

Abstract
The mechanisms underlying the progression of prostate cancer (PCa) to neuroendocrine prostate cancer (NEPC), an aggressive PCa variant, are largely unclear. Two prominent NEPC phenotypes are elevated NE marker expression and heightened angiogenesis. Identifying the still elusive direct molecular links connecting angiogenesis and neuroendocrine differentiation (NED) is crucial for our understanding and targeting of NEPC. Here we found that histone deacetylase 2 (HDAC2), whose role in NEPC has not been reported, is one of the most upregulated epigenetic regulators in NEPC. HDAC2 promotes both NED and angiogenesis. G protein-coupled receptor kinase 3 (GRK3), also upregulated in NEPC, is a critical promoter for both phenotypes too. Of note, GRK3 phosphorylates HDAC2 at S394, which enhances HDAC2’s epigenetic repression of potent anti-angiogenic factor Thrombospondin 1 (TSP1) and master NE-repressor RE1 Silencing Transcription Factor (REST). Intriguingly, REST suppresses angiogenesis while TSP1 suppresses NE marker expression in PCa cells, indicative of their novel functions and their synergy in cross-repressing the two phenotypes. Furthermore, the GRK3-HDAC2 pathway is activated by androgen deprivation therapy and hypoxia, both known to promote NED and angiogenesis in PCa. These results indicate that NED and angiogenesis converge on GRK3-enhanced HDAC2 suppression of REST and TSP1, which constitutes a key missing link between two prominent phenotypes of NEPC.
Keywords: Neuroendocrine differentiation, angiogenesis, Histone Deacetylase 2 HDAC2, epigenetic regulation, G protein-coupled receptor kinase 3 GRK3, REST, TSP1, androgen deprivation, hypoxia
INTRODUCTION
Androgen deprivation therapy (ADT) that targets androgen receptor (AR) is the mainstay treatment for prostate cancer (PCa). ADT has been initially effective, but the majority of tumors invariably relapse and progress to become castration resistant prostate cancer (CRPC). Approximately 20% of lethal CRPCs have a neuroendocrine phenotype, and thus are called neuroendocrine prostate cancer (NEPC, CRPC-NE) (1–3). NEPC is characterized by loss of AR signaling in the majority of them, resistance to ADT, and elevated levels of neuroendocrine (NE) markers, such as neuron-specific enolase (ENO2), synaptophysin (SYP), chromogranin A (CHGA), and chromogranin B (CHGB) (2,4–6). NEPCs only transiently respond to chemotherapy (1,7–11), and there is no effective therapy. With common use of new generation ADT in the clinic, the incidence of NEPC has increased, as a mechanism of resistance to potent ADT (6,12–15). The signaling events involved in NEPC development are largely unclear (1,6,10).
NEPC is known to be highly vascularized (16–19). Elevated expression of NE markers and increased angiogenesis are two prominent phenotypes of NEPC (16–19), and thus are expected to be linked. However, direct molecular links between these two phenotypes are largely unclear, whose elucidation will substantially expand our knowledge in NEPC and enable the development of effective treatments for NEPC. Epigenetic regulation has been implicated in ADT resistance, neuroendocrine differentiation (NED) and immune regulation. In this study, to identify potential epigenetic drivers of NEPC, we first examined 147 epigenetic regulators in several prostate cancer datasets (20–24). We found that histone deacetylase 2 (HDAC2), whose role in NEPC has never been explored, is one of the most upregulated epigenetic regulators in NE+ vs NE− PCa. We speculated that HDAC2 contributes to one or both critical NEPC phenotypes, i.e. NED and angiogenesis.
Through unbiased shRNA and cDNA screenings of human kinases, we previously uncovered that G protein-coupled receptor kinase 3 (GRK3) is a critical regulator of PCa progression, in part through inducing angiogenesis (25). GRK3, a Ser/Thr protein kinase, is a member of the G protein-coupled receptor kinase (GRK) subfamily, whose assumed primary roles are to regulate and mediate G protein-coupled receptor (GPCR) signaling (26,27). We recently reported that GRK3 promotes ADT resistance and NE marker expression of PCa cells (28). These results define a critical role of GRK3 in controlling both NED and angiogenesis in NEPC progression, although the mechanism underlying GRK3’s role in these processes is largely unknown.
In this study, our objectives are to investigate critical mechanisms underlying NED and angiogenesis in PCa progression and to uncover their missing molecular links. We found that GRK3-mediated phosphorylation of HDAC2 enhances HDAC2’s epigenetic activity in repressing REST and TSP1, two proteins that cooperate in repressing NED and angiogenesis in prostate cancer.
MATERIAL AND METHOD
Cell culture
LNCaP, 22RV1, VCaP, and C4–2 cells were purchased from ATCC. 144–13 and PC3 cells were generously provided by Ana Aparicio (29) and Isaiah J. Fidler (30), respectively. LNCaP, VCaP, 22RV1, 144–13, LN3, C4–2, and PC3 cells were cultured in RPMI 1640 media (Corning), supplemented with 10% FBS (Gibco), 2% L-Glutamine, and 1% penicillin-streptomycin. NE1.3 cells, a generous gift from Ming-Fong Lin, were derived from LNCaP cells after long-term culturing in media with charcoal-stripped serum (CSS) (31). 293T cells and endothelial cells SVEC4–10, from ATCC, were cultured in DMEM media (Corning), supplemented with 10% FBS and 1% penicillin-streptomycin. All cell lines were cultured in a 5% CO2 humid incubator at 37°C. All cell lines were matched to their corresponding ATCC profile by DNA STR fingerprinting (Biosynthesis Inc). All cell lines were frequently tested to ensure mycoplasma-free using the Lonza MycoAlert Detection kit (LT07–218).
cDNA/shRNA transduction and transfection
All Lentiviral Knockout (LKO)-based constitutively-expressed shRNA lentiviral vectors were purchased from Sigma. Inducible shRNAs were cloned in CW39 lentiviral vector using the effective shRNA sequences from their corresponding LKO construct (25). Each plasmid was packaged into viruses using 293T cells, as we previously described (32). Cells carrying the following vectors were selected with appropriate selection markers to make a stable cell line: GFP/GRK3 cDNA (blasticidine), CW39 vectors (G418), pLKO (puromycin), and TSP1 cDNA (hygromycin B). For doxycycline-inducible shGRK3 in CW39 vector, cells were treated with doxycycline (400–600 ng/ml) for 4–5 days to obtain GRK3 knockdown.
Mutagenesis of HDAC2 S394
In order to determine the functionality of HDAC2 phosphorylation at S394, we mutated 394aa from Serine to Alanine, using Q5® Site-Directed Mutagenesis Kit (NEB) according to the instruction provided by the supplier. The primers used for point mutation are forward: tcatgaagacgctggagatgaagatggag; and reverse: acagcatcttctggaatag.
Cell viability
Cell proliferation in 96-well plates was measured using Alamar Blue assay, as we previously described (25).The fluorescence intensity was measured using a TECAN Infinite plate reader at excitation and emission wavelengths of 535 and 595 nm, respectively.
Western blots
We used the same western blotting procedure as we previously described (33). The primary antibodies used in this research have been listed in Supplementary Table S1.
Nuclear/cytosol fraction preparation
Nuclear and cytosolic fractions of cells were extracted as per supplier standard protocol (nuclear/Cytosol Fractionation Kit, BioVision K266 fractionation kit). The distribution patterns of each protein were determined by western blotting.
Reverse transcription and qPCR
RNA samples have been extracted using TRIzol™ Reagent (Thermo Scientific™) based on the protocol described by the supplier. We then used the RT-PCR procedure as we previously described (33). Primers for qPCR are listed in Supplementary Table S2.
Chromatin Immunoprecipitation (CHIP)
DNA cross-linking was performed with 1% formaldehyde at room temperature for 15 minutes with gentle mixing. For HDAC2 and pHDAC2 ChIP, cross-linking was performed with 2 mM disuccinimidyl glutarate (DSG) for 35 minutes and then 1% formaldehyde for further 10 minutes of incubation (34). The remaining steps in ChIP procedure were similar to what we previously described (33).
The following antibodies were used for ChIP in this study: HDAC2 (D6S5P) (Cell Signaling Technology, 57156); pHDAC2 S394 (Genscript, A01169); Acetyl-Histone H3 (Lys27) (D5E4) (Cell Signaling Technology, 8173); Tri-Methyl-Histone H3 (Lys27) (C36B11) antibody (Cell Signaling Technology; 9733S) and normal rabbit control IgG antibody (Cell Signaling Technology, 2729S). The qPCR primers for measuring enrichments of the binding of abovementioned specific antibody (vs IgG controls) to TSP1 and REST promoter sequences are as following: TSP1 (F: gtcatacaacactcccacgc; R: agccagcatgactctagctt); REST (F: ccgcgagttggtgtgtaaat; R: cacgaattcctccgtgctg).
Immunoprecipitation (IP)
One mg of protein lysate was subjected to pre-clear for 2–4 hours using pre-washed Protein A/G beads. Then each supernatant was divided into three tubes for input, primary and IgG antibodies. 500 ul of samples were incubated with 1 ug recombinant GRK3 rabbit antibody (Abcam, ab109303) and GRK3 rabbit antibody (C-14) (Santa Cruz Biotechnology, sc-563) or V5-Tag (E9H8O) mouse antibody (Cell Signaling Technology, 80076) overnight at 4 °C. Corresponding IgG antibodies, mouse (G3A1) IgG1 Isotype Control (Cell Signaling Technology, 5415), and normal rabbit IgG (Cell Signaling Technology, 2729S) were also used as a control. The next day, samples were transferred to pre-washed Protein A/G beads and incubated for 4 hours while rocking. After three washing steps, samples were eluted in Laemmli SDS Sample Buffer (Alfa Aesar) at 100 °C for 10 minutes. Interaction of proteins was probed by western blotting.
In vitro thio-phosphorylation kinase assay
HDAC2 phosphorylation by GRK3 was detected by following the kinase reaction and alkylation protocol from Abcam, which is based on this publication (35). Briefly, HDAC2-FLAG recombinant protein (Active Motif, 31505) with or without GRK3-GST recombinant protein (LifeTech, PV3827) were incubated with ATP-gamma-S (Abcam, ab138911) in kinase reaction buffer at 30°C for one hour. The kinase reaction was stopped by adding EDTA, followed by alkylation with PNBM (p-nitrobenzyl mesylate, Abcam, ab138910) for two hours at 30°C. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) loading buffer was then added to the reaction and the samples were analyzed by western blotting using the Thiophosphate Ester Specific Rab MAb (Abcam, ab92570) to detect thiophosphate-labelled GRK3 protein (auto-phosphorylation) and HDAC2 protein (phosphorylation by GRK3). To detect GRK3’s phosphorylation of endogenous HDAC2 proteins from NEPC cells, HDAC2 proteins were IPed (immunoprecipitated) from NEPC cells 144–13 using anti-HDAC2 Ab. The IPed HDAC2 on beads was then washed once with GRK3 kinase buffer, followed by similar thio-phosphorylation and western blotting procedures as described above.
Mass Spectrometry (MS)
For identifying phosphorylation site on HDAC2 through mass spectrometry, similar kinase reaction was carried out as the above thio-phosphorylation assay, except using regular ATP and without the PNBM alkylation step. The kinase reaction mixtures were run on SDS-PAGE gel briefly for <1 cm. The gel portions containing the protein samples were cut out and provided to the mass spectrometry core lab at our Institute, following a standard label-free MS sample preparation protocol. About 1 microgram of tryptic digest peptides was analyzed by LC/MS/MS (Liquid Chromatography with tandem mass spectrometry) on an Orbitrap Fusion™ Tribrid™ mass spectrometer (Thermo Scientific™) interfaced with a Dionex UltiMate 3000 Binary RSLCnano System. The raw data files were processed using Thermo Scientific™ Proteome Discoverer™ software version 1.4, spectra were searched against the Uniprot Mus musculus plus DT database using Sequest HT search engine. Search results were trimmed to a 1% False Discovery Rate (FDR) using Percolator.
Wound healing of endothelial cells
To determine the impacts on angiogenesis from modulating our pathway proteins in cancer cells, we employed a commonly used in vitro angiogenesis assay, i.e. wound healing of endothelial cells in the conditioned medium from cancer cells (36,37). At day 1, we seeded 650,000 SVEC cells per well into a 6-well plate and 400,000 cancer cells into 60 ml tissue culture dishes with 10% FBS medium. At day 2, cancer and endothelial cells were starved with serum-free medium for 20 and 12 hours, respectively. At day 3, after discarding the medium on SVEC cells, a uniform scratch was scraped by a 200-ul sterile pipette tip, and cells were washed with serum-free medium to remove cells scraped off. Then conditioned medium from cancer cells was collected and added to endothelial cells. Wound healing of endothelial cells was monitored, and pictures were captured from 0 hr up to 72 hr after scratching. Wound healing of endothelial cells was calculated by measuring and comparing width of the gaps at 0 hr after scratch and the indicted time point of each experiment. Each experiment was repeated 3 times.
