

Abstract
The difficulty of faithfully recapitulating malarial protein complexes in heterologous expression systems has long impeded structural study for much of the Plasmodium falciparum proteome. However, recent advances in single-particle cryo electron microscopy (cryoEM) enable structure determination to atomic resolutions with significantly reduced requirements for both sample quantity and purity. Combined with recent developments in gene editing, these advances open the door for structure determination and structural proteomics of macromolecular complexes enriched directly from P. falciparum parasites. Furthermore, the combination of cryoEM with the rapidly emerging use of in situ cryo electron tomography (cryoET) to directly visualize ultrastructures and protein complexes in the native cellular context will yield exciting new insights into the molecular machinery underpinning malaria parasite biology and pathogenesis.
Keywords: cryoEM, malaria, in situ cryoET, endogenous structure determination
The malaria parasite Plasmodium falciparum poses specific challenges to structural study
Malaria (see Glossary) exacts a devastating toll on global public health, with an estimated 3.8 billion people, half the world’s population, currently at risk [1]. The recent rise of drug-resistant malaria parasites poses an urgent need to identify new targets for the development of anti-malarial treatments with novel modes of action [2–7]. Furthermore, a large number of the biological pathways driving parasite biology and pathogenesis remain enigmatic. For instance, the molecular mechanisms by which parasite effector proteins are trafficked to key subcellular compartments such as the host cell membrane are largely unknown. The limited insight into these mechanisms hinders understanding of malaria parasite biology and impacts efforts to combat the pathogen.
Structure determination plays an important role in addressing these unknowns, often revealing previously unidentified interactions and pathways, and subsequently providing key insights into both the functions and molecular mechanisms of potential therapeutic targets. Unfortunately, there are several difficulties in recapitulating proper folding and assembly of malarial protein complexes in heterologous systems that have precluded structural and biochemical study of many important P. falciparum protein complexes using conventional approaches [8, 9]. For example, the P. falciparum genome is exceptionally AT rich, with an average AT content of 80.6% and a heavily skewed codon usage bias, making cloning into heterologous expression systems challenging [10, 11]. Although this challenge is now mitigated with the use of codon optimization algorithms [12], much of the proteome is highly aggregation prone, littered with low complexity regions and extensive charged residue repeats, posing a major obstacle to heterologous expression of many P. falciparum proteins [13, 14].
As evidenced by the severe paucity of high resolution P. falciparum structures in the Protein Data Bank (PDB)i relative to other organisms [8, 9], these obstacles have hampered structural studies of the P. falciparum proteome using conventional methods such as X-ray crystallography (XRC) and nuclear magnetic resonance (NMR), which depend heavily on the production of large amounts of highly purified protein via recombinant overexpression [9, 15–18] (see also below). These challenges have hindered efforts to build a better understanding of novel malaria parasite biology. Consequently, many of the molecular mechanisms underlying the ability of the parasite to hijack human erythrocytes have remained enigmatic (Box 1).
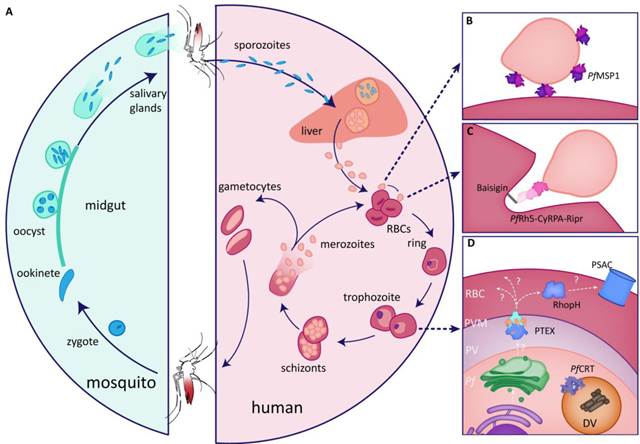
Box 1. Malarial life cycle and erythrocyte invasion.
