

Abstract
LY6E is an antiviral restriction factor that inhibits coronavirus spike-mediated fusion, but the cell types in vivo that require LY6E for protection from respiratory coronavirus infection are unknown. Here we used a panel of seven conditional Ly6e knockout mice to define which Ly6e-expressing cells confer control of airway infection by murine coronavirus and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Loss of Ly6e in Lyz2-expressing cells, radioresistant Vav1-expressing cells and non-haematopoietic cells increased susceptibility to murine coronavirus. Global conditional loss of Ly6e expression resulted in clinical disease and higher viral burden after SARS-CoV-2 infection, but little evidence of immunopathology. We show that Ly6e expression protected secretory club and ciliated cells from SARS-CoV-2 infection and prevented virus-induced loss of an epithelial cell transcriptomic signature in the lung. Our study demonstrates that lineage confined rather than broad expression of Ly6e sufficiently confers resistance to disease caused by murine and human coronaviruses.
Host resistance to viral pathogens depends in part on constitutively expressed and infection-induced effector proteins that selectively target distinct steps of the viral replication cycle. In concert with humoral and cellular immune responses, cell-intrinsic antiviral effectors limit viral burden and consequently constrain viral pathogenesis. Intracellular viral detection triggers rapid production and extracellular release of type I and III interferons that act in an autocrine and paracrine manner to induce the expression of cytokines, chemokines and cell-intrinsic effector proteins1. While the expression of a subpopulation of effectors proteins are undetectable in the absence of interferons, several proteins categorized as interferon-stimulated genes (ISGs) are prominently expressed without infectious stimuli. These basally expressed cell-intrinsic effectors may serve as an early barrier against viral infection that precedes production of inflammatory cytokines and recruitment of immune cells.
Lymphocyte antigen 6 complex, locus E (LY6E, previously called retinoic acid-inducible gene E (RIG-E), thymic shared antigen 1 (TSA-1) and stem cell antigen 2 (SCA-2)) is constitutively expressed across multiple tissues in humans2 and mice3–5 and is also induced by type I interferon6. LY6E belongs to the LY6/uPAR superfamily of secreted or plasma membrane-associated proteins that are characterized by a conserved, three-finger folding motif that is maintained through disulfide bonds7. LY6E modulates immune response signalling pathways8–10 and may be involved in myeloid and lymphoid cell development11–13. The murine ortholog of LY6E was also identified as the cognate receptor for mouse syncytin-A3, which mediates cell-to-cell fusion during formation of the syncytiotrophoblast layer I of the fetomaternal placenta. Genetic deletion of germline Ly6e is incompatible with embryonic development14 due to disrupted placental architecture15.
The role of LY6E in modulating infection by enveloped RNA viruses has been an active area of investigation over the past decade. LY6E was originally identified through high-throughput screening as an ISG that enhances infection by flaviviruses, a subset of alphaviruses, and influenza A virus16–18. In following studies, LY6E was found to enhance post-attachment viral entry of flaviviruses and influenza A virus6,19 as well as human immunodeficiency virus 1 (ref. 20). In more recent screening efforts, LY6E was identified as a restriction factor for coronaviruses21–24 including members of the betacoronavirus genus: mouse hepatitis virus (MHV) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). However, our understanding of how LY6E controls coronavirus pathogenesis in vivo has only been demonstrated with MHV21, which is effectively restricted by LY6E in the immune compartment.
In this Article, we sought to address two principal knowledge gaps: (1) the contribution of Ly6e in distinct immune cell subsets to controlling MHV infection in mice, and (2) the role of Ly6e in an immunocompetent murine model of non-mouse-adapted SARS-CoV-2 infection without ectopic expression of human ACE2. For the MHV studies, we used comparative models of intraperitoneal versus intranasal administration in (1) new mouse lines with Ly6e knocked out in distinct immune cells, (2) bone-marrow chimaeric mice and (3) Ly6e whole-body knockouts. We found that Ly6e-dependent control of MHV-induced pathogenesis is cumulatively conferred by Lyz2-positive cells, by a radioresistant Vav1-positive cell population, and by non-haematopoietic Vav1-negative cells. We additionally found that whole-body Ly6e knockout mice are more susceptible to SARS-CoV-2, with pathogenic outcomes that mirror mild to moderate coronavirus disease 2019 (COVID-19) in humans. From transcriptomic analysis of lungs of SARS-CoV-2-infected animals, we found a gene signature consistent with loss of secretory cell-associated transcripts. Furthermore, we used orthologous approaches to demonstrate that global loss of Ly6e expression increases SARS-CoV-2 infection of ciliated cells and secretory club cells, which may underly the observed clinical disease.
Myeloid Ly6e protects the airways from MHV infection
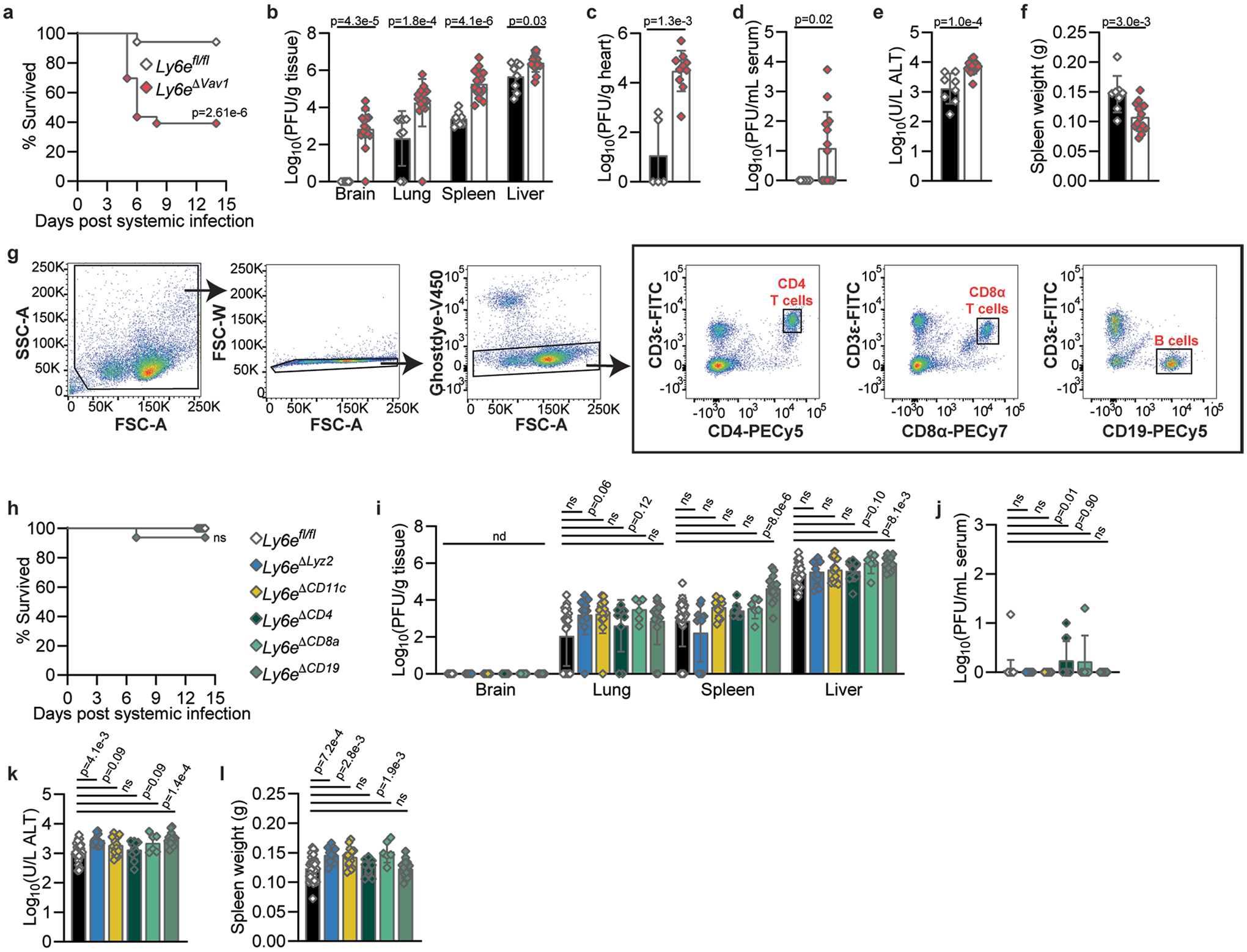
Control of intraperitoneal infection by mouse hepatitis virus A59 (MHV), a murine coronavirus, is mediated in part by LY6E in immune cells21. To investigate whether haematopoietic expression of Ly6e contributes to airway defence against intranasal coronavirus infection, we intranasally infected Ly6eΔVav1 mice, which lack Ly6e in all immune cells, and Ly6efl/fl littermates with MHV. In this intranasal infection model, Ly6eΔVav1 had 100% lethality with a viral dose that was sublethal in Ly6efl/fl littermates (Fig. 1a). Intraperitoneal infection with the same dose, which is ten-fold lower than was used previously21, caused less morbidity (Extended Data Fig. 1a), confirming a role for haematopoietic LY6E during respiratory coronavirus infection.
Fig. 1 |. Control of respiratory murine coronavirus infection is conferred in part by Ly6e in myeloid cells.
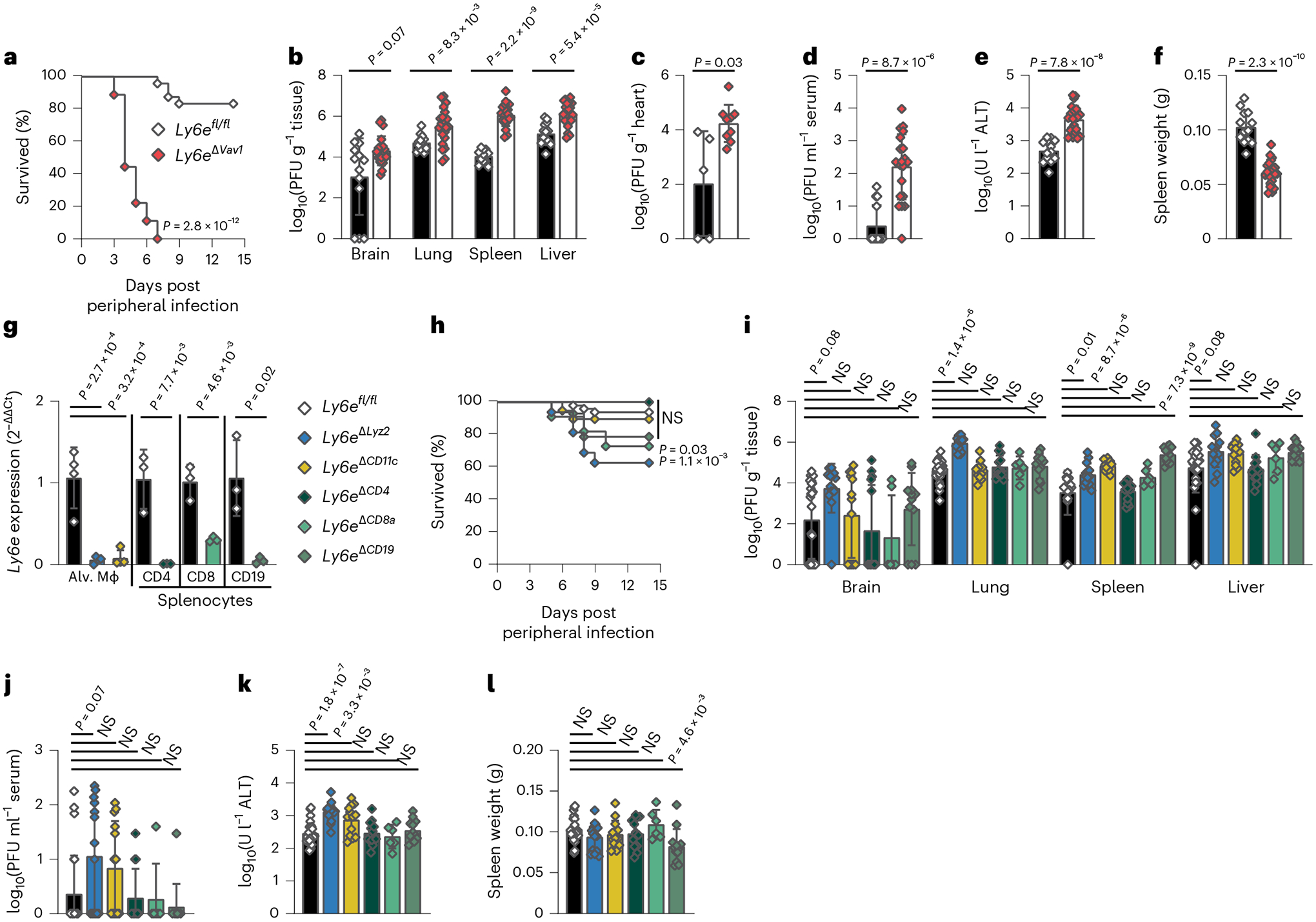
a–f, Mice were intranasally infected with 5,000 plaque-forming units (PFU) MHV-A59 and assessed for survival (a), viral burden in brain, lung, spleen and liver (b), viral burden in heart (c), viral burden in serum (d), serum ALT (e) and post-mortem spleen weight (f). g, Relative Ly6e mRNA levels in lung alveolar macrophages (Alv. Mϕ, n = 4 Ly6efl/fl, n = 4 Ly6eΔLyz2, n = 4 Ly6eΔCD11c), splenic CD3ε+CD4+ T cells (n = 3 Ly6efl/fl and n = 3 Ly6eΔCD4), splenic CD3ε+CD8α+ T cells (n = 3 Ly6efl/fl and n = 3 Ly6eΔCD8a) and splenic CD19+CD3ε− B cells (n = 3 Ly6efl/fl and n = 3 Ly6eΔCD19). h–l, Mice were intranasally infected with 5,000 PFU MHV-A59 and assessed for survival (n = 48 Ly6efl/fl, n = 16 Ly6eΔLyz2, n = 19 Ly6eΔCD11c, n = 13 Ly6eΔCD4, n = 11 Ly6eΔCD8a and n = 14 Ly6eΔCD19) (h), viral burden in brain, lung, spleen and liver (i), viral burden in serum (j), serum ALT (k) and post-mortem spleen weight (l). In a, data represent the mean of n = 24 Ly6efl/fl and n = 18 Ly6eΔVav1; in b, n = 13 Ly6efl/fl and n = 21 Ly6eΔVav1; in c, n = 5 Ly6efl/fl and n = 9 Ly6eΔVav1; in d–f, n = 13 Ly6efl/fl and n = 21 Ly6eΔVav1; in i–l, n = 26 Ly6efl/fl, n = 14 Ly6eΔLyz2, n = 13 Ly6eΔCD11c, n = 13 Ly6eΔCD4, n = 6 Ly6eΔCD8a and n = 12 Ly6eΔCD19. Male and female mice were used at an approximately 1-to-1 ratio for these experiments. Statistical significance was determined by log-rank (Mantel–Cox) tests (a and h), two-sided Mann–Whitney test (b–d), two-sided unpaired t-test (e–g), Kruskal–Wallis test with Dunn’s multiple comparisons test (i and j) and one-way analysis of variance with Holm–Šídák’s multiple comparisons test (k and l). Error bars represent mean ± standard deviation. NS, not significant.
To evaluate how Ly6e in Vav1-expressing cells controls viral replication and pathogenesis, we examined viral burden and disease outcomes in MHV-infected mice. Viral burden in the lungs, spleen, liver (Fig. 1b and Extended Data Fig. 1b), heart (Fig. 1c and Extended Data Fig. 1c) and serum (Fig. 1d and Extended Data Fig. 1d) was higher in Ly6eΔVav1 mice after either intranasal or intraperitoneal infection. MHV infection of the brain was higher in intraperitoneally infected Ly6eΔVav1 mice (Extended Data Fig. 1b) but not after intranasal infection (Fig. 1b). Serum alanine aminotransferase (ALT), indicating hepatic damage, was higher (Fig. 1e and Extended Data Fig. 1e) and post-mortem spleen size (Fig. 1f and Extended Data Fig. 1f) was lower in Ly6eΔVav1 mice irrespective of infection route.
To identify subsets of cells that protect MHV-infected Ly6eΔVav1 mice from disease we generated conditional knockout mouse strains using distinct Cre-drivers and confirmed targeted gene ablation (Fig. 1g and Extended Data Fig. 1g). Ly6eΔLyz2 mice, which are predicted to have loss of Ly6e in subsets of macrophages, monocytes, neutrophils and epithelial cells25,26, partially recapitulated the susceptibility of Ly6eΔVav1 mice to intranasal MHV infection (Fig. 1h). Ly6eΔCD8a mice, which are predicted to have loss of Ly6e in CD8α+ T cells and CD8α+ dendritic cells, exhibited a modest susceptibility to intranasal MHV infection, while Ly6eΔCD11c, Ly6eΔCD4 and Ly6eΔCD19 mice were resistant to coronavirus-induced disease. In stark contrast to Ly6eΔVav1 mice, all conditional mutant strains survived intraperitoneal infection (Extended Data Fig. 1h).