Migration of endothelial cells
Migration of endothelial cells toward conditioned media from cancer cells is another common in vitro angiogenesis assay (33). PC3-GFP, PC3-GRK3, PC3-GRK3-shHDAC2, and SVEC endothelial cells were seeded. The next day, cells were starved overnight. Then 600 ul of conditioned medium from cancer cells were added into the lower chamber of Corning™ Transwell™ multiple well plate, and 65,000 SVEC cells were seeded in serum-free medium into the upper chambers of the permeable polycarbonate membrane inserts. The migration of SVEC cells toward conditioned medium lasted for 10 hours. The chamber was fixed with cold 4% paraformaldehyde and then stained with crystal violet solution. Pictures were captured using Olympus DP26, and quantitative analysis was performed using optimized color thresholding tools with Fiji Image J software.
GST pulldown assay
Recombinant proteins FLAG-HDAC2 (Active Motif, 31505, 1ug) and GST-GRK3 (Life Tech, PV3827, 2ug), alone or mixed, were incubate at 4°C for 2hr. Washed glutathione sepharose 4B slurry (Millipore Sigma, 17-0756-01) were then added to the proteins, incubated at 4°C for 3hr, washed twice with IP dilution buffer, then 35ul of lysate buffer and 7ul of 6x Laemmli sample buffer were added to the proteins and beads, boiled 10min at 95°C and run on SDS-PAGE gels, followed by western blotting using anti-FLAG (CST, 2368) and anti-GST (SCBT, sc-138) antibodies.
Animal experiments
NOD/SCID mice were purchased from Charles River Laboratories. Half million of NE1.3 cells labeled with luciferase and expressing control shRNA (shControl) or shGRK3 were mixed with Matrigel (1:1) and implanted into both flanks of NOD/SCID male mice (6–8 mice per group, two implantations per mouse). Tumor growth signals were captured by bioluminescence imaging using an IVIS Lumina II platform (Caliper Life Sciences). All mice were sacrificed 70 days after implantation, at which the s.c. tumors were extracted, weighted and preserved. These animal experimental procedures were performed in accordance with all relevant ethical regulations and protocols approved by the Animal Welfare Committee at University of Texas Health Science Center at Houston.
Genomics data collections and analyses
All non-TCGA and TCGA genomics datasets as indicated were downloaded from cBioPortal (38), GEO database (http://www.ncbi.nlm.nih.gov/gds), or directly from the supplementary data of the corresponding papers. All expression values from the datasets were transformed and normalized by the authors of each dataset. Expression values for the indicated genes were extracted from these datasets and used in our analysis and statistical calculation.
Statistical analysis
Data are presented as the mean and standard deviation (SD) from three independent repeats. P-value was calculated using a two-tailed Student’s t-test to compare the differences between the conditions. A P-value lower than 0.05 was considered statistically significant.
RESULTS
NEPC cells upregulate NE markers and enhance angiogenesis of endothelial cells
To study the connections between angiogenesis and neuroendocrine phenotypes, we first examined these two phenotypes in Androgen-Dependent Prostate Cancer (ADPC) cell line LNCaP, and NEPC cell line 144–13 (29). As expected, NE markers SYP, tubulin beta 3 class III (TUBB3), and CHGA are substantially higher in 144–13 than in LNCaP cells (Fig. 1A). To measure angiogenesis in vitro, we used would healing assay that measured both proliferation and migration of endothelial cells, two key factors in angiogenesis (36,37). We found that the speeds of wound healing (closure of scratch) of monolayers of endothelial cells in the presence of conditioned media from 144–13 cells were faster than in conditioned media from LNCaP cells (Fig. 1B).
Figure 1. Overexpressed in NEPC, HDAC2 promotes NE marker expression and angiogenesis.
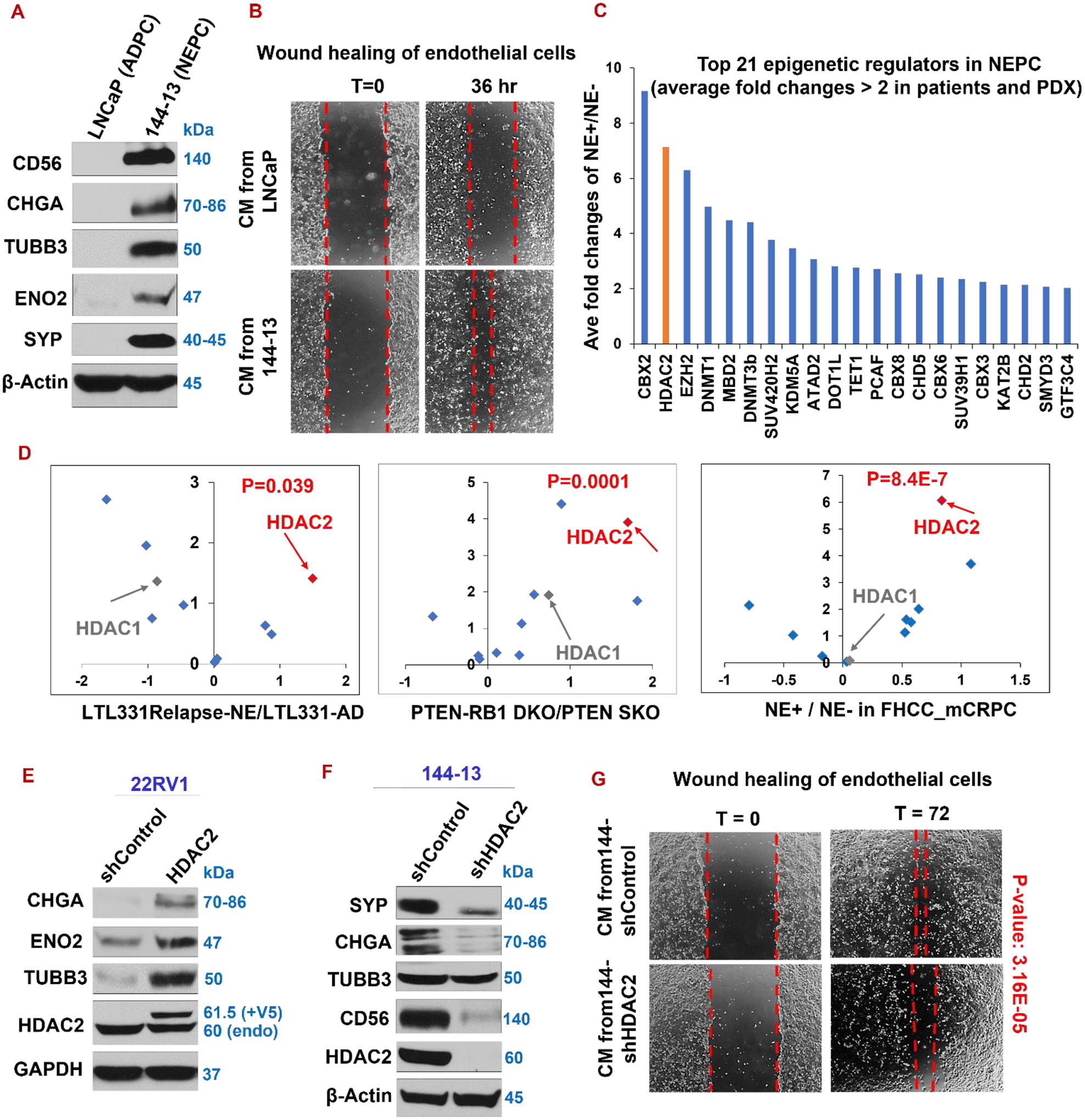
(A) Western blotting of indicated NE markers, comparing LNCaP and 144–13 cell lines. (B) Wound healing assay of endothelial cells in conditioned medium (CM) from LNCaP and 144–13 cancer cells. (C) Top 21 (out of 147) epigenetic regulators with biggest average fold changes of NE+ vs NE− prostate cancers in patient samples and a PDX model. The data for this plot were obtained from Supplemental Table S1 in a study by Clermont et al. (20), which was based on the datasets of Beltran_2011_Cancer Discovery patients (7)and the LTL331 PDX model (43). (D) Analyses of GSE59984, GSE90891, and GSE77930 prostate cancer datasets indicate that HDAC2, but not its closely related HDAC1, is the most upregulated HDAC in NEPC (relapsed NE+ LTL331R PDX vs LTL331 adenocarcinoma, PTEN-RB1 double knockout vs PTEN single knockout GEM tumors, NE+ CRPC than NE− CRPC in patients). Volcano plots: X-axis: log2 of NE/non-NE fold changes; Y-axis: - log10 of P values of NE vs. non-NE. (E-F) Western blotting of NE markers in 22RV1-GUS (vector control) and -HDAC2-cDNA cell lines (E), as well as 144–13 shControl and shHDAC2 (F). (G) Wound healing assay of endothelial cells in the conditioned media from 144–13-shControl or -shHDAC2 cells.
HDAC2 is among the most upregulated epigenetic regulators in NEPC, and it positively regulates NE marker expression and angiogenesis
We next investigated the molecular mechanisms underlying the two NEPC phenotypes. Epigenetic regulation is implicated in ADT resistance, cellular plasticity, and NE phenotype (21–24). To identify novel epigenetic drivers for NEPC, we examined 147 epigenetic regulators in several well-cited public NEPC datasets (20,23,24,39–42). We found that histone deacetylase 2 (HDAC2) is the second most upregulated epigenetic regulators and the most-induced HDAC in NE+ versus non-NE tumors in mouse and patient prostate tumors (Fig. 1C–D). HDAC acts as an epigenetic enzyme for the de-acetylation of histone, suppressing gene expression. Interestingly, expression of HDAC1, the closest sibling of HDAC2, is not significantly elevated in NE+ versus NE− CRPC (Fig. 1D).
We speculated that HDAC2 contributes to aggressive phenotypes of NEPC, such as NED and/or angiogenesis. Indeed, overexpressing HDAC2 upregulates NE markers in CRPC cells 22RV1 (Fig. 1E). On the other hand, silencing HDAC2 in NEPC cells 144–13 represses NE markers SYP, TUBB3, and ENO2 (Fig. 1F). We observed similar result in another NEPC cell line NE1.3 (Supplementary Fig. S1A). In addition, wound healing of endothelial cells exposed to conditioned medium from NEPC cells 144–13 is decreased upon silencing HDAC2 (Fig. 1G). Together, these results indicate that HDAC2 is critical in regulating angiogenesis and NE phenotypes in prostate cancer cells.
REST and TSP1 are expressed lower in NEPC and are direct HDAC2-repressed targets
We next investigated HDAC2 mechanisms in regulating NE and angiogenesis. Since it is mainly a transcriptional repressor, we focused on genes downregulated in NEPC, from which we nominate master NE repressor REST and potent anti-angiogenic protein TSP1 as potential HDAC2 targets in NEPC. We previously showed that TSP1 expression is lower in NE+CRPC than in NE-CRPC (33). Here we showed that REST is also expressed at lower level in NE+CRPC than in NE-CRPC (Fig. 2A), which is consistent with reports by others (44,45).
Figure 2. Suppressed in NEPC, REST and TSP1 are direct HDAC2-repressed genes.
