

Abstract
INTRODUCTION
The “A/T/N” (amyloid/tau/neurodegeneration) framework provides a biological basis for Alzheimer's disease (AD) diagnosis and can encompass additional changes such as inflammation (“I”). A spectrum of T/N/I imaging and plasma biomarkers was acquired in a phase 2 clinical trial of rasagiline in mild to moderate AD patients. We evaluated these to understand biomarker distributions and relationships within this population.
METHODS
Plasma biomarkers of pTau‐181, neurofilament light chain (NfL), glial fibrillary acidic protein (GFAP), other inflammation‐related proteins, imaging measures including fluorodeoxyglucose (FDG) positron emission tomography (PET), flortaucipir PET, and volumetric magnetic resonance imaging (MRI), and cognitive endpoints were analyzed to assess characteristics and relationships for the overall population (N = 47 at baseline and N = 21 for longitudinal cognitive comparisons) and within age‐decade subgroups (57‐69, 70‐79, 80‐90 years).
RESULTS
Data demonstrate wide clinical and biomarker heterogeneity in this population influenced by age and sex. Plasma pTau‐181 and GFAP correlate with tau PET, most strongly in left inferior temporal cortex (p = 0.0002, p = 0.0006, respectively). In regions beyond temporal cortex, tau PET uptake decreased with age for the same pTau‐181 or GFAP concentrations. FDG PET and brain volumes correlate with tau PET in numerous regions (such as inferior temporal: p = 0.0007, p = 0.00001, respectively). NfL, GFAP, and all imaging modalities correlate with baseline MMSE; subsequent MMSE decline is predicted by baseline parahippocampal and lateral temporal tau PET (p = 0.0007) and volume (p = 0.0006). Lateral temporal FDG PET (p = 0.006) and volume (p = 0.0001) are most strongly associated with subsequent ADAS‐cog decline. NfL correlates with FDG PET and baseline MMSE but not tau PET. Inflammation biomarkers are intercorrelated but correlated with other biomarkers in only the youngest group.
DISCUSSION
Associations between plasma biomarkers, imaging biomarkers, and cognitive status observed in this study provide insight into relationships among biological processes in mild to moderate AD. Findings show the potential to characterize AD patients regarding likely tau pathology, neurodegeneration, prospective clinical decline, and the importance of covariates such as age.
Highlights
Plasma pTau‐181 and GFAP correlated with regional and global tau PET in mild to moderate AD.
NfL correlated with FDG PET and cognitive endpoints but not plasma pTau‐181 or tau PET.
Volume and FDG PET showed strong relationships to tau PET, one another, and cognitive status.
Temporal volumes most strongly predicted decline in both MMSE and ADAS‐cog.
Volume and plasma biomarkers can enrich for elevated tau PET with age a significant covariate.
Keywords: A/T/N, Alzheimer's disease, FDG PET, flortaucipir, GFAP, Inflammation, NfL, plasma biomarkers, pTau‐181, Tau PET, volumetric MRI
1. BACKGROUND
Alzheimer's disease (AD) is characterized by progressive amyloid plaque aggregation (“A”), neurofibrillary tangle (NFT) accumulation (“T”), and neurodegeneration (“N”) associated with cognitive decline and functional disability. The “A/T/N” framework 1 provides a biological basis for AD diagnosis based on hallmark changes and includes clinical staging that supplements the biological definition. Additional aspects have been identified, including neuroinflammation (“I”). 2 More broadly, the ATX(N) framework is flexible and can encompass other key AD pathogenic features with identifiable biomarkers. 3 Biomarkers reflecting AD pathophysiology are measurable using positron emission tomography (PET) imaging, magnetic resonance imaging (MRI), cerebrospinal fluid (CSF) assays, and plasma assays. Within a Phase 2 clinical trial of the drug rasagiline in mild to moderate AD patients, 4 a broad set of T/N/I biomarkers was acquired, providing an opportunity to explore their relationships in this clinical severity range. Our work focused on understanding relationships toward the goal of efficiently characterizing patients for clinical trials and eventual clinical care.
1.1. Biomarkers of hallmark AD pathology
Our study measured tau NFT burden using tau PET and plama pTau‐181 (both “T”). Tau PET and plasma pTau‐181 correlate across the AD spectrum with some discordance primarily in plasma‐positive PET‐negative cases. 5 , 6 , 7 , 8 Plasma pTau‐181 levels differentiate AD from other dementias similar to tau PET, indicate amyloid and tau positivity, and are associated with cognitive decline. 5 , 7 , 8 , 9 , 10 Age and sex influence merit consideration given observations of greater frontoparietal tau NFT burden in younger cohorts 11 , 12 and higher burden in females. 13
1.2. Biomarkers of function and neurodegeneration
Several neurodegenerative imaging and plasma markers were included. Regional cerebral glucose hypometabolism (FDG PET), reflecting synaptic and neuronal dysfunction and degeneration, and atrophy (volumetric MRI) (“N”) have shown spatial consistency with NFT aggregation and phenotype 14 and correlate with clinical decline. 9 , 15 , 16 , 17 Plasma neurofilament light (NfL), an etiologically nonspecific protein subunit released from damaged axons, 10 , 18 , 19 correlates with atrophy, hypometabolism, 9 and cognitive decline in A+T+ cohorts, 18 , 20 predicts more rapid cognitive decline, 7 , 10 and increases with age. 21 , 22 Glial fibrillary acidic protein (GFAP, “N”, “I”) found in astrocytes surrounding synapses and expressed with reactive astrogliosis 23 increases with amyloid positivity 24 and disease stage, and correlates with tau PET. 25 While not AD‐specific, 24 higher GFAP predicts progression to AD dementia and accelerated cognitive decline. 26 Levels increase with age, with higher levels observed in females. 24 , 26 , 27
1.3. Biomarkers of inflammation
Inflammation (“I”) is associated with AD through genome‐wide association studies and elevated inflammatory markers in AD patients’ blood samples. 28 , 29 , 30 Markers in our study included: proinflammatory cytokines (IL [interleukin] ‐1β, IL‐6, IL‐7, IL‐12, IL‐18, TNF‐α (tumor necrosis factor), FGF‐Basic (fibroblast growth factor), GM‐CSF (granulocyte‐macrophage colony‐stimulating factor), MIF (macrophage migratory inhibiting factor)); proteins that can act as cytokines (HGF (hepatocyte growth factor)); anti‐inflammatory cytokines (IL [interleukin] ‐1ra, IL‐4, IL‐13, G‐CSF [granulocyte colony‐stimulating factor], M‐CSF [macrophage colony‐stimulating factor]); chemokines (Eotaxin, IP‐10, CTAK [T‐cell attracting chemokine], MCP‐1 [monocyte chemotactic protein‐1], SDF‐1alpha [stromal cell derived factor], RANTES); and cytokines associated with apoptosis (TRAIL [tumor necrosis factor‐related apoptosis‐inducing ligand], PDGF‐BB [platelet‐derived growth factor]). While their roles are not fully elucidated in AD, inflammatory cytokine increases relate to a decreased ability of microglia to clear Aβ, and to cognitive decline. 2 IL‐1α, IL‐1β, and TNF‐α are released by reactive astrocytes and activated microglia in the presence of Aβ, creating feedback amplifying amyloid plaque formation. 31
1.4. Objectives of this work
Using the biomarkers collected in the rasagiline clinical trial (NCT02359552), objectives of this post hoc exploratory analysis were to: (1) characterize the relationship between imaging and plasma biomarker levels in this mild to moderate AD clinical trial population, considering age, sex, and apolipoprotein E (APOE) genotype; (2) test hypotheses that: (a) pTau‐181 correlates with tau PET in this population, (b) NfL correlates with FDG PET, and (c) baseline tau PET and NfL predict subsequent cognitive change; (3) explore relationships between GFAP and other biomarkers; and (4) evaluate the potential for plasma biomarkers, volumetric, and/or FDG measures to predict NFT burden as screening support in AD.
2. METHODS
2.1. Rasagiline study design
FDG and tau PET imaging, MRI, plasma biomarkers, and clinical endpoints (Table 1) were acquired from patients with a clinical diagnosis of mild to moderate AD who participated in a double‐blind, placebo‐controlled trial of rasagiline (results described previously 4 ). Data were acquired under Institutional Review Board approval with informed patient consent at the Cleveland Clinic Lou Ruvo Center for Brain Health in Nevada and two Cleveland Clinic sites in Ohio. Primary inclusion criteria in the original study were a clinical diagnosis of probable AD (NINDS‐ADRDA criteria), age 50‐90, Mini‐Mental State Examination (MMSE) 12‐26, and FDG PET pattern of hypometabolism consistent with AD. Exclusion criteria included neurologic, radiologic, or laboratory indications of non‐AD dementia; medications that might interact with rasagiline; and factors that might preclude study completion. Patients on stable doses of cholinesterase inhibitors and memantine for at least 3 months prior to randomization were permitted.
TABLE 1.
