

Abstract
Introduction
Fatty‐acid oxidation disorders (FAODs) are recessive genetic diseases.
Materials and methods
We report here clinical and paraclinical data from a retrospective study of 44 adults with muscular FAODs from six French reference centers for neuromuscular or metabolic diseases.
Results
The study cohort consisted of 44 adult patients: 14 with carnitine palmitoyl transferase 2 deficiency (32%), nine with multiple acyl‐CoA deficiency (20%), 13 with very long‐chain acyl‐CoA dehydrogenase deficiency (30%), three with long‐chain 3‐hydroxyacyl‐CoA dehydrogenase deficiency (7%), and five with short‐chain acyl‐CoA dehydrogenase deficiency (11%). Disease onset occurred during childhood in the majority of patients (59%), with a mean age at onset of 15 years (range = 0.5–35) and a mean of 12.6 years (range = 0–58) from disease onset to diagnosis. The principal symptoms were acute muscle manifestations (rhabdomyolysis, exercise intolerance, myalgia), sometimes associated with permanent muscle weakness. Episodes of rhabdomyolysis were frequent (84%), with a mean creatinine kinase level of 68,958 U/L (range = 660–300,000). General metabolic complications were observed in 58% of patients, respiratory manifestations in 18% of cases, and cardiological manifestations in 9% of cases. Fasting acylcarnitine profile was used to orient genetic explorations in 65% of cases. After a mean follow‐up of 10 years, 33% of patients were asymptomatic and 56% continued to display symptoms after exercise. The frequency of rhabdomyolysis decreased after diagnosis in 64% of cases.
Conclusion
A standardized register would complete this cohort description of muscular forms of FAODs with exhaustive data, making it possible to assess the efficacy of therapeutic protocols in real‐life conditions and during the long‐term follow‐up of patients.
Keywords: acylcarnitine profile, beta‐oxidation, exercise intolerance, fatty acids, genetic analyses, long‐term improvement, muscle weakness, rhabdomyolysis
INTRODUCTION
Fatty‐acid oxidation disorders (FAODs) are rare genetic diseases with a recessive mode of inheritance. Most fatty‐acid oxidation occurs via mitochondrial beta‐oxidation (90%), which involves several enzymes [1]. Each of these enzymes may be dysfunctional, resulting in the existence of a number of different diseases. However, all these diseases have similar clinical characteristics. Figure 1 summarizes mitochondrial fatty acid oxidation and the enzymes involved.
FIGURE 1.

Mitochondrial fatty‐acid oxidation. FABPpm, fatty‐acid binding protein; FADH2, reduced flavin‐adenine‐dinucleotide; CACT, carnitine acylcarnitine translocase; CPT, carnitine‐palmitoyl transferase; ETF, electron transfer flavoprotein; FAD, flavin–adenine–dinucleotide; LCAD, long‐chain acyl‐CoA dehydrogenase; LCEH, long‐chain enoyl‐CoA hydratase; LCFA, long‐chain fatty acid; LCHAD, long‐chain 3‐hydroxyacyl‐CoA dehydrogenase; LKCAT, long‐chain 3‐ketoacyl‐CoA thiolase; M/SCHAD, medium‐/short‐chain 3‐hydroxyacyl‐CoA dehydrogenase; MCAD, medium‐chain acyl‐CoA dehydrogenase; MCFA, medium‐chain fatty acid; MKCAT, medium‐chain 3‐ketoacyl‐CoA thiolase; NAD, nicotinamide‐adenine‐dinucleotide; NADH2, reduced nicotinamide‐adenine‐dinucleotide; OCTN2, organic cation transporter type 2; SCAD, short‐chain acyl‐CoA dehydrogenase; SCFA, short‐chain fatty acid; TFP, trifunctional protein; VLCAD, very long‐chain acyl‐CoA dehydrogenase.
The most frequent forms have a childhood onset. The neonatal form begins during the first month of life. It is a severe multivisceral disease, with a predominance of cardiac (heart failure), metabolic (vomiting, severe nonketotic hypoglycemia, lactic acidosis, hyperammonemia), and central nervous system (seizure, encephalopathy, coma) involvement. The childhood form begins during the first 10 years of life. Cardiac, metabolic, and respiratory (respiratory failure) involvement also predominate in this form. Both these pediatric forms are life‐threatening. Neonatal diagnostic techniques have been developed for the rapid identification of these forms, so that treatment can be initiated quickly [2, 3, 4].
The muscular form starts later, typically during adolescence or adulthood, with a predominance of acute muscle episodes (myalgia, myoglobinuria, rhabdomyolysis, and exercise intolerance). There may be an associated permanent muscle weakness and, more rarely, involvement of other organs. This form is also potentially life‐threatening, mostly due to rhabdomyolysis with renal failure, but also due to cardiac events. The diagnosis of these myopathies remains difficult, and the interval between symptom onset and diagnosis remains long. However, diagnostic methods have improved in recent years, with the training of myologists concerning the utility of acylcarnitine profiles and the development of genetic and functional analyses [5, 6].
We describe here clinical and paraclinical data for an adult population with muscular FAODs. This population of patients presented various different FAODs: carnitine palmitoyl transferase 2 (CPTII) deficiency, multiple acyl‐CoA (MAD) deficiency, very‐long‐chain acyl‐CoA dehydrogenase (VLCAD) deficiency, long‐chain 3‐hydroxy‐acyl‐CoA (LCHAD) deficiency, and short‐chain acyl‐CoA dehydrogenase (SCAD) deficiency. The aim was to improve both our knowledge of the natural course of these diseases and diagnostic performance based on noninvasive explorations, and to define optimal care for these patients.
MATERIALS AND METHODS
We retrospectively collected data for patients from six French reference centers for neuromuscular or metabolic diseases. Patients were included in this study if they had a genetically diagnosed FAOD, were >18 years old, had at least one muscle symptom (myalgia, rhabdomyolysis, exercise intolerance, or permanent muscle deficit), and did not oppose the collection of their data.
We collected data for demographic characteristics, symptoms, the results of clinical examinations, the results of evaluations (diagnostic and organ involvement), and the long‐term clinical course of the disease.