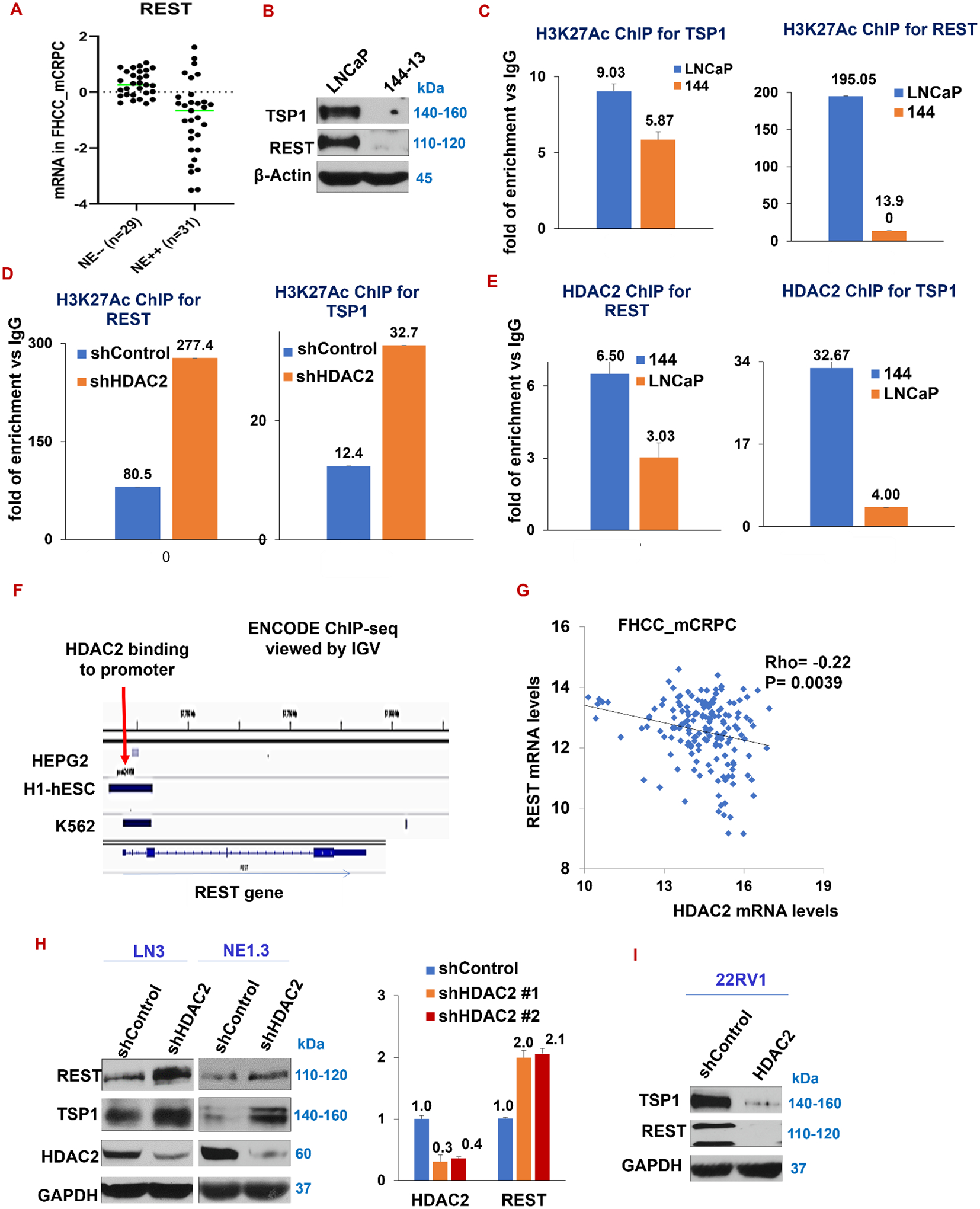
(A) Expression of REST is lower in NE++ (high) patients compared to NE−− (low) patients. A prostate tumor is called NE++ if its average expression of 3 NE makers (CHGA, TUBB3 and ENO2) are at least one SD above the average of these 3 NE markers in all tumors. A NE−− tumor is that with at least one SD below the average (FHCC_mCRPC_GSE77930 dataset). (B) TSP1 and REST protein expression in LNCaP versus 144–13. (C-D) The levels of H3K27 acetylation marks on the promoters of REST and TSP1 were measured using H3K27Ac ChIP assay on LNCaP and 144–13 cells (C), and in 144–13 shControl cells vs shHDAC2 cells (D). The levels of H3K27Ac on REST and TSP1 promoters were calculated and plotted as folds of enrichment over the IgG control. (E) The levels of HDAC2’s binding on promoter regions of REST and TSP1 in LNCaP and 144–13 cells using ChIP assay. (F) Analysis of the ENCODE ChIP-seq database reveals different human cell lines with HDAC2 binding at the REST promoter region. (G) Expression of REST and HDAC2 negatively correlates in FHCC_mCRPC (P=0.0039) (data downloaded from cBioPortal). (H) Western blotting (left panel) and RT-qPCR (right panel) show upregulation of REST and TSP1 in HDAC2-silenced NE1.3, LN3, and VCaP prostate cancer cells. In RT-qPCR, GAPDH was the loading control. (I) Western blotting of REST and TSP1 in 22RV1 cells upon overexpression of HDAC2 wild type cDNA. Representative DNA gel images for ChIP-PCR herein are in Supplementary Fig. S7.
As expected, compared with ADPC cells LNCaP, the expression of REST and TSP1 is reduced in NEPC cells 144–13 (Fig. 2B). To determine a potential involvement of epigenetic repression of REST and TSP1 mRNA expression in NEPC, we measured levels of the most commonly examined histone mark for transcriptional activation, H3K27Ac, in the vicinity of promoters of REST and TSP1 in ADPC and NEPC cell models. Chromatin immunoprecipitation (ChIP) using anti-H3K27Ac antibody followed by qPCR of TSP1 and REST promoter sequences indicated that H3K27Ac mark is lower at REST and TSP1 promoters in NEPC cells 144–13 than in ADPC cells LNCaP (Fig. 2C). H3K27Ac level is regulated by several HDACs and other epigenetic regulators (46). In order to determine a specific contribution of HDAC2 on the reduction of H3K27Ac levels in NEPC cells, we silenced HDAC2 in 144–13 cells, followed by the same H3K27Ac ChIP-qPCR procedure. Supporting our hypothesis that HDAC2 represses REST and TSP1 expression, H3K27 acetylation around their promoters increased upon silencing HDAC2 in NEPC cells (Fig. 2D).
Using anti-HDAC2 antibody in ChIP-PCR, we found that the direct binding of HDAC2 binding to promoter regions of TSP1 and REST increases in NEPC cells 144–13 (Fig. 2E), suggesting that REST and TSP1 are HDAC2 targets in NE+ prostate cancer cells. In line with this result, we further found that HDAC2 binds to promoter region of REST in several human cell lines in the ENCODE ChIP-seq database (Fig. 2F). Moreover, expression of HDAC2 and REST negatively correlate with each other in mCRPC patient samples (FHCC_mCRPC_GSE77930) (Fig. 2G).
Further, silencing HDAC2 in NEPC cell line NE1.3 and CRPC cell line LN3 induces REST and TSP1 (Fig. 2H). mRNA expression of REST is significantly increased upon silencing HDAC2 with two different shRNAs (Fig. 2H and Supplementary Fig. S1B for 144–13 cell). On the other hand, overexpressing HDAC2 in 22RV1 cells reduces TSP1 and REST expression (Fig. 2I). Collectively, these data indicate that suppression of TSP1 and REST expression in NEPC cells probably occurs through epigenetic regulation with H3K27 de-acetylation by the elevated level of HDAC2 in NEPC cells. We then asked what may promote HDAC2’s suppression of TSP1 and REST in NEPC. We previously reported in two separate studies that G protein-coupled receptor kinase 3 (GRK3) as a new critical kinase in prostate cancer, inducing angiogenesis, NE marker expression, and ADT resistance (25,28). The mechanism underlying GRK3’s role in these processes was largely unknown.
We have also shown, in different biological contexts, that CREB1 activation is critical for ADT-induced NE differentiation and chronic stress-induced angiogenesis, respectively (33,47). Importantly, CREB1 activation induces GRK3 and HDAC2, in these two separate contexts(33,47). GRK3 and HDAC2 both repress TSP1 and promote angiogenesis (25,47), as well as induce NE marker expression (28) (Fig. 1–2 for HDAC2). We therefore hypothesized that GRK3 and HDAC2 are functionally linked in NEPC, and GRK3 may promote HDAC2’s functions.
Induced by ADT and in NEPC, GRK3 is essential for NEPC cells, especially in ADT condition
Indeed, GRK3 expression is higher in NE positive CRPC compared to NE negative CRPC (FHCC_mCRPC dataset (40) (Fig. 3A). Moreover, GRK3 expression is higher, while REST and TSP1 expression are lower, in relapsed NEPC PDX tumor compared to pre-relapsed ADPC PDX tumor in the LTL331 PDX model(39)(Fig. 3B). In line with these genomics data, GRK3 expression is upregulated in NEPC cells 144–13 compared to ADPC cells LNCaP (Fig. 3C).
Figure 3. Induced in NEPC, GRK3 is essential for survival and NED in prostate cancer cells and for in vitro angiogenesis.
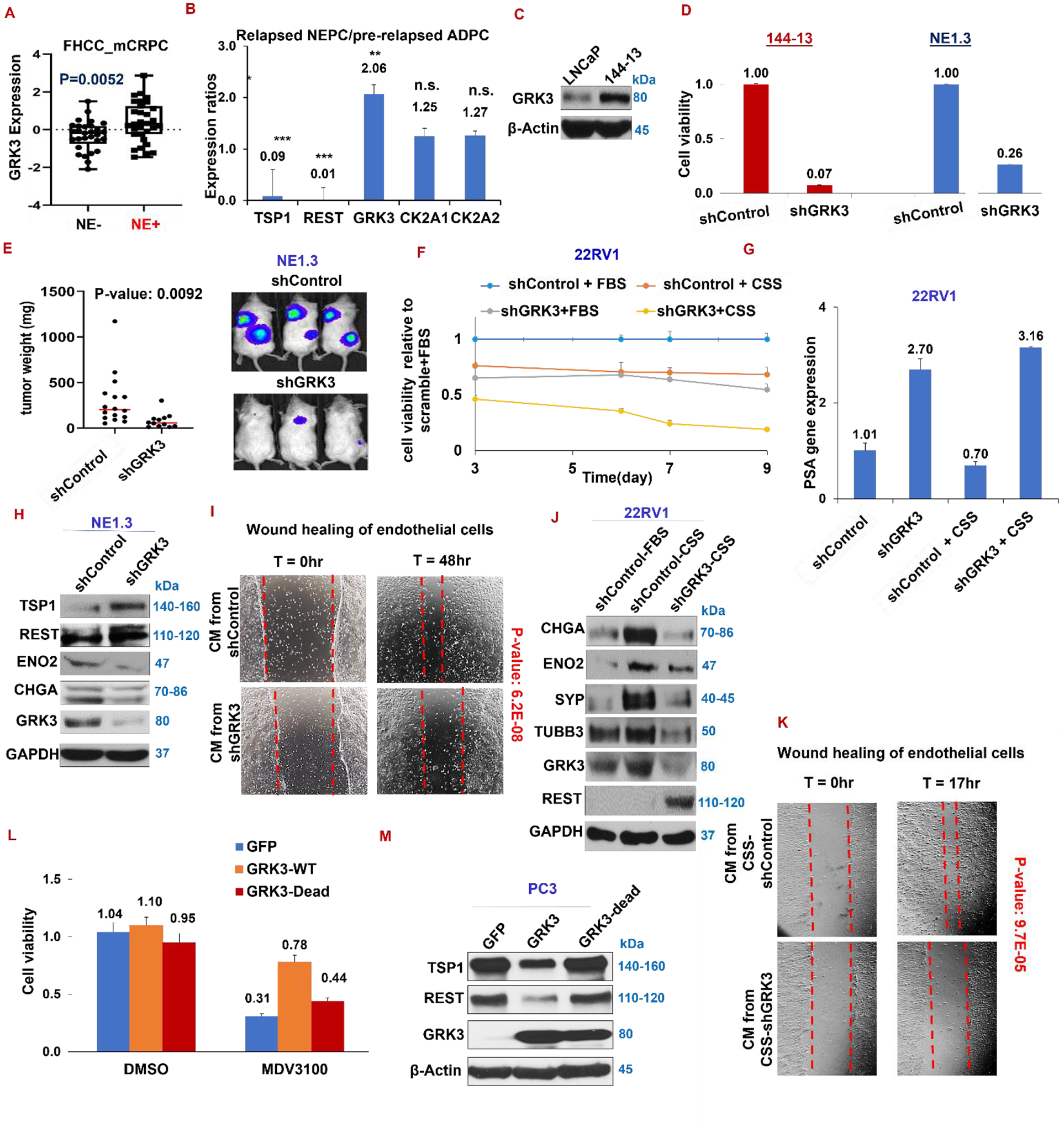
(A) Expression of GRK3 in NE+ and NE− prostate cancer, P=0.0052 (FHCC_mCRPC). (B) Expression of GRK3, two isoforms of CK2, REST and TSP1 in relapsed NEPC LTL331R PDX versus pre-relapsed ADPC PDX LTL331, from mining genomics dataset of LTL331 model (39). (C) Western blotting for GRK3 in ADPC cells LNCaP and NEPC cells 144–13. (D) Cell viability of NEPC cells 144–13 and NE1.3 expressing either shControl or shGRK3. (E) NE1.3 cells labelled with luciferase and expressing shControl or shGRK3 were implanted into flanks of NOD/SCID mice (6–8 mice per group, two implantations per mouse). (F) Cell viability of 22RV1 cells expressing either doxycycline (DOX)-inducible shControl or shGRK3 with 9 days in CSS and in DOX treatment (0.4 μg/ml). Medium was changed every 2–3 days to replenish DOX. (G) qPCR for PSA mRNA levels in 22RV1 cells with and without GRK3 and CSS. (H) Western blotting of TSP1 and REST in NE1.3 shControl and shGRK3. (I) Wound healing of endothelial cells treated with conditioned medium from 144–13 expressing shControl or shGRK3. (J) Western blotting of indicated proteins in 22RV1 cells expression shControl or shGRK3 in FBS or CSS media. (K) Wound healing of endothelial cells in conditioned medium from 22RV1 shControl or shGRK3 cells treated with CSS. (L) Cell viability of PC3 cells expressing either GFP, GRK3 wild-type, or GRK3 kinase-dead cDNA after treatment in DMSO or MDV3100 (enzalutamide, 10uM) for 4 days. (M) Western blotting of TSP1 and REST in PC3 cells expressing GRK3 wild-type and GRK3 kinase-dead cDNA.
