

Abstract
We have identified a region within the ectodomain of the fusogenic human immunodeficiency virus type 1 (HIV-1) gp41, different from the fusion peptide, that interacts strongly with membranes. This conserved sequence, which immediately precedes the transmembrane anchor, is not highly hydrophobic according to the Kyte-Doolittle hydropathy prediction algorithm, yet it shows a high tendency to partition into the membrane interface, as revealed by the Wimley-White interfacial hydrophobicity scale. We have investigated here the membrane effects induced by NH2-DKWASLWNWFNITNWLWYIK-CONH2 (HIVc), the membrane interface-partitioning region at the C terminus of the gp41 ectodomain, in comparison to those caused by NH2-AVGIGALFLGFLGAAGSTMGARS-CONH2 (HIVn), the fusion peptide at the N terminus of the subunit. Both HIVc and HIVn were seen to induce membrane fusion and permeabilization, although lower doses of HIVc were required for comparable effects to be detected. Experiments in which equimolar mixtures of HIVc and HIVn were used indicated that both peptides may act in a cooperative way. Peptide-membrane and peptide-peptide interactions underlying those effects were further confirmed by analyzing the changes in fluorescence of peptide Trp residues. Replacement of the first three Trp residues by Ala, known to render a defective gp41 phenotype unable to mediate both cell-cell fusion and virus entry, also abrogated the HIVc ability to induce membrane fusion or form complexes with HIVn but not its ability to associate with vesicles. Hydropathy analysis indicated that the presence of two membrane-partitioning stretches separated by a collapsible intervening sequence is a common structural motif among other viral envelope proteins. Moreover, sequences with membrane surface-residing residues preceding the transmembrane anchor appeared to be a common feature in viral fusion proteins of several virus families. According to our experimental results, such a feature might be related to their fusogenic function.
The human immunodeficiency virus type 1 (HIV-1) relies on the fusogenic activity of the gp120/41 glycoprotein at the virion surface to enter and infect the CD4+ host cells (7, 24). Activation of the HIV-1 envelope protein occurs after the binding of the surface gp120 subunit to CD4 and human chemokine receptors (4, 7, 8, 24). Subsequently, the transmembrane gp41 subunit supports structural rearrangements involving a coiled-coil domain (5, 12, 25, 39) that bring about the eventual exposure to the aqueous medium of the fusion peptide at its N terminus (12, 13, 24, 25). The fusion peptide is likely to insert into the target lipid bilayer, making gp41 capable of interacting simultaneously with the viral and cell membranes (5, 8, 12, 13, 25, 39). The involvement of this conserved hydrophobic segment of about 25 amino acids in triggering fusion is supported by prediction (13, 15, 40), mutational analysis (2, 10, 11, 35), and in vitro studies carried out using synthetic peptides and model membranes (16, 20, 23, 27–31, 36). Fusion peptides inserted into the target membrane might drive the aggregation of gp41 trimers at initial sites of fusion as well (39). It has been argued that a higher-order envelope glycoprotein complex is involved in HIV-1 fusion as well as in analogous viral fusion processes (17, 19, 39).
Recent resolution of the atomic structure of the gp41 ectodomain demonstrates that this region is organized into a very stable helical bundle or “hairpin” (4, 5, 39). The core of this crystallized structure is constituted by a triple-stranded coiled coil. Three α-helices oriented obliquely in an antiparallel sense pack hydrophobically against the core coiled coil. The resulting trimeric structure would locate both fusion peptides and transmembrane anchors at the same end of the molecule. Two main models have been developed to explain the involvement of the hairpin structure in the gp41-mediated membrane fusion process. In one model (4, 5, 12, 25, 39, 43) a transient intermediate exists between the native nonfusogenic and the fusogenic six-helix complex, termed by Chan and Kim (4) as the “prehairpin.” The alternative model postulates that the hairpin is already present in the prefusion state (38). Binding to CD4 and chemokine receptors would expose the hairpin in close vicinity to the target membrane. Both molecular models propose that this structure represents the fusion-active version of gp41. Thus, formation of a hairpin could be coupled to the apposition of the target cell and viral membranes provided that gp41 trimers interacted simultaneously with both membranes.
The hypothesis of the hairpin structure as a fusion mediator has gained support from studies indicating that synthetic peptides corresponding to helical regions in the gp41 core structure are inhibitors of HIV-1 fusion. The DP-178 synthetic peptide representing the hydrophobic α-helix that interacts with the coiled-coil core of the hairpin completely inhibits virus-mediated cell-cell fusion and reduces infectivity (43). DP-178-induced inhibition occurs after receptor binding-induced activation and blocks the fusion process at a hemifusion state (12, 25). This peptide (also called T-20) has already been tested in clinical trials and has been shown to be an effective suppressor of HIV-1 replication in infected individuals (19a). In addition, based on the knowledge of this functional structure new strategies for the development and identification of new anti-HIV compounds have recently been delineated (8a, 9a).
At this point it is unclear how hairpin-apposed membranes can fuse (4). Several findings indicate that the fusion peptide inserted into the target membrane would cause the distortion of the bilayer organization necessary for merging (6, 30). Hydropathy-at-interface plots elaborated according to the hydrophobicity scale developed by Wimley and White (33, 42, 45) identify, in addition to the fusion peptide, a second region within the ectodomain of the fusogenic HIV-1 gp41 that shows high tendency to partition into the membrane interface. This tryptophan-rich region is located proximal to the viral membrane overlapping the DP-178 region at its C terminus and preceding the transmembrane anchor of gp41. Recent mutational analysis has revealed its importance for gp41-mediated fusion and infectivity. Salzwedel et al. (34) identified several mutations indicating that this gp41 stretch is dispensable for the normal maturation, transport, and receptor-binding ability of the protein but required for membrane fusion. Later functional characterization in cell-cell fusion assays by Muñoz-Barroso et al. (26) revealed three different phenotypes among the studied gp41 mutants: wild-type-like phenotypes showing reduced activity, defective variants unable to mediate fusion, and mutants able to assemble nonexpanding fusion pores.
Our results here demonstrate that, as for the N-terminal fusion peptide, HIVc, a sequence representing the pretransmembrane region partitions into membranes and induces the same type of perturbations. Moreover, the fusion peptide stimulates the activity of this second membrane-interacting sequence. These observations would be consistent with the existence of concerted action by both sequences that could simultaneously destabilize gp41-apposed membranes at the points of fusion. A mutation shown to inhibit cell-cell fusion and virus entry without affecting the maturation, transport, or CD4-binding ability of HIV-1 glycoprotein (34) caused the abrogation of both membrane fusion by the membrane-bound peptide and the ability to form complexes with the N-terminal fusion peptide. This finding provides a direct correlation between the activity of the HIVc peptide in vitro and the activity of gp41 in the virus and the transfected-cell context. Hydropathy analysis of several X-ray-solved ectodomain structures further indicates that, following the intervening protein sequences involved in the hairpin formation and preceding the transmembrane anchor, an additional membrane-interacting domain is also present. Sequence analysis extended to other viral fusion proteins indicates that an unusual concentration of aromatic residues, just preceding the transmembrane anchor, is commonly found among several virus families including retroviruses, filoviruses, orthomyxoviruses, rhabdoviruses, alphaviruses, and flaviviruses.