We determined prevalence for qualitative variables and position and distribution parameters for quantitative variables. Considering this small cohort, we made group comparisons only for variables for which data were available for at least 80% of the patients. Chi‐squared tests were used to compare qualitative data; Kruskal–Wallis tests followed by Mann–Whitney tests were used to compare quantitative data. As this study was retrospective in nature, express consent was not required from patients and their nonopposition to the use of their data was considered sufficient, in accordance with French law. On inclusion, each patient received an information note concerning the processing of their personal data and their rights, including, in particular, the right to oppose the use of their data.
RESULTS
Patients and muscle symptoms/inaugural features
Clinical presentation is summarized in Figure 2.
FIGURE 2.
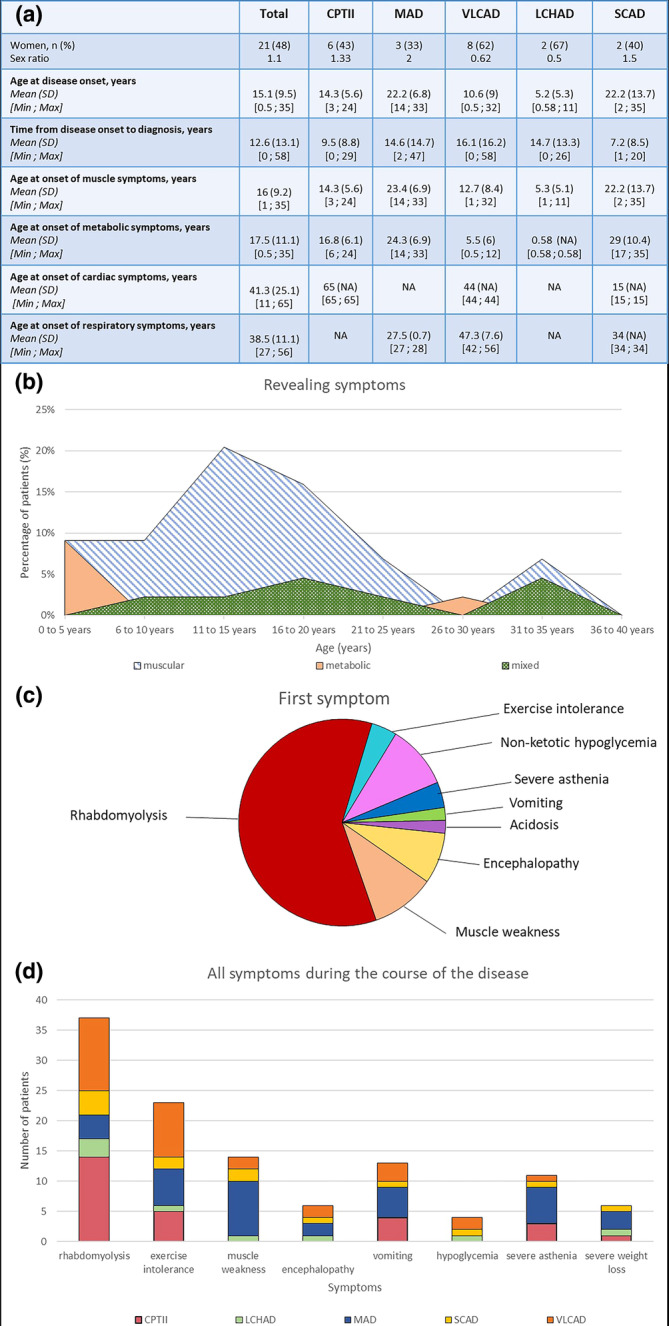
Clinical presentation of the cohort. (a) Age at the onset of symptoms and time to diagnosis. (b) Type of revealing symptoms by age group. (c) Inaugural symptom revealing a fatty‐acid oxidation disorder. (d) All muscular and metabolic symptoms occurring during the course of the disease. CPTII, carnitine palmitoyl transferase 2; LCHAD, long‐chain 3‐hydroxy‐acyl‐CoA; MAD, multiple acyl‐CoA; NA, not available; SCAD, short‐chain acyl‐CoA dehydrogenase; VLCAD, very‐long‐chain acyl‐CoA dehydrogenase.
This cohort included 44 patients: 14 with CPTII deficiency, nine with MAD deficiency, 13 with VLCAD deficiency, three with LCHAD deficiency, and five with SCAD deficiency. Twenty‐one of the patients were female (48%). Mean age at disease onset was 15 years (±9.5, range = 0.5–35), and the time from symptom onset to diagnosis was 12.6 years (±13.1, range = 0–58). Symptoms began during childhood in 26 patients (59%), 10 of whom were diagnosed during childhood (23%), the other 16 not being diagnosed until adulthood (36%). The disease began during adulthood in 18 patients (41%; Figure 2a).
The first symptoms were muscular (rhabdomyolysis, muscle weakness, exercise intolerance, 68%) or metabolic (non‐ketotic hypoglycemia, severe asthenia, vomiting, encephalopathy, acidosis, 16%), with a mixture of metabolic and muscular symptoms in 16% of cases. The revealing symptoms were predominantly metabolic for cases with an onset in early childhood (0.5–5 years) but also between 26 and 30 years for a few patients. They were predominantly muscular in cases with an onset later in childhood or in early adulthood (5–30 years). A mixture of muscular and metabolic revealing symptoms occurred in patients of all age groups, but at a higher frequency in patients with a disease onset during adolescence or early adulthood (15–25 years) or after the age of 30 years (Figure 2b).
Muscle symptoms began at a mean age of 16 years (±9.2, range = 1–35), and were the symptoms that revealed the disease in patients aged between 5 and 30 years. The most frequent muscle symptom was rhabdomyolysis (84%), which was triggered by exercise (75%), fasting (50%), fever (32%), or infection (25%). Most patients (75%) had a mean of ≤2 rhabdomyolysis episodes per year. The other muscle symptoms were exercise intolerance (52%) and permanent muscle deficit (32%), predominantly proximal limb deficits. Permanent muscle weakness was observed in all patients with MAD deficiency (Figure 2c,d). Twenty‐five patients had metabolic symptoms (58%), with a mean age at onset of 17 years (±11, range = 0.5–35). The following metabolic symptoms were reported: severe vomiting (52%), severe asthenia (44%), encephalopathy or coma (24%), severe weight loss (24%), and nonketotic hypoglycemia (16%).
