

Summary
Current studies in early cancer detection based on liquid biopsy data often rely on off-the-shelf models and face challenges with heterogeneous data, as well as manually designed data preprocessing pipelines with different parameter settings. To address those challenges, we present AutoCancer, an automated, multimodal, and interpretable transformer-based framework. This framework integrates feature selection, neural architecture search, and hyperparameter optimization into a unified optimization problem with Bayesian optimization. Comprehensive experiments demonstrate that AutoCancer achieves accurate performance in specific cancer types and pan-cancer analysis, outperforming existing methods across three cohorts. We further demonstrated the interpretability of AutoCancer by identifying key gene mutations associated with non-small cell lung cancer to pinpoint crucial factors at different stages and subtypes. The robustness of AutoCancer, coupled with its strong interpretability, underscores its potential for clinical applications in early cancer detection.
Subject areas: Health sciences, Cancer systems biology, Cancer, Computing methodology, Machine learning
Graphical abstract
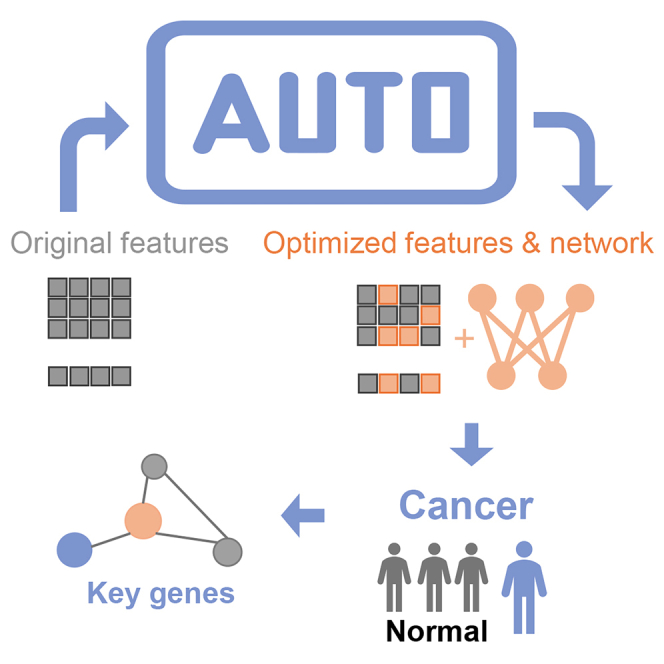
Highlights
-
•
An automated, multimodal, and interpretable framework for early cancer detection
-
•
Unify feature selection, neural architecture search, and hyperparameter optimization
-
•
Evaluated in both specific cancer types and pan-cancer analysis
-
•
Identifying key gene mutations associated with different cancer stages and subtypes
Health sciences; Cancer systems biology; Cancer; Computing methodology; Machine learning
Introduction
According to the latest report1 from the International Agency for Research on Cancer (IARC), cancer is the leading cause of premature deaths worldwide. The Office of the National Statistics highlights the importance of early detection for improved survival rates.2 As a potential solution, liquid biopsy, a non-invasive technique involving the sampling of non-solid specimens,3,4 offers the possibility for early cancer detection and longitudinal tracking. This technique analyzes circulating tumor cells (CTCs), extracellular vesicles (EVs), cell-free DNA (cfDNA), and circulating tumor DNA (ctDNA) from fluids like blood, urine, and saliva. Despite its promise, early cancer screening based on liquid biopsy remains as an emerging field with research questions to be addressed.
Firstly, the diversity of liquid biopsy components contributes to data complexity, heterogeneity, poor annotation, and unstructured nature.5,6 However, the standardization and unification of features are challenging due to the inherent data multimodality, such as methylation, single nucleotide variants (SNVs), copy number variations (CNVs), protein levels,7,8 and even other data types such as fragmentomics and multiple analytes.6,9 Secondly, biomarker selection is a formidable challenge due to the multifaceted and intricate mechanisms underlying cancer progressions.10 Given the massive analytes with over 20,000 genes and more than 50,000 protein isoforms, identifying cancer-related biomarkers presents a huge feature selection challenge. Thirdly, the development of automated workflow is crucial for achieving rapid and accurate analysis in cancer detection.11,12 Such workflow can minimize human intervention and lower the technical barriers of machine learning for non-specialist medical practitioners. Lastly, interpretability is essential for the successful integration of deep learning models into clinical practice.5,13 Ensuring these models with interpretabilities allows medical professionals to understand the underlying decision-making processes and validate the detections.
Computational approaches, including statistical analysis, traditional machine learning, and deep learning, show promises in identifying cancer-specific signatures from liquid biopsies. Statistical analysis select relevant biomarkers with strong correlations to target (outcome) variables (such as disease status or phenotypes) from large-scale medical data and determine the thresholds for these biomarkers, enabling cancer detection or analysis based on liquid biopsies.14,15,16 These methods efficiently handle large-scale data, but their effectiveness depends on data quality, as biased data can lead to inaccurate results. Traditional machine learning algorithms, including linear models, decision trees, and SVMs, are frequently employed for early cancer detection due to their simplicity and robustness. Notable applications include lung-CLiP,17 which combines multiple algorithms to estimate cancer likelihood from blood cfDNA using multigenomic features, GEMINI,18 which employs logistic regression to analyze early-stage genome-wide mutational profiles of cfDNA, and CancerSEEK,19 which uses logistic regression and random forest to classify cancers based on ctDNA mutations and protein biomarkers. Machine learning techniques are also emerging in the analysis of other liquid components, such as extracellular vesicles.20,21 In the field of fragmentomics, the methods such as DELFI9,22 have demonstrated the feasibility in recognizing fragmentation patterns for early cancer detection. Liu et al.23 developed an Adaptive SVM to enhance early cancer screening accuracy based CNVs and fragmentomics. Deep learning-based methods have shown potential in early cancer detection. Wong et al.24 constructed and introduced AnDE classifiers based on blood test records from 1,817 patients for cancer detection. Li et al.25 introduced DISMIR, a model integrating Convolutional Neural Networks and Long Short-Term Memory (LSTM) to differentiate whether a sequencing read originates from cancerous or normal tissue. Li et al.26 developed cfSort, a neural network-based model designed to quantify tissue composition in cfDNA using a supervised approach. Deep learning methods offer advantages such as high accuracy and the ability to model complex patterns in large datasets, suggesting a potential area for exploration further.
In consideration of the challenges, we propose AutoCancer, an automated, multimodal, and Transformer-based framework that is both interpretable and versatile. By integrating feature selection (FS), neural architecture search (NAS), and hyperparameter optimization (HPO) within a unified workflow, AutoCancer addresses the need for human intervention in deploying early cancer detection models and provides the related users with a simplified pipeline. We demonstrate the efficacy of AutoCancer in facilitating the early detection of specific cancer types as well as pan-cancer analysis. Furthermore, we leverage the interpretability of AutoCancer to pinpoint key gene mutations associated with non-small cell lung cancer (NSCLC). Our findings concur with the gene mutations reported in existing literature and reveal mutations and mutation pairs that may be relevant to specific tumor stages and subtypes. By harnessing the state-of-the-art deep learning techniques and incorporating multimodal data sources, we hope that this work adds significant values to ongoing efforts in early cancer research, providing a valuable tool for both clinicians and biomedical researchers. The source code of AutoCancer is publicly available at https://github.com/ElaineLIU-920/AutoCancer.git.
Results
Overview of AutoCancer
Figure 1A presents an overview of AutoCancer, a versatile framework that integrates automated deep learning, disease diagnosis, gene screening, and gene pair discovery into a comprehensive framework for early cancer detection. This framework is designed to handle multimodal inputs and is capable of simultaneously performing feature selection (FS) and neural network design (incorporating NAS and HPO). The attention mechanism within AutoCancer plays a crucial role in enhancing its functionality. By leveraging this mechanism, attention scores can be utilized to identify genes with significant contributions to cancer detection. Additionally, the Transformer, as a context-aware model, allows for the deep examination of gene pair interactions, providing insights into the unique roles of co-mutated gene pairs underlying the complex processes within cancer development.
