

Abstract
Background
SerpinA1, a serine protease inhibitor, is involved in the modulation of microglial‐mediated inflammation in neurodegenerative diseases. We explored SerpinA1 levels in cerebrospinal fluid (CSF) and serum of amyotrophic lateral sclerosis (ALS) patients to understand its potential role in the pathogenesis of the disease.
Methods
SerpinA1, neurofilament light (NfL) and heavy (NfH) chain, and chitinase‐3‐like protein‐1 (CHI3L1) were determined in CSF and serum of ALS patients (n = 110) and healthy controls (n = 10) (automated next‐generation ELISA), and correlated with clinical parameters, after identifying three classes of progressors (fast, intermediate, slow). Biomarker levels were analyzed for diagnostic power and association with progression and survival.
Results
SerpinA1serum was significantly decreased in ALS (median: 1032 μg/mL) compared with controls (1343 μg/mL) (p = 0.02). SerpinA1CSF was elevated only in fast progressors (8.6 μg/mL) compared with slow (4.43 μg/mL, p = 0.01) and intermediate (4.42 μg/mL, p = 0.03) progressors. Moreover, SerpinA1CSF correlated with neurofilament and CHI3L1 levels in CSF. Contrarily to SerpinA1CSF, neurofilament and CHI3L1 concentrations in CSF correlated with measures of disease progression in ALS, while SerpinA1serum mildly related with time to generalization (rho = 0.20, p = 0.04). In multivariate analysis, the ratio between serum and CSF SerpinA1 (SerpinA1 ratio) and NfHCSF were independently associated with survival.
Conclusions
Higher SerpinA1CSF levels are found in fast progressors, suggesting SerpinA1 is a component of the neuroinflammatory mechanisms acting upon fast‐progressing forms of ALS. Both neurofilaments or CHI3L1CSF levels outperformed SerpinA1 at predicting disease progression rate in our cohort, and so the prognostic value of SerpinA1 alone as a measure remains inconclusive.
Keywords: ALS, biomarker, serpinA1
INTRODUCTION
Biological markers in amyotrophic lateral sclerosis (ALS) research may help accelerate diagnosis or even predict disease onset, constituting an important current subject of investigation. Longitudinal studies on presymptomatic ALS‐mutation carriers showed that neurofilament (Nf) levels rise before disease diagnosis is reached, reinforcing their role as possible predictors of phenoconversion [1, 2] or informing about the pace of degeneration in ALS individuals. Neurofilaments are acknowledged as rather non‐specific, still efficient, biomarkers of neuroinflammation‐driven neuroaxonal degeneration that can be employed mainly in research settings [3]. Although their role as efficacy endpoints in clinical trials is still questioned [4], neurofilament measurements may be helpful during stratification in trials design increasing the statistical power with a reduced sample size [5].
Different mechanisms contribute to ALS etiopathology and progression, with neuroinflammation driven by microglial dysregulation gaining increasing attention [6]. Chronic or sustained activation of microglia can be detrimental for neuronal survival: upon central nervous system (CNS) injury and in neurodegenerative conditions, microglia acquire a proinflammatory signature, contributing to impaired neuronal plasticity and functionality [7]. In ALS patients the increased expression of the proinflammatory molecules chitotriosidase‐1 (CHIT1), chitinase‐3‐like protein‐1 (CHI3L1, or YKL‐40), and monocyte chemoattractant protein‐1 (MCP‐1) has been suggested as a possible prognostic instrument complementary to other specific neurodegenerative biomarkers [8, 9, 10].
Amongst the molecules that may regulate the microglia inflammatory signature is the SerpinA1 protein, a serine protease inhibitor displaying immunoregulatory activities. SerpinA1 is mainly expressed by hepatocytes, gastrointestinal epithelial cells, monocytes, and macrophages [11]. Although genetic mutations of the SERPINA1 gene were associated with increased risk of lung disease and/or liver disease [12], recent evidence highlights the potential protective role of SerpinA1 in the modulation of microglial‐mediated inflammation, with implications for several neurodegenerative diseases [13, 14]. In particular, SerpinA1 regulates proinflammatory cytokine release [15], binding to complement C3 [16] and modulating neutrophil functions [17], playing a decisive role during the acute phase of systemic inflammation. CSF SerpinA1 levels were found increased or differentially regulated in its charge isoforms in Alzheimer's disease (AD) [18], Parkinson's disease (PD), Parkinson's disease dementia, dementia with Lewy bodies, Creutzfeldt–Jakob disease (CJD), and frontotemporal lobar degeneration (FTLD), in comparison with controls [19, 20, 21, 22]. In addition, in AD models SerpinA1 protects microglial cells from amyloid‐β‐induced toxicity [13], at least in part via inhibition of NLRP‐3 inflammasome [14].
Only a few studies specifically focused on CSF SerpinA1 determination in ALS patients with different conclusions [23, 24]. In this context, we quantitatively determined the levels of SerpinA1 in serum and CSF specimens from ALS patients and healthy controls (HC), to explore its prognostic power during the early phases of disease. Moreover, we looked for correlations between SerpinA1 and other known players in neuroinflammation, along with the levels of currently established biomarkers of neurodegeneration and clinical ALS features.
METHODS
Study population
Clinical features and sample collection were obtained from Neurobiobank of Modena, which consists of specimens collected from patients admitted at the Neurology Unit of Modena University Hospital. Both cases and controls were recruited between January 1, 2015 and December 31, 2021. Samples from HC were stored after performing lumbar puncture (LP) as part of the diagnostic workup, in the suspicion, later unconfirmed, of an acute neurological disease. All controls were subsequently discharged from hospital without the diagnosis of a major disease (mostly with diagnosis of an idiopathic headache). Eligible ALS participants include patients who received a diagnosis of definite or probable ALS according to the El Escorial revised criteria [25], who underwent LP, and still had at least 0.5 mL of CSF and serum available for analysis.
Standard protocol approvals, registrations, and patients' consent
Approval from the Ethical Committee of Area Vasta Emilia Nord (file number: 0015974/22) was obtained. All participants gave informed consent for LP procedure, biobanking, and research studies.
Clinical measures
The following demographic and clinical variables were collected for ALS patients: sex, age, site of onset (bulbar, upper limb or lower limb, respiratory), phenotype (classic, bulbar, upper motor neuron predominant, flail arm and flail leg, respiratory ALS) [26, 27], genotype, presence of dementia or family history, diagnostic latency, respiratory function as assessed by forced vital capacity (FVC), body mass index (BMI), Amyotrophic Lateral Sclerosis Functional Rating Scale‐Revised (ALSFRS‐R), time to non‐invasive (NIV) and invasive ventilation (IV), and time to percutaneous endoscopic gastrostomy. Comorbidities were categorized as hypertension, dyslipidemia, chronic obstructive pulmonary disease (COPD), diabetes mellitus, psychiatric disorders and depression, autoimmune diseases, and oncological history. Past or actual tobacco use was also recorded. We dated the onset of disease based on patients' referral of distinct motor weakness, excluding prodromal signs and symptoms as cramps, fasciculations, or vague motor impairment [28]. Genetic analysis was available for 84 patients for at least SOD1, FUS, TARDBP mutations, and C9orf72 expansion [29], while for a subset of 34 patients a next‐generation sequencing (NGS) panel of 78 genes was performed as described elsewhere [30]. Cognitive and behavioral impairment in the frontotemporal dementia (FTD) disease spectrum were evaluated according to Strong criteria [31].