Demographic, cognitive, and biomarker values by total population and age group.
| All participants | Model with predictors age, sex, APOE, MMSE a | By age group | Difference between age groups (p‐value) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameter | Age 57‐90 | Age p‐value | MMSE p‐value |
1: Age 57‐69 |
2: Age 70‐79 |
3: Age 80‐89 |
1:2 | 2:3 | 1:3 |
| Number (N) | 47 | 12 | 26 | 9 | |||||
| Demographic | |||||||||
| Age | 73.9 (6.9) | 65.0 (3.8) | 75.0 (3.0) | 82.8 (3.4) | p < 0.00001 | p < 0.00001 | p < 0.00001 | ||
| Sex (F/M) | 47%/53% | 50%/50% | 46%/54% | 44%/56% | ‐ | ‐ | ‐ | ||
| Education (years) | 14.2 (2.6) | 14.9 (2.2) | 14.0 (2.3) | 13.8 (3.7) | ‐ | ‐ | ‐ | ||
| APOE genotype %E4 + | 76% | 83% | 85% | 44% | ‐ | 0.02 | 0.04 | ||
| Cognitive | |||||||||
| MMSE | 20.1 (3.5) | 18.7 (4.0) | 20.4 (3.2) | 21.4 (3.2) | 0.10 | ‐ | 0.06 | ||
| MMSE age‐adjusted | 18.7 (4.0) | 20.9 (3.2) | 22.4 (3.2) | 0.07 | ‐ | 0.03 | |||
| ADAS‐cog | 24.9 (7.9) | ‐ | <0.0001 | 25.9 (9.7) | 25.6 (7.5) | 21.8 (6.6) | ‐ | ‐ | ‐ |
| Digit Span | 12.5 (3.1) | ‐ | 0.02 | 12.3 (3.6) | 12.5 (3.2) | 12.9 (2.3) | ‐ | ‐ | ‐ |
| COWAT | 24.4 (13.2) | ‐ | 0.0004 | 22.5 (17.9) | 23.9 (12.3) | 28.7 (8.1) | ‐ | ‐ | ‐ |
| NPI | 7.72 (8.27) | ‐ | ‐ | 7.33 (10.2) | 7.50 (7.28) | 9.00 (9.21) | ‐ | ‐ | ‐ |
| Tau PET SUVRs | |||||||||
| MetaTemporal | 1.74 (0.38) | 0.013 | 0.13 | 1.96 (0.42) | 1.69 (0.37) | 1.59 (0.18) | 0.05 | ‐ | 0.04 |
| Braak Stage I | 1.56 (0.21) | 0.18 | ‐ | 1.64 (0.25) | 1.54 (0.21) | 1.52 (0.14) | ‐ | ‐ | ‐ |
| Braak Stage III | 1.56 (0.28) | 0.05 | 0.10 | 1.71 (0.35) | 1.52 (0.26) | 1.46 (0.11) | ‐ | ‐ | 0.08 |
| Braak Stage IV | 1.52 (0.30) | 0.011 | ‐ | 1.71 (0.32) | 1.49 (0.29) | 1.38 (0.15) | 0.05 | ‐ | 0.01 |
| Braak Stage V | 1.41 (0.34) | 0.001 | 0.03 | 1.65 (0.37) | 1.37 (0.33) | 1.19 (0.15) | 0.07 | ‐ | 0.01 |
| Braak Stage VI | 1.11 (0.17) | 0.002 | 0.02 | 1.21 (0.16) | 1.09 (0.17) | 1.01 (0.10) | 0.04 | ‐ | 0.01 |
| Lateral temporal | 1.81 (0.45) | 0.012 | 0.09 | 2.07 (0.12) | 1.75 (0.08) | 1.60 (0.14) | 0.04 | ‐ | 0.03 |
| Parietal (angular) | 1.67 (0.54) | 0.004 | 0.11 | 1.97 (0.55) | 1.65 (0.55) | 1.34 (0.27) | 0.07 | ‐ | 0.01 |
| Precuneus | 1.62 (0.50) | 0.001 | ‐ | 1.88 (0.50) | 1.60 (0.52) | 1.29 (0.21) | 0.09 | ‐ | 0.01 |
| Middle frontal | 1.43 (0.48) | 0.0003 | 0.02 | 2.05 (1.12) | 1.70 (1.21) | 0.73 (0.94) | ‐ | 0.06 | 0.01 |
| Total cortical | 1.40 (0.30) | 0.001 | 0.03 | 1.61 (0.31) | 1.36 (0.28) | 1.21 (0.13) | 0.03 | ‐ | 0.01 |
| FDG PET SUVRs (ref pons) | |||||||||
| Medial temporal | 0.84 (0.09) | 0.013 | ‐ | 0.82 (0.07) | 0.82 (0.09) | 0.90 (0.08) | ‐ | 0.03 | 0.06 |
| Lateral temporal | 0.97 (0.11) | 0.09 | 0.03 | 0.91 (0.13) | 0.97 (0.10) | 1.04 (0.11) | ‐ | ‐ | 0.05 |
| Isthmus cingulate | 1.10 (0.15) | 0.001 | 0.03 | 1.03 (0.20) | 1.09 (0.11) | 1.23 (0.09) | ‐ | 0.00 | 0.03 |
| Parietal (angular) | 0.96 (0.16) | 0.015 | 0.001 | 0.90 (0.20) | 0.96 (0.14) | 1.06 (0.11) | ‐ | 0.06 | 0.03 |
| Precuneus | 1.21 (0.16) | 0.018 | 0.01 | 1.14 (0.21) | 1.20 (0.14) | 1.29 (0.07) | ‐ | 0.08 | 0.03 |
| Middle frontal | 1.14 (0.16) | 0.029 | 0.02 | 1.06 (0.19) | 1.15 (0.15) | 1.22 (0.12) | ‐ | 0.11 | 0.05 |
| Volume (MRI) z‐scores | |||||||||
| Hippocampus | −2.12 (1.53) | ‐ | 0.07 | −2.12 (1.27) | −2.13 (1.78) | −2.07 (1.14) | ‐ | ‐ | ‐ |
| Lateral temporal | −1.69 (0.94) | ‐ | 0.007 | −1.96 (0.99) | −1.69 (0.98) | −1.32 (0.70) | ‐ | ‐ | 0.11 |
| Parietal (angular) | −2.16 (1.18) | 0.005 | 0.09 | −2.57 (1.50) | −2.27 (0.91) | −1.28 (1.09) | ‐ | 0.03 | 0.05 |
| Precuneus | −1.71 (1.30) | ‐ | 0.11 | −1.82 (1.30) | −1.95 (1.20) | −0.88 (1.40) | ‐ | 0.03 | ‐ |
| Middle frontal | −1.58 (1.25) | 0.005 | ‐ | −2.07 (1.17) | −1.70 (1.21) | −0.58 (0.39) | ‐ | 0.03 | 0.01 |
| Total cortical | −2.65 (1.36) | 0.005 | 0.03 | −3.30 (0.36) | −2.96 (0.24) | −1.33 (0.41) | ‐ | 0.002 | 0.003 |
| Plasma biomarkers | |||||||||
| pTau‐181 pg/mL | 6.5 (3.1) | 0.002 | ‐ | 8.0 (2.8) | 6.2 (2.7) | 5.5 (1.0) | 0.06 | ‐ | 0.01 |
| GFAP pg/mL | 392 (158) | 0.004 | 0.03 | 371 (169) | 407 (168) | 377 (119) | ‐ | ‐ | ‐ |
| NfL b pg/mL | 26.6 (12.2) | <0.001 | 0.001 | 36.0 (10.0) | 25.1 (12.0) | 18.1 (7.1) | 0.004 | ‐ | 0.0004 |
Since covariates are combined with MMSE as an initial view of relationships to biomarkers, p‐values for MMSE will differ somewhat from those in Table 3, where covariates were combined with the biomarker to predict MMSE or ADAS‐cog; variables are adjusted for normal aging.
NfL values were not adjusted for creatinine clearance or renal function.
RESEARCH IN CONTEXT
Systematic review: The authors performed an extensive review of the literature using PubMed, additional internet searches, and conference abstracts and presentations in order to develop hypotheses and to compare the findings of this study to other published findings. Prior relevant publications are cited.
Interpretation: Our findings demonstrated significant relationships among plasma biomarkers, imaging measures, and cognitive status. Data also illustrated the heterogeneity present in mild to moderate Alzheimer's disease (AD) populations and covariate influences to be considered when designing clinical trials or using plasma and imaging biomarkers to predict tau positron emission tomography (PET) levels for patient selection and analysis.
Future directions: Future work would involve (a) confirmation in a larger study toward use in patient characterization for clinical trials and care, (b) work toward reducing variability in plasma biomarkers of neuroinflammation, and (c) further establishment of normative references for plasma biomarkers.
Of 96 patients screened, 50 patients, randomized 1:1 rasagiline to placebo, were enrolled. Our analyses used baseline data from all participants for cross‐sectional comparisons and from placebo‐treated participants to explore relationships to cognitive change. After excluding those with missing plasma biomarker data and follow‐up data (for longitudinal analyses), our analyses included 47 participants at baseline and 21 placebo‐treated participants for longitudinal comparisons.
