and genetics of retinoblastoma. In: Alkatan HM, editor. Retinoblastoma - Past, Present and Future. IntechOpen; 2019. Available from: https://www.intechopen.com/chapters/68882 . [Google Scholar]
- 3.Dimaras H, Kimani K, Dimba EA, Gronsdahl P, White A, Chan HS, et al. Retinoblastoma. Lancet. 2012;379:1436–46. doi: 10.1016/S0140-6736(11)61137-9. [DOI] [PubMed] [Google Scholar]
- 4.Ortiz MV, Dunkel IJ. Retinoblastoma. J Child Neurol. 2016;31:227–36. doi: 10.1177/0883073815587943. [DOI] [PubMed] [Google Scholar]
- 5.Rao R, Honavar SG. Retinoblastoma. Indian J Pediatr. 2017;84:937–44. doi: 10.1007/s12098-017-2395-0. [DOI] [PubMed] [Google Scholar]
- 6.Kivelä T. The epidemiological challenge of the most frequent eye cancer: Retinoblastoma, an issue of birth and death. Br J Ophthalmol. 2009;93:1129–31. doi: 10.1136/bjo.2008.150292. [DOI] [PubMed] [Google Scholar]
- 7.Chawla B, Hasan F, Azad R, Seth R, Upadhyay AD, Pathy S, et al. Clinical presentation and survival of retinoblastoma in Indian children. Br J Ophthalmol. 2016;100:172–8. doi: 10.1136/bjophthalmol-2015-306672. [DOI] [PubMed] [Google Scholar]
- 8.Kaliki S, Srinivasan V, Gupta A, Mishra DK, Naik MN. Clinical features predictive of high-risk retinoblastoma in 403 Asian Indian patients: A case-control study. Ophthalmology. 2015;122:1165–72. doi: 10.1016/j.ophtha.2015.01.018. [DOI] [PubMed] [Google Scholar]
- 9.Beniwal V, Maheshwari G, Beniwal S, Dhanawat A, Tantia P, Adlakha P. Retinoblastoma: A review of clinical profile at a regional cancer center in Northwest India. J Cancer Res Ther. 2022;18:1623–8. doi: 10.4103/jcrt.JCRT_1263_20. [DOI] [PubMed] [Google Scholar]
- 10.Naseripour M, Nazari H, Bakhtiari P, Modarres-zadeh M, Vosough P, Ausari M. Retinoblastoma in Iran: Outcomes in terms of patients’ survival and globe survival. Br J Ophthalmol. 2009;93:28–32. doi: 10.1136/bjo.2008.139410. [DOI] [PubMed] [Google Scholar]
- 11.Bowman RJ, Mafwiri M, Luthert P, Luande J, Wood M. Outcome of retinoblastoma in east Africa. Pediatr Blood Cancer. 2008;50:160–2. doi: 10.1002/pbc.21080. [DOI] [PubMed] [Google Scholar]
- 12.Melamud A, Palekar R, Singh A. Retinoblastoma. Am Fam Physician. 2006;73:1039–44. [PubMed] [Google Scholar]
- 13.Dimaras H, Corson TW, Cobrinik D, White A, Zhao J, Munier FL, et al. Retinoblastoma. Nat Rev Dis Primer. 2015;1:15021. doi: 10.1038/nrdp.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mallipatna A, Marino M, Singh AD. Genetics of retinoblastoma. Asia-Pac J Ophthalmol Phila Pa. 2016;5:260–4. doi: 10.1097/APO.0000000000000219. [DOI] [PubMed] [Google Scholar]
- 15.Xu XL, Singh HP, Wang L, Qi DL, Poulos BK, Abramson DH, et al. Rb suppresses human cone-precursor-derived retinoblastoma tumours. Nature. 2014;514:385–8. doi: 10.1038/nature13813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soliman SE, Racher H, Zhang C, MacDonald H, Gallie BL. Genetics and molecular diagnostics in retinoblastoma—An update. Asia Pac J Ophthalmol (Phila) 2017;6:197–207. doi: 10.22608/APO.201711. [DOI] [PubMed] [Google Scholar]
- 17.Rootman DB, Gonzalez E, Mallipatna A, Vandenhoven C, Hampton L, Dimaras H, et al. Hand-held high-resolution spectral domain optical coherence tomography in retinoblastoma: Clinical and morphologic considerations. Br J Ophthalmol. 2013;97:59–65. doi: 10.1136/bjophthalmol-2012-302133. [DOI] [PubMed] [Google Scholar]
