

Abstract
Background and purpose
Adamantanes were listed as an interesting option as an early intervention against COVID‐19. We aimed to evaluate the effectiveness of amantadine in preventing the progression of COVID‐19 and its neurological sequelae.
Methods
Unvaccinated patients with confirmed SARS‐CoV‐2 infection within 5 days were enrolled. Subjects were randomized (50:50) to amantadine (AMD; 100 mg twice daily) or placebo (PLB) for 14 days. The Ordinal Scale for Clinical Improvement of the World Health Organization (OSCI‐WHO) was the primary measure. Secondary endpoints included assessment for fatigue; depression, disorders of smell and taste, and sleepiness on Days 1 and 15.
Results
We enrolled 99 patients (49 AMD and 50 PLB). Disease progression (OSCI‐WHO = 4) was observed in 6% (AMD) and 8% (PLB) patients (p > 0.05) with further deterioration (OSCI‐WHO〉4) in 0% (AMD) and 8% (PLB) patients (p > 0.05). Complete recovery on Day 15 was 60% higher in the AMD compared with the PLB group (p = 0.025). There was improvement in taste (AMD: p = 0.003; PLB: p = 0.0001) and smell (AMD: p = 0.005; PLB: p = 0.0004) but not in fatigue in both groups. Improvement was observed in the AMD (p = 0.010) but not in the PLB group (p = 0.058) when assessing depression as well as sleepiness (AMD: p = 0.0002; PLB: p = 0.341). There was one death in the PLB group (2.0%) and none in the AMD group (p > 0.05) until Day 210. Overall, the drug was well tolerated.
Conclusion
The central effects of amantadine on the nervous system with reduction of sleepiness and depression might have had a supportive effect on faster recovery in early COVID‐19 patients.
Keywords: amantadine, COVID‐19, neurological and psychiatric complications, outcome, trial
Amantadine enhanced recovery from mild to moderate COVID‐19 in unvaccinated patients. Amantadine improved patients' neurological functions reducing sleepiness and improving mood. Amantadine treatment may alleviate neuro‐COVID‐19 signs and symptoms.
INTRODUCTION
COVID‐19 pandemia has become a global health problem with devastating short‐ and long‐term consequences [1]. Adamantanes were listed as an interesting option in the therapeutic armamentarium among other compounds [2, 3]. In particular, amantadine gained attention as a drug with a complex mechanism of action [4].
Amantadine was widely used as a prophylactic agent against influenza A, and later become a neurological drug. It has been successfully repurposed to Parkinson's disease (PD), and is offered off‐label to multiple sclerosis (MS) patients to alleviate fatigue [4, 5, 6].
A questionnaire‐based study performed during the early phase of the COVID‐19 pandemic provided the first clinical evidence on amantadine's potential protective activity against SARS‐COV‐2 infection progression. All amantadine‐treated subjects with PD and MS who had known exposure to the virus tested positive for SARS‐COV‐2 infection. Interestingly none of them developed overt COVID‐19 despite the burden of neurological comorbidity [7].
This prompted us to review amantadine's pharmacology in the context of possible repurposing for COVID‐19 [8, 9]. We found circumstantial evidence for a few mechanisms whereby this drug could prevent the development of the severe stage of SARS‐CoV‐2 infection. Amantadine may display some antiviral activity against SARS‐COV‐2 virus [10, 11, 12], but even more importantly might be its central nervous system (CNS) effects including neurotransmitter modulation and anti‐inflammatory activity [8]. We assumed that in order to effectively block the vicious cycle leading to life‐threating complications of COVID‐19, treatment with amantadine should be implemented shortly after virus infection occurs. Further studies provided more evidence [13, 14, 15, 16, 17], strengthening the rationale for a proper clinical trial. Considering the above, we designed a study that aimed to evaluate the safety and clinical efficacy of amantadine among unvaccinated participants in the early phase of SARS‐COV‐2 infection.
PATIENTS AND METHODS
Study design and participants
The study protocol was approved by the Ethics Committee of the Medical University of Lublin and subsequently the institutional review boards of the participating hospitals. Trial registration: www.clinicaltrials.gov identification no. NCT04854759; Eudra CT number: 2021‐001144‐98 (dated 27 February 2021).
The full protocol has been published previously [18]. Briefly, this was a prospective, multicenter, randomized, double‐blind, placebo‐controlled trial conducted at seven clinical centers in Poland, coordinated by University Hospital SPSK No. 4 of the Medical University of Lublin. The study included a double‐blinded phase (from Day 1 to Day 15 or optionally Day 30, depending on the patients' clinical status). Inclusion and exclusion criteria are presented in Table S1. We planned on recruiting 200 patients. The study was designed and initiated in the period when vaccinations were not available for the broad population and thus unvaccinated patients were enrolled in the study.
Study participants were randomized (50:50) to treatment with amantadine (study arm A) or placebo (study arm B). The drug was administered orally at a dose of 100 mg twice daily (morning and noon) for a period of 14 days. The dose could be modified in the event of poor tolerance (up to 1 × 1 daily 100 mg) or worsening of clinical symptoms (maximum 4 × 1 per 100 mg for no longer than 2 days, as a loading dose). This treatment was added to the standard care and treatment recommended in the early phase of infection.
Outcomes
Primary outcome measure (from Day 1 to Days 15–30 in double‐blind phase):
Ordinal Scale for Clinical Improvement (OSCI) of the World Health Organization (WHO) [19].
Uninfected – WHO = 0: no clinical or virological evidence of infection.
Ambulatory – OSCI‐WHO = 1: no limitation of activities; WHO = 2 limitation of activities.
Hospitalized – mild disease – WHO = 3: no oxygen therapy; WHO = 4: oxygen by mask or nasal prongs.
Hospitalized – severe disease – WHO = 5: non‐invasive ventilation or high‐flow oxygen; WHO = 6: intubation and mechanical ventilation; WHO = 7: ventilation+additional organ support‐pressors, renal replacement therapy (RRT), extracorporeal membrane oxygenation (ECMO); WHO = 8: death.