To determine whether GRK3 is essential for NEPC cells, we silenced GRK3 in two NEPC cell lines (NE1.3 and 144–13 cells), where its silencing significantly reduces cell viability (Fig. 3D). Of note, silencing GRK3 also inhibits the growth of NE1.3 cells in subcutaneous xenograft tumors (Fig. 3E). Silencing GRK3 in combination with CSS significantly inhibits cell growth in CRPC cells 22RV1 (Fig. 3F). Interestingly, the mRNA expression of Prostate Specific Antigen (PSA), a classic AR target and an AR signaling readout, was considerably enhanced after silencing GRK3, while reduced after growing in media with CSS (ADT-like) (Fig. 3G). Molecularly, silencing GRK3 increases TSP1 and REST expression in NE1.3 cells, as expected (Fig. 3H).
We also found that conditioned medium from 144–13-shGRK3 cells had worse wound healing effect of endothelial cells than that from 144–13- shControl cells (Fig. 3I). Moreover, GRK3 silencing prevents NE marker induction by CSS (ADT-like) (Fig. 3J for 22RV1 cells and Supplementary Fig. S2A for C4–2). GRK3 silencing in 22RV1 cancer cells also reduces cancer cells’ ability to enhance endothelial in vitro angiogenesis (Fig. 3K in CSS media and Supplementary Fig. S2B in FBS media). All these results indicate that GRK3 is essential for NEPC cell survival in culture, growth in mouse xenograft, as well as ADT-induction of NED and angiogenesis.
We then examined whether GRK3 overexpression is sufficient to promote ADT resistance, NED and angiogenesis, and whether its kinase activity is required. We found that overexpression of GRK3 wild-type cDNA, but not GRK3 kinase-dead cDNA, promotes resistance to ADT drug MDV3100 in LNCaP cells (Fig. 3L). Molecularly, GRK3 wild-type cDNA represses REST and TSP1 in PC3 cells that express adequate amount of these two proteins, which is not the case when overexpressing GRK3 kinase-dead cDNA (Fig. 3M). These results indicate that GRK3 overexpression alone is sufficient to induce NED and angiogenesis in prostate cancer cells, and, importantly, its actions largely depend on its kinase activity.
HDAC2 is essential for GRK3’s repressing TSP1 and REST and promoting angiogenesis and NE phenotypes
GRK3’s mechanism and its kinase substrates in prostate cancer are unknown. We hypothesized that GRK3 suppresses the two NEPC repressors REST and TSP1 by activating HDAC2, and this pathway is a key missing link between neuroendocrine phenotype and angiogenesis. We first showed that the expression of GRK3 positively correlates with HDAC2 expression in TCGA_PCa (Spearman correlation coefficient Rho=0.27, P=4.82E-10), and in Taylor_PCa (48) with primary and metastatic tumors (Rho = 0.28, P =0.00065) (Fig. 4A). Secondly, in FHCC_mCRPC dataset, NE markers ENO2 and TUBB3 positively correlate with HDAC2 and GRK3, but not with HDAC1, the closest relative of HDAC2 in the HDAC family (Fig. 4B).
Figure 4. HDAC2 is critical for GRK3’s functions in PCa cells.
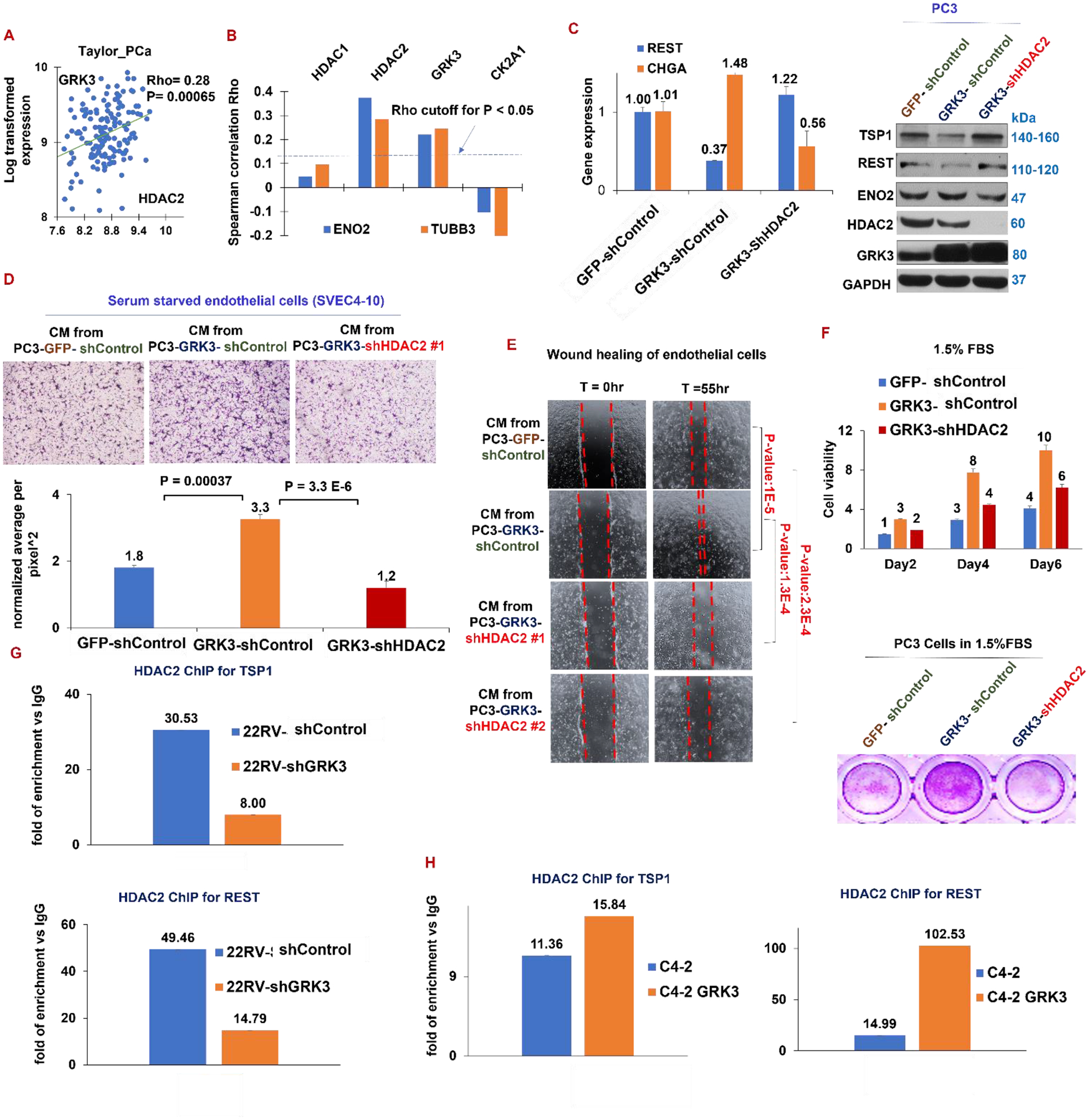
(A) Expression of GRK3 and HDAC2 positively correlates in prostate cancers (Taylor_PCa dataset GSE21034). (B) Plotted on Y axis are the Spearman correlation coefficients Rho for the indicated genes with NE markers ENO2 and TUBB3 in FHCC_mCRPC GSE77930 dataset. The dash line indicated the minimal Rho value that reaches statistical significance of P<0.05. (C) qPCR of indicated genes (left) and western blotting of indicated proteins (right) for PC3-GFP, PC3-GRK3 cDNA and PC3-GRK3 cDNA + shHDA2 cell lines. (D) Top panel: endothelial cell migration assay toward conditioned medium from the three PC3 cell lines. Bottom panel: quantitative analysis of migration assay using ImageJ. (E) Endothelial cell wound healing assay in conditioned medium from the three PC3 cell lines. (F) Six-day proliferation assay of the indicated PC3 cell lines in RPMI medium containing 1.5% FBS. (G-H) HDAC2 ChIP followed by qPCR of REST and TSP1 promoters on 22RV1 shControl and shGRK3 cells (G), and on C4–2 parental cells and C4–2-GRK3 cDNA cells (H). Representative DNA gel images for ChIP-PCR herein are in Supplementary Fig. S7.
Thirdly, induction of NE markers and reduction of TSP1 and REST by GRK3 cDNA was reversed after silencing HDAC2 in GRK3-overexpressing cells (Fig. 4C). In endothelial cell migration and wound healing assays for in vitro angiogenesis, migration of endothelial cells toward conditioned medium from PC3-GRK3 cDNA cells is higher than that from PC3-GFP cells, which is reversed by conditioned medium from PC3-GRK3-shHDAC2 cells (Fig. 4D). Same conclusions could be drawn from wound healing of endothelial cells grown in these conditioned media (Fig. 4E). Moreover, PC3 cell proliferation was enhanced by overexpressing GRK3 cDNA and then decreased with silencing HDAC2, when the cells grow in 1.5% or 6% FBS for 6 days (Fig. 4F and Supplementary Fig. S3A).
We next genetically modulated GRK3, by cDNA or shRNA, followed by HDAC2-ChIP and qPCR of promoter sequences of REST and TSP1. As showed in Fig. 4G, HDAC2’s binding to the promoters of TSP1 and REST in 22RV1 cells is reduced after silencing GRK3. We had similar observations in 144–13 cells (Supplementary Fig. S3B). On the other hand, HDAC2’s binding to the two promoters is elevated by overexpressing GRK3 in C4–2 cells (Fig. 4H). These observations indicate that GRK3 dictates HDAC2’s repression of REST and TSP1, and HDAC2 is critical for GRK3’s functions in regulating REST and TSP1 expression, as well as NE markers and angiogenesis.
GRK3 interacts with HDAC2 and phosphorylates HDAC2 at S394 to enhance its epigenetic activity
We next set out to further dissect the mechanism underlying GRK3’s regulation of HDAC2 suppression of REST and TSP1 expression. Using immunoprecipitation (IP), we first found that GRK3 physically interacts with HDAC2 in PC3 cells overexpressing GRK3, but not with its closest sibling protein, HDAC1 (Fig. 5A), and GRK3’s interaction with HDAC2 is increased upon HDAC2 overexpression (Fig. 5B). Moreover, endogenous GRK3’s interaction with endogenous HDAC2 is increased in NEPC cells 144–13, compared to ADPC cells LNCaP (Fig. 5C).
Figure 5. GRK3 phosphorylates HDAC2 at S394 to suppress TSP1 and REST expression.