- 18.Kooi IE, Mol BM, Massink MPG, de Jong MC, de Graaf P, van der Valk P, et al. A Meta-Analysis of retinoblastoma copy numbers refines the list of possible driver genes involved in tumor progression. PLoS One. 2016;11:e0153323. doi: 10.1371/journal.pone.0153323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rushlow DE, Mol BM, Kennett JY, Yee S, Pajovic S, Thériault BL, et al. Characterisation of retinoblastomas without RB1 mutations: Genomic, gene expression, and clinical studies. Lancet Oncol. 2013;14:327–34. doi: 10.1016/S1470-2045(13)70045-7. [DOI] [PubMed] [Google Scholar]
- 20.McEvoy J, Nagahawatte P, Finkelstein D, Richards-Yutz J, Valentine M, Ma J, et al. RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget. 2014;5:438–50. doi: 10.18632/oncotarget.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kooi IE, Mol BM, Massink MPG, Ameziane N, Meijers-Heijboer H, Dommering CJ, et al. Somatic genomic alterations in retinoblastoma beyond RB1 are rare and limited to copy number changes. Sci Rep. 2016;6:25264. doi: 10.1038/srep25264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy SR, Kaliki S. Retinoblastoma: A major review. Mymensingh Med J MMJ. 2021;30:881–95. [PubMed] [Google Scholar]
- 23.Shields JA, Shields CL, Suvarnamani C, Schroeder RP, DePotter P. Retinoblastoma manifesting as orbital cellulitis. Am J Ophthalmol. 1991;112:442–9. doi: 10.1016/s0002-9394(14)76255-5. [DOI] [PubMed] [Google Scholar]
- 24.Tomar AS, Finger PT, Gallie B, Kivelä TT, Mallipatna A, Zhang C, et al. High-risk pathologic features based on presenting findings in advanced intraocular retinoblastoma: A Multicenter, International Data-Sharing American joint committee on cancer study. Ophthalmology. 2022;129:923–32. doi: 10.1016/j.ophtha.2022.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rao R, Honavar SG. Recent developments in retinoblastoma. Delhi J Ophthalmol. 2016;27:50. [Google Scholar]
- 26.Traine PG, Schedler KJ, Rodrigues EB. Clinical presentation and genetic paradigm of diffuse infiltrating retinoblastoma: A review. Ocul Oncol Pathol. 2016;2:128–32. doi: 10.1159/000441528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jijelava KP, Grossniklaus HE. Diffuse anterior retinoblastoma: A review. Saudi J Ophthalmol. 2013;27:135–9. doi: 10.1016/j.sjopt.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cruz-Gálvez CC, Ordaz-Favila JC, Villar-Calvo VM, Cancino-Marentes ME, Bosch-Canto V. Retinoblastoma: Review and new insights. Front Oncol. 2022;12:963780. doi: 10.3389/fonc.2022.963780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rishi P, Sharma U, Sharma T. Cavitary retinoblastoma: Clinical observations. Eye. 2020;34:704–10. doi: 10.1038/s41433-019-0581-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gündüz K, Müftüoglu O, Günalp I, Unal E, Taçyildiz N. Metastatic retinoblastoma clinical features, treatment, and prognosis. Ophthalmology. 2006;113:1558–66. doi: 10.1016/j.ophtha.2006.03.039. [DOI] [PubMed] [Google Scholar]
- 31.Cozza R, De Ioris MA, Ilari I, Devito R, Fidani P, De Sio L, et al. Metastatic retinoblastoma: Single institution experience over two decades. Br J Ophthalmol. 2009;93:1163–6. doi: 10.1136/bjo.2008.148932. [DOI] [PubMed] [Google Scholar]
- 32.Ali MJ, Honavar SG, Reddy VA. Distant metastatic retinoblastoma without central nervous system involvement. Indian J Ophthalmol. 2013;61:357–9. doi: 10.4103/0301-4738.97077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poulaki V, Mukai S. Retinoblastoma: Genetics and pathology. Int Ophthalmol Clin. 2009;49:155–64. doi: 10.1097/IIO.0b013e3181924bc2. [DOI] [PubMed] [Google Scholar]
- 34.Mallipatna AC, Gallie BL, Chévez-Barrios P, Lumbroso-Le Rouic L, Chantada GL, Doz F, et al. Retinoblastoma. In: Amin MB, Edge SB, Greene FL, Byrd DR, Brookland RK, Washington MK, et al., editors. AJCC Cancer Staging Manual. 8th. New York: Springer; 2017. pp. 819–31. [Google Scholar]
