

Abstract
Background
Alpha‐synuclein (α‐Syn) oligomers and fibrils have been shown to augment the aggregation of TAR DNA‐binding Protein 43 (TDP‐43) monomers in vitro, supporting the idea that TDP‐43 proteinopathies such as ALS may be modulated by the presence of toxic forms of α‐Syn. Recently, parkinsonian features were reported in a study of European patients and Lewy bodies have been demonstrated pathologically in a similar series of patients. Based on these and other considerations, we sought to determine whether seed‐competent α‐Syn can be identified in spinal fluid of patients with ALS including familial, sporadic, and Guamanian forms of the disease.
Methods
Based on the finding that α‐Syn has been found to be a prion‐like protein, we have utilized a validated α‐Synuclein seed amplification assay to determine if seed‐competent α‐Syn could be detected in the spinal fluid of patients with ALS.
Results
Toxic species of α‐Syn were detected in CSF in 18 of 127 ALS patients, 5 of whom were from Guam. Two out of twenty six samples from patients with C9orf72 variant ALS had positive seed‐amplification assays (SAAs). No positive tests were noted in superoxide dismutase type 1 ALS subjects (n = 14). The SAA was negative in 31 control subjects.
Conclusions
Our findings suggest that a sub‐group of ALS occurs in which self‐replicating α‐Syn is detectable and likely contributes to its pathogenesis. This finding may have implications for the diagnosis and treatment of this disorder.
Keywords: alpha‐synuclein, amyotrophic lateral sclerosis, neurodegeneration, seed amplification assay, self‐replicating proteins
INTRODUCTION
Alpha‐synuclein (α‐Syn) is an intracellular protein that, in its fibrillar form, is the major component of Lewy bodies (LBs) and Lewy neurites, which are pathologic hallmarks of Parkinson's disease (PD), LB dementia (LBD), pure autonomic failure, and multiple system atrophy (MSA). Synuclein, first identified in the electric organs of squid, was subsequently isolated from bovine, rat, and human brain [1]. The normal physiological roles of α‐Syn remain unclear. However, among its unique properties is its propensity to sense the curvature of membranes such as synaptic vesicles. This feature may account for its effect on the trafficking of these organelles, and likely its role in the fragmentation or fusion of mitochondria [2, 3, 4].
The native structure of α‐Syn, an intrinsically disordered monomer, is soluble and the most common form resident in the cytoplasm [5]. Although the mechanism underlying aggregation of monomers is not fully understood, the result is the formation of oligomers that assume various morphologies such as beta sheet‐rich oligomers, porelike structures, and amyloid fibrils (Figure 1), the configuration of which may explain their pathologic potential [6].
FIGURE 1.
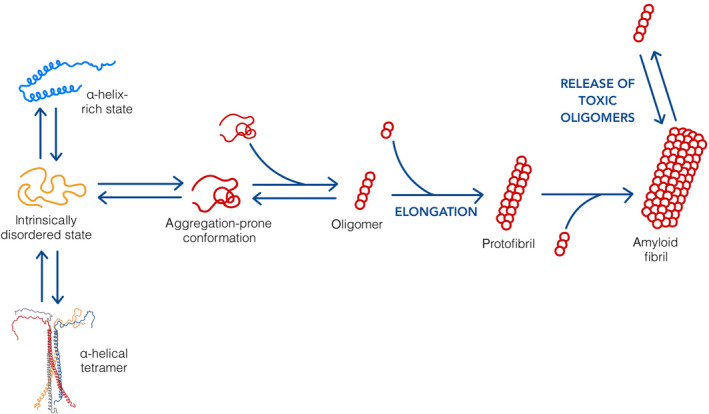
Alpha‐synuclein (α‐Syn) structural dynamics. α‐Syn is an intrinsically disordered protein that can adopt many different conformations. As a monomer, it can form various aggregation‐prone structures that polymerize to form toxic oligomers. These structures elongate into protofibrils and amyloid fibrils via the addition of aggregation‐prone monomers or oligomers. Soluble oligomers may be released from the surface of fibrils, thus causing the harmful cellular effects associated with α‐Syn toxicity.
Current evidence suggests that α‐Syn contributes to disease through a gain of toxic function. Merely a decline in the physiological function of the protein is not enough to induce significant neurodegeneration [3]. Soluble α‐Syn oligomers have been suggested to be more toxic than larger fibrils [7, 8, 9]. It is unknown whether the latter are protective or disruptive in the instance of human disease, albeit in experimental settings oligomers and amyloid fibrils have been observed to induce cellular damage including the disruption of cellular membranes [10]. Notable effects of this on cells and mitochondria include aberrant ion influx/efflux leading to the production of reactive oxygen species and cell death [11].
In 1993, α‐Syn was determined to be a constituent of amyloid plaques in the frontal cortex of Alzheimer's disease (AD) patients [12]. Similar studies in AD utilizing an anti‐tau antibody established that α‐Syn was associated with neurofibrillary tangles [13]. These and other studies led to the notion that α‐Syn is involved in “cross‐seeding,” a process that additionally facilitates aggregation of other seed‐competent proteins such as TAR DNA‐binding Protein 43 (TDP‐43) and FUsed in Sarcoma (FUS).
Considering the time it took for prion proteins to be accepted as the basis for neurodegenerative diseases such as Kuru and Creutzfeldt–Jakob disease (CJD), it is remarkable how readily prion‐like proteins such as α‐Syn have been accepted as etiologic agents in the instance of central nervous system disorders. Following the identification of α‐Syn in LBs, several observations over the next few years led to the conclusion that α‐Syn propagates through the nervous system in a prion‐like manner. A common staging scheme of LB pathology in PD has been suggested: LB pathology first appears in the enteric nervous system and/or the olfactory bulb. In later stages of the disease, it appears in the brainstem, limbic areas, and finally, the cortex. This coincides with the onset of motor, psychiatric, and cognitive signs [14]. In 2008, two independent groups observed LB pathology in fetal graft tissue in the striatum of PD patients, suggesting pathologic spread of the protein to the graft [15, 16]. Following up these observations, Lee et al. [17] conducted a seminal experiment that has broad implications; they demonstrated that α‐Syn fibrils, injected into the striatum and cortex of mice, induced aggregation of native protein that ultimately spread in a prion‐like fashion through the nervous system, leading to the occurrence of pathologic signatures characteristic of PD [18].