The following clinical variables were considered as surrogates of disease progression: time to generalization (the time between onset and spreading of clinical signs from spinal or bulbar localization to both [32]), disease progression rate (DPR) at sampling (DPRSA) and at last observation (DPRLO) obtained as previously described [33], and the monthly decline of the ALSFRS‐R from sampling to last observation (DPRLO‐SA) [34].
Patients were categorized in different classes of progressors based on DPRLO. Slow progressors were defined as those with a DPRLO < 25th percentile (0.5 points/month) in our regional registry [35], fast progressors had a DPRLO > 75th percentile (1.4 points/month), and intermediate progressors had DPRLO values in between these values.
Sample collection and laboratory essays
Serum and CSF samples were obtained by venipuncture and LP, respectively, and processed following standard procedures. After sample centrifugation for 10 min at 1300× g, the supernatant was divided into aliquots and stored in polypropylene tubes at −80°C until analysis. SerpinA1, neurofilament light chain (NfL), neurofilament heavy chain (NfH), and chitinase‐3‐like protein‐1 (CHI3L1) were determined using an automated next‐generation enzyme‐linked immunosorbent assay (ELISA), via Ella Simple Plex assay technology (BioTechne, ProteinSimple) as previously described [36]. In this immunoassay, samples run through a channel each composed of three glass nano reactors (GNRs) coated with a capture antibody. In this way the sample is automatically read in triplicate. The samples were loaded into the cartridge with the following dilutions: 1:200,000 for serum and 1:10,000 for CSF for SerpinA1 cartridge; 1:2 for both serum and CSF for NfL cartridge; 1:2 for serum and 1:4 for CSF for NfH cartridge; and 1:10 for serum and 1:100 for CSF for CHI3L1 cartridge. Intra‐assay and inter‐assay variability are evaluated by the manufacturer: for intra‐assay each control was tested 16 times in one assay, while for inter‐assay the replicates of each control were tested in multiple assays performed by at least three technicians using two batches of reagents (Supplementary Table S1). Data on blood–brain barrier (BBB) damage were also collected by albumin quantification in CSF and serum (normal value <0.7).
Statistical methods
Continuous variables were reported as means (standard deviations, SD) or medians (interquartile ranges, IQR), according to the distribution, and categorical variables as absolute numbers (percentages, %). We evaluated clinical and demographic features by performing two‐tailed t‐tests and analysis of variance (ANOVA) for continuous variables between two‐groups or multiple groups, respectively, and chi‐square test for categorical variables. A p‐value < 0.05 was considered as statistically significant. Since the distributions of all the biomarker concentrations were extremely left‐skewed and non‐normal, nonparametric tests were employed for analysis in which normality could not be achieved.
For comparisons between two or multiple groups we employed the Mann–Whitney U test and Kruskal–Wallis test with post‐hoc Dunn correction, respectively, with statistical significance set at p < 0.05 for each biomarker. Influence of demographic variables on biomarker concentrations were assessed by ANOVA and Pearson correlation test after normalization. Correlations between each biomarker and other clinical variables were assessed using Spearman's test. Receiver‐operating characteristic (ROC) curves were used to evaluate the single and combined performance of each biomarker to discriminate between ALS patients versus controls, and fast‐progressing ALS patients versus all other ALS patients. Overall accuracy of biomarkers' sensitivity and specificity were assessed as areas under the curve (AUC); cut‐off values for sensitivity and specificity evaluations were chosen according to the highest Youden index.
Survival (months from onset to death/tracheostomy, and alive patients censored) was assessed first with univariate analysis using a Cox proportional hazards regression model, Kaplan–Meier curves, and log‐rank test. More specifically, univariate Cox regression analysis was initially used to assess the prognostic role of each clinical variable and biomarker in our battery. The following variables were examined: age at onset and at sampling, sex, cognitive or behavioral changes, FTD, diagnostic latency, site of onset, genotype (wild‐type/C9orf72/other genes), BMI and weight at sampling, DPRSA, SerpinaA1, and neurofilament and CHI3L1 levels in both CSF and serum. A multivariate Cox proportional hazard model was next run applying the stepwise backward method and including those variables that were found significant with a retention criterion of 0.1. Hazard ratios (HR) were calculated for each variable with 95% confidence intervals (CI). Data analysis was performed using the STATA statistical package 17 (StataCorp.2017; StataCorp LLC).
RESULTS
Participant characteristics
Serum and CSF samples were collected from 110 ALS patients (42 females/68 males) and 10 HC (6 females/4 males). Patients' features are summarized in Table 1. Controls were on average younger than cases: 57.25 (±12) years versus 46.38 (±20.24) years for patients and controls, respectively.
TABLE 1.
Characteristics of amyotrophic lateral sclerosis patients and controls.
| Variable | Patients (n = 110), n (%), mean [SD] | Controls (n = 10), n (%), mean [SD] | P‐value |
|---|---|---|---|
| Sex, males | 68 (61.82) | 4 (40) | 0.17 |
| Age at sampling, years | 57.25 [11.25] | 46.38 [20.24] | 0.01 |
| Tobacco use, never/ex/actual | 73 (66.36)/19 (17.27)/18 (16.36) | 5 (50.00)/3 (30.00)/2 (20.00) | 0.53 |
| Age at onset, years | 56.1 [11.79] | NA | NA |
| Diagnostic delay, months | 9.89 [9.06] | NA | NA |
| Mutational status a , c9/other/WT | 10 (11.90)/5 (5.95)/69 (82.14) | NA | NA |
|
Site of onset Bulbar/spinal |
29 (26.36)/81 (73.64) | NA | NA |
| ALSFRS‐R total score at sampling, points | 39.99 [6.77] | NA | NA |
| Time to generalization, months | 15.61 [17.50] | NA | NA |
| DPRSA, points/month | 0.95 [1.05] | NA | NA |
| BMI at sampling, kg/m2 | 24.70 [4.26] | NA | NA |
| FVC at sampling, % | 91.32 [22.59] | NA | NA |
|
FTD ALS‐ci/ALS‐bi |
12 (10.91) | NA | NA |
| 14 (12.73)/17 (15.45) | |||
| Time to NIV b , months | 27.96 [19.49] | NA | NA |
| Time to PEG c , months | 33.05 [29.08] | NA | NA |
| Time to IV/death d , months | 46.51 [40.23] | NA | NA |
| DPRLO, points/month | 1.09 [0.90] | NA | NA |
| DPRLO‐SA, points/month | 1.55 [2.48] | NA | NA |
Note: Means with standard deviations [SD] are reported or absolute numbers with percentages (%), according to distribution. See Methods section for calculation of clinical variables. P values inferior to 0.05 were presented in bold character to denote statistical significance.
Abbreviations: ALSFRS‐R, Amyotrophic Lateral Sclerosis Functional Rating Scale‐Revised; B, bulbar; bi‐ALS, ALS with behavioral involvement; BMI, body mass index; ci‐ALS, ALS with cognitive involvement; DPRLO, disease progression rate at last observation; DPRLO‐SA, monthly decline in ALSFRS‐R calculated between baseline visit and last observation; DPRSA, disease progression rate at sampling; FTD, frontotemporal dementia; FVC, forced vital capacity; IV, invasive ventilation; LL, lower limbs; NA, not available; NIV, non‐invasive ventilation; PEG, percutaneous endoscopic gastrotomy; R, respiratory; SD, standard deviation; UL, upper limbs; WT, wild‐type.
Genetic analysis available for 84 patients.
56 patients.
46 patients.
81 patients.