- 35.Callaway NF, Mruthyunjaya P. Widefield imaging of retinal and choroidal tumors. Int J Retina Vitreous. 2019;5(Suppl 1):49. doi: 10.1186/s40942-019-0196-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim JW, Ngai LK, Sadda S, Murakami Y, Lee DK, Murphree AL. Retcam fluorescein angiography findings in eyes with advanced retinoblastoma. Br J Ophthalmol. 2014;98:1666–71. doi: 10.1136/bjophthalmol-2014-305180. [DOI] [PubMed] [Google Scholar]
- 37.Seider MI, Grewal DS, Mruthyunjaya P. Portable optical coherence tomography detection or confirmation of ophthalmoscopically invisible or indeterminate active retinoblastoma. Ophthalmic Surg Lasers Imaging Retina. 2016;47:965–8. doi: 10.3928/23258160-20161004-12. [DOI] [PubMed] [Google Scholar]
- 38.Soliman SE, VandenHoven C, MacKeen LD, Héon E, Gallie BL. Optical coherence tomography-guided decisions in retinoblastoma management. Ophthalmology. 2017;124:859–72. doi: 10.1016/j.ophtha.2017.01.052. [DOI] [PubMed] [Google Scholar]
- 39.de Graaf P, Göricke S, Rodjan F, Galluzzi P, Maeder P, Castelijns JA, et al. Guidelines for imaging retinoblastoma: Imaging principles and MRI standardization. Pediatr Radiol. 2012;42:2–14. doi: 10.1007/s00247-011-2201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moulin AP, Gaillard MC, Balmer A, Munier FL. Ultrasound biomicroscopy evaluation of anterior extension in retinoblastoma: A clinicopathological study. Br J Ophthalmol. 2012;96:337–40. doi: 10.1136/bjophthalmol-2011-300051. [DOI] [PubMed] [Google Scholar]
- 41.Razek AAKA, Elkhamary S. MRI of retinoblastoma. Br J Radiol. 2011;84:775–84. doi: 10.1259/bjr/32022497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hiasat JG, Saleh A, Al-Hussaini M, Al Nawaiseh I, Mehyar M, Qandeel M, et al. The predictive value of magnetic resonance imaging of retinoblastoma for the likelihood of high-risk pathologic features. Eur J Ophthalmol. 2019;29:262–8. doi: 10.1177/1120672118781200. [DOI] [PubMed] [Google Scholar]
- 43.Chawla B, Sharma S, Sen S, Azad R, Bajaj MS, Kashyap S, et al. Correlation between clinical features, magnetic resonance imaging, and histopathologic findings in retinoblastoma: A prospective study. Ophthalmology. 2012;119:850–6. doi: 10.1016/j.ophtha.2011.09.037. [DOI] [PubMed] [Google Scholar]
- 44.Song KD, Eo H, Kim JH, Yoo SY, Jeon TY. Can preoperative MR imaging predict optic nerve invasion of retinoblastoma? Eur J Radiol. 2012;81:4041–5. doi: 10.1016/j.ejrad.2012.03.034. [DOI] [PubMed] [Google Scholar]
- 45.Chiranthan M, Meel R, Sharma S, Lomi N, Kashyap S, Singh Bajaj M. Can enhancement pattern in normal-sized optic nerves on magnetic resonance imaging better predict tumor invasion in retinoblastoma eyes? Ocul Oncol Pathol. 2023;9:107–14. doi: 10.1159/000531354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jansen RW, de Bloeme CM, Brisse HJ, Galluzzi P, Cardoen L, Göricke S, et al. MR Imaging features to differentiate retinoblastoma from Coats’ Disease and persistent fetal vasculature. Cancers (Basel) 2020;12:3592. doi: 10.3390/cancers12123592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bakhshi S, Meel R, Kashyap S, Sharma S. Bone marrow aspirations and lumbar punctures in retinoblastoma at diagnosis: Correlation with IRSS staging. J Pediatr Hematol Oncol. 2011;33:e182–5. doi: 10.1097/MPH.0b013e3182103f5c. [DOI] [PubMed] [Google Scholar]