Primary endpoint:
Occurrence of clinical worsening defined as any of the following:
Moderate or severe dyspnoea.
Drop in O2 saturation (<92% with patient exposure to room air) and/or additional oxygen demand to maintain O2 saturation ≥92%).
Achievement of ≥4 points on the WHO [OSCI‐WHO] scale (nine‐point clinical status assessment scale).
Meeting the above criteria qualified the patient for further treatment in hospital, in accordance with the current recommendations.
Secondary outcome measures:
Fatigue (Fatigue Severity Scale, FSS) [20].
Depression (Beck Depression Inventory, BDI) [21]
Disorders of smell and taste (visual analogue scales: S‐VAS, T‐VAS) [22].
Sleep disorders (Epworth Sleepiness Scale, ESS) [23].
Secondary endpoints:
Time to clinical deterioration (from Day 1 to Days 15–30).
Time of survival until Day 210.
Statistical analysis
The details are described in a previous publication [18].
Before the database was closed, a statistical analysis plan (SAP) was created describing in detail the planned statistical analysis. Unless otherwise noted, the tests used were two‐sided.
Demographics and baseline characteristics analysis
Demographics and baseline characteristics were analyzed using descriptive methods and compared between the study arms. The descriptive statistics for categorical variables included the number and percentage of occurrences. The descriptive statistics for continuous variables included the mean and standard deviation (SD), the median with the 25th and 75th percentiles (Q1 and Q3), and the minimum and maximum. The distribution of categorical variables was compared between subgroups using either a Fisher test or the chi‐square test, depending on the expected size of the categories. Continuous variables with a normal distribution (the normality of the distribution tested with the Shapiro–Wilk test) were compared between the subgroups using Student's t‐test. Otherwise, the Mann–Whitney test was used.
Primary endpoint analysis
The primary endpoint was assessed on study Day 15. No data imputation methods were applied and data were analyzed as available [18].
Secondary endpoints analysis
Secondary efficacy endpoints of the study were: time to clinical deterioration (assessed up to Days 15–30), overall survival (assessed up to Day 210 of extended follow‐up), and neurological assessment of fatigue, depression, smell and taste, sleep disturbance, and quality of life (assessed on study Days 1 and Days 15–30). Data on secondary endpoints were analyzed using descriptive methods and compared between the study arms (see Demographics and Baseline Characteristics Analysis section for details of the descriptive methods used). Change from baseline for neurological assessment of fatigue, depression, smell and taste, sleep disturbance, and quality of life at consecutive time points were reported and compared between the study arms. In the case of assessing changes in relation to baseline values, Student's t‐test or Wilcoxon's test for dependent groups was used, depending on the normality of the variable distribution.
The percentage of patients cured (WHO = 0) during 15 days of observation (recorded on the 15th day of observation) was assessed. This endpoint was observed within 15 days of observation in patients included in the study. For the 15‐day cure (WHO score 0 on Day 15), the Mantel–Haenszel method calculated the relative benefit (RB) as well as the absolute parameter number needed to treat (NNT). The impact of the intervention on the chance of cure within 15 days of observation from randomization (defined as obtaining a WHO score of 0 on Day 15) taking into account factors such as baseline severity of symptoms (WHO scale), age, gender and risk factors was assessed using multivariate regression model logistics. An exploratory analysis was also carried out in subgroups distinguished on the basis of selected demographic and clinical risk factors. The results were reported using a 95% confidence interval (95% CI) and a statistical significance threshold of 0.05 was assumed.
Randomization and allocation concealment
A secure, web‐based randomization system was used to allocate treatment assignments.
The test was fully blinded (arm A or arm B). The medication in both arms was identically packed to prevent unblinding. All patients meeting the inclusion criteria and none of the exclusion criteria were randomized 50:50 to the study using the central Interactive Web Response System (IWRS). A randomization list was constructed according to a flowchart at each site. Each patient was identified by an individual number. Each patient's number was assigned by the IWRS system. The individual number was assigned to the patient during the consent signing process and could not be used again.
Safety analysis
The analysis of the safety profile was based on the prevalence of adverse effects (AEs) and serious adverse effects (SAEs) and the analysis of safety parameters (vital signs, electrocardiogram [ECG], laboratory parameters). The analysis was carried out using descriptive methods, by the measurement time point. The results were compared between the study arms. Additionally, descriptive statistics for the change in the value of selected safety parameters relative to the baseline value were presented.
RESULTS
Between 3 April 2021 and 15 February 2022, 500 patients were assessed for eligibility and 99 patients were included in the per protocol population and were qualified for final analysis (Figure 1). The study enrollment was stopped in response to the increasing number of vaccinated subjects in the Polish population. The protective effect of vaccinations had primary importance. Thus we did not modify the protocol during the course of the study to include vaccinated patients and this had an impact on the number of enrolled subjects. We reached our pre‐planned criteria for the interim analysis (50% enrollment with randomization) and hereby present the obtained results. Patients were recruited in seven clinical centers with separate multidisciplinary medical care professionals. The study group included infectious diseases, emergency medicine, surgery, neurology and internal medicine specialists engaged to work in the designated COVID‐19 care units in the respective medical centers as part of local medical teams. Typically, patients were admitted in accordance with standard clinical practice and were consulted by a respective specialist after receiving a positive SARS‐CoV‐2 test within 5 days. Patients were invited to enter the clinical trial with the intervention added to standard clinical care. Based on their clinical status, the decision was made whether to admit the patient to the ward or to maintain them in an ambulatory state. We did not test in order to identify virus variants in individual patients. In the period of the trial conductance, SARS‐CoV‐2 variants alpha (trial initiation) and delta (trial completion) were predominant in the Polish population.
FIGURE 1.