Correlations between demographic or laboratory variables and biomarker concentrations
Gender or BMI were not correlated with SerpinA1, CHI3L1, NfL, and NfH concentrations both in serum and CSF, except for BMI that was negatively associated with NfLserum (r = −0.23, p = 0.05). CHI3L1 concentrations in serum and CSF were associated with age at sampling (r = 0.45, p < 0.001; r = 0.25, p = 0.010, respectively). SerpinA1, CHI3L1, or neurofilament levels were not different among tobacco actual users, ex‐smokers, or ever‐smokers.
Correlations among biomarkers
We found significant correlations between CSF and serum concentrations of NfL (rho = 0.80, p < 0.001) and NfH (rho = 0.49, p < 0.001), CHI3L1(rho = 0.36, p = 0.002), but not for SerpinA1 (rho = −0.02, p = 0.836). SerpinA1CSF correlated with NfLCSF (rho = 0.33, p = 0.003), NfHCSF (rho = 0.32, p = 0.001), and CHI3L1CSF (rho = 0.46, p < 0.0001); CHI3L1CSF correlated with Nf in CSF as well (with NfLCSF, rho = 0.36, p = 0.002; with NfHCSF, rho = 0.20, p = 0.043). CHI3L1serum and SerpinA1serum levels did not correlate with NfHserum and NfLserum, which in turn strongly related with each other (rho = 0.65, p < 0.0001). SerpinA1serum was not correlated with markers of peripheral inflammation (C‐reactive protein [CRP], white blood ceslls [WBC], neutrophils, or lymphocytes count), while CHI3L1serum slightly increased with CRP (rho = 0.25, p = 0.007). A CSF/serum ratio multiplied by 100 was calculated for each biomarker and correlated with BBB damage index, showing SerpinA1, CHI3L1, or neurofilaments did not correlate with this index.
Biomarkers in ALS patients versus controls
We compared each biomarker concentration between ALS patients and HC, confirming both NfH and NfL, in CSF as well as in serum, were significantly higher in ALS (Table 2). SerpinA1serum was significantly decreased in ALS patients compared with HC (p = 0.017), while there were no differences for SerpinA1CSF and CHI3L1CSF. ROC analysis supported the high accuracy of NfHCSF, NfLCSF, and NfLserum in discriminating ALS from HC (AUC = 0.97, 95% CI: 0.93–1.00; AUC = 0.97, 95% CI: 0.93–1.00; AUC = 0.998, 95% CI: 0.993–1.00, respectively), while NfHserum was less precise (AUC = 0.94, 95% CI: 0.86–1.00). ROC analysis for SerpinA1serum showed an AUC of 0.73 (95% CI: 0.57–0.88); at the cut‐off of 900.46 μg/mL, a sensitivity of 100% and a specificity of 41.7% were obtained.
TABLE 2.
Comparison of biomarker levels between amyotrophic lateral sclerosis patients and controls.
| Biomarker | N | Patients median (IQR) | N | Controls median (IQR) | P‐value |
|---|---|---|---|---|---|
| SerpinA1CSF, μg/mL | 95 | 4.84 (3.2–6.9) | 9 | 3.64 (2.8–4.8) | 0.291 |
| SerpinA1serum, μg/mL | 110 | 1032 (658.9–1467.9) | 10 | 1343 (1008.1–3047.8) | 0.017 |
| NfLCSF, pg/mL | 73 | 6454 (3261–10,872) | 3 | 220 (109–351) | 0.045 |
| NfLserum, pg/mL | 77 | 111 (73.7–153) | 7 | 5.3 (3.8–8.9) | <0.001 |
| NfHCSF, pg/mL | 110 | 4092.5 (2494–7354) | 10 | 426 (259–598) | <0.001 |
| NfHserum, pg/mL | 108 | 1272 (610.5–2497) | 10 | 118.2 (66.3–191) | <0.001 |
| CHIT3L1CSF, ng/mL | 95 | 73.1 (48.9–131) | 10 | 86.4 (45.6–95) | 0.540 |
| CHIT3L1serum, ng/mL | 108 | 36 (27.3–62.8) | 10 | 39 (32.3–75.4) | 0.389 |
| BBB damage index, % | 49 | 0.49 (0.36–0.65) | 9 | 0.51 (0.44–0.6) | 0.855 |
Note: Data are presented as median (IQR). P‐values inferior to 0.05 were presented in bold character to denote statistical significance.
Abbreviations: BBB damage index, blood–brain barrier damage index, calculated by the ratio of AlbuminCSF/Albuminserum, multiplied per 100; CHI3L1, chitinase‐3‐like protein‐1; IQR, interquartile range; NfL, neurofilament light chain; NfH, neurofilament heavy chain.
Correlation between biomarkers and clinical features and disease progression
When accounting for genotype, neither stratifying C9orf72 expansion carriers against overall non‐mutated and other genes in mutated patients nor by multiple‐groups comparison between differently mutated patients revealed different levels of SerpinA1 or CHI3L1 in serum and CSF (Supplementary Table S2). No differences in investigated biomarkers were found across patients with different disease onset and cognitive decline (Supplementary Table S2). Overall, Nf levels, especially in CSF, correlated with variables of disease progression (diagnostic latency, DPRSA, DPRLO, time to generalization, ALSFRS‐R at sampling, DPRLO‐SA) as shown in Table 3.
TABLE 3.
Correlations between biomarkers and clinical variables of disease progression in amyotrophic lateral sclerosis.
| Biomarker | Diagnostic latency | DPRSA | DPRLO | ALSFRS‐R at sampling | FVC | Time to generalization | DPRLO‐SA | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spearman rho correlation | P‐value | Spearman rho correlation | P‐value | Spearman rho correlation | P‐value | Spearman rho correlation | P‐value | Spearman rho correlation | P‐value | Spearman rho correlation | P‐value | Spearman rho correlation | P‐value | |
| NfLCSF | −0.44 | <0.001 | 0.38 | <0.001 | 0.66 | <0.001 | −0.29 | 0.01 | −0.05 | 0.67 | −0.40 | <0.001 | 0.61 | <0.001 |
| NfLserum | −0.36 | 0.001 | 0.47 | <0.001 | 0.67 | <0.001 | −0.42 | <0.001 | −0.19 | 0.11 | −0.34 | 0.003 | 0.60 | <0.001 |
| NfHCSF | −0.41 | <0.001 | 0.41 | <0.001 | 0.60 | <0.001 | −0.32 | 0.002 | −0.05 | 0.62 | −0.38 | <0.001 | 0.52 | <0.001 |
| NfHserum | −0.07 | 0.50 | 0.15 | 0.11 | 0.36 | 0.001 | −0.10 | 0.32 | −0.20 | 0.04 | −0.34 | <0.001 | 0.37 | <0.002 |
| SerpinA1CSF | −0.20 | 0.06 | 0.10 | 0.31 | 0.14 | 0.18 | −0.12 | 0.25 | −0.001 | 0.96 | −0.13 | 0.21 | −0.11 | 0.30 |
| SerpinA1serum | 0.18 | 0.06 | −0.09 | 0.37 | −0.13 | 0.18 | −0.04 | 0.96 | 0.01 | 0.95 | 0.20 | 0.04 | −0.09 | 0.37 |
| CHI3L1CSF | −0.18 | 0.08 | 0.11 | 0.27 | 0.20 | 0.05 | −0.15 | 0.16 | 0.004 | 0.97 | −0.02 | 0.88 | 0.26 | 0.01 |
| CHI3L1serum | 0.18 | 0.06 | −0.09 | 0.37 | −0.11 | 0.25 | −0.02 | 0.80 | −0.20 | 0.04 | 0.16 | 0.09 | −0.10 | 0.28 |
| BBB damage index | −0.22 | 0.12 | −0.09 | 0.52 | 0.03 | 0.83 | 0.13 | 0.38 | −0.12 | 0.44 | −0.10 | 0.5 | 0.10 | 0.47 |
| SerpinA1 ratio | −0.25 | 0.02 | 0.14 | 0.17 | 0.13 | 0.22 | −0.18 | 0.09 | −0.02 | 0.83 | −0.19 | 0.08 | 0.10 | 0.54 |
Note: See Methods section for calculation of clinical variables. P‐values inferior to 0.05 were presented in bold character to denote statistical significance.