- 48.Berry JL, Xu L, Murphree AL, Krishnan S, Stachelek K, Zolfaghari E, et al. Potential of aqueous humor as a surrogate tumor biopsy for retinoblastoma. JAMA Ophthalmol. 2017;135:1221–30. doi: 10.1001/jamaophthalmol.2017.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reese AB, Ellsworth RM. The evaluation and current concept of retinoblastoma therapy. Trans Am Acad Ophthalmol Otolaryngol. 1963;67:164–72. [PubMed] [Google Scholar]
- 50.Linn Murphree A. Intraocular retinoblastoma: The case for a new group classification. Ophthalmol Clin N Am. 2005;18:41–53. doi: 10.1016/j.ohc.2004.11.003. viii. [DOI] [PubMed] [Google Scholar]
- 51.Shields CL, Mashayekhi A, Au AK, Czyz C, Leahey A, Meadows AT, et al. The International classification of retinoblastoma predicts chemoreduction success. Ophthalmology. 2006;113:2276–80. doi: 10.1016/j.ophtha.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 52.Novetsky DE, Abramson DH, Kim JW, Dunkel IJ. Published international classification of retinoblastoma (ICRB) definitions contain inconsistencies--an analysis of impact. Ophthalmic Genet. 2009;30:40–4. doi: 10.1080/13816810802452168. [DOI] [PubMed] [Google Scholar]
- 53.Chantada G, Doz F, Antoneli CBG, Grundy R, Clare Stannard FF, Dunkel IJ, et al. A proposal for an international retinoblastoma staging system. Pediatr Blood Cancer. 2006;47:801–5. doi: 10.1002/pbc.20606. [DOI] [PubMed] [Google Scholar]
- 54.Maitray A, Khetan V. Classification of retinoblastoma: Evolution with time and the need for uniformity. Oman J Ophthalmol. 2017;10:133–4. doi: 10.4103/0974-620X.216017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tomar AS, Finger PT, Gallie B, Kivelä TT, Mallipatna A, Zhang C, et al. Metastatic death based on presenting features and treatment for advanced intraocular retinoblastoma: A multicenter registry-based study. Ophthalmology. 2022;129:933–45. doi: 10.1016/j.ophtha.2022.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shields CL, Lally SE, Leahey AM, Jabbour PM, Caywood EH, Schwendeman R, et al. Targeted retinoblastoma management: When to use intravenous, intra-arterial, periocular, and intravitreal chemotherapy. Curr Opin Ophthalmol. 2014;25:374–85. doi: 10.1097/ICU.0000000000000091. [DOI] [PubMed] [Google Scholar]
- 57.Ancona-Lezama D, Dalvin LA, Shields CL. Modern treatment of retinoblastoma: A 2020 review. Indian J Ophthalmol. 2020;68:2356–65. doi: 10.4103/ijo.IJO_721_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shields CL, Meadows AT, Shields JA, Carvalho C, Smith AF. Chemoreduction for retinoblastoma may prevent intracranial neuroblastic malignancy (trilateral retinoblastoma) Arch Ophthalmol Chic Ill 1960. 2001;119:1269–72. doi: 10.1001/archopht.119.9.1269. [DOI] [PubMed] [Google Scholar]
- 59.Turaka K, Shields CL, Meadows AT, Leahey A. Second malignant neoplasms following chemoreduction with carboplatin, etoposide, and vincristine in 245 patients with intraocular retinoblastoma. Pediatr Blood Cancer. 2012;59:121–5. doi: 10.1002/pbc.23278. [DOI] [PubMed] [Google Scholar]
- 60.Shields CL, Fulco EM, Arias JD, Alarcon C, Pellegrini M, Rishi P, et al. Retinoblastoma frontiers with intravenous, intra-arterial, periocular, and intravitreal chemotherapy. Eye. 2013;27:253–64. doi: 10.1038/eye.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chan H, Gallie B, Munier F, Beckpopovic M. Chemotherapy for retinoblastoma. Ophthalmol Clin N Am. 2005;18:55–63. doi: 10.1016/j.ohc.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 62.Kaliki S, Shields CL, Shah SU, Eagle RC, Shields JA, Leahey A. Postenucleation adjuvant chemotherapy with vincristine, etoposide, and carboplatin for the treatment of high-risk retinoblastoma. Arch Ophthalmol Chic Ill 1960. 2011;129:1422–7. doi: 10.1001/archophthalmol.2011.289. [DOI] [PubMed] [Google Scholar]