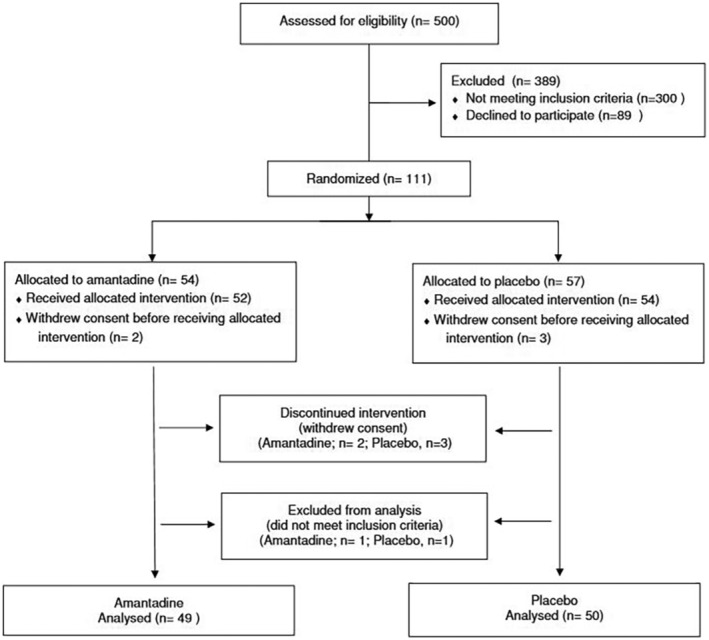
Participant flow diagram.
Participants' baseline characteristics were similar across the amantadine and placebo groups (Table 1). There were 49 in the AMD group (24/25 F/M; mean age 49.9 years) and 50 in the PLB group (23/27 F/M; mean age 46.7 years). On entry into the study (Day 1), 47% of patients randomized to AMD and 45% of patients randomized to PLB required hospitalization due to comorbidities, while the remaining participants were under outpatient observation. A mild course of the disease was observed in the majority of patients in both groups as presented in Table 1. In the AMD group, the disease progression criterion WHO = 4 was observed in three patients (6%), and in the PLB group in four patients (8%) (p > 0.05). In these patients, the administration of the study product was discontinued and standard hospital treatment procedures were implemented, as decided by the attending physician, and clinical follow‐up was continued using the WHO scale based on available medical records until Day 15. Due to the small number of disease progression events described above in the WHO scale, the differences in the probability of recovery including neuropsychiatric symptoms (WHO = 0) on Day 15 were assessed. The probability of recovery on Day 15 was significantly 60% higher in the AMD group compared to the PLB group: RB = 1.58 (95% CI: 1.06, 2.36), p = 0.025, NNT = 5 (95% CI: 3, 25).
TABLE 1.
Demographic and clinical characteristics at randomization – population per protocol (N = 99) and outcome. Disease progression/death (WHO≥4) or recovery (WHO = 0) from COVID‐19 in observations between Day 1 and Day 15 and survival in extended follow‐up until Day 210.
| Characteristic | Amantadine (N = 49) | Placebo (N = 50) | Difference |
|---|---|---|---|
| Gender | |||
| Female | 24 (49.0%) | 23 (46.0%) | 0.767 |
| Male | 25 (51.0%) | 27 (54.0%) | |
| Age (years) | |||
| Mean (SD) [range] | 49.9 (SD 13.9) [18–80] | 46.7 (SD 12.5) [20–79] | 0.305 |
| Median (IQR) | 48 (IQR 62) | 45 (IQR 59) | |
| BMI (kg/m2) | |||
| Mean (SD) [range] | 27.2 (SD 4.5) [20.3–38.8] | 28.0 (SD 4.4) [20.8–42.4] | 0.322 |
| Median (IQR) | 26.6 (IQR 18.4) | 28.1 (IQR 21.5) | |
| Comorbidities and risk factors | |||
| 1 | 25 (51.0%) | 23 (46.0%) | 0.658 |
| 2 | 13 (26.5%) | 17 (34.0%) | |
| 3 | 10 (20.4%) | 10 (20.0%) | |
| 4 | 1 (2.0%) | 0 | |
| Mean (SD) [range] | 1.73 (SD 0.86) [1, 2, 3, 4] | 1.74 (SD 0.78) [1, 2, 3] | 0.832 |
| Median (IQR) | 1 (IQR 3) | 2 (IQR 2) | |
| Comorbidities | >0.05 | ||
| Hypertension | 10 (20%) | 12 (24%) | |
| COPD | 3 (6%) | 4 (8%) | |
| Hypothyroidism | 2 (4%) | 4 (8%) | |
| Diabetes | 5 (10%) | 6 (12%) | |
| Multiple sclerosis | 1 (2%) | – | |
| Rheumatoid arthritis | 1 (2%) | – | |
| Glaucoma | – | 1 (2%) | |
| Dyslipidemia | 2 (4%) | 3 (6%) | |
| Anaemia | – | 1 (2%) | |
| Cardiac insufficiency | 3 (6%) | – | |
| Hospitalization at randomization | |||
| No | 26 (53.1%) | 27 (54.0%) | 0.925 |
| Yes | 23 (46.9%) | 23 (46.0%) | |
| Systolic pressure (mmHg) | |||
| Mean (SD) [range] | 130.0 (SD 15.8) [100.0–170.0] | 128.9 (SD 14.6) [100.0–162.0] | 0.774 |
| Median (IQR) | 130.0 (IQR 70.0) | 126.0 (IQR 62.0) | |
| Diastolic pressure (mmHg) | |||
| Mean (SD) [range] | 81.7 (SD 8.8) [58.0–102.0] | 81.2 (SD 10.3) [55.0–109.0] | 0.450 |
| Median (IQR) | 80.0 (IQR 44.0) | 80.0 (IQR 54.0) | |
| Body temperature (°C) | |||
| Mean (SD) [range] | 36.7 (SD 0.5) [35.8–37.8] | 36.9 (SD 0.5) [36.0–38.4] | 0.215 |
| Median (IQR) | 36.6 (IQR 2.0) | 36.7 (IQR 2.4) | |
| Heart rate (per min) | |||
| Mean (SD) [range] | 81.6 (SD 14.7) [60.0–125.0] | 81.3 (SD 11.8) [54.0–110.0] | 0.750 |
| Median (IQR) | 80.0 (IQR 65.0) | 80.0 (IQR 56.0) | |
| Respiration (per min) | |||
| Mean (SD) [range] | 15.1 (SD 2.1) [12.0–22.0] | 15.3 (SD 2.7) [10.0–24.0] | 0.900 |
| Median (IQR) | 16.0 (IQR 10.0) | 15.0 (IQR 14.0) | |
| Overweight (BMI ≥25 kg/m2) | |||
| No | 19 (38.8%) | 14 (28.0%) | 0.255 |
| Yes | 30 (31.2%) | 36 (72.0%) | |
| Obesity (BMI ≥30 kg/m2) | |||
| No | 36 (73.5%) | 36 (72.0%) | 0.870 |
| Yes | 13 (26.5%) | 14 (28.0%) | |
| Concomitant treatment – PPI | |||
| No | 46 (93.9%) | 45 (60.0%) | 0.479 |
| Yes | 3 (6.1%) | 5 (10.0%) | |
| Concomitant treatment – LMWHs | |||
| No | 31 (63.3%) | 35 (70.0%) | 0.477 |
| Yes | 18 (36.7%) | 15 (30.0%) | |
| Concomitant treatment – antibiotics | |||