Figure 1.

Overview of the AutoCancer methodology
(A) Functional specifications of AutoCancer.
(B) Workflow steps of AutoCancer.
Figure 1B illustrates the workflow of AutoCancer. In the specific early cancer detection task of NSCLC, the employed features of samples are heterogeneous data. During the FS process, essential features are extracted from the original input, ensuring that only the most relevant information is retained. These selected features are then introduced into an automatically designed Transformer block, where they undergo a feature fusion process. This step effectively combines distinct features, enabling the model to capture complex relationships and patterns within given data. After feature fusion, the feature embeddings are incorporated into an automatically designed multilayer perceptron (MLP) block, which is responsible for executing cancer detection. Bayesian optimization (BO) is employed to co-optimize the processes of FS and neural network design by maximizing the model performance and minimizing the fraction of selected features. Such optimization stratgey streamlines the model development process and ensures robust performance in cancer detection tasks.
AutoCancer enables effective early cancer detection
In this study, we initially evaluated the performance of AutoCancer and the state-of-the-arts (SOTA) method, Lung-CLiP17 on an NSCLC dataset. As shown in Table 1, our proposed AutoCancer significantly outperformed Lung-CLiP by improving the accuracy from 0.780 to 0.833 on the in-sample test set. Furthermore, we evaluated the performance of our model on a completely independent external cohort, referred as the out-of-sample test set, to ensure its effectiveness and generalizability. AutoCancer outperformed Lung-CLiP by improving the accuracy from 0.656 to 0.703 on this independent test set. This comparison was performed under ten repetitions, each with a random data splitting and parameter initialization. Previously, Lung-CLiP achieved the best performance for early cancer detection on the dataset by integrating five conventional machine learning classifiers and analyzing the features after conducting complex statistical scoring. It is worth noting that the Lung-CLiP model normalized the prediction likelihood across the entire test set after obtaining the final prediction result. In contrast, our model does not utilize the information of the test set before reporting its performance, yet it still demonstrates better results across almost all metrics, as tabulated in Table 1 and Figures 2A and 2B.
Table 1.
Performance comparison of AutoCancer and Lung-CLiP
| Metric | In-sample test set |
Out-of-sample test set |
||
|---|---|---|---|---|
| AutoCancer | Lung-CLiP | AutoCancer | Lung-CLiP | |
| Accuracy | 0.833 0.015 | 0.780 0.020 | 0.703 0.007 | 0.656 0.006 |
| ROC-AUC | 0.915 0.014 | 0.871 0.017 | 0.761 0.002 | 0.731 0.003 |
| PR-AUC | 0.959 0.007 | 0.928 0.011 | 0.812 0.004 | 0.786 0.003 |
| Precision | 0.930 0.011 | 0.861 0.021 | 0.738 0.002 | 0.640 0.008 |
| Recall | 0.803 0.020 | 0.792 0.020 | 0.620 0.011 | 0.684 0.007 |
∗mean s.d under 10 repeats with different random seeds.
Figure 2.

Comparison of AutoCancer and SOTA in terms of different metrics
(A) Comparison with Lung-CLiP on the in-sample test set for NSCLC.
(B) Comparison with Lung-CLiP on the out-of-sample test set for NSCLC.
(C) Comparison with nine optimized methods on the out-of-sample test set of NSCLC.
(D) Comparison with GENIMI and DELFI on the test set for LUCAS cohort.
(E) Comparison with DELFI and ASVM on the test set for pan-cancer cohort. n = 10 repeats with different random seeds for data splitting and model initialization. The error bars indicate mean ± s.d.
To validate the performance of AutoCancer on the detection of NSCLC further, we conducted a comprehensive comparison with several baseline methods, including bi-directional LSTM (Bi-LSTM),27 MLP, Extra Trees,28 Random Forest,29 stochastic gradient descent (SGD),30 gradient boosting,31 SVM,32 AdaBoost,33 and Gaussian process.34 In order to ensure a fair comparison between AutoCancer and other classifiers, we applied HPO on each method. The comparative results are presented in Figure 2C, which clearly demonstrates that AutoCancer significantly surpasses traditional methods in terms of accuracy, PR-AUC, and ROC-AUC. It is worth noting that the performance of these learning methods is varied along with the chosen metric of interest; for instance, Extra Trees was ranked second when accuracy was selected as the metric of interest; gradient boosting was ranked second when PR-AUC was chosen; and AdaBoost was ranked second in terms of ROC-AUC. In contrast, AutoCancer consistently maintains superior and stable performance across all metrics, achieving the best results with relatively small variances. The comparison results reveal that the performance of conventional methods is not satisfactory, as the average accuracy is mostly around 0.6.
Subsequently, we tested the performance of AutoCancer against SOTA methods DELFI and GENIMI using the LUCAS cohort. This lung cancer dataset, consisting of over 500 features, served as a stringent test for AutoCancer’s FS capabilities. As demonstrated in the LUCAS cohort study (Figure 2D), AutoCancer exhibited exceptional performance compared to DELFI and GENIMI. In terms of PR-AUC, precision, accuracy, and ROC-AUC, AutoCancer outperformed both methods with 0.861, 0.809, 0.764, and 0.843, respectively. While DELFI displayed a marginally higher recall rate (0.697 vs. 0.690), it fell short in other metrics compared to AutoCancer. These results clearly illustrate the robustness of AutoCancer’s FS capabilities, even when handling datasets with over 500 features.
Finally, we evaluated the performance of AutoCancer against SOTA methods DELFI and ASVM using the pan-cancer cohort. As depicted in Figure 2E, although AutoCancer’s recall is lower, its overall accuracy and ROC-AUC remain competitive when compared to both ASVM and DELFI. In terms of precision and PR-AUC, AutoCancer surpasses both ASVM and DELFI, with 0.911 (precision) and 0.937 (PR-AUC), compared to 0.852 (precision) and 0.928 (PR-AUC) for ASVM, and 0.846 (precision) and 0.913 (PR-AUC) for DELFI. This enhanced performance allows for more accurate identification of cancer cases, thereby minimizing the need for subsequent diagnostic procedures for patients. Furthermore, AutoCancer’s robust interpretability distinguishes it from these two SOTA methods, offering an additional advantage in the field of cancer detection.
These comprehensive comparisons not only highlight the effectiveness of AutoCancer but also demonstrate its robustness and consistency in performance, further solidifying its potential as a reliable tool for early cancer detection.
Analysis of data modality and feature selection results
To demonstrate the importance of integrating multiple data modalities and explore the potential benefits of multimodal approaches in cancer detection, we conducted two modality ablation experiments on LUCAS and NSCLC cohorts with different combinations of data modalities.
In the first experiment, as illustrated in Figure 3A, the performance of models with different combinations of data modalities (mutational, mutational+clinical, mutational+Fragmentome, and mutational+Fragmentome+clinical) are varied substantially in the LUCAS cohort. Specifically, the model incorporating all three modalities (Mutation+Fragmentome+Clinic) demonstrated the highest accuracy, F1 score, PR-AUC, and ROC-AUC, indicating the advantages of multimodal data integration. The results suggest that combining genetic mutation information with fragmentomic data and clinical data can lead to a more comprehensive understanding of the underlying mechanisms, thereby improving the model’s ability to detect NSCLC. Moreover, the standard deviation of models with more data modalities was generally lower than those with fewer modalities, suggesting that the integration of multiple data sources not only enhances the performance but also contributes to the stability and reliability in model performance.
Figure 3.

Data modality ablation and feature selection results
(A) Bar charts of modality ablation experiment on LUCAS cohort.
(B) Bar charts of modality ablation experiment on NSCLC cohort. n = 10 repeats with different random seeds for data splitting and model initialization. The error bars indicate mean ± s.d.
(C) Violin plots on the statistical differences in 1D features between non-small cell lung cancer and normal samples.