Most of the patients in the cohort had already had general anesthesia (63%), and complications were observed in only a small proportion (7%). These complications included fever after halogen‐based anesthesia (VLCAD deficiency) and vomiting episodes after curare (CPTII deficiency). Many of the patients had practiced sports during childhood (73%), frequently with limitation during exercise (72%) and abnormal muscle aches (83%). Maternal muscle complications were observed during childbirth in 21% of cases (n = 3 VLCAD deficiency and n = 1 SCAD deficiency). Two episodes of pancreatitis were reported (4%), including one after rhabdomyolysis (VLCAD deficiency) and one with no identified etiology (CPTII deficiency).
Diagnostic examinations
Figure 3 summarizes the results of key muscle explorations for FAOD diagnosis.
FIGURE 3.

Biological tests. (a) Creatine phosphokinase (CK) level at rest and during rhabdomyolysis. (b) Results of urinary explorations. CPTII, carnitine palmitoyl transferase 2; LCHAD, long‐chain 3‐hydroxy‐acyl‐CoA; MAD, multiple acyl‐CoA; SCAD, short‐chain acyl‐CoA dehydrogenase; VLCAD, very‐long‐chain acyl‐CoA dehydrogenase.
Mean creatine phosphokinase (CK) level at rest was 660 U/L (±1573, range = 64–8039) and did not differ significantly between groups. CK levels at rest were normal in most cases, but high in 14 patients (n = 2 CPTII deficiency, n = 4 MAD deficiency, n = 6 VLCAD deficiency, and n = 2 SCAD deficiency). Mean CK level during rhabdomyolysis was 68,958 U/L (±78,573, range = 660–300,000; Figure 3a).
Fasting acylcarnitine profile (ACP) was abnormal for 31 patients (79%) during the first exploration. Only one of the patients with SCAD deficiency (20%) had an abnormal initial ACP, with high levels of short‐chain acylcarnitines (C4). Six of the patients with CPTII deficiency had high levels of long‐chain acylcarnitines (46%, [C16 + C18:1]/C2). All patients with MAD deficiency had high levels of all acylcarnitines. Only one of the patients with LCHAD deficiency had an abnormal initial ACP, with high levels of long‐chain acylcarnitines (C14OH, C16OH, C18OH, C18:1OH). ACP was abnormal in all the patients with VLCAD deficiency, and 11 of them (92%) had high levels of long‐chain acylcarnitines (C14:1, C14:1/C12:1). At some centers, fluxomic analyses were performed on whole‐blood samples from all patients with abnormal ACP, to distinguish between normal and disrupted β‐oxidation. These analyses confirmed the diagnosis and led to requests for genetic analysis, but were not available at all centers.
Initial fasting ACP was normal in 10 patients (n = 5 CPTII deficiency, n = 1 LCHAD deficiency, and n = 4 SCAD deficiency). For three of these 10 patients, ACP became abnormal during follow‐up (n = 2 CPTII deficiency and n = 1 SCAD deficiency). For patients with persistent normal fasting ACP, or if ACP was not performed (n = 5 patients), if the diagnostic was strong, diagnosis was made directed by genetic analyses given the clinical history of repeated rhabdomyolysis with urinary excretion of ethylmalonic acid.
Urinary organic acid excretion was observed in seven patients (32%), and the proportion of patients with this sign was greater among those with MAD deficiency (p = 0.46; Figure 3b).
Muscle biological and neurophysiological assessments
Muscle biopsy
Twenty‐nine patients (66%) underwent muscle biopsy. The results were normal for 11 of these patients (38%); increase of fatty‐content was present in 38% of cases (n = 5 MAD deficiency, n = 2 SCAD deficiency, and n = 4 VLCAD deficiency) and mitochondrial respiratory chain involvement was observed in 10% of cases (n = 3 MAD deficiency).
Electromyogram
An electromyogram was performed for 31 patients (70%), and the results were normal for 21 of these patients (68%). The abnormal electromyograms revealed myogenic syndrome in 60% of cases (n = 3 MAD deficiency, n = 2 SCAD deficiency, and n = 1 VLCAD deficiency) and axonal neuropathy in 40% of cases (n = 2 LCHAD deficiency, n = 1 MAD deficiency, and n = 1 VLCAD deficiency).
Involvement of other organs during the course of the disease
Figure 4 summarizes the other organ manifestations reported.
FIGURE 4.

Involvement of other organs. (a) Other symptoms. (b) Results of cardiac, respiratory, and abdominal explorations. CPTII, carnitine palmitoyl transferase 2; LCHAD, long‐chain 3‐hydroxy‐acyl‐CoA; MAD, multiple acyl‐CoA; SCAD, short‐chain acyl‐CoA dehydrogenase; VLCAD, very‐long‐chain acyl‐CoA dehydrogenase.
Cardiac manifestations
Four patients had cardiac symptoms (9%; n = 1 SCAD deficiency, n = 1 CPTII deficiency, and n = 2 VLCAD deficiency), and the mean age at onset of these symptoms was 41 years (±25, range = 11–65). The cardiac symptoms observed were syncope (75%) and dyspnea (50%). One patient had heart failure revealed by dyspnea on exercise and confirmed by echocardiography (CPTII deficiency). Cardiac explorations revealed heart conduction disorders in 5% of cases (n = 1 SCAD deficiency and n = 1 MAD deficiency), heart rate disorders in 5% of cases (n = 1 CPTII deficiency and n = 1 MAD deficiency), and cardiopathy with normal cardiac function in one patient (MAD deficiency).
Respiratory manifestations
Eight patients had respiratory symptoms (18%); the mean age at onset of these symptoms was 38 years (±11, range = 27–56). The respiratory symptoms observed were exercise dyspnea (88%), rest dyspnea (38%), and sleep apnea syndrome (25%). No episodes of respiratory failure were reported. Pulmonary function tests (PFT) were performed for 18 patients (41%), for 15 of whom (83%) the results were normal. These explorations revealed two cases of obstructive syndrome (11%, n = 1 SCAD deficiency and n = 1 VLCAD deficiency), one case of restrictive syndrome (5%, VLCAD deficiency), and one case of diaphragmatic insufficiency (5%, VLCAD deficiency).