We next investigated how ablation of Ly6e in distinct immune lineages affected MHV replication and pathogenesis. In intranasally infected mice, viral titres in the brain, liver (Fig. 1i), and serum (Fig. 1j) were unaffected by Ly6e deletion in any compartment. Ly6eΔLyz2 mice had higher viral burden in the lung; Ly6eΔLyz2, Ly6eΔCD11c and Ly6eΔCD19 mice had increased viral burden of the spleen (Fig. 1i). Serum ALT was higher in Ly6eΔLyz2 and Ly6eΔCD11c mice (Fig. 1k). Ly6eΔCD19 mice exhibited splenic hypoplasia after intranasal infection that was not observed in other strains (Fig. 1l). In intraperitoneally infected mice, viral burden in brain and lung were similar across all strains (Extended Data Fig. 1i). Only Ly6eΔCD19 mice had elevated viral titres in spleen and liver, consistent with Ly6e-deficient splenic B cells being highly permissive to MHV21. Several mice infected intraperitoneally were viraemic, with a modest elevation in viral titres in Ly6eΔCD8a mice (Extended Data Fig. 1j). Serum ALT levels were only higher in Ly6eΔLyz2 and Ly6eΔCD19 mice after intraperitoneal infection (Extended Data Fig. 1k). In contrast to the splenic hypoplasia observed in intraperitoneally infected Ly6eΔVav1 mice, Ly6eΔLyz2, Ly6eΔCD11c and Ly6eΔCD8a mice had enlarged spleens relative to Ly6efl/fl littermates (Extended Data Fig. 1l).
Ly6e in radioresistant cells protects against MHV infection
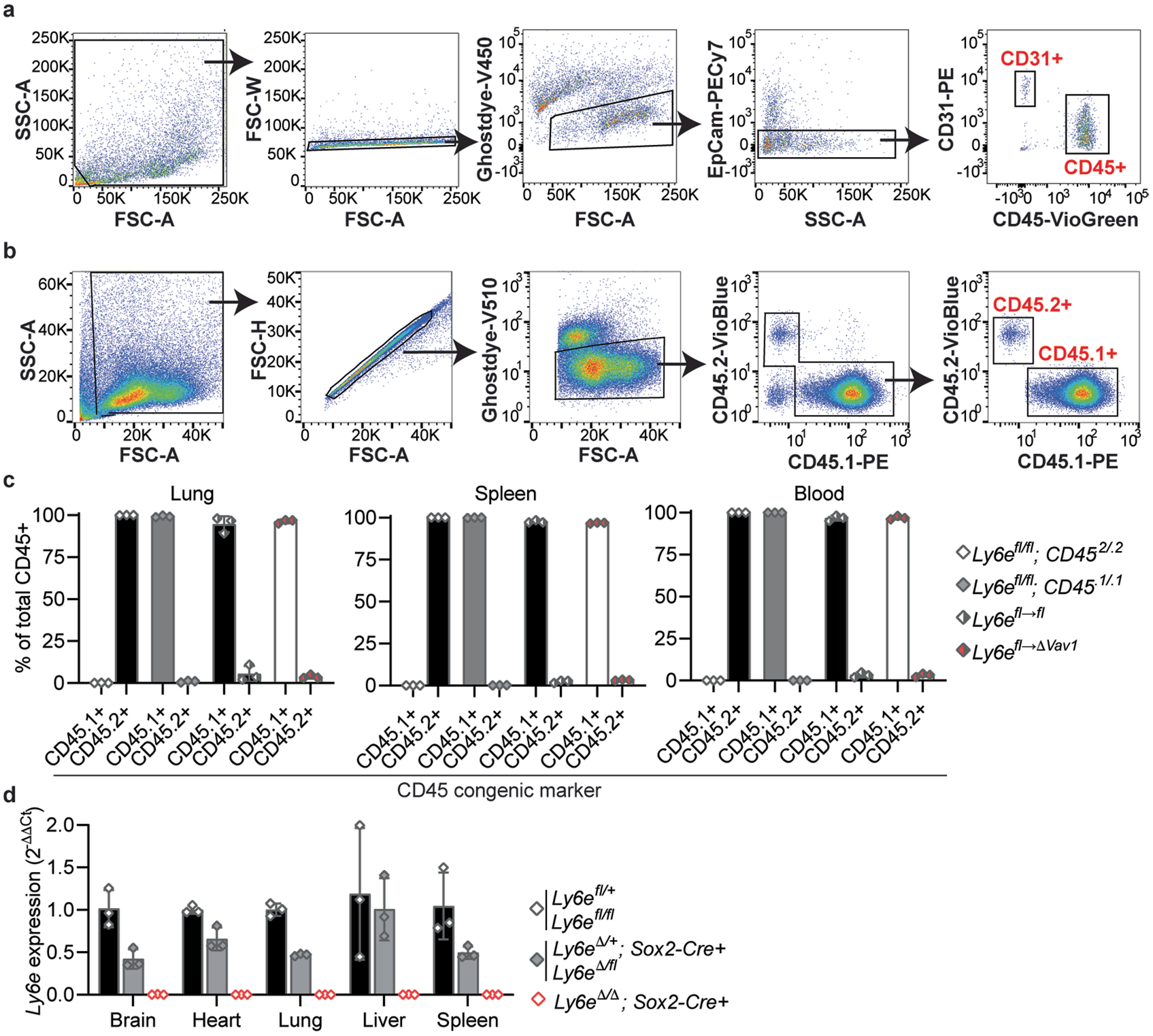
None of the conditional knockout strains fully recapitulated the pathogenesis phenotypes of Ly6eΔVav1 mice. Accordingly, we hypothesized that a Ly6e-deficient non-immune cell compartment generated by the Vav1-iCre transgene may contribute to the phenotype5,27. To test this model, we generated bone marrow chimaeric mice (Ly6efl→ΔVav1) to reconstitute Ly6e expression in the radiosensitive haematopoietic compartment of Ly6eΔVav1 mice (Fig. 2a). We confirmed restoration of Ly6e expression in the haematopoietic compartment of Ly6efl→ΔVav1 chimaeras by donor cells (Fig. 2b,c and Extended Data Fig. 2a–c). We found that Vav1-iCre did not affect Ly6e expression in lung CD31+CD45−EpCam− endothelial cells (Fig. 2d and Extended Data Fig. 2a). Ly6efl→ΔVav1 mice succumbed at a greater rate to intranasal MHV infection than Ly6efl→fl littermates (Fig. 2e). This suggests that Ly6e expression in a radioresistant Vav1-expressing compartment contributes to resistance to MHV. In contrast to Ly6eΔVav1 mice, Ly6efl→ΔVav1 chimaeras exhibited similar viral burden in lung, spleen, liver (Fig. 2f) and serum (Fig. 2g) as Ly6efl→fl littermates (Fig. 2f). Strikingly, virus was detected in the brains of six of nine Ly6efl→ΔVav1 mice, but only in two of ten Ly6efl→fl littermates. Ly6e-sufficient immune cells rescued hepatic damage (Fig. 2h) and splenic hypoplasia (Fig. 2i). This suggests that Ly6e expression in radioresistant Vav1-expressing cells reduces MHV infection of the brain but is dispensable for controlling viral replication and pathogenesis in peripheral organs. Collectively, our studies indicate that Ly6e in Lyz2-expressing cells and radioresistant Vav1-expressing cells confers resistance to MHV.
Fig. 2 |. Ly6e in radioresistant cells contributes to protection against murine coronavirus infection.
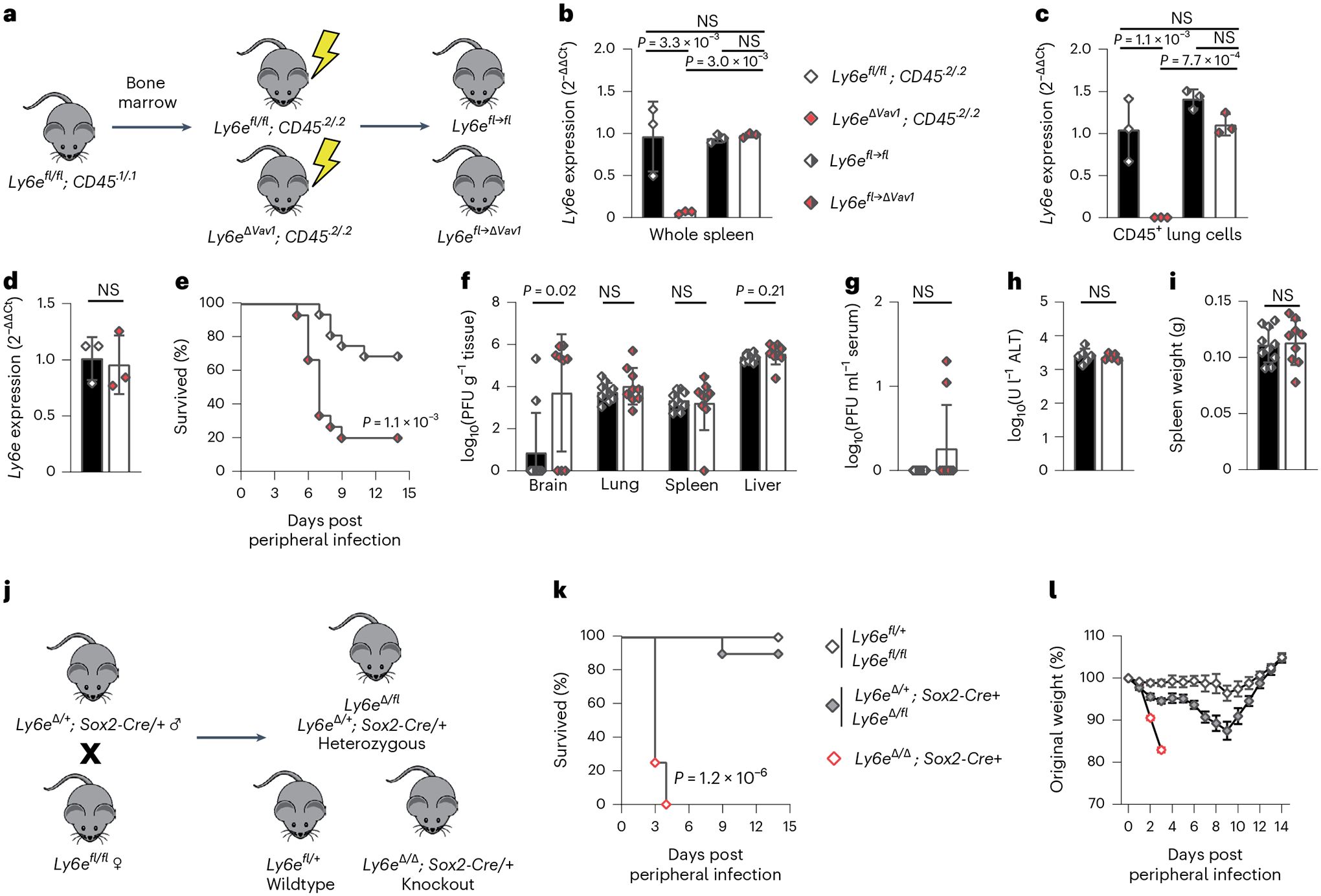
a, Experimental scheme to generate bone marrow chimaeric mice. b–d, Relative Ly6e mRNA levels in whole spleen (b), lung CD45+ immune cells (c) and lung CD31+CD45−EpCam− endothelial cells (d). e–i, Ly6efl→fl and Ly6efl→ΔVav1 mice were intranasally infected with 1,000 plaque-forming units (PFU) MHV-A59 and assessed for survival (e), viral burden in brain, lung, spleen and liver (f), viral burden in serum (g), serum ALT (h) and post-mortem spleen weight (i). j, Breeding scheme to generate whole-body Ly6e heterozygous and knockout mice. k,l, Ly6e wild-type, heterozygous and knockout mice were intranasally infected with 5,000 PFU MHV-A59 and assessed daily for survival (k) and weight loss (l). In b and c, data represent means from n = 3 Ly6efl/fl, n = 3 Ly6eΔVav1, n = 3 Ly6efl→fl and n = 3 Ly6efl→ΔVav1; in d, n = 3 Ly6efl/fl and n = 3 Ly6eΔVav1; in e, n = 16 Ly6efl→fl and n = 15 Ly6efl→ΔVav1; in f–i, n = 10 Ly6efl→fl and n = 9 Ly6efl→ΔVav1; in k and l, n = 6 Ly6e wild type, n = 10 Ly6e heterozygous and n = 10 Ly6e knockout. Male and female mice were used at an approximately 1-to-1 ratio for these experiments. Statistical significance was determined by log-rank (Mantel–Cox) tests (e and k), two-sided Mann–Whitney test (f and g), one-way analysis of variance (b and c) and two-sided unpaired t-test (d and h–l). Error bars represent mean ± standard deviation in all panels except for l, where error bars indicate mean ± standard error of the mean. NS, not significant.
The contribution of Ly6e in non-haematopoietic cells to defence against MHV is unclear. To address this gap, we used Sox2-Cre transgenic mice to generate Ly6e whole-body knockout mice (Fig. 2j and Extended Data Fig. 2d). This strategy bypasses the embryonic lethal effects of germline Ly6e deletion14 by preserving gene expression in the placenta15. Ly6e knockout mice (Ly6eΔ/Δ; Sox2-Cre+) succumbed rapidly to intranasal MHV infection (Fig. 2k). Ly6e heterozygous mice exhibited transient weight loss (Fig. 2l) but recovered. The additional loss of Ly6e in Vav1-negative cell types accelerated MHV-driven lethality compared with genetic ablation in Vav1-expressing cells alone, demonstrating a major contribution of the non-haematopoietic compartment to surviving MHV challenge.
Ly6e protects mice from a SARS-CoV-2 variant of concern
Ancestral strains of SARS-CoV-2 could not infect mice without transgenic overexpression of human ACE2 (ref. 28) or reverse genetic manipulation and passaging in mice29,30. This hindered our ability to examine infection of clinical isolates of SARS-CoV-2 in Ly6e-deficient mice in our previous study21. At the end of 2020 through early 2021, several SARS-CoV-2 variants of concern with the N501Y mutation in the receptor binding domain of the spike protein, which has been associated with increased affinity for murine ACE2 (ref. 31), began to circulate globally. The SARS-CoV-2 Gamma variant (P.1), which was first detected in January 2021, replicates efficiently in the respiratory tracts of young C57BL/6J mice without causing clinically observable disease such as weight loss32. Viral antigen was detected in the respiratory bronchial and alveolar epithelial cells of infected mice, indicating that P.1 replicates in non-haematopoietic cells in the airways. The molecular and cellular pathways that confer murine resistance to SARS-CoV-2 variants such as P.1 remain undefined.