Malaria is a disease caused by single-cell, eukaryotic parasites from the genus Plasmodium. Of the human-infecting species of Plasmodium, P. falciparum is associated with the most severe forms of the disease and the highest mortality. The definitive host of P. falciparum is the Anopheles mosquito, which transmits parasites to the human host during a blood feed. Within a newly infected human, parasites traverse to the liver, where they rapidly replicate before egressing into the bloodstream to initiate the asexual replication cycle that occurs within erythrocytes (red blood cells, RBCs; Figure I). All the clinical manifestations of malaria are associated with the asexual, intraerythrocytic replication of the parasites, highlighting the need to study the asexual lifecycle. Within the erythrocyte, the parasite feeds on host cell hemoglobin to support its own growth before replicating into 16–32 daughter parasites known as merozoites (Figure IA). The merozoites egress from the erythrocyte, remaining briefly extracellular while merozoite surface proteins interact with receptors on a new erythrocyte host (Figure IB). After finding its new host cell, the merozoite reorients such that its apical end is in contact with the erythrocyte, and the parasite forces itself into the host cell (Figure IC). After entering the erythrocyte, the cycle begins again: the parasite exports hundreds of proteins into the host to create its niche (Figure ID) and imports hemoglobin to consume and prepare for a new round of replication. A subset of asexual parasites exit this replication cycle to develop into gametocytes that are taken up into a new mosquito host for sexual development and propagation to a new human host.
Fortunately, the explosion of recent and ongoing advances in determining the structures [19–35] and dynamics [36–40] of macromolecular complexes using cryoelectron microscopy (cryoEM) have set in motion a paradigm shift in structural biology. This shift has particularly significant implications for the structural study of previously intractable biological systems like P. falciparum, as there are no known homologs outside the Plasmodium genus for up to one-third of the proteome [41], and only 5% of the proteome has been structurally characterized [8, 9]. For comparison, high resolution structural information exists for more than 23% of the human proteome [9]. In this Review, we first discuss how cryoEM has enabled structure determination of previously intractable, recombinantly expressed P. falciparum proteins. We then highlight how several key advantages of the cryoEM technique have been leveraged to enable structure determination of endogenously expressed protein complexes enriched directly from native malaria parasites. Looking forward, we envision an integrated view of malarial protein complexes in their native cellular contexts, combining high resolution single-particle cryoEM structures with in situ cryo electron tomography (cryoET) imaging of protein complexes at subnanometer resolutions in intact parasites.
CryoEM enables structure determination of previously intractable P. falciparum protein complexes overexpressed in recombinant systems
CryoEM is now routinely used to determine high resolution structures of challenging proteins and complexes that were refractory to structure determination using XRC, as they could only be produced in small quantities, even when recombinantly overexpressed [42]. Indeed, the amounts of protein required to achieve an atomic resolution structure using cryoEM can be up to 3–4 orders of magnitude less than those typically required for XRC. Recently, cryoEM coupled with heterologous expression has enabled the structure determination of key P. falciparum protein complexes that play important roles in anti-malarial drug resistance and parasite invasion (Box 1).
For instance, a 3.2 Å single-particle cryoEM structure of the P. falciparum chloroquine resistance transporter (PfCRT), a drug/metabolite transporter [43] responsible for conferring resistance to the anti-malaria drugs chloroquine and piperaquine [44, 45], was determined using PfCRT expressed recombinantly in HEK293 cells and bound to a PfCRT-specific antigen binding fragment to overcome current cryoEM size limitations [46]. The structure reveals a cluster of mutations in a central cavity known to function in transport of physiological substrates across the digestive vacuole (DV) membrane (Figure 1A). These mutations are known to affect the binding and export of chloroquine or piperaquine from the DV by PfCRT, and the study provides a structural framework for understanding the molecular basis of parasite resistance to these drugs [46].
Figure 1 |. P. falciparum cryoEM protein structures from recombinant expression systems.