We propose that the existence of a membrane interface-residing sequence close to the transmembrane domain can be a general motif for a considerable number of viral fusion proteins and may be related to their fusion activity. Our study of the gp41 pretransmembrane region provides a mechanistic basis for understanding its role in viral fusion and infection and might be important for devising new inhibitors based on this sequence.
MATERIALS AND METHODS
Materials.
Dioleoylphosphatidylcholine (DOPC), dioleoylphosphatidylethanolamine (DOPE), cholesterol (CHOL), and the fluorescent probes N-(7-nitro-benz-2-oxa-1,3-diazol-4-yl)phosphatidylethanolamine (N-NBD-PE) and N-(Lissamine-rhodamine B-sulfonyl)phosphatidylethanolamine (N-Rh-PE) were purchased from Avanti Polar Lipids (Birmingham, Ala.). 8-Aminonaphthalene-1,3,6-trisulfonic acid sodium salt (ANTS) and p-xylenebis(pyridinium)bromide (DPX) were from Molecular Probes (Junction City, Oreg.). Octaethyleneglycol monododecyl ether (C12E8) and Triton X-100 were obtained from Sigma (St. Louis, Mo.). All other reagents were of analytical grade. The sequences NH2-DKWASLWNWFNITNWLWYIK-CONH2 (HIVc), DKAASLANAFNITNWLWYIK-CONH2 [HIVW(1–3)A], and NH2-AVGIGALFLGFLGAAGSTMGARS-CONH2 (HIVn) were synthesized as their C-terminal carboxamides and purified (estimated homogeneity, >90%) by Quality Controlled Biochemicals, Inc. (Hopkinton, Mass.). Peptide stock solutions were prepared in dimethyl sulfoxide (spectroscopy grade).
Vesicle preparation.
Large unilamellar vesicles (LUV) consisting of DOPC, DOPE, and CHOL (molar ratio, 1:1:1) were prepared according to the extrusion method of Hope et al. (18) in 5 mM HEPES–100 mM NaCl (pH 7.4). Lipid concentrations of liposome suspensions were determined by phosphate analysis (3). The mean diameter of DOPC-DOPE-CHOL vesicles was 130 nm as estimated by quasielastic light scattering using a Malvern Zeta-Sizer instrument.
Fluorimetric assays for vesicle destabilization.
All fluorescence measurements were conducted in thermostatically controlled cuvettes (37°C) using a Perkin-Elmer LS50-B spectrofluorimeter. The medium in the cuvettes was continuously stirred to allow the rapid mixing of peptide and vesicles.
Membrane lipid mixing was monitored using the resonance energy transfer (RET) assay described by Struck et al. (37). The assay is based on the dilution of N-NBD-PE and N-Rh-PE. Dilution due to membrane mixing results in an increase in N-NBD-PE fluorescence. Vesicles containing 0.6 mol% of each probe were mixed with unlabeled vesicles at a 1:4 ratio (final lipid concentration, 0.1 mM). The 7-nitro-benz-2-oxa-1,3-diazol-4-yl NBD emission at 530 nm was monitored, with the excitation wavelength set at 465 nm. A cutoff filter at 515 nm was used between the sample and the emission monochromator to avoid scattering interferences. The fluorescence scale was calibrated such that the zero level corresponded to the initial residual fluorescence of the labeled vesicles and the 100% value corresponded to complete mixing of all the lipids in the system. The latter value was set by the fluorescence intensity of vesicles labeled with 0.12 mol% of each fluorophore at the same total lipid concentration as in the fusion assay.
Release of vesicular contents to the medium was monitored by the ANTS-DPX assay. LUV containing 12.5 mM ANTS, 45 mM DPX, 20 mM NaCl, and 5 mM HEPES (9) were obtained by separating the unencapsulated material by gel filtration in a Sephadex G-75 column eluted with 5 mM HEPES–100 mM NaCl (pH 7.4). Osmolarities were adjusted to 200 mosM in a cryoscopic osmometer (Osmomat 030; Gonotec, Berlin, Germany). Fluorescence measurements were performed by setting the ANTS emission at 520 nm and the excitation at 355 nm. A cutoff filter (470 nm) was placed between the sample and the emission monochromator. The 0% leakage corresponded to the fluorescence of the vesicles at time zero; 100% leakage was the fluorescence value obtained after the addition of Triton X-100 (0.5% [vol/vol]).
Peptide-membrane and peptide-peptide interactions.
Peptide-vesicle and peptide-peptide interactions were investigated by monitoring the change in emitted Trp fluorescence. In order to produce an intrinsically fluorescent sequence, Phe8 was replaced by Trp within the HIVn peptide. Control experiments demonstrated that the resulting fluorescent analog was equally active at inducing membrane destabilization (data not shown). The peptide-vesicle mixtures were incubated for 10 min at 37°C and then for 1 h at room temperature before data acquisition. Corrected spectra were recorded in a Perkin-Elmer MPF-66 spectrofluorimeter with excitation set at 280 nm and slits of 2.5 nm (excitation) and 10 nm (emission). The signal was further corrected for inner-filter effects.
Peptide partitioning into DOPC-DOPE-CHOL LUV was estimated by physically separating unbound and bound peptides. Unbound peptides were removed from the mixtures by gel filtration in a Sephadex G-75 column. Vesicles (1 ml; 1 mM total lipid) were incubated at 37°C for 15 min with peptides prior to gel filtration. Chromatographed peptide-lipid samples containing bound peptides were subsequently solubilized with 2 mM C12E8 in order to minimize the scattering contribution to Trp fluorescence. The Trp signal at 333 nm (excitation at 280 nm) was normalized to the amount of coeluted lipid that was simultaneously quantified also by means of spectrofluorimetry. To that end, we used liposomes labeled with 0.1 mol% of N-NBD-PE and N-Rh-PE. In addition to lipid quantification, NBD and rhodamine fluorescence measurements confirmed complete vesicle solubilization by C12E8. With [L] and [W] as the molar concentrations of peptides in lipid and water, respectively, the apparent mole fraction partition coefficient was estimated as described previously (41, 42) using the following expression: Kapp = {([PT − [PF])/[L]}/([PF]/[W]) where [PT] is the molar concentration of peptide added to samples before chromatography and [PF] is the peptide concentration in aqueous solution after incubation with vesicles. The latter can be determined from
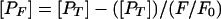 |
where F0 and F represent Trp fluorescence emission intensities in the peptide-vesicle mixtures before and after chromatography, respectively.
RESULTS
A classical way of detecting amino acid stretches bearing hydrophobic character in proteins is to plot their so-called average hydropathies (21, 42). As shown in Fig. 1, for gp41 ectodomains in primate lentiviruses, such a plot based on the widely used hydropathy scale of Kyte and Doolittle (21) clearly identified two conspicuous areas on the hydrophobic side above the zero midpoint line. The N-terminal hydrophobic regions consisted of the fusion peptides, whereas the regions at the C terminus represented the transmembrane anchors.
FIG. 1.