Abbreviations: ALSFRS‐R, Amyotrophic Lateral Sclerosis Functional Rating Scale‐Revised; BBB damage index, blood–brain barrier damage index; CHI3L1, chitinase‐3‐like protein‐1; DPRLO at last observation, disease progression rate at last observation; DPRLO‐SA, monthly decline in ALSFRS‐r calculated between baseline visit and last observation; DPRSA at sampling, disease progression rate at sampling; FVC, forced vital capacity; NfL, neurofilament light chain; NfH, neurofilament heavy chain.
We therefore investigated any correlation between less explored biomarkers such as SerpinA1 and CHI3L1 and ALS clinical features (Table 3), revealing a mild correlation between CHI3L1CSF and DPRLO (rho = 0.20, p = 0.05), and DPRLO‐SA (rho = 0.26, p = 0.01). SerpinA1serum correlated with time to generalization (the longer the time to generalization, the higher the SerpinA1serum) (rho = 0.20, p = 0.04). The BBB damage index did not correlate with any clinical variable of ALS severity or progression.
Biomarkers status in slow and fast progressing patients
We assessed whether differences existed between slow (SP), intermediate (IP), and fast (FP) progressors according to each of the biomarker levels (Table 4). Nf were increasingly elevated according to the category of progression (Figure 1), while SerpinA1CSF was elevated only in FP compared with both SP and IP (p‐value for post‐hoc Dunn correction FP vs. SP: 0.03; for FP vs. IP: 0.01, while SP vs. IP p = 0.44; Supplementary Figure S1). SP patients showed instead slightly increased levels of SerpinA1serum (p‐value SP vs. IP: 0.04; p‐value SP vs. FP: 0.06). Given the trend towards an opposite behavior of SerpinA1 between the two compartments, we also considered the ratio between SerpinA1CSF/SerpinA1serum (SerpinA1 ratio) in different classes of progressors (Bonferroni corrected p‐value FP vs. SP: 0.03; p‐value FP vs. IP: 0.02). ROC analysis for discrimination between fast versus all other patients showed good discriminatory performance of SerpinA1CSF (AUC: 0.78, 95% CI: 0.58–0.98), though NfLCSF and NfHCSF had better AUC (for NFLCSF, AUC: 0.91, 95% CI: 0.82–0.99, p‐value vs. SerpinA1CSF: 0.41; for NfHCSF AUC 0.81, 95% CI: 0.68–0.95, p‐value vs. SerpinA1CSF: 0.65). SerpinA1CSF combined with NfL and NfH in CSF in ROC analysis did not increase the AUC substantially (data not shown).
TABLE 4.
Comparison of biomarker levels among slow, intermediate, and fast progressors.
| Biomarker | N | Slow progressors (n = 24) | N | Intermediate progressors (n = 67) | N | Fast progressors (n = 12) | P‐value |
|---|---|---|---|---|---|---|---|
| SerpinA1CSF, μg/mL | 16 | 4.91 (3.04–6.55) | 60 | 4.46 (3.19–6.61) | 10 | 8.62 (4.67–13.31) | 0.058 |
| SerpinA1serum, μg/mL | 24 | 1174.62 (896.8–1841.8) | 65 | 1003.3 (618.6–1304.5) | 12 | 788.6 (581.1–1350.1) | 0.160 |
| SerpinA1 ratio | 16 | 0.004 (0.003–0.006) | 58 | 0.004 (0.003–0.008) | 10 | 0.007 (0.006–0.2) | 0.098 |
| NfLCSF, pg/mL | 9 | 2821 (1879–3249) | 50 | 7152.1 (3906–13,124) | 8 | 10963.5 (8777.5–25726.5) | <0.001 |
| NfLserum, pg/mL | 11 | 53.6 (43.1–68.5) | 52 | 117 (75.9–152.5) | 9 | 206 (191–268) | <0.001 |
| NfHCSF, pg/mL | 16 | 2565.5 (1873–3557.5) | 60 | 4180.5 (2561–7330.5) | 10 | 8325.5 (7601–12,401) | <0.001 |
| NfHserum, pg/mL | 24 | 816 (536–1352.5) | 65 | 1467 (611–2715) | 12 | 2005.5 (1336.5–3293.5) | 0.012 |
| CHIT3L1CSF, ng/mL | 16 | 50.20 (41.30–71.06) | 60 | 95.46 (56.10–149) | 10 | 91.5 (45.5–190) | 0.021 |
| CHIT3L1serum, ng/mL | 24 | 38.59 (27.71–77.17) | 65 | 36.56 (27.97–62.73) | 12 | 34.26 (22.88–60) | 0.684 |
| BBB damage index, % | 12 | 0.53 (0.36–0.66) | 25 | 0.49 (0.37–0.65) | 6 | 0.49 (0.35–1.12) | 0.961 |
Note: Data are presented as median (IQR). P‐values inferior to 0.05 were presented in bold character to denote statistical significance.
Abbreviations: BBB damage index, blood–brain barrier damage index; CHI3L1, chitinase‐3‐like protein‐1; IQR, interquartile range; NfL, neurofilament light chain; NfH, neurofilament heavy chain.
FIGURE 1.
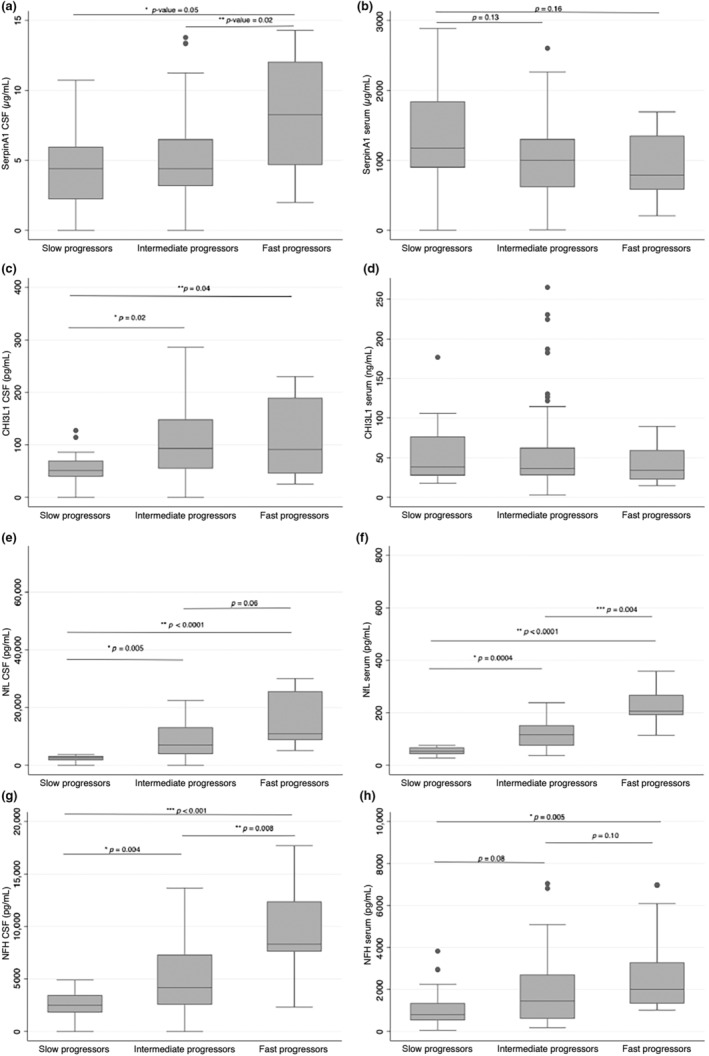
Comparisons of biomarker levels across different classes of progressors. (a, b) Box plots of SerpinA1 concentrations in cerebrospinal fluid (CSF) and serum, respectively, according to slow, intermediate, and fast progressors. (c, d) Box plots of chitinase‐3‐like protein‐1 concentrations in CSF and serum, respectively, according to slow, intermediate, and fast progressors. (e, f) Box plots of neurofilament light chain concentrations in CSF and serum, respectively, according to slow, intermediate, and fast progressors. (g, h) Box plots of neurofilament light chain concentrations in CSF and serum, respectively, according to slow, intermediate, and fast progressors. All boxes display medians with interquartile ranges, outliers are shown as separate dots. CHI3L1, chitinase‐3‐like protein‐1; CSF, cerebrospinal fluid; NfL, neurofilament light chain; NfH, neurofilament heavy chain.