2.2. Data acquisition and measurement
Clinical endpoints acquired at screening or baseline and at timepoints up to 26 weeks included MMSE, Alzheimer's Disease Assessment Scale–Cognitive Subscale (ADAS‐cog), Digit Span, Controlled Oral Word Association Test (COWAT), and Neuropsychiatric Inventory (NPI). 4 Volumetric MR (T1‐weighted) images were acquired at screening, FDG PET at screening and 24 weeks, and flortaucipir PET images at baseline and 24 weeks. Plasma pTau‐181, NfL, and GFAP were collected at baseline and at week 24; a 48‐plex panel of inflammatory biomarkers was collected at baseline (methods in Supplement).
2.3. Analyses
2.3.1. Baseline characterization
Baseline demographic, cognitive, imaging, and plasma biomarker distributions were characterized for the study population and three decade‐based age subgroups (57‐69, 70‐79, 80‐90 years; see Supplement). Tau PET standardized uptake value ratios (SUVRs) referenced to cerebellar cortex (eroded to reduce spill‐in) were measured in a MetaTemporal composite, 32 Braak stage composites, 33 total cortex, and individual regions. FDG PET and brain volumes were measured in the same regions as tau PET, including regions known to exhibit progressive hypometabolism and atrophy in AD.
Tau PET and FDG PET measures were adjusted for normal aging using data acquired using the same tracers and protocols in amyloid‐negative cognitively unimpaired individuals as previously described. 4 Volumetric measures were adjusted for intracranial volume, age, sex, and scanner using a normative reference of amyloid‐negative cognitively unimpaired individuals as implemented in ADM Diagnostics’ CorInsights MRI® software. Plasma pTau‐181, NfL, and GFAP were adjusted for normal aging using slopes derived from amyloid‐negative cognitively impaired individuals in published studies using the same assays. 18 , 22 , 24 , 34 Normative aging data were not available for inflammatory biomarkers (Supplement).
2.3.2. Relationships between biomarkers
Inter‐region tau PET relationships were first examined as influenced by age and other covariates, focusing on relationships with inferior temporal cortex as it is an early region of NFT spread, used in criteria for clinical trial participation. 35 To test our predefined hypotheses in this primarily tau PET+ (98%) population, we examined relationships between (a) plasma pTau‐181 and tau PET by region and cortical average, (b) plasma NfL and FDG PET in AD‐associated regions (medial temporal, lateral temporal, isthmus cingulate, inferior parietal, middle frontal), and (c) all biomarkers versus cognitive endpoints at baseline and 24‐week change. We similarly evaluated relationships between tau PET and NfL, GFAP, FDG PET, brain volumes, and combined plasma and FDG PET or volumetric biomarkers. As part of these investigations, we compared left‐right hemispheric imaging asymmetries as these ratios remove reference region effects, reducing technical variability.
The above relationships were evaluated using model fitting (standard least squares) in JMP v16 statistical software (SAS). Distributions were examined for normality and variables not meeting this condition (Shapiro‐Wilk and Anderson‐Darling tests, significance p < 0.05) were log10 transformed (all plasma variables were transformed). Covariates of age, sex, APOE genotype; education for cognitive endpoint comparisons; and cognitive baseline values when evaluating 24‐week cognitive change were included in models when significant or trend level. Models were also evaluated within each age decade to minimize age effects (particularly for inflammatory markers where normal aging data were not available) and age correction effects. Nonlinear models and variable interactions were also investigated. Pearson correlation was applied except for non‐normal distributions or group sizes below fifteen, where Spearman's Rank Order (rho) or nonparametric Wilcoxon tests were used. Given the exploratory nature of this work, numerous comparisons were made and significance was considered p < 0.05, uncorrected for multiple comparisons; p‐values <0.0001 are likely candidates to survive Bonferroni correction. To further illustrate relationship patterns among the imaging biomarkers, we derived voxel‐based multivariate machine learning classifiers as described and shown in the Supplement.
For cross‐correlation heat maps, plasma variables with a coefficient of variation (standard deviation/mean) greater than 20% after log10 transformation were excluded. Spearman's Rank Order (rho) correlation was used to generate the younger and oldest age group maps due to their lower subject numbers.
We also examined whether pTau‐181 could have been used as a screen for tau PET positivity. Participants were considered tau PET+ if their bilateral MetaTemporal SUVR (without partial volume correction) exceeded 1.29 32 , 36 or other target region SUVRs were positive. A cutoff was estimated as the value above which all tau PET+ scans were found and compared to values reported for A‐ and A+ MCI subjects in a study using the same assay. 5
3. RESULTS
3.1. Baseline characteristics
Table 1 and Supplemental Table S1 list baseline values for demographic, cognitive, imaging, and plasma measures overall and by age group, with examples in Figure 1. p‐values are listed for age and MMSE (representing clinical severity), which were included as model inputs along with sex and APOE 4 status.
FIGURE 1.
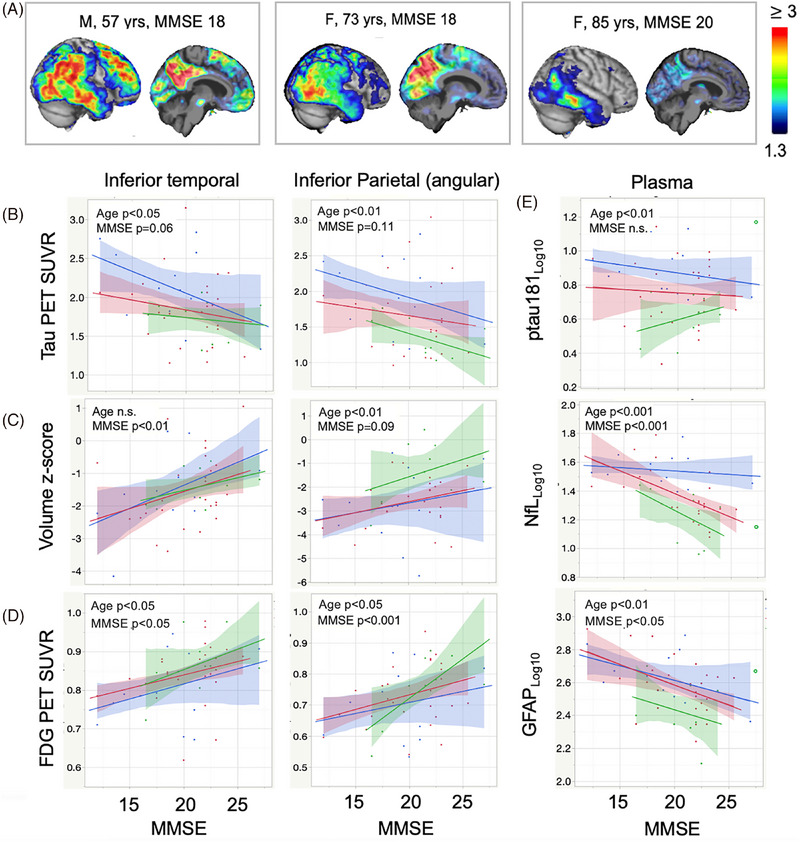
Imaging and plasma biomarker distributions and relationships to MMSE and age. (A) Flortaucipir SUVR images for three subjects of varying age and tau spatial extent. (B‐D) Relationships between tau PET, volume, and FDG PET with MMSE score, stratified by age‐decade (57‐69, blue; 70‐79, red; 80‐90, green). (E) Relationships of pTau‐181, NfL, and GFAP to MMSE stratified by age‐decade (same color table). Overall correlations are apparent as well as an age effect within the biomarker‐clinical severity relationships despite adjustment for normal aging. The unfilled green circle is for one participant in the 80‐ to 90‐year group whose plasma values were outliers and are not included in the line of fit. “n.s.” = not significant (p‐values between 0.05 and 0.12 listed as a trend).
Participants (N = 47) ranged from age 57–90 years (74 6.9); 54% were female and 78% were APOE4 carriers. Baseline cognitive scores varied as: MMSE (20.1 3.5, range 12‐27), ADAS‐cog (24.9 8.0, range 9‐50), COWAT (24.4 13.2, 0‐60), Digit Span (12.5 3.1, 6‐21), and NPI (7.7 8.3, 0‐32). Cognitive scores were unaffected by age, sex, and APOE genotype except MMSE scores were slightly higher in older participants (p = 0.04). APOE4 carrier percentage was lower in participants aged 80–90 (44%) versus 57–69 (83%) and 70–79 (85%).
Tau PET patterns were consistent with Braak staging 33 , 37 and heterogeneous in intensity, extent, and asymmetry. Uptake was most pervasive in younger participants, variable at age 70–79, more limited in participants age 80–90 (Figure 1A), and inversely related to MMSE (Table 1, Figure 1). Age‐related differences were observed increasingly in Braak Stages IV and later with greatest disparity in anterior cingulate and frontal regions such as middle frontal cortex (R = 0.58, p = 0.00002). Temporoparietal and middle frontal glucose metabolism and volumes characteristic of AD were reduced compared to cognitively unimpaired, amyloid‐negative reference values, and more pronounced with younger age (Table 1, Figure 1C,D) in frontoparietal regions (individual examples in Supplemental Figure S1).
Age‐adjusted pTau‐181, NfL, and GFAP (Figure 1E) were elevated with higher concentrations in younger participants. Sex was significant only for GFAP and CTAK (females > males, p = 0.0005, p = 0.008). APOE genotype was significant for SDF‐1alpha (e4 Noncarriers > Heterozygous > Homozygous, p = 0.005).