Hydropathy plots corresponding to sequences spanning the ectodomains of several Env transmembrane (TM) subunits of primate lentiviruses (top, HIV-1, BH10 isolate, residues 500 to 720 of the Env precursor; middle, HIV-2, SBLISY isolate, residues 490 to 710 of the Env precursor; bottom, simian immunodeficiency virus (SIV), Mm142-83 isolate, residues 510 to 730 of the Env precursor). The stretches plotted include the fusion peptide and the TM anchor regions. Hydropathy plots (mean values for a window of 11 amino acids) were elaborated using the Kyte-Doolittle (KD) hydropathy index (21) (thin lines) and Wimley-White (WW) interfacial hydrophobicity (45) (thick lines) scales for individual residues.
The hydropathy index for each amino acid adopted by Kyte and Doolittle (21) was an estimate based on bulk phase partitioning of side chain hydrophobicity alone. As recently proposed by White and Wimley (42), other contributions, specifically those arising from the peptide bonds and the bilayer effect, must be taken into account in order to compose a hydrophobicity scale that could be useful to detect membrane-interacting amino acid sequences. Thus, these authors elaborated a whole-residue scale based on the water-to-membrane interface transfer free energies for each amino acid (45). This “interface scale” was determined for 1-palmitoyl-2-oleoylphosphatidylcholine bilayer interfaces using two types of oligopeptides, which allowed the evaluation of both side chain and peptide bond hydrophobicities.
When average interfacial hydrophobicity was plotted for gp41 ectodomains of primate lentiviruses, two main hydrophobic regions were also detected (Fig. 1). The region at the N terminus was coincident with that detected by the Kyte-Doolittle hydropathy plot, meaning that the interfacial hydrophobicity plot identified the fusion peptide as well. However, at the C terminus a clear segregation between the hydrophobic region detected by the Kyte-Doolittle plot and that detected by Wimley-White interfacial hydrophobicity occurred. The latter was shifted closer to the N terminus. This finding is further illustrated by the data displayed in Table 1. For the three analyzed lentiviruses, the positions of the hydropathy-positive peaks at the gp41 N terminus coincided in both types of plots, the only difference being that the hydrophobic regions detected by the Kyte-Doolittle plot were somewhat wider. In contrast, the positive maxima detected by Wimley-White within the ectodomain C-terminal region were shifted 15 to 20 residues toward the N terminus.
TABLE 1.
Positions and lengths of hydrophobic regions within ectodomains of envelope transmembrane subunits of primate lentiviruses as detected by hydropathy analysisa
| Viruse | C-terminal region
|
N-terminal region
|
||||
|---|---|---|---|---|---|---|
| Maximum intensityb (peak range [aa]c) by:
|
Positiond | Maximum intensity (peak range [aa]) by:
|
Position | |||
| W-W | K-D | W-W | K-D | |||
| HIV-1 | 0.73 (666–692) | 2.6 (681–707) | −15 | 0.37 (516–528) | 2.4 (515–538) | +1 |
| HIV-2 | 0.44 (655–678) | 2.8 (671–692) | −20 | 0.39 (507–519) | 2.5 (507–528) | 0 |
| SIV | 0.44 (680–716) | 3.0 (696–719) | −19 | 0.44 (532–544) | 2.6 (532–553) | −1 |
Hydropathy plots were elaborated according to Wimley-White (W-W) or Kyte-Doolittle (K-D) scales using a window of 11 residues.
Maximum intensity of the hydrophobic peaks.
Peak range and position (aa, amino acids) determined by cutting off peaks at the level corresponding to 10% of maximum hydrophobicity.
Position of the peak maximum detected by W-W plot relative to the peak maximum detected by K-D plot. Negative values indicate that peaks detected by W-W plots are located closer to the N terminus of the protein sequence.
The position of the hydrophobic region at the C terminus in the Kyte-Doolittle plot (residues 681 to 707 in HIV-1) is almost coincident with that of the putative transmembrane segment of gp41 (residues 684 to 706 in HIV-1). We conclude that, at the C terminus of the gp41 ectodomain, there exists a wide nonpolar region (approximately 40 residues long) which is segmented into two domains: one located somewhat farther from the protein C terminus (residues 664 to 683 in HIV-1) showing high propensity to reside within the membrane interface and another one closer to the C terminus, which constitutes the membrane-spanning anchor. The hydrophobic-at-interface region (residues 664 to 683) detected within the C terminus of the gp41 ectodomain does not merely reflect the common preferential localization of aromatic residues within the interfacial boundary regions that precede transmembrane segments (22, 32). Both the peak range and the intensity of the hydrophobic maximum indicate that, for this fusogenic viral subunit, the stretch of membrane surface-residing amino acids that precedes the transmembrane domain is uniquely elongated (compare Tables 1 and 3 and see Discussion below).
TABLE 3.
Positions and lengths of transmembrane hydrophobic regions of several human type I membrane proteins as detected by hydropathy analysisa
| Proteind | Maximum intensity of hydrophobic peak (peak range [aa]b) by:
|
Positionc | |
|---|---|---|---|
| W-W | K-D | ||
| EGFR | 0.30 (151–166) | 3.4 (147–167) | 0 |
| CD4 | 0.57 (401–419) | 2.9 (395–419) | 0 |
| GLPA | 0.27 (96–113) | 2.8 (91–114) | +10 |
| ICA1 | 0.24 (495–501) | 3.2 (477–498) | +12 |
| ITB | 0.45 (723–745) | 3.2 (717–745) | +1 |
| PVR | 0.63 (343–369) | 3.5 (342–366) | +9 |
Hydropathy plots were elaborated according to Wimley-White (W-W) and Kyte-Doolittle (K-D) scales using a window of 11 residues.
Peak range and position (aa, amino acids) determined by cutting off peaks at the level corresponding to 10% of maximum hydrophobicity.
Position of the peak maximum detected by W-W plot relative to the peak maximum detected by K-D plot. Negative values indicate that peaks detected by W-W plot are located closer to the N terminus of the protein sequence.
Protein sequences were randomly selected from reference 22. EGFR, epidermal growth factor receptor precursor; CD4, T-cell surface glycoprotein CD4 precursor; GLPA, glycophorin A precursor; ICA1, intercellular adhesion molecule 1 precursor; ITB, platelet membrane glycoprotein IIIA precursor; PVR, poliovirus receptor precursor.
Figure 1 identifies a second membrane-partitioning domain within the gp41 ectodomain (residues 664 to 683) in addition to the fusion peptide (residues 512 to 534). Since the whole-residue interfacial hydrophobicity scale was composed on the basis of the partitioning into zwitterionic membrane interfaces (42, 45), we characterized the membrane-interacting capabilities of that second C-terminal region by using a representative synthetic sequence, HIVc, and electrically neutral LUV as targets. Previous work by our group (28, 29) has shown that the partition of a sequence representing the fusion peptide at the N terminus of HIV-1 gp41 (HIVn) has the capacity to destabilize neutral liposomes composed of DOPC, DOPE, and CHOL (molar ratio, 1:1:1). These vesicles select for an extended peptide structure that becomes fusogenic in a dose-dependent fashion. Morphological data reveal the formation of nonlamellar lipidic aggregates during the time course of lipid mixing (30). At subfusogenic doses the fusion peptide also causes the release of trapped contents from liposomes, indicating the existence of a peptide-mediated permeabilization process prior to and independent from the development of fusion (28, 29). For comparative purposes we explored the ability of HIVc and HIVn to perturb DOPC-DOPE-CHOL membranes. Assuming that both sequences would be present at an equimolar ratio within a potential fusogenic complex during gp41-mediated fusion, we also analyzed the membrane-perturbing abilities of a HIVc-HIVn mixture (1:1 mole ratio).