Biomarkers and survival
Complete results of univariate and next multivariate analysis are shown in Table 5. Besides FVC and DPR at sampling, NfHCSF was confirmed as a good predictor of survival; SerpinA1 ratio was retained in the model suggesting its role as an independent prognostic factor for survival. When stratifying the two biomarkers according to the cut‐off values of 7354 pg/mL for NfHCSF and of 0.017 for SerpinA1 ratio, NfHCSF was confirmed as a good predictor of survival (median survival 26.7 months, 95% CI: 13.7–32.1, for NfHCSF above 7354 pg/mL, and 37.4 months, 95% CI: 31.5–46.6 for NfHCSF below 7354 pg/mL; log‐rank test p = 0.002) as well as SerpinA1 ratio (median survivals for SerpinA1 ratio above 0.017: 20 months, 95% CI: 9–n.a; for SerpinA1 ratio below 0.017: 35.3 months, 95% CI: 30.4–42.3; p = 0.02) (Figure 2).
TABLE 5.
Cox proportional hazard regression at univariate and multivariate analysis.
| Variable | Observations (n) | Univariate analysis | Multivariate analysis (n = 71) | ||
|---|---|---|---|---|---|
| HR (95% CI) | P‐value | HR (95% CI) | P‐value | ||
| Sex (male as reference) | 109 | 0.8 (0.5–1.27) | 0.35 | ||
| Age at onset | 109 | 1.01 (1–1.03) | 0.18 | ||
| Diagnostic delay | 109 | 0.94 (0.91–0.97) | <0.001 | ||
| BMI at sampling | 109 | 0.94 (0.89–0.99) | 0.03 | ||
| Mutation (WT as reference) | 84 | 1 | 0.50 | ||
| c9orf72 | 1.7 (0.72–4) | 0.23 | |||
| Other | 1.26 (0.4–4.1) | 0.70 | |||
| FTD | 109 | 1.62 (0.83–3.15) | 0.16 | ||
| Cognitive changes | 110 | 1.94 (1.07–3.53) | 0.03 | ||
| Behavioral changes | 110 | 1.65 (0.92–2.95) | 0.09 | ||
| Site of onset (spinal as reference) | 109 | 1.1 (0.67–1.82) | 0.71 | ||
| FVC at sampling | 106 | 0.99 (0.98–1) | 0.01 | 0.97 (0.96–0.97) | 0.001 |
| ALSFRS‐R at sampling | 109 | 0.98 (0.96–1.01) | 0.16 | ||
| Time to generalization | 108 | 0.97 (0.95–0.99) | <0.001 | ||
| DPRSA | 109 | 1.31 (1.16–1.48) | <0.001 | 2.02 (1.49–2.74) | < 0.001 |
| NfLserum | 76 | 1.004 (1.002–1.01) | <0.001 | ||
| NfLCSF | 72 | 1 (1–1.00004) | 0.002 | ||
| NfHserum | 107 | 1 (1–1.0002) | 0.03 | ||
| NfHCSF | 93 | 1 (1–1.0002) | <0.001 | 1.000 (1.000–1.000) | < 0.001 |
| SerpinA1serum | 107 | 1 (0.99–1.02) | 0.45 | ||
| SerpinACSF | 94 | 1.05 (1.02–1.09) | 0.003 | ||
| SerpinA1ratio | 92 | 3.68 (1.73–7.83) | 0.001 | 5.16 (2.34–11.36) | < 0.001 |
| CHI3L1serum | 107 | 1 (0.996–1.004) | 0.97 | ||
| CHI3L1CSF | 94 | 1.01 (1–1.008) | 0.003 | ||
| BBB damage index | 49 | 1.22 (0.33–4.53) | 0.76 | ||
Note: Hazard ratios are presented with 95% confidence interval and relative p‐value. Figures in bold type denote statistical significance. See Methods section for calculation of clinical variables.
Abbreviations: ALSFRS‐R, Amyotrophic Lateral Sclerosis Functional Rating Scale‐Revised; BBB damage index, blood–brain barrier damage index, calculated by the ratio of AlbuminCSF/Albuminserum, multiplied per 100; BMI, body mass index; CHI3L1, chitinase‐3‐like protein‐1; CI, confidence interval; CSF, cerebrospinal fluid; DPRSA, disease progression rate at sampling; FTD, frontotemporal dementia; FVC, forced vital capacity; HR, hazard ratio; NfL, neurofilament light chain; NfH, neurofilament heavy chain; WT, wild‐type.
FIGURE 2.
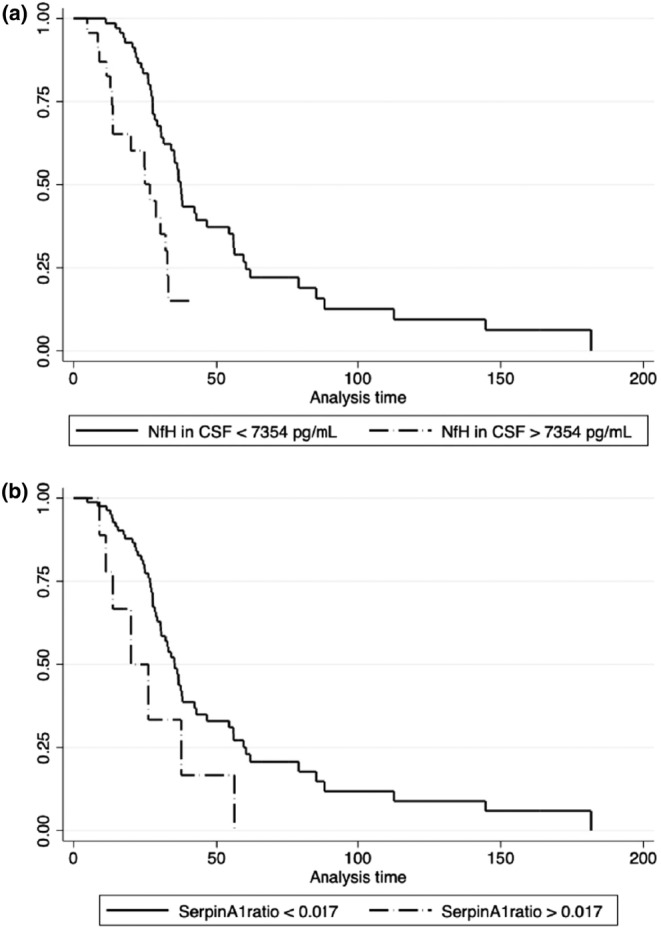
Kaplan–Meyer curves of patients stratified by cerebrospinal fluid neurofilament heavy chain (NfHCSF) and SerpinA1 ratio. (a) Kaplan–Meyer survival curves for values above and below the cut‐off value of 7354 pg/mL of NfHCSF in the amyotrophic lateral sclerosis (ALS) cohort. (b) Kaplan–Meyer survival curves for values above and below the cut‐off value of 0.0083 of SerpinA1 ratio in the ALS cohort. CSF, cerebrospinal fluid; NfH, neurofilament heavy chain.