A, Surface representation of PfCRT cryoEM structure (PDB ID: 6UKJ, [46]). Substituted amino acids in cavity, resulting from genetic mutations that enable chloroquine (CQ) and piperaquine (PPQ) binding, are colored light blue. DV: digestive vacuole. B, CryoEM structure of PfMSP1 dimer [47]. Left protomer shows reconstruction of the six different monomer conformations found in sample, with arrows indicating flexibility (EMD-11150–55). The right protomer is shown as a cylindrical representation of the atomic model (PDB ID: 6ZBJ). Asterisk indicates interface between two protomers, which is highlighted in light pink. C, CryoEM reconstruction of Rh5-CyRPA-Ripr-Basigin complex ([48], EMD-9192). Cylindrical representation for Rh5 (light pink), CyRPA (coral), Ripr (salmon), and Basigin binding site is indicated with an arrow (PDB ID: 6MPV). Representations for all reconstructions and models were created in ChimeraX [103].
Additionally, two recent cryoEM studies of proteins on the merozoite surface have helped expand the field’s understanding of merozoite invasion [47–49] (Box 1). The Merozoite Surface Protein (MSP) family form a fibrillar coat on the surface of the parasite and play roles in invasion. The most abundant MSP – PfMSP1 – binds the erythrocyte surface proteins glycophorin A and band 3, and is crucial for successful invasion [50–53]. CryoEM was used to determine a 3.1 Å structure of recombinantly expressed PfMSP1 monomer (Figure 1B), as well as two homodimeric forms and five other subtly different monomeric confirmations, highlighting the flexibility of the PfMSP1 structure [47]. PfMSP1 interacts with many other MSPs and erythrocyte proteins, and its inherent flexibility may allow for the diversity of those interactions [50–52, 54–58]. Importantly, as PfMSP1 is a candidate for vaccine development, the cryoEM structure enabled mapping of immunogenic epitopes [59–61].
Following initial interaction with the erythrocyte, successful invasion requires surface-exposed PfRh5, in a complex with the proteins PfCyRPA and PfRipr, to bind erythrocyte basigin [56, 62, 63]. Furthermore, the complex is likely involved in the formation of a pore between parasite and host, enabling an influx of Ca2+ ions that is essential to complete invasion into the erythrocyte [63–66]. Despite its importance for invasion, the structure of the ternary complex remained unknown until the recent publication of two cryoEM structures of the PfCyRPA-Ripr and PfRh5-CyRPA-Ripr complexes, recombinantly expressed, at overall resolutions of 5.07 Å and 7.17 Å, respectively [48] (Figure 1C). Importantly, the ternary structure revealed the three subunits assembled in a 1:1:1 ratio, with PfCyRPA acting as a core joining PfRh5 and PfRipr. Fitting a previously published PfRh5-basigin crystal structure [67] into the ternary complex cryoEM density yielded insights into the possible mechanism of PfRh5-Ripr insertion into the erythrocyte membrane; briefly, the N-terminus of PfRh5 binds to basigin, orienting the PfRh5-Ripr complex parallel to the erythrocyte membrane. This positions the amphipathic PfRh5 C-terminal helical bundle close to the membrane, potentially initiating insertion into the membrane. The cryoEM structure of the ternary complex provides an important step forward in the field’s understanding of PfRh5-CyRPA-Ripr’s role in parasite invasion and enables future studies seeking to block invasion by interfering with the complex.