3.2. Tau PET inter‐region relationships
Tau PET values correlated between regions, particularly in the same hemisphere, diminishing with later Braak stages and where uptake varied with age. Left inferior temporal SUVRs correlated with left middle temporal (R = 0.95), fusiform (R = 0.92), superior temporal (R = 0.91), inferior parietal (R = 0.87), isthmus cingulate (R = 0.87), middle frontal (R = 0.82), precuneus (R = 0.81), and lateral occipital regions (R = 0.67), p‐values <0.00001. Average inferior temporal SUVRs correlated with MetaTemporal (R = 0.99) followed by Braak Stage IV (R = 0.97), Cortical Average (R = 0.92), Braak Stage V (R = 0.88), and Braak Stage VI (R = 0.75) (p‐values <0.00001).
3.3. Relationships of plasma and imaging biomarkers to tau PET
3.3.1. pTau‐181 association with tau PET
Plasma pTau‐181 levels correlated positively with tau PET (Table 2, Figure 2A) in regions including Inferior Temporal (left p = 0.0002, bilateral p = 0.0008), MetaTemporal (p = 0.01), Cortical Average (p = 0.04), and Braak Stage III (p = 0.01), IV (p = 0.01), and V (p = 0.04). Except for medial and inferior temporal regions, for the same pTau‐181 concentrations regional tau PET was greater with younger age (Figure 2). Strongest correlations between pTau‐181 and tau PET were in temporal cortex in older age groups and in Cortical Average and Braak Stage V regions in the youngest group (Figure 2A). One 84‐year‐old had a high pTau‐181 value corresponding only with their lateralized left inferior temporal tau and was otherwise an outlier (examples in Supplemental Figure S1).
TABLE 2.
Relationships between tau PET and plasma biomarkers, FDG PET, and volume (MRI).
| pTau‐181 | GFAP | FDG PET | Volume | Volume + pTau‐181 | Volume + GFAP | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | R a | p b | R a | p b | R a | p b | R a | p b | R a | R a |
| Inferior temporal: L | 0.52 | 0.0002 | 0.48 | 0.0006 | 0.48 | 0.0008 | 0.70 | p < 0.00001 | 0.76 | 0.75 |
| Inferior temporal | 0.47 | 0.0008 | 0.44 | 0.002 | 0.48 | 0.0007 | 0.68 | p < 0.00001 | 0.74 | 0.73 |
| Middle temporal | 0.57 | 0.01 | 0.53 | 0.04 | 0.56 | 0.00004 | 0.73 | p < 0.00001 | 0.80 | 0.77 |
| Meta temporal | 0.53 | 0.01 | 0.50 | 0.03 | 0.50 | 0.0003 | 0.71 | 0.00002 | 0.74 | n.i. |
| Cortical average | 0.63 | 0.04 | 0.60 | 0.009 | 0.68 | 0.002 | 0.72 | 0.00004 | 0.75 | 0.73 |
| Braak III | 0.52 | 0.01 | 0.46 | 0.01 | 0.55 | 0.004 | 0.72 | 0.00002 | 0.73 | n.i. |
| Braak IV | 0.53 | 0.01 | 0.50 | 0.03 | 0.52 | 0.001 | 0.67 | p < 0.00001 | 0.68 | n.i. |
| Braak V | 0.57 | 0.04 | 0.60 | 0.008 | 0.68 | 0.002 | 0.71 | p < 0.00001 | 0.75 | 0.74 |
| Braak VI | ‐ | ‐ | ‐ | ‐ | 0.57 | 0.01 | 0.60 | 0.01 | n.i. | n.i. |
| Amygdala | 0.39 | 0.04 | ‐ | ‐ | 0.37 | 0.10 | 0.46 | 0.004 | 0.49 | 0.48 |
| Inferior parietal | 0.56 | 0.02 | 0.54 | 0.03 | 0.65 | 0.002 | 0.75 | p < 0.00001 | n.i. | n.i. |
| Isthmus cingulate | 0.57 | 0.07 | 0.59 | 0.04 | 0.64 | 0.003 | 0.69 | 0.0001 | 0.72 | 0.71 |
| Precuneus | ‐ | ‐ | 0.53 | 0.07 | 0.68 | 0.001 | 0.73 | 0.00001 | n.i. | n.i. |
| Middle frontal c | 0.61 | 0.11 | 0.64 | 0.003 | 0.75 | p < 0.00001 | 0.75 | p < 0.00001 | n.i. | 0.78 |
| Lateral occipital | ‐ | ‐ | ‐ | ‐ | 0.62 | 0.0003 | 0.60 | 0.0005 | n.i. | n.i. |
| Asymmetry inf temp | 0.68 | p < 0.00001 | 0.60 | 0.00001 | ||||||
| Asymmetry inf parietal | 0.60 | p < 0.00001 | 0.63 | 0.00001 | ||||||
R is an absolute square root of R‐squared value for model, including age, sex, and APOE genotype where significant or trend.
p‐value is the value associated with the biomarker after correction for age, sex, and APOE genotype, and therefore does not always correspond to the overall R‐value for each model; “‐” means neither significant nor trend; “n.i.” means that the addition of the plasma marker did not improve Model R‐value as compared to volume only; analyses are limited to univariate relationships and do not reflect prediction that may be obtained with multivariate models.
Correlation values are for FDG PET and volumetric regions that had greatest correspondence to the tau PET region listed.
FIGURE 2.
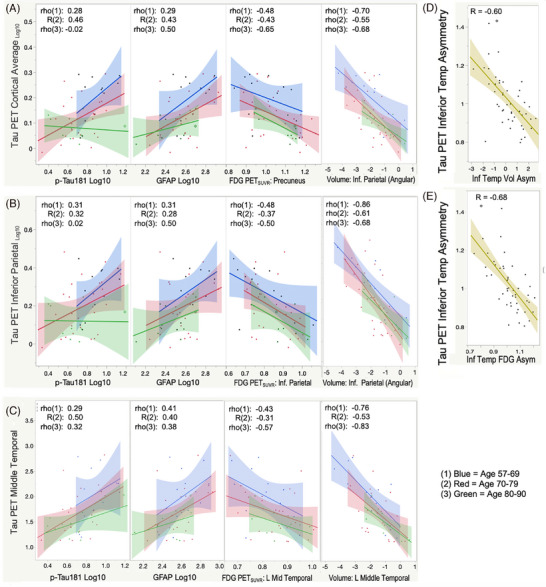
Relationships between tau PET and other imaging and plasma biomarkers. Relationship between tau PET in (A) total cortical, (B) inferior parietal, and (C) left inferior temporal regions as compared to pTau‐181log10, GFAPlog10, regional FDG SUVR, and regional volumes. Correlation coefficients are shown for the three age‐decade groups: (1) 57–69 years (blue), (2) 70–79 years (red), (3) 80–90 years (green). (D, E) The relationships between hemispheric asymmetry in the inferior iemporal region for tau PET as compared to volume and FDG PET, respectively.
In this population, pTau‐181 values could have been used to exclude one tau PET‐negative participant. Using a cutoff of 1.29 for inferior temporal tau SUVR that included all tau PET+ individuals, 1 of 46 would have been a false positive.
3.3.2. GFAP association with tau PET
Plasma GFAP concentrations correlated positively with tau PET, strongest in inferior temporal cortex (left p = 0.0006, bilateral p = 0.002), similar significance to pTau‐181 (Table 2, Figure 2B). Association slopes in later Braak stage regions were more consistent across age groups than for pTau‐181 but for the same GFAP values, tau PET was greater with lower age.
3.3.3. Other plasma associations with tau PET
Plasma NfL associations with tau PET were limited to regions including Braak stage V (p = 0.05), middle frontal (p = 0.03), precuneus (p = 0.07), and inferior parietal (p = 0.09). Most inflammatory plasma biomarkers did not correlate with tau PET, but negative correlations were observed in the youngest group for Eotaxin in MetaTemporal (R = −0.75, p = 0.003) and Cortical Average (R = −0.60, p = 0.03), and for IL‐7 in MetaTemporal (R = −0.57, p = 0.04).
3.3.4. Imaging associations with tau PET
FDG PET and volumetric MRI measures correlated negatively with tau PET (Table 2, Figure 2, Supplemental Figure S2), significant after correction for multiple comparisons in several regions. Tau PET left‐right hemisphere asymmetries in inferior temporal and inferior parietal regions correlated with asymmetries observed in volume and glucose metabolism (R = 0.60–0.68, p‐values <0.00001) (image association patterns in Supplemental Figure S3).
3.3.5. Combined imaging and plasma biomarkers
Combining temporal volumetric measures with either pTau‐181 or GFAP further increased model association with tau PET (R = 0.80, p < 0.00001) over the variables individually (pTau‐181 R = 0.53, volume R = 0.73) (Table 2). For prediction of tau PET uptake in later‐stage regions (e.g., parietal, middle frontal), plasma biomarkers did not add further to volumetric models.