In the second experiment, there was a noticeable difference in performance between a single modality and two modalities in the NSCLC cohort. As illustrated in Figure 3B, the models incorporating both SNV and clinical data consistently outperformed those solely utilizing SNVs across all metrics. This indicates that the inclusion of clinical data with SNVs improves the models’ performance in NSCLC detection. Similarly, the standard deviation of the models with both SNVs and clinical data is lower than those with SNVs only for all metrics, suggesting that the former provides stable and reliable performance.
These results underscore the importance of multimodal approaches in cancer detection. By integrating different data modalities, such as genetic mutations, fragmentomic data, and clinical data, we can leverage the complementary information to enhance the accuracy, reliability, and robustness of cancer detection models. This highlights the potential of such integrative approaches in improving early cancer detection and personalized treatment strategies.
Upon analyzing the results of multiple FS iterations for the NSCLC cohort, we observed that among all 1D features of the input, the number of SNVs () was consistently chosen, while other 1D features such as CNVs, plasma cfDNA concentration, and age were often excluded. We further investigated the differences between these features in non-small cell lung cancer and normal samples, as depicted in Figure 3C. Our analysis revealed that, although some of the unselected 1D features also exhibit statistical significance, the feature stands out as the most significant among them as ranked by p-value. This observation provides evidence, to a certain extent, that the features selected by AutoCancer are reliable and effective in distinguishing non-small cell lung cancer from normal samples. The results also underscore the capability of the proposed framework to identify and prioritize relevant features, thereby reducing the dimensionality of the input and potentially enhancing the performance of subsequent analyses.
Evaluation of framework components and optimization robustness
Figure 4A and Table 2 present an ablation study comparing two settings derived from the AutoCancer framework: Feature Selection (FS) combined with NAS (as in Table 3) and HPO, and NAS combined with HPO alone. The table displays the mean and standard deviation of several evaluation metrics across 10 repetitions with different random seeds. The FS+NAS+HPO approach consistently outperforms the NAS+HPO approach on both in-sample and out-of-sample test sets. In particular, FS+NAS+HPO attains higher accuracy, with the means of 0.833 and 0.703 on the two test sets, as opposed to 0.723 and 0.643 from NAS+HPO. Likewise, the ROC-AUC and PR-AUC metrics exhibit substantial improvement with FS+NAS+HPO, presenting the means of 0.915 and 0.959 on the in-sample test set, and 0.761 and 0.812 on the out-of-sample test set. In contrast, NAS+HPO yields the means of 0.860 and 0.930 for ROC-AUC and PR-AUC on the in-sample test set, and 0.714 and 0.749 on the out-of-sample test set. Moreover, FS+NAS+HPO also demonstrates superior performance in terms of precision and recall. These findings demonstrate that incorporating FS into the NAS and HPO process significantly enhances model performance across various evaluation metrics, indicating that FS+NAS+HPO is a more effective method for automated deep learning deployment.
Figure 4.

Evaluation of AutoCancer framework components and robustness of optimization
(A) Bar charts comparing ablation experiments with and without feature selection on the NSCLC dataset.
(B) Bar charts comparing Transformer-based fusion block with basic vector operations on the NSCLC dataset. n = 10 repeats with different random seeds for data splitting and model initialization. The error bars indicate mean ± s.d.
(C) Evolution process of the objective function under Bayesian optimization.
Table 2.
Ablation study results
| Metric | In-sample test set |
Out-of-sample test set |
||
|---|---|---|---|---|
| FS+NAS+HPO | NAS+HPO | FS+NAS+HPO | NAS+HPO | |
| Accuracy | 0.833 0.015 | 0.723 0.033 | 0.703 0.007 | 0.643 0.022 |
| ROC-AUC | 0.915 0.014 | 0.860 0.023 | 0.761 0.002 | 0.714 0.016 |
| PR-AUC | 0.959 0.007 | 0.930 0.013 | 0.812 0.004 | 0.749 0.007 |
| Precision | 0.930 0.011 | 0.872 0.037 | 0.738 0.002 | 0.707 0.044 |
| Recall | 0.803 0.020 | 0.723 0.076 | 0.620 0.011 | 0.600 0.077 |
∗mean s.d under 10 repeats with different random seeds.
Table 3.
The search space of network architecture
| Decision variables | Type | NSCLC | LUCAS | Pan-cancer |
|---|---|---|---|---|
| Transformer dropout | Real | [1e-1, 0.3] | [1e-1, 0.3] | \ |
| Number of encoder layers | Integer | [1, 5] | [1, 2] | \ |
| Number of attention heads | Categorical | [2, 4, 6, 8] | [2, 4] | \ |
| Dimension of each hidden | Categorical | [16, 32, 64] | [2, 4, 6, 8] | \ |
| MLP dropout | Real | [1e-1, 0.3] | [1e-1, 0.3] | [1e-1, 0.3] |
| Number of MLP layers | Integer | [1, 5] | [1, 5] | [1, 5] |
| Learning rate | Real | [1e-4, 1e-1] | [1e-4, 1e-1] | [1e-4, 1e-1] |
| Patience | Integer | [1, 10] | [1, 10] | [1, 10] |
To explore the benefits of adopting the Transformer as a fusion module, we conducted an experiment comparing the performance of Transformer-based feature fusion with different basic vector operations (addition, dot product, and subtraction) for NSCLC detection. This experiment aimed to assess whether these simpler methods, which are computationally less demanding, could provide comparable or even better performance than the Transformer-based method. The results of this experiment are visualized in Figure 4B, demonstrating that the Transformer-based feature fusion method consistently outperforms the other three vector operations across all metrics. Specifically, the Transformer-based method achieved an accuracy of 0.703, which is notably higher than the accuracies obtained by the dot product (0.660), subtraction (0.588), and addition (0.569) methods. Similar trends can be observed for the F1 score, PR-AUC, and ROC-AUC metrics, further emphasizing the performance of the Transformer-based approach as depicted in Figure 4B. These results highlight the effectiveness of the Transformer-based feature fusion method in the context of NSCLC detection. By leveraging the representation learning capabilities of the Transformer architecture, the proposed method can lead to good performance in distinguishing cancerous cases from control cases.
In Figure 4C, we present the evolution process of the objective function values under different experimental settings, revealing the convergence behavior of BO. Upon observing Figure 4C, it is evident that the objective function values experience a rapid decrease during the initial few calls. Subsequently, the rate of reduction gradually slows down, with the objective function values eventually stabilizing at around 0.8. The depicted trends demonstrate the ability of BO to converge under different conditions, highlighting its robustness and adaptability in optimizing the objective function. Furthermore, the rapid initial decrease in the objective function values suggests that BO can efficiently identify promising regions in search space, thus contributing to the overall effectiveness of optimization.
Key gene mutation associated with non-small cell lung cancer
It is important to consider that each patient’s single nucleotide variant (SNV) length varies. Lung-CLiP transformed informative SNV features into a numerical score. Consequently, the contribution of these features to the final prediction of cancer or normal status cannot be observed directly in the Lung-CLiP model. This limitation highlights the necessity for our subsequent interpretability analysis of the AutoCancer model, which aims to provide a more transparent understanding of the feature contributions and prediction process.
For the non-small cell lung cancer dataset utilized in this study, the Transformer block optimized by the AutoCancer framework comprises four heads. By visualizing the attention matrix of each head (Figure S1), we observed that Head 1 and Head 4 jointly focus on the interactions between genes, while Head 2 and Head 3 primary focus on the relationships between the number of SNVs for each gene. We calculated the attention scores of mutations for all patients to identify the top 50 genetic mutations associated with non-small cell lung cancer. The attention relationships between these top 50 genes are illustrated in Figure 5A, where the horizontal axis coordinate of the heatmap arranges the genes in descending order based on their attention scores. Notably, our analysis identified several well-studied genes associated with NSCLC, such as TP53, ARID1A, FGFR1, PIK3CA, KRAS, ALK, CDKN2A, NF1, and others.35,36,37,38,39,40,41,42 Additionally, we revealed a few reported but not widely confirmed potential genes, including top-ranking FAT3, FAM135B, ZNF536, SLC8A1, and others.43,44 These findings underscore the AutoCancer framework’s potential to uncover novel genetic factors, contributing to NSCLC development and progression, warranting further investigation in future studies.