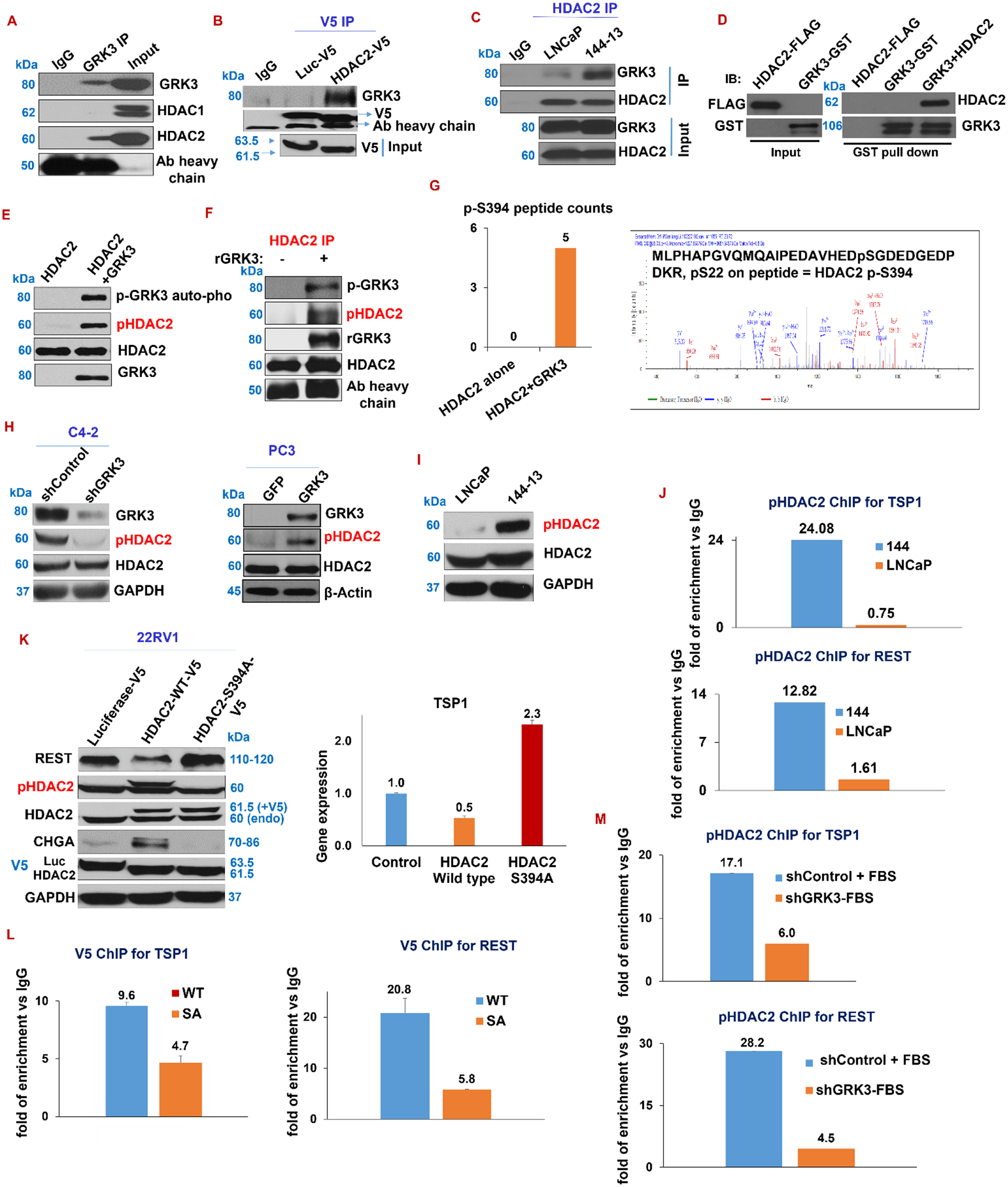
(A-C) Immunoprecipitation (IP) assays: A) GRK3 IP on PC3-GRK3 cells. B) IP with V5 antibody against V5 fusion with luciferase (control) or HDAC2 cDNA in 22RV1 cells. C) HDAC2 IP on 144–13 and LNCaP cell lysates, followed by western blotting. To diminish inference from the factor of higher GRK3 protein level in 144–13 cells than in LNCaP cells in the interaction strength of HDAC2-GRK3 in the two cell lines, ~25% less lysate from 144–13 than from LNCaP cells was used in this immunoprecipitation. (D) GST-pull down assay of recombinant HDAC2 and GRK3 proteins, followed by western blotting. (E-F) Kinase assay with GRK3 recombinant proteins with either HDAC2 recombinant proteins (E), or HDAC2 proteins immunoprecipitated from NEPC cells 144–13 (F). (G) Mass spectrometry: Left panel: Y-axis is the number of peptides containing p-S394 phosphorylation from Mass Spec, comparing HDAC2 protein alone and HDAC2 protein admixed with GRK3 protein, followed by kinase assay and mass spectrometry. Right panel: the spectrum contains the p-S394-HDAC2 peptide from HDAC2 + GRK3 protein mixture. (H) Western blotting of indicated protein after silencing and overexpressing GRK3 in C4–2 and PC3 cells, respectively. (I) Western blotting of HDAC2 and p-S394-HDAC2 (referred as pHDAC2 in this study) in LNCaP and 144–13 cells. (J) ChIP with p-S394-HDAC2 Ab, followed by qPCR on REST and TSP1 promoter sequences from LNCaP and 144–13 cells. (K) Measure the expression of REST, TSP1, p-S394-HDAC2, CHGA, V5, and HDAC2 in 22RV1 cells expressing V5-tagged Luciferase (control), HDAC2 wild-type, and HDAC2 S394A mutant cDNA. (L-M) ChIP and qPCR on TSP1 and REST promoters: L) V5 ChIP on NE1.3 cells overexpressing V5 tagged wild-type HDAC2, and S394A mutated HDAC2; M) p-S394-HDAC2 ChIP on 22RV1 expressing shControl or shGRK3. Representative DNA gel images for ChIP-PCR herein are in Supplementary Fig. S7.
Since GRK3’s kinase activity and HDAC2 are required for GRK3’s actions in repressing REST and TSP1, as well as in inducing NE markers, we speculated that GRK3 phosphorylates and activates HDAC2. Firstly, through GST-pull down assay of purified recombinant FLAG-HDAC2 and GST-GRK3 proteins, we showed that HDAC2 directly interacts with GRK3 protein (Fig. 5D). Secondly, through in vitro kinase assay with the two recombinant proteins, we demonstrated that GRK3 protein phosphorylates HDAC2 protein (Fig. 5E). Using a similar kinase assay, we further showed that GRK3 protein phosphorylate endogenous HDAC2 protein that was IPed from NEPC cells 144–13 (Fig. 5F). Thirdly, through mass spectrometry analyses, we consistently found that only S394 of HDAC2 (Uniprot ID Q92769) is phosphorylated when HDAC2 proteins are co-incubated with GRK3 in kinase assays (Fig. 5G). Fourthly, using commercial antibodies for p-S394-HDAC2 (referred as pHDAC2 in this study), we showed that GRK3 overexpression increases, while its silencing reduces, pHDAC2 level in PCa cells (Fig. 5H). Finally, pS394-HDAC2 protein level and its binding to the two promoters are higher in NEPC 144–13 cells than in LNCaP cells (Fig. 5I–J), implying that suppression of TSP1 and REST in NEPC is, at least in part, mediated by S394-phosphorylated HDAC2.
GRK3 suppresses HDAC2’s target gene through phosphorylation
To further investigate the functionality of HDAC2 phosphorylation of S394 site by GRK3, we performed point mutation for the 394-amino acid from serine to alanine (S394A, SA, non-phophorylatable) (49,50) to study “loss of function” on HDAC2 S394 phosphorylation. TSP1 and REST are decreased, while pHDAC2 is increased, by overexpressing HDAC2-WT with V5 tag, which did not occur when overexpressing HDAC2-SA with V5 tag (Fig. 5K and Supplementary Fig. S4A). As shown earlier in Fig. 4C, phosphorylation of HDAC2 is increased by overexpressing GRK3, which is then reduced by silencing HDAC2 on top of GRK3 overexpression (Fig. 4C). This result is in line with results from V5 ChIP, which shows that binding of V5-HDAC2 to the promoter of TSP1 and REST is lower when overexpressing HDAC2-SA mutant than overexpressing HDAC2-WT protein in NE1.3 (Fig. 5L). Furthermore, binding of pHDAC2 to TSP1 and REST promoters is significantly reduced after silencing GRK3 (Fig. 5M and Supplementary Fig. S7), while this binding is increased by GRK3-overexpression, but not by overexpressing GRK3 kinase-dead cDNA (Supplementary Fig. S4B).
GRK3 is the major kinase phosphorylating HDAC2 and regulating angiogenesis and NE in advanced PCa
Casein kinase II (CK2) is the kinase previously reported in the literature to phosphorylate HDAC2 at S394, such as in heart, Hela cells, and bronchial epithelial cells (49–52). We next asked whether GRK3 or CK2A1 (the main isoform of CK2) is the major kinase phosphorylating HDAC2 and regulating angiogenesis and neuroendocrine differentiation in prostate cancer. In contrast to GRK3, CK2A1 expression is not increased in metastatic prostate tumors compared to primary prostate tumors in patients’ samples (Fig. 6A). Similarly, GRK3 is, while CK2A1 is not, expressed higher in prostate tumors from NEPC GEMM TRAMP mice than in ADPC GEMM Hi-Myc mice or wild-type mice (Fig. 6B) (53). Consistent with these data, CK2A1 expression negatively correlates with the expression of NE markers ENO2 and TUBB3 in FHCC_mCRPC (40) (Fig. 4B).
Figure 6. GRK3, not CK2, suppresses TSP1 and REST through HDAC2 phosphorylation.
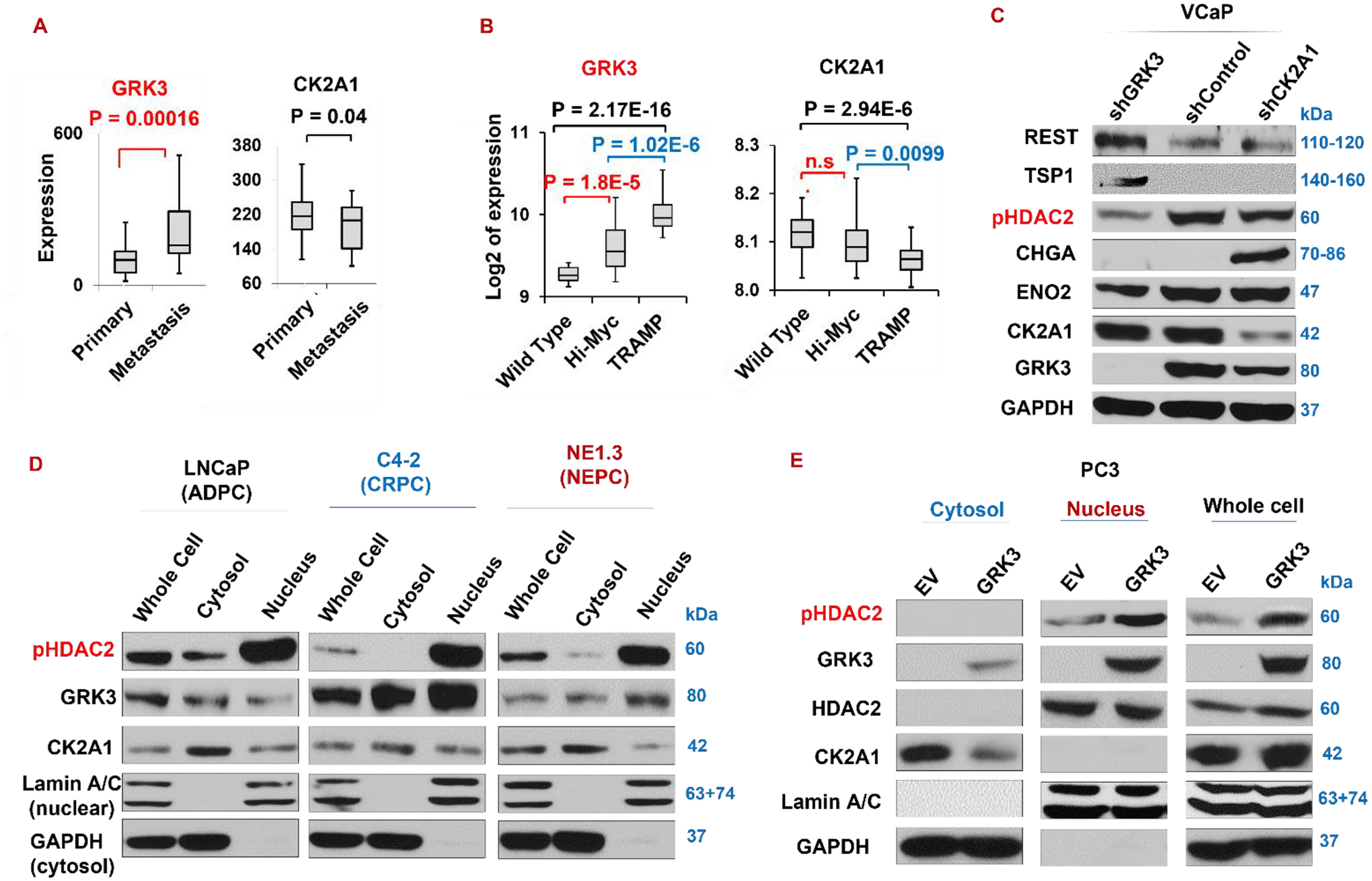
(A) Expression of GRK3, but not CK2’s main isoform CK2A1, is significantly higher in metastasis than in primary prostate cancer (Yu_Prostate, GSE6919). (B) GRK3 and CK2A1 expression in NEPC TRAMP prostate tumors, ADPC Hi-Myc prostate tumors, and normal prostate tissues from wild-type mice (Abate-Shen_GEM_Prostate, GSE92721). (C) Western blotting of indicated proteins on VCaP cells silencing either GRK3 or CK2A1. (D-E) Nuclear and cytosol localizations of GRK3, CK2A1, and p-S394-HDAC2 proteins in three prostate cancer cell lines LNCaP, NE1.3, and C4–2 (D), and in PC3-empty vector (EV) and PC3-GRK3 cells (E).