The HIVc peptide induced membrane fusion in the DOPC-DOPE-CHOL LUV system employed in this study, as evidenced by the occurrence of membrane lipid mixing. Figure 2A presents comparative results of membrane lipid mixing for the three peptide samples, HIVc, HIVn, and HIVc-HIVn, as monitored with the RET assay. The final extents of fusion were dependent on the peptide-to-lipid ratios in the three cases. However, data displayed in Fig. 2 readily demonstrate that, in terms of peptide added per lipid, the HIVc sequence was more efficient at inducing lipid mixing than HIVn. For HIVc we consistently found that the peptide-to-lipid ratio required to induce 20 to 30% lipid mixing was 1:1,000. In comparison, HIVn induced 10% lipid mixing at a 10-times-higher peptide-to-lipid ratio of 1:100. The mixture HIVc-HIVn did not achieve better results than HIVc by itself. These data strongly support the idea of HIVc being capable of destabilizing membranes and inducing them to merge. Moreover, they suggest that this sequence is a more potent perturbing agent than HIVn.
FIG. 2.

Final extents of fusion (A) and leakage (B) as a function of increasing concentrations of HIVc (●), HIVn (○), and HIVc-HIVn (▴). The peptide was added to DOPC-DOPE-CHOL LUV at different peptide-to-lipid ratios (Ri) and incubated for 1 h at 37°C before measuring the extents of lipid mixing and leakage. Lipid concentration, 100 μM.
Analogous results were obtained when the three peptide preparations were assayed for their ability to release the ANTS-DPX probes from the aqueous interiors of the DOPC-DOPE-CHOL vesicles (Fig. 2B). Peptide-induced leakage was activated in all cases at peptide-to-lipid ratios lower than those that activated lipid mixing. In addition, HIVc was again more potent than HIVn at lower peptide-to-lipid ratios. At a peptide-to-lipid ratio of 1:5,000 the extent of leakage induced by HIVc consistently amounted to more than 30%, whereas, to obtain a comparable extent of fusion, the ratio had to be increased 10-fold. HIVn induced 10% leakage at a peptide-to-lipid ratio of 1:1,000, whereas a ratio of 1:100 was necessary to detect a comparable amount of lipid mixing. The mixture HIVc-HIVn paralleled the behavior of HIVc alone.
Taken together, results in Fig. 2 would be consistent with the existence of membrane perturbations caused by HIVc partitioning into DOPC-DOPE-CHOL vesicles that are qualitatively similar to those caused by HIVn partitioning. However, compared to HIVn, 10-times-less HIVc was necessary to promote comparable lipid mixing or leakage. This fact may reflect an inherently higher potency to perturb membranes for the HIVc sequence or else more peptide associated with vesicles in HIVc-treated samples. To clarify this issue, the peptide binding to vesicles was investigated next.
In Fig. 3 we show the fluorescence spectra obtained utilizing HIVc and the fluorescent F8W analog of HIVn. At a peptide-to-lipid ratio of 1:100 levels of intrinsic fluorescence emission in HIVc and in HIVn changed significantly in the presence of DOPC-DOPE-CHOL LUV. Emission intensity was enhanced, and the maxima shifted to lower wavelengths: from 346 to 342 nm for HIVc and from 354 to 347 nm for HIVn. Both effects suggest that Trp residues in the presence of vesicles sense a less polar environment which is indicative of partition and the subsequent penetration of both peptides into the bilayer millieu. We observed that the levels of Trp fluorescence of both peptides in solution decreased with time, an effect that may be related to self-aggregation due to their hydrophobic character. This process may compete with membrane association and, therefore, precludes correct determination of real partition coefficients (41). With this caveat in mind, and just for comparative purposes, we determined apparent mole fraction partition coefficients (Kapp) for HIVc and HIVn (F8W analog). For a peptide-to-lipid ratio of 1:500, we obtained under our experimental conditions (see Materials and Methods) 93% of HIVc coeluting with vesicles whereas that value was reduced to 35% for HIVn. Therefore the Kapp values were 7.3 × 105 for HIVc and 3.0 × 104 for HIVn. Again, these values should not be taken as quantitatively reflecting the energetics of the peptide-membrane interaction process, but rather as descriptive of the ability of these peptides to associate with membranes under our particular experimental conditions. They further indicate that, after correcting for the difference in bound peptide, membrane-associated HIVn is as effective as HIVc at perturbing membranes (Fig. 2). Thus, in our in vitro assays, the activity displayed by HIVn is actually limited by the ability of the peptide to partition into vesicles, but this activity in the membrane-bound form would be comparable to that of HIVc.
FIG. 3.

Fluorescence emission spectra of HIVc and HIVn (F8W analog) in buffer (dotted lines) and incubated with DOPC-DOPE-CHOL LUV (solid lines). The peptide concentration was 1 μM, and, in vesicle samples, the peptide-to-lipid ratio was 1:100. au, arbitrary units.
The extents of fusion and leakage seem to indicate that the mixture HIVc-HIVn followed a membrane-perturbing pattern similar to that exhibited by HIVc alone. However those measurements were carried out after 1 h of incubation of peptides and vesicles. It is possible that the initial rates of membrane perturbation might be altered by the combination of both peptides. Thus, in search for a possible synergism in the mode of action of HIVc and HIVn, we carried out a kinetic characterization of the initial stages of the perturbing processes induced by each of the peptides and by the HIVc-HIVn mixture. Initial rates of HIVc-induced lipid mixing increased with peptide concentration (Fig. 4). We investigated the effect of coadding HIVn on the former process under conditions where this peptide alone did not induce lipid mixing or induced it just marginally (i.e., at peptide-to-lipid ratios ranging from 1:1,000 to 1:100). As shown in Fig. 4, the initial rates of lipid mixing induced by the mixture HIVc-HIVn were always higher than those induced by HIVc alone. At a ratio at which HIVn induced extensive lipid mixing by itself, i.e., 1:25, no additive effects of the HIVc-HIVn mixture were found. This indicates that, under most circumstances, synergism between these peptides in the mode of inducing fusion may occur. Similar results were obtained for the leakage assay, and again potentiation of peptide effects was more evident at lower peptide-to-lipid ratios (Fig. 5).
FIG. 4.

Kinetics of membrane lipid mixing of LUV of DOPC-DOPE-CHOL (1:1:1) induced by HIVc (●), HIVn (○), and HIVc-HIVn (▴). The peptide was added at the peptide-to-lipid ratios indicated. Lipid concentration was 100 μM in all cases.
FIG. 5.

Kinetics of leakage of ANTS-DPX from LUV of DOPC-DOPE-CHOL (1:1:1) induced by HIVc (●), HIVn (○), and HIVc-HIVn (▴). Conditions otherwise were as described for Fig. 4.