| No | 35 (71.4%) | 38 (76.0%) | 0.605 |
| Yes | 14 (28.6%) | 12 (24.0%) | |
| Concomitant treatment – inhaled or systemic corticosteroids | |||
| No | 46 (93.9%) | 43 (86.0%) | 0.193 |
| Yes | 3 (6.1%) | 7 (14.0%) | |
| Concomitant treatment – systemic corticosteroids | |||
| No | 47 (95.9%) | 47 (94.0%) | 0.663 |
| Yes | 2 (4.1%) | 3 (6.0%) | |
| FSS score, mean (SD) median (IQR) [range] | 4.27 (SD 1.49) 5 (IQR 6) [1–7] | 4.13 (SD 1.82) 4 (IQR 6) [1–7] | 0.769 |
| BDI score, mean (SD) median (IQR) [range] | 8.37 (SD 6.58) 8 (IQR 28) [0–28] | 7.57 (SD 6.16) 5 (IQR 23) [0–23] | 0.606 |
| Smell VAS score, mean (SD) median (IQR) [range] | 55.10 (SD 39.07) 70 (IQR 100) [0–100] | 56.10 (SD 40.67) 70 (IQR 100) [0–100] | 0.812 |
| Taste VAS score, mean (SD) median (IQR) [range] | 57.24 (SD 37.04), 70 (IQR 100), [0–100] | 64.90 (SD 37.04) 80 (IQR 100) [0–100] | 0.232 |
| ESS score, mean (SD) median (IQR) [range] | 7.49 (SD 4.61) 7 (IQR 18) [1–19] | 7.18 (SD 4.33) 7 (IQR 20) [0–20] | 0.811 |
| Outcome on Day 15 | Amantadine (N = 49) | Placebo (N = 50) | RB (95% CI), p‐value |
| Progression WHO = 4 | 3 (6.1%) | 4 (8.0%) | 0.77 (95% CI: 0.18; 3.24), p = 0.717 |
| Progression WHO >4 | 0 | 4 (8.0%) | 0.11 (95% CI: 0.01; 2.05), p = 0.1405 |
| Recovery WHO = 0 | 31 (63.3%) | 20 (40.0%) | 1.58 (95% CI: 1.06; 2.36), p = 0.025 |
| Death WHO = 8 | 0 | 1 (2.0%) | 0.34 (95% CI: 0.01; 8.15), p = 0.506 |
Abbreviations: BDI, Beck Depression Inventory; BMI, body mass index; CI, confidence interval; COPD, chronic obstructive pulmonary disease; ESS, Epworth Sleepiness Scale; FSS, Fatigue Severity Scale; IPP, define; IQR, interquartile range; LMWH, low molecular weight heparin; PPI, proton pump inhibitors; RP, relative benefit; SD, standard deviation; VAS, visual analogue scale. WHO, Ordinal Scale for Clinical Improvement of the World Health Organization (OSCI‐WHO).
In the next stage of the analysis, a comparison of disease course indices was performed in patients in both groups based on WHO score on Day 15, assessing the chances of full recovery (WHO = 0) in the logistic regression model in correlation with demographic and clinical characteristics and therapeutic intervention. The analysis also included those patients who met the criterion of disease progression and had investigational product (IP) discontinuation (all received at least two doses of the IP).
In the logistic regression model, the main significant factor related to outcome was the therapeutic intervention (p = 0.028), increasing the chance of recovery. Conversely, being overweight (body mass index [BMI] ≥25 kg/m2) significantly reduced the chance of this endpoint (p = 0.046). Other variables were non‐significant (NS), but overall age ≥45 years reduced the chance of recovery on Day 15 (NS), as did male gender (NS) and hospitalization at study entry (WHO score 3 at Day 1), while the number of the risk factors = 3–4 (NS) chance was slightly higher (but when analyzing the probability, it is very similar, namely 51.2% vs. 52.3% in subgroups 1–2).
Next, an exploratory post hoc analysis of subgroups was performed, taking into account demographic and clinical factors in correlation with the effect of therapeutic intervention.
The subgroup analysis showed results consistent with the main analysis, with all subgroups showing an increase in the likelihood of recovery in the amantadine group. In a subgroup analysis by gender (men vs. women), age (≥45 vs. <45 years), risk of comorbidities (score 3–4 vs. 1–2), presence of overweight (BMI ≥25 kg/m2) and hospitalization or outpatient treatment at randomization (WHO 3 vs. 1–2) results were consistent with the main analysis, with all subgroups showing an increase in recovery rate in the amantadine group (Figure 2a). A stronger effect of amantadine on increasing the probability of recovery on Day 15 was observed in the subgroup of subjects aged ≥45 years (64% vs. 24%; increase in probability 2.7‐fold, p = 0.01), males (68% vs. 30%; increase of 2.3 times, p = 0.001), with a higher risk of comorbidities (3–4) (82% vs. 20%; 4‐fold increase, p = 0.03) and hospitalization at randomization (WHO = 3 on Day1) (65% vs. 30%, 2.1‐fold increase, p = 0.029). The overweight subgroup also had a numerically superior efficacy compared to no risk factors, but the differences were not statistically significant (Figure 2).