- 63.Honavar SG, Singh AD, Shields CL, Meadows AT, Demirci H, Cater J, et al. Postenucleation adjuvant therapy in high-risk retinoblastoma. Arch Ophthalmol. 2002;120:923–31. doi: 10.1001/archopht.120.7.923. [DOI] [PubMed] [Google Scholar]
- 64.Gobin YP, Dunkel IJ, Marr BP, Brodie SE, Abramson DH. Intra-arterial chemotherapy for the management of retinoblastoma: Four-year experience. Arch Ophthalmol Chic Ill 1960. 2011;129:732–7. doi: 10.1001/archophthalmol.2011.5. [DOI] [PubMed] [Google Scholar]
- 65.Yamane T, Kaneko A, Mohri M. The technique of ophthalmic arterial infusion therapy for patients with intraocular retinoblastoma. Int J Clin Oncol. 2004;9:69–73. doi: 10.1007/s10147-004-0392-6. [DOI] [PubMed] [Google Scholar]
- 66.Manjandavida FP, Stathopoulos C, Zhang J, Honavar SG, Shields CL. Intra-arterial chemotherapy in retinoblastoma – A paradigm change. Indian J Ophthalmol. 2019;67:740–54. doi: 10.4103/ijo.IJO_866_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Magan T, Khoo CTL, Jabbour PM, Shields CL. Intraarterial chemotherapy for retinoblastoma in a 2-month-old infant. Retin Cases Brief Rep. 2017;11:24–6. doi: 10.1097/ICB.0000000000000279. [DOI] [PubMed] [Google Scholar]
- 68.Shields CL, Kaliki S, Shah SU, Bianciotto CG, Liu D, Jabbour P, et al. Minimal exposure (One or two cycles) of intra-arterial chemotherapy in the management of retinoblastoma. Ophthalmology. 2012;119:188–92. doi: 10.1016/j.ophtha.2011.06.036. [DOI] [PubMed] [Google Scholar]
- 69.Abramson DH, Dunkel IJ, Brodie SE, Kim JW, Gobin YP. A Phase I/II Study of direct intraarterial (Ophthalmic Artery) chemotherapy with melphalan for intraocular retinoblastoma: Initial results. Ophthalmology. 2008;115:1398–404.e1. doi: 10.1016/j.ophtha.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 70.Shields CL, Manjandavida FP, Lally SE, Pieretti G, Arepalli SA, Caywood EH, et al. Intra-arterial chemotherapy for retinoblastoma in 70 eyes: Outcomes based on the international classification of retinoblastoma. Ophthalmology. 2014;121:1453–60. doi: 10.1016/j.ophtha.2014.01.026. [DOI] [PubMed] [Google Scholar]
- 71.Shields CL, Bianciotto CG, Jabbour P, Ramasubramanian A, Lally SE, Griffin GC, et al. Intra-arterial chemotherapy for retinoblastoma: Report No. 1, Control of retinal tumors, subretinal seeds, and vitreous seeds. Arch Ophthalmol. 2011;129:1399–406. doi: 10.1001/archophthalmol.2011.150. [DOI] [PubMed] [Google Scholar]
- 72.Shields CL, Say EA, Pointdujour-Lim R, Cao C, Jabbour PM, Shields JA. Rescue intra-arterial chemotherapy following retinoblastoma recurrence after initial intra-arterial chemotherapy. J Fr Ophtalmol. 2015;38:542–9. doi: 10.1016/j.jfo.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 73.Gobin YP, Dunkel IJ, Marr BP, Francis JH, Brodie SE, Abramson DH. Combined, sequential intravenous and intra-arterial chemotherapy (Bridge Chemotherapy) for young infants with retinoblastoma. PLoS One. 2012;7:e44322. doi: 10.1371/journal.pone.0044322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rodriguez A, Zugbi S, Requejo F, Deu A, Sampor C, Sgroi M, et al. Combined high-dose intra-arterial and intrathecal chemotherapy for the treatment of a case of extraocular retinoblastoma. Pediatr Blood Cancer. 2018;65:e27385. doi: 10.1002/pbc.27385. [DOI] [PubMed] [Google Scholar]