Figure 5.

Key gene mutations identified by AutoCancer
(A) Attention matrix of top 50 focused gene mutations in all patients.
(B) Functional enrichment results of top 50 genes.
(C) Top 20 genes with highest attention scores inferred by our model across different stages.
(D) Top 20 genes with highest attention scores inferred by our model across different subtypes of NSCLC.
We performed functional enrichment analysis on the top 50 genes and identified the relevant pathways associated with NSCLC, as presented in Figure 5B. Our model pinpointed several KEGG pathways (Figure 5B, first panel). Some of them are evidently involved in cancer, such as ‘non-small cell lung cancer’, ‘central carbon metabolism in cancer’, ‘proteoglycans in cancer’, and ‘microRNAs in cancer’. We also identified other validated pathways associated with NSCLC, such as the Ras signaling pathway, a well-established oncogenic pathway regulating cell proliferation, differentiation, and survival.45,46 Aberrant activation of this pathway, often due to mutations in the Ras family of genes, has been implicated in lung cancer development and progression.47,48 The Ras signaling pathway also influences the EGFR pathway, which contributes to the pathogenesis of various tumors, including NSCLC.49 RAP1 is essential for cell growth and, in conjunction with cAMP, plays a key mediating role in developing platinum resistance in NSCLC.50,51
The WP terms (Figure 5B, second panel) link the top genes identified by our model (TP53, PIK3CA, KRAS, CDKN2A, and ALK) to non-small cell lung cancer. In addition, TP53 network and DNA damage response (only ATM dependent)52 are also enriched.
The GO:MF terms (Figure 5B, third panel) identified by our model include MDM2/MDM4 family protein binding, glutamate-gated calcium ion channel activity, and NMDA glutamate receptor activity. MDM2 and MDM4 proteins are known to be involved in the regulation of the tumor suppressor p53,53 crucial for cancer prevention.54 Abnormalities in the binding and regulation of these proteins could contribute to NSCLC development and progression.55 Furthermore, glutamate-gated calcium ion channels and NMDA glutamate receptors, although primarily associated with the nervous system, have been implicated in regulating cell proliferation and apoptosis. Dysregulation of these channels and receptors has also been validated to influence lung cancer initiation and progression.56,57
Lastly, the HP terms directly link the top genes (TP53, PIK3CA, KRAS, and CDKN2A) identified by our model to the lung adenocarcinoma and non-small cell lung carcinoma. This association further validates the potential relevance of our model’s findings to NSCLC development and progression. More details about the pathways identified by AutoCancer are listed in Table S1. The identified pathways and functional terms provide valuable insights into the molecular mechanisms behind NSCLC and may serve as potential therapeutic targets for future research.
Key gene mutation across different stages and subtypes in NSCLC
As depicted in Figures 5C and S2A, among the top 20 most focused genes across the three stages of NSCLC, some top genes are shared by different stages. FAT3 and TP53 are shared by all stages. ARID1A and GRIN2B are shared between Stage I and Stage II, while FAM135B and ANK2 are shared between Stage I and Stage III. SPHKAP, ZNF536, and NPAP1 are shared between Stage II and Stage III. This observation suggests that these shared genes may play crucial roles in the development and progression of NSCLC across different stages. However, more attention is given to stage-specific genes. The details of stage-specific genes are listed in Table S2.
In stage I, our attention model shows particular interest in 14 genes. Three of these genes belong to the ZNF family, including ZNF521, ZNF831, and ZNF423. As transcription factors, ZNFs comprise the largest family of sequence-specific DNA binding proteins, orchestrating a wide range of differentiation, development, metabolism, apoptosis, autophagy, and stemness maintenance.58 Other genes also involve in various biological processes, such as cell cycle regulation (CDKN2A), oxidative stress response (KEAP1), and cell adhesion (SLITRK3). The presence of these genes in Stage I suggests that their dysregulation may contribute to the initiation of tumorigenesis and early-stage NSCLC development.
In stage II, our model infers that there are thirteen genes that should be focused. These genes are implicated in various cellular processes, such as cell proliferation (NF1, RB1), cell adhesion (PCDH17, CDH10), and signal transduction (GRIN2A, EPHA6). Their presence in Stage II may indicate a role in the progression of NSCLC from early to more advanced stages, possibly through promoting cell growth, invasion, and metastasis.
For Stage III, thirteen genes of particular interest are reported by our model. These genes involve in more advanced stages of NSCLC, contributing to processes such as cell survival (TP63, KRAS, PIK3CA), angiogenesis (FGFR1), and immune response (DCSTAMP). Their involvement in Stage III NSCLC suggests that they may play a role in driving disease progression and resistance to therapy.
In Figures 5D and S2B, among the top 20 most focused genes in the three histological subtypes, TP53, FAT3, and FAM135B are shared by all subtypes. This suggests that these genes may play a crucial role in the pathogenesis of NSCLC, regardless of the cancer subtype. Based on the descriptions in GeneCards, TP53 is a well-known tumor suppressor gene. FAT3 yields a higher mutation rate in NSCLC patients. On the other hand, FAM135B are less studied and warrant for further investigation to understand their roles in NSCLC. ARID1A, FGFR1, and TP63 as shared by adenocarcinoma and squamous cell carcinoma, indicating that these genes may be involved in common molecular pathways between these two subtypes. ARID1A is a chromatin remodeler, and its mutations have been linked to various cancers. FGFR1 is a receptor tyrosine kinase, and its aberrant activation has been implicated in tumorigenesis. TP63 is a member of the p53 family and plays a role in epithelial development and differentiation. LRFN2 is shared by adenocarcinoma and large cell carcinoma, suggesting a potential role in the development of these subtypes. LRFN2 is a synaptic adhesion molecule, and its function in cancer remains largely unknown. SPHKAP, LRFN5, and CSMD3 are shared by squamous cell carcinoma and large cell carcinoma, indicating that these genes may be involved in the molecular mechanisms common to these subtypes. The functions of these genes in cancer are not well understood and require further investigation. In addition to shared genes, our analysis also identified subtype-specific genes of interest. The details of subtype-specific genes are listed in Table S3.
In adenocarcinoma, genes of particular interest include thirteen items. These genes are involved in various biological processes, such as cell adhesion (PCDH17, CDH10), signal transduction (GRIN2A, GPR112), and cell migration (SLITRK3, LRRC7). Their presence in adenocarcinoma suggests that their dysregulation may contribute to the development and progression of this specific histological subtype of NSCLC.
In squamous cell carcinoma, 12 genes are selected with particular interest. These genes are implicated in various cellular processes, such as cell-cell communication (NRXN1, GRIN2B), signal transduction (TSHZ2, ZIC1), and cellular stress response (RNF216, LPPR4). Their presence in squamous cell carcinoma may indicate a role in the development and progression of this histological subtype of NSCLC, possibly through promoting cell growth, invasion, and metastasis.
For large cell carcinoma, genes of particular interest include thirteen items. These genes may be involved in various cellular processes, such as cell cycle regulation (RB1, PTEN), cell migration (NAV3, KIF2B), and signal transduction (GPR158, DUSP27). Their involvement in large cell carcinoma suggests that they may play a role in driving disease progression and resistance to therapy in this histological subtype of NSCLC.