FIGURE 2.
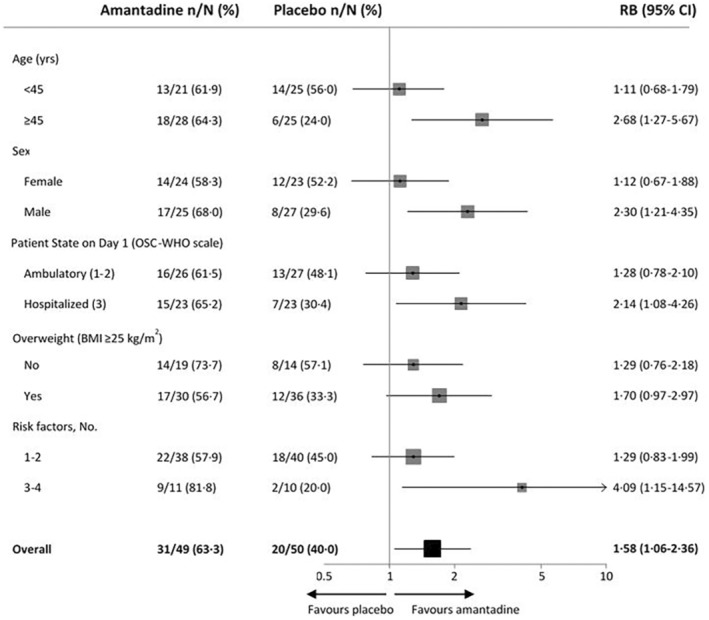
Clinical recovery on Day 15 in adult patients with SARS‐CoV‐2 infection. BMI, body mass index; CI, confidence interval; OSCI‐WHO, Ordinal Scale for Clinical Improvement of the World Health Organization; RB, relative benefit.
In addition, median and mean values for WHO scores on Days 1, 6 and 10 were also compared. There was a significant difference between AMD versus PLB on Day 15 (p = 0.014). The distribution of individual prognostic categories in the two arms of the study was also compared: amantadine versus placebo on Days 1, 6 and 15.
Statistically significant differences in the distribution of clinical disease progression categories in the WHO scale between the groups were shown, indicating a higher probability of improvement of clinical status in the amantadine group (p = 0.028) compared to placebo.
Assessment of secondary endpoints in the course of the disease was performed with comparison of the scores on individual scales for the studied patient populations on Day 1 and Day 15. It should be noted that, according to the protocol of the COV‐PREVENT study, the results obtained on Day 1, in the scales listed, were not the basis for randomization (Table 1).
Only those patients who completed the questionnaires on Day 1 and Day 15 participated in the further analysis (data from several patients were unavailable due to their severe health condition or failure to provide the questionnaires). Overall, there were no significant differences between the groups in terms of scores on individual scales on Day 1 and Day 15. Next, we compared the range of change between Day 15 and Day 1 in each study arm based on the scale results.
A statistically significant improvement in taste scale scores (N = 46; −20.22 [SD 43.23] [95% CI: 7.38, 33.06], p = 0.003; PLB: N = 45; −20.47 [SD 29.64] [95% CI: 11.56, 29.37], p = 0.0001) (Figure 3a) and olfaction (AMD: N = 47; 19.15 [SD 44.62] [95% CI: 6.05, 32.25], p = 0.005; placebo: N = 44; 20.11 [SD 33.89] [95% CI: 9.81, 30.42], p = 0.0004) (Figure 3b) was demonstrated in both groups. There was no significant difference between Day 1 and Day 15 in FSS in both groups (AMD: N = 47 change −0.32 [SD 1.60] [95% CI: −0.79, 0.15], p = 0.123; PLB: N = 45; −0.35 [SD 1.57] [95% CI: −0.82, 0.12], p = 0.166) (Figure 3c). For the BDI score, a statistically significant improvement was observed between Day 15 and Day 1 in the AMD group (N = 47–1.91 [SD 4.98] [95% CI: −3.38; −0, 45], p = 0.010) while not in the PLB group (N = 43; −1.35 [SD 4.92] [95% CI: −2.86, 0.16], p = 0.058) (Figure 3d). Similarly, a significant improvement in the sleepiness scores between Day 15 and Day 1 was seen in the AMD group (N = 47; −2.04 [SD 3.46], [95% CI: −3.06, −1.03], p = 0.0002) but not in the PLB group (N = 45; −0.49 [SD 4.12] [95% CI: −1.73, 0.75], p = 0.341) (Figure 3e).
FIGURE 3.