Additional studies demonstrating that injection of α‐Syn fibrils into the sciatic nerve or the hind limb of mice can induce pathologic features of PD led to the conclusion that aggregated α‐Syn can travel along peripheral neural pathways [19, 20]. Analogous effects were seen when α‐Syn fibrils were instilled into the tail vein, peritoneum, or gut of mice [21].
Although the presence of LBs is a pathological hallmark of PD, LBD, and MSA, neurodegenerative disorders that are not considered to be synucleinopathies, such as AD, may exhibit LB pathology [22, 23]. This variant of AD displays a faster rate of cognitive decline and shorter survival time when compared to the usual course of the disease [24, 25].
Heretofore, it has been inferred that a subgroup of ALS patients exist in whom α‐Syn may be an etiologic factor. In a study of 108 ALS patients, 11% were found to have LBs at autopsy [22]. Studies of protein interactions point to a link between α‐Syn and misfolded proteins typically associated with ALS [26]. The injection of α‐Syn preformed fibrils derived from recombinant protein into the cortex of superoxide dismutase type 1 (SOD1) G93A mice has been shown to increase the number of SOD1 aggregates in the facial nerve, albeit this procedure did not alter survival. Furthermore, α‐Syn oligomers and fibrils have been shown to augment the aggregation of TDP‐43 monomers in vitro, supporting the idea that TDP‐43 proteinopathies such as amyotrophic lateral sclerosis (ALS) may be modulated by the presence of toxic forms of α‐Syn [27].
In addition to overlapping pathology, clinical associations between ALS and PD have long been observed. The best example is Guamanian ALS Parkinson dementia complex (ALS‐PDC), a neurodegenerative disorder once endemic to islands in the Western Pacific, including the Kii Peninsula in Japan and West New Guinea [28]. Its incidence in the recent past was 50–100 times that of ALS in the United States [29]. Based on its rapid decline over the past several decades [30, 31], the endemic has been attributed to an environmental cause rather than a genetic one [28, 32]. Thus, ALS‐PDC has long been considered a potential “Rosetta Stone” for understanding the pathogenesis of neurodegenerative disorders.
More recently, in a study of 101 European ALS patients, 33% exhibited parkinsonian features: bradykinesia associated with either resting tremor, rigidity, or both. Conceptually, these cases are included under the rubric “ALS‐plus syndrome,” a term that has been used to describe ALS cases that demonstrate clinical symptoms not typically associated with motor neuron disease [33].
Following up on the above considerations, we sought to determine whether seed‐competent α‐Syn can be identified in spinal fluid of patients with ALS including both familial and sporadic forms of the disease as well as cerebrospinal fluid (CSF) obtained from Guamanian ALS patients approximately 50 years ago utilizing a seed amplification assay (SAA) developed at Amprion Inc. This assay has been used to distinguish Parkinson's patients from those with multisystem atrophy and can detect prodromal Parkinson's individuals before diagnosis [34].
METHODS
Samples
One hundred sixty‐two CSF samples were analyzed in this study, 127 of which were obtained from patients with ALS. Four samples were collected from patients with Guamanian PD‐dementia. The remaining 31 samples were collected from patients with non‐neurodegenerative illnesses. Table 1 provides an overview of the clinical diagnoses and demographic data associated with these samples. No significant difference was found in terms of age (p = 0.658) or gender (p = 0.875) between the case group and control group tested in this study. Specific diagnoses of non‐neurodegenerative control samples are provided in Table S1.
TABLE 1.
Demographic data for samples.
| Male | Female | Total | Age, years | |
|---|---|---|---|---|
| Amyotrophic lateral sclerosis | 66 | 61 | 127 | 62.9 ± 10.9 |
| Sporadic | 37 | 40 | 77 | 66.7 ± 10.0 |
| Guamanian | 7 | 3 | 10 | 52.8 ± 10.7 |
| C9orf72 | 15 | 11 | 26 | 60.4 ± 7.53 |
| SOD1 | 7 | 7 | 14 | 53.7 ± 10.9 |
| Controls | 15 | 16 | 31 | 62.4 ± 11.4 |
| Nonneurodegenerative illnesses a | 15 | 16 | 31 | 62.4 ± 11.4 |
See Table S1 for specific control diagnoses.
This study was conducted in a blinded manner. Diagnoses associated with samples from the National ALS Biorepository; University of California, San Diego; Binghamton University; Barrow Neurological Institute; and the Bay Area Lyme Disease Foundation were provided to our researchers after analysis of the samples was complete. The diagnosis associated with samples from Ulm were available upon receipt, requiring us to subsequently blind them before they were sent to Amprion.
α‐Syn SAA
The version of the assay utilized for these studies was validated in the Amprion Clinical Laboratory. It differs from a previously reported research version [34, 35]. In its current iteration, a 2.45‐mm‐diameter borosilicate glass bead is added to each test well of a COSTAR 96‐well plate. The reaction mixture, comprised of 0.5 M PIPES buffer, pH 6.5, 5 mM thioflavin T (fluorescent dye), nuclease‐free water, 5 M NaCl, and recombinant C‐terminal His‐Tagged monomeric human α‐Syn, is subsequently added to each test well. Following addition of the reaction mixture, each CSF sample is added to three wells. The plate is then sealed with an optical adhesive film.