In Figures 5C and 5D, the top 20 gene mutations focused on by the model in different stages and histological subtypes of NSCLC are presented, respectively. Interestingly, TP53, a widely focused and studied gene in cancer, is also a major focus of our proposed AutoCancer. It is the primary gene considered by our model in any cancer stage and histological subtype. Nonetheless, the degree of attention varies, with an increased emphasis on TP53 as the stage advances. In different subtypes, the attention given to TP53 by the model is comparable in adenocarcinoma and large cell carcinoma, while it is particularly concentrated on TP53 mutations in squamous cell carcinoma. As a tumor suppressor gene, TP53 plays a pivotal role in regulating cell cycle progression, apoptosis, and DNA repair.59
We further examined the top-ranking genes and their mutual focus across various stages and subtypes in Figures S2C and S2D. The visualization of attention among these targeted gene mutations unveils a variety of focus patterns for each stage and subtype, emphasizing their unique characteristics. By investigating the top 20 most focused genes, we can uncover key biological pathways and networks in non-small cell lung cancer, potentially identifying therapeutic targets and biomarkers for diagnosis and prognosis. In conclusion, our study of the top 20 most focused genes with SNV mutations in NSCLC stages and histological subtypes has revealed potential stage- and subtype-specific as well as shared molecular mechanisms underlying disease development and progression. Further research on these genes’ functions and interactions may offer valuable insights into NSCLC’s molecular basis and lead to the identification of diagnostic and therapeutic targets for patients with different stages and histological subtypes of this disease.
Gene mutation combinations associated with NSCLC
Here, we discuss some of the notable gene pairs and their potential implications in lung cancer development and progression. Figure 6A represents top 50 gene pairs associated with NSCLC that exhibit SNVs, as identified by AutoCancer in all patients. The information of these gene pairs can be found in Table S4.
Figure 6.

Combination gene mutation associated with non-small cell lung cancer
(A) Top 50 gene pairs in all samples.
(B) Top 20 gene pairs in different stages.
(C) Top 20 gene pairs in different subtypes.
The AutoCancer has identified several gene pairs involving well-known cancer-related genes such as TP53, NF1, and CDKN2A. For instance, TP53, a well-established tumor suppressor gene, is found in multiple gene pairs, including TP53-SPHKAP, TP53-FAM135B, TP53-CDH10, TP53-ZNF423, TP53-TAS2R1, TP53-LPPR4, TP53-HTR5A, TP53-GPR158, and TP53-NPAP1. The frequent appearance of TP53 in these pairs highlights its critical role in NSCLC. Similarly, the NF1 gene, a known tumor suppressor, is found in gene pairs NF1-ITGB3, NF1-GPR112, and SLC8A1-NF1. This suggests a possible association between these genes and NSCLC, and further research into their functional roles and interactions could provide valuable insights. The CDKN2A gene, another well-known tumor suppressor, appears in the CDKN2A-ARID1A and ZNF521-CDKN2A pairs.
Another interesting observation is the presence of genes involved in synaptic transmission and neuronal signaling, such as GRIN2A and LRRC7. The NPAP1-GRIN2A and NCKAP5-GRIN2A pairs suggest a possible association between neuronal signaling and NSCLC. The RIMS2-LRRC7 and PEG3, ZIM2-LRRC7 pairs also hint at a potential link between synaptic transmission and cancer development.
Interestingly, the gene pairs PCDH17-FAT3, ZNF536-FAT3, and SLITRK4-FAT3 all involve the FAT3 gene. FAT3 is a member of the protocadherin family, which plays a role in cell adhesion and signaling. This gene’s involvement in multiple pairs indicates its potential significance in NSCLC, possibly through its role in tumor cell invasion and metastasis.
As depicted in Figures 6B and 6C, a diverse set of gene pairs is identified across various stages and subtypes of NSCLC. In the gene-pair graph, the size of a node represents its degree, which indicates the number of edges connected to it. This measure also reflects the importance of a gene in a given context, as it demonstrates the frequency of a gene’s appearance among the top gene pairs. The central nodes of each subgraph correspond to the three nodes with the highest degree. By examining Figures 6B and 6C, it is evident that each stage and subtype displays unique combinations of gene pairs, and the central node genes differ accordingly. These distinctions underscore the discrete molecular mechanisms involved in the development and progression of each NSCLC stage and histological subtype. Detailed information regarding these gene pairs across different situations can be found in Tables S5 and S6.
In conclusion, the AutoCancer model has successfully identified several gene pairs with SNV mutations associated with NSCLC. Our analysis of these gene pairs has unveiled potential molecular mechanisms and therapeutic targets for this disease, thereby highlighting the intricate genetic landscape of NSCLC and emphasizing the necessity for further investigation into these gene pairs and their functional consequences in lung cancer development and progression.
Discussion
To conclude, we introduce AutoCancer, an automated, interpretable, and multimodal framework that utilizes metaheuristic optimization and deep learning methodologies for early cancer detection. To the best of our knowledge, this is the inaugural application of the Transformer model to liquid biopsy-based cancer detection, enabling our framework to handle both well-structured and heterogeneous data across various dimensions. Furthermore, we consolidate FS, NAS, and HPO into a comprehensive optimization framework, concurrently addressing these three challenges via BO.
The comparison between AutoCancer and SOTA methods highlights the performance of AutoCancer, indicating its potential to significantly impact clinical applications. The identification of key gene mutations and their combinations associated with NSCLC, as well as the pinpointing of crucial factors at different stages and subtypes, demonstrates the interpretability of AutoCancer. This interpretability is essential for clinical applications, as it allows for a better understanding of the underlying biological mechanisms and the development of targeted therapies.
A critical aspect of cancer research that warrants emphasis is the complex and multifactorial nature of cancer etiology. Cancer arises from a combination of genetic, epigenetic, and environmental factors that interact in intricate ways to drive disease progression. Consequently, it is crucial for researchers to adopt a multi-omics or multimodal approach in their investigations, integrating diverse data types such as genomics, transcriptomics, proteomics, and metabolomics. AutoCancer’s ability to handle heterogeneous data inputs is aligned well with this perspective, highlighting its potential to contribute significantly to the field of cancer research.
Limitations of the study
One of the primary limitations in the development and validation of AutoCancer is the current scarcity of public multimodal liquid biopsy data. This paucity of available data restricts the opportunities to comprehensively assess the full potential of our proposed framework and may inadvertently lead to overfitting or underestimation of its performance. Additionally, the limited data may not encompass the entire spectrum of cancer types and stages, potentially hindering the generalizability of AutoCancer to a wider range of clinical scenarios. Addressing these concerns and establishing secure data-sharing platforms will be essential for facilitating the advancement of AutoCancer and similar frameworks. Despite these limitations, the development of AutoCancer represents a significant step forward in the field of cancer detection and diagnosis. As more multimodal liquid biopsy data become available, it will be essential to further evaluate and refine AutoCancer, ensuring its effectiveness and applicability in a broad range of cancer detection tasks. This will ultimately contribute to the development of more accurate, efficient, and personalized diagnostic tools for cancer patients.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| NSCLC cohort | Lung-CLiP | https://doi.org/10.1038/s41586-020-2140-0 |
| LUCAS cohort (fragmentome+clinic) | DELFI | https://doi.org/10.1038/s41467-021-24994-w |
| LUCAS cohort (mutation) | GEMINI | https://doi.org/10.1038/s41588-023-01446-3 |
| Pan-cancer cohort | ASVM | https://doi.org/10.1093/bioinformatics/btab236 |
| Software and algorithms | ||
| traditional machine learning models | scikit-learn | https://scikit-learn.org/stable/ |
| Lung-Clip model | Nature | https://doi.org/10.1038/s41586-020-2140-0 |
| DELFI | Nature | https://doi.org/10.1038/s41467-021-24994-w |
| GEMINI | Nature Genetics | https://doi.org/10.1038/s41588-023-01446-3 |
| ASVM model | Bioinfomatics | https://doi.org/10.1093/bioinformatics/btab236 |
| AutoCancer | This paper | https://github.com/ElaineLIU-920/AutoCancer.git |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Prof. Ka-Chun Wong (kc.w@cityu.edu.hk).
Materials availability
This study did not generate new biological data.