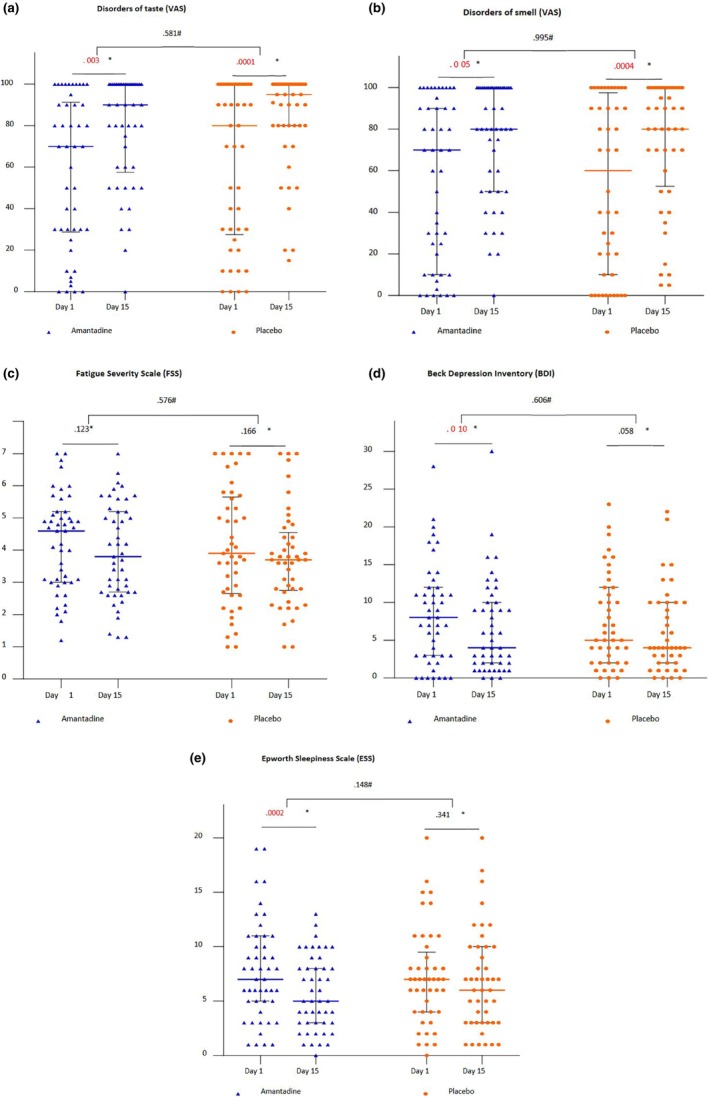
Neurological outcomes. The range of changes between Day 15 and Day 1 in each study arm based on the results of various scales: (a) disorder of taste (VAS); (b) disorder of smell (VAS); (c) fatigue (FSS); (d) depression (BDI); and (e) sleepiness (ESS). *Intra‐group change (Wilcoxon test). #Difference in change between groups (Mann–Whitney test). Lines with error bars represent medians with interquartile ranges. BDI, Beck Depression Inventory; ESS, Epworth Sleepiness Scale; FSS, Fatigue Severity Scale; VAS, visual analogue scale.
Finally, the safety reports were compared between study arms. Overall the drug was well tolerated and there was no statistical difference between AMD and PLB in the safety analysis measures until Day 15. Mortality rate did not change until Day 210. Table 2 presents reported events in different categories of AEs and SAEs between the study groups in the blinded phases of the study. The detailed list of AEs and SAEs is presented in Table S2.
TABLE 2.
Safety reports in study groups in blinded phase of the study (Day 1–Day 15).
| Parameter | Total | Placebo | Amantadine |
|---|---|---|---|
| Safety reports | 96 (100%) | 51 (53%) | 45 (47%) |
| Related to the IP | |||
| Yes | 11 (11%) | 6 | 5 |
| No | 82 (85%) | 42 | 40 |
| Unknown | 3 (4%) | 3 | 0 |
| Severity | |||
| Mild | 74 (77%) | 41 | 33 |
| Moderate | 20 (21%) | 8 | 12 |
| Severe | 2 (2%) | 2 | 0 |
| SAE (serious) | |||
| Yes | 7 (8%) | 4 | 3 |
| No | 87 (90%) | 47 | 40 |
| Unknown | 2 (2%) | 0 | 2 |
Abbreviations: IP, investigional product; SAE, serious adverse event.
DISCUSSION
In this study, patients infected with the SARS‐CoV‐2 virus received standard medical care at an early stage of the disease and the investigational product (amantadine or placebo) was added. Generally, a fairly mild course of the disease was observed in this population. The number of patients included in the study was too small to demonstrate the effect of therapeutic intervention on the risk of disease progression. The results of the extended analysis provided further findings. Administration of amantadine in the early stages of COVID‐19 was found to increase the likelihood of complete recovery from COVID‐19 as assessed on Days 15–30. The above results are consistent with the research hypothesis and data from the literature indicating a pleiotropic effect of the drug. Amantadine hydrochloride was first approved as a prophylactic against Asian flu in 1966. In subsequent years, both prophylactic and therapeutic use of this drug was widely recommended during influenza A epidemics, although its effectiveness was rather moderate and subsequent viral resistance was observed [24]. Its potential usefulness in this indication has been suggested, although due to its action independent of its antiviral activity. An observation on peripheral airway dysfunction in influenza A and its accelerated recovery after taking amantadine has been highlighted [24, 25]. Interestingly, the administration of amantadine significantly reduced the risk of pneumonia in acute stroke patients as the main cause of mortality in this disease group [26]. Our results are also consistent with the work of another group [15] that retrospectively analyzed the course of COVID‐19 in patients with neurological diseases (PD and MS). Patients taking amantadine had a milder COVID‐19 severity compared to controls, which confirmed our first report [7]. It is noteworthy that the drug given too late in more advanced stages of COVID‐19 may be ineffective as described in a recent study [16] that, however, has biases such as not controlling for age and previous exposure to vaccinations in the enrolled population. In another study with a similar design [27], vaccinated patients were included that did not have disease progression and thus the authors were unable to assess the effect of amantadine on the course of COVID‐19. This can be explained by the strong protective effect of vaccination, which obviously diluted any possible activity of amantadine in a cohort of COVID‐19 patients. On the contrary, another article provided important data on the effect of intravenous amantadine added to standard therapy (remdesivir, tocilizumab) on the course of COVID‐19 in patients during ventilator therapy [17]. Interestingly, the addition of amantadine reduced mortality in these critically ill patients (76% amantadine vs. 91% control).