Thioflavin T fluorescence is measured at baseline and once per day for the following 9 days using a FLUOStar Ω Microplate Reader with an excitation wavelength of 440 nm and emission wavelength of 490 nm. When fluorescence values are not being collected, the plate is kept on a microplate shaker (Edmund Buhler TiMix‐5) that is stored within a 37°C incubator. The microplate shaker produces orbital shaking at 800 rpm for 1 min followed by a pause for 29 min, at the conclusion of which fluorescence is measured after each cycle for 150 h.
Following the final measurement, the maximum relative fluorescence units (RFU) of each well are determined. If at least two out of three of the wells for each sample yield a fluorescent value greater than or equal to 25,000 RFU, the sample is classified as “detected,” meaning that it is positive for misfolded α‐Syn (Figure 2).
FIGURE 2.
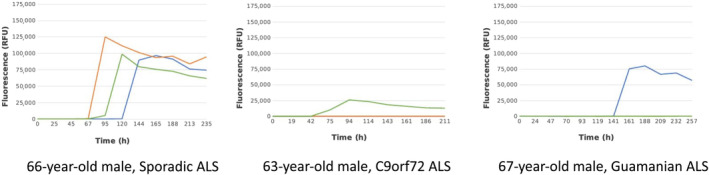
Graphs illustrating a positive and two negative seed amplification assays. ALS, amyotrophic lateral sclerosis; RFU, relative fluorescence units.
Statistical analysis
Proportions (n positive results within group/total n group results) for selected group pairs were compared using the MedCalc chi‐squared calculator. Comparisons were considered significant at p‐values of 0.05 or less.
RESULTS
Demographics
A total of 127 ALS patients were studied, more than half of whom were classified as sporadic ALS. Ten Guamanian ALS cases and 40 familial cases were assessed. The controls (n = 31) were represented by subjects without known neurodegenerative conditions.
Seed amplification assay
Of the 127 ALS CSF samples analyzed in this study, 14% (n = 18) were positive for seed‐competent, fibrillar species of α‐Syn. This included eleven sporadic, five Guamanian, and two C9orf72 ALS subjects. Of the 31 subjects without known neurodegenerative disease, none of them tested positive for α‐Syn. Table 2 gives an overview of the results. The incidence of positive SAA tests in sporadic ALS patients, compared to nonneurodegenerative subjects, was significant (p = 0.0271). Furthermore, the degree of positivity in the small sample of Guamanian patients was notable in comparison to the degree of positivity in the sporadic subgroup (p = 0.0064). Additionally, the difference in positivity between sporadic and Guamanian ALS and the familial cases (C9orf27 and SOD1) was significant (p = 0.0453).
TABLE 2.
α‐Syn seed amplification assay results.
| Positive for toxic α‐Syn | Total | |
|---|---|---|
| Amyotrophic lateral sclerosis | 18 | 127 |
| Sporadic | 11 | 77 |
| Guamanian | 5 | 10 |
| C9orf72 | 2 | 26 |
| SOD1 | 0 | 14 |
| Controls | 0 | 31 |
| Nonneurodegenerative illnesses | 0 | 31 |
Abbreviation: α‐Syn, alpha‐synuclein.
Neuropathologic confirmation
Postmortem brain tissue was available and analyzed for eight of the 10 Guamanian ALS patients (see Table S3). Three of the Guam cases with ALS exhibited clinical and/or pathologic features of PD. The SAA was positive in the instance of only one of these.
The subset of samples (n = 28) obtained from the Barrow Neurological Institute were accompanied by postmortem tissue that was stained for α‐Syn, tau, and amyloid and were examined for LBs and Lewy neurites (see Figure 3).
FIGURE 3.
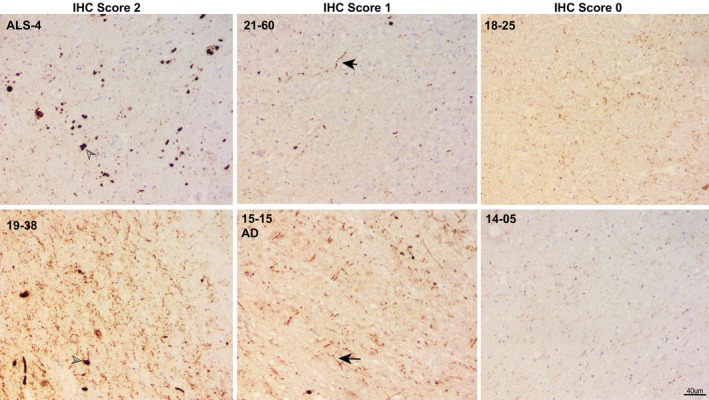
Alpha‐synuclein immunohistochemistry (IHC) staining of midbrain regions of select amyotrophic lateral sclerosis (ALS) cases listed in Table 3 and an additional Alzheimer disease (AD) case, showing examples of the scoring system used. Arrowheads point to Lewy bodies, and arrows show Lewy neurites. Images were captured at 20× magnification (scale bar = 40 μm).
Scoring of tissues for α‐Syn pathology
A subset of ALS cases (n = 28) scored for α‐Syn pathology was provided by the Barrow Neurological Institute. Six of the 28 samples were positive for the presence of either LBs or Lewy neurites. In four of these, self‐replicating α‐Syn was detected utilizing the SAA (Tables 4 and 5).
TABLE 4.