3.4. Relationships of plasma and imaging biomarkers to FDG PET
Plasma NfL correlated with FDG PET as hypothesized, with significant relationships in precuneus (R = −0.51, p = 0.0003), inferior parietal (R = −0.40, p = 0.0005), isthmus cingulate (R = −0.59, p = 0.02), middle frontal (R = −0.53, p = 0.0002), and lateral temporal regions (R = −0.36, p = 0.01), similar across age groups. GFAP correlated with FDG PET in lateral temporal (R = −0.37, p = 0.009) and middle frontal regions (R = −0.57, p = 0.003). Plasma pTau‐181 correlated with FDG PET only in left inferior temporal cortex (R = −0.47, p = 0.001), where the relationship between pTau‐181 and tau PET was also most significant. Volumes correlated with FDG PET for all regions tested (R‐values 0.60–0.51, p‐values 0.00004–0.003). Volumetric left‐right hemispheric asymmetries correlated with left‐right asymmetries in FDG PET in inferior temporal (R = 0.74, p < 0.00001) and inferior parietal cortices (R = 0.77, p < 0.00001). (Supplemental Table S2, Figure S2)
3.5. Relationships between plasma biomarkers
Associations were found for the overall population between GFAP and NfL (R = 0.64, p = 0.0001), GFAP and pTau‐181 (R = 0.62, p = 0.0001), and NfL and pTau‐181 (R = 0.46, p = 0.001). Age influenced the GFAP to NfL relationship with younger participant values positively (Figure 3A). GFAP correlated with pTau‐181 most strongly in the youngest group (Figure 3B). PTau‐181 correlated with NfL in the youngest group (R = 0.71), and somewhat in the middle age group (R = 0.40), but not in the oldest group.
FIGURE 3.
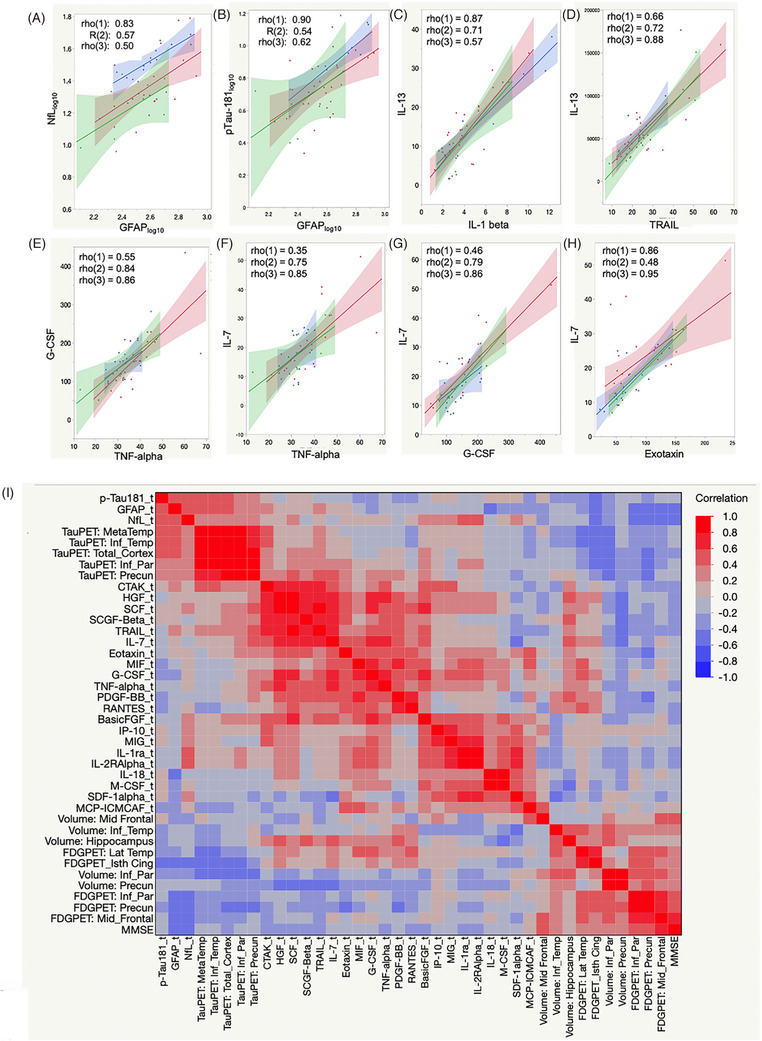
Plasma biomarker relationships. Plasma biomarker relationships are shown by age group (A‐H) and in an overall comparison that includes imaging biomarkers shown for the 70‐ to 79‐year age group (I). (“_t” in variable names indicates log10 transformation).
Figure 3C‐H shows inflammatory plasma biomarker relationships that were consistent across all age groups: IL‐13 versus IL‐1 beta and TRAIL, TNF‐alpha versus G‐CSF and IL‐7, and IL‐7 versus G‐CSF and Exotaxin. Figure 3I presents a heat map showing relationships between all plasma and selected imaging biomarkers and MMSE for the 70‐ to 79‐year‐old age group (all groups in Supplemental Figure S4). Within all age groups, relationships are most prominent among NfL, GFAP, pTau‐181 and imaging biomarkers, and among inflammatory biomarkers, which also exhibit subclusters (Figure 3I). Correlations between tau PET and inflammatory biomarkers Eotaxinlog10, IL‐7, and MCP‐1 were noted only in the youngest group.
3.6. Relationships to cognitive endpoints
Several biomarkers correlated with cognitive endpoints (Table 3), including: NfL and GFAP with baseline MMSE (R = 0.50, p = 0.0005; R = 0.39, p = 0.007) and ADAS‐cog (R = 0.30, p = 0.04; R = 0.37, p = 0.01); Tau PET with baseline MMSE in regions including total cortical (R = 0.37, p = 0.009) and middle frontal (R = 0.42, p = 0.002), which correlated with ADAS‐cog (R = 0.40, p = 0.006); FDG PET with MMSE and ADAS‐cog in regions including precuneus (R = 0.41, p = 0.004; R = 0.40, p = 0.005) and caudal middle frontal (R = 0.45, p = 0.002; R = 0.37, p = 0.01); and volumes with MMSE in lateral temporal (R = 0.42, p = 0.005) and temporoparietal regions. Combining NfL or GFAP with volume resulted in the strongest relationships with MMSE (R = 0.58, p = 0.0002; GFAP not significant when NfL included).
TABLE 3.
Relationships between biomarkers and cognitive endpoints at baseline and 24‐week change.
| Parameter | Baseline (N = 47) | 24‐Week change (N = 21) | ||||||
|---|---|---|---|---|---|---|---|---|
| MMSE | ADAS‐cog | MMSE | ADAS‐cog | |||||
| R | p * | R | p * | R | p * | R | p * | |
| Plasma biomarkers | ||||||||
| pTau‐181 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| NfLlog10 | −0.50 | 0.0005 | 0.30 | 0.04 | ‐ | ‐ | ‐ | ‐ |
| GFAPlog10 | −0.39 | 0.007 | 0.37 | 0.01 | ‐ | ‐ | ‐ | ‐ |
| Eotaxin log10 | ‐ | ‐ | ‐ | ‐ | 0.58 | 0.01 | ‐ | ‐ |
| G‐CSF1 log10 | ‐ | ‐ | ‐ | ‐ | 0.68 | 0.008 | ‐ | ‐ |
| HGF log10 | ‐ | ‐ | ‐ | ‐ | 0.66 | 0.007 | ‐ | ‐ |
| IL‐7 log10 | ‐ | ‐ | ‐ | ‐ | 0.65 | 0.008 | ‐ | ‐ |
| TNF‐alpha log10 | ‐ | ‐ | ‐ | ‐ | 0.57 | 0.006 | ‐ | ‐ |
| Tau PET | ||||||||
| Parahippocampus | ‐ | ‐ | ‐ | ‐ | 0.68 | 0.0007 | ‐ | ‐ |
| Meta temporal | −0.28 | 0.05 | ‐ | ‐ | 0.62 | 0.003 | ‐ | 0.10 |
| Total corticallog 10 | −0.37 | 0.009 | 0.28 | 0.05 | 0.46 | 0.04 | ‐ | ‐ |
| Middle frontallog10 | −0.44 | 0.002 | 0.40 | 0.006 | ‐ | ‐ | ‐ | ‐ |
| Braak Stage III | −0.33 | 0.02 | ‐ | 0.10 | 0.71 | 0.0004 | ‐ | ‐ |
| Braak Stage IV | ‐ | ‐ | ‐ | ‐ | 0.62 | 0.002 | ‐ | 0.10 |
| Braak Stage Vlog 10 | −0.37 | 0.009 | 0.32 | 0.03 | ‐ | ‐ | ‐ | ‐ |
| FDG PET (reference pons) | ||||||||
| Medial temporal | ‐ | ‐ | ‐ | ‐ | 0.52 | 0.02 | ‐ | ‐ |
| Lateral temporal | 0.36 | 0.01 | ‐ | 0.07 | 0.42 | 0.05 | 0.69 | 0.006 |
| Isthmus cingulate | 0.39 | 0.008 | ‐ | 0.09 | ‐ | ‐ | ‐ | ‐ |
| Caudal middle frontal | 0.45 | 0.002 | −0.37 | 0.01 | ‐ | ‐ | 0.60 | 0.01 |
| Precuneus | 0.41 | 0.004 | −0.40 | 0.005 | ‐ | ‐ | ‐ | ‐ |
| Inf. parietal (angular) | 0.42 | 0.003 | −0.36 | 0.01 | ‐ | ‐ | 0.57 | 0.05 |
| Volume | ||||||||
| Hippocampus | ‐ | ‐ | ‐ | ‐ | 0.62 | 0.01 | ‐ | ‐ |
| Parahippocampus | 0.33 | 0.08 | ‐ | ‐ | 0.78 | 0.0006 | 0.61 | 0.03 |
| Lateral temporal | 0.42 | 0.005 | 0.32 | 0.03 | 0.66 | 0.0002 | 0.81 | 0.0001 |
| Middle frontal | ‐ | ‐ | ‐ | 0.09 | ‐ | ‐ | ‐ | 0.07 |
| Inferior parietal | 0.32 | 0.03 | ‐ | ‐ | ‐ | ‐ | 0.63 | 0.01 |
| TemporoParietal combination | 0.40 | 0.005 | 0.30 | 0.04 | 0.62 | 0.005 | 0.67 | 0.008 |
| TempParFrontal combination | 0.42 | 0.003 | 0.33 | 0.02 | 0.54 | 0.02 | 0.76 | 0.0007 |
| Total cortical | 0.37 | 0.01 | 0.30 | 0.04 | ‐ | ‐ | 0.71 | 0.003 |
| Volume + Plasma biomarker | ||||||||
| Lateral temporal + NfL | 0.58 | 0.0002 | 0.41 | 0.02 | n.i. | n.i. | n.i. | n.i. |
| Lateral temporal + GFAP | 0.51 | 0.001 | 0.46 | 0.02 | n.i. | n.i. | n.i. | n.i. |
| TempParFront + NfL | 0.57 | 0.0002 | 0.49 | 0.02 | n.i. | n.i. | n.i. | n.i. |
| TempParFront + GFAP | 0.50 | 0.002 | 0.49 | 0.02 | n.i. | n.i. | n.i. | n.i. |
p‐values are after adjustment for covariates including age, sex, education, APOE e4 carrier status, and (for longitudinal cognitive change) baseline cognitive values; R‐values are for the model including the biomarker(s) and covariates; “‐” is neither significant nor trend; TempPar comb. = combination of temporal and parietal volumes; TempParFront combination = combination of temporal, parietal, and frontal volumes; “n.i.” means no improvement to combination as compared to variables alone.