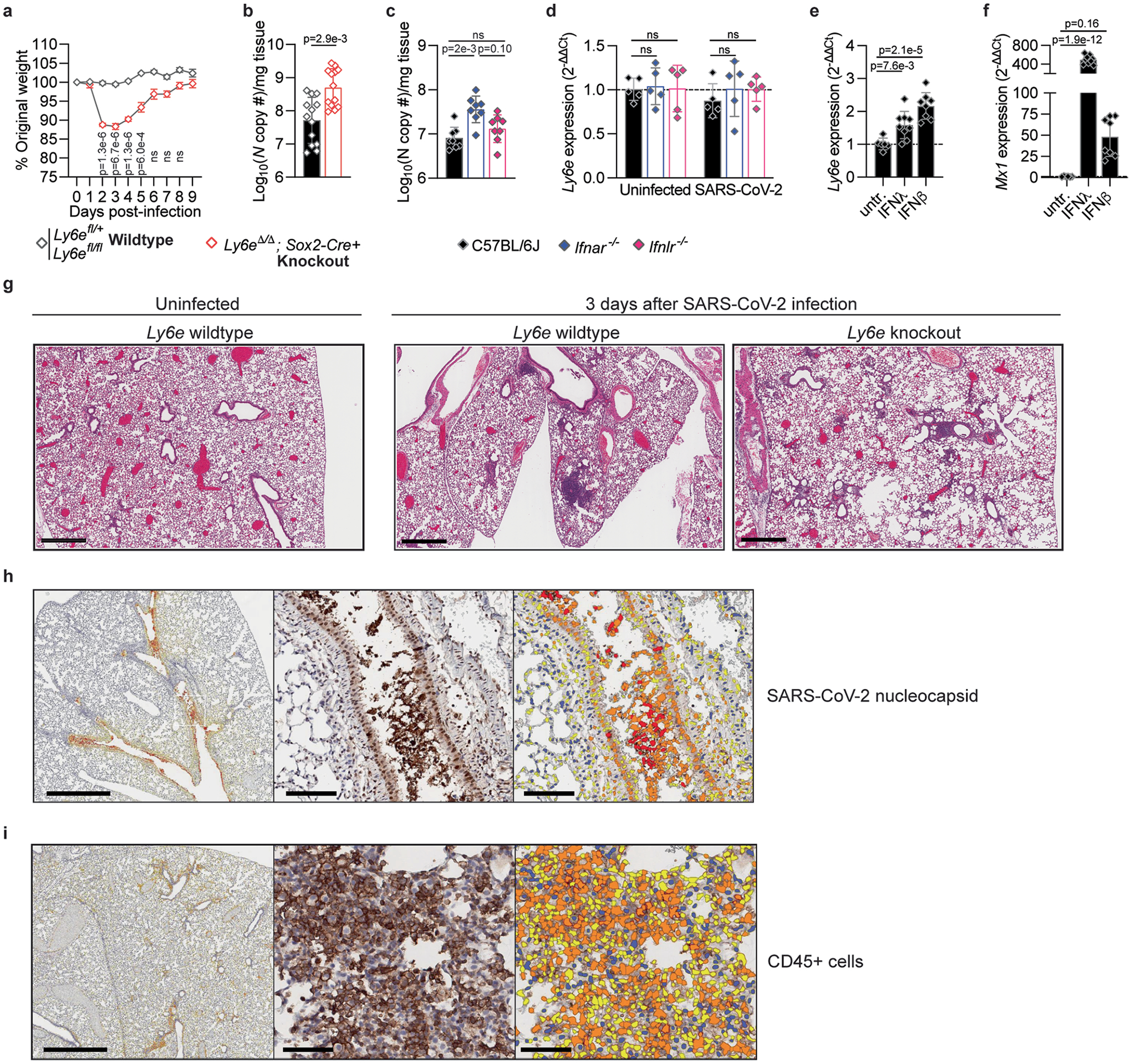
We hypothesized that Ly6e, which is constitutively expressed in most tissues5 and lung cell types (www.lungendothelialcellatlas.com), contributes to murine resistance to SARS-CoV-2 infection. To test this, we infected Ly6e wild-type, heterozygous and knockout mice with P.1 SARS-CoV-2. Ly6e knockout mice on average lost 10% of their original body weight by 2 days post-infection and began to regain weight by day 4. Ly6e wild-type and heterozygous mice exhibited little to no weight loss (Fig. 3a and Extended Data Fig. 3a). Lung viral burden was elevated in Ly6e knockout mice compared with wild-type mice (Fig. 3b and Extended Data Fig. 3b). To compare the contributions of Ly6e and the interferon response to murine resistance to SARS-CoV-2, we infected wild-type mice or mice lacking type I interferon (Ifnar−/−) and type III interferon (Ifnlr−/−) signalling. Strikingly, Ifnar−/− and Ifnlr−/− mice displayed similar resistance as wild-type mice to SARS-CoV-2-induced weight loss (Fig. 3c) despite elevated viral burden (Fig. 3d and Extended Data Fig. 3c). This contrasted with the weight loss observed in Ly6e knockout mice. As Ly6e is an ISG6, we examined its expression before and after SARS-CoV-2 infection or IFNβ or IFNλ treatment. SARS-CoV-2 did not modulate lung Ly6e expression (Extended Data Fig. 3d). IFNλ or IFNβ resulted in a modest 1.5- to 2-fold induction of Ly6e, as compared with a robust 50- to 500-fold induction of Mx1 (Extended Data Fig. 3e,f). Collectively, these data suggest that interferons contribute to limiting SARS-CoV-2 replication in the lungs, while Ly6e helps to control early viral pathogenesis before other components of the antiviral immune response clear infection and accelerate host recovery.
Fig. 3 |. Ly6e knockout mice have higher SARS-CoV-2 burden and infection-associated pathology.
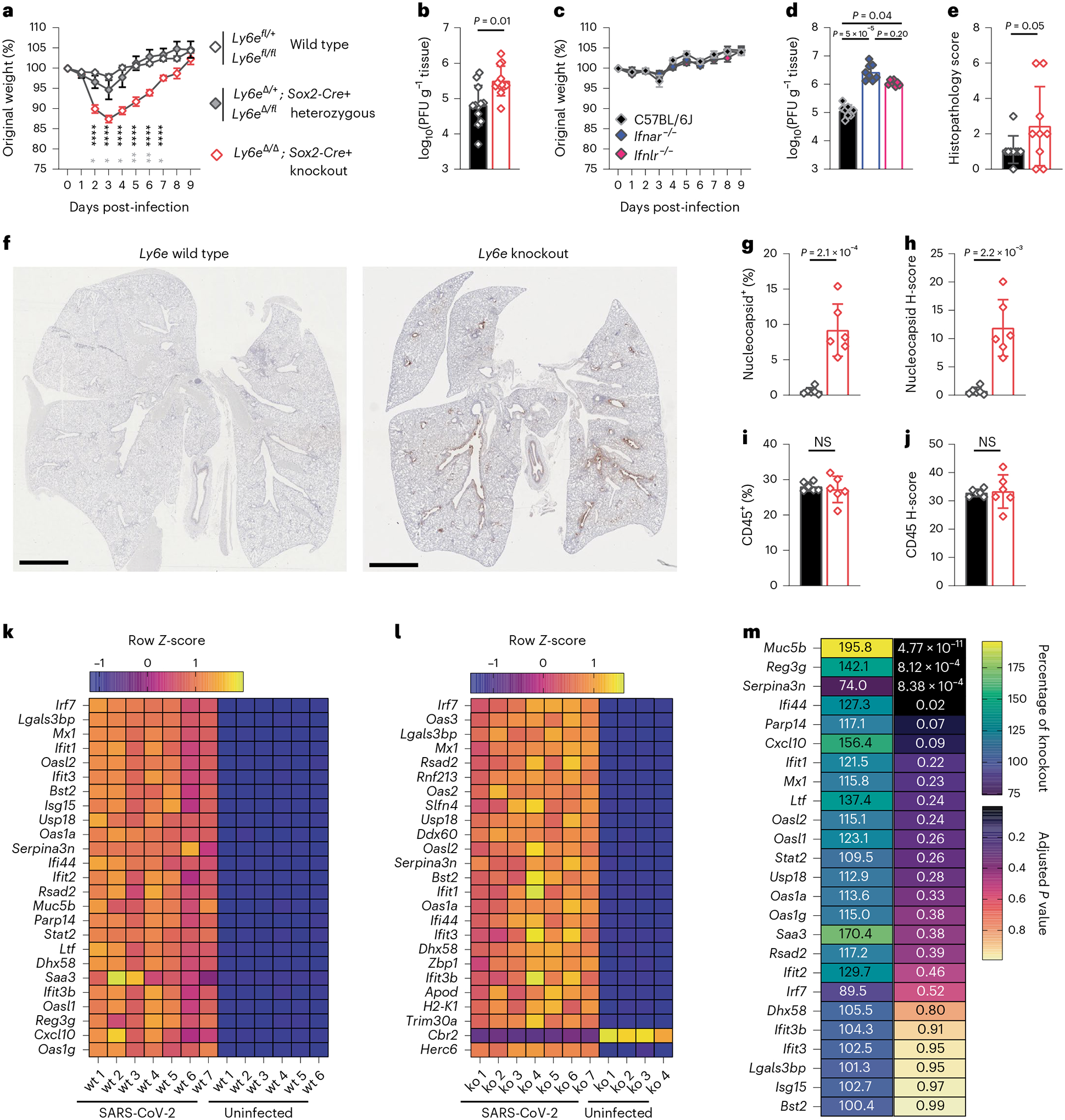
Mice were intranasally infected with 60,000 plaque-forming units (PFU) P.1 SARS-CoV-2 or administered PBS (uninfected). For b and d–m, mice were killed 3 days post-infection. a, Daily weight (n = 7 Ly6e wild type, n = 4 Ly6e heterozygous and n = 7 Ly6e knockout). b, Viral burden in lung (n = 12 Ly6e wild type and n = 12 Ly6e knockout). c, Daily weight (n = 10 C57BL6/J, n = 10 Ifnar−/− and n = 10 Ifnlr−/−). d, Viral burden in lung (n = 8 C57BL6/J, n = 8 Ifnar−/− and n = 8 Ifnlr−/−). e, Histopathological scoring for features of acute pneumonia in lung sections (n = 9 Ly6e wild type and n = 9 Ly6e knockout). f, Representative lung sections stained for SARS-CoV-2 nucleocapsid (DAB) and a haematoxylin counterstain. Quantitative analyses of lung sections from n = 6 Ly6e wild-type and n = 6 Ly6e knockout mice: percentage of SARS-CoV-2 nucleocapsid-positive cells of total lung cells (g), nucleocapsid stain H-score that integrates staining intensity and proportion of total cells (h), percentage of CD45-positive cells of total lung cells (i) and CD45 stain H-score (j). k,l, Z-score heat map of the top 25 most differentially expressed genes between SARS-CoV-2-infected and uninfected Ly6e wild-type mice (k) and SARS-CoV-2-infected and uninfected Ly6e knockout mice (l) based on RNA-seq. m, Differential gene expression of the top 25 genes induced by SARS-CoV-2 infection in Ly6e wild-type mice (as in k) shown relative to average expression levels in Ly6e knockout mice. Statistical significance was determined by two-sided unpaired t-tests (a, g and i), two-sided Mann–Whitney test (b, d, h and j), and Kolmogorov–Smirnov test (e). In a, black text indicates statistical comparison between wild type and knockout (days 2–7, left to right: P = 1 × 10−5, P = 6 × 10−6, P = 4 × 10−6, P = 6.1 × 10−5, P = 1.2 × 10−5 and P = 8.6 × 10−5), and grey symbols (days 2–7, left to right: P = 0.02, P = 0.03, P = 0.02, P = 0.01, P = 2.8 × 10−3 and P = 0.02) indicate comparison between heterozygous and knockout. Error bars represent mean ± standard deviation in all panels except for a and c, where error bars indicate mean ± standard error of the mean. Scale bars: 2 mm (f). NS, not significant.
To gain further insight into how Ly6e controls early viral pathogenesis, we performed histological and immunohistochemical analyses of lungs from SARS-CoV-2-infected mice at 3 days post-infection. SARS-CoV-2 infection caused mild perivascular inflammation in most Ly6e wild-type mice, whereas Ly6e knockout mice exhibited moderate perivascular inflammation, mild pleural and alveolar inflammation, and signs of haemorrhage (Fig. 3e, Extended Data Fig. 3g and Supplementary Table 1). We detected SARS-CoV-2 nucleocapsid in the epithelium of the main airways of all Ly6e knockout mice, particularly in cells sloughed off into the airway (Fig. 3f–h, Extended Data Fig. 3h and Supplementary Table 2). Infiltration of CD45+ immune cells in response to SARS-CoV-2 infection was similar between infected Ly6e wild-type and Ly6e knockout mice (Fig. 3i,j, Extended Data Fig. 3i and Supplementary Table 2). These data suggest that Ly6e expression helps reduce SARS-CoV-2-induced pathology independent of immune cell recruitment.
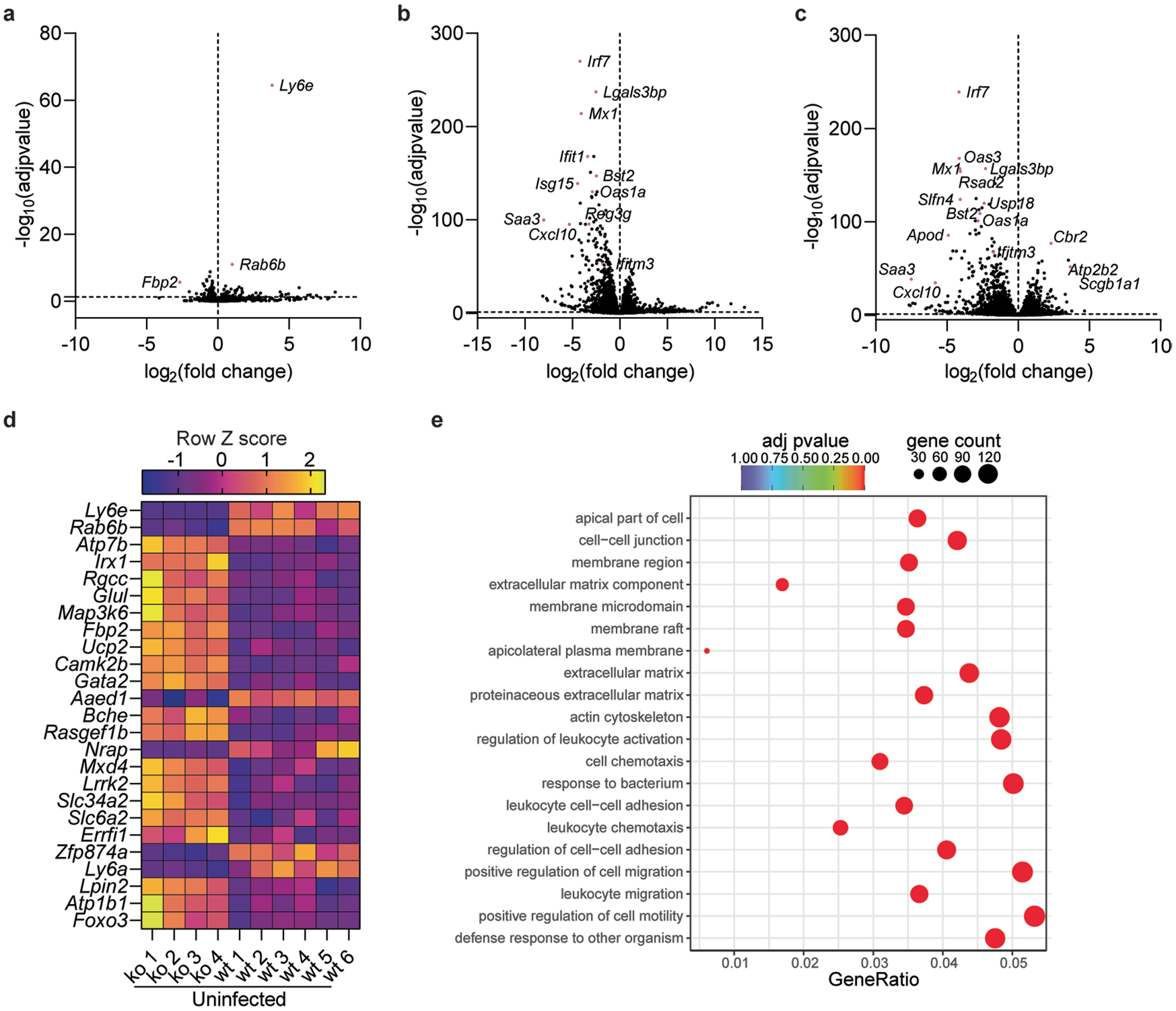
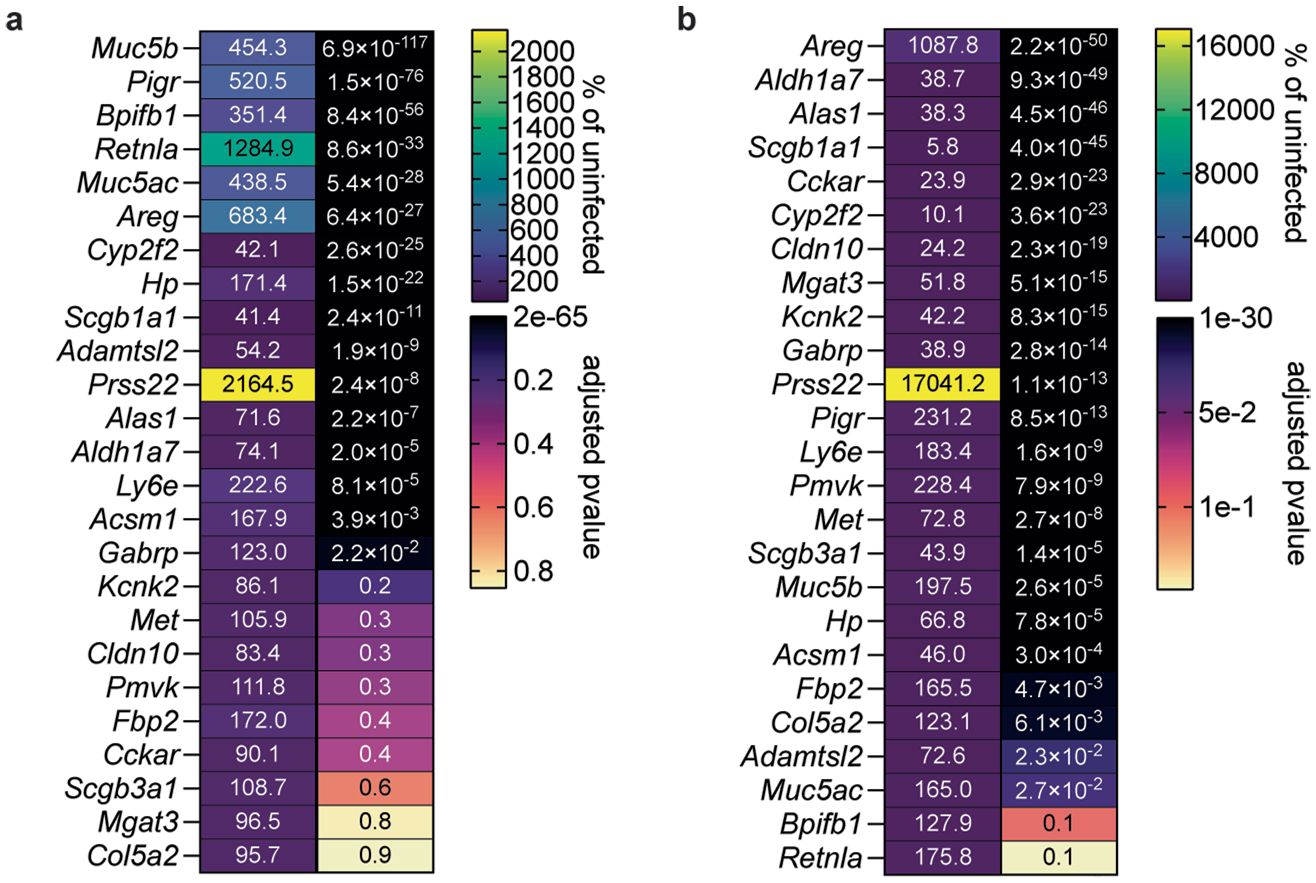
We next investigated the cause of clinical signs of disease in SARS-CoV-2-infected Ly6e knockout mice by transcriptomic analyses of lung tissue. Expression of 30 genes differed more than two-fold between uninfected wild-type and Ly6e knockout mice (Extended Data Fig. 4a,d). SARS-CoV-2 infection induced a similar antiviral ISG signature irrespective of genotype (Fig. 3k,l and Extended Data Fig. 4b,c). Of the top 25 genes induced by SARS-CoV-2 in wild-type mice (Fig. 3k), we found that infected Ly6e knockout mice had reduced upregulation of two mucosa-associated genes: Reg3g and Muc5b (Fig. 3m). Gene Ontology (GO) enrichment analysis of the total transcriptome revealed that gene sets associated with leukocyte motility and adhesion were enriched in wild-type mice compared with Ly6e knockout mice (Extended Data Fig. 4e). Collectively, these analyses indicate that increased disease in Ly6e knockout mice was not due to impaired antiviral gene response but was instead potentially associated with reduced leukocyte mobility and dampened upregulation of several mucosal genes induced by SARS-CoV-2 infection.