The existence of HIVc-HIVn interactions underlying their mutual synergism was confirmed by detecting changes of HIVc Trp fluorescence in the presence of the nonfluorescent HIVn. The data displayed in Fig. 6 reflect an increase in the emitted fluorescence intensity of HIVc in solution and a shift of the wavelength of maximum emission (λmax) from 346 to 340 nm when HIVc was premixed with HIVn at a 1:10 HIVc-to-HIVn mole ratio. Results in this figure further indicate that the Trp emission of HIVc premixed with HIVn (curve 4) also increased and was slightly blue shifted from 342 to 338 nm when associated with vesicles. This effect was clearly larger than the one detected for the fluorescent HIVc alone (curve 3). According to the estimated partition coefficients, under the experimental conditions in Fig. 6, ≈60% of HIVc was bound to vesicles. Thus, the polarity change caused by premixing with HIVn might be due to an increase in binding or might originate from a different bilayer localization of HIVc when interacting together with HIVn and/or from the formation of HIVc-HIVn complexes within the vesicle membranes.
FIG. 6.

Trp fluorescence emission spectra of HIVc alone (0.2 μM) and premixed with HIVn (HIVc-to-HIVn ratio, 1:10) in buffer and incubated with DOPC-DOPE-CHOL LUV (HIVc-to-lipid ratio, 1:500). Curves: 1, HIVc in buffer; 2, HIVc-HIVn in buffer; 3, HIVc in the presence of vesicles; 4, HIVc-HIVn in the presence of vesicles. au, arbitrary units.
In order to confirm the relevance of the previous observations, we also explored the effect of a sequence substitution on the ability of HIVc to perturb membranes and associate with HIVn (Fig. 7). The HIVW(1–3)A sequence with the first three Trp residues of HIVc replaced by Ala represents the pretransmembrane region of a mutant gp41 described by Salzwedel et al. (34) as reproducing the phenotype of deleting the sequence spanning residues 665 to 682. This phenotype consists of the specific abolition of cell-cell fusion and virus entry mediated by gp41 without affecting normal maturation and transport to the cell surface or CD4-binding ability of the envelope glycoprotein. Our results in Fig. 7A show that, in contrast to HIVc, the HIVW(1–3)A peptide was unable to induce membrane fusion at peptide-to-lipid molar ratios ranging from 1:1,000 to 1:100. Since the absence of an effect might be caused by the existence of defective binding, we also measured the partitioning of this sequence into vesicles. At a 1 mM lipid concentration approximately 50% of the added HIVW(1–3)A peptide partitioned into DOPC-DOPE-CHOL membranes yielding a Kapp of ≈55,000. This means that for the fusion experiments in Fig. 7A the 1:100 HIVW(1–3)A/lipid ratio condition is comparable to that of the 1:500 HIVc/lipid ratio. We conclude that HIVW(1–3)A bound to membranes was unable to induce the type of perturbations induced by the HIVc parental sequence.
FIG. 7.

(A) Kinetics of membrane lipid mixing of LUV of DOPC-DOPE-CHOL (1:1:1) induced by HIVc (●) and HIVW(1–3)A (○). The peptides were added at the peptide-to-lipid ratios of 1:500 (HIVc) and 1:100 [HIVW(1–3)A]. Lipid concentration was 100 μM. (B) Trp fluorescence emission spectra of HIVW(1–3)A (1 μM) in buffer (dotted line) and incubated with DOPC-DOPE-CHOL LUV [HIVW(1–3)A-to-lipid ratio, 1:100] (solid lines). au, arbitrary units.
The ability of HIVW(1–3)A to associate with membranes may also be inferred from the emission spectra displayed in Fig. 7B. As for HIVc and HIVn (Fig. 3) the intrinsic fluorescence of HIVW(1–3)A increased in the presence of vesicles, indicating that Trp residues must be sensing a less polar environment. In contrast to the observed effects for HIVc (Fig. 6), under comparable conditions the emission of HIVW(1–3)A did not experience any change in the presence of the nonfluorescent HIVn (data not shown). The latter result seems to reflect the absence of HIVW(1–3)A-HIVn interactions.
The existence of a membrane interface-residing sequence preceding the transmembrane anchor might represent a particular characteristic of primate lentiviruses or else a structural motif shared by other retroviruses and other families. To explore this hypothesis, we first performed on several viral sequences a hydropathy analysis analogous to those described in Table 1 for primate lentiviruses. The selected fusogenic sequences (or close homologous relatives) have been either predicted or formally demonstrated to form hairpin structures. Data in Table 2 indicate that, with the single exception of Sendai virus F protein, there exists within the analyzed sequences a hydrophobic-at-interface region preceding the transmembrane segment. Paramyxovirus-mediated membrane fusion does not require the sequences connecting the coiled coil and the transmembrane anchor and might take place according to a mechanism distinct from that proposed for the rest of the virus families analyzed (1) (see Discussion). As a control, a similar hydropathy analysis of transmembrane regions of several human type I membrane proteins (Table 3) revealed that segregation of the hydrophobic-at-interface domain from the transmembrane segment is not found in those proteins. The preferential occurrence of aromatic residues at the cytoplasmic boundary of transmembrane segments in the human proteins (22, 32) does not account for the position and length of the interfacially hydrophobic sequences detected in viral envelope proteins.
TABLE 2.
Positions and lengths of C-terminal hydrophobic regions at ectodomains of precursors of envelope fusogenic subunits of different viruses as detected by hydropathy analysisa
| Virusd | Maximum intensity of hydrophobic peak (peak range [aa]b) by:
|
Positionc | |
|---|---|---|---|
| W-W | K-D | ||
| MoMLV | 0.35 (598–614) | 3.3 (609–637) | −12 |
| RSV | 0.29 (538–549) | 3.8 (546–579) | −25 |
| Ebola | 0.50 (644–654) | 3.5 (656–677) | −17 |
| Influenza | 0.54 (523–549) | 2.9 (526–551) | −12 |
| Sendai | 0.25 (505–523) | 4.3 (501–526) | +2 |
Hydropathy plots were elaborated according to Wimley-White (W-W) and Kyte-Doolittle (K-D) scales using a window of 11 residues.
Peak range and position (aa, amino acids) determined by cutting off peaks at the level corresponding to 10% of maximum hydrophobicity.
Position of the peak maximum detected by W-W plot relative to the peak maximum detected by K-D plot. Negative values indicate that peaks detected by W-W plot are located closer to the N terminus of the protein sequence.
Protein sequences for the viruses are as follows: MoMLV (Moloney murine leukemia virus), Env protein, GenBank accession no. J02255; RSV (Rous sarcoma virus), glycoprotein, Prague C strain, GenBank accession no. V01197; Ebola virus, glycoprotein, Zaire strain, GenBank accession no. U31033; influenza virus, hemagglutinin, strain A/Turkey/Oregon/71, GenBank accession no. M31689; Sendai virus, fusion protein, HVJ strain, GenBank accession no. M12396.