Data and code availability
All relevant data are public data and also have been deposited on Github. The DOIs are listed in the key resources table. All original code has been deposited at the Github and is publicly available as of the date of publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
This paper analyzes existing, publicly available data. The study does not use experimental models typical in life sciences.
Method details
Datasets collection and splitting
The NSCLC cohort was collected from.17 The first dataset of NSCLC cohort, from which the in-sample test set is derived, consists of a discovery cohort of 104 patients with early-stage NSCLC and 56 risk-matched controls from four cancer centers. The second dataset, constituting the out-of-sample test set, was collected from an independent institution, involving 46 patients with early-stage NSCLC and 48 risk-matched controls. The data includes nine 1D clinical features and 37 features describing each SNV (the number of SNVs varied among patients; this finally form 2D feature). The LUCAS cohort, consisting of genome-wide mutation, fragmentome, and clinical data, was collected from9 and.18 The dataset includes 387 patients with lung cancer. The pan-cancer cohort, involving fragmentome, genome-wide CNV, and clinical data, was collected from.22 The dataset comprises 423 patients across eight cancer types.
For all datasets, we employed k-fold nested cross-validation to partition the datasets. The inner cross-validation was used for selecting the optimal model parameters, while the outer cross-validation was employed to evaluate model performance. The choice of k-value depends on the selection in the original SOTA comparison methods: k = 5 for the NSCLC and LUCAS cohorts, and k = 10 for the pan-cancer cohort.
Model backbone
In our framework, we employ the Transformer encoder60 combined with a MLP as the backbone of AutoCancer. The Transformer block is utilized to encode and integrate the multi-modal features, while the MLP block serves as a classifier.
The Transformer is a context-aware architecture that incorporates attention mechanisms61 to establish global dependencies. The encoder of the Transformer consists of N identical layers, each containing a multi-head attention sub-layer and a position-wise feedforward network sub-layer. Residual connections and layer normalization are applied around each sub-layer. We represent the 1D features as and the 2D features as . First, we project the 1D features onto the dimensions using linear projection (LP) denoted by:
| (Equation 1) |
Next, we concatenate and along the second dimension denoted by:
| (Equation 2) |
The input embedding for the Transformer is then given by . In this case, and . The queries, keys, and values are intermediate representations obtained from inputs through linear transformations: , , . The regular bidirectional dot-product attention, a key component in the Transformer, has the following form, where is attention matrix:
| (Equation 3) |
The feature embedding after applying Transformer is denoted as . By utilizing MLP, we obtained the final classification according to:
| (Equation 4) |
Finally, cross-entropy loss (Equation 5) was used to update the entire model based on supervisory signals:
| (Equation 5) |
Here, y represents the true label, denotes the predicted probabilities, C is the number of classes, and and are the true and predicted probabilities of class i, respectively.
Feature selection and neural network design via Bayesian optimization
Bayesian optimization (BO)62 is a probability based, highly efficient, and robust metaheuristic optimization technique. The main advantages of Bayesian optimization are its ability to handle high-dimensional and complex functions, as well as its robustness to noise and uncertainty. In our framework, Bayesian optimization implements the co-optimization of feature selection and neural network design. The search space of features is all available features obtained from public datasets, and the search space of neural network design is listed in Table 3.
In this optimization problem, we minimize the fraction of selected features and maximize the model performance, as shown in the objective function (Equation 6).
| (Equation 6) |
Here, is the denotation of optimized features, F is the original features, and is the optimized model. The dimension of features is denoted by , and the performance metric of a model is denoted by . α and β are the weights to balance the multi-objective optimization problem of minimizing the fraction of selected features by and maximizing the model performance by . In this manuscript, we set α and β to be 0.03 and 1, respectively. Here, assigning a small weight to α implies that the model is more inclined to impose a minor penalty on redundant features. Our purpose of implementing Bayesian optimization is to find the optimal subset of features and model architecture that minimize Equation 6. Consequently, this optimization problem should be further formulated as Equation 7, where and represent the optimal solution combination.
| (Equation 7) |
Clearly, the exact functional form of this optimization problem is unknown, making it impossible to compute an analytical solution using gradient methods. For complex black-box optimization problems with the aforementioned characteristics, Bayesian optimization serves as an effective solution. This is because it relies on Bayes’ theorem to estimate the probability distribution of optimization objectives and decision variables while actively selecting the most promising solutions based on the fitted results.
The core components of Bayesian optimization consist of a surrogate model and an acquisition function, which balances the trade-off between exploration and exploitation in the search for the global optimum. Specifically, this method seeks to optimize an unknown objective function by constructing a probabilistic surrogate model that approximates the underlying function. The acquisition function focuses on regions where the objective function is expected to yield minimum values. In the iterative process of Bayesian optimization, the surrogate model is updated with each new objective function evaluation, and the acquisition function is subsequently optimized to determine the next point to be evaluated. This iterative procedure continues until a predetermined stopping criterion is met, such as reaching a maximum number of iterations or achieving a satisfactory level of convergence.
In AutoCancer, we adopt Gaussian processes63 as surrogate model. As the Gaussian process is a non-parametric model,64 it is less prone to overfitting while possessing extensible flexibility.65
A Gaussian process consists of a mean function and a positive semi-definite covariance function as Equation 8, where , and .
| (Equation 8) |
For acquisition function, we utilize Expected Improvement66 to realize the active strategy for selecting the next evaluation point. Let represents the observed set, represents the decision vector, represents the observation value, and represents the observation error. The acquisition (Equation 9) function is constructed from the posterior distribution obtained from the observed dataset , and guides the selection of the next evaluation point by maximizing it.
| (Equation 9) |
In Equation 9, is the current optimal function value, is the standard normal distribution probability density function.
Biological analysis tools
For gene function without a citation in this paper, we referred https://www.genecards.org/. We implemented enrichment analysis on top-ranked genes using the method on the g:Profiler67 Website https://biit.cs.ut.ee/gprofiler.
Quantification and statistical analysis
In this manuscript, Mann-Whitney-Wilcoxon tests (M.W.W. in figures, also known as rank-sum tests) were conducted to compare the difference between non-small cell lung cancer and normal samples.
Acknowledgments
This research was substantially sponsored by the research project (Grant No. 32170654) supported by the National Natural Science Foundation of China and was substantially supported by the Shenzhen Research Institute, City University of Hong Kong. The work described in this paper was substantially supported by the grant from the Research Grants Council of the Hong Kong Special Administrative Region [CityU 11203723]. The work described in this paper was partially supported by the grants from City University of Hong Kong (2021SIRG036, CityU 9667265, CityU 11203221) and Innovation and Technology Commission (ITB/FBL/9037/22/S).
Author contributions
L.L. and K.-C.W. conceived the study; L.L. and Y.X. designed and implemented the algorithms; L.L., Z.Z., and L.H. conducted the result analysis; L.L. wrote and revised the manuscript; Y.X., Z.Z., L.H., J.S., Q.L., B.T., and K.-C.W. proofread the manuscript; K.-C.W. funded and supervised the study.
Declaration of interests
The authors declare no competing interest.
Published: June 5, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110183.