Ly6e protects ciliated and club cells from SARS-CoV-2 infection
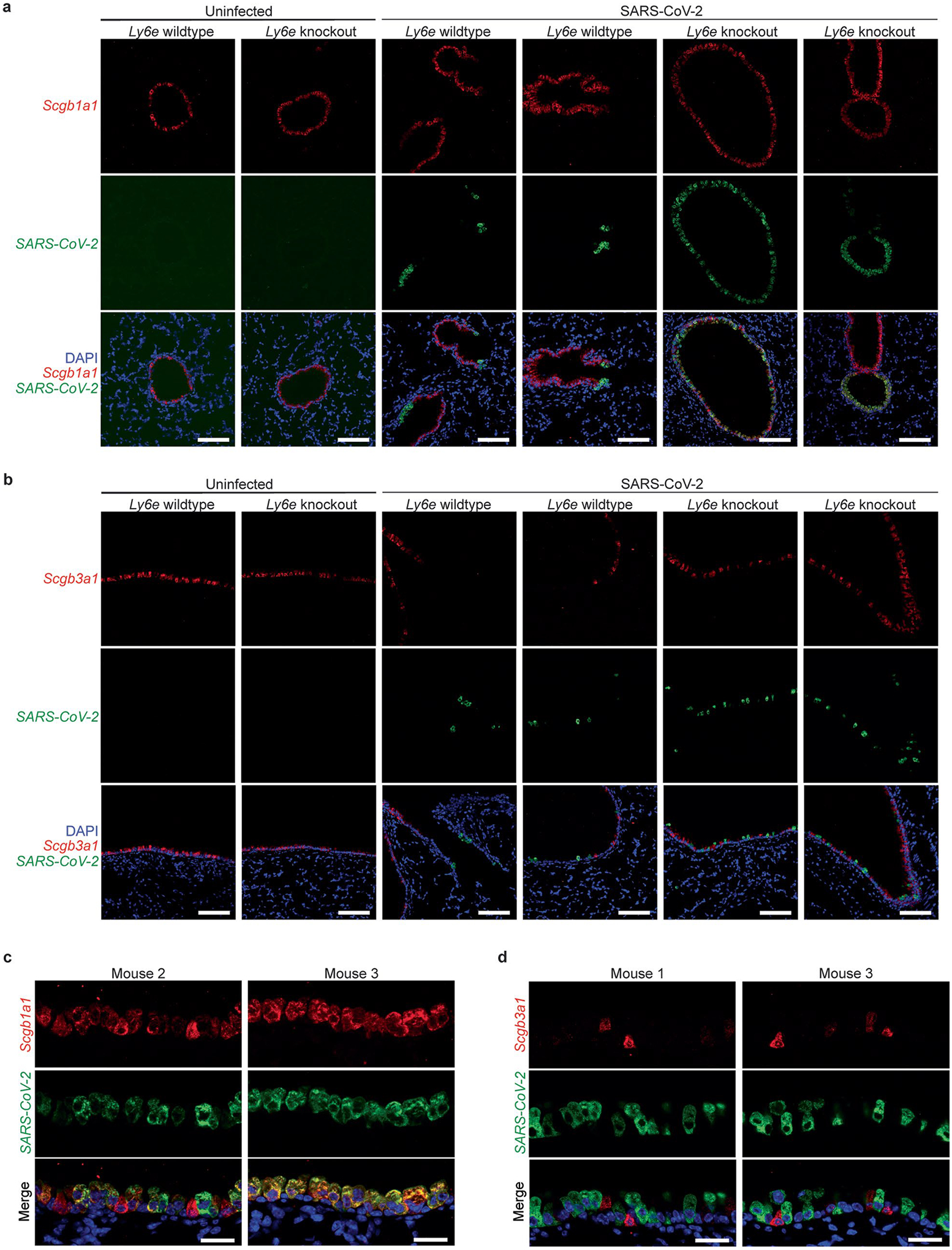
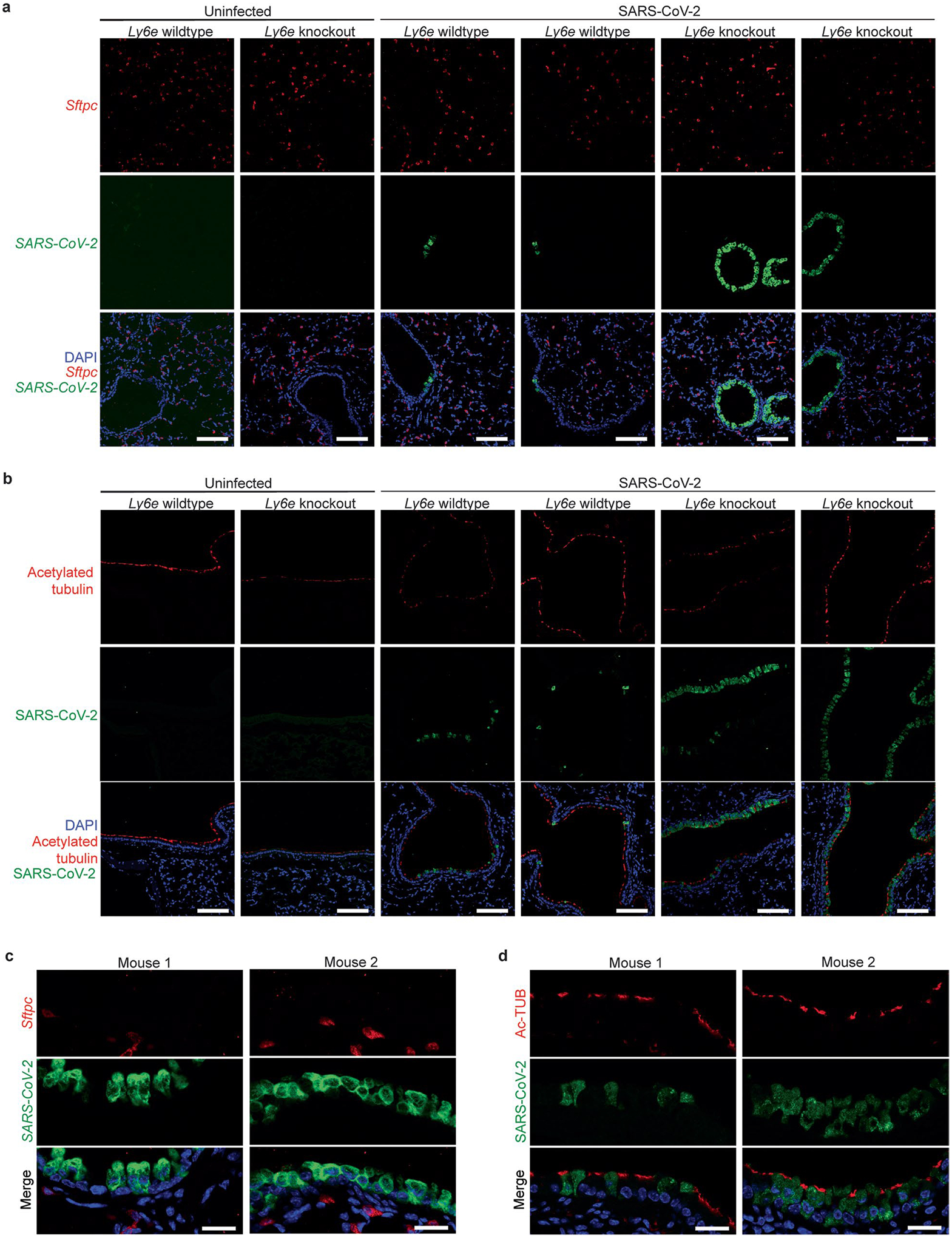
We hypothesized that basal Ly6e expression protects a specific subset of pulmonary epithelial cells from SARS-CoV-2 infection, which minimizes coronavirus-induced disease. By comparing the top 25 differentially expressed genes between SARS-CoV-2-infected wild-type and Ly6e knockout mice, as opposed to only virus-induced genes (Fig. 3m), we found that genes associated with secretory and ciliated cells (www.lungendothelialatlas.com) were more highly expressed in infected wild-type mice (Fig. 4a and Extended Data Fig. 5a,b). Using hybridization chain reaction RNA fluorescence in situ hybridization (HCR-RNA-FISH) confocal microscopy, we found that Scgb1a1-expressing cells in the large airways, classically defined as secretory club and goblet cells but also encompasses ciliated cells, were the primary target of SARS-CoV-2 infection in Ly6e knockout mice (Fig. 4b,c and Extended Data Fig. 6a,c). Secretory goblet cells (Scgb3a1+) and alveolar type II epithelial cells (Sftpc+) were not targeted by SARS-CoV-2 in Ly6e wild-type or knockout animals (Fig. 4d,e and Extended Data Figs. 6b,d and 7a,c). We also detected SARS-CoV-2 infection near ciliated cells, using acetylated tubulin as a marker. However, due to the unique distribution of acetylated tubulin relative to viral nucleocapsid, it was unclear whether these two signals co-localized in the same cells (Fig. 4f and Extended Data Fig. 7b,d). To overcome this limitation, we assessed the relative infectivity of enzymatically dissociated lung cells by flow cytometry. We found that both SCGB1A1-positive cells and acetylated tubulin-positive cells were more susceptible to SARS-CoV-2 infection in the absence of Ly6e (Fig. 4g,h and Extended Data Fig. 8a), further validating the HCR-FISH results. In corroboration of our mouse model, we found that human LY6E contributes to control of SARS-CoV-2 infection in a human bronchial epithelial cell line (Extended Data Fig. 9a,b). Collectively, these data suggest that Ly6e expression protects secretory club cells and ciliated cells from SARS-CoV-2 infection, which in turn limits clinical disease.
Fig. 4 |. Ly6e protects lung secretory club cells and ciliated cells from SARS-CoV-2 infection.
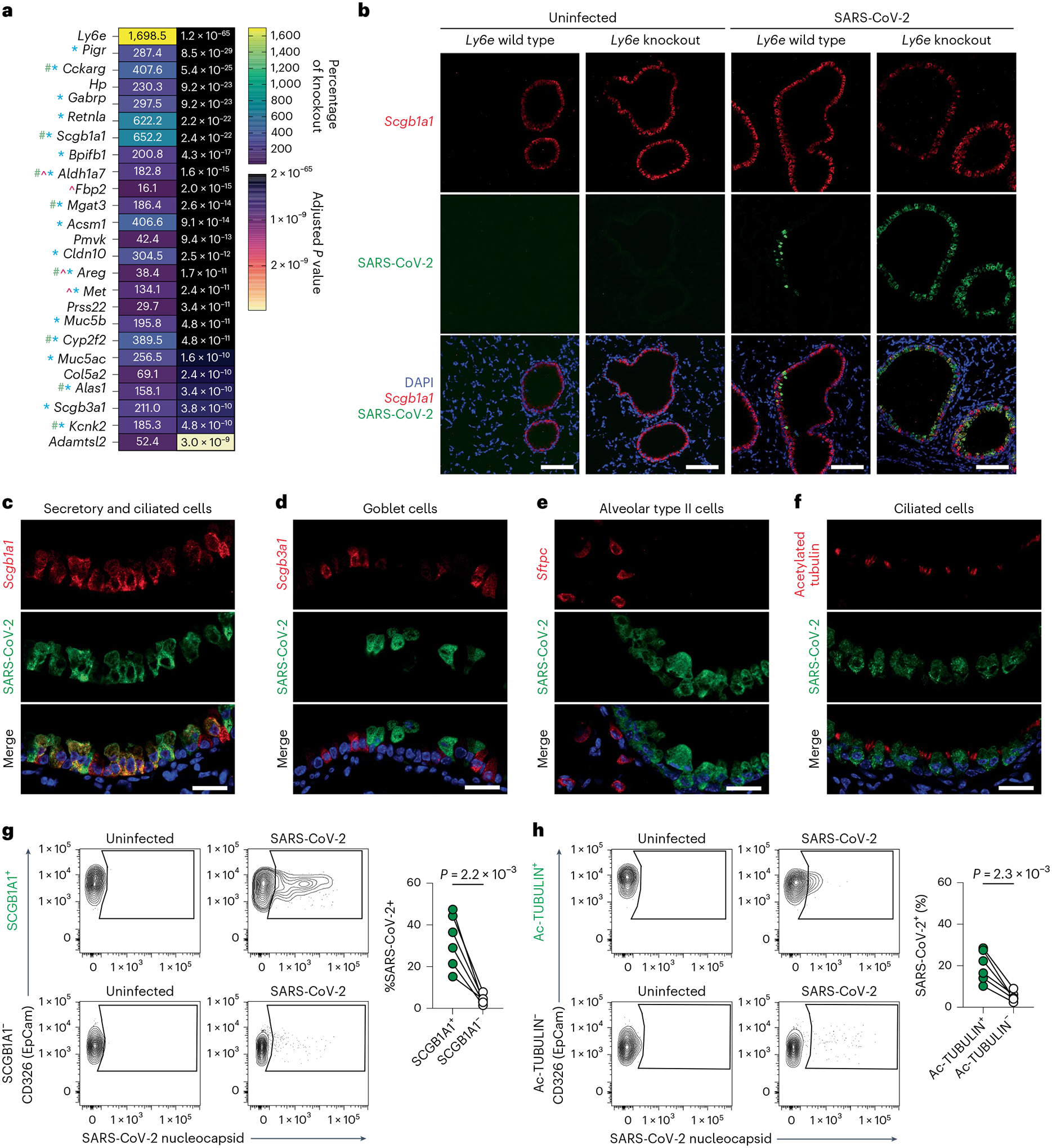
a, Mice were intranasally infected with 60,000 plaque-forming units (PFU) P.1 SARS-CoV-2 and killed 3 days post-infection. The heat map illustrates the top 25 most differentially expressed genes between SARS-CoV-2-infected Ly6e wild-type mice (n = 7) and Ly6e knockout mice (n = 7) based on RNA-seq. Genes enriched in secretory cells (blue asterisk), ciliated cells (green pound sign) or alveolar type II epithelial cells (pink caret) are labelled according to public single-cell RNA-seq data (lungendothelialcellatlas.com). b–h, Mice were intranasally infected with 8,700 PFU P.1 SARS-CoV-2 and killed 1 day post-infection. Lung sections were probed by HCR-RNA-FISH for SARS-CoV-2 RNA (green) and Scgb1a1 mRNA (red) (b and c), Scgb3a1 mRNA (red) (d) and Sftpc mRNA (red) (e), or by IHC for SARS-CoV-2 nucleocapsid (green) and acetylated tubulin (red) (f). Representative images were selected from n = 3 Ly6e knockout mice; remaining images from other two mice are shown in Extended Data Figs. 6 and 7. g,h, Representative contour plots of CD45−EpCam+ cells from lungs of SARS-CoV-2-infected or uninfected Ly6e knockout mice stained for SARS-CoV-2 nucleocapsid and SCGB1A1 (g) or acetylated tubulin (h) and quantification of infection (n = 6 Ly6e knockout mice). Male and female mice were used at an approximately 1-to-1 ratio for these experiments. Statistical significance was determined by two-sided paired t-test (g and h). Scale bars, 100 μM (b) and 25 μM (c–f).