As expected, silencing GRK3 in VCaP cells substantially reduces S394 phosphorylation of HDAC2, however, silencing CK2A1 does not reduce S394 phosphorylation of HDAC2 as much. Of note, TSP1 and REST are induced, while NE marker ENO2 is reduced, by shGRK3, but not by shCK2A1 (Fig. 6C and Supplementary Fig. S5).
We next set out to check the cytoplasmic and nuclear distributions of pHDAC2, GRK3 and CK2. We found that pHDAC2 is localized mainly in nucleus (the site of its epigenetic functions) in ADPC (LNCaP) and CRPC cells (NE1.3 and C4–2) that we examined. Interestingly, some GRK3 proteins were found in nucleus, and its nuclear proportion is increased in CRPC cells vs ADPC cell LNCaP (Fig. 6D). Unlike GRK3, CK2A1 protein level in nuclear is evidently lower than in cytosol in all three cell lines. In addition, when GRK3 is overexpressed in CRPC cell line PC3, pHDAC2 level in the nucleus is increased (Fig. 6E), suggesting that GRK3 increases pHDAC2 levels in nucleus, either by promoting pHDAC2 nuclear translocation upon phosphorylation or enhancing pHDAC2 phosphorylation directly in the nucleus.
REST and TSP1 synergize in repressing angiogenesis and NE phenotypes
From above experiments, we have been observing direct connections of NED and angiogenesis in prostate cancer through the GRK3-HDAC2 axis. Since REST and TSP1 are master repressors for NED and angiogenesis, respectively, we suspected that they have a close relationship too. Firstly, in TCGA_PCa dataset, we found that expression of REST and TSP1 positively correlates (P = 9.1E-20) (Fig. 7A). Positive correlation was also observed in other prostate cancer genomics dataset, e.g. Taylor_PCa (Fig. 7B). Previously, we showed that silencing anti-angiogenic protein TSP1 induces NE markers in PC3 cells (33). Here we further showed that overexpressing TSP1 reduces NE marker expression in 22RV1 cells (Fig. 7C).
Figure 7. REST inhibits angiogenesis by inducing TSP1 which represses NE markers in prostate cancer cells.
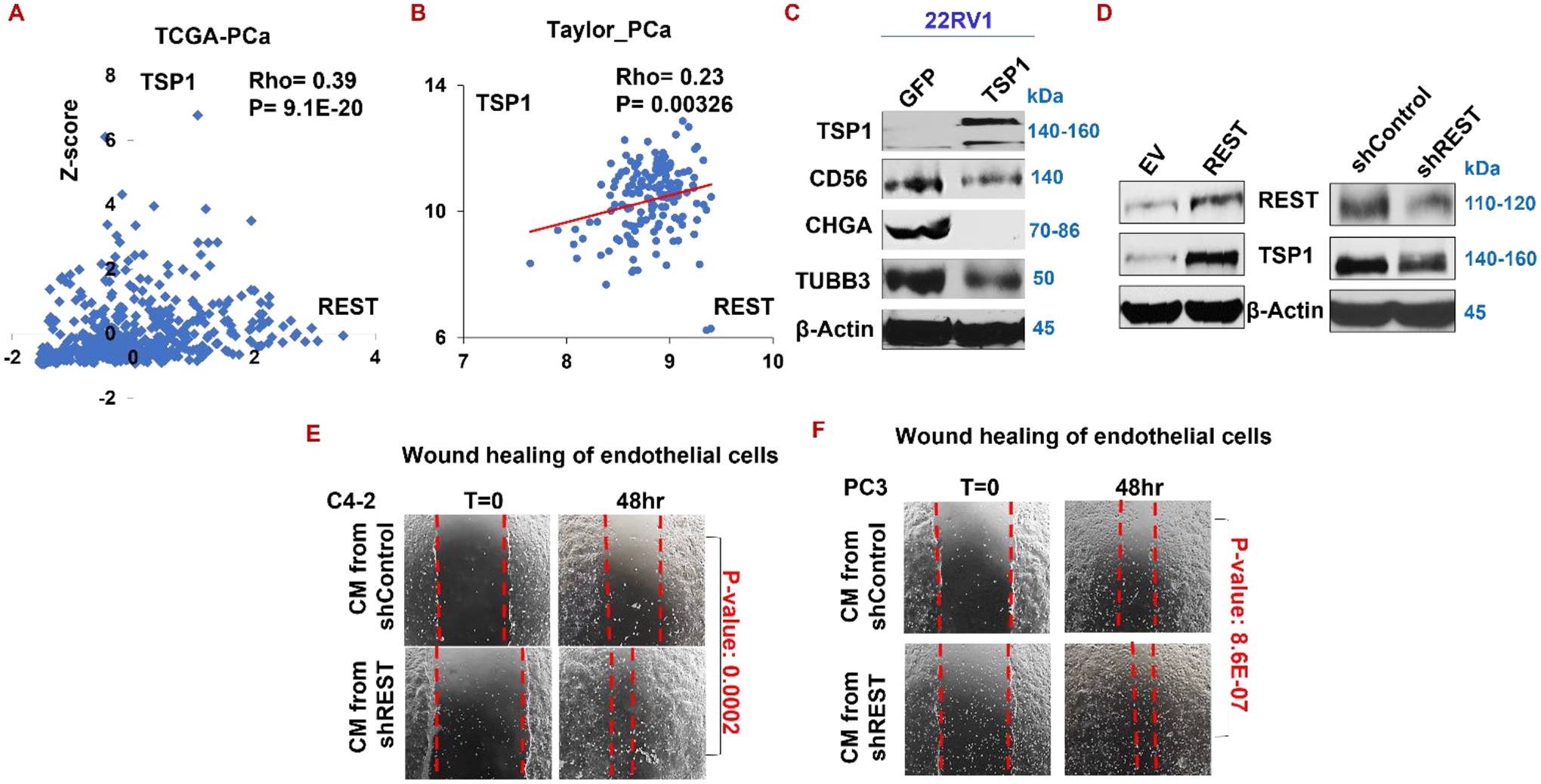
(A-B) Analyzing TCGA_PCa (A) and Taylor_PCa (B) patient dataset reveals that expression of REST and TSP1 positively correlates. (C) Western blotting of TSP1 and NE markers in 22RV1 cell overexpressing TSP1. (D) Western blotting of REST, TSP1, and NE markers upon silencing or overexpressing REST. (E-F) Wound healing of endothelial cells in conditioned medium from C4–2 (E) and PC3 (F) expressing shControl or shREST.
Interestingly, silencing REST downregulates TSP1, while overexpressing REST induced TSP1 in PC3 cells, as shown in Fig. 7D. Concordantly, silencing REST in prostate cancer cells C4–2 and PC3 significantly increases wound healing capability of endothelial cells (Fig. 7E–F), indicating that REST represses angiogenesis. These data establish a novel role of REST in angiogenesis and indicate that REST positively regulates TSP1, constituting a positive feedback loop in regulating NE and angiogenesis.
The GRK3-HDAC2 axis is activated under stress conditions
The literature indicates that pathobiologic stresses, such as ADT and hypoxia, promote NEPC progression (6,12,54). We next determine whether these stresses activate GRK3-HDAC2 signaling. As expected, ADT (by growing cells in media with charcoal stripped serum), hypoxia (by growing cells for 24 hours in a hypoxia chamber with no oxygen) or low FBS induced NE markers (Fig. 8A–B, and Supplementary Fig. S6). We also found that ADT and hypoxia increased GRK3 and pHDAC2 protein levels, decreased the level of REST and TSP1 (Fig. 8A–B and Supplementary Fig. S6). Interestingly, based on our ChIP result, ADT induces binding of HDAC2 to the promoter regions of REST and TSP1 (Fig. 8C). Altogether, these data suggest that GRK3-HDAC2 pathway is a critical pathway activated during NEPC progression induced by ADT and hypoxia.
Figure 8. GRK3-HDAC2 axis is activated under hypoxia and ADT conditions.
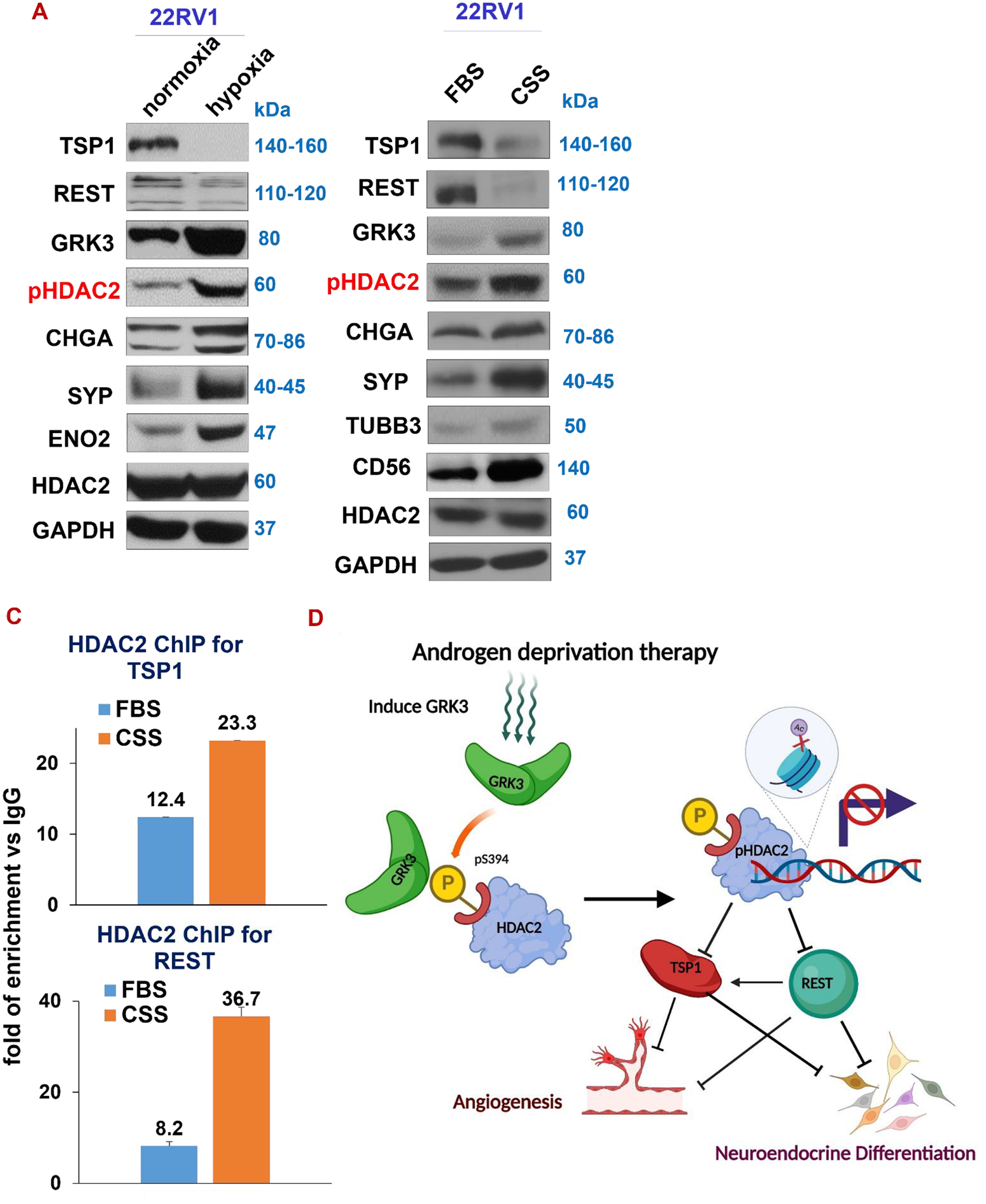
(A) Western blotting of indicated proteins in cells cultured in a hypoxia chamber with 0% oxygen for 24hr. (B) Western blotting for cells growing in CSS-containing media for 72hr. (C) HDAC2 ChIP, followed by qPCR of REST and TSP1 promoter DNAs in 22RV1 cells cultured in FBS or CSS. (D) Illustrative model for the mechanism underlying GRK3-HDAC2 axis in linking angiogenesis and neuroendocrine in prostate cancer cells. The schematic diagram has been draw using BioRender.
DISCUSSION
In this study, we discovered a key missing link connecting neuroendocrine differentiation and angiogenesis, two aggressive phenotypes in prostate cancer progression, as depicted in Fig. 8D. In this study, we reveal that NED and angiogenesis converge on GRK3-mediated phosphorylation and activation of HDAC2 which epigenetically represses REST and TSP1, two proteins synergizing in repressing the two phenotypes.