TDP‐43, tau, Aβ, and α‐Syn staining of ALS cases with a positive SAA.
| Case | Location | TDP‐43 | Tau | Aβ | α‐Syn |
|---|---|---|---|---|---|
| ALS3 | FCTX | 0 | NE | 0 | 0 |
| MCTX | 1 | NE | NE | NE | |
| Hippocampus | NE | 1 | 0 | NE | |
| Amygdala | 0 | 1 | NE | NE | |
| Midbrain | NE | NE | NE | 2 | |
| ALS11 | FCTX | 0 | 0 | 2 | 0 |
| MCTX | 2 | NE | NE | NE | |
| Sup and mid temporal | NE | NE | 2 | NE | |
| Hippocampus | 0 | 0 | 2 | NE | |
| Amygdala | NE | 0 | NE | NE | |
| Inf parietal lobule | NE | NE | 2 | NE | |
| OccCTX | NE | NE | 1 | NE | |
| Midbrain | NE | NE | NE | 0 | |
| Cerebellum | 0 | NE | 0 | NE | |
| ALS13 | FCTX | 0 | 0 | 0 | 0 |
| MCTX | 2 | NE | NE | NE | |
| Sup and mid temporal | NE | NE | 0 | NE | |
| Hippocampus | 0 | 0 | 0 | NE | |
| Amygdala | 1 | 2 | NE | NE | |
| Inf parietal lobule | NE | NE | 0 | NE | |
| OccCTX | NE | NE | 0 | NE | |
| Midbrain | NE | NE | 2 | NE | |
| Cerebellum | 0 | NE | 0 | NE | |
| ALS19 a | FCTX | 0 | 0 | 0 | 0 |
| MCTX | 0 | 0 | NE | 0 | |
| Sup and mid temporal | NE | 0 | 0 | 0 | |
| Hippocampus | 0 | 0 | 0 | 0 | |
| Amygdala | NE | 1 | NE | NE | |
| Inf parietal lobule | NE | NE | NE | NE | |
| Basal ganglia/basal forebrain | NE | 1 | NE | 2 | |
| OccCTX | NE | 0 | 0 | NE | |
| Midbrain | NE | NE | NE | 2 | |
| Pons | NE | NE | NE | 2 | |
| Medulla | NE | NE | NE | 2 | |
| Cerebellum | 0 | NE | 0 | NE | |
| ALS20 | FCTX | 2 | 1 | 2 | 0 |
| MCTX | 3 | NE | NE | NE | |
| Sup and mid temporal | NE | NE | 2 | NE | |
| Hippocampus | 1 | 2 | 1 | NE | |
| Amygdala | NE | 2 | NE | NE | |
| Inf parietal lobule | NE | NE | 2 | NE | |
| OccCTX | NE | NE | 2 | NE | |
| Midbrain | NE | NE | 2 | 1 | |
| Cerebellum | 0 | NE | 0 | NE | |
| ALS25 | FCTX | 0 | 0 | 0 | 0 |
| MCTX | 1 | NE | NE | NE | |
| Sup and mid temporal | NE | 0 | 0 | NE | |
| Hippocampus | 1 | 0 | 0 | NE | |
| Amygdala | NE | 0 | NE | NE | |
| Inf parietal lobule | NE | NE | NE | NE | |
| Basal ganglia/basal forebrain | NE | 0 | NE | NE | |
| OccCTX | NE | 0 | 0 | NE | |
| Midbrain | NE | NE | NE | 1 | |
| Cerebellum | 0 | NE | 0 | NE |
Note: See Table S2 for list of antibodies used.
Abbreviations: ALS, amyotrophic lateral sclerosis; Aβ, beta‐amyloid; FCTX, Frontal Cortex; Inf, inferior; MCTX, Middle Cerebral Cortex; NE, not examined; OccCTX, Occipital Cortex; SAA, seed amplification assay; Sup, superior; α‐Syn, alpha‐synuclein.
In respect to ALS19, motor neuorn loss and the presence of p62 inclusions suggests that this case resulted from an FUS or SOD1 mutation.
TABLE 5.
Age comparison for positive and negative samples by group.
| Group | Average age, years |
|---|---|
| Amyotrophic lateral sclerosis | |
| α‐Syn positive | 65.9 ± 10.2 |
| α‐Syn negative | 62.4 ± 11.0 |
| Controls | |
| α‐Syn positive | N/A |
| α‐Syn negative | 62.4 ± 11.4 |
Abbreviations: N/A, not applicable; α‐Syn, alpha‐synuclein.
TABLE 3.
Scoring of tissues for α‐Syn pathology.
| Case | Diagnosis | α‐Syn SAA results | Midbrain synuclein score | C9 +/− | Gender | Age, years |
|---|---|---|---|---|---|---|
| ALS1 | ALS | Negative | 0 | − | F | 76 |
| ALS2 | ALS | Negative | 0 | − | F | 69 |
| ALS3 | ALS | Positive | 2 | + | M | 67 |
| ALS4 | ALS | Negative | 0 | − | F | 66 |
| ALS5 | ALS | Negative | 0 | − | F | 81 |
| ALS6 | ALS | Negative | 0 | − | M | 89 |
| ALS7 | ALS | Negative | 0 | − | F | 61 |
| ALS8 | ALS | Negative | 0 | − | F | 68 |
| ALS9 | ALS | Negative | 0 | − | M | 67 |
| ALS10 | ALS | Negative | 0 | − | M | 74 |
| ALS11 | ALS | Positive | 0 | − | F | 60 |
| ALS12 | ALS | Negative | 0 | − | M | 39 |
| ALS13 | ALS | Positive | 0 | − | M | 83 |
| ALS14 | ALS | Negative | 0 | + | F | 51 |
| ALS15 | ALS, CTE | Negative | 0 | − | M | 44 |
| ALS16 | ALS, AGD | Negative | 0 | − | F | 69 |
| ALS17 | ALS, AGD, mental delay | Negative | 0 | − | F | 61 |
| ALS18 | ALS, meningitis | Negative | 0 | − | M | 78 |
| ALS19 | ALS | Positive | 2 | F | 58 | |
| ALS20 | ALS, FTD‐TDP | Positive | 1 | + | F | 62 |
| ALS21 | ALS, FTD‐TDP, tauopathy | Negative | 0 | − | M | 40 |
| ALS22 | ALS | Negative | 0 | M | 73 | |
| ALS23 | ALS | Negative | 0 | − | M | 67 |
| ALS24 | ALS, FTD | Negative | 0 | F | 82 | |
| ALS25 | ALS | Positive | 1 | M | 69 | |
| ALS26 | ALS | Negative | 0 | M | 62 | |
| ALS27 | ALS | Negative | 0 | F | 56 | |
| ALS28 | ALS | Negative | 0 | M | 54 |
Abbreviations: AGD, argyrophilic grain disease; ALS, amyotrophic lateral sclerosis; CTE, chronic traumatic encephalopathy; F, female; FCTX, frontal cortex; FTD, frontal temporal dementia; FTD‐TDP, frontal temporal dementia subtype characterized by accumulation of TDP‐43; MCTX, middle cerebral cortex; OccCTX, occipital cortex; M, male; SAA, seed amplification assay; α‐Syn, alpha‐synuclein.