Supplemental information
References
- 1.Soerjomataram I., Bray F. Planning for tomorrow: Global cancer incidence and the role of prevention 2020-2070. Nat. Rev. Clin. Oncol. 2021;18:663–672. doi: 10.1038/s41571-021-00514-z. [DOI] [PubMed] [Google Scholar]
- 2.John S., Broggio J. Newport: Office for National Statistics; 2019. Cancer Survival in England: National Estimates for Patients Followed up to 2017. [Google Scholar]
- 3.Crowley E., Di Nicolantonio F., Loupakis F., Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013;10:472–484. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- 4.Liu L., Chen X., Petinrin O.O., Zhang W., Rahaman S., Tang Z.-R., Wong K.-C. Machine learning protocols in early cancer detection based on liquid biopsy: a survey. Life. 2021;11:638. doi: 10.3390/life11070638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miotto R., Wang F., Wang S., Jiang X., Dudley J.T. Deep learning for healthcare: review, opportunities and challenges. Brief. Bioinform. 2018;19:1236–1246. doi: 10.1093/bib/bbx044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song P., Wu L.R., Yan Y.H., Zhang J.X., Chu T., Kwong L.N., Patel A.A., Zhang D.Y. Limitations and opportunities of technologies for the analysis of cell-free DNA in cancer diagnostics. Nat. Biomed. Eng. 2022;6:232–245. doi: 10.1038/s41551-021-00837-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen K., Sun J., Zhao H., Jiang R., Zheng J., Li Z., Peng J., Shen H., Zhang K., Zhao J., et al. Non-invasive lung cancer diagnosis and prognosis based on multi-analyte liquid biopsy. Mol. Cancer. 2021;20:23–27. doi: 10.1186/s12943-021-01323-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pantel K., Alix-Panabières C. Liquid biopsy and minimal residual disease—latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019;16:409–424. doi: 10.1038/s41571-019-0187-3. [DOI] [PubMed] [Google Scholar]
- 9.Mathios D., Johansen J.S., Cristiano S., Medina J.E., Phallen J., Larsen K.R., Bruhm D.C., Niknafs N., Ferreira L., Adleff V., et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes. Nat. Commun. 2021;12:5060. doi: 10.1038/s41467-021-24994-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W., Liu J.-B., Hou L.-K., Yu F., Zhang J., Wu W., Tang X.-M., Sun F., Lu H.-M., Deng J., et al. Liquid biopsy in lung cancer: significance in diagnostics, prediction, and treatment monitoring. Mol. Cancer. 2022;21:25. doi: 10.1186/s12943-022-01505-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moser T., Kühberger S., Lazzeri I., Vlachos G., Heitzer E. Bridging biological cfDNA features and machine learning approaches. Trends Genet. 2023;39:285–307. doi: 10.1016/j.tig.2023.01.004. [DOI] [PubMed] [Google Scholar]
- 12.Thomasian N.M., Kamel I.R., Bai H.X. Machine intelligence in non-invasive endocrine cancer diagnostics. Nat. Rev. Endocrinol. 2022;18:81–95. doi: 10.1038/s41574-021-00543-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holzinger A., Langs G., Denk H., Zatloukal K., Müller H. Causability and explainability of artificial intelligence in medicine. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 2019;9:e1312. doi: 10.1002/widm.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lennon A.M., Buchanan A.H., Kinde I., Warren A., Honushefsky A., Cohain A.T., Ledbetter D.H., Sanfilippo F., Sheridan K., Rosica D., et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science. 2020;369:eabb9601. doi: 10.1126/science.abb9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qvick A., Stenmark B., Carlsson J., Isaksson J., Karlsson C., Helenius G. Liquid biopsy as an option for predictive testing and prognosis in patients with lung cancer. Mol. Med. 2021;27:68. doi: 10.1186/s10020-021-00331-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keup C., Suryaprakash V., Hauch S., Storbeck M., Hahn P., Sprenger-Haussels M., Kolberg H.-C., Tewes M., Hoffmann O., Kimmig R., Kasimir-Bauer S. Integrative statistical analyses of multiple liquid biopsy analytes in metastatic breast cancer. Genome Med. 2021;13:85. doi: 10.1186/s13073-021-00902-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chabon J.J., Hamilton E.G., Kurtz D.M., Esfahani M.S., Moding E.J., Stehr H., Schroers-Martin J., Nabet B.Y., Chen B., Chaudhuri A.A., et al. Integrating genomic features for non-invasive early lung cancer detection. Nature. 2020;580:245–251. doi: 10.1038/s41586-020-2140-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruhm D.C., Mathios D., Foda Z.H., Annapragada A.V., Medina J.E., Adleff V., Chiao E.J., Ferreira L., Cristiano S., White J.R., et al. Single-molecule genomewide mutation profiles of cell-free DNA for non-invasive detection of cancer. Nat. Genet. 2023;55:1301–1310. doi: 10.1038/s41588-023-01446-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen J.D., Li L., Wang Y., Thoburn C., Afsari B., Danilova L., Douville C., Javed A.A., Wong F., Mattox A., et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359:926–930. doi: 10.1126/science.aar3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoshino A., Kim H.S., Bojmar L., Gyan K.E., Cioffi M., Hernandez J., Zambirinis C.P., Rodrigues G., Molina H., Heissel S., et al. Extracellular vesicle and particle biomarkers define multiple human cancers. Cell. 2020;182:1044–1061.e18. doi: 10.1016/j.cell.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei R., Chen L., Qin D., Guo Q., Zhu S., Li P., Min L., Zhang S. Liquid biopsy of extracellular vesicle-derived miR-193a-5p in colorectal cancer and discovery of its tumor-suppressor functions. Front. Oncol. 2020;10:1372. doi: 10.3389/fonc.2020.01372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cristiano S., Leal A., Phallen J., Fiksel J., Adleff V., Bruhm D.C., Jensen S.Ø., Medina J.E., Hruban C., White J.R., et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570:385–389. doi: 10.1038/s41586-019-1272-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu L., Chen X., Wong K.-C. Early cancer detection from genome-wide cellfree DNA fragmentation via shuffled frog leaping algorithm and support vector machine. Bioinformatics. 2021;37:3099–3105. doi: 10.1093/bioinformatics/btab236. [DOI] [PubMed] [Google Scholar]
- 24.Wong K.-C., Chen J., Zhang J., Lin J., Yan S., Zhang S., Li X., Liang C., Peng C., Lin Q., et al. Early cancer detection from multianalyte blood test results. iScience. 2019;15:332–341. doi: 10.1016/j.isci.2019.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J., Wei L., Zhang X., Zhang W., Wang H., Zhong B., Xie Z., Lv H., Wang X. Dismir: D eep learning-based noninvasive cancer detection by i ntegrating dna s equence and methylation information of i ndividual cell-free dna r eads. Brief. Bioinform. 2021;22:bbab250. doi: 10.1093/bib/bbab250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li S., Zeng W., Ni X., Liu Q., Li W., Stackpole M.L., Zhou Y., Gower A., Krysan K., Ahuja P., et al. Comprehensive tissue deconvolution of cell-free DNA by deep learning for disease diagnosis and monitoring. Proc. Natl. Acad. Sci. USA. 2023;120 doi: 10.1073/pnas.2305236120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sak H., Senior A., Beaufays F. Long short-term memory based recurrent neural network architectures for large vocabulary speech recognition. arXiv. 2014 doi: 10.48550/arXiv.1402.1128. Preprint at. [DOI] [Google Scholar]
- 28.Ahmad M.W., Reynolds J., Rezgui Y. Predictive modelling for solar thermal energy systems: A comparison of support vector regression, random forest, extra trees and regression trees. J. Clean. Prod. 2018;203:810–821. [Google Scholar]
- 29.Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- 30.Zhang T. Proceedings of the twenty-first international conference on Machine learning. 2004. Solving large scale linear prediction problems using stochastic gradient descent algorithms; p. 116. [Google Scholar]
- 31.Friedman J.H. Greedy function approximation: a gradient boosting machine. Ann. Statist. 2001;29:1189–1232. [Google Scholar]
- 32.Chang C.-C., Lin C.-J. LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011;2:1–27. [Google Scholar]