CryoEM enables structure determination of endogenous P. falciparum protein complexes
In the previously described studies, the reduced sample requirement associated with cryoEM was leveraged to enable structure determination of noteworthy protein complexes that eluded characterization by XRC, leading to important observations about the parasite’s biology. However, there is another implication of the reduced sample requirement that should not be overlooked: the requirement is low enough to enable cryoEM structure determination of endogenously derived complexes [68–72]. Additionally, unlike XRC, which requires highly purified samples to allow for billions of identical protein molecules to pack together into a highly ordered crystal lattice, protein molecules in cryoEM samples are preserved in a frozen-hydrated state [73, 74], thereby capturing the full range of conformations and orientations sampled by the molecules while in aqueous solution. The ramifications for P. falciparum and other non-model organisms with proteomes that resist expression in recombinant systems are significant. CryoEM structure determination of endogenous protein complexes enriched directly from the parasite presents an exciting path forward for the many challenging protein complexes that are not amenable to expression in heterologous systems, enabling structure determination of these important complexes in near-native states. Further, this approach ensures the presence of biologically relevant post-translation modifications and potentially allows for identification of previously undiscovered native substrates or binding partners, and even the discovery and characterization of previously unknown protein complexes.
CRISPR-assisted tagging of endogenous proteins enables structure determination of complexes enriched directly from P. falciparum parasites using cryoEM
We feel that combined with advances in CRISPR/Cas9-assisted parasite gene editing that enable insertion of affinity tags onto proteins of interest for purification [71, 75–77], the endogenous approach to cryoEM structure determination is made all the more powerful. For example, this approach was first used to determine the structure and mechanism of an essential malarial membrane protein complex known as the Plasmodium Translocon of Exported Proteins (PTEX) [71]. PTEX is the sole gateway for export of hundreds of effector proteins into the host cell, a process essential to the malaria parasite’s ability to inhabit and reproduce within human erythrocytes (Box 1). To obtain the long-sought structure of this novel translocon, endogenous PTEX was enriched directly from P. falciparum parasites via an epitope tag inserted into the locus of a PTEX subunit using CRISPR/Cas9. Single-particle cryoEM was then used to determine structures of the native PTEX complex in two distinct functional states, yielding the first reported near-atomic resolution cryoEM structures of protein(s) isolated directly from a native source using an epitope tag inserted into the endogenous locus with CRISPR/Cas9 gene editing [71].
Remarkably, native cargo protein peptides were observed still bound in the central channel of the PTEX translocon in both structures (Figure 2), a direct consequence of the fact that the PTEX complex was enriched from parasites harvested at a point in the life cycle when they were actively exporting hundreds of effector proteins [71]. This serendipitous outcome provided a key insight into the molecular mechanism of effector protein translocation by the PTEX translocon (Figure 2) that would have been missed had the complex been artificially reconstituted in a heterologous system.
Figure 2 |. CryoEM of endogenous PTEX complex reveals native cargo.

CryoEM density maps of HSP101 from PTEX translocon in transparent white with cargo (pink) and interdigitating pore loop tyrosines (purple, stick representation) in the engaged and resetting states (EMD-8951, EMD-8952). Maps are bisected to show the endogenous cargo and pore loop tyrosines in the protein-unfolding channel of the HSP101 unfoldase. Comparing the positioning of these pore loops relative to the cargo between the two states suggests a model for the mechanisms by which HSP101 unfolds the cargo protein and threads it through the transmembrane channel of the translocon. Full PTEX complex structures are shown in the insets for context (PDB IDs: 6E10, 6E11).
This work demonstrates that by enabling structure determination of endogenously derived samples, cryoEM not only provides a path forward for many previously intractable malarial macromolecular complexes, but also presents the possibility of observing as yet undiscovered native substrates, binding partners, or post-translational modifications [78], none of which would be found in samples produced in heterologous systems. With similar unanticipated observations reported in many of a growing number of endogenous structures [71, 78–83], it is increasingly clear that the potential for discovering native binding partners, substrates, or modifications represents a major advantage of endogenous cryoEM that should be capitalized on.