Parahippocampal and lateral temporal tau PET and volumes were most predictive of 24‐week decline in MMSE (R = 0.72, p = 0.0005; R = 0.73, p = 0.0006). Lateral temporal FDG PET and volume were most predictive of 24‐week decline in ADAS‐cog (R = 0.69 p = 0.006; R = 0.81, p = 0.0002). Inflammatory biomarkers listed in Table 3 showed relationships to subsequent cognitive change but did not relate to baseline cognition.
4. DISCUSSION
This post hoc exploratory study examined biomarkers of pathology, function, neurodegeneration, and inflammation within the same mild to moderate AD population, providing several insights. It illustrates the substantial diversity across all aspects of the “A/T/N + I” framework and clinical status present in a mild to moderate AD dementia population. Findings supported our hypotheses that pTau‐181 would correlate with tau PET and that NfL would correlate with FDG PET in this population. GFAP related to tau PET similarly to pTau‐18, and to cognition. Strong regional relationships were found between volumes, FDG PET, and tau PET, corroborating findings in other studies. 14 , 15 , 16 , 17 , 38 Age‐related differences beyond those attributable to normal aging were observed within and between biomarkers, which may underlie different clinical trajectories. 39
One novel aspect of our work was the comprehensive set of biomarkers, including inflammatory markers, acquired at the same timepoint. In addition, while relationships between pTau‐181, GFAP, and NfL plasma biomarkers and imaging biomarkers have been demonstrated across broad ranges of clinical severity and longitudinal progression, 9 , 17 our results show that these relationships hold within this later stage of mild to moderate AD. The correlation between plasma pTau‐181 and tau PET is of note because it was within a primarily tau‐positive, demented population rather than a spectrum of unimpaired or early prodromal individuals and tau‐positive dementia patients.
Our work also sought an understanding of implications for tau spatial distribution and burden versus plasma biomarkers as influenced by age. The findings that for different age groups, the same pTau‐181 concentration predicted different local tau burden in later‐stage Braak regions and correlated most strongly with different spatial patterns suggest that age, in addition to AD variants, 14 , 38 should be considered if attempting to predict NFT distribution from plasma values. Further, the strong relationships between regional volume and tau PET suggest that plasma‐based pathology confirmation can be combined with volumetric measurement to provide greater insight to NFT distribution. The participant with high lateralized temporal tau PET uptake, low cortical uptake, and highest pTau‐181 values, if not an outlier, raised questions regarding pTau and NFT burden relationships to be addressed by studying a larger number of similar cases.
Plasma GFAP correlated with tau PET, and did so more consistently in oldest participants than pTau‐181. Although GFAP is associated with amyloid, it also correlates with cognitive decline, which is associated with tau, in amyloid‐positive patients. 40 NfL was not associated with tau PET but correlated with FDG PET, and with pTau‐181 in the youngest age group. NfL is a nonspecific marker of neurodegeneration, and while FDG PET patterns reflect disease type and phenotype, 41 hypometabolism also reflects overall neuronal damage arising from comorbidities. 42
The distinct clusters observed for (a) neurodegenerative (pTau‐181, GFAP, NfL) and (b) inflammation plasma biomarkers were consistent with a study of a subset of these markers in other AD cohorts. 30 This clustering was consistent across age groups, with inflammatory subclusters that varied by age group. Inflammation biomarkers (Eotaxin, IL‐7, MCP‐1) related inversely to tau PET only in the youngest subgroup. While four inflammation biomarkers (including Eotaxin and IL‐7, Table 3) correlated with subsequent change in MMSE, directionality was not consistent with literature associating higher levels with greater clinical decline and greater pathology. 43 , 44 Further study is warranted, including earlier disease stages.
Findings suggested that in this later‐stage population, volumetric measures were more useful in predicting cognitive decline than plasma markers. FDG and tau PET showed predictive association and may have greater sensitivity in earlier stages of disease, and with alternate approaches to tau PET measurement. 45 Plasma biomarkers may be predictive with longer follow‐up, given short‐term cognitive measure variability.
Our work corroborated and extended upon prior intermodality relationship findings, focusing on this later‐stage population. The spatial variation and age association of tau PET were consistent with previous studies 10 , 21 , 46 , 47 as was the correspondence between tau PET, FDG PET, and volumetric MRI. 14 , 17 , 38 Our results suggest that, within this population, volumetric measures can be combined with pTau‐181 and GFAP, with consideration to age, to increase prediction of likely tau spatial distribution. Given its potential benefit in clinical trial and diagnostic screening, this is an area for further research, using multivariate machine learning approaches.
There are limitations in this exploratory study. The sample size was small with 47 participants available for baseline comparisons and 21 participants to compare cognitive change. Amyloid PET was not available; however, based on other studies, positivity may be inferred in those who are tau PET positive. 48 Since there were no amyloid‐negative cognitively unimpaired controls, reference data were used to establish normal aging adjustments; these were not available for inflammatory markers, mitigated by comparisons within age subgroups. Since most participants were tau positive, demonstration of plasma‐based selection was limited. While a commercial platform was used for plasma measurements, cross‐site and cross‐platform variability are undergoing industry refinement and comparisons to other studies require caution. Ptau‐217, not measured, has advantages over pTau‐181 including amyloid sensitivity and tau specificity 49 though in this later stage, population results may be similar. 10 , 21 , 46 , 47 Renal function can affect pTau‐181 and NfL measures and could be evaluated next. 50 Our results apply to a mild to moderate AD population and findings may differ in earlier stages with less NFT burden and neurodegeneration.
The associations between pTau‐181, GFAP, FDG, and regional atrophy as compared to tau PET and cognitive status observed in this study demonstrate the potential to efficiently characterize and stratify both pathology and neurodegenerative effects in AD patients for clinical trials and care. The inflammatory biomarker clustering observed provides a basis for further investigation of the role of “I” in the A/T/N/X framework.
AUTHOR CONTRIBUTIONS
J. Cummings designed the rasagiline study, served as Principal Investigator, and provided manuscript input and editing. J. Kinney collaborated on plasma analysis study design, performed the plasma biomarker and APOE genotyping assays, and provided manuscript input. A. Ritter served as rasagiline study co‐investigator and provided manuscript review and input. R. Andrews performed quality control, processing, and analyses, and provided manuscript review. L. Koenig provided manuscript review and assistance. E Toledano Strom assisted with the plasma biomarker assays. A. Lukic provided the software for data processing and analysis, and manuscript input. K. Zhong provided input to the study protocol and manuscript review. J. Leverenz and B. Tousi served as clinical investigators. Howard Feldman, and Carolyn Revta provided study data coordination by the ADCS during the trial and contributed to editing and critical feedback on all versions of the manuscript. H. Fillit provided input to study design and manuscript review.