Discussion
How tissue-specific expression of cell-intrinsic antiviral effectors contributes to controlling respiratory coronavirus pathogenesis in mice has not been previously investigated. Our study expands our understanding of Ly6e-mediated control of MHV infection by identifying specific cellular compartments that require Ly6e to establish host resistance. We also discovered a protective role for Ly6e in pulmonary secretory club cells and ciliated cells using a SARS-CoV-2 variant that infects mice without ectopic overexpression of human ACE2, a strategy that augments SARS-CoV-2 tropism in a non-physiological manner. By using different beta-coronaviruses with distinct host receptors, we establish that LY6E is a pan-coronavirus restriction factor in respiratory tract infection.
Using MHV, we found demonstrated organ-specific contributions of Ly6e in distinct cellular compartments. Ly6e in Lyz2-expressing cells limited viral replication in lung and spleen only in the intranasal infection model, suggesting a role for LY6E in constraining respiratory tract replication and dissemination beyond the airways. In both intranasal and intraperitoneal infection models, serum ALT in Ly6eΔLyz2 mice was higher, indicating that Ly6e may protect a Lyz2-expressing cell type that is crucial for limiting MHV-driven hepatic damage. Our bone marrow chimaera studies suggest that LY6E in a radioresistant Vav1-expressing cell type contributes to host defence against MHV. Microglia in the brain and endothelial cells are two radioresistant cell types that may be targeted by Vav1-iCre expression5,27. We speculate that microglia, which are not uniformly targeted by the LysM-Cre transgene33 but highly express Vav1, are more likely to be a relevant cell type in Ly6eΔVav1 and Ly6efl→ΔVav1 mice than brain endothelial cells, which are poor expressors of Vav1 (ref. 5).
We propose that Ly6e-deficient mice might be useful for modelling ‘mild to moderate’ COVID-19 in humans or for testing vaccines and anti-virals. This model exhibits lower respiratory tract disease34 and recapitulates a more common form of human disease than the more severe and lethal models of mouse-adapted SARS-CoV-2 (refs. 29–31,35–37) or SARS-CoV-2 infection in K18-hACE2 mice38–40. Infection of Ly6e knockout mice with a natural SARS-CoV-2 variant of concern may also be a useful tool for understanding how increased susceptibility of club and ciliated cells results in clinical disease. Indeed, SARS-CoV-2 has been previously shown to exhibit tropism for lower respiratory tract secretory club cells41–44 and was associated with decreased expression of Scgb1a1 in a mouse-adapted SARS-CoV-2 model30. On the basis of our observation of a striking reduction in virus-induced pulmonary Muc5b expression in Ly6e-deficient mice relative to SARS-CoV-2-infected wild-type mice, we also propose that Ly6e knockout mice may useful for understanding the potential protective role of MUC5B against COVID-19, as has been implied by the results of multiple meta-analyses45–48.
Using conditional knockout and bone marrow chimaeric mice, we identified the cellular compartments in which expression of Ly6e confers resistance to MHV. However, we cannot conclude whether Ly6e expression by a single cell type is required for host survival. Also we do not provide direct evidence that Ly6e-deficient club or ciliated cells are killed by SARS-CoV-2 infection. Further mechanistic studies are required to understand the precise cellular compartments from which Ly6e restricts coronavirus pathogenesis.
Methods
Viruses
Generation of an infectious clone of mouse hepatitis virus, polytropic strain A59 (MHV) has been described previously49. MHV stocks used for infections were prepared by propagation of viral seed stocks in 17CL1 cells. In brief, a 100% confluent 175 cm2 tissue culture flask of 17Cl1 cells was inoculated with a concentrated stock of MHV at a multiplicity of infection of 0.4 in 10 ml of 10% foetal bovine serum (FBS, Gibco)/Minimum Essential Medium (MEM, Gibco) for 3–5 h at 33 °C. Viral inoculum was replaced with 0.22 μM filter-sterilized pH 6.8 growth medium (1× non-essential amino acids (Gibco)/20 mM HEPES, pH 6.8 (Gibco)/5 mM sodium bicarbonate (Sigma-Aldrich)/1× penicillin–streptomycin (Gibco)/1% tryptose phosphate broth (Gibco)/10% FBS (Gibco)/Dulbecco’s modified Eagle medium (DMEM) #12100–046 (Gibco)), to limit syncytial formation. Cells were incubated overnight at 33 °C, then the entire flask was frozen at −20 °C. After thawing the flask at room temperature, the supernatant was clarified by centrifugation at 1,000g for 5 min before aliquoting and storage at −80 °C. Viral titre was determined by plaque assay on L929 cells. In brief, L929 were plated on 24-well plates at a density of 165,000 cells per well the day before inoculation with 12 dilutions from a 10-fold dilution series. The infection was incubated for 2–3 h at 37 °C and then the viral inoculum was removed and replaced with a pH 7.2 methyl cellulose overlay medium (1% methyl cellulose (Sigma-Aldrich)/5% FBS (Gibco)/1× penicillin–streptomycin (Gibco)/44 mM sodium bicarbonate (Sigma-Aldrich)/DMEM #12100–046 (Gibco)). After overnight incubation at 37 °C, overlay medium was removed, and cells were washed twice with distilled water, and stained for at least 0.5 h with crystal violet (0.5% crystal violet (Sigma-Aldrich)/20% methanol (Sigma-Aldrich)) before removal and syncytium enumeration.
SARS-CoV-2, isolate hCoV-19/Japan/TY7–503/2021 (P.1) of an unknown titre was obtained (BEI Resources) and inoculated onto Vero-E6-C1008 (ATCC) plated at a density of 7 × 106 cells in a 175 cm2 tissue culture flask in 2% FBS (Gibco)/1× non-essential amino acids (Gibco)/MEM (Gibco) for 45 min at 37 °C. Viral inoculum was removed and replaced with 2% FBS (Gibco)/1× non-essential amino acids (Gibco)/MEM (Gibco) and cells were incubated for 3 days at 37 °C before supernatants were clarified by centrifugation at 1,000g for 5 min before aliquoting and storage at −80 °C. Viral titre was determined by plaque assay on Vero-E6-C1008 cells (ATCC). In brief, cells were plated at 650,000 cells per well of a six-well plate and infected with six dilutions from a ten-fold dilution series in 1% FBS (Gibco)/1× non-essential amino acids (Gibco)/MEM (Gibco) for 30 min at 37 °C. Inoculum was then removed and replaced with Avicel overlay medium (5% FBS (Gibco)/1× penicillin–streptomycin (Gibco)/1× GlutaMAX (Gibco)/1× modified Eagle medium, Temin’s Modification #11935–046 (Gibco)/1.2% Avicel RC-591 (DuPont)). Cells were incubated for 3 days at 37 °C before overlay medium was removed and cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) for 30 min at room temperature. Fixative was then removed, and cells were stained for at least 0.5 h with crystal violet (0.2% crystal violet (Sigma-Aldrich)/20% ethanol (Sigma-Aldrich)) before removal and plaque enumeration. To generate virus from DNA-based infectious clone pCC1–4K-SARS-CoV-2-Wuhan-Hu-1-ZsGreen (a gift from S. Wilson)50, 3 μg of plasmid was transfected into two individual six wells of 4 × 105 BHK-21J cells using X-treme Gene9 Transfection Reagent (Sigma-Aldrich). Three days post transfection, medium was aspirated from a T75 flask containing 3 × 106 VeroE6-C1008-TMPRSS2 cells and roughly 4 ml of supernatant from two individual six wells was combined and transferred. Supernatant was incubated for 30 min, followed by an add-back of 6 ml 3% FBS (Gibco)/1× non-essential amino acids (Gibco)/MEM (Gibco). After 4 days, supernatant was collected, cleared by centrifugation and aliquoted. Viral titre was determined by flow cytometry.
Mice
Generation of Ly6efl/fl (#036173, The Jackson Laboratory) and Ly6eΔVav1 mice was described previously21. Ly6efl/fl mice were crossed with LysM-Cre transgenic mice (a kind gift from Lora Hooper, University of Texas Southwestern Medical Center (UTSW)) to generate Ly6eΔLyz2 mice. Ly6efl/fl mice were crossed with CD11c-Cre transgenic mice (a kind gift from Lora Hooper, UTSW) to generate Ly6eΔCD11c mice. Ly6efl/fl mice were crossed with CD4-Cre transgenic mice (#022071, The Jackson Laboratory) to generate Ly6eΔCD4 mice. Ly6efl/fl mice were crossed with CD8α-Cre transgenic mice (#017562, The Jackson Laboratory) to generate Ly6eΔCD8a mice. Ly6efl/fl mice were crossed with CD19-Cre transgenic mice (#006785, The Jackson Laboratory) to generate Ly6eΔCD19 mice. Ly6efl/fl mice (originally generated on CD45.2/.2 background) were crossed with CD45.1/.1 mice (#002014, The Jackson Laboratory) to generate Ly6efl/fl; CD45.1/.1 mice. Whole-body Ly6e knockout mice were generated employing a previously described strategy15. In brief, a male Ly6efl/fl mouse was crossed to a Sox2-Cre female mouse (#008454, The Jackson Laboratory) to generate Ly6eΔ/+; Sox2-Cre+ mice. Ly6eΔ/+; Sox2-Cre+ male mice were crossed to Ly6efl/fl female mice to obtain Ly6eΔ/Δ; Sox2-Cre+ mice at a 1:3 Mendelian ratio. Ly6eΔ/Δ; Sox2-Cre+ male mice were also crossed to Ly6efl/fl female mice to obtain Ly6eΔ/Δ; Sox2-Cre+ mice at a 1:1 Mendelian ratio. Ifnar−/− and Ifnlr−/− mice on a C57BL/6J background were a kind gift from Julie Pfeiffer (UTSW) and Megan Baldridge (Washington University in St. Louis (WUSTL)). C57BL/6J mice (#000664, The Jackson Laboratory) were purchased and delivered at least 1 week before infection to allow for acclimation. Genotyping was outsourced to Transnetyx. All Cre recombinase-expressing strains were maintained by crossing a Cre hemizygous mouse to a Cre non-carrier mouse to ensure the possibility of littermate controls. Every effort was made to design experiments that used age-, sex- and litter-matched controls when available. Male and female mice were used at an equal ratio for all experiments when available between the age of 6 and 15 weeks. Animal studies were carried out in specific-pathogen-free barrier facilities managed and maintained by the UTSW Animal Resource Center. MHV infections were performed in animal biosafety level 2 containment. SARS-CoV-2 infections were performed in animal biosafety level 3 containment. Facilities were maintained at an acceptable range of 68–79 °F at a humidity of 30–70% on a 12 h dark/12 h light cycle. All procedures used in this study complied with federal and institutional guidelines enforced by the UTSW Institutional Animal Care and Use Committee (IACUC) and the UTSW Institutional Biosafety Committee and were granted institutional approval after veterinary and committee review (protocols #2016–101828 and #2020–102987). All infected mice were monitored daily for weight, clinical appearance and mortality. Animals that lost more than 20% of their original body weight were killed per IACUC guidelines.
Bone marrow chimaeras
Five- to 10-week-old Ly6efl/fl; CD45.2/.2 and Ly6eΔVav1; CD45.2/.2 littermates were selected as bone marrow recipients. Six- to 12-week-old Ly6efl/fl; CD45.1/.1 mice were used as sex-matched donors. Recipient CD45.2/.2 mice were started on 0.4 mg ml−1 enrofloxacin drinking water 1 week before irradiation and continued treatment 3 weeks after irradiation. Recipient animals were subject to lethal total body irradiation with 900–950 cGy in a SARRP Small Animal Irradiator (Xstrahl) in the UTSW Preclinical Radiation Core Facility and rested for 6–7 h. During that time, femurs and tibias were collected from killed donor mice and flushed with 1× phosphate-buffered saline (PBS, Gibco) to isolate bone marrow. Red blood cells (RBCs) were lysed by treatment with 1× RBC lysis buffer (Tonbo Biosciences). Cell density was determined with Invitrogen Countess 3 Automated Cell Counter (Thermo Fisher Scientific). Six hours after irradiation, recipient animals were lightly anaesthetized with isoflurane and retro-orbitally injected with 2–5 million live donor bone marrow cells resuspended in 50 μl of 1× PBS (Gibco). Donor animals were rested for a minimum of 6 weeks before MHV infection or up to 11 weeks for assessing Ly6e expression and degree of chimaerism.
In vivo infections, interferon treatment, viral titring and serum ALT
For intranasal MHV and SARS-CoV-2 infections, 6–15-week-old non-chimaeric mice were weighed, then intraperitoneally injected with an anaesthetic cocktail of 80 mg kg−1 ketamine, 6 mg kg−1 xylazine, and 1× PBS. Anaesthetized mice were inoculated intranasally with 30 μl of virus diluted in chilled 1× PBS (MHV) or administered undiluted in 2% FBS (Gibco)/1× non-essential amino acids (Gibco)/MEM (Gibco) (SARS-CoV-2) via the left nostril and then placed on heat until ambulatory. For intraperitoneal MHV infections, mice were weighed then intraperitoneally injected with 100 μl of virus diluted in 1× PBS. Infected mice were monitored daily for weight and mortality. For viral burden studies, intranasally infected mice were killed 1 day earlier (4 days post-infection) than intraperitoneally infected mice (5 days post-infection) due to earlier onset of lethal disease. Bone marrow chimaeric mice were infected with a five-fold lower viral inoculum due to increased sensitivity to infection. Viral burden in bone marrow chimaeric mice was assessed at 6 days post-infection due to delayed disease onset with the lower viral inoculum. Animals that lost more than 20% of their original body weight were killed per IACUC guidelines.
For quantification of gene expression after interferon treatment, C57BL/6J mice were anaesthetized with ketamine/xylazine cocktail as above and intranasally treated with 3 × 104 U of human interleukin-29/interferon lambda 1 or retro-orbitally injected with 1 × 106 units of mouse interferon β (PBL Assay Science) diluted in 0.1% bovine serum albumin (BSA)/PBS. Mice were killed 6 h post-treatment and lungs were collected directly into TRIzol and frozen for RNA isolation.
For quantifying MHV viral load, whole lungs, an approximately 2 cm3 section of liver, whole spleen, and heart were homogenized in 600 μl 2% FBS (Gibco)/MEM (Gibco) and the right hemisphere of the brain, including cerebellum and olfactory bulb, was homogenized in 800 μl 2% FBS (Gibco)/MEM (Gibco) with a single 3.5 mm stainless steel UFO bead (NextAdvance) in a Bullet Blender Lite (NextAdvance). All tissues were weighed before homogenization. Tissue homogenates were clarified of debris by centrifugation at 4 °C at 800g for 5 min, and aliquots for plaque assay were frozen at −80 °C. Blood was collected by terminal cardiac puncture immediately after CO2 asphyxiation and transferred to serum gel blood tubes (Thermo Fisher Scientific). After a minimum of 20 min at room temperature, coagulated blood was removed by centrifugation at 4,000g for 5 min, and serum aliquots for plaque assay were frozen at −80 °C. Plaque assay for MHV samples was performed as described in the above section titled ‘Viruses’ with four dilutions from a ten-fold dilution series. ALT was measured in fresh, unfrozen serum using VITROS MicroSlide Technology by the UTSW Metabolic Phenotyping Core.