Seed amplification results in association with age
Age is thought to be a primary risk factor for the development of synucleinopathies [36]. In this study, the average age of ALS patients with positive SAA results does not differ significantly from ALS patients with a negative SAA result (p = 0.444), suggesting that our detection of pathologic α‐Syn in ALS spinal fluid samples is not a byproduct of normal aging.
DISCUSSION
Our data extend the observations made by others suggesting that there is a subgroup of ALS patients in whom there is clinical and pathologic evidence consistent with the involvement of α‐Syn in the pathogenesis of ALS. This is not totally unexpected, given the involvement of α‐Syn in other neurodegenerative diseases, namely, AD, LBD, and MSA. In our study, including ALS and Guam patients, but excluding those with familial disease, 18% of patients were positive for toxic species of α‐Syn. Among the 40 familial cases of ALS included in our study, only two of them (C9orf72) were found to have pathologic species of α‐Syn, suggesting a lesser role for this protein in the pathogenesis of familial disease. However, C9orf72 mutations have been observed occasionally in PD and MSA, two signature alpha‐synucleopathies.
In the subset of 28 subjects provided by Barrow Neurological Institute, 27 in whom the diagnosis was confirmed pathologically, LB pathology was detected in six subjects (21%). Recently, Robinson et al. published a clinical study involving 108 ALS patients, in whom LBs were noted at autopsy in 11% of subjects [22]. Considering the six Barrow's ALS cases positive for α‐Syn, staining for neurofibrillary tangles (tau) was noted in four of these and in half of the samples amyloid plaques were apparent (beta‐amyloid stain). In two instances (ALS13 and ALS20) both neurofibrillary tangles and amyloid plaques were present.
It is noteworthy that there may be a disconnect between the results of the SAA and the clinical and pathologic features [35]. This was, more or less, the conclusion reached in a recent analysis of Guamanian ALS‐PDC, in which only self‐replicating beta‐amyloid and tau were detected in tissue extracts despite patients exhibiting features of parkinsonism. Furthermore, toxic species of α‐Syn were not isolated from these tissues despite the clinical presentation. A possible explanation is that the assays used have been shown to detect misfolded synuclein aggregates associated with MSA, but not those associated with PD and LBD, which are far more prevalent.
Despite the evidence that misfolded α‐Syn plays a role in the pathogenesis of synucleinopathies, it is controversial whether LBs are protective or disruptive [37]. In regard to ALS, one could conjecture that the protein is interfering with transit of molecules in and out of the nuclear pore via the sequestration of Ras‐related nuclear protein, a recently proposed notion that conceivably explains the mislocation of TDP‐43 that is associated with all but SOD1 ALS [38]. Realistically, an almost unlimited number of deleterious effects could result from misfolding or aggregation of α‐Syn leading to disruption of cellular membranes, mitochondria, Golgi bodies, et cetera [2, 39]. In fact, one cannot exclude that the occurrence of seed‐competent α‐Syn may be an incidental finding without notable consequences.
As previously noted, CSF samples obtained from Target ALS were collected postmortem. We cannot be certain about the collection status of the other samples. Based on current information, this is not expected to affect the results of the SAA (R. Lebovitz, personal communication). Similarly, ordinary storage and use conditions should not affect the assays (i.e., long term storage at −80°C or repeated use of previously frozen CSF).
Considering that the pathogenesis of familial ALS is still not understood years after identification of genetic causes of the disease, we consider it unlikely that the role of α‐Syn will be elucidated anytime soon. The importance of our finding may be best demonstrated in a treatment trial, either by downregulating the expression of α‐Syn or by preventing its aggregation.
Whereas mutations of the α‐Syn gene have never been implicated in the pathogenesis of ALS, it is tempting to attribute our results to an environmental insult. Our finding that some Guamanian CSF samples contain a toxic species of α‐Syn supports this notion, as ALS‐PDC was likely due to consumption of cycad [29]. Daily oral administration of β‐N‐methylamino‐L‐alanine, one of several neurotoxicity constituents of cycad, has been shown to deplete regulatory gut bacteria in mice leading to the formation of α‐Syn aggregates in the ileum, the vagus nerve, and the mesencephalon [40]. If correct, α‐Syn may well prove to be the sentinel equivalent of a canary in a coal mine. Although this idea appeals to us, it cannot be excluded that some, possibly all, of our α‐Syn‐positive samples were due to the coexistence of a mutation in Parkinson‐associated genes such as synuclein alpha (SNCA) or PTEN‐induced putative kinase 1 (PINK1) [41, 42]. If this were the case in the instance of Guamanian ALS, one would not have expected the disease to disappear unless this variant of ALS is an example of a unique interaction between a genetic susceptibility and an environmental toxin [43]. This cannot be excluded since current residents of Guam have been reported to harbor a medley of gene mutations associated with familial PD [44]. For the moment it is unresolved whether mutations of the SNCA or PINK1 genes are responsible for our findings.