CONFLICT OF INTEREST STATEMENT
Dawn Matthews is CEO of ADM Diagnostics, Inc., Scientific Advisor to the Alzheimer's Drug Development Foundation, and Co‐Chair of the Radiological Society of North America PET Tau Profile Working Group and PET Amyloid Profile Working Group, and reports that ADM Diagnostics received grant funding (2R44AG060861‐02) from the National Institutes of Aging, University of Arizona subaward (R01 AG063826), and University of Arizona subaward (R01 AG075122‐01 during the conduct of this work. She reports support for attending meetings and/or travel hotel and registration fees from Alzheimer's Drug Discovery Foundation, and stock or stock options in ADM Diagnostics and Abiant. Jefferson Kinney reports a grant from the National Institutes of General Medical Sciences (NIGMS) COBRE grant #P20GM109025, along with grants NIH NIA R01 AG074392‐01, NIH NIA P20 AG068053‐01, and R01AG062762‐01A1. Aaron Ritter reports payment from Corium Pharmaceuticals and Lundbeck/Otsuka for speaker board and advertisement board. He also has a leadership role in Alzheimer's Association Orange County Board and Alzheimer's Family Center. Ana Lukic, Randolph Andrews, and Lauren Koenig are employees of ADM Diagnostics, Inc. Carolyn Revta has nothing to disclose. Babak Tousi received a grant from the Alzheimer's Drug Discovery Foundation during the study, and has received consulting fees from Eisai, Biogen, Lilly, GE Healthcare, Kisbee, and Otsuka. He received honoraria from Eisai and Lily, support for attending meetings or travel from Novo Nordisk, and participated in a Data Safety Monitoring Board or Advisory Board at Eisai and Lilly. James Leverenz reports the grant P30AG072959, a Scientific Advisory leadership role for the Lewy Body Dementia Association, and receipt of ligands and software for imaging from GE Healthcare. Howard Fillit founding Executive Director and Chief Science Officer of the Alzheimer's Drug Discovery Foundation, which funded the rasagiline clinical trial, and has provided consulting support to the following pharmaceutical companies: Otsuka, LifeWorx, and Alector. He reports royalties from Icahn School of Medicine at Mount Sinai, and participation in a DSMB at Alector. He reports stock or stock options at CarePredict, and other non‐financial interests with the Lilly GERAS study and Roche Genentech Claims study. Howard Feldman reports a service agreement through UCSD with the Cleveland Clinic for data management and biostatistics during the conduct of the original study; grants from Annovis (QR Pharma), Vivoryon (Probiodrug), AC Immune, Biohaven Pharmaceuticals, and LuMind Foundation, and service agreements through UCSD for consulting with LuMind Foundation, Novo Nordisk, Arrowhead Pharmaceuticals, and Axon Neurosciences Roche/Genentech Pharmaceuticals. He reports support for attending meetings and/or travel for Novo Nordisk, and Royal Society of Canada; participation in DMC and DSMB for Tau Consortium, Rooche/Genentech Pharmaceuticals, and Janssen Research and Development; and philanthropic support for Alzheimer therapeutic research. Jeffrey Cummings reports grants from the National Institutes of General Medical Sciences (NIGMS) COBRE grant #P20GM109025; NINDS grant U01NS093334; NIA grant R01AG053798; NIA grant P30AG072959; NIA grant R35AG71476; NIA R25 AG083721‐01; Alzheimer's Disease Drug Discovery Foundation (ADDF); Ted and Maria Quirk Endowment; Joy Chambers‐Grundy Endowment during the conduct of the study. He reports consulting fees from Acadia, Actinogen, Acumen, AlphaCognition, ALZpath, Aprinoia, AriBio, Artery, Biogen, Biohaven, BioVie, BioXcel, Bristol‐Myers Squib, Cassava, Cerecin, Diadem, Eisai, GAP Foundation, GemVax, Janssen, Jocasta, Karuna, Lighthouse, Lilly, Lundbeck, LSP/eqt, Mangrove Therapeutics, Merck, NervGen, New Amsterdam, Novo Nordisk, Oligomerix, ONO, Optoceutics, Otsuka, Oxford Brain Diagnostics, Prothena, ReMYND, Roche, Sage Therapeutics, Signant Health, Simcere, sinaptica, Suven, TrueBinding, Vaxxinity, and Wren pharmaceutical, assessment, and investment companies. He reports participation in Data Safety Monitoring Board or Advisory Board for Acadia, Biogen, Genentech, Grifols, Janssen, Karuna, Otsuka, reMYND, Roche, Signant Health and stock or stock options for Artery, Vaxxinity, Behrens, Alzheon, MedAvante‐Prophase, and Acumen. Finally, outside the submitted work he is Chief Scientific Advisor to CNS Innovations, LLC and owns the copyright for the Neuropsychiatric Inventory (NPI). Kate Zhong and Erin Toledano Strom have nothing to disclose. Author disclosures are available in the supporting information.
CONSENT FOR PUBLICATION
All appropriate author consents have been obtained. Other consent is not applicable.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We are grateful to the patients, families, and caregivers who participated in the study, and whose involvement makes these learnings possible. We thank Avid Radiopharmaceuticals (Eli Lilly & Company) for providing the flortaucipir PET tracer, and the Alzheimer's Drug Discovery Foundation (ADDF) for providing funding for the original study. We thank the following persons for their contributions to data acquisition in the study: Cleveland Clinic: Yolande Mucharbach, ChristineWhitman, Hilda Sosic, Elaine Pienschke, Tami Kaczur, Sagar Patel, Nelson Rubina, and others who supported efforts; Alzheimer's Disease Coordiating Study: Jennifer Mason; Avid Radiopharmaceuticals: Michael Pontecorvo, Amanda Potasnik, Elisabeth DePardo. The Alzheimer's Drug Discovery Foundation provided funding for the original study, and the flortaucipir PET tracer was provided by Avid Radiopharmaceuticals (Eli Lilly & Company).
Matthews DC, Kinney JW, Ritter A, et al. Relationships between plasma biomarkers, tau PET, FDG PET, and volumetric MRI in mild to moderate Alzheimer's disease patients. Alzheimer's Dement. 2024;10:e12490. 10.1002/trc2.12490
DATA AVAILABILITY STATEMENT
The original study data were published in clinicaltrials.gov, and additional results generated in this work can be available upon reasonable request.
REFERENCES
- 1. Jack CR, Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. doi: 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Taipa R, das Neves SP, Sousa AL, et al. Proinflammatory and anti‐inflammatory cytokines in the CSF of patients with Alzheimer's disease and their correlation with cognitive decline. Neurobiol Aging. 2019;76:125‐132. doi: 10.1016/j.neurobiolaging.2018.12.019 [DOI] [PubMed] [Google Scholar]
- 3. Hampel H, Cummings J, Blennow K, Gao P, Jack CR, Vergallo A. Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat Rev Neurol. 2021;17:580‐589. doi: 10.1038/s41582-021-00520-w [DOI] [PubMed] [Google Scholar]
- 4. Matthews DC, Ritter A, Thomas RG, et al. Rasagiline effects on glucose metabolism, cognition, and tau in Alzheimer's dementia. Alzheimers Dement (N Y). 2021;7:e12106. doi: 10.1002/trc2.12106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mielke MM, Hagen CE, Xu J, et al. Plasma phospho‐tau181 increases with Alzheimer's disease clinical severity and is associated with tau‐ and amyloid‐positron emission tomography. Alzheimers Dement. 2018;14:989‐997. doi: 10.1016/j.jalz.2018.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tissot C, Therriault J, Kunach P, et al. Comparing tau status determined via plasma pTau181, pTau231 and [18F]MK6240 tau‐PET. eBioMedicine. 2022;76:103837. doi: 10.1016/j.ebiom.2022.103837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thijssen EH, Verberk IMW, Kindermans J, et al. Differential diagnostic performance of a panel of plasma biomarkers for different types of dementia. Alzheimers Dement (Amst). 2022;14:e12285. doi: 10.1002/dad2.12285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coomans EM, Verberk IMW, Ossenkoppele R, et al. A head‐to‐head comparison between plasma pTau181 and Tau PET along the Alzheimer's disease continuum. J Nucl Med. 2023;64:437‐443. doi: 10.2967/jnumed.122.264279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moscoso A, Grothe MJ, Ashton NJ, et al. Longitudinal associations of blood phosphorylated Tau181 and neurofilament light chain with neurodegeneration in Alzheimer disease. JAMA Neurol. 2021;78:396‐406. doi: 10.1001/jamaneurol.2020.4986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smirnov DS, Ashton NJ, Blennow K, et al. Plasma biomarkers for Alzheimer's disease in relation to neuropathology and cognitive change. Acta Neuropathol. 2022;143:487‐503. doi: 10.1007/s00401-022-02408-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tetzloff KA, Graff‐Radford J, Martin PR, et al. Regional distribution, asymmetry, and clinical correlates of tau uptake on [18F]AV‐1451 PET in atypical Alzheimer's disease. JAD. 2018;62:1713‐1724. doi: 10.3233/JAD-170740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ottoy J, Kang MS, Savard M, et al. The effect of age on tau burden is dependent on amyloid status in late‐onset Alzheimer's disease: Neuroimaging /Multi‐modal comparisons. Alzheimer Dement. 2020;16:e044202. doi: 10.1002/alz.044202 [DOI] [Google Scholar]