- 33.Hastie T., Rosset S., Zhu J., Zou H. Multi-class adaboost. Stat. Interface. 2009;2:349–360. [Google Scholar]
- 34.Williams C.K., Rasmussen C.E. Vol. 2. MIT Press; 2006. Gaussian Processes for Machine Learning. [Google Scholar]
- 35.Mogi A., Kuwano H. TP53 Mutations in Nonsmall Cell Lung Cancer. BioMed Research International. 2011;2011:583929. doi: 10.1155/2011/583929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manceau G., Letouzé E., Guichard C., Didelot A., Cazes A., Corté H., Fabre E., Pallier K., Imbeaud S., Le Pimpec-Barthes F., et al. Recurrent inactivating mutations of ARID2 in non-small cell lung carcinoma. Int. J. Cancer. 2013;132:2217–2221. doi: 10.1002/ijc.27900. [DOI] [PubMed] [Google Scholar]
- 37.Dutt A., Ramos A.H., Hammerman P.S., Mermel C., Cho J., Sharifnia T., Chande A., Tanaka K.E., Stransky N., Greulich H., et al. Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PLoS One. 2011;6:e20351. doi: 10.1371/journal.pone.0020351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto H., Shigematsu H., Nomura M., Lockwood W.W., Sato M., Okumura N., Soh J., Suzuki M., Wistuba I.I., Fong K.M., et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008;68:6913–6921. doi: 10.1158/0008-5472.CAN-07-5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riely G.J., Marks J., Pao W. KRAS mutations in non-small cell lung cancer. Proc. Am. Thorac. Soc. 2009;6:201–205. doi: 10.1513/pats.200809-107LC. [DOI] [PubMed] [Google Scholar]
- 40.Gerber D.E., Minna J.D. ALK inhibition for non-small cell lung cancer: from discovery to therapy in record time. Cancer Cell. 2010;18:548–551. doi: 10.1016/j.ccr.2010.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gutiontov S.I., Turchan W.T., Spurr L.F., Rouhani S.J., Chervin C.S., Steinhardt G., Lager A.M., Wanjari P., Malik R., Connell P.P., et al. CDKN2A loss-offunction predicts immunotherapy resistance in non-small cell lung cancer. Sci. Rep. 2021;11:20059. doi: 10.1038/s41598-021-99524-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayashi T., Desmeules P., Smith R.S., Drilon A., Somwar R., Ladanyi M. RASA1 and NF1 are preferentially co-mutated and define a distinct genetic subset of smoking-associated non-small cell lung carcinomas sensitive to MEK inhibition. Clin. Cancer Res. 2018;24:1436–1447. doi: 10.1158/1078-0432.CCR-17-2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu X. Proceedings of the 2019 11th International Conference on Bioinformatics and Biomedical Technology. 2019. Analysis of the Target Genes of Transcription Factor ZNF536 in Lung Adenocarcinoma; pp. 81–85. [DOI] [Google Scholar]
- 44.Feng Z., Yin Y., Liu B., Zheng Y., Shi D., Zhang H., Qin J. Prognostic and immunological role of FAT family genes in non-small cell lung cancer. Cancer Control. 2022;29 doi: 10.1177/10732748221076682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alam M., Hasan G.M., Eldin S.M., Adnan M., Riaz M.B., Islam A., Khan I., Hassan M.I. Investigating regulated signaling pathways in therapeutic targeting of non-small cell lung carcinoma. Biomed. Pharmacother. 2023;161:114452. doi: 10.1016/j.biopha.2023.114452. [DOI] [PubMed] [Google Scholar]
- 46.Han J., Liu Y., Yang S., Wu X., Li H., Wang Q. MEK inhibitors for the treatment of non-small cell lung cancer. J. Hematol. Oncol. 2021;14:1–12. doi: 10.1186/s13045-020-01025-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Downward J. Targeting RAS and PI3K in lung cancer. Nat. Med. 2008;14:1315–1316. doi: 10.1038/nm1208-1315. [DOI] [PubMed] [Google Scholar]
- 48.Brose M.S., Volpe P., Feldman M., Kumar M., Rishi I., Gerrero R., Einhorn E., Herlyn M., Minna J., Nicholson A., et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997–7000. [PubMed] [Google Scholar]
- 49.Cooper W.A., Lam D.C.L., O’Toole S.A., Minna J.D. Molecular biology of lung cancer. J. Thorac. Dis. 2013;5:S479–S490. doi: 10.3978/j.issn.2072-1439.2013.08.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao L., Lan X., Shi X., Zhao K., Wang D., Wang X., Li F., Huang H., Liu J. Cytoplasmic RAP1 mediates cisplatin resistance of non-small cell lung cancer. Cell Death Dis. 2017;8:e2803. doi: 10.1038/cddis.2017.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park J.-Y., Juhnn Y.-S. cAMP signaling increases histone deacetylase 8 expression via the Epac2-Rap1A-Akt pathway in H1299 lung cancer cells. Exp. Mol. Med. 2017;49:e297. doi: 10.1038/emm.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lundholm L., Hååg P., Zong D., Juntti T., Mörk B., Lewensohn R., Viktorsson K. Resistance to DNA-damaging treatment in non-small cell lung cancer tumorinitiating cells involves reduced DNA-PK/ATM activation and diminished cell cycle arrest. Cell Death Dis. 2013;4:e478. doi: 10.1038/cddis.2012.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toledo F., Wahl G.M. MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int. J. Biochem. Cell Biol. 2007;39:1476–1482. doi: 10.1016/j.biocel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duffy M.J., Synnott N.C., McGowan P.M., Crown J., O’Connor D., Gallagher W.M. p53 as a target for the treatment of cancer. Cancer Treat Rev. 2014;40:1153–1160. doi: 10.1016/j.ctrv.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 55.Arnoff T.E., El-Deiry W.S. MDM2/MDM4 amplification and CDKN2A deletion in metastatic melanoma and glioblastoma multiforme may have implications for targeted therapeutics and immunotherapy. Am. J. Cancer Res. 2022;12:2102–2117. [PMC free article] [PubMed] [Google Scholar]
- 56.Pollock P.M., Cohen-Solal K., Sood R., Namkoong J., Martino J.J., Koganti A., Zhu H., Robbins C., Makalowska I., Shin S.-S., et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat. Genet. 2003;34:108–112. doi: 10.1038/ng1148. [DOI] [PubMed] [Google Scholar]
- 57.Deutsch S.I., Tang A.H., Burket J.A., Benson A.D. NMDA receptors on the surface of cancer cells: target for chemotherapy? Biomed. Pharmacother. 2014;68:493–496. doi: 10.1016/j.biopha.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jen J., Wang Y.-C. Zinc finger proteins in cancer progression. J. Biomed. Sci. 2016;23:53–59. doi: 10.1186/s12929-016-0269-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang X., Simpson E.R., Brown K.A. p53: protection against tumor growth beyond effects on cell cycle and apoptosis. Cancer Res. 2015;75:5001–5007. doi: 10.1158/0008-5472.CAN-15-0563. [DOI] [PubMed] [Google Scholar]
- 60.Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A.N., Kaiser Ł., Polosukhin I. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017;30 [Google Scholar]
- 61.Lin Z., Feng M., Santos C.N.d., Yu M., Xiang B., Zhou B., Bengio Y. A structured self-attentive sentence embedding. arXiv. 2017 doi: 10.48550/arXiv.1703.03130. Preprint at. [DOI] [Google Scholar]
- 62.Frazier P.I. Informs; 2018. Bayesian Optimization”. Recent Advances in Optimization and Modeling of Contemporary Problems; pp. 255–278. [Google Scholar]
- 63.MacKay D.J. Introduction to Gaussian processes. NATO ASI series F computer and systems sciences. 1998;168:133–166. [Google Scholar]
- 64.Seeger M. Gaussian processes for machine learning. Int. J. Neural Syst. 2004;14:69–106. doi: 10.1142/S0129065704001899. [DOI] [PubMed] [Google Scholar]
- 65.Ghahramani Z. Probabilistic machine learning and artificial intelligence. Nature. 2015;521:452–459. doi: 10.1038/nature14541. [DOI] [PubMed] [Google Scholar]
- 66.Benassi R., Bect J., Vazquez E. International Conference on Learning and Intelligent Optimization. Springer; 2011. Robust Gaussian Process-Based Global Optimization Using a Fully Bayesian Expected Improvement Criterion; pp. 176–190. [Google Scholar]
- 67.Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. g: Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are public data and also have been deposited on Github. The DOIs are listed in the key resources table. All original code has been deposited at the Github and is publicly available as of the date of publication. DOIs are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.