Other manifestations
Forty patients (90%) underwent liver tests, which revealed high levels of hepatic enzymes (aspartate aminotransferase and alanine aminotransferase) away from rhabdomyolysis episodes in 10% of cases, hepatomegaly in 15% of cases, and splenomegaly in 7% of cases.
Retinopathy was detected in one of the three patients with LCHAD deficiency.
Genetic results
The results of the genetic analyses are summarized in Table 1.
TABLE 1.
Genetic analyses.

Most of the patients with CPTII deficiency were compound heterozygous (57%), with c.338C>T (p.Ser113Leu) the most frequent variant (72% of alleles). All the patients with LCHAD deficiency were compound heterozygous, with no predominance of any particular allele. The patients with MAD deficiency had mutations of the ETFDH gene only. All patients with SCAD deficiency were homozygous for the same variant (c.625G>A, p.Gly209Ser), but one patient (20%) was both homozygous for the c.625G>A mutation and heterozygous for the c.1147C>T (p.Arg359Cys) mutation. Most of the patients with VLCAD deficiency were compound heterozygous (92%), with no predominance of any particular allele.
Long‐term follow‐up
The mean duration of follow‐up was 10.5 years (±11.3, range = 0–53; Figure 5a). Most patients had symptoms only after exercise (53%), 30% were asymptomatic, 16% had a permanent muscle weakness without handicap, and one had a permanent muscle weakness with handicap (VLCAD deficiency; Figure 5b).
FIGURE 5.

Long‐term follow‐up of the patients. (a) Duration of follow‐up. (b) Change in muscle symptoms. (c) Change in the frequency of rhabdomyolysis. (d) Changes in cardiac, respiratory, and metabolic symptoms. CPT II, carnitine palmitoyl transferase 2; LCHAD, long‐chain 3‐hydroxy‐acyl‐CoA; MAD, multiple acyl‐CoA; SCAD, short‐chain acyl‐CoA dehydrogenase; VLCAD, very‐long‐chain acyl‐CoA dehydrogenase.
The mean frequency of rhabdomyolysis before diagnosis was approximately 1.92 episodes per year per patient (CPTII deficiency: 1.71, MAD deficiency: 0.25, VLCAD deficiency: 4.9, LCHAD deficiency: 2.25, SCAD deficiency: 0.5). After diagnosis, the mean frequency of rhabdomyolysis was 0.82 episodes per year per patient (CPTII deficiency: 1.21, MAD deficiency: 0, VLCAD deficiency: 2.5, LCHAD deficiency: 0, SCAD deficiency: 0.4). Following diagnosis, the frequency of rhabdomyolysis decreased in 63% of cases, stabilized in 30% of cases, and increased in 4.5% of cases (two patients: CPTII deficiency and SCAD deficiency; Figure 5c). There was only one new admission to intensive care after diagnosis (VLCAD deficiency). Cardiac, respiratory, and metabolic manifestations generally improved (Figure 5d), with only one patient (VLCAD deficiency) presenting an aggravation of respiratory signs after diagnosis.
DISCUSSION
In this cohort, the age of the patients at disease onset was variable (0.5–35 years), and more than half the patients (59%) experienced their first symptoms during childhood without onset during the neonatal period. Thus, muscle symptoms, including rhabdomyolysis in particular, should be carefully monitored in patients with onset of an FAOD during childhood.
In this cohort, 14 patients had high CK levels at rest (>300 IU/L). All diseases are concerned except for patients with LCHAD deficiency. In FAODs, CK levels at rest are usually normal. High CK levels at rest have already been described in patients with long‐chain FAODs with no associated symptoms [7].
ACP analysis is the most cost‐effective exploration. Provided that it is performed correctly (after 14 h of fasting), it can rapidly identify the deficient enzyme; high levels of specific long‐chain acylcarnitines are seen in patients with LCHAD (C14OH, C16OH, C18OH, C18:1OH), VLCAD (C14:1, C14:1/C12:1), and CPTII ((C16 + C18:1)/C2) deficiencies, high total acylcarnitines levels (C4–C18) are observed in patients with MAD deficiency, and high levels of short‐chain acylcarnitines (C4) are characteristic of SCAD deficiency. In cases in which an FAOD is strongly suspected, the ACP should be repeated, with checks to ensure that the fasting constraints are respected. In cases in which the ACP and urinary organic acid excretion data are not informative for diagnosis, a muscle biopsy may prove useful. In some cases with a suggestive clinical history of repeated rhabdomyolysis, genetic analyses should be performed even if the ACP remains normal.
A large proportion of patients underwent a muscle biopsy because they were diagnosed when ACP and genetic analyses were less available. Muscle biopsy is no longer essential for diagnosis. It can be useful in cases of a nonevocative presentation, in which specific staining may reveal an increase muscle fat content. Biopsies are not always conclusive, because increased muscle fat content may be missed or have a misleading appearance, resembling muscle necrosis (in two patients: MAD and SCAD deficiencies) or glycogen infiltration associated with fatty infiltration (CPTII deficiency). A presentation resembling muscle necrosis has already been described in a patient with MAD deficiency [8].
Electroneuromyogram abnormalities are not specific for this type of myopathy. However, sensorimotor axonal length‐dependent neuropathy was observed in all patients with LCHAD deficiency and in 25% of those with MAD deficiency. This type of neuropathy has already been described in patients with LCHAD deficiency [9, 10] and MAD deficiency [11].
Cardiorespiratory symptoms are rare in muscular forms, with only 9% and 18% of the patients from our cohort presenting cardiac and respiratory involvement, respectively. Only three cardiac or respiratory episodes were reported in a Chinese cohort of 90 patients with MAD deficiency [12]. The symptoms of such involvement can be severe. Cardiac and respiratory monitoring should be performed by recording an electrocardiogram, echocardiogram, and PFT at the beginning of patient management, and regularly thereafter with the frequency of monitoring adapted according to initial disease severity. Metabolic involvement is more frequent, occurring in all the diseases described here. In this cohort, we found only one case of retinopathy, in a patient with LCHAD deficiency. This frequency is lower than that in the Austrian and Swedish cohorts [9, 13], probably due to the smaller number of patients with LCHAD deficiency included (n = 3) and unsystematic searches for retinopathy.