Epigenetic regulation is implicated in drug resistance, cellular plasticity and neuroendocrine differentiation in prostate and other cancer types (21,55,56). For example, Ku et al. reported that EZH2, an epigenetic regulator that is induced in NEPC and by loss of tumor suppressors Rb and p53, critically contributes to these phenotypes in prostate cancer (24). We also reported that EZH2 activity is enhanced by ADT-activated CREB1 signaling, which induces angiogenesis and NED in prostate cancer cells (33). It is still unknown how CREB1 signaling enhances EZH2 activity and how EZH2 induces NED in prostate cancer cells, two questions that we are actively investigating. Our current study reveals a key direct molecular link underlying the intimate relationships between NED and angiogenesis, and between two NEPC master suppressors REST and TSP1.
HDACs play crucial roles in a number of cellular processes and pathological conditions (57,58). Weichert et al. reported that HDAC2 is the most upregulated class I HDACs during PCa progression (59). We previously showed that HDAC2 is critical for prostate cancer progression induced by chronic bio-behavioral stress (47). Here we further showed that HDAC2 upregulation in NE versus non-NE tumors is the most pronounced among HDACs, and that its silencing reduces NE marker expression (Fig. 1), which suggests that HDAC2 is particularly important for NEPC cells. We therefore focused on HDAC2 for our study of HDACs in NEPC. However, some other HDACs may also contribute critically to NEPC progression, besides HDAC2. It is still important and necessary to determine the roles of each HDACs in NEPC progression, maybe through silencing each HDAC individually and specifically.
The majority of studies on the regulation of REST expression in cancer have been on its protein degradation or alternative splicing (44,60,61). RNA splicing factor serine/arginine repetitive matrix 3 and 4 (SRRM3 and SRRM4) regulates REST splicing and NE marker expression in prostate cancer (60). REST is also regulated through degradation by ubiquitin-mediated proteolysis (62). Few studies have shown transcriptional regulations responsible for its downregulation. Jin et al reported that KDM1A promotes small cell lung cancer (also NE tumor) progression by transcriptionally silencing REST (63). In this study, we reveal a somewhat surprising epigenetic regulator for REST’s downregulation in NEPC. It is conceivable that multiple of abovementioned mechanisms are in play to repress REST expression in NEPC. In NE-negative cells, REST and HDAC2 are usually considered partners in gene repression by recruiting CoREST and HDAC1 to genes with REST binding sites, such as CHGA, CHGB, and SYP (50,64). Our work uncovered that HDAC2 epigenetically suppresses REST in NEPC (Fig. 2). This result thus introduces a new concept in the HDAC2-REST relationship.
Endothelial cell migration, tube formation and wound healing assays are three commonly used in vitro angiogenesis assays. In our previous papers (33,47), we have used endothelial cell migration and tube formation assays to study the impact of prostate cancer cells on endothelial cells through regulating TSP1 expression in cancer cells. Here in the present study, we chose to demonstrate in vitro angiogenesis using one more assay, i.e. endothelial cell wound healing assay. Wound healing assay is a relatively simple and cost-effective in vitro model for studying some aspects of endothelial cell behavior during angiogenesis. It has been utilized in various research studies and has contributed to our understanding of angiogenesis and potential therapeutic targets (36). While it is not a direct measurement of angiogenesis (the formation of new blood vessels), it can provide valuable insights into the cellular processes involved in the early stages of angiogenesis, such as endothelial cell migration and proliferation.
GRK3 is considered a cytoplasmic protein in the literature, due to its canonical function in regulating GPCR signaling (65). From the literature, we could only find another study showing a nuclear localization of GRK3, where its nuclear abundance increased in NRK49F rat kidney fibroblasts during prolonged hypoxia (1% O2 for 12 hours) (66). Since GRK3 does not contain a DNA-binding nuclear localization sequence (NLS) (65), our data imply that GRK3 protein translocates to the nucleus with aid of some unknown protein(s). Given GRK3’s roles in regulating NEPC progression as showed in this study and in regulating GPCR signaling as its conventional function, it is of high interest to further study the mechanism and implications of its nuclear localization in the near future.
We provided compelling evidence that HDAC2 phosphorylation at S394 is functional and essential in HDAC2’s suppression of TSP1 and REST expression (Fig. 5). pHDAC2 level and NE markers are increased, while TSP1 and REST expression is decreased, after overexpressing HDAC2 wild-type cDNA, but not after overexpression S394 non-phosphorylatable mutant cDNA (S394A). In addition, binding of exogenous HDAC2-V5 fusion protein to the promoter region of REST and TSP1, assessed via anti-V5 ChIP, is lower in cancer cells expressing HDAC2-S394A-V5 than in cells expressing HDAC2-WT-V5. The roles of REST in repressing NE markers and TSP1 in suppressing angiogenesis are well established (67–70). Surprisingly, we found that REST positively regulates TSP1 and represses angiogenesis. On the other hand, TSP1 suppresses NE marker expression in prostate cancer. These results uncover novel functions of REST and TSP1. Our results suggest that REST and TSP1 synergize in repressing NED and angiogenesis. It is still unknown how TSP1 represses NE markers in prostate cancer cells, which warrants further investigation.
Hypoxia and drug resistance are significant obstacles in our tackle of epithelial tumors (71–73). Better understandings of them will not only expand our knowledge in epithelial tumor progression, but also offer valuable drug targets and therapeutics candidates to abate tumor progression. Therefore, in this study we also examined impacts of hypoxia and ADT on the GRK3-HDAC2 pathway that we uncovered. GRK3-HDAC2 pathway is activated in prostate cancer cells under hypoxia and ADT conditions, and binding of HDAC2’s to the promoters of TSP1 and REST increased under ADT. Secondly, silencing GRK3 is essential to prevent NE and angiogenesis induced by ADT. These data further highlight the importance of this pathway in aggressive phenotypes of prostate cancer, which suggests a potential combining of ADT with GRK3 silencing or inhibition to re-sensitize CRPC cells to ADT and to prevent/block NEPC progression.
While there have been some successes and FDA approvals of HDAC inhibitors (HDACi) for some types of blood cancers (74), HDAC inhibitors largely failed in the clinic for most major solid tumors (75). This is mainly because there is few subtype-specific HDACi while different HDACs may have different or even opposing functions in tumor biology (76–82). The roles of different HDACs in prostate cancer tumorigenesis and progression are still poorly understood (76). It is very intriguing that HDAC2 is the HDAC consistently having most increase in NEPC. Some other HDACs are downregulated in NEPCs as compared to adenocarcinomas, as shown in the volcano plots in Fig. 1D. For example, HDAC1, one of the closest HDAC to HDAC2, was downregulated in the LTL331R CRPC/NEPC PDX as compared to its pre-castration adenocarcinoma PDX model LTL331. In this study, we highlight the importance of specific targeting HDAC2, especially for NEPC. Unfortunately, no potent HDAC2-specific inhibitors are currently available. Targeting its upstream regulators may be an excellent alternative, such as inhibiting GRK3, a kinase belonging to a family of proteins that are known to be druggable. Thus, developing a potent and specific GRK3 inhibitor, alone or in combinations with ADT, is a promising approach to prevent or treat NEPC, and ultimately reduce or eliminate deaths from prostate cancer in patients.
Supplementary Material
Highlights:
Established novel roles of HDAC2, REST and TSP1 in prostate cancer progression.
GRK3 phosphorylates HDAC2 which promotes neuroendocrine phenotype and angiogenesis.
GRK3-HDAC2 pathway is a key missing link between the two prominent cancer phenotypes.
The GRK3 pathway mediates cancer progression by androgen deprivation and hypoxia.
Acknowledgement
We are grateful to Dr. Hyun Kook and Dr. Edward Seto for their generous gifts of HDAC2 cDNA plasmid (data are not included in this manuscript). We also would like to thank other members of the Li lab and Texas Therapeutics Institute for helpful discussions and assistance. We are also thankful to thesis committee members, Drs. Daniel Frigo, Mikhail Kolonin, Guangwei Du, Wenbo Li, and Rebecca Berdeaux, for guidance. We apologize to the colleagues whose works were not cited due to space constraint. This work was supported by a Rising STARS Award from University of Texas System, as well as awards from the Cancer Prevention and Research Institute of Texas (RP170330) and American Cancer Society (RSG-17-062-01), and NIH/NCI (1R01CA270096) to W. Li.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
No potential conflicts of interest were disclosed.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- 1.Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, Ayala G, et al. Aggressive variants of castration-resistant prostate cancer. Clinical Cancer Research. 2014;20(11):2846–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aparicio A, Logothetis CJ, Maity SN. Understanding the lethal variant of prostate cancer: power of examining extremes. Cancer Discov. 2011;1(6):466–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas G v, et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. Journal of Clinical Oncology. 2018;36(24):2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirano D, Okada Y, Minei S, Takimoto Y, Nemoto N. Neuroendocrine differentiation in hormone refractory prostate cancer following androgen deprivation therapy. Eur Urol. 2004;45(5):586–92. [DOI] [PubMed] [Google Scholar]
- 5.Papandreou CN, Daliani DD, Thall PF, Tu SM, Wang X, Reyes A, et al. Results of a phase II study with doxorubicin, etoposide, and cisplatin in patients with fully characterized small-cell carcinoma of the prostate. Journal of clinical oncology. 2002;20(14):3072–80. [DOI] [PubMed] [Google Scholar]
- 6.Beltran H, Tagawa ST, Park K, MacDonald T, Milowsky MI, Mosquera JM, et al. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J Clin Oncol. 2012;30(36):e386–9. [DOI] [PubMed] [Google Scholar]
- 7.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1(6):487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jongsma J, Oomen MH, Noordzij MA, van Weerden WM, Martens GJM, van der Kwast TH, et al. Kinetics of neuroendocrine differentiation in an androgen-dependent human prostate xenograft model. Am J Pathol. 1999;154(2):543–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Terry S, Beltran H. The many faces of neuroendocrine differentiation in prostate cancer progression. Front Oncol. 2014;4:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vlachostergios PJ, Papandreou CN. Targeting neuroendocrine prostate cancer: molecular and clinical perspectives. Front Oncol. 2015;5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conteduca V, Aieta M, Amadori D, de Giorgi U. Neuroendocrine differentiation in prostate cancer: current and emerging therapy strategies. Crit Rev Oncol Hematol. 2014;92(1):11–24. [DOI] [PubMed] [Google Scholar]
- 12.Wang HT, Yao YH, Li BG, Tang Y, Chang JW, Zhang J. Neuroendocrine prostate cancer (NEPC) progressing from conventional prostatic adenocarcinoma: factors associated with time to development of NEPC and survival from NEPC diagnosis—a systematic review and pooled analysis. Journal of Clinical Oncology. 2014;32(30):3383–90. [DOI] [PubMed] [Google Scholar]
- 13.Goodman OB, Flaig TW, Molina A, Mulders PFA, Fizazi K, Suttmann H, et al. Exploratory analysis of the visceral disease subgroup in a phase III study of abiraterone acetate in metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2014;17(1):34–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. New England Journal of Medicine. 2012;367(13):1187–97. [DOI] [PubMed] [Google Scholar]
- 15.Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clinical cancer research. 2014;20(4):890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grobholz R, Bohrer MH, Siegsmund M, Jünemann KP, Bleyl U, Woenckhaus M. Correlation between neovascularisation and neuroendocrine differentiation in prostatic carcinoma. Pathology-Research and Practice. 2000;196(5):277–84. [DOI] [PubMed] [Google Scholar]
- 17.Heinrich E, Trojan L, Friedrich D, Voß M, Weiss C, Michel MS, et al. Neuroendocrine tumor cells in prostate cancer: Evaluation of the neurosecretory products serotonin, bombesin, and gastrin–impact on angiogenesis and clinical follow‐up. Prostate. 2011;71(16):1752–8. [DOI] [PubMed] [Google Scholar]
- 18.Yazdani S, Kasajima A, Tamaki K, Nakamura Y, Fujishima F, Ohtsuka H, et al. Angiogenesis and vascular maturation in neuroendocrine tumors. Hum Pathol. 2014;45(4):866–74. [DOI] [PubMed] [Google Scholar]