- 13. Cho H, Mundada NS, Apostolova LG, et al. Amyloid and tau‐PET in early‐onset AD: baseline data from the longitudinal early‐onset Alzheimer's Disease Study (LEADS). Alzheimers Dement. 2023;19 Suppl 9:S98‐114. doi: 10.1002/alz.13453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ossenkoppele R, Schonhaut DR, Schöll M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. 2016;139:1551‐1567. doi: 10.1093/brain/aww027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. La Joie R, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer's disease correlates with the intensity and topography of baseline tau‐PET. Sci Transl Med. 2020;12:eaau5732. doi: 10.1126/scitranslmed.aau5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chételat G, Arbizu J, Barthel H, et al. Amyloid‐PET and 18F‐FDG‐PET in the diagnostic investigation of Alzheimer's disease and other dementias. Lancet Neurol. 2020;19:951‐962. doi: 10.1016/S1474-4422(20)30314-8 [DOI] [PubMed] [Google Scholar]
- 17. Strom A, Iaccarino L, Edwards L, et al. Cortical hypometabolism reflects local atrophy and tau pathology in symptomatic Alzheimer's disease. Brain. 2022;145:713‐728. doi: 10.1093/brain/awab294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lewczuk P, Ermann N, Andreasson U, et al. Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer's disease. Alzheimers Res Ther. 2018;10:71. doi: 10.1186/s13195-018-0404-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gaiottino J, Norgren N, Dobson R, et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS One. 2013;8:e75091. doi: 10.1371/journal.pone.0075091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Teitsdottir UD, Jonsdottir MK, Lund SH, Darreh‐Shori T, Snaedal J, Petersen PH. Association of glial and neuronal degeneration markers with Alzheimer's disease cerebrospinal fluid profile and cognitive functions. Alzheimers Res Ther. 2020;12:92. doi: 10.1186/s13195-020-00657-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thijssen EH, La Joie R, Strom A, et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer's disease and frontotemporal lobar degeneration: a retrospective diagnostic performance study. Lancet Neurol. 2021;20:739‐752. doi: 10.1016/S1474-4422(21)00214-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khalil M, Pirpamer L, Hofer E, et al. Serum neurofilament light levels in normal aging and their association with morphologic brain changes. Nat Commun. 2020;11:812. doi: 10.1038/s41467-020-14612-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388‐405. doi: 10.1016/S1474-4422(15)70016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cicognola C, Janelidze S, Hertze J, et al. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimers Res Ther. 2021;13:68. doi: 10.1186/s13195-021-00804-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Janelidze S, Christian BT, Price J, et al. Detection of brain tau pathology in down syndrome using plasma biomarkers. JAMA Neurol. 2022;79:797‐807. doi: 10.1001/jamaneurol.2022.1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Verberk IMW, Laarhuis MB, Van Den Bosch KA, et al. Serum markers glial fibrillary acidic protein and neurofilament light for prognosis and monitoring in cognitively normal older people: a prospective memory clinic‐based cohort study. Lancet Healthy Longev. 2021;2:e87‐95. doi: 10.1016/S2666-7568(20)30061-1 [DOI] [PubMed] [Google Scholar]
- 27. Sass D, Guedes VA, Smith EG, et al. Sex differences in behavioral symptoms and the levels of circulating GFAP, Tau, and NfL in patients with traumatic brain injury. Front Pharmacol. 2021;12:746491. doi: 10.3389/fphar.2021.746491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement (N Y). 2018;4:575‐590. doi: 10.1016/j.trci.2018.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morgan AR, Touchard S, Leckey C, O'Hagan C, Nevado‐Holgado AJ, Consortium NIMA, et al. Inflammatory biomarkers in Alzheimer's disease plasma. Alzheimers Dement. 2019;15:776‐787. doi: 10.1016/j.jalz.2019.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Elahi FM, Casaletto KB, La Joie R, et al. Plasma biomarkers of astrocytic and neuronal dysfunction in early‐ and late‐onset Alzheimer's disease. Alzheimers Dement. 2020;16:681‐695. doi: 10.1016/j.jalz.2019.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. González‐Reyes RE, Nava‐Mesa MO, Vargas‐Sánchez K, Ariza‐Salamanca D, Mora‐Muñoz L. Involvement of astrocytes in Alzheimer's disease from a neuroinflammatory and oxidative stress perspective. Front Mol Neurosci. 2017;10:427. doi: 10.3389/fnmol.2017.00427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jack CR, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement. 2017;13:205‐216. doi: 10.1016/j.jalz.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schöll M, Lockhart SN, Schonhaut DR, et al. PET Imaging of Tau Deposition in the Aging Human Brain. Neuron. 2016;89:971‐982. doi: 10.1016/j.neuron.2016.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tissot C, L Benedet A, Therriault J, et al. Plasma pTau181 predicts cortical brain atrophy in aging and Alzheimer's disease. Alzheimers Res Ther. 2021;13:69. doi: 10.1186/s13195-021-00802-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer's Disease. N Engl J Med. 2021;384:1691‐1704. doi: 10.1056/NEJMoa2100708 [DOI] [PubMed] [Google Scholar]
- 36. Weigand AJ, Maass A, Eglit GL, Bondi MW. What's the cut‐point?: A systematic investigation of tau PET thresholding methods. Alzheimer's Research & Therapy. 2022;14:49. doi: 10.1186/s13195-022-00986-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389‐404. doi: 10.1007/s00401-006-0127-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ossenkoppele R, Lyoo CH, Sudre CH, et al. Distinct tau PET patterns in atrophy‐defined subtypes of Alzheimer's disease. Alzheimers Dement. 2020;16:335‐344. doi: 10.1016/j.jalz.2019.08.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bernick C, Cummings J, Raman R, Sun X, Aisen P. Age and rate of cognitive decline in Alzheimer disease: implications for clinical trials. Arch Neurol. 2012;69:901‐905. doi: 10.1001/archneurol.2011.3758 [DOI] [PubMed] [Google Scholar]
- 40. Alawode DOT, Fox NC, Zetterberg H, Heslegrave AJ. Alzheimer's disease biomarkers revisited from the amyloid cascade hypothesis standpoint. Front Neurosci 2022;16:837390. doi: 10.3389/fnins.2022.837390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Qiao Z, Wang G, Zhao X, et al. Neuropsychological performance is correlated with tau protein deposition and glucose metabolism in patients with Alzheimer's disease. Front Aging Neurosci. 2022;14:841942. doi: 10.3389/fnagi.2022.841942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beach TG, Malek‐Ahmadi M. Alzheimer's disease neuropathological comorbidities are common in the younger‐old. JAD. 2021;79:389‐400. doi: 10.3233/JAD-201213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sirivichayakul S, Kanchanatawan B, Thika S, Carvalho AF, Maes M. Eotaxin, an endogenous cognitive deteriorating chemokine (ECDC), is a major contributor to cognitive decline in normal people and to executive, memory, and sustained attention deficits, formal thought disorders, and psychopathology in schizophrenia patients. Neurotox Res. 2019;35:122‐138. doi: 10.1007/s12640-018-9937-8 [DOI] [PubMed] [Google Scholar]
- 44. Torres‐Acosta N, O'Keefe JH, O'Keefe EL, Isaacson R, Small G. Therapeutic potential of TNF‐α inhibition for Alzheimer's disease prevention. JAD. 2020;78:619‐626. doi: 10.3233/JAD-200711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gérard T, Colmant L, Malotaux V, et al. The spatial extent of tauopathy on [18F]MK‐6240 tau PET shows stronger association with cognitive performances than the standard uptake value ratio in Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2024;51(6):1662‐1674. doi: 10.1007/s00259-024-06603-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Benedet AL, Milà‐Alomà M, Vrillon A, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurology. 2021;78:1471‐1483. doi: 10.1001/jamaneurol.2021.3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Maass A, Landau S, Baker SL, et al. Comparison of multiple tau‐PET measures as biomarkers in aging and Alzheimer's disease. Neuroimage. 2017;157:448‐463. doi: 10.1016/j.neuroimage.2017.05.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hammes J, Bischof GN, Bohn KP, et al. One‐stop shop: 18F‐Flortaucipir PET differentiates amyloid‐positive and ‐negative forms of neurodegenerative diseases. J Nucl Med. 2021;62:240‐246. doi: 10.2967/jnumed.120.244061 [DOI] [PubMed] [Google Scholar]
- 49. Barthélemy NR, Bateman RJ, Hirtz C, et al. Cerebrospinal fluid phospho‐tau T217 outperforms T181 as a biomarker for the differential diagnosis of Alzheimer's disease and PET amyloid‐positive patient identification. Alz Res Therapy. 2020;12:26. doi: 10.1186/s13195-020-00596-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang B, Zhang C, Wang Y, et al. Effect of renal function on the diagnostic performance of plasma biomarkers for Alzheimer's disease. Front Aging Neurosci. 2023;15:1150510. doi: 10.3389/fnagi.2023.1150510 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Data Availability Statement
The original study data were published in clinicaltrials.gov, and additional results generated in this work can be available upon reasonable request.