The long‐term follow‐up data for this cohort showed the prognosis to be reasonably good for these myopathies, not only for muscle involvement, but also for cardiac, respiratory, and metabolic involvement. We observed an improvement in severity parameters, such as hospitalization in intensive care (no new admissions after diagnosis) and the annual frequency of rhabdomyolysis episodes, after diagnosis and appropriate management. These data suggest that patients adapt their lifestyles, based on a better identification and understanding of the triggers of rhabdomyolysis and other causes of decompensation. Patients try to avoid fasting and heat, treat fever from its onset, and adapt their effort levels (reducing intensity and duration of effort and sometimes increasing treatment intake before major effort). This low frequency of severity manifestations in adults suggests that such manifestations are not an appropriate outcome measure for future therapeutic trials for muscular forms. It might be useful to develop new assessment scales based on quality of life or the functional impact of symptoms.
The main treatments used in this cohort were L‐carnitine, riboflavin, triheptanoin, and fibrates. The main diets used were medium‐chain triglyceride oil, a low‐fat diet, and a carbohydrate‐rich diet. This retrospective study precludes any firm conclusions about treatment or regimen efficacy for a specific disease. In the Appendix, we propose recommendations for treatment, dietary management, and adaptation for these patients based on literature and our own experience.
All the patients with SCAD deficiency in this cohort were homozygous for the c.625G>A (p.Gly209Ser) variant of the ACADS gene. The pathogenicity of the c.625G>A (p.Gly209Ser) variant is currently a matter of debate, as some consider this mutation to correspond to a “biochemical variant.” We included these patients with SCAD deficiency because they all had muscle symptoms at diagnosis; all presented myalgia, four patients had a single episode of rhabdomyolysis, and two patients displayed permanent muscle weakness. Currently, two of five patients have persistent symptoms distant from diagnosis: a patient with muscle weakness and a patient with myalgia. One patient was heterozygous for another pathogenic mutation (c.1147C>T) and also homozygous for the c.625G>A variant, with a more severe clinical presentation: delayed psychomotor development, exercise intolerance, seizure, severe conduction disorder, and urinary excretion of ethyl‐malonic acid. In our cohort, muscle symptoms were observed in patients homozygous for the c.625G>A variant only, but these symptoms were more severe when it was associated with another pathological mutation, consistent with the finding of biochemical studies [14]. This may explain why muscle symptoms were not systematically detected in patients homozygous for the c.625G>A variant, particularly those without extramuscular symptoms. The patients with SCAD deficiency in our cohort appeared to have a higher CK rate at rest than patients with other diseases, and their ACP was normal in 80% of cases. SCAD deficiency has a broad clinical presentation, with a predominance of metabolic decompensation during the first year of life and seizures [15]. The prevalence of muscle symptoms is unknown. The pathophysiology of SCAD deficiency involved toxic mechanisms related to the misfolding of the enzyme (accumulation of various metabolites), leading to a mitochondrial dysfunction and a chronic increase in oxidative stress [16]. Individual antioxidant reserves may provide the first element of an explanation for the great variability of phenotypic expression of SCAD deficiency [17]. Considering these data, SCAD deficiency can be considered as a disease, including in a muscular form.
More than half the patients had already had general anesthesia (65%), with a low rate of complications with moderate severity [4]. One patient with VLCAD deficiency had a fever after halogen‐based anesthesia. This episode may correspond to minimal malignant hyperthermia, which has been described mostly in core myopathies, particularly those due to RYR1 and CACNA1S mutations [18, 19]. One patient with CPTII deficiency suffered vomiting after the administration of curare. Postoperative monitoring does not, therefore, appear to require closer surveillance than for other myopathies, although some precautions would be wise in cases of halogen‐based anesthesia.
A few studies have focused on pregnancy and childbirth in patients with FAODs [20, 21, 22, 23, 24]. We noted only a few complications during pregnancy and childbirth in our patients. The most frequent complications were maternal muscle complications (21%), with myalgia or rhabdomyolysis occurring during or just after the delivery. The postpartum period is a high‐risk period for rhabdomyolysis in patients with VLCAD and CPTII deficiencies, due to the catabolic state prevailing during this period. None of the patients from our cohort was admitted to intensive care, with intensive rehydration proving sufficient in all cases. Our data suggest that it would be prudent to monitor the mother closely during childbirth and the postpartum period, by measuring capillary glycemia, CK levels, and the occurrence of muscle symptoms.
The main strength of this study is the long‐term nature of follow‐up (mean = 10 years) and the relatively large numbers of patients given the rarity of these diseases. Its principal limitation is its retrospective nature, with the many biases and the issues of missing data inherent to all retrospective studies.
CONCLUSIONS
Fatty‐acid oxidation disorders are rare diseases, and the forms in which muscle symptoms predominate are even rarer.
It would be interesting to build on this study by developing a national standardized registry for these myopathies. The establishment of such a registry would facilitate the exhaustive collection of clinical data, the results of systematic explorations at initial consultations and during follow‐up, inclusions in therapeutic protocols, and long‐term follow‐up. All these data would improve our understanding of these muscular forms of fatty‐acid oxidation disorders and drive improvements in medical treatment.
AUTHOR CONTRIBUTIONS
Alice Rouyer: Conceptualization; investigation; writing – original draft; methodology; writing – review and editing; data curation. Céline Tard: Writing – review and editing; validation; supervision. Anne‐Frédérique Dessein: Writing – review and editing. Marco Spinazzi: Writing – review and editing. Dalia Dimitri‐Boulos: Writing – review and editing. Aleksandra Nadaj‐Pakleza: Writing – review and editing. Jean‐Baptiste Chanson: Writing – review and editing. Guillaume Nicolas: Writing – review and editing. Claire Douillard: Validation; writing – review and editing; supervision. Pascal Laforêt: Conceptualization; investigation; methodology; visualization; writing – review and editing; supervision.