- 19.Villaume K, Blanc M, Gouysse G, Walter T, Couderc C, Nejjari M, et al. VEGF secretion by neuroendocrine tumor cells is inhibited by octreotide and by inhibitors of the PI3K/AKT/mTOR pathway. Neuroendocrinology. 2010;91(3):268–78. [DOI] [PubMed] [Google Scholar]
- 20.Clermont PL, Lin D, Crea F, Wu R, Xue H, Wang Y, et al. Polycomb-mediated silencing in neuroendocrine prostate cancer. Clin Epigenetics. 2015;7(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ge R, Wang Z, Montironi R, Jiang Z, Cheng M, Santoni M, et al. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Annals of Oncology. 2020;31(4):470–9. [DOI] [PubMed] [Google Scholar]
- 22.Sheahan A v, Ellis L. Epigenetic reprogramming: A key mechanism driving therapeutic resistance. In: Urologic Oncology: Seminars and Original Investigations. Elsevier; 2018. p. 375–9. [DOI] [PubMed] [Google Scholar]
- 23.Davies A, Zoubeidi A, Selth LA. The epigenetic and transcriptional landscape of neuroendocrine prostate cancer. Endocr Relat Cancer. 2020;27(2):R35–50. [DOI] [PubMed] [Google Scholar]
- 24.Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science (1979). 2017;355(6320):78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li W, Ai N, Wang S, Bhattacharya N, Vrbanac V, Collins M, et al. GRK3 is essential for metastatic cells and promotes prostate tumor progression. Proceedings of the National Academy of Sciences. 2014;111(4):1521–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willets JM, Challiss RAJ, Nahorski SR. Non-visual GRKs: are we seeing the whole picture? Trends Pharmacol Sci. 2003;24(12):626–33. [DOI] [PubMed] [Google Scholar]
- 27.Watari K, Nakaya M, Kurose H. Multiple functions of G protein-coupled receptor kinases. J Mol Signal. 2014;9(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sang M, Hulsurkar M, Zhang X, Song H, Zheng D, Zhang Y, et al. GRK3 is a direct target of CREB activation and regulates neuroendocrine differentiation of prostate cancer cells. Oncotarget. 2016;7(29):45171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleb B, Estécio MRH, Zhang J, Tzelepi V, Chung W, Jelinek J, et al. Differentially methylated genes and androgen receptor re-expression in small cell prostate carcinomas. Epigenetics. 2016;11(3):184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pettaway CA, Pathak S, Greene G, Ramirez E, Wilson MR, Killion JJ, et al. Selection of highly metastatic variants of different human prostatic carcinomas using orthotopic implantation in nude mice. Clinical cancer research. 1996;2(9):1627–36. [PubMed] [Google Scholar]
- 31.Yuan TC, Veeramani S, Lin FF, Kondrikou D, Zelivianski S, Igawa T, et al. Androgen deprivation induces human prostate epithelial neuroendocrine differentiation of androgen-sensitive LNCaP cells. Endocr Relat Cancer. 2006;13(1):151–67. [DOI] [PubMed] [Google Scholar]
- 32.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124(6):1283–98. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Zheng D, Zhou T, Song H, Hulsurkar M, Su N, et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat Commun. 2018;9(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamakawa H, Cheng J, Penney J, Gao F, Rueda R, Wang J, et al. The transcription factor Sp3 cooperates with HDAC2 to regulate synaptic function and plasticity in neurons. Cell Rep. 2017;20(6):1319–34. [DOI] [PubMed] [Google Scholar]
- 35.Allen JJ, Li M, Brinkworth CS, Paulson JL, Wang D, Hübner A, et al. A semisynthetic epitope for kinase substrates. Nat Methods. 2007;4(6):511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nareshkumar RN, Sulochana KN, Coral K. Inhibition of angiogenesis in endothelial cells by Human Lysyl oxidase propeptide. Sci Rep. 2018;8(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seo HR, Jeong HE, Joo HJ, Choi SC, Park CY, Kim JH, et al. Intrinsic FGF2 and FGF5 promotes angiogenesis of human aortic endothelial cells in 3D microfluidic angiogenesis system. Sci Rep. 2016;6(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akamatsu S, Wyatt AW, Lin D, Lysakowski S, Zhang F, Kim S, et al. The placental gene PEG10 promotes progression of neuroendocrine prostate cancer. Cell Rep. 2015;12(6):922–36. [DOI] [PubMed] [Google Scholar]
- 40.Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22(4):369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruggero K, Farran-Matas S, Martinez-Tebar A, Aytes A. Epigenetic regulation in prostate cancer progression. Curr Mol Biol Rep. 2018;4(2):101–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ge R, Wang Z, Montironi R, Jiang Z, Cheng M, Santoni M, et al. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Annals of Oncology. 2020;31(4):470–9. [DOI] [PubMed] [Google Scholar]
- 43.Lin D, Wyatt AW, Xue H, Wang Y, Dong X, Haegert A, et al. High Fidelity Patient-Derived Xenografts for Accelerating Prostate Cancer Discovery and Drug DevelopmentNext-Generation Models of Prostate Cancer. Cancer Res. 2014;74(4):1272–83. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Coleman IM, Brown LG, True LD, Kollath L, Lucas JM, et al. SRRM4 Expression and the Loss of REST Activity May Promote the Emergence of the Neuroendocrine Phenotype in Castration-Resistant Prostate CancerNeuroendocrine Phenotype in Castration-Resistant Prostate Cancer. Clinical Cancer Research. 2015;21(20):4698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lapuk A v, Wu C, Wyatt AW, McPherson A, McConeghy BJ, Brahmbhatt S, et al. From sequence to molecular pathology, and a mechanism driving the neuroendocrine phenotype in prostate cancer. J Pathol. 2012;227(3):286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sadoul K, Boyault C, Pabion M, Khochbin S. Regulation of protein turnover by acetyltransferases and deacetylases. Biochimie. 2008;90(2):306–12. [DOI] [PubMed] [Google Scholar]
- 47.Hulsurkar M, Li Z, Zhang Y, Li X, Zheng D, Li W. Beta-adrenergic signaling promotes tumor angiogenesis and prostate cancer progression through HDAC2-mediated suppression of thrombospondin-1. Oncogene. 2017;36(11):1525–36. [DOI] [PubMed] [Google Scholar]
- 48.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adenuga D, Rahman I. Protein kinase CK2-mediated phosphorylation of HDAC2 regulates co-repressor formation, deacetylase activity and acetylation of HDAC2 by cigarette smoke and aldehydes. Arch Biochem Biophys. 2010;498(1):62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khan DH, He S, Yu J, Winter S, Cao W, Seiser C, et al. Protein kinase CK2 regulates the dimerization of histone deacetylase 1 (HDAC1) and HDAC2 during mitosis. Journal of Biological Chemistry. 2013;288(23):16518–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsai SC, Seto E. Regulation of histone deacetylase 2 by protein kinase CK2. Journal of Biological Chemistry. 2002;277(35):31826–33. [DOI] [PubMed] [Google Scholar]
- 52.Eom GH, Cho YK, Ko JH, Shin S, Choe N, Kim Y, et al. Casein kinase-2α1 induces hypertrophic response by phosphorylation of histone deacetylase 2 S394 and its activation in the heart. Circulation. 2011;123(21):2392–403. [DOI] [PubMed] [Google Scholar]
- 53.Zou M, Toivanen R, Mitrofanova A, Floch N, Hayati S, Sun Y, et al. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Discov. 2017;7(7):736–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Labrecque MP, Takhar MK, Nason R, Santacruz S, Tam KJ, Massah S, et al. The retinoblastoma protein regulates hypoxia-inducible genetic programs, tumor cell invasiveness and neuroendocrine differentiation in prostate cancer cells. Oncotarget. 2016;7(17):24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carceles-Cordon M, Kelly WK, Gomella L, Knudsen KE, Rodriguez-Bravo V, Domingo-Domenech J. Cellular rewiring in lethal prostate cancer: the architect of drug resistance. Nat Rev Urol. 2020;17(5):292–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tiwari R, Manzar N, Ateeq B. Dynamics of cellular plasticity in prostate cancer progression. Front Mol Biosci. 2020;7:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abbas A, Gupta S. The role of histone deacetylases in prostate cancer. Epigenetics. 2008;3(6):300–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kelly TK, de Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28(10):1069–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer. 2008;98(3):604–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Y, Donmez N, Sahinalp C, Xie N, Wang Y, Xue H, et al. SRRM4 drives neuroendocrine transdifferentiation of prostate adenocarcinoma under androgen receptor pathway inhibition. Eur Urol. 2017;71(1):68–78. [DOI] [PubMed] [Google Scholar]
- 61.Labrecque MP, Brown LG, Coleman IM, Lakely B, Brady NJ, Lee JK, et al. RNA splicing factors SRRM3 and SRRM4 distinguish molecular phenotypes of castration-resistant neuroendocrine prostate cancer. Cancer Res. 2021;81(18):4736–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Westbrook TF, Hu G, Ang XL, Mulligan P, Pavlova NN, Liang A, et al. SCFβ-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature. 2008;452(7185):370–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jin Y, Ma D, Gramyk T, Guo C, Fang R, Ji H, et al. Kdm1a promotes SCLC progression by transcriptionally silencing the tumor suppressor Rest. Biochem Biophys Res Commun. 2019;515(1):214–21. [DOI] [PubMed] [Google Scholar]
- 64.Wagoner MP, Gunsalus KTW, Schoenike B, Richardson AL, Friedl A, Roopra A. The transcription factor REST is lost in aggressive breast cancer. PLoS Genet. 2010;6(6):e1000979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson LR, Scott MGH, Pitcher JA. G protein-coupled receptor kinase 5 contains a DNA-binding nuclear localization sequence. Mol Cell Biol. 2004;24(23):10169–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shakib K, Norman JT, Fine LG, Brown LR, Godovac‐Zimmermann J. Proteomics profiling of nuclear proteins for kidney fibroblasts suggests hypoxia, meiosis, and cancer may meet in the nucleus. Proteomics. 2005;5(11):2819–38. [DOI] [PubMed] [Google Scholar]
- 67.Hwang JY, Zukin RS. REST, a master transcriptional regulator in neurodegenerative disease. Curr Opin Neurobiol. 2018;48:193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Negrini S, Prada I, D’Alessandro R, Meldolesi J. REST: an oncogene or a tumor suppressor? Trends Cell Biol. 2013;23(6):289–95. [DOI] [PubMed] [Google Scholar]
- 69.Agah A, Kyriakides TR, Lawler J, Bornstein P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am J Pathol. 2002;161(3):831–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nör JE, Mitra RS, Sutorik MM, Mooney DJ, Castle VP, Polverini PJ. Thrombospondin-1 induces endothelial cell apoptosis and inhibits angiogenesis by activating the caspase death pathway. J Vasc Res. 2000;37(3):209–18. [DOI] [PubMed] [Google Scholar]
- 71.Teicher BA. Hypoxia and drug resistance. Cancer and Metastasis Reviews. 1994;13(2):139–68. [DOI] [PubMed] [Google Scholar]
- 72.Goldie JH. Drug resistance in cancer: a perspective. Cancer and Metastasis Reviews. 2001;20(1):63–8. [DOI] [PubMed] [Google Scholar]
- 73.Morin PJ. Drug resistance and the microenvironment: nature and nurture. Drug Resistance Updates. 2003;6(4):169–72. [DOI] [PubMed] [Google Scholar]
- 74.San José-Enériz E, Gimenez-Camino N, Agirre X, Prosper F. HDAC inhibitors in acute myeloid leukemia. Cancers (Basel). 2019;11(11):1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu W, Xu L, Chen B, Ou S, Muzzarelli KM, Hu D, et al. Targeting prostate cancer cells with enzalutamide‐HDAC inhibitor hybrid drug 2‐75. Prostate. 2019;79(10):1166–79. [DOI] [PubMed] [Google Scholar]
- 76.Rana Z, Diermeier S, Hanif M, Rosengren RJ. Understanding failure and improving treatment using HDAC inhibitors for prostate cancer. Biomedicines. 2020;8(2):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park D, Park H, Kim Y, Kim H, Jeoung D. HDAC3 acts as a negative regulator of angiogenesis. BMB Rep. 2014;47(4):227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jamaladdin S, Kelly RDW, O’Regan L, Dovey OM, Hodson GE, Millard CJ, et al. Histone deacetylase (HDAC) 1 and 2 are essential for accurate cell division and the pluripotency of embryonic stem cells. Proceedings of the National Academy of Sciences. 2014;111(27):9840–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zimberlin CD, Lancini C, Sno R, Rosekrans SL, McLean CM, Vlaming H, et al. HDAC1 and HDAC2 collectively regulate intestinal stem cell homeostasis. The FASEB Journal. 2015;29(5):2070–80. [DOI] [PubMed] [Google Scholar]
- 80.Kong D, Ahmad A, Bao B, Li Y, Banerjee S, Sarkar FH. Histone deacetylase inhibitors induce epithelial-to-mesenchymal transition in prostate cancer cells. 2012; [DOI] [PMC free article] [PubMed]
- 81.Ray A, Alalem M, Ray BK. Loss of epigenetic Kruppel-like factor 4 histone deacetylase (KLF-4-HDAC)-mediated transcriptional suppression is crucial in increasing vascular endothelial growth factor (VEGF) expression in breast cancer. Journal of Biological Chemistry. 2013;288(38):27232–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dressel U, Bailey PJ, Wang SCM, Downes M, Evans RM, Muscat GEO. A dynamic role for HDAC7 in MEF2-mediated muscle differentiation. Journal of Biological Chemistry. 2001;276(20):17007–13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.