Elongation of sequences containing aromatic residues at the N termini of membrane-spanning regions is common in viral envelope proteins implicated in viral fusion. Figure 8 shows sequences from six virus families aligned with respect to the membrane-spanning hydrophobic region rich in aliphatic amino acids. The Retroviridae have been further subdivided into their principal subfamilies, Lentivirinae, types B, C, and D, avian Oncovirinae, and Spumaretrovirinae. A high degree of clustering of aromatic amino acids, most prominently tryptophan, is found in the primate and ovine lentiviruses, mammalian oncoviruses of groups B, C, and D, the filoviruses Ebola virus and Marburg virus, the rhabdovirus vesicular stomatitis virus G protein, and the alphavirus Sindbis virus E1 protein. A lower level of clustered aromatics, but higher than expected for type I glycoproteins, is found in avian retroviruses, spumaretroviruses, influenza A virus HA2, and the hepatitis C flavivirus E2 protein. Such an unusual concentration of aromatic amino acids exists against a background of considerable sequence diversity and in different positions in the linear sequence, even among closely related viruses. There are notable exceptions to the pattern, however. As noted in Table 2, the frequency of preinsertion aromatic amino acids in Sendai paramyxovirus F glycoprotein does not exceed that expected for type I glycoproteins. Likewise, Arenavirus GP2 (not shown) lacks any clustering of aromatics. Therefore, while aromatic and especially tryptophan-rich regions prior to membrane insertion have membrane-distorting properties that could be involved in fusion by many viruses, such regions would not appear to be indispensable.
FIG. 8.

Clustering of aromatic amino acids amino-terminal to the membrane-spanning region of the transmembrane (TM) superfamily of glycoproteins. Sequences of TM glycoproteins implicated in fusion are hand aligned by their membrane-spanning regions, using the 20-amino-acid span of hydrophobicity indicated at the top and bottom by vertical lines and any apparently conserved amino acids wherever possible, without introducing any gaps. Selected sequences and GenBank accession numbers of TM superfamily glycoproteins are as described in Tables 1 and 2 with the following additional sequences: SA-OMVV, ovine lentivirus, M13646; EIAV, equine infectious anemia virus, M16575; MMTV, mouse mammary tumor virus, M15122; BLV, bovine leukemia virus, K02120; HumFoamy, human foamy spumaretrovirus, U21247; Marburg filovirus GP2, U31033; VSV, vesicular stomatitis virus, Indiana strain, G protein, J02428; Sindbis virus E2 protein, J02363; HepC, hepatitis C virus, strain 1b, flavivirus E2 protein, AJ238800; MPMV, Mason-Pfizer monkey virus coat protein GP20, M12349.
DISCUSSION
Viral fusion proteins are designed to interact simultaneously with two membranes during the fusion reaction cycle. Interaction with the virion membrane is primarily mediated by the transmembrane anchor. In addition protruding ectodomains contain a conserved hydrophobic stretch of amino acids, the fusion peptide, which is thought to insert into the target membrane upon fusion activation (4, 13, 14, 17, 19, 33, 40). By using a hydropathy analysis based on the hydrophobicity-at-interface scale recently proposed by Wimley and White (45), we have confirmed the high propensity of the gp41 fusion peptide to partition into membranes (Fig. 1). According to our results (Table 1), within the HIV-1 fusion peptide region, the sequence IGALFLGFLGAAG containing the conserved FLG motif (13), shows a high probability to localize at the bilayer interface. Despite the experimental limitations inherent to the hydrophobicity-at-interface scale (45), the Wimley-White hydropathy analysis appears to be clearly superior to others for detecting potential membrane-interacting sequences within overall soluble polypeptide domains (42). The most important advantage in comparison with bulk phase scales is that the Wimley-White scale takes into account the effect of the membrane interface on partitioning. This bilayer region consists of a complex mixture of water and chemically heterogeneous phospholipid groups (polar head groups, glyceryl, phosphoryl, carbonyl, and methylenes) in which significant changes in polarity occur at short range (42). This results in the main difference between the Wimley-White scale and scales such as the one proposed by Kyte and Doolittle (21): even in the absence of solid physical foundations (46), aromatic residues, namely, Phe, Tyr, and Trp, appear to be the most hydrophobic ones when located at interfaces.
An illustrative example of the differences between scales can be observed for the C-terminal region within the gp41 ectodomain (Fig. 1). The Wimley-White hydrophobicity-at-interface scale identifies the region preceding the gp41 transmembrane anchor as having a high tendency to partition into the membrane interface. Its pronounced hydrophobic-at-interface character is based on the presence of 1 Phe residue, 1 Tyr residue, and 5 Trp residues closely located within a 20-residue spanning sequence. The preference of Trp and Tyr residues for boundary regions that flank the transmembrane domains is a known feature shared by virtually all known membrane proteins (22, 32). The C-terminal region detected within the gp41 ectodomain appears to be an elongation tail of membrane surface-residing domains. To test the functional significance that such a membrane interface-residing domain could have for gp41-mediated HIV-1 fusion, we characterized it as a membrane-interacting sequence. During the course of our studies, Salzwedel et al. (34) reported on the effect of mutations targeted to the same tryptophan-rich domain. More recently, Muñoz-Barroso et al. (25) have evaluated the effect of those mutations on the various stages of fusion. The mutational and functional analyses have confirmed its crucial role during gp41-mediated fusion.
Proposed molecular models for viral fusion processes highlight a role for coiled-coil structures, which were first observed for influenza virus HA2 (44) and which have been extended to the retroviruses (15) and filoviruses (14). Formation of those stable structures could be energetically coupled to membrane apposition (4, 39). It is generally accepted that both anchoring and apposition of the viral and target membranes are mediated by the ectodomains at the transmembrane subunits of envelope proteins such as the HIV-1 gp41. Following gp41-induced apposition, a membrane perturbation is likely to take place within the points of fusion, causing both membranes to merge (4, 39). Within this framework, the fusion peptide is postulated to anchor the fusion protein to the target membrane and perturb subsequently apposed membranes. Our data indicate that the HIV-1 fusion peptide would be capable of both functions, given its tendency to partition into membrane interfaces and to perturb their architecture (29, 30). In addition, our experimental data demonstrate that the sequence preceding the gp41 transmembrane anchor could be implicated in perturbing the apposed membranes as well. According to the leakage, lipid mixing, and binding assays, this sequence was even more potent at inducing membrane destabilization than the fusion peptide. The physiological relevance of our in vitro approach was supported by the fact that a mutation known to block gp41 activity in vivo completely impaired the ability of HIVc to perturb membranes (Fig. 7). Similarly, the in vitro membrane activity of the synthetic N-terminal fusion peptide has been also reported to be sensitive to sequence substitutions affecting gp41-mediated cell-cell fusion and viral infectivity (20, 29). Within the hairpin structure postulated to represent the fusion-competent version of gp41, both sequences would be closely located at the same end of the molecule. Moreover, if a higher-order envelope glycoprotein complex is involved in HIV-1 fusion, membrane perturbations induced by both sequences might be confined to the point of fusion (25, 26).
The presence of two membrane-partitioning sequences separated by a collapsible intervening sequence is a common motif shared by other viral envelope products. Results summarized in Table 2 demonstrate that, with the single exception of those of paramyxoviruses, known hairpin structures would locate two membrane interface-partitioning sequences in close proximity. Baker et al. (1) demonstrated that paramyxovirus-mediated membrane fusion does not require the flexible sequence connecting the coiled coil and the transmembrane anchor. These authors suggested that conformational changes in paramyxovirus F proteins may involve flexibility in other regions of the protein such as the intervening sequences connecting the N- and C-terminal heptad repeats. Thus, in contrast to what is found for the rest of the analyzed cases, flexibility between membrane-anchoring regions of the F protein would not suffice to allow hairpin formation.