For quantifying SARS-CoV-2 viral load, whole lungs from infected mice were weighed then homogenized in 600 μl 2% FBS (Gibco)/MEM (Gibco) with 3.2 mm stainless steel beads (NextAdvance) in a Bullet Blender Storm Pro (NextAdvance). Lung homogenates were clarified of debris by centrifugation at 800g for 5 min, then aliquots were frozen at −80 °C neat for plaque assay or diluted at a 1:4 ratio in TRIzol (Sigma-Aldrich) for RNA isolation and quantitative polymerase chain reaction (PCR). Plaque assay for SARS-CoV-2 samples was performed as described in the above section titled ‘Viruses’. RNA was isolated using the Direct-zol RNA miniprep kit following the manufacturer’s instructions (Zymo Research). A 20 μl reaction contained 5 μl RNA (1/10th of elution), 5 μl TaqMan Fast Virus 1-Step Master Mix and 1.8 μl SARS-CoV-2 or Rpl32 primer/probe set containing 6.7 μM each primer/1.7 μM probe (final concentration of primer/probe were 600 nM/150 nM probe). SARS-CoV-2 primers and probe were designed as recommended by the Center for Disease Control and are as follows: forward (SARS-CoV-2_N1-F), 5′ GACCCCAAAATCAGCGAAAT 3′/reverse (SARS-CoV-2_N1-R), 5′ TCTGGTTACTGCCAGTTGAATCTG 3′/probe (SARS-CoV-2_N1-P), 5′ FAM-ACCCCGCATTACGTTTGGTGGACC-BHQ1 3′. Rpl32 primers were previously described51 and are as follows: forward, 5′ CACCAGTCAGACCGATATGTGAAAA 3′/reverse, 5′ TGTTG TCAATGCCTCTGGGTTT 3′/probe, 5′ CCGCCAGTTTCGCTTAA 3′. All oligonucleotides for SARS-CoV-2 and positive control (Rpl32) gene amplification were synthesized by LGC Biosearch Technologies. In vitro transcribed RNA was used to generate a standard curve for detection of SARS-CoV-2 from a ten-fold dilution series starting at 5 × 1010 copies of RNA. SARS-Cov-2 nucleocapsid (N) gene was amplified by PCR from a synthesized N gene fragment (Integrated DNA Technologies) with primers (Integrated DNA Technologies) that introduced a T7 promoter sequence on the 3′ end: forward, 5′ TAATACGACTCAC TATAGGGATGTCTGATAATGGACCCCAAAATCAGC 3′/reverse, 5′ CT AATTGCGGCCGCTTAGGCCTGAGTTGAGTCAGCAC 3′. PCR product was purified using QIAquick PCR Purification Kit (Qiagen). In vitro transcription was performed using T7 RiboMAX Express Large Scale RNA Production System following the manufacturer’s protocol (Promega). RNA was quantitated by nanodrop on DS-11 FX Spectrophotometer (DeNovix). Reverse transcription was performed at 50 °C for 5 min, followed by inactivation at 95 °C for 2 min and 40 cycles of PCR (95 °C for 3 s, 60 °C for 30 s) on a QuantStudio 3 (Applied Biosystems).
Gene expression in purified cells and whole tissue
To confirm loss of Ly6e expression in Ly6eΔCD4, Ly6eΔCD8a and Ly6eΔCD19 conditional knockout mice relative to Ly6efl/fl littermates, spleens from uninfected mice were pulverized on a 70 μM cell strainer with a plunger from a sterile 3 ml syringe. Cells were rinsed from the strainer with 1× penicillin–streptomycin (Gibco)/Roswell Park Memorial Institute medium (Gibco) and pelleted at 450g for 4–5 min at 4 °C. RBCs were lysed by treatment with 1× RBC lysis buffer (Tonbo Biosciences) and removed by centrifugation. Cell density was determined with Invitrogen Countess 3 Automated Cell Counter (Thermo Fisher Scientific). For sorting experiments, 20 × 106 splenocytes were processed and stained in 15 ml conical tubes as follows: 30 min incubation with 1:1,000 Ghost Dye Violet 450 per the manufacturer’s protocol (Tonbo Biosciences), 10 min incubation with 1:100 anti-CD16/CD32 (70–0161-U500, Tonbo Biosciences) and 20 min incubation with primary antibody cocktail (Ly6eΔCD4: 1:800 anti-CD4-PECy5 (GK1.5, 50-201-474, Tonbo Biosciences)/1:400 anti-CD3ε-FITC (145–2C11, 35–0031, Tonbo Biosciences); Ly6eΔCD8a: 1:200 anti-CD8α-PECy7 (53–6.7, 60–0081-U025, Tonbo Biosciences)/1:400 anti-CD3ε-FITC (145–2C11, 35–0031, Tonbo Biosciences); Ly6eΔCD19: 1:800 anti-CD19-PECy5 (1D3, 60–0193-U025, Tonbo Biosciences)/1:400 anti-CD3ε-FITC (145–2C11, 35–0031, Tonbo Biosciences)). Splenocytes were washed then sorted for viability and cell surface markers by the UTSW Immunology Flow Cytometry Core. After sorting, cells were pelleted and lysed for RNA isolation with RNAaqueous-Micro Total RNA Isolation Kit (Thermo Fisher Scientific) per the manufacturer’s protocol.
To confirm loss of Ly6e expression in Ly6eΔLyz2 and Ly6eΔCD11c conditional knockout mice relative to Ly6efl/fl littermates, bronchoalveolar lavage fluid was collected in 5 mM ethylenediaminetetraacetic acid/1× PBS (Gibco) as previously described via the trachea52 and plated on a poly-lysine-coated 12-well plate in PBS++ (Gibco) in alveolar macrophage medium (1× penicillin–streptomycin (Gibco) and 10% FBS (Gibco)/0.1% β-mercaptoethanol (Sigma-Aldrich)). After a 2 h incubation at 37 °C, wells were rinsed twice with warm PBS++ (Gibco) to remove non-adherent cells. Cells were lysed 4 h later for RNA isolation with RNAaqueous-Micro Total RNA Isolation Kit (Thermo Fisher Scientific) per the manufacturer’s protocol. Alveolar macrophage purity of >95% (based on positive staining for F4/80 and CD11c) was previously determined by flow cytometry during the optimization of this protocol.
To measure Ly6e expression in CD45+ and CD31+ lung cells in Ly6eΔVav1 and bone marrow chimaeric mice relative to Ly6efl/fl mice, whole lungs were excised, rinsed once in 1× PBS, and mechanically homogenized following the manufacturer’s instructions using a gentleMACS Octo Dissociator (Miltenyi Biotec) with the Lung Dissociation Kit, mouse (Miltenyi Biotec). Lung homogenates were filtered through a 70 μM cell strainer, centrifuged at 450g for 5 min, then subject to treatment with 1× RBC lysis buffer (Tonbo Biosciences). Cell density was determined with Invitrogen Countess 3 Automated Cell Counter (Thermo Fisher Scientific). Up to 20 × 106 lung cells were stained in 15 ml conical tubes as follows: 30 min incubation with 1:1,000 Ghost Dye Violet 450 per the manufacturer’s protocol (Tonbo Biosciences), 10 min incubation with 1:100 anti-CD16/CD32 (70–0161-U500, Tonbo Biosciences) and 20 min incubation with primary antibody cocktail (CD45+ immune cells: 1:50 anti-CD45-VioGreen (30F11, 130-123-900, Miltenyi Biotec); CD31+ endothelial cells: 1:2,000 anti-CD31-PE (390, 102407, BioLegend)/1:50 anti-CD45-VioGreen (30F11, 130-123-900, Miltenyi Biotec)/1:100 anti-EpCam-PECy7 (G8.8, 118215, BioLegend)). Lung cells were washed then sorted for viability and cell surface markers by the UTSW Immunology Flow Cytometry Core. After sorting, cells were pelleted and lysed for RNA isolation with RNAaqueous-Micro Total RNA Isolation Kit (Thermo Fisher Scientific) per the manufacturer’s protocol.
For Ly6e expression in spleen, brain, heart, lung and liver, tissues were homogenized in 600 μl or 800 μl (brain only) TRIzol (Sigma-Aldrich) with a single 3.5 mm stainless steel UFO bead (NextAdvance) in a Bullet Blender Lite (NextAdvance). Unclarified tissue homogenates were diluted fresh in TRIzol (Sigma-Aldrich) at a 1:9 ratio, and frozen at −80 °C. For RNA isolation, chloroform was added to thawed samples at a 1:5 ratio, shaken for 15 s, then centrifuged for 15 min at 12,000g at 4 °C. The aqueous phase was transferred to a fresh tube, mixed with 1 equal volume of 70% ethanol, then transferred to a RNeasy Mini Kit filter column (Qiagen). Samples were processed per the manufacturer’s instructions with on-column DNase I treatment (Qiagen).
RNA was quantitated by NanoDrop on DS-11 FX Spectrophotometer (DeNovix). Ly6e expression (forward, 5′ atcttcggggcctcttcac 3′/reverse, 5′ atgagaagcacatcagggaat 3′) or Mx1 expression (forward 5′ GACCATAGGGGTCTTGACCAA 3′/reverse, 5′ AGACTTGCTCTTTCTG AAAAGCC 3′) was quantified relative to the housekeeping gene Rpl32 (forward, 5′ aagcgaaactggcggaaac 3′/reverse, 5′ taaccgatgtt gggcatcag 3′) using one-step quantitative reverse transcription PCR with either QuantiFast SYBR Green RT-PCR Kit (Qiagen) or QuantiNova SYBR Green RT-PCR Kit (Qiagen) per the manufacturer’s protocol on a QuantStudio 3 (Applied Biosystems).
Flow cytometric quantification of CD45.1+ and CD45.2+ cells in spleen, lung and blood
Spleens from uninfected mice were pulverized on a 70 μM cell strainer with a plunger from a sterile 3 ml syringe. Cells were rinsed from the strainer with 1× penicillin–streptomycin (Gibco)/Roswell Park Memorial Institute medium (Gibco), pelleted at 450g for 4–5 min at 4 °C, then subjected to treatment with 1× RBC lysis buffer (Tonbo Scientific). Whole lungs were excised, rinsed once in 1× PBS and mechanically homogenized following the manufacturer’s instructions using a gentleMACS Octo Dissociator (Miltenyi Biotec) with the Lung Dissociation Kit, mouse (Miltenyi Biotec). Lung homogenates were filtered through a 70 μM cell strainer, centrifuged at 450g for 5 min, then subject to treatment with 1× RBC lysis buffer (Tonbo Biosciences). Blood was collected by cardiac puncture using syringes coated internally with 50 μl of 0.5 M ethylenediaminetetraacetic acid (Gibco), then subjected to three sequential treatments with 1× RBC lysis buffer (Tonbo Scientific) to isolate mononuclear cells. Cell density was determined with Invitrogen Countess 3 Automated Cell Counter (Thermo Fisher Scientific). About 500,000 cells were stained as follows: 30 min incubation with 1:1,000 Ghost Dye Violet 510 per the manufacturer’s protocol (Tonbo Biosciences), 10 min incubation with 1:100 anti-CD16/CD32 (70-0161-U500, Tonbo Biosciences) and 20 min incubation with primary antibody cocktail (1:50 anti-CD45.1-PE, REAfinity (REA1179, 130-121-214, Miltenyi Biotec)/1:50 anti-CD45.2-VioBlue, REAfinity (REA1223, 130-124-082, Miltenyi Biotec)). Samples were analysed using a S1000 Flow Cytometer with an A600 96-well plate high-throughput extension (Stratedigm) and compensated using CellCapture v5 (Stratedigm). Data were analysed with FlowJo v9 (Tree Star).
Histopathology
SARS-CoV-2-infected mice were killed by CO2 asphyxiation and lungs were inflated to full tidal volume with 10% neutral buffered formalin (Sigma-Aldrich) via the trachea, which was then clamped shut. Pleural cavity organs were removed and placed in 20 volumes 10% neutral buffered formalin and agitated on an orbital shaker for a minimum of 72 h. Heart, thymus, trachea and lymph nodes were removed, and formalin-fixed lungs were submitted in 70% ethanol to the UTSW Histo Pathology Core for paraffin embedding, sectioning and staining with haematoxylin and eosin. Scoring of lungs from SARS-CoV-2-infected mice for acute pneumonia were graded on a 4-point system (0, not seen; 1, mild; 2, moderate; 3, severe) for the following criteria: perivascular inflammation, alveolar inflammation, pleural inflammation, haemorrhage and oedema, with a maximum possible score of 15. Pathological scoring was performed by an independent UTSW pathologist who was blinded to the experimental conditions (Supplementary Table 1). The scoring criteria were adapted from a published rubric53. Immunostaining was performed at HistoWiz as follows. Immunohistochemistry (IHC) was performed on a Bond Rx autostainer (Leica Biosystems) with citric-acid-based retrieval buffer with a pH of 6.0 and heat-induced epitope retrieval for 20 min. Lung sections were stained with primary antibodies to detect either SARS-CoV-2 nucleocapsid (GTX635686, GeneTex) or CD45-positive cells (ab25836, Abcam) then developed with 3,3′-diaminobenzidine (DAB), which was followed by a haematoxylin counterstain. The slides were scanned and digitized by Leica AT2 scanner (Leica Biosystems). The digitized whole slide images were analysed by Halo software (V3.3, Indica Labs) at Histowiz. The Multiplex IHC module was used to calculate the number of positive cells per mm2 of tissue and the H score (Supplementary Table 2). The histopathological features of target findings were analysed and quantified. The H score was generated following the convention below: weak positive pixels (1+) are highlighted in yellow, moderate positive pixels (2+) are highlighted in orange and strong positive pixels (3+) are in red. The positivity threshold of weak, moderate and strong staining was 0.42, 0.78 and 1.3 respectively in Halo. The minimum tissue optical density (OD) is 0.037. The same segmentation threshold of positive staining in nucleus and cytoplasm of target proteins decided by visual inspection were utilized throughout the whole image sets to maintain consistency in evaluation. An example of the image analysis is shown in Extended Data Fig. 3h,i.
RNA sequencing
Whole lungs were isolated from SARS-CoV-2-infected mice and homogenized in 600 μl TRIzol (Sigma-Aldrich) with 3.2 mm stainless steel beads (NextAdvance) in a Bullet Blender Storm Pro (NextAdvance). Lung homogenates were clarified of debris by centrifugation at 800g for 5 min, diluted into fresh TRIzol (Sigma-Aldrich) at a 1:9 ratio, and frozen at −80 °C. For RNA isolation, chloroform was added to thawed samples at a 1:5 ratio, shaken for 15 s, then centrifuged for 15 min at 12,000g at 4 °C. The aqueous phase was transferred to a fresh tube, mixed with 1 equal volume of 70% ethanol, then transferred to a RNeasy Mini Kit filter column (Qiagen). Samples were processed per the manufacturer’s instructions with on-column DNase I treatment (Qiagen). SARS-CoV-2 N gene levels were also quantified in these samples by quantitative PCR as described in the above section titled ‘In vivo infections, interferon treatment, viral titring and serum ALT’. Purified RNA was submitted to Novogene in DNA/RNA shield (Zymo Research) for library preparation and RNA sequencing (RNA-seq) analysis. Messenger RNA was purified from total RNA using poly-T oligo-attached magnetic beads. After fragmentation, the first-strand complementary DNA was synthesized using random hexamer primers, followed by the second-strand cDNA synthesis using dTTP for generation of a non-directional library. The library was checked with Qubit and real-time PCR for quantification and bioanalyzer for size distribution detection. The clustering of the index-coded samples was performed according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina platform and paired-end reads were generated. Raw reads of fastq format were first processed through in-house perl scripts. In this step, clean reads were obtained by removing reads containing adapter and low-quality reads from raw data. Index of the reference genome was built and paired-end clean reads were aligned to the reference genome using Hisat2 v2.0.5. featureCounts v1.5.0-p3 was used to count the reads numbers mapped to each gene and fragments per kilobase of transcript per million (FPKM) mapped reads of each gene was calculated on the basis of the length of the gene and reads count mapped to this gene. Differential expression analysis was performed using the DESeq2 R package (1.20.0). The resulting P values were adjusted using the Benjamini and Hochberg’s approach for controlling the false discovery rate. GO enrichment analysis of differentially expressed genes was implemented by the clusterProfiler R package, in which gene length bias was corrected. GO terms with corrected P value less than 0.05 were considered significantly enriched by differentially expressed genes. Volcano plots and heat maps were prepared using GraphPad Prism version 9.1.0.