APPENDIX 1. RECOMMENDATIONS FOR THERAPEUTICS, DIETARY MANAGEMENT, AND LIFESTYLE ADAPTATION
(i) When to start treatment?
The treatment can be introduced at diagnostic suspicion.
The timing of the initial assessment following the initiation of treatment depends on the severity of the initial presentation:
It is close to the date of diagnosis for fatty‐acid oxidation disorders (FAODs) concerning the metabolism of long chains (carnitine palmitoyl transferase 2 [CPTII], multiple acyl‐CoA [MAD], very‐long‐chain acyl‐CoA dehydrogenase [VLCAD], and long‐chain 3‐hydroxy‐acyl‐CoA [LCHAD]), that is to say, most often weekly the first month following diagnosis in adulthood (and the patient has an emergency certificate in the event of decompensation or a situation at risk of decompensation).
It is only one per year for short‐chain acyl‐CoA dehydrogenase (SCAD) the year following diagnosis. Then, monitoring is renewed only if necessary (the patient is nevertheless provided with a certificate to avoid any period of very prolonged fasting).
(ii) Treatment and regimens used:
The goals of initial treatment are:
To stop intoxication by excluding lipid intake and by restarting the metabolism through a caloric intake based on carbohydrates (which will correct possible hypoglycemia).
To correct rhabdomyolysis, acute renal failure, hyperammonemia, hepatocellular failure, heart failure, or other acute decompensations.
In the acute phase of decompensation, L‐carnitine is offered in small doses, but in the event of cardiac failure and/or rhythm disturbance, it is necessary to wait for the biological results (carnitine) and to know the diagnostic suspicion (acylcarnitine profile) before starting supplementation.
The goals of chronic treatment are to maintain metabolic balance and if possible prevent metabolic decompensation.
This table lists treatments and regimens that can be used. Fibrates were studied in CPTII and VLCAD deficiency and showed no efficacy (Ørngreen et al. 2014, Bastin et al. 2015) [25, 26].
| Treatment and regimen | CPTII | MAD | VLCAD | LCHAD | SCAD | References |
|---|---|---|---|---|---|---|
| Triheptanoin (= triglyceride of medium‐chain fatty acids esterified with heptanoate) | X | Contraindicated | X | X | Vockley et al. (2015), Gillingham et al. (2017), Vockley et al. (2017) [27, 28, 29] | |
| Riboflavin | X | X (symptomatic only) | Jethva et al. (2008), Béhin et al. (2016), Macchione et al. (2020), Nochi et al. (2017) [16, 30, 31, 32] | |||
| L‐Carnitine | X | X | X | X | Arnold et al. (2009), Spiekerkoetter et al. (2009), Knottnerus et al. (2018), Almannai et al. (2019) [33, 34, 35, 36] | |
| Sodium beta‐hydroxybutyrate (in severe FAODs as an energy supplement) | X | X | X | X | Van Hove et al. (2003), Knottnerus et al. (2018), Bhattacharya et al. (2020) [35, 37, 38] | |
| Restricting periods of fasting a | X | X | X | X | X | PNDS b |
| DCI: normal | X | X | X | X | X | PNDS b |
| High carbohydrate diet (50%–55% DCI) | X | X | X | X | PNDS b | |
| Low‐fat diet (LCFA + MCFA = 25%–30% of the DCI, i.e., 10% of DCI for LCFA + 20%–25% of DCI for MCFA) | X | X | X | Knottnerus et al. (2018), Van Calcar et al. (2020) [35, 39] | ||
| Very low‐fat diet (10% DCI and only with LCFA) | X b | PNDS b | ||||
| Medium‐chain triglycerides (not necessary if there is triheptanoin intake) | X | Contraindicated | X | X | Knottnerus et al. (2018), Van Calcar et al. (2020) [35, 39] |
Abbreviation: DCI, daily caloric intake; LCFA, long‐chain fatty acid; MCFA, medium‐chain fatty acid.
The duration of tolerated fasting varies depending on the age of the patient, the severity of the enzyme deficiency and the response to treatment. This duration varies from one patient to another but generally from 14h in adults in long chain faod (here: CPTII, MAD VLCAD, LCHAD).
Restrictive diet would be less restrictive if initial presentation is mild or if MAD is responsive to Riboflavin. Abbreviation: DCI, daily caloric intake; PNDS, Protocole National de Diagnostic et de Soins (PNDS) Déficit en MCAD (Acyl‐CoA déshydrogénase des acides gras à chaine moyenne) et autres déficits de la β‐oxydation mitochondriale des acides gras, Filière de santé maladies rares G2M (https://www.filiere‐g2m.fr).
(iii) Lifestyle adaptation recommendations:
Situations leading either to a lack of food intake (fasting, vomiting, surgical intervention, etc.) or to an increase in energy needs (infection, intercurrent disease, physical exercise, stress, etc.) must be the subject of preventive measures to prevent the onset of decompensation of the disease. Therapeutic education of patients is essential in the following situations:
Eviction of prolonged fasting: duration of tolerated fasting varies depending on the age of the patient, the severity of the enzyme deficiency, and the response to treatment. This duration varies from one patient to another but generally from 14 h in adults in long‐chain FAODs (here: CPTII, MAD, VLCAD, LCHAD).
Limit the intensity and duration of effort.
Adapt therapies according to the degree of physical activity: be careful not to start physical activity while fasting (taking complex carbohydrates and/or medium‐chain triglycerides (MCT) before exercise), make sure to regularly consume sugary foods or drinks during physical exercise (Orngreen et al. 2007, Behrend et al. 2012, Vissing et al. 2016)[40, 41, 42].
Eviction of intense heat.
Treat fever and/or infection early.
(iv) The patient must always have an emergency certificate (also accessible online: https://www.filiere‐g2m.fr). This certificate indicates the biological assessment to be carried out and the initial emergency treatment, adapted according to the signs of seriousness.