Another consideration is the possibility that an unchecked inflammatory process promotes α‐Syn misfolding [45]. Because inflammation is a common feature in ALS and other neurodegenerative disorders, it is possible that the occurrence of misfolded α‐Syn in ALS is a consequence of this. Because inflammation is present in all types of ALS, one would expect the incidence of seed‐competent α‐Syn to be similar between familial and sporadic disease groups. The fact that seed‐competent α‐Syn was identified in C9orf72 familial ALS but not in SOD1 familial ALS could be explained by C9orf72 having well‐recognized immunological functions that may promote inflammation and by extension α‐Syn misfolding. Our data suggest otherwise, but naturally occurring antibodies to both monomeric and oligomeric α‐Syn have been detected in the aged and in patients with PD, suggesting an immune response to aggregation of this protein [46].
Other explanations for our results include the possibility that aggregated SOD1 or TDP‐43 molecules present in ALS spinal fluid could have cross‐seeded the aggregation of α‐Syn monomer in the seed amplification reaction, thus causing a fluorescent signal that would erroneously lead to a positive result. However, this does not appear to be the case, because familial ALS cases showed a lower frequency of misfolded α‐Syn than sporadic ALS. TDP‐43 is present in all ALS cases excluding SOD1 ALS [47]. If cross‐seeding occurred during the assay, we would expect to see an equal frequency of positive results among sporadic and C9orf72 ALS cases. Furthermore, the SOD1 ALS samples did not yield positive results, so it is unlikely that the presence of misfolded SOD1 affected the assay.
It is probable that some of the samples we tested were obtained from patients whose diagnosis was incorrect, because they had not been autopsied. The misdiagnosis rate of ALS by either a primary care provider or specialist has been reported to be 7%–8% [48]. However, this would not explain the seed‐competent α‐Syn positivity in the C9orf72 cases, because these patients were identified by genetic testing as well as clinical phenotypes.
CONCLUSIONS
Our data lead us to conclude that a subgroup of ALS patients harbor seed‐competent α‐Syn that may play a role in the pathogenesis of the disease. The implications for clinical research are obvious, namely, it may be necessary to stratify this group of patients in future trials. Furthermore, if spinal fluid is available from past trials, it may be judicious to determine whether these study results were compromised by failure to exclude this subgroup of patients from the trial analysis. The recent recognition that many cases of neurodegenerative diseases present pathologically as mixed forms with multiple misfolded proteins is consistent with our findings. Because mixed forms may have different clinical presentations and outcomes, a complete biomarker analysis including biomarkers for beta‐amyloid, tau, α‐Syn, and TDP‐43 may become standard diagnostic practice for all patients suspected of having an underlying neurodegenerative disease.
If confirmed, the subgroup of ALS patients we have identified might well be treated by lowering α‐Syn levels with an antisense oligonucleotide [49]. Similarly, the use of a monoclonal antibody targeting α‐Syn, recently shown to reduce α‐Syn pathology and motor impairments in a mouse model, could be a therapeutic advance if successful in a clinical trial [50]. In the instance of PD, experimental treatment with monoclonal antibodies that target α‐Syn failed to demonstrate meaningful clinical results. Conceivably, a therapeutic strategy that is limited to patients in whom a seed‐competent assay was positive at the outset might be more informative.
CONFLICT OF INTEREST STATEMENT
R.L. is the founder and CEO of Amprion. All other authors have no conflicts of interest to report.
ETHICS STATEMENT
No ethics board was needed, because first person consent was obtained for both biospecimen and postmortem collections and the identity of subjects was not provided to us.
LICENSING
The seed amplification methodology utilized by Amprion has been licensed from the University of Texas. The statistical tool employed was acquired from the internet and does not require a license.
Supporting information
TABLE S1
TABLE S2
TABLE S3
ACKNOWLEDGMENTS
The authors acknowledge the editorial assistance of Kathleen Myers, PhD, the statistical assistance of John Middleton, and the logistical support of Cherryl Goldstein. Further, this manuscript is dedicated to Fred Fisher, whose tireless efforts have advanced the care of ALS patients and promoted fruitful research collaborations.