Leveraging cryoEM to discover previously unknown protein complexes within heterogeneous samples enriched from P. falciparum cell lysate
With cryoEM, not only are the protein molecules more likely to be captured in near-native, biologically relevant states, but the resulting 2D projections of individual protein molecules in a dataset of cryoEM micrographs, known as particles (Figure 3A), are extensively classified before distinct subsets of self-similar particles are averaged to yield the final high resolution 3D reconstructions (Figure 3B, C) [26, 28, 34, 84, 85]. During these powerful 2D and 3D classification steps, the sample is subjected to what is in essence an additional purification step, in silico. Thanks to this extra step, sample heterogeneity that would be prohibitive for XRC is not only tolerated, but can sometimes be leveraged to achieve multiple high resolution structures of a single protein complex in different conformational states (Figure 3D) [71, 86, 87], or even several distinct structures of completely unrelated protein complexes from a single cryoEM dataset [70].
Figure 3 |. Single-particle cryoEM data processing workflow.

A, cryoEM micrographs of a protein sample. Particles are identified, or “picked,” then extracted from the micrographs. B, Extracted particles are sorted into classes containing self-similar views, and then averaged to produce 2D class averages. C, Particles from high resolution 2D class averages are then used to calculate 3D reconstructions. D, 3D reconstructions are further classified and refined to yield one or more final high resolution cryoEM density maps.
We recently developed a cryoEM-enabled endogenous structural proteomics approach that leverages this capacity for in silico purification to determine multiple structures from coarsely fractionated parasite lysate containing a heterogeneous mixture of protein complexes. (Figure 4A, Key Figure) [70]. Parasite lysates are subjected to sucrose gradient fractionation, and the protein complexes in the resulting fractions are identified using a combination of cryoEM, mass spectrometry, and cryoID, a program that identifies proteins in cryoEM density maps of unknown protein complexes at better than 4.0 Å, with no prior knowledge of their primary sequence(s) (Figure 4A–C) [70]. This approach was used to identify and determine the structures of three P. falciparum protein complexes, glutamine synthetase, M18 aspartyl aminopeptidase, and the 20S proteasome. from images of a single sucrose gradient fraction in a proof of principle study [70]. To further demonstrate the power of this approach, the endogenous structural proteomics approach was used in a second study to determine a structure of the P. falciparum RhopH complex [72].
Figure 4, Key Figure |. CryoEM, FIB-SEM, and in situ cryoET workflow.

A, Tag-free sample preparation for endogenous structural proteomics. Protein complexes are enriched from malaria parasite lysates using sucrose gradient fractionation. Mass spectrometry and negative stain electron microscopy are used to identify fractions containing protein complexes of interest, which are then plunge frozen on cryoEM grids for B, single-particle cryoEM imaging in a high resolution 300kV transmission electron microscope (TEM). C, CryoEM analysis yields near-atomic resolution cryoEM density maps. CryoID is used to identify the protein(s) in the maps, enabling model building of atomic resolution structures. D, Sample preparation for endogenous CRISPR-tagged single-particle cryoEM. Affinity tags are inserted into the endogenous loci of proteins of interest in malaria parasites using CRISPR/Cas9 gene editing. Tagged proteins are affinity purified from parasite lysates, plunge frozen on cryoEM grids, and used for single-particle cryoEM imaging (B). E, CryoEM analysis yields near-atomic resolution cryoEM density maps, enabling atomic model building. F, Sample preparation for in situ cryoET. Proteins of interest are fluorescently tagged using CRISPR/Cas9 gene editing. The resulting transgenic P. falciparum parasites are grown in synchronous culture, and parasite-infected red blood cells (iRBCs) are isolated and plunge frozen directly on cryoEM grids. G, Vitrified iRBCs are then thinned in a dual-beam cryo focused ion beam scanning electron microscope (cryoFIB-SEM), yielding 100–200nm thick sections, called lamellae. Vitrified grids can be visualized in a cryo correlative light and electron microscope (cryoCLEM) to identify promising sites for cryoFIB milling. H, Tilt series are collected on lamella using a dose-symmetric tilt scheme ranging from −60° to +60° on a 300 kV TEM. I, Tilt series are aligned and reconstructed into 3D volumes called tomograms. 3D segmentation and subtomogram averaging (STA) are then used to reveal subcellular details at subnanometer resolutions. J, Integrating atomic resolution information from single-particle cryoEM with the cellular context from in situ cryoET provides further insights in to the molecular mechanisms underlying parasite biology and pathogenesis. High resolution reconstructions from cryoEM can be inserted into lower resolution subtomogram averages for context on immediate environment. Reconstructions can also be mapped back to the original 3D segmentation for cellular context.