Rouyer A, Tard C, Dessein A‐F, et al. Long‐term prognosis of fatty‐acid oxidation disorders in adults: Optimism despite the limited effective therapies available. Eur J Neurol. 2024;31:e16138. doi: 10.1111/ene.16138
Claire Douillard and Pascal Laforêt contributed equally to this study.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. de Lonlay P, Djouadi F, Bonnefont JP, Saudubray JM, Bastin J. [Mitochondrial beta‐oxidation of fatty acids: an essential metabolic pathway of muscular function]. Arch Pediatr. 2002;9(Suppl 2):175s‐178s. [DOI] [PubMed] [Google Scholar]
- 2. Vargas CR, Ribas GS, da Silva JM, et al. Selective screening of fatty acids oxidation defects and organic acidemias by liquid chromatography/tandem mass spectrometry acylcarnitine analysis in Brazilian patients. Arch Med Res. 2018;49(3):205‐212. [DOI] [PubMed] [Google Scholar]
- 3. Landau YE, Waisbren SE, Chan LMA, Levy HL. Long‐term outcome of expanded newborn screening at Boston children's hospital: benefits and challenges in defining true disease. J Inherit Metab Dis. 2017;40(2):209‐218. [DOI] [PubMed] [Google Scholar]
- 4. Janeiro P, Jotta R, Ramos R, et al. Follow‐up of fatty acid β‐oxidation disorders in expanded newborn screening era. Eur J Pediatr. 2019;178(3):387‐394. [DOI] [PubMed] [Google Scholar]
- 5. Laforêt P, Vianey‐Saban C, Vissing J. 162nd ENMC International Workshop: disorders of muscle lipid metabolism in adults 28‐30 November 2008, Bussum, The Netherlands. Neuromuscul Disord. 2010;20(4):283‐289. [DOI] [PubMed] [Google Scholar]
- 6. Laforêt P, Vianey‐Saban C. Disorders of muscle lipid metabolism: diagnostic and therapeutic challenges. Neuromuscul Disord. 2010;20(11):693‐700. [DOI] [PubMed] [Google Scholar]
- 7. Lund AM, Skovby F, Vestergaard H, Christensen M, Christensen E. Clinical and biochemical monitoring of patients with fatty acid oxidation disorders. J Inherit Metab Dis. 2010;33(5):495‐500. [DOI] [PubMed] [Google Scholar]
- 8. Vengalil S, Preethish‐Kumar V, Polavarapu K, et al. Fatty acid oxidation defects presenting as primary myopathy and prominent dropped head syndrome. Neuromuscul Disord. 2017;27(11):986‐996. [DOI] [PubMed] [Google Scholar]
- 9. Karall D, Brunner‐Krainz M, Kogelnig K, et al. Clinical outcome, biochemical and therapeutic follow‐up in 14 Austrian patients with long‐chain 3‐hydroxy acyl CoA dehydrogenase deficiency (LCHADD). Orphanet J Rare Dis. 2015;10:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Immonen T, Ahola E, Toppila J, Lapatto R, Tyni T, Lauronen L. Peripheral neuropathy in patients with long‐chain 3‐hydroxyacyl‐CoA dehydrogenase deficiency – a follow‐up EMG study of 12 patients. Eur J Paediatr Neurol. 2016;20(1):38‐44. [DOI] [PubMed] [Google Scholar]
- 11. Wang Z, Hong D, Zhang W, et al. Severe sensory neuropathy in patients with adult‐onset multiple acyl‐CoA dehydrogenase deficiency. Neuromuscul Disord. 2016;26(2):170‐175. [DOI] [PubMed] [Google Scholar]
- 12. Xi J, Wen B, Lin J, et al. Clinical features and ETFDH mutation spectrum in a cohort of 90 Chinese patients with late‐onset multiple acyl‐CoA dehydrogenase deficiency. J Inherit Metab Dis. 2014;37(3):399‐404. [DOI] [PubMed] [Google Scholar]
- 13. Fahnehjelm KT, Liu Y, Olsson D, et al. Most patients with long‐chain 3‐hydroxyacyl‐CoA dehydrogenase deficiency develop pathological or subnormal retinal function. Acta Paediatr. 2016;105(12):1451‐1460. [DOI] [PubMed] [Google Scholar]
- 14. Nagan N, Kruckeberg KE, Tauscher AL, Bailey KS, Rinaldo P, Matern D. The frequency of short‐chain acyl‐CoA dehydrogenase gene variants in the US population and correlation with the C(4)‐acylcarnitine concentration in newborn blood spots. Mol Genet Metab. 2003;78(4):239‐246. [DOI] [PubMed] [Google Scholar]
- 15. Corydon MJ, Vockley J, Rinaldo P, et al. Role of common gene variations in the molecular pathogenesis of short‐chain acyl‐CoA dehydrogenase deficiency. Pediatr Res. 2001;49(1):18‐23. [DOI] [PubMed] [Google Scholar]
- 16. Nochi Z, Olsen RKJ, Gregersen N. Short‐chain acyl‐CoA dehydrogenase deficiency: from gene to cell pathology and possible disease mechanisms. J Inherit Metab Dis. 2017;40(5):641‐655. [DOI] [PubMed] [Google Scholar]
- 17. Dessein AF, Fontaine M, Joncquel‐Chevalier Curt M, et al. Fluxomic evidence for impaired contribution of short‐chain acyl‐CoA dehydrogenase to mitochondrial palmitate β‐oxidation in symptomatic patients with ACADS gene susceptibility variants. Clin Chim Acta. 2017;471:101‐106. [DOI] [PubMed] [Google Scholar]
- 18. Rosenberg H, Pollock N, Schiemann A, Bulger T, Stowell K. Malignant hyperthermia: a review. Orphanet J Rare Dis. 2015;10:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaur H, Katyal N, Yelam A, Kumar K, Srivastava H, Govindarajan R. Malignant hyperthermia. Mo Med. 2019;116(2):154‐159. [PMC free article] [PubMed] [Google Scholar]
- 20. Laforêt P, Acquaviva‐Bourdain C, Rigal O, et al. Diagnostic assessment and long‐term follow‐up of 13 patients with very long‐chain acyl‐coenzyme a dehydrogenase (VLCAD) deficiency. Neuromuscul Disord. 2009;19(5):324‐329. [DOI] [PubMed] [Google Scholar]
- 21. Slater PM, Grivell R, Cyna AM. Labour management of a woman with carnitine palmitoyl transferase type 2 deficiency. Anaesth Intensive Care. 2009;37(2):305‐308. [DOI] [PubMed] [Google Scholar]