Smith R, Hovren H, Bowser R, et al. Misfolded alpha‐synuclein in amyotrophic lateral sclerosis: Implications for diagnosis and treatment. Eur J Neurol. 2024;31:e16206. doi: 10.1111/ene.16206
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Nakajo S, Omata K, Aiuchi T, et al. Purification and characterization of a novel brain‐specific 14‐kDa protein. J Neurochem. 1990;55(6):2031‐2038. doi: 10.1111/j.1471-4159.1990.tb05792.x [DOI] [PubMed] [Google Scholar]
- 2. Schaser AJ, Osterberg VR, Dent SE, et al. Alpha‐synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci Rep. 2019;9(1):10919. doi: 10.1038/s41598-019-47,227-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sulzer D, Edwards RH. The physiological role of alpha‐synuclein and its relationship to Parkinson's disease. J Neurochem. 2019;150(5):475‐486. doi: 10.1111/jnc.14810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. West A, Brummel BE, Braun AR, Rhoades E, Sachs JN. Membrane remodeling and mechanics: experiments and simulations of alpha‐synuclein. Biochim Biophys Acta. 2016;1858(7 Pt B):1594‐1609. doi: 10.1016/j.bbamem.2016.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eliezer D, Kutluay E, Bussell R Jr, Browne G. Conformational properties of alpha‐synuclein in its free and lipid‐associated states. J Mol Biol. 2001;307(4):1061‐1073. doi: 10.1006/jmbi.2001.4538 [DOI] [PubMed] [Google Scholar]
- 6. Bernal‐Conde LD, Ramos‐Acevedo R, Reyes‐Hernández MA, et al. Alpha‐synuclein physiology and pathology: a perspective on cellular structures and organelles. Front Neurosci. 2020;13:1399. doi: 10.3389/fnins.2019.01399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alam P, Bousset L, Melki R, Otzen DE. α‐Synuclein oligomers and fibrils: a spectrum of species, a spectrum of toxicities. J Neurochem. 2019;150(5):522‐534. doi: 10.1111/jnc.14808 [DOI] [PubMed] [Google Scholar]
- 8. Cascella R, Bigi A, Cremades N, Cecchi C. Effects of oligomer toxicity, fibril toxicity and fibril spreading in synucleinopathies. Cell Mol Life Sci. 2022;79(3):174. doi: 10.1007/s00018-022-04166-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Winner B, Jappelli R, Maji SK, et al. In vivo demonstration that alpha‐synuclein oligomers are toxic. Proc Natl Acad Sci USA. 2011;108(10):4194‐4199. doi: 10.1073/pnas.1100976108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fusco G, Chen SW, Williamson PTF, et al. Structural basis of membrane disruption and cellular toxicity by α‐synuclein oligomers. Science. 2017;358(6369):1440‐1443. doi: 10.1126/science.aan6160 [DOI] [PubMed] [Google Scholar]
- 11. Cascella R, Chen SW, Bigi A, et al. The release of toxic oligomers from alpha‐synuclein fibrils induces dysfunction in neuronal cells. Nat Commun. 2021;12(1):1814. doi: 10.1038/s41467-021-21937-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ueda K, Fukushima H, Masliah E, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA. 1993;90(23):11282‐11286. doi: 10.1073/pnas.90.23.11282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garruto RM, Fukatsu R, Yanagihara R, Gajdusek DC, Hook G, Fiori CE. Imaging of calcium and aluminum in neurofibrillary tangle‐bearing neurons in parkinsonism‐dementia of Guam. Proc Natl Acad Sci USA. 1984;81(6):1875‐1879. doi: 10.1073/pnas.81.6.1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Braak H, Del Tredici K, Bratzke H, Hamm‐Clement J, Sandmann‐Keil D, Rüb U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson's disease (preclinical and clinical stages). J Neurol. 2002;249(Suppl 3):III/1‐III/5. doi: 10.1007/s00415-002-1301-4 [DOI] [PubMed] [Google Scholar]
- 15. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body‐like pathology in long‐term embryonic nigral transplants in Parkinson's disease. Nat Med. 2008;14(5):504‐506. doi: 10.1038/nm1747 [DOI] [PubMed] [Google Scholar]
- 16. Li JY, Englund E, Holton JL, et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host‐to‐graft disease propagation. Nat Med. 2008;14(5):501‐503. doi: 10.1038/nm1746 [DOI] [PubMed] [Google Scholar]
- 17. Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological α‐synuclein initiates a rapidly progressive neurodegenerative α‐synucleinopathy in mice. J Exp Med. 2012;209(5):975‐986. doi: 10.1084/jem.20112457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim S, Kwon SH, Kam TI, et al. Transneuronal propagation of pathologic α‐synuclein from the gut to the brain models Parkinson's disease. Neuron. 2019;103(4):627‐641.e7. doi: 10.1016/j.neuron.2019.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ayers JI, Riffe CJ, Sorrentino ZA, et al. Localized induction of wild‐type and mutant alpha‐synuclein aggregation reveals propagation along neuroanatomical tracts. J Virol. 2018;92(18):e00586‐18. doi: 10.1128/JVI.00586-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sacino AN, Brooks M, Thomas MA, et al. Intramuscular injection of alpha‐synuclein induces CNS alpha‐synuclein pathology and a rapid‐onset motor phenotype in transgenic mice. Proc Natl Acad Sci USA. 2014;111(29):10732‐10737. doi: 10.1073/pnas.1321785111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manfredsson FP, Luk KC, Benskey MJ, et al. Induction of alpha‐synuclein pathology in the enteric nervous system of the rat and non‐human primate results in gastrointestinal dysmotility and transient CNS pathology. Neurobiol Dis. 2018;112:106‐118. doi: 10.1016/j.nbd.2018.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robinson JL, Lee EB, Xie SX, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age‐related and APOE4‐associated. Brain. 2018;141(7):2181‐2193. doi: 10.1093/brain/awy146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Visanji NP, Lang AE, Kovacs GG. Beyond the synucleinopathies: alpha synuclein as a driving force in neurodegenerative comorbidities. Transl Neurodegener. 2019;8:28. doi: 10.1186/s40035-019-0172-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arnold MR, Coughlin DG, Brumbach BH, et al. α‐Synuclein seed amplification in CSF and brain from patients with different brain distributions of pathological α‐synuclein in the context of Co‐pathology and non‐LBD diagnoses. Ann Neurol. 2022;92(4):650‐662. doi: 10.1002/ana.26453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chung EJ, Babulal GM, Monsell SE, Cairns NJ, Roe CM, Morris JC. Clinical features of Alzheimer disease with and without Lewy bodies. JAMA Neurol. 2015;72(7):789‐796. doi: 10.1001/jamaneurol.2015.0606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koch Y, Helferich AM, Steinacker P, et al. Aggregated α‐synuclein increases SOD1 oligomerization in a mouse model of amyotrophic lateral sclerosis. Am J Pathol. 2016;186(8):2152‐2161. doi: 10.1016/j.ajpath.2016.04.008 [DOI] [PubMed] [Google Scholar]