In the current study, we also focused on CNS complications in the course of the disease. Interestingly, there was a statistically significant improvement in mood and arousal between Day 15 and Day 1 in the amantadine‐treated COVID‐19 patients. The aforementioned data indicate that amantadine may have a central effect on the CNS in patients in the early stages of COVID‐19 and act symptomatically by reducing excessive sleepiness and improving mood. This is consistent with the characteristics of the drug and is used clinically in other acute brain injury conditions. The abovementioned effects correlated with the clinical assessment results on WHO with a higher proportion of patients with full recovery on Day 15. This is the first study demonstrating the supportive effects of amantadine to ameliorate neuropsychiatric COVID manifestations in relation to acute infection. Indeed, neuro‐COVID is already well defined as a complex syndrome including headache, fatigue, malaise, anosmia, dysgeusia, somnolence, cognitive dysfunction and psychiatric symptoms during the acute and subacute phase of the disease [28]. The analysis of a 2‐year retrospective cohort showed that the increased incidence of mood and anxiety disorders was an early phenomenon. In addition, the increased risk of psychotic disorder, cognitive deficit, dementia, and epilepsy or seizures persisted throughout [29]. Currently, there are no evidence‐based therapeutic recommendations regarding nervous system complications, although there is ongoing discussion on the possible use of adamantanes and amantadine in particular to treat CNS‐related signs and symptoms as an important component of COVID‐19 complex pathogenesis [30, 31]. Further studies are needed to further investigate the potential efficacy of adamantanes to prevent and/or alleviate CNS‐related neuropsychiatric complications in the course of COVID‐19 and other infectious diseases affecting the nervous system with a long‐term approach and based on larger populations.
CONCLUSIONS
In conclusion, this clinical trial included unvaccinated patients in the early stages of SARS‐COV‐2 infection. These patients remained under standard medical care, and a relatively mild course of the disease was observed. Amantadine treatment enhanced the recovery in these patients to reach asymptomatic status after a 2‐week observation and was safe during the prolonged follow‐up. These observations were explained by amantadine's effects on CNS‐related signs and symptoms.
AUTHOR CONTRIBUTIONS
Konrad Rejdak, Piotr Fiedor, Robert Bonek, Agnieszka Gala‐Błądzińska, Jacek Łukasiak, Ewa Papuć, Adriana Zasybska and Paweł Grieb contributed to the study design. Konrad Rejdak, Piotr Fiedor, Robert Bonek, Agnieszka Gala‐Błądzińska, Jacek Łukasiak, Ewa Papuć, Adriana Zasybska, Paweł Grieb and Marcin Kaczor contributed to data analysis and interpretation. Konrad Rejdak, Piotr Fiedor, Robert Bonek, Agnieszka Gala‐Błądzińska, Waldemar Chełstowski, Sławomir Kiciak and Mateusz Dec led clinical conduct as principal investigators of the clinical sites. Konrad Rejdak, Piotr Fiedor, Agnieszka Gala‐Błądzińska, Robert Bonek, Waldemar Chełstowski, Jacek Łukasiak, Sławomir Kiciak, Piotr Dąbrowski, Mateusz Dec and Adriana Zasybska participated in clinical assessment and data collection. Konrad Rejdak, Paweł Grieb and Paweł Grieb contributed to study bioanalysis. Konrad Rejdak, Piotr Fiedor and Paweł Grieb contributed to study management and execution. The manuscript was written by all the authors, with Konrad Rejdak and Paweł Grieb as the overall lead authors. No person not named as an author contributed to writing the manuscript. Konrad Rejdak, Paweł Grieb and Marcin Kaczor directly accessed and verified the underlying data reported in this manuscript. All authors had full access to all the data in the study and had final responsibility for the decision to submit the manuscript for publication.
FUNDING INFORMATION
The study was funded by Grant No. 2020/ABM/COVID19/SPSK4 from the Medical Research Agency in Poland.
CONFLICT OF INTEREST STATEMENT
K.R. has received speaking honoraria and travel expenses for participation in scientific meetings, and participated in advisory boards previously with Bayer, Biogen, Merck, Novartis, Roche, Sanofi‐Genzyme and Teva Pharmaceuticals. R.B. has received speaking honoraria and travel expenses for participation in scientific meetings, and participated in advisory boards previously with Biogen, CSL Behring, Merck Healthcare KGaA, Novartis, Roche, Sanofi‐Genzyme and Takeda. All the other authors declare no competing interests.
Supporting information
Table S1.
Table S2.
ACKNOWLEDGMENTS
COV‐PREVENT Study Group: Katarzyna Stelmasiak, Aleksandra Cieniuch‐Paciejewska, Marcin Wieczorski, Jan Siwiec, Dorota Szubińska, Ewa Betscher, Piotr Błaszak, Aleksander Goch, Katarzyna Gronowska, Agnieszka Witowska, Agnieszka Wdowczyk, Kamil Bończak, Małgorzata Adamczyk, Adam Pieniążek, Marcelina Grzybowska and Monika Mikołajczyk.
The authors thank Dr. Paweł Pinkosz and SPSK No. 4 team for assistance with site selection and contracting and administrative support.
Rejdak K, Fiedor P, Bonek R, et al. Amantadine in unvaccinated patients with early, mild to moderate COVID‐19: A randomized, placebo‐controlled, double‐blind trial. Eur J Neurol. 2024;31:e16045. doi: 10.1111/ene.16045
The COV‐PREVENT Study Group members are listed in the Appendix.