- 22. Lilker S, Kasodekar S, Goldszmidt E. Anesthetic management of a parturient with carnitine palmitoyltransferase II deficiency. Can J Anaesth. 2006;53(5):482‐486. [DOI] [PubMed] [Google Scholar]
- 23. Ramsey PS, Biggio JR. Carnitine palmitoyltransferase deficiency in pregnancy. J Matern Fetal Neonatal Med. 2005;18(5):357‐359. [DOI] [PubMed] [Google Scholar]
- 24. van Eerd DCD, Brussé IA, Adriaens VFR, et al. Management of an LCHADD patient during pregnancy and high intensity exercise. JIMD Rep. 2017;32:95‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ørngreen MC, Vissing J, Laforét P. No effect of bezafibrate in patients with CPTII and VLCAD deficiencies. J Inherit Metab Dis. 2014;38(2):373‐374. doi: 10.1007/s10545-014-9779-3 [DOI] [PubMed] [Google Scholar]
- 26. Bastin J, Bonnefont J, Djouadi F, Bresson J. Should the beneficial impact of bezafibrate on fatty acid oxidation disorders be questioned? J Inherit Metab Dis. 2014;38(2):371‐372. doi: 10.1007/s10545-014-9775-7 [DOI] [PubMed] [Google Scholar]
- 27. Vockley J, Marsden D, McCracken E, et al. Long‐term major clinical outcomes in patients with long chain fatty acid oxidation disorders before and after transition to triheptanoin treatment—A retrospective chart review. Mol Genet Metab. 2015;116(1–2):53‐60. doi: 10.1016/j.ymgme.2015.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gillingham MB, Heitner SB, Martin J, et al. Triheptanoin versus trioctanoin for long‐chain fatty acid oxidation disorders: a double blinded, randomized controlled trial. J Inherit Metab Dis. 2017;40(6):831‐843. doi: 10.1007/s10545-017-0085-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vockley J, Burton B, Berry GT, et al. UX007 for the treatment of long chain‐fatty acid oxidation disorders: Safety and efficacy in children and adults following 24 weeks of treatment. Mol Genet Metab. 2017;120(4):370‐377. doi: 10.1016/j.ymgme.2017.02.005 [DOI] [PubMed] [Google Scholar]
- 30. Jethva R, Bennett MJ, Vockley J. Short‐chain acyl‐coenzyme A dehydrogenase deficiency. Mol Genet Metab. 2008;95(4):195‐200. doi: 10.1016/j.ymgme.2008.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Béhin A, Acquaviva‐Bourdain C, Souvannanorath S, et al. Multiple acyl‐CoA dehydrogenase deficiency (MADD) as a cause of late‐onset treatable metabolic disease. Rev Neurol. 2016;172(3):231‐241. doi: 10.1016/j.neurol.2015.11.008 [DOI] [PubMed] [Google Scholar]
- 32. Macchione F, Salviati L, Bordugo A, et al. Multiple acyl‐COA dehydrogenase deficiency in elderly carriers. J Neurol. 2020;267(5):1414‐1419. doi: 10.1007/s00415-020-09729-z [DOI] [PubMed] [Google Scholar]
- 33. Arnold GL, Van Hove J, Freedenberg D, et al. A Delphi clinical practice protocol for the management of very long chain acyl‐CoA dehydrogenase deficiency. Mol Genet Metab. 2009;96(3):85‐90. doi: 10.1016/j.ymgme.2008.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spiekerkoetter U, Lindner M, Santer R, et al. Treatment recommendations in long‐chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis. 2009;32(4):498‐505. doi: 10.1007/s10545-009-1126-8 [DOI] [PubMed] [Google Scholar]
- 35. Knottnerus SJG, Bleeker JC, Wüst RCI, Ferdinandusse S, IJlst L., Wijburg F. A., Wanders R. J. A., Visser G., & Houtkooper R. H. Disorders of mitochondrial long‐chain fatty acid oxidation and the carnitine shuttle. Rev Endocr Metab Disord. 2018;19(1):93‐106. doi: 10.1007/s11154-018-9448-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Almannai M, Alfadhel M, El‐Hattab AW. Carnitine Inborn Errors of Metabolism. Molecules. 2019;24(18):3251. doi: 10.3390/molecules24183251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Van Hove JL, Grünewald S, Jaeken J, et al. D,L‐3‐hydroxybutyrate treatment of multiple acyl‐CoA dehydrogenase deficiency (MADD). The Lancet. 2003;361(9367):1433‐1435. doi: 10.1016/s0140-6736(03)13105-4 [DOI] [PubMed] [Google Scholar]
- 38. Bhattacharya K, Matar W, Tolun AA, et al. The use of sodium DL‐3‐Hydroxybutyrate in severe acute neuro‐metabolic compromise in patients with inherited ketone body synthetic disorders. Orphanet J Rare Dis. 2020;15(1):53. doi: 10.1186/s13023-020-1316-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van Calcar SC, Sowa M, Rohr F, et al. Nutrition management guideline for very‐long chain acyl‐CoA dehydrogenase deficiency (VLCAD): An evidence‐ and consensus‐based approach. Mol Genet Metab. 2020;131(1–2):23‐37. doi: 10.1016/j.ymgme.2020.10.001 [DOI] [PubMed] [Google Scholar]
- 40. Orngreen MC, Norgaard MG, van Engelen BGM, Vistisen B, Vissing J. Effects of IV glucose and oral medium‐chain triglyceride in patients with VLCAD deficiency. Neurology. 2007;69(3):313‐315. doi: 10.1212/01.wnl.0000265854.41013.84 [DOI] [PubMed] [Google Scholar]
- 41. Behrend AM, Harding CO, Shoemaker JD, et al. Substrate oxidation and cardiac performance during exercise in disorders of long chain fatty acid oxidation. Mol Genet Metab. 2012;105(1):110‐115. doi: 10.1016/j.ymgme.2011.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vissing J. Exercise training in metabolic myopathies. Rev Neurol. 2016;172(10):559‐565. doi: 10.1016/j.neurol.2016.08.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.