HCR-RNA-FISH
HCR-RNA-FISH was performed following the Molecular Instruments HCR v3.0 protocol for FFPE human tissue sections54,55. Briefly, tissue sections were deparaffinized with xylene and rehydrated with decreasing concentrations of ethanol (100%, 95% and 80%) followed by molecular grade water. Antigen unmasking was performed with slides submerged in 10 mM citrate buffer, pH 6.0 (ab64236, Abcam) and heated in a steamer for 20 min at ~90 °C. Slides were cooled to room temperature in the citrate buffer for approximately 30 min. Sections were outlined with a hydrophobic barrier pap pen (H4000, Vector Labs) followed by treatment with 10 μg ml−1 proteinase K (25-530-049, Invitrogen) for 10 min at 37 °C in a humidified chamber. Sections were washed with RNase-free water to remove proteinase K. Samples were incubated for 10 min at 37 °C in hybridization buffer (Molecular Instruments). Sections were incubated overnight in a humidified chamber at 37 °C with either 3 pmol (SARS-CoV2) or 6 pmol (cell markers) of initiator probes in hybridization buffer. We designed probes for Scgb1a1 (Supplementary Table 4) and Sftpc (Supplementary Table 5) in house. Custom probes for SARS-CoV2 (Lot RTB659) and Scgb3a1 (Lot RTB657) were designed by Molecular Instruments. The next day, slides were washed in probe wash buffer and 5× saline-sodium citrate with 0.1% Tween (SSCT) (BP1325–1, Fisher Scientific) four times for 15 min, according to the manufacturer’s instructions. Following washing, samples were incubated in a humidified chamber for 30 min in amplification buffer (Molecular Instruments). Fluorescent hairpins were heated to 95 °C for 90 s in a thermocycler and snap cooled in the dark at room temperature for 30 min. Hairpins and amplification buffer were added to the sample and incubated overnight at room temperature in a humidified chamber in the dark. Hairpins were washed off with 5× SSCT for 5 min, 15 min, 15 min and 5 min followed by a wash with PBS containing 1 μg ml−1 4′,6-diamidino-2-phenylindole (DAPI) (EN62248, Fisher Scientific). Samples were then treated with True View autofluorescence quenching kit (SP-8400–15, Vector Labs), according to the manufacturer’s instructions, followed by mounting in VECTASHIELD with DAPI (Vector Labs). Slides were imaged on a Zeiss 780 inverted confocal microscope in the UTSW Quantitative Light Microscopy Core Facility.
Immunofluorescence
Tissue sections were deparaffinized with xylene and rehydrated with decreasing concentrations of ethanol (100%, 95% and 80%) followed by distilled water. Antigen unmasking was performed with slides submerged in 10 mM citrate buffer (pH 6.0) (ab64236, Abcam) and heated in a steamer for 20 min at ~90 °C. Slides were cooled to room temperature in the citrate buffer for approximately 30 min. Sections were outlined with a hydrophobic barrier pap pen (H4000, Vector Labs) followed by two washes in PBS. Sections were stained with primary antibodies, anti-acetyl-α tubulin Alexa Fluor 488, clone 6–11B-1 (sc-23950 AF488, Santa Cruz Biotechnology) and anti-SARS-CoV2 nucleocapsid Alexa Fluor 647 (ab302552, Abcam), in 0.5% BSA in PBS overnight. The following day, sections were washed four times for 10 min with PBS followed by a wash with PBS containing 1 μg ml−1 DAPI (EN62248, Fisher Scientific). Samples were then treated with True View autofluorescence quenching kit (SP-8400–15), according to the manufacturer’s instructions, followed by mounting in VECTASHIELD with DAPI (Vector Labs). Slides were imaged on a Zeiss 780 inverted confocal microscope in the UTSW Quantitative Light Microscopy Core Facility.
Flow cytometric quantification of SARS-CoV-2 infection in epithelial cells
Ly6eΔ/Δ; Sox2-Cre+ mice were intraperitoneally injected with an anaesthetic cocktail of 100 mg kg−1 ketamine, 10 mg kg−1 xylazine and 1× PBS followed by a second intraperitoneal injection with 0.01 U g−1 heparin sodium (Sigma-Aldrich) diluted in 1× PBS. Mice were perfused with 25 U ml−1 heparin sodium diluted in 1× PBS and whole lungs were excised, rinsed once in 1× PBS and mechanically homogenized using a gentleMACS Octo Dissociator (Miltenyi Biotec). For dissociation, the following enzyme mix was used: 30 U neutral protease (LS02104, Worthington), 2,500 U Collagenase I (LS004196, Worthington) and 500 U DNase I (4716728001, Sigma-Aldrich) in a total volume of 5 ml 1× PBS with calcium and magnesium. A published dissociation protocol was used: temperature, 37 °C; loop 6× (spin 300 rpm, 10′′; spin −300 rpm, 10′′); loop 2× (spin 150 rpm, 5′′; spin −150 rpm, 5′′); loop 2× (spin 20 rpm, 5′ 0′′; spin −20 rpm, 5′ 0′′); loop 6× (ramp 360 rpm, 15′′; ramp −360 rpm, 15′′) (ref. 37). Lung homogenates were filtered through a 70 μM cell strainer, centrifuged for 5 min, then subject to treatment with 1× RBC lysis buffer (Tonbo Biosciences). Lung cell suspensions (about 1 × 106 cells) were stained with 1:1,000 Live/Dead Orange dye per the manufacturer’s protocol (Thermo Fisher Scientific) for 30 min, then cells were washed with fluorescence-activated cell sorting (FACS) buffer (1× PBS and 3% FBS), and incubated for 10 min with 1:100 anti-CD16/CD32 (70–0161-U500, Tonbo Biosciences) and then fixed in 4% paraformaldehyde for at least 30 min. Cells were washed with FACS buffer and incubated for 20 min with primary antibody cocktail (1:100 anti-CD326-Brilliant Violet 421, 563214 and 1:100 anti-CD45pan-Brilliant Violet 786, 654225, BD Biosciences). Cells were washed and resuspended in 1× Fix/Perm solution using BD Biosciences Cytofix/Cytoperm kit (BD Biosciences) for 10 min at 4 °C. Cells were stained for intracellular markers (1:50 anti-SARS-CoV-2 nucleocapsid-AlexaFluor 647, ab302552, Abcam, UK), 1:25 anti-SCGB1A1-AlexaFluor 488 (NBP2–90517AF488, Novus Biologicals) and 1:25 anti-acetylated tubulin-AlexaFluor 488 (sc-23950 AF488, Santa Cruz Biotechnologies). Cells were washed twice with FACS buffer and analysed by S1000 Flow Cytometer (Stratedigm). Data were compensated and analysed with FlowJo v10 (Tree Star). All centrifugations were performed at 500g for 5 min.
Cells
The 16HBE14o- human bronchial epithelial cell line (HBE, SCC150, Sigma-Aldrich) were grown in 10% FBS/DMEM and transduced with SCRBBL lentivirus vector56 expressing human ACE2 (HBE-ACE2) to generate a stable population of cells. HBE-ACE2 cells were transduced with lentiCRISPRv2-puro (a gift from Feng Zhang, 52961, Addgene) or lentiCRISPRv2-blast (a gift from Brett Stringer, 98293, Addgene) expressing two independent non-targeting sgRNAs or two independent sgRNAs targeting human LY6E. sgRNA sequences (Supplementary Table 3) for targeting LY6E were cloned into the lentiCRISPRv2 plasmid as detailed by the GeCKO protocol57,58. Non-targeting sequences were previously described56. Cells were selected in 10 μg ml−1 blasticidin (Gibco)/3 μg ml−1 puromycin (Gibco)/10% FBS/DMEM and passaged several times before infecting with SARS-CoV-2.
SARS-CoV-2 infection of LY6E knockout cells
HBE-ACE2 NT1/NT2 and LY6E sg4/sg12 cells were plated at a density of 2 × 105 cells ml−1 in 12-well plates. Cells were infected with a titration of SARS-CoV-2-Wuhan-Hu-1-zsGreen for 30 min at 37 °C, followed by addition of 1 ml 10% FBS/DMEM. After 24 h, cells were dislodged from plate by addition of Accumax (Sigma-Aldrich), followed by fixation in 4% paraformal-dehyde, and resuspension in FACs buffer. Cells were run on S1000EON Flow Cytometer (Stratedigm) and analysed by FlowJo (Tree Star).
Western blotting
Cells were washed once in 1× PBS, resuspended in RIPA buffer, incubated for 30 min on ice, then sheared with a syringe and needle. Samples were centrifuged to remove cellular debris. The protein concentration of the remaining supernatants was quantified by bicinchoninic acid assay (Pierce), normalized to a BSA standard curve, on a Synergy/LX Multi-mode reader (BioTek). Tris-tricine sample loading buffer (200 mM Tris–HCl pH 6.8/40% glycerol/2% sodium dodecyl sulfate/0.04% Coomassie blue G-250/2% β-mercaptoethanol (Sigma-Aldrich)) was added, and samples were boiled at 95 °C for 5 min, then frozen at −80 °C. Lysates (20 μg) were thawed and loaded into a low-molecular-weight tricine gel6. Protein was transferred to polyvinylidene difluoride (PVDF) membrane (Bio-Rad) using Trans-Blot TurboTransfer System (Bio-Rad). Membranes were blocked in 5% milk/TBST overnight at 4 °C. Membrane was incubated for 1 h in 1:300 anti-human LY6E (mouse monoclonal clone 4D8.6.7, a gift from Genentech)/1:20,000 anti-β-actin (clone 2D4H5, 6609–1-Ig, Proteintech) in TBST. Membranes were washed 3 × 5 min in TBST then incubated for 30 min at room temperature with 1:5,000 donkey anti-mouse IgG conjugated to HRP (A16011, Invitrogen) in TBST. Membranes were again washed 5 × 5 min in TBST before enhanced chemiluminescence (ECL, Pierce) and preparation for exposure and development using ChemiDoc Imaging System (Bio-Rad).
Statistical analysis
Statistical analyses for all data except for RNA-seq data were performed using GraphPad Prism versions 9.1.0 and 9.4.1. Individual statistical tests are specified within the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. a-f, Ly6efl/fl and Ly6eΔVav1 mice were intraperitoneally infected with 5,000 PFU MHV-A59 and assessed for survival (a), viral burden in brain, lung, spleen, and liver (b), viral burden in heart (c), viral burden in serum (d), serum alanine aminotransferase (e), and post-mortem spleen weight (f).
In a, data represents means from n = 34 Ly6efl/fl and n = 23 Ly6eΔVav1; b, d-f, n = 8 Ly6efl/fl and n = 15 Ly6eΔVav1; c, n = 5 Ly6efl/fl and n = 10 Ly6eΔVav1. g, Flow cytometry gating strategy for sorting lymphocytes from the spleen for examining Ly6e gene expression in Ly6eΔCD4, Ly6eΔCD8a, and Ly6eΔCD19 mice relative to Ly6efl/fl littermates in Fig. 1g. h–l, mice were intraperitoneally infected with 5,000 PFU MHV-A59 and assessed for (h) survival (n = 47 Ly6efl/fl, n = 15 Ly6eΔLyz2, n = 14 Ly6eΔCD11c, n = 14 Ly6eΔCD4, n = 5 Ly6eΔCD8a, and n = 16 Ly6eΔCD19), viral burden in brain, lung, spleen, and liver (i), viral burden in serum (j), serum alanine aminotransferase (k), and post-mortem spleen weight (l). In i-l, data represents means from n = 30 Ly6efl/fl, n=13 Ly6eΔLysM, n = 14 Ly6eΔCD11c, n = 10 Ly6eΔCD4, n = 6 Ly6eΔCD8a, and n = 14 Ly6eΔCD19. Male and female mice were used at an approximately 1 to 1 ratio for these experiments. Statistical significance was determined by log-rank (Mantel-Cox) tests (a, h), two-sided Mann-Whitney test (b-d), two-sided unpaired t-test (e-f), Kruskal-Wallis test (i-j), and one-way ANOVA (k-l). Error bars represent mean ± standard deviation.
Extended Data Fig. 2 |. a, Flow cytometry gating strategy for sorting lung CD45+ cells and CD31+ cells from Ly6eΔVav1 mice for determining Ly6e gene expression as shown in Fig. 2c,d.
b, Flow cytometry gating strategy for identifying donor (CD45.1+) and recipient (CD45.2+) immune cells. c, Relative composition of CD45.1+ and CD45.2+ immune cells of compartment total CD45+ cells in lung, spleen, and blood. d, Relative Ly6e mRNA levels in brain, heart, lung, liver, and spleen from Ly6e wildtype, heterozygous, and knockout mice. In c, data represents means from n = 3 Ly6efl/fl; CD45.2/.2, n = 3 Ly6efl/fl; CD45.1/.1, n = 3 Ly6efl→fl, n = 3 Ly6efl→ΔVav1; d, n = 3 Ly6e wildtype, n = 3 Ly6e heterozygous, n = 3 Ly6e knockout. Error bars represent mean ± standard deviation.
Extended Data Fig. 3 |. a, Ly6e wildtype and knockout mice were intranasally infected with 8,700 PFU SARS-CoV-2 and monitored daily for weight loss.
Lung viral burden measured by quantitative PCR for Ly6e wildtype and knockout mice (b) or C57BL/6J, Ifnar−/−, and Ifnlr−/− mice (c) that were infected with 60,000 PFU SARS-CoV-2 and euthanized 3 days post-infection. d, Lung Ly6e expression from C57BL/6J, Ifnar−/−, and Ifnlr−/− mice that were mock treated with PBS or infected with 8,700 PFU SARS-CoV-2 and euthanized the next day. Expression is shown relative to PBS-treated C57BL/6J mice. Expression of Ly6e (e) and Mx1 (f) in lungs from C57BL/6J mice treated intranasally with recombinant human IFNλ or retro-orbitally with recombinant murine IFNβ relative to untreated (untr). In a, data represents means from n = 5 Ly6e wildtype, n = 5 Ly6e knockout mice; b, n = 13 Ly6e wildtype, n = 13 Ly6e knockout mice; c, n = 8 C57BL6/J, n = 8 Ifnar−/−, and n=8 Ifnlr−/−; d, n = 5 for each group and genotype; e-f, n = 6 untreated, n = 9 IFNλ-treated, n = 8 IFNβ-treated. g, Representative hematoxylin and eosinstained lung sections used for analysis shown in Fig. 3e from Ly6e wildtype (n = 6) and Ly6e knockout (n = 6) mice euthanized 3 days after treated with PBS or intranasal infection with 60,000 PFU SARS-CoV-2. Example of automated image analysis of SARS-CoV-2 infected cells for Fig. 3g–h (h) and of CD45+ cells for Fig. 3i, j (i). In the corresponding markup images, cells without DAB marker (for example positive stain) are colored blue, and positive cells identified as weak positive, moderate positive, and strong positive for the DAB marker are colored as yellow, orange, and red respectively. Statistical significance was determined by two-sided unpaired t-test (a), two-sided Mann-Whitney test (b-c), one way ANOVA with Holm-Šídák′s multiple comparisons test (d–f). Error bars represent mean ± SEM in a and mean ± SD in b-f. Scale bars: 600 μm (g), 1 mm and 100 μm (h-i).
Extended Data Fig. 4 |. mRNA sequencing of lungs from Ly6e wildtype and Ly6e knockout mice that were intranasally infected with 60,000 PFU P.1 SARS-CoV-2 and euthanized 3 days post-infection.
Volcano plot summarization of differentially expressed genes (DEGs) between uninfected Ly6e wildtype (n = 6) and Ly6e knockout (n = 4) mice (a), uninfected wildtype (n = 6) and SARS-CoV-2 infected wildtype (n = 7) mice (b), and uninfected knockout (n = 4) and SARS-CoV-2 infected knockout (n=7) mice (c). d, Z-score heatmap of the top 25 DEGs for data summarized in a. e, Gene Ontology (GO) enrichment analysis of transcriptomic data from SARS-CoV-2 infected Ly6e wildtype and Ly6e knockout mice. The abscissa is the ratio of the number of differential genes linked with the GO term to the total number of differential genes. The size of a point represents the number of genes annotated to a specific GO term, and the color from red to purple represents the significant level of the enrichment. Male and female mice were used at an approximately 1 to 1 ratio for these experiments. For a-c, P-values were determined using the negative binomial distribution model and adjusted for multiple hypothesis testing using the Benjamini and Hochberg’s approach for controlling the false discovery rate. P-values for e were determined using hypergeometric distribution and adjusted testing using the Benjamini and Hochberg’s approach for controlling the false discovery rate.
Extended Data Fig. 5 |.
Differential expression of select genes highlighted in Fig. 4a between SARS-CoV-2-infected Ly6e wildtype mice and uninfected Ly6e wildtype mice (a) and SARS-CoV-2-infected Ly6e knockout mice and uninfected Ly6e knockout mice (b)
Extended Data Fig. 6 |. Mice were infected with 8,700 PFU P.1 SARS-CoV-2 and euthanized 1 day post-infection.
Lung sections were probed for SARS-CoV-2 RNA (green) and Scgb1a1 mRNA (red) (a, c) or Scgb3a1 mRNA (red) (b, d). Lung sections from n = 3 Ly6e knockout mice were stained and imaged. Images shown are from two different mice than in Fig. 4. Scale bars: 100 μM (a-b), 25 μM (c-d).
Extended Data Fig. 7 |. Mice were infected with 8,700 PFU SARS-CoV-2 and euthanized 1 day post-infection.