DISCUSSION
In this study we explored how CSF and serum SerpinA1 levels could vary in ALS and provide valuable information about intrinsic pathomechanisms and disease course.
Without underestimating neurofilaments' value as robust prognostic biomarker in ALS, our results could suggest that SerpinA1 may represent an independent and complementary indicator of ALS progression, being more representative of microglial activation. Indeed, in our patients' cohort SerpinA1 CSF concentrations were significantly increased only in fast progressors. This is at variance with neurofilament concentrations, which directly correlate with clinical measures of disease progression across all rates of progression [3]. With the necessary caution related to the low sample size, we may speculate a SerpinA1 CSF “threshold effect” of microglial activation in pathological state [37] that, after an initial phenotype adjustment towards an increased immune vigilance, runs out in a toxic phase, failing to reverse the damage already accumulated.
SerpinA1 might therefore play an anti‐inflammatory role by attenuating microglial activity, and the increase in SerpinA1CSF in fast progressors could be considered a stronger early attempt to balance the developing neuroinflammation/neurodegeneration when it progresses faster. Possibly, SerpinA1 immunomodulation may attenuate during a slower disease course as already reported for CJD [22]. Chronic stimulation as it occurs in neurodegenerative diseases has been suggested to lead to exhaustion of phagocytosis in microglia and dysfunction. Alternatively, the increased activation of SerpinA1, as well as its increased expression in the motor cortex of ALS patients [38], may occur as a consequence of an exhausted microglia [39].
Another explanation for SerpinA1 raised concentrations being limited to fast progressors could be related to distinct pathogenic mechanisms with a peculiar impact on neuroinflammation‐mediated pathways, particularly in aggressive ALS forms. In vitro and in vivo studies on SerpinA1 expression might be important to understand whether SerpinA1 really mitigates the pathological hallmarks through the modulation of neuroinflammation in some patients or whether its dysregulation is only one of several downstream effects of the pathological process characterizing ALS. To this extent, besides similar observations in other neurodegenerative conditions [19, 20, 22, 40], more recently different SerpinA1 isoforms were found to be prevalent in different dementia groups, underlying the complexity of these proteases and the importance of further studies focused on possible ALS‐related isoforms.
As well as being reduced in faster progressors compared with slower forms, SerpinA1serum was significantly decreased in ALS with respect to controls, suggesting a generally protective role of this protease. Accordingly, sustained levels of circulating SerpinA1 have been correlated with increased regulatory T cells (Tregs) [41], that likely contribute to disease progression in ALS where high frequency of activated Tregs and high ratio between activated and resting Tregs in blood are associated with better survival [42]. Decreased serum levels of SerpinA1 may mirror the already described Tregs reduction and dysfunction in ALS patients [43].
In this hypothetical explanation, SerpinA1 may exert a protective immune action also by inhibiting neutrophil‐released elastase and proteinase‐3 inside and outside the cell, in this way controlling inflammation deleterious effects, such as activation of IL‐1β. SerpinA1 has been found to have independent anti‐inflammatory/immunomodulatory properties, protecting against cell apoptosis, inhibiting superoxide production, and inducing IL‐1 receptor antagonist expression [44]. Conversely, in the CNS, SerpinA1 upregulation has already been described, among other neurodegenerative diseases, in FTLD [22, 38].
Our study highlighted that, in contrast to the other biomarkers such as neurofilaments for example, SerpinA1 concentrations in CSF and serum did not correlate. Although SerpinA1 can be easily quantified in the CSF, the source of CSF serpinA1 is still not clear [18], as well as what cell types express SerpinA1 in the CNS, and which isoforms of SerpinA1 are contained in the CSF. The protein might diffuse out of venous blood and be released from the brain tissue into the CSF [19]. Alternatively, but not mutually exclusive, SerpinA1CSF levels may result from an intrathecal production of SerpinA1, as already reported for AD patients [20]. Future investigations are required to understand the pathological significance of the lack of correlation between serum and CSF SerpinA1 as well as the lack of correlation with the albumin quotient.
While neurofilaments appeared to be influenced by BMI and CHI3L1 by age of sampling, as already reported [45, 46], SerpinA1 was not related to any demographic variable, nor with the inflammatory markers, or “proinflammatory” habits or comorbidity (i.e., COPD, tobacco use) [47] measured in our study population. Further larger studies should also assess these interesting and possibly clinically useful points.
The present study has some strengths and limitations. All ALS patients were intensely clinically and genetically characterized and followed up, and a large set of comorbidities were taken into account. The association with survival data, the simultaneous determination in serum and CSF, and the correlation with systemic inflammation indicators and more solid ALS biomarkers constitute significant added value to our work. However, when ALS patients were stratified based on clinical variables, the sample size for each subgroup became smaller with decreased power. Furthermore, the sample size of the control group was not age‐matched and rather small, due to the difficulty of obtaining CSF for diagnostic purposes from HC; this highlights the need to find and include larger and better‐matched HC, along with other neurodegenerative diseases, in order to draw a more robust conclusion about the significance of the observed behavior of SerpinA1 levels in ALS patients. Conversely, the ability to distinguish between HC and ALS with possible new biomarkers may be of secondary importance given the known good performance of Nf that was confirmed also in this study. Overall, while shedding light on possible mechanisms related to aggregates accumulation in the CNS and serine proteases upregulation, our study does not support the use of SerpinA1 for clinical purposes. A critical point which limits the prognostic utility of SerpinA1 is the necessity to obtain CSF by LP, in contrast to Nf levels whose levels linearly correlate in serum and CSF and reliably inform about disease progression across all stages. Finally, the cross‐sectional nature of this study does not allow information to be obtained on the longitudinal trajectory of SerpinA1 during ALS course.
In conclusion, the novel finding of this study is the correlation between SerpinA1 levels and patients with a faster rate of disease progression, which might be related to the pace of neurodegeneration and glial activation, as supported by the correlation with Nf and CHI3L1 levels.
Should our results be confirmed, this might be useful in clinical practice to identify patients with a fast evolution and thus inform their management, counselling, and treatment choices. Fast progressors are an even more fragile subgroup of individuals affected by ALS, as they are frequently excluded from research studies and clinical trials due to their devastating condition, requiring the greatest efforts from ALS multidisciplinary teams that try to deal with in time the complex and various needs resulting from the rapid disease progression in these individuals.
Conversely, being aware of disease progression could allow a more precise stratification of patients in clinical trials, and fast disease evolution may offer the possibility to monitor the effect of promising drugs or treatments in a shorter time with respect to slow or normal progressing patients.
However, both neurofilament or CHI3L1 CSF levels outperformed SerpinA1 at predicting the rate of disease progression in our cohort of patients, and so the prognostic value of SerpinA1 alone as a measure remains unconclusive.