The endogenous structural proteomics approach will be particularly impactful in organisms like P. falciparum, for which dissection of important protein-protein interactions is often laborious due to the genetic intractability of the parasite.
In situ cryoET holds potential to resolve cellular ultrastructures of intact, parasite-infected erythrocytes at subnanometer resolution
The single-particle cryoEM studies described thus far have all made significant contributions toward our understanding of various aspects of malaria parasite biology and pathogenesis. However, one major drawback of single-particle cryoEM studies is their dependence on purified protein complexes. During the purification process, red blood cells and their resident parasites must be broken open, destroying the cellular context within which macromolecular complexes exist, along with many important clues to the function and mechanism of the proteins in their native environments.
The loss of cellular context can be addressed using cryo electron tomography (cryoET), a technique whereby a series of images are taken of a vitrified, intact cell from many angles and then aligned and combined to produce a 3D reconstruction of the cell, called a tomogram, to directly visualize intracellular features within their preserved context. For example, earlier this year, a cryoET study of the Trypanosoma brucei flagellum revealed the architecture underlying the parasite’s flagellum-driven, non-planar helical motility, elucidating how the specific manner in which the axoneme and the paraflagellar rod are connected enables the typical movement of T. brucei essential for infection [88]. Within Apicomplexan parasites, cryoET has been used to investigate the apex of Toxoplasma parasites [89]; the membrane structure of the Plasmodium apicoplast [90]; and the nuclear pore complexes and microtubule structures within Plasmodium sporozoites [91, 92].
However, sample thickness is one of the major determinants of achievable resolution in cryoET and imposes a limit on the types of samples that can be investigated with this method. Consequently, cryoET studies, like the ones listed above, have been limited to analyses of isolated organelles, the thinnest areas of cells like the parasite apex, or life stages that are naturally thinner, such as Plasmodium sporozoites.
Recently, the development of an approach known as cryo focused ion beam (cryoFIB) milling, using a cryoFIB-scanning electron microscope (cryoFIB-SEM) to create 100–200nm thin sections, called lamellae, of vitrified, unfixed cells has enabled cryoET beyond naturally thin cell areas [93]. This combination of cryoFIB milling with cryoET, known as in situ cryoET (Figure 4F–H), enables the collection of tilt series anywhere in any cell, and with significantly improved contrast, yielding tomographic reconstructions in which supramolecular features and even individual protein complexes are resolved at unprecedented subnanometer resolutions [94, 95]. Further averaging of repeated particles within a tomogram, using a method called subtomogram averaging, can yield 3D reconstructions of the particles at resolutions approaching sub-4 Å [30]. These reconstructions can then be mapped back into the full cellular tomogram to capture the cellular context (Figure 4I) [96–98].
While the potential advances enabled by in situ cryoET are significant, it is still a relatively new technique, subject to a fair share of limitations and challenges (see Outstanding Questions). Even with recent innovations in automation, cryoFIB milling remains a laborious, time-consuming, and low throughput technique. The achievable resolution for subtomogram averaging depends heavily on the quality of the sample and the number of protein complexes that can be extracted from a tomogram [99]. Ribosomes and proteasomes are highly abundant proteins in cells and can therefore be found and targeted with relative ease, as can repetitive ultrastructures. However, many proteins of interest are harder to pinpoint in a crowded cellular environment, and targeting specific areas of the cell is still challenging. Using cryo correlative light electron microscopy (cryoCLEM), light microscopy images of cells expressing fluorescently tagged proteins can be mapped onto SEM images of the same sample, enabling reasonable targeting of specific regions in the x-y direction for FIB milling [100]. However, for accurate mapping in z direction, specialized confocal [101] or super-resolution microscopes [100, 102] are needed in combination with correlative software programs. To avoid ice contamination from transferring devices and improve ease of correlation, high resolution light objectives and detectors that can be integrated into the cryoFIB-SEM instrument are currently under development, but the resolution in z remains a significant obstacle. Despite current limitations, in situ cryoET provides a window into cells at an unprecedented resolution.