Lung sections were probed for SARS-CoV-2 RNA (green) and Sftpc mRNA (red) (a, c) or for SARS-CoV-2 nucleocapsid (green) and acetylated tubulin (red) (b, d). Lung sections from n = 3 Ly6e knockout mice were stained and imaged. Images shown are from different mice than in Fig. 4. Scale bars: 100 μM (a-b), 25 μM (c-d).
Extended Data Fig. 8 |.
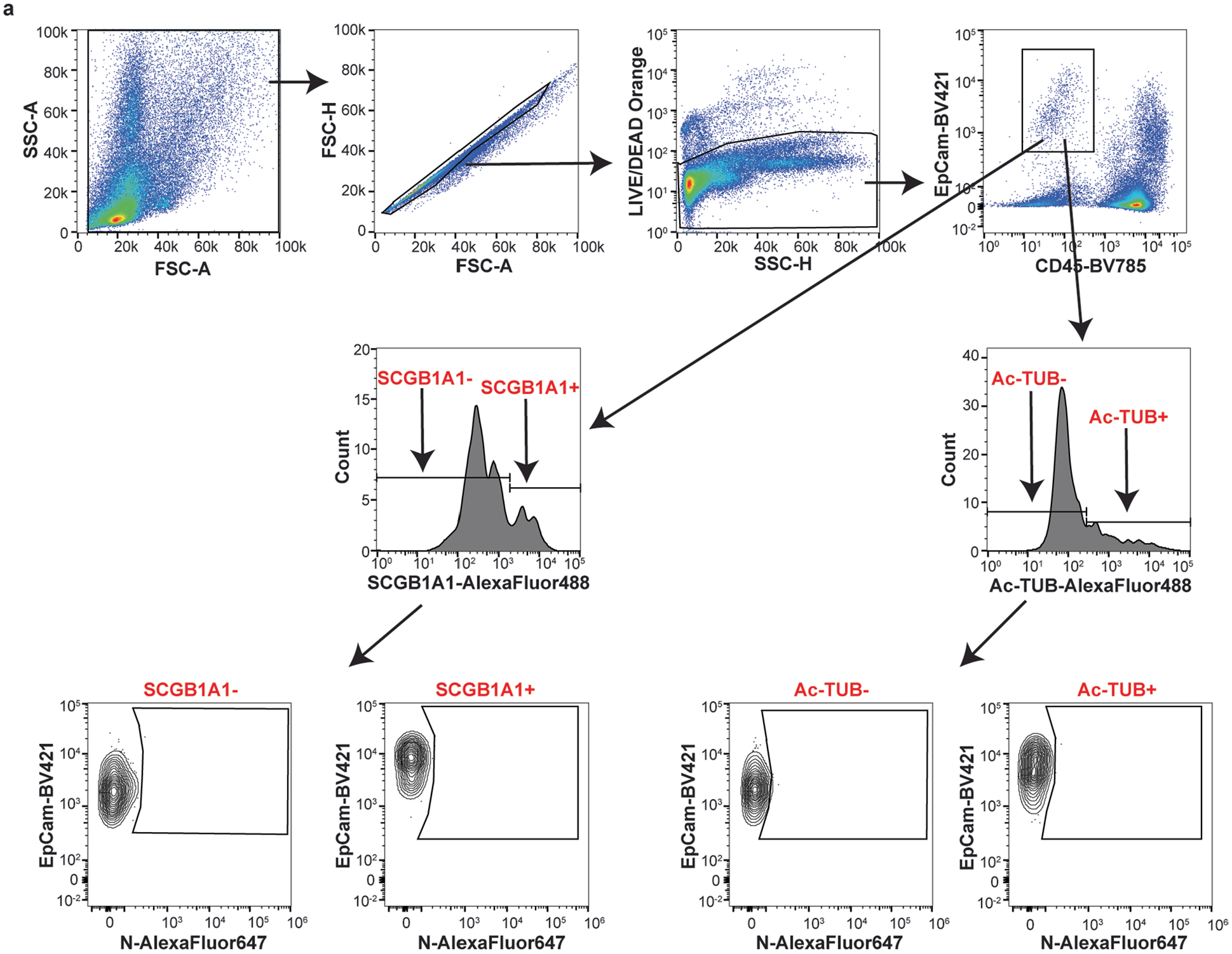
a, Flow cytometry gating strategy used in Fig. 4g–h for detecting SARS-CoV-2 nucleocapsid (N) in SCGB1A1-positive and acetylated tubulin-positive pulmonary epithelial cells. Representative plots are from uninfected Ly6e knockout mice.
Extended Data Fig. 9 |. a, Area under the curve analysis of SARS-CoV-2 infectivity in HBE-ACE2 cells from n = 3 independent experiments, b, Western blot of two independent preparations of control and LY6E knockout cells used in a.
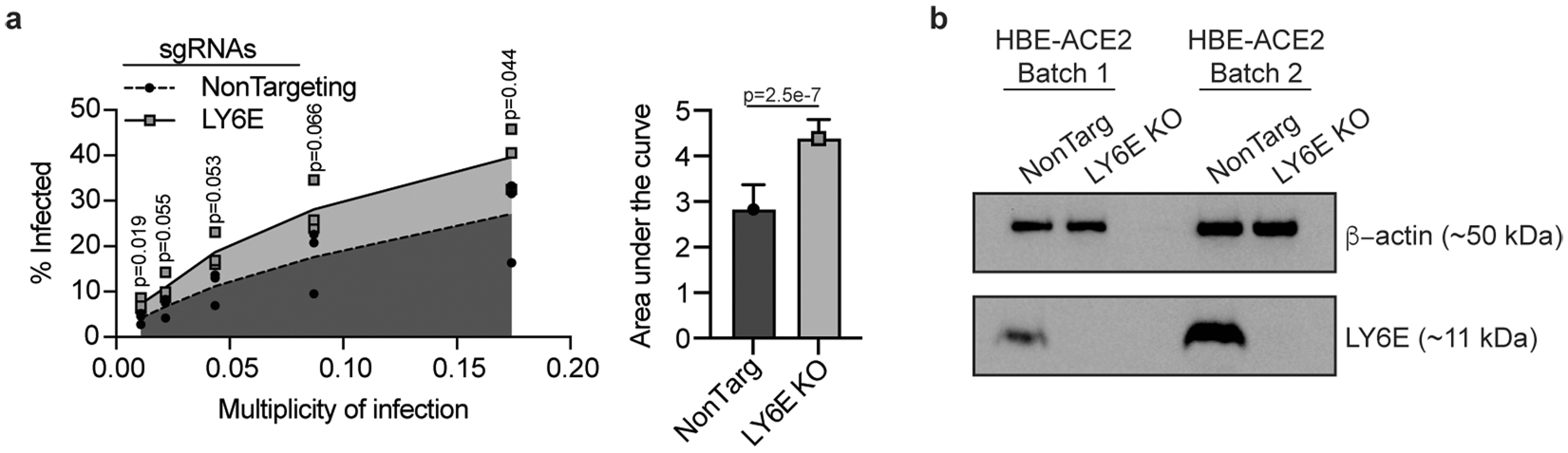
Statistical significance was determined by two-sided unpaired t-test (a). Error bars represent mean ± standard deviation.
Supplementary Material
Acknowledgements
We thank members of the Schoggins lab for useful discussions. We also thank M. Aufiero and T. Hohl (Memorial Sloan Kettering Cancer Center) for advice and feedback for bone marrow chimera studies, the lab of L. Hooper (University of Texas Southwestern Medical Center (UTSW)) for CD11c-Cre and LysM-Cre transgenic mice, the labs of J. Pfeiffer (UTSW) and M. Baldridge (Washington University in St. Louis) for Ifnar−/− and Ifnlr−/− mice, respectively, the UTSW Animal Resource Center for training and animal husbandry, the UTSW Metabolic Phenotyping Core for analysis of serum samples for ALT levels and expertise, the UTSW Histo Pathology Core, the UTSW Preclinical Radiation Core Facility (supported by funding from CPRIT grant RP180770) and the UTSW Immunology Flow Cytometry Core. The authors also acknowledge the Quantitative Light Microscopy Core, a Shared Resource of the Harold C. Simmons Cancer Center, supported in part by an NCI Cancer Center Support Grant, 1P30 CA142543-01, and 1S10 RR029731-01 to K. Luby-Phelps.
Footnotes
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41564-023-01431-w.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41564-023-01431-w.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files or are available on request. The RNA-seq data discussed in this publication have been deposited in the Gene Expression Omnibus database (GSE209974). Source data are provided with this paper.
References
- 1.Schoggins JW Interferon-stimulated genes: what do they all do? Annu. Rev. Virol 6, 567–584 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Mao M et al. RIG-E, a human homolog of the murine Ly-6 family, is induced by retinoic acid during the differentiation of acute promyelocytic leukemia cell. Proc. Natl Acad. Sci. USA 93, 5910–5914 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bacquin A et al. A cell fusion-based screening method identifies glycosylphosphatidylinositol-anchored protein Ly6e as the receptor for mouse endogenous retroviral envelope syncytin-A. J. Virol 91, e00832–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schupp JC et al. Integrated single-cell atlas of endothelial cells of the human lung. Circulation 144, 286–302 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabula Muris C et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mar KB et al. LY6E mediates an evolutionarily conserved enhancement of virus infection by targeting a late entry step. Nat. Commun 9, 3603 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee PY, Wang JX, Parisini E, Dascher CC & Nigrovic PA Ly6 family proteins in neutrophil biology. J. Leukoc. Biol 94, 585–594 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Saitoh S et al. Modulation of TCR-mediated signaling pathway by thymic shared antigen-1 (TSA-1)/stem cell antigen-2 (Sca-2). J. Immunol 155, 5574–5581 (1995). [PubMed] [Google Scholar]
- 9.Mao W, Hunt HD & Cheng HH Cloning and functional characterization of chicken stem cell antigen 2. Dev. Comp. Immunol 34, 360–368 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Xu X et al. IFN-stimulated gene LY6E in monocytes regulates the CD14/TLR4 pathway but inadequately restrains the hyperactivation of monocytes during chronic HIV-1 infection. J. Immunol 193, 4125–4136 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Classon BJ & Coverdale L Mouse stem cell antigen Sca-2 is a member of the Ly-6 family of cell surface proteins. Proc. Natl Acad. Sci. USA 91, 5296–5300 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu L et al. Mouse thymus dendritic cells: kinetics of development and changes in surface markers during maturation. Eur. J. Immunol 25, 418–425 (1995). [DOI] [PubMed] [Google Scholar]
- 13.Noda S, Kosugi A, Saitoh S, Narumiya S & Hamaoka T Protection from anti-TCR/CD3-induced apoptosis in immature thymocytes by a signal through thymic shared antigen-1/stem cell antigen-2. J. Exp. Med 183, 2355–2360 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zammit DJ et al. Essential role for the lymphostromal plasma membrane Ly-6 superfamily molecule thymic shared antigen 1 in development of the embryonic adrenal gland. Mol. Cell. Biol 22, 946–952 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langford MB, Outhwaite JE, Hughes M, Natale DRC & Simmons DG Deletion of the Syncytin A receptor Ly6e impairs syncytiotrophoblast fusion and placental morphogenesis causing embryonic lethality in mice. Sci. Rep 8, 3961 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krishnan MN et al. RNA interference screen for human genes associated with West Nile virus infection. Nature 455, 242–245 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schoggins JW et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schoggins JW et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505, 691–695 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hackett BA & Cherry S Flavivirus internalization is regulated by a size-dependent endocytic pathway. Proc. Natl Acad. Sci. USA 115, 4246–4251 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu J, Liang C & Liu SL Interferon-inducible LY6E protein promotes HIV-1 infection. J. Biol. Chem 292, 4674–4685 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfaender S et al. LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat. Microbiol 5, 1330–1339 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wickenhagen A et al. A prenylated dsRNA sensor protects against severe COVID-19. Science 374, eabj3624 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Danziger O, Patel RS, DeGrace EJ, Rosen MR & Rosenberg BR Inducible CRISPR activation screen for interferon-stimulated genes identifies OAS1 as a SARS-CoV-2 restriction factor. PLoS Pathog 18, e1010464 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mac Kain A et al. Identification of DAXX as a restriction factor of SARS-CoV-2 through a CRISPR/Cas9 screen. Nat. Commun 13, 2442 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abram CL, Roberge GL, Hu Y & Lowell CA Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods 408, 89–100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCubbrey AL, Allison KC, Lee-Sherick AB, Jakubzick CV & Janssen WJ Promoter specificity and efficacy in conditional and inducible transgenic targeting of lung macrophages. Front. Immunol 8, 1618 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joseph C et al. Deciphering hematopoietic stem cells in their niches: a critical appraisal of genetic models, lineage tracing, and imaging strategies. Cell Stem Cell 13, 520–533 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Bao L et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 583, 830–833 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Dinnon KH 3rd et al. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature 586, 560–566 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leist SR et al. A mouse-adapted SARS-CoV-2 induces acute lung injury and mortality in standard laboratory mice. Cell 183, 1070–1085 e1012 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu H et al. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science 369, 1603–1607 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imai M et al. Characterization of a new SARS-CoV-2 variant that emerged in Brazil. Proc. Natl Acad. Sci. USA 118, e2106535118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orthgiess J et al. Neurons exhibit Lyz2 promoter activity in vivo: Implications for using LysM-Cre mice in myeloid cell research. Eur. J. Immunol 46, 1529–1532 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Gandhi RT, Lynch JB & Del Rio C Mild or moderate Covid-19. N. Engl. J. Med 383, 1757–1766 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Huang K et al. Q493K and Q498H substitutions in Spike promote adaptation of SARS-CoV-2 in mice. EBioMedicine 67, 103381 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun S et al. Characterization and structural basis of a lethal mouse-adapted SARS-CoV-2. Nat. Commun 12, 5654 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muruato A et al. Mouse-adapted SARS-CoV-2 protects animals from lethal SARS-CoV challenge. PLoS Biol 19, e3001284 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winkler ES et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol 21, 1327–1335 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng J et al. COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature 589, 603–607 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fumagalli V et al. Administration of aerosolized SARS-CoV-2 to K18-hACE2 mice uncouples respiratory infection from fatal neuroinvasion. Sci. Immunol 7, eabl9929 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salahudeen AA et al. Progenitor identification and SARS-CoV-2 infection in human distal lung organoids. Nature 588, 670–675 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ravindra NG et al. Single-cell longitudinal analysis of SARS-CoV-2 infection in human airway epithelium identifies target cells, alterations in gene expression, and cell state changes. PLoS Biol 19, e3001143 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fiege JK et al. Single cell resolution of SARS-CoV-2 tropism, antiviral responses, and susceptibility to therapies in primary human airway epithelium. PLoS Pathog 17, e1009292 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peng Y et al. Angiotensin-converting enzyme 2 in peripheral lung club cells modulates the susceptibility to SARS-CoV-2 in chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell. Mol. Physiol 322, L712–L721 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fadista J et al. Shared genetic etiology between idiopathic pulmonary fibrosis and COVID-19 severity. EBioMedicine 65, 103277 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Moorsel CHM et al. The MUC5B promoter polymorphism associates with severe COVID-19 in the European population. Front. Med 8, 668024 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verma A et al. A MUC5B gene polymorphism, rs35705950-T, confers protective effects against COVID-19 hospitalization but not severe disease or mortality. Am. J. Respir. Crit. Care Med 206, 1220–1229 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Initiative C-HG A first update on mapping the human genetic architecture of COVID-19. Nature 608, E1–E10 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coley SE et al. Recombinant mouse hepatitis virus strain A59 from cloned, full-length cDNA replicates to high titers in vitro and is fully pathogenic in vivo. J. Virol 79, 3097–3106 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rihn SJ et al. A plasmid DNA-launched SARS-CoV-2 reverse genetics system and coronavirus toolkit for COVID-19 research. PLoS Biol 19, e3001091 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brattelid T et al. Reference gene alternatives to Gapdh in rodent and human heart failure gene expression studies. BMC Mol. Biol 11, 22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jhingran A, Kasahara S & Hohl TM Flow cytometry of lung and bronchoalveolar lavage fluid cells from mice challenged with fluorescent Aspergillus reporter (FLARE) conidia. Bio Protoc 6, e1927 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dietert K et al. Spectrum of pathogen- and model-specific histopathologies in mouse models of acute pneumonia. PLoS ONE 12, e0188251 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choi HM et al. Mapping a multiplexed zoo of mRNA expression. Development 143, 3632–3637 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi HMT et al. Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145, dev165753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hurskainen M et al. Single cell transcriptomic analysis of murine lung development on hyperoxia-induced damage. Nat. Commun 12, 1565 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Richardson RB et al. A CRISPR screen identifies IFI6 as an ER-resident interferon effector that blocks flavivirus replication. Nat. Microbiol 3, 1214–1223 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shalem O et al. Genome-scale CRISPR–Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files or are available on request. The RNA-seq data discussed in this publication have been deposited in the Gene Expression Omnibus database (GSE209974). Source data are provided with this paper.