- 75.Manjandavida FP, Shields CL. The role of intravitreal chemotherapy for retinoblastoma. Indian J Ophthalmol. 2015;63:141–5. doi: 10.4103/0301-4738.154390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu MD, Dalvin LA, Welch RJ, Shields CL. Precision intravitreal chemotherapy for localized vitreous seeding of retinoblastoma. Ocul Oncol Pathol. 2019;5:284–9. doi: 10.1159/000491432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stathopoulos C, Beck-Popovic M, Moulin AP, Munier FL. Ten-year experience with intracameral chemotherapy for aqueous seeding in retinoblastoma: long-term efficacy, safety and toxicity. Br J Ophthalmol. 2023;108:124–30. doi: 10.1136/bjo-2022-322492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Munier FL, Moulin A, Gaillard MC, Bongiovanni M, Decembrini S, Houghton S, et al. Intracameral chemotherapy for globe salvage in retinoblastoma with secondary anterior chamber invasion. Ophthalmology. 2018;125:615–7. doi: 10.1016/j.ophtha.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 79.Chawla B, Jain A, Azad R. Conservative treatment modalities in retinoblastoma. Indian J Ophthalmol. 2013;61:479–85. doi: 10.4103/0301-4738.119424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abramson DH, Schefler AC. Transpupillary thermotherapy as initial treatment for small intraocular retinoblastoma. Ophthalmology. 2004;111:984–91. doi: 10.1016/j.ophtha.2003.08.035. [DOI] [PubMed] [Google Scholar]
- 81.Shields CL, Shields JA, Cater J, Othmane I, Singh AD, Micaily B. Plaque radiotherapy for retinoblastoma: Long-term tumor control and treatment complications in 208 tumors. Ophthalmology. 2001;108:2116–21. doi: 10.1016/s0161-6420(01)00797-7. [DOI] [PubMed] [Google Scholar]
- 82.Kim JY, Park Y. Treatment of retinoblastoma: The role of external beam radiotherapy. Yonsei Med J. 2015;56:1478–91. doi: 10.3349/ymj.2015.56.6.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Honavar S, Singh A. Management of advanced retinoblastoma. Ophthalmol Clin N Am. 2005;18:65–73. doi: 10.1016/j.ohc.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 84.Shields CL, Palamar M, Sharma P, Ramasubramanian A, Leahey A, Meadows AT, et al. Retinoblastoma regression patterns following chemoreduction and adjuvant therapy in 557 tumors. Arch Ophthalmol. 2009;127:282–90. doi: 10.1001/archophthalmol.2008.626. [DOI] [PubMed] [Google Scholar]
- 85.Schaiquevich P, Francis JH, Cancela MB, Carcaboso AM, Chantada GL, Abramson DH. Treatment of retinoblastoma: What is the latest and what is the future. Front Oncol. 2022;12:822330. doi: 10.3389/fonc.2022.822330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kothari P, Marass F, Yang JL, Stewart CM, Stephens D, Patel J, et al. Cell‐free DNA profiling in retinoblastoma patients with advanced intraocular disease: An MSKCC experience. Cancer Med. 2020;9:6093–101. doi: 10.1002/cam4.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cassoux N, Lumbroso L, Levy-Gabriel C, Aerts I, Doz F, Desjardins L. Retinoblastoma: Update on current management. Asia Pac J Ophthalmol (Phila) 2017;6:290–5. doi: 10.22608/APO.201778. [DOI] [PubMed] [Google Scholar]
- 88.Zhao J, Li Q, Feng ZX, Zhang J, Wu S, Jin L, et al. Tylectomy safety in salvage of eyes with retinoblastoma. Cancers. 2021;13:5862. doi: 10.3390/cancers13225862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Targeted Therapy Technologies, LLC A Phase I Study of sequestered transscleral, controlled-release topotecan delivered from an episcleral reservoir in retinoblastoma eyes. clinicaltrials.gov. 2023 Available from: https://clinicaltrials.gov/study/NCT04156347. [Last accessed on 2023 Jul 13] [Google Scholar]