Taken together, our findings warrant further confirmatory studies in larger cohorts, in which exploring SerpinA1 isoforms could be a fascinating field of study. To obatin a more complete picture, larger collaborative longitudinal studies will be necessary, including different disease stages and multiple microglial biomarkers together with neurofilaments.
AUTHOR CONTRIBUTIONS
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article. Jessica Mandrioli, Elisabetta Zucchi, and Ilaria Martinelli formulated the initial idea of the study. Cecilia Simonini and Roberta Bedin conducted the laboratory tests. Andrea Ghezzi, Giulia Gianferrari, Chiara Biral, Nicola Fini, and Ilaria Martinelli collected the clinical data. Elisabetta Zucchi and Jessica Mandrioli performed the statistical analyses. Ilaria Martinelli and Elisabetta Zucchi wrote the draft of the manuscript and prepared the tables and figures with Cecilia Simonini. Serena Carra and Jessica Mandrioli supervised and reviewed the manuscript draft.
All authors had full access to all the study data and take complete responsibility for the integrity of the data and accuracy of the data analysis. All named authors take responsibility for the integrity of the work as a whole. All authors read and approved the final manuscript.
FUNDING INFORMATION
This study was supported by Agenzia Italiana del Farmaco (grant number 2016‐02364678), by the Italian Agency for the Research on Amyotrophic Lateral Sclerosis (AriSLA, bando per la ricerca clinica 2015), by the Italian Ministry of Health (bando per la ricerca finalizzata 2016, grant number RF‐2016‐02361616), by the University of Modena and Reggio Emilia (bando FAR 2021, Progetti di ricerca Interdisciplinari Mission Oriented, NEURALS project), and by Fondazione Cassa di Risparmio di Modena (Neurobiobanca di Modena).
CONFLICT OF INTEREST STATEMENT
None of the authors have any conflicts of interest to report.
Supporting information
Supplementary Figure 1
Supplementary Table 1
Supplementary Table 2
ACKNOWLEDGMENTS
The authors would like to thank all the patients and their families for their support and understanding. The authors also wish to thank the Modena Neurobiobank for providing the samples.
Martinelli I, Zucchi E, Simonini C, et al. SerpinA1 levels in amyotrophic lateral sclerosis patients: An exploratory study. Eur J Neurol. 2024;31:e16054. doi: 10.1111/ene.16054
DATA AVAILABILITY STATEMENT
Anonymized data are available upon request from any qualified investigator.
REFERENCES
- 1. Bjornevik K, O'Reilly EJ, Molsberry S, et al. Prediagnostic neurofilament light chain levels in amyotrophic lateral sclerosis. Neurology. 2021;97(15):e1466‐e1474. doi: 10.1212/WNL.0000000000012632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saracino D, Dorgham K, Camuzat A, et al. Plasma NfL levels and longitudinal change rates in C9orf72 and GRN‐associated diseases: from tailored references to clinical applications. J Neurol Neurosurg Psychiatry. 2021;92(12):1278‐1288. doi: 10.1136/jnnp-2021-326914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zucchi E, Bonetto V, Sorarù G, et al. Neurofilaments in motor neuron disorders: towards promising diagnostic and prognostic biomarkers. Mol Neurodegener. 2020;15(1):58. doi: 10.1186/s13024-020-00406-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van den Berg LH, Sorenson E, Gronseth G, et al. Revised Airlie House consensus guidelines for design and implementation of ALS clinical trials. Neurology. 2019;92(14):e1610‐e1623. doi: 10.1212/WNL.0000000000007242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benatar M, Zhang L, Wang L, et al. Validation of serum neurofilaments as prognostic and potential pharmacodynamic biomarkers for ALS. Neurology. 2020;95(1):e59‐e69. doi: 10.1212/WNL.0000000000009559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol. 2012;237(1):147‐152. doi: 10.1016/j.expneurol.2012.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Azam S, Haque ME, Kim IS, Choi DK. Microglial turnover in ageing‐related neurodegeneration: therapeutic avenue to intervene in disease progression. Cells. 2021;10(1):150. doi: 10.3390/cells10010150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Masrori P, De Schaepdryver M, Floeter MK, et al. Prognostic relationship of neurofilaments, CHIT1, YKL‐40 and MCP‐1 in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2022;93(6):681‐682. doi: 10.1136/jnnp-2021-327877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gille B, De Schaepdryver M, Dedeene L, et al. Inflammatory markers in cerebrospinal fluid: independent prognostic biomarkers in amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry. 2019;90(12):1338‐1346. doi: 10.1136/jnnp-2018-319586 [DOI] [PubMed] [Google Scholar]
- 10. Illán‐Gala I, Alcolea D, Montal V, et al. CSF sAPPβ, YKL‐40, and NfL along the ALS‐FTD spectrum. Neurology. 2018;91(17):e1619‐e1628. doi: 10.1212/WNL.0000000000006383 [DOI] [PubMed] [Google Scholar]
- 11. https://www.proteinatlas.org/ENSG00000197249‐SERPINA1/immune+cell. Accessed March 1, 2023.
- 12. Foil KE. Variants of SERPINA1 and the increasing complexity of testing for alpha‐1 antitrypsin deficiency. Ther Adv Chronic Dis. 2021;12_suppl:20406223211015954. doi: 10.1177/20406223211015954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gold M, Dolga AM, Koepke J, et al. α1‐antitrypsin modulates microglial‐mediated neuroinflammation and protects microglial cells from amyloid‐β‐induced toxicity. J Neuroinflammation. 2014;11:165. doi: 10.1186/s12974-014-0165-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ebrahimi T, Rust M, Kaiser SN, et al. α1‐antitrypsin mitigates NLRP3‐inflammasome activation in amyloid β1‐42‐stimulated murine astrocytes. J Neuroinflammation. 2018;15(1):282. doi: 10.1186/s12974-018-1319-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pott GB, Chan ED, Dinarello CA, Shapiro L. Alpha‐1‐antitrypsin is an endogenous inhibitor of proinflammatory cytokine production in whole blood. J Leukoc Biol. 2009;85(5):886‐895. doi: 10.1189/jlb.0208145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O'Brien ME, Murray G, Gogoi D, et al. A review of alpha‐1 antitrypsin binding partners for immune regulation and potential therapeutic application. Int J Mol Sci. 2022;23(5):2441. doi: 10.3390/ijms23052441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bergin DA, Reeves EP, Hurley K, et al. The circulating proteinase inhibitor α‐1 antitrypsin regulates neutrophil degranulation and autoimmunity. Sci Transl Med. 2014;6(217):217ra1. doi: 10.1126/scitranslmed.3007116 [DOI] [PubMed] [Google Scholar]
- 18. Barba L, Halbgebauer S, Paolini Paoletti F, et al. Specific cerebrospinal fluid SerpinA1 isoform pattern in Alzheimer's disease. Int J Mol Sci. 2022;23(13):6922. doi: 10.3390/ijms23136922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Halbgebauer S, Nagl M, Klafki H, et al. Modified serpinA1 as risk marker for Parkinson's disease dementia: analysis of baseline data. Sci Rep. 2016;6:26145. doi: 10.1038/srep26145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nielsen HM, Minthon L, Londos E, et al. Plasma and CSF serpins in Alzheimer disease and dementia with Lewy bodies. Neurology. 2007;69(16):1569‐1579. doi: 10.1212/01.wnl.0000271077.82508.a0 [DOI] [PubMed] [Google Scholar]