Influenza virus hemagglutinin also appears to be unique in the sense that although hydrophobic-at-interface and hydrophobic peaks are located at different positions, both regions span almost the same sequence of approximately 25 to 27 residues, i.e., they are not segregated. Clearly, in order to find a common mechanistic role for these membrane interface-seeking regions during fusion, more mutational and functional studies will be necessary. We believe that the new Wimley-White hydropathy scale will be a powerful predictive tool that will lead to the detection of functional domains implicated in protein-membrane interactions during viral fusion events.
The existence of the hydrophobic-at-interface region preceding the transmembrane anchor appears to be a specific attribute of certain viral fusion glycoproteins rather than a general feature of type I membrane proteins (Table 3). In viral proteins, such a region appears to be an elongation of the short sequences containing aromatic residues at the cytoplasmic boundary of transmembrane anchors (22, 32). A survey of such regions in a number of virus families indicates that they are common and especially prominent in the retrovirus and filovirus families (Fig. 8). While it is difficult to accurately align highly divergent sequences, the aromatic amino acids do not occur with any reproducible periodicity, as one would expect in the α-helical configuration suggested by Salzwedel et al. (34), even in closely related viruses. Any mechanism by which these regions participate in fusion must account for the positional variability.
An alternative explanation for the clustering of tryptophans in retroviruses relates to the genetic code, in that only the triplet UGG codes for tryptophan. Any G-to-A hypermutation by reverse transcriptase at any of five clustered UGG codons could generate a nonsense codon and premature stop in translation just prior to membrane insertion. This could generate a population of truncated glycoproteins accumulating during chronic infections, along with other defective mutants, as soluble glycoproteins that might function as cellular and immunological decoys, as does sGP in filoviruses. However, this explanation would not apply to those viruses such as murine leukemia virus preferentially rich in phenylalanine. The more likely general explanation for such preinsertion aromatic clusters is that they promote fusion through their membrane-distorting properties.
Taken together, these observations point to the possibility that a number of viral fusogenic subunits might operate by putting into close contact within the fusion locus two perturbed membranes, that of the virus and that of the target cell, and/or two interacting perturbing sequences, the fusion peptide and the surface-residing tail that precedes the transmembrane anchor.
ACKNOWLEDGMENTS
We are grateful to Maier Lorizate for technical assistance.
T.S. and A.A. are recipients of predoctoral fellowships from the Basque Government. This work was supported by DGCYT (grant PB96-0171), the Basque Government (PI 96-46; EX-1998-28; PI-1998-32), and the University of the Basque Country (UPV 042.310-EA085/97; UPV 042.310-G03/98). Support to W.R.G. includes senior fellowship 1F32AI08549 and grant 1R01DE10862 from the National Institutes of Health of the United States.
REFERENCES
- 1.Baker K A, Dutch R E, Lamb R A, Jardetzky T S. Structural basis for paramyxovirus-mediated membrane fusion. Mol Cell. 1999;3:309–319. doi: 10.1016/s1097-2765(00)80458-x. [DOI] [PubMed] [Google Scholar]
- 2.Bosch M L, Earl P L, Fargnoli K, Picciafuoco S, Giombini F, Wong-Staal F, Franchini G. Identification of the fusion peptide of primate immunodeficiency viruses. Science. 1989;244:694–697. doi: 10.1126/science.2541505. [DOI] [PubMed] [Google Scholar]
- 3.Böttcher C S F, van Gent C M, Fries C. A rapid and sensitive sub-micro phosphorus determination. Anal Chim Acta. 1961;24:203–204. [Google Scholar]
- 4.Chan D C, Kim P S. HIV entry and its inhibition. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 5.Chan D C, Fass D, Berger J M, Kim P S. Core structure of gp41 from the HIV-1 envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 6.Colotto A, Epand R M. Structural study of the relationship between the rate of membrane fusion and the ability of the fusion peptide of influenza virus to perturb bilayers. Biochemistry. 1997;36:7644–7651. doi: 10.1021/bi970382u. [DOI] [PubMed] [Google Scholar]
- 7.Dalgleish A G, Beverley P C L, Clapham P R. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312:763–767. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 8.Doms R W, Peiper S C. Unwelcomed guests with master keys: how HIV uses chemokine receptors for cellular entry. Virology. 1997;235:179–190. doi: 10.1006/viro.1997.8703. [DOI] [PubMed] [Google Scholar]
- 8a.Eckert D M, Malashkevich V N, Hong L H, Carr P A, Kim P S. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- 9.Ellens H, Bentz J, Szoka F C. H+- and Ca2+-induced fusion and destabilization of liposomes. Biochemistry. 1985;24:3099–3106. doi: 10.1021/bi00334a005. [DOI] [PubMed] [Google Scholar]
- 9a.Ferrer M, Kapoor T M, Strassmaier T, Weissenhorn W, Skehel J J, Oparian D, Schreiber S L, Wiley D C, Harrison S C. Selection of gp41-mediated HIV-1 cell entry inhibitors from biased combinatorial libraries of non-natural binding elements. Nat Struct Biol. 1999;6:953–960. doi: 10.1038/13324. [DOI] [PubMed] [Google Scholar]
- 10.Freed E O, Myers D J, Risser R. Characterization of the fusion domain of the human immunodeficiency virus type 1 envelope glycoprotein gp41. Proc Natl Acad Sci USA. 1990;87:4650–4654. doi: 10.1073/pnas.87.12.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freed E O, Delwart E L, Buchschacher G L, Panganiban A T. A mutation in the human immunodeficiency virus type 1 transmembrane glycoprotein gp41 dominantly interferes with fusion and infectivity. Proc Natl Acad Sci USA. 1992;89:70–74. doi: 10.1073/pnas.89.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furuta R A, Wild C T, Weng Y, Weiss C D. Capture of an early fusion-active conformation of HIV-1 gp41. Nat Struct Biol. 1998;5:276–279. doi: 10.1038/nsb0498-276. [DOI] [PubMed] [Google Scholar]
- 13.Gallaher W R. Detection of a fusion peptide sequence in the transmembrane protein of the human immunodeficiency virus. Cell. 1987;50:327–328. doi: 10.1016/0092-8674(87)90485-5. [DOI] [PubMed] [Google Scholar]
- 14.Gallaher W R. Similar structural models of the transmembrane proteins of Ebola and avian sarcoma viruses. Cell. 1996;85:477–478. doi: 10.1016/s0092-8674(00)81248-9. [DOI] [PubMed] [Google Scholar]
- 15.Gallaher W R, Ball J M, Garry R F, Griffin M C, Montelaro R C. A general model for the transmembrane proteins of HIV and other retroviruses. AIDS Res Hum Retroviruses. 1989;5:431–440. doi: 10.1089/aid.1989.5.431. [DOI] [PubMed] [Google Scholar]