- 27. Dhakal S, Wyant CE, George HE, Morgan SE, Rangachari V. Prion‐like C‐terminal domain of TDP‐43 and α‐synuclein interact synergistically to generate neurotoxic hybrid fibrils. J Mol Biol. 2021;433(10):166953. doi: 10.1016/j.jmb.2021.166953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Spencer PS. Parkinsonism and motor neuron disorders: lessons from Western Pacific ALS/PDC. J Neurol Sci. 2022;433:120021. doi: 10.1016/j.jns.2021.120021 [DOI] [PubMed] [Google Scholar]
- 29. Spencer PS, Palmer VS, Kisby GE. Western Pacific ALS‐PDC: evidence implicating cycad genotoxins. J Neurol Sci. 2020;419:117185. doi: 10.1016/j.jns.2020.117185 [DOI] [PubMed] [Google Scholar]
- 30. Garruto RM, Yanagihara R, Gajdusek DC. Disappearance of high‐incidence amyotrophic lateral sclerosis and parkinsonism‐dementia on Guam. Neurology. 1985;35(2):193‐198. doi: 10.1212/wnl.35.2.193 [DOI] [PubMed] [Google Scholar]
- 31. Plato CC, Garruto RM, Galasko D, et al. Amyotrophic lateral sclerosis and parkinsonism‐dementia complex of Guam: changing incidence rates during the past 60 years. Am J Epidemiol. 2003;157(2):149‐157. doi: 10.1093/aje/kwf175 [DOI] [PubMed] [Google Scholar]
- 32. Bradley WG, Cox PA. Beyond Guam: cyanobacteria, BMAA and sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10(Suppl 2):5‐6. doi: 10.3109/17482960903268676 [DOI] [PubMed] [Google Scholar]
- 33. McCluskey L, Vandriel S, Elman L, et al. ALS‐plus syndrome: non‐pyramidal features in a large ALS cohort. J Neurol Sci. 2014;345(1–2):118‐124. doi: 10.1016/j.jns.2014.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Siderowf A, Concha‐Marambio L, Lafontant DE, et al. Assessment of heterogeneity among participants in the Parkinson's progression markers initiative cohort using α‐synuclein seed amplification: a cross‐sectional study. Lancet Neurol. 2023;22(5):407‐417. doi: 10.1016/S1474-4422(23)00109-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Condello C, Ayers JI, Dalgard CL, et al. Guam ALS‐PDC is a distinct double‐prion disorder featuring both tau and Aβ prions. Proc Natl Acad Sci USA. 2023;120(13):e2220984120. doi: 10.1073/pnas.2220984120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Collier TJ, Kanaan NM, Kordower JH. Ageing as a primary risk factor for Parkinson's disease: evidence from studies of non‐human primates. Nat Rev Neurosci. 2011;12(6):359‐366. doi: 10.1038/nrn3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chartier S, Duyckaerts C. Is Lewy pathology in the human nervous system chiefly an indicator of neuronal protection or of toxicity? Cell Tissue Res. 2018;373(1):149‐160. doi: 10.1007/s00441-018-2854-6 [DOI] [PubMed] [Google Scholar]
- 38. Aizawa H, Yamashita T, Kato H, Kimura T, Kwak S. Impaired nucleoporins are present in sporadic amyotrophic lateral sclerosis motor neurons that exhibit Mislocalization of the 43‐kDa TAR DNA‐binding protein. J Clin Neurol. 2019;15(1):62‐67. doi: 10.3988/jcn.2019.15.1.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vicario M, Cieri D, Brini M, Cali T. The close encounter between alpha‐synuclein and mitochondria. Front Neurosci. 2018;12:388. doi: 10.3389/fnins.2018.00388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Esteves AR, Munoz‐Pinto MF, Nunes‐Costa D, et al. Footprints of a microbial toxin from the gut microbiome to mesencephalic mitochondria. Gut. 2023;72(1):73‐89. doi: 10.1136/gutjnl-2021-326,023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cresto N, Gardier C, Gubinelli F, et al. The unlikely partnership between LRRK2 and α‐synuclein in Parkinson's disease. Eur J Neurosci. 2019;49(3):339‐363. doi: 10.1111/ejn.14182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Flagmeier P, Meisl G, Vendruscolo M, et al. Mutations associated with familial Parkinson's disease alter the initiation and amplification steps of α‐synuclein aggregation. Proc Natl Acad Sci USA. 2016;113(37):10328‐10333. doi: 10.1073/pnas.1604645113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Garruto RM. A commentary on neuronal degeneration and cell death in Guam ALS and PD: an evolutionary process of understanding. Curr Alzheimer Res. 2006;3(4):397‐401. doi: 10.2174/156720506778249425 [DOI] [PubMed] [Google Scholar]
- 44. Sieh W, Choi Y, Chapman NH, et al. Identification of novel susceptibility loci for Guam neurodegenerative disease: challenges of genome scans in genetic isolates. Hum Mol Genet. 2009;18(19):3725‐3738. doi: 10.1093/hmg/ddp300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lema Tomé CM, Tyson T, Rey NL, Grathwohl S, Britschgi M, Brundin P. Inflammation and α‐synuclein's prion‐like behavior in Parkinson's disease – is there a link? Mol Neurobiol. 2013;47(2):561‐574. doi: 10.1007/s12035-012-8267-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pandey MK. The role of alpha‐synuclein autoantibodies in the induction of brain inflammation and neurodegeneration in aged humans. Front Aging Neurosci. 2022;14:902191. doi: 10.3389/fnagi.2022.902191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Suk TR, Rousseaux MWC. The role of TDP‐43 mislocalization in amyotrophic lateral sclerosis. Mol Neurodegener. 2020;15(1):45. doi: 10.1186/s13024-020-00397-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol. 2020;27(10):1918‐1929. doi: 10.1111/ene.14393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith RA, Miller TM, Yamanaka K, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116(8):2290‐2296. doi: 10.1172/JCI25424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tran HT, Chung CH, Iba M, et al. Alpha‐synuclein immunotherapy blocks uptake and templated propagation of misfolded alpha‐synuclein and neurodegeneration. Cell Rep. 2014;7(6):2054‐2065. doi: 10.1016/j.celrep.2014.05.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1
TABLE S2
TABLE S3
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.