- 21. Jesse S, Lehnert S, Jahn O, et al. Differential sialylation of serpin A1 in the early diagnosis of Parkinson's disease dementia. PLoS ONE. 2012;7(11):e48783. doi: 10.1371/journal.pone.0048783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abu‐Rumeileh S, Halbgebauer S, Steinacker P, et al. CSF SerpinA1 in Creutzfeldt–Jakob disease and frontotemporal lobar degeneration. Ann Clin Transl Neurol. 2020;7(2):191‐199. doi: 10.1002/acn3.50980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brettschneider J, Mogel H, Lehmensiek V, et al. Proteome analysis of cerebrospinal fluid in amyotrophic lateral sclerosis (ALS). Neurochem Res. 2008;33(11):2358‐2363. doi: 10.1007/s11064-008-9742-5 [DOI] [PubMed] [Google Scholar]
- 24. Wormser U, Mandrioli J, Vinceti M, et al. Reduced levels of alpha‐1‐antitrypsin in cerebrospinal fluid of amyotrophic lateral sclerosis patients: a novel approach for a potential treatment. J Neuroinflammation. 2016;13(1):131. doi: 10.1186/s12974-016-0589-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brooks BR, Miller RG, Swash M, Munsat TL. World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293‐299. doi: 10.1080/146608200300079536 [DOI] [PubMed] [Google Scholar]
- 26. Calvo A, Moglia C, Lunetta C, et al. Factors predicting survival in ALS: a multicenter Italian study. J Neurol. 2017;264(1):54‐63. doi: 10.1007/s00415-016-8313-y [DOI] [PubMed] [Google Scholar]
- 27. Schito P, Ceccardi G, Calvo A, et al. Clinical features and outcomes of the flail arm and flail leg and pure lower motor neuron MND variants: a multicentre Italian study. J Neurol Neurosurg Psychiatry. 2020;91(9):1001‐1003. doi: 10.1136/jnnp-2020-323542 [DOI] [PubMed] [Google Scholar]
- 28. Benatar M, Granit V, Andersen PM, et al. Mild motor impairment as prodromal state in amyotrophic lateral sclerosis: a new diagnostic entity. Brain. 2022;145(10):3500‐3508. doi: 10.1093/brain/awac185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chiò A, Calvo A, Mazzini L, et al. Extensive genetics of ALS: a population‐based study in Italy. Neurology. 2012;79(19):1983‐1989. doi: 10.1212/WNL.0b013e3182735d36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martinelli I, Zucchi E, Pensato V, et al. G507D mutation in FUS gene causes familial amyotrophic lateral sclerosis with a specific genotype‐phenotype correlation. Neurobiol Aging. 2022;118:124‐128. doi: 10.1016/j.neurobiolaging.2022.05.006 [DOI] [PubMed] [Google Scholar]
- 31. Strong MJ, Abrahams S, Goldstein LH, et al. Amyotrophic lateral sclerosis ‐ frontotemporal spectrum disorder (ALS‐FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(3–4):153‐174. doi: 10.1080/21678421.2016.1267768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tortelli R, Copetti M, Panza F, et al. Time to generalisation as a predictor of prognosis in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2016;87(6):678‐679. doi: 10.1136/jnnp-2014-308478 [DOI] [PubMed] [Google Scholar]
- 33. Lu CH, Macdonald‐Wallis C, Gray E, et al. Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis [published correction appears in Neurology. 2015;85(10):921]. Neurology. 2015;84(22):2247‐2257. doi: 10.1212/WNL.0000000000001642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mandrioli J, Biguzzi S, Guidi C, et al. Heterogeneity in ALSFRS‐R decline and survival: a population‐based study in Italy. Neurol Sci. 2015;36(12):2243‐2252. doi: 10.1007/s10072-015-2343-6 [DOI] [PubMed] [Google Scholar]
- 35. Gianferrari G, Martinelli I, Zucchi E, et al. Epidemiological, clinical and genetic features of ALS in the last decade: a prospective population‐based study in the Emilia Romagna region of Italy. Biomedicine. 2022;10(4):819. doi: 10.3390/biomedicines10040819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Simonini C, Zucchi E, Bedin R, et al. CSF heavy neurofilament may discriminate and predict motor neuron diseases with upper motor neuron involvement. Biomedicine. 2021;9(11):1623. doi: 10.3390/biomedicines9111623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deczkowska A, Amit I, Schwartz M. Microglial immune checkpoint mechanisms. Nat Neurosci. 2018;21(6):779‐786. doi: 10.1038/s41593-018-0145-x [DOI] [PubMed] [Google Scholar]
- 38. Ebbert MTW, Ross CA, Pregent LJ, et al. Conserved DNA methylation combined with differential frontal cortex and cerebellar expression distinguishes C9orf72‐associated and sporadic ALS, and implicates SERPINA1 in disease. Acta Neuropathol. 2017;134(5):715‐728. doi: 10.1007/s00401-017-1760-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nikodemova M, Small AL, Smith SM, et al. Spinal but not cortical microglia acquire an atypical phenotype with high VEGF, galectin‐3 and osteopontin, and blunted inflammatory responses in ALS rats. Neurobiol Dis. 2014;69:43‐53. doi: 10.1016/j.nbd.2013.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Puchades M, Hansson SF, Nilsson CL, Andreasen N, Blennow K, Davidsson P. Proteomic studies of potential cerebrospinal fluid protein markers for Alzheimer's disease. Brain Res Mol Brain Res. 2003;118(1–2):140‐146. doi: 10.1016/j.molbrainres.2003.08.005 [DOI] [PubMed] [Google Scholar]
- 41. Subramanian S, Shahaf G, Ozeri E, et al. Sustained expression of circulating human alpha‐1 antitrypsin reduces inflammation, increases CD4+FoxP3+ Treg cell population and prevents signs of experimental autoimmune encephalomyelitis in mice. Metab Brain Dis. 2011;26(2):107‐113. doi: 10.1007/s11011-011-9239-9 [DOI] [PubMed] [Google Scholar]
- 42. Yazdani S, Seitz C, Cui C, et al. T cell responses at diagnosis of amyotrophic lateral sclerosis predict disease progression. Nat Commun. 2022;13(1):6733. doi: 10.1038/s41467-022-34526-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Henkel JS, Beers DR, Wen S, et al. Regulatory T‐lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med. 2013;5(1):64‐79. doi: 10.1002/emmm.201201544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aggarwal N, Korenbaum E, Mahadeva R, et al. α‐Linoleic acid enhances the capacity of α‐1 antitrypsin to inhibit lipopolysaccharide induced IL‐1β in human blood neutrophils. Mol Med. 2016;22:680‐693. doi: 10.2119/molmed.2016.00119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pichet Binette A, Janelidze S, Cullen N, et al. Confounding factors of Alzheimer's disease plasma biomarkers and their impact on clinical performance. Alzheimers Dement. 2022;19:1403‐1414. doi: 10.1002/alz.12787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bojesen SE, Johansen JS, Nordestgaard BG. Plasma YKL‐40 levels in healthy subjects from the general population. Clin Chim Acta. 2011;412(9–10):709‐712. doi: 10.1016/j.cca.2011.01.022 [DOI] [PubMed] [Google Scholar]
- 47. Kelly‐Robinson GA, Reihill JA, Lundy FT, et al. The serpin superfamily and their role in the regulation and dysfunction of serine protease activity in COPD and other chronic lung diseases. Int J Mol Sci. 2021;22(12):6351. doi: 10.3390/ijms22126351 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Table 1
Supplementary Table 2
Data Availability Statement
Anonymized data are available upon request from any qualified investigator.