Outstanding Questions Box.
The rapidly evolving repertoire of single-particle cryo electron microscopy (cryoEM) tools are making structural biology increasingly accessible to the previously disparate cell biology community. However, significant barriers to entry remain for in situ cryo electron tomography (cryoET), even for those within the cryoEM community. What key innovations would help to alleviate this problem?
What additional steps can the cryoEM community take remove barriers to entry to structural biology for the cell biology community and the parasitology community in particular?
How can transient interactions and biological processes such as those mediating invasion and exflagellation during the P. falciparum life-cycle be addressed with cryoEM and in situ cryoET?
How can cryoEM and in situ cryoET be leveraged for structural study of malaria parasites in the liver and vector-borne stages of the life cycle?
Concluding Remarks
The use of cryoEM is quickly advancing our understanding of malaria parasite structural biology and thereby the mechanisms underpinning P. falciparum invasion and subjugation of human erythrocytes. By combining the subnanometer to near-atomic resolution cellular context attainable using in situ cryoET and subtomogram averaging with the atomic resolution structural information from single-particle cryoEM (Figure 4J; see Outstanding Questions), we will be able to directly visualize the molecular machinery underlying novel parasite biology and host-pathogen interactions at atomic resolution. These high-resolution insights into P. falciparum biology will be invaluable tools in the continuous need to develop new strategies for combatting the parasite.
Highlights.
With half the world’s population currently at risk, malaria remains a significant global health burden.
Difficulty expressing many malarial protein complexes in heterologous systems has precluded structural and biochemical study, impeding efforts to elucidate the functions and molecular mechanisms of many important but poorly understood biological pathways, including potential therapeutic targets.
Recent and ongoing advances in structure determination of macromolecular complexes using cryo electron microscopy (cryoEM) provide new avenues for structural study of the P. falciparum proteome, much of which has previously resisted structure determination.
CryoEM of endogenously derived macromolecular complexes, enabled by significantly reduced sample requirements of cryoEM studies, will lead to the discovery of as yet unknown native substrates, binding partners, and modifications.
Acknowledgements
We thank Michael Reese, Vasant Muralidharan, and Anthony W. P. Fitzpatrick for helpful comments regarding the manuscript. We regret that the diversity of the topics discussed in this review prevented us from being able to cite many more excellent papers. CMH is supported by a NIH Director’s Early Independence Award (DP5OD029613) and a Precision Medicine Pilot Grant from the Columbia Precision Medicine Initiative. LA is supported by a Postdoctoral Mobility Fellowship from the Swiss National Science Foundation (200205).
Glossary
- Amphiphatic
a molecule that has both hydrophilic and hydrophobic properties
- Apicomplexa
a phylum of unicellular parasites
- Apicoplast
an organelle found in most apicomplexan parasites with four membranes and is the site of many metabolic pathways
- Codon usage bias
the use of specific codons during translation of genes versus other synonymous codons
- Cryo electron tomography (cryoET)
a technique in which a series of two dimensional images, known as a tilt series, is collected on a sample as it is tilted
- Cryo focused ion beam (cryoFIB) milling
the process of ablating frozen cell material using the ion beam in a cryoFIB- scanning electron microscope (cryoFIB-SEM) for the purpose of creating a thin cross-section of the cell for cryo electron tomography (cryoET) imaging
- Effector proteins
proteins of a pathogen affecting the function and metabolism of the host cell
- Immunogenic epitopes
molecular patterns of a pathogen that can elicit an immune response
- Lamella
a thin cross-section of a frozen cell created in a cryoFIB-SEM for cryoET imaging