- 16.Gordon L M, Curtain C C, Zhong Y C, Kirkpatrick A, Mobley P W, Waring A J. The amino terminal peptide of HIV-1 glycoprotein 41 interacts with human erythrocyte membranes: peptide conformation, orientation and aggregation. Biochim Biophys Acta. 1992;1139:257–274. doi: 10.1016/0925-4439(92)90099-9. [DOI] [PubMed] [Google Scholar]
- 17.Hernández L D, Hoffman L R, Wolfsberg T G, White J M. Virus-cell and cell-cell fusion. Annu Rev Cell Dev Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- 18.Hope M J, Bally M B, Webb G, Cullis P R. Production of large unilamellar vesicles by a rapid extrusion procedure. Characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim Biophys Acta. 1985;812:55–65. doi: 10.1016/0005-2736(85)90521-8. [DOI] [PubMed] [Google Scholar]
- 19.Hughson F M. Enveloped viruses: a common mode of membrane fusion? Curr Biol. 1997;7:565–569. doi: 10.1016/s0960-9822(06)00283-1. [DOI] [PubMed] [Google Scholar]
- 19a.Kilby J M, Hopkins S, Venetta T M, et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med. 1998;4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 20.Kliger Y, Aharon A, Rapaport D, Jones P, Blumenthal R, Shai Y. Fusion peptides derived from the HIV type I glycoprotein 41 associate within phospholipid membranes and inhibit cell-cell fusion. J Biol Chem. 1997;272:13496–13505. doi: 10.1074/jbc.272.21.13496. [DOI] [PubMed] [Google Scholar]
- 21.Kyte J, Doolittle R F. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 22.Landolt-Marticorena C, Williams K A, Deber C M, Reithmeier R A F. Non-random distribution of amino acids in the transmembrane segments of human type I single span membrane proteins. J Mol Biol. 1993;229:602–608. doi: 10.1006/jmbi.1993.1066. [DOI] [PubMed] [Google Scholar]
- 23.Martin I, Defrise-Quertain F, Decroly E, Vandenbranden M, Brasseur R, Ruysschaert J M. Orientation and structure of the NH2-terminal HIV-1 gp41 peptide in fused and aggregated liposomes. Biochim Biophys Acta. 1993;1145:124–133. doi: 10.1016/0005-2736(93)90389-h. [DOI] [PubMed] [Google Scholar]
- 24.Moore J P, Bradford A J, Weiss R, Sattentau Q J. The HIV-cell fusion reaction. In: Bentz J, editor. Viral fusion mechanisms. Boca Raton, Fla: CRC Press Inc.; 1993. pp. 233–289. [Google Scholar]
- 25.Muñoz-Barroso I, Durell S, Sakaguchi K, Appella E, Blumenthal R. Dilation of the human immunodeficiency virus-1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41. J Cell Biol. 1998;140:315–323. doi: 10.1083/jcb.140.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muñoz-Barroso I, Salzwedel K, Hunter E, Blumenthal R. Role of the membrane-proximal domain in the initial stages of human immunodeficiency virus type 1 envelope glycoprotein-mediated membrane fusion. J Virol. 1999;73:6089–6092. doi: 10.1128/jvi.73.7.6089-6092.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nieva J L, Nir S, Muga A, Goñi F M, Wilschut J. Interaction of the HIV-1 fusion peptide with phospholipid vesicles: different structural requirements for leakage and fusion. Biochemistry. 1994;33:3201–3209. doi: 10.1021/bi00177a009. [DOI] [PubMed] [Google Scholar]
- 28.Nieva J L, Nir S, Wilschut J. Destabilization and fusion of zwitterionic large unilamellar lipid vesicles induced by a β-type structure of the HIV-1 fusion peptide. J Liposome Res. 1998;8:165–182. [Google Scholar]
- 29.Pereira F B, Goñi F M, Muga A, Nieva J L. Permeabilization and fusion of uncharged lipid vesicles induced by the HIV-1 fusion peptide adopting an extended conformation: dose and sequence effects. Biophys J. 1997;73:1977–1986. doi: 10.1016/S0006-3495(97)78228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pereira F B, Valpuesta J M, Basañez G, Goñi F M, Nieva J L. Interbilayer lipid-mixing induced by the HIV-1 fusion peptide on large unilamellar vesicles: the nature of the nonlamellar intermediates. Chem Phys Lipids. 1999;103:11–20. doi: 10.1016/s0009-3084(99)00087-0. [DOI] [PubMed] [Google Scholar]
- 31.Rafalski M, Lear J, DeGrado W. Phospholipid interactions of synthetic peptides representing the N-terminus of HIV gp41. Biochemistry. 1990;29:7917–7922. doi: 10.1021/bi00486a020. [DOI] [PubMed] [Google Scholar]
- 32.Reithmeier R A F. Characterization and modeling of membrane protein using sequence analysis. Curr Opin Struct Biol. 1995;5:491–500. doi: 10.1016/0959-440x(95)80034-4. [DOI] [PubMed] [Google Scholar]
- 33.Ruiz-Argüello M B, Goñi F M, Pereira F B, Nieva J L. Phosphatidylinositol-dependent membrane fusion induced by a putative fusogenic sequence of Ebola virus. J Virol. 1998;72:1775–1781. doi: 10.1128/jvi.72.3.1775-1781.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salzwedel K, West J T, Hunter E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J Virol. 1999;73:2469–2480. doi: 10.1128/jvi.73.3.2469-2480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schaal H, Klein M, Gehrmann P, Adams O, Scheid A. Requirement of N-terminal amino acid residues of gp41 for human immunodeficiency virus type 1-mediated cell fusion. J Virol. 1995;69:3308–3314. doi: 10.1128/jvi.69.6.3308-3314.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slepushkin V A, Andreev S M, Sidorova M V, Melikyan G B, Grigoriev V B, Chumakov V M, Grinfeldt A E, Manukyan R A, Karamov E V. Investigation of human immunodeficiency virus fusion peptides. Analysis of interrelations between their structure and function. AIDS Res Hum Retroviruses. 1992;8:9–18. doi: 10.1089/aid.1992.8.9. [DOI] [PubMed] [Google Scholar]
- 37.Struck D K, Hoekstra D, Pagano R E. Use of resonance energy transfer to monitor membrane fusion. Biochemistry. 1981;20:4093–4099. doi: 10.1021/bi00517a023. [DOI] [PubMed] [Google Scholar]
- 38.Turner B G, Summers M F. Structural biology of HIV. J Mol Biol. 1999;285:1–32. doi: 10.1006/jmbi.1998.2354. [DOI] [PubMed] [Google Scholar]
- 39.Weissenhorn W, Dessen A, Harrison S C, Skehel J J, Wiley D C. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–428. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 40.White J. Membrane fusion. Science. 1992;258:917–923. doi: 10.1126/science.1439803. [DOI] [PubMed] [Google Scholar]
- 41.White S, Wimley W C, Ladokhin A S, Hristova K. Protein folding in membranes: determining energetics of peptide-bilayer interactions. Methods Enzymol. 1998;295:62–87. doi: 10.1016/s0076-6879(98)95035-2. [DOI] [PubMed] [Google Scholar]
- 42.White S H, Wimley W C. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 43.Wild C, Shugars D C, Greenwell T, McDanal C, Matthews T. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type-1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci USA. 1994;91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson I A, Skehel J J, Wiley D C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 45.Wimley W, White S H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 46.Yau W, Wimley W C, Gawrisch K, White S H. The preference of tryptophan for membrane interfaces. Biochemistry. 1998;37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]