DATA AVAILABILITY STATEMENT
All data generated in this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Petersen E, Koopmans M, Go U, et al. Comparing SARS‐CoV‐2 with SARS‐CoV and influenza pandemics. Lancet Infect Dis. 2020;20:e238‐e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brenner SR. The potential of memantine and related adamantanes such as amantadine, to reduce the neurotoxic effects of COVID‐19, including ARDS and to reduce viral replication through lysosomal effects. J Med Virol. 2020;92:2341‐2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tipton PW, Wszolek ZK. What can Parkinson's disease teach us about COVID‐19? Neurol Neurochir Pol. 2020;54:204‐206. [DOI] [PubMed] [Google Scholar]
- 4. Nisar T, Sutherland‐Foggio H, Husar W. Antiviral amantadine. Lancet Neurol. 2019;18:1080. [DOI] [PubMed] [Google Scholar]
- 5. Danysz W, Dekundy A, Scheschonka A, Riederer P. Amantadine: reappraisal of the timeless diamond‐target updates and novel therapeutic potentials. J Neural Transm (Vienna). 2021;128:127‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pahwa R. Amantadine: an old drug reborn. Lancet Neurol. 2021;20:975‐977. [DOI] [PubMed] [Google Scholar]
- 7. Rejdak K, Grieb P. Adamantanes might be protective from COVID‐19 in patients with neurological diseases: multiple sclerosis, parkinsonism and cognitive impairment. Mult Scler Relat Disord. 2020;42:102163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grieb P, Rejdak K. Are central nervous system drugs displaying anti‐inflammatory activity suitable for early treatment of COVID‐19? Folia Neuropathol. 2021;59:113‐120. [DOI] [PubMed] [Google Scholar]
- 9. Grieb P, Swiatkiewicz M, Prus K, Rejdak K. Hypoxia may be a determinative factor in COVID‐19 progression. Curr Res Pharmacol Drug Discov. 2021;2:100030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smieszek SP, Przychodzen BP, Polymeropoulos MH. Amantadine disrupts lysosomal gene expression: a hypothesis for COVID19 treatment. Int J Antimicrob Agents. 2020;55:106004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Toft‐Bertelsen TL, Jeppesen MG, Tzortzini E, et al. Amantadine has potential for the treatment of COVID‐19 because it inhibits known and novel ion channels encoded by SARS‐CoV‐2. Commun Biol. 2021;4:1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fink K, Nitsche A, Neumann M, Grossegesse M, Eisele KH, Danysz W. Amantadine inhibits SARS‐CoV‐2 in vitro. Viruses. 2021;13:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mancilla‐Galindo J, García‐Méndez JÓ, Márquez‐Sánchez J, et al. All‐cause mortality among patients treated with repurposed antivirals and antibiotics for COVID‐19 in Mexico City: a real‐world observational study. EXCLI J. 2021;20:199‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aranda‐Abreu GE, Aranda‐Martínez JD, Araújo R, Hernández‐Aguilar ME, Herrera‐Covarrubias D, Rojas‐Durán F. Observational study of people infected with SARS‐Cov‐2, treated with amantadine. Pharmacol Rep. 2020;72:1538‐1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kamel WA, Kamel MI, Alhasawi A, Elmasry S, AlHamdan F, al‐Hashel JY. Effect of pre‐exposure use of amantadine on COVID‐19 infection: a hospital‐based cohort study in patients with Parkinson's disease or multiple sclerosis. Front Neurol. 2021;12:704186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barczyk A, Czajkowska‐Malinowska M, Farnik M, et al. Efficacy of oral amantadine among patients hospitalised with COVID‐19: a randomised, double‐blind, placebo‐controlled, multicentre study. Respir Med. 2023;212:107198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chober D, Czajkowski Z, Aksak‐Wąs B, et al. Improved survival in intensive care unit in severe COVID‐19 associated with amantadine use – retrospective study. Int J Infect Dis. 2022;124:143‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rejdak K, Fiedor P, Bonek R, et al. The use of amantadine in the prevention of progression and treatment of COVID‐19 symptoms in patients infected with the SARS‐ CoV‐2 virus (COV‐ PREVENT): study rationale and desing. Contemp Clin Trials. 2022;116:106755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. World Health Organization . WHO R&D Blueprint. Novel coronavirus. COVID‐19 therapeutic trial synopsis. https://www.who.int/docs/default‐source/blue‐print/covid‐19‐therapeutic‐trial‐synopsis.pdf
- 20. Krupp LB, LaRocca NG, Muir‐Nash J, Steinberg AD. The fatigue severity scale. Application to patients with multiple sclerosis and systemic lupus erythematosus. Arch Neurol. 1989;46:1121‐1123. [DOI] [PubMed] [Google Scholar]
- 21. Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:561‐571. [DOI] [PubMed] [Google Scholar]
- 22. Haxel BR, Bertz‐Duffy S, Fruth K, Letzel S, Mann WJ, Muttray A. Comparison of subjective olfaction ratings in patients with and without olfactory disorders. J Laryngol Otol. 2012;126:692‐697. [DOI] [PubMed] [Google Scholar]
- 23. Johns MW. A new method for measuring daytime sleepiness: the Epworth Sleepiness Scale. Sleep. 1991;14:540‐545. [DOI] [PubMed] [Google Scholar]
- 24. Little JW, Hall WJ, Douglas RG Jr, et al. Amantadine effect on peripheral airways abnormalities in influenza. A study in 15 students with natural influenza a infection. Ann Intern Med. 1976;85:177‐182. [DOI] [PubMed] [Google Scholar]
- 25. Cunha BA. Amantadine may be lifesaving in severe influenza A. Clin Infect Dis. 2006;43:1372‐1373. [DOI] [PubMed] [Google Scholar]
- 26. Nakagawa T, Wada H, Sekizawa K, Arai H, Sasaki H. Amantadine and pneumonia. Lancet. 1999;353:1157. [DOI] [PubMed] [Google Scholar]
- 27. Weis N, Bollerup S, Sund JD, et al. Amantadine for COVID‐19 treatment (ACT) study: a randomized, double‐blinded, placebo‐controlled clinical trial. Clin Microbiol Infect. 2023; S1198‐743X(23)00301‐4. doi: 10.1016/j.cmi.2023.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dangayach NS, Newcombe V, Sonnenville R. Acute neurologic complications of COVID‐19 and postacute sequelae of COVID‐19. Crit Care Clin. 2022;38:553‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taquet M, Sillett R, Zhu L, et al. Neurological and psychiatric risk trajectories after SARS‐CoV‐2 infection: an analysis of 2‐year retrospective cohort studies including 1 284 437 patients. Lancet Psychiatry. 2022;9:815‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Butterworth RF. Potential for the repurposing of adamantane antivirals for COVID‐19. Drugs R D. 2021;21:267‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Müller T, Riederer P, Kuhn W. Aminoadamantanes: from treatment of Parkinson's and Alzheimer's disease to symptom amelioration of long COVID‐19 syndrome? Expert Rev Clin Pharmacol. 2023;16:101‐107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Table S2.
Data Availability Statement
All data generated in this study are available from the corresponding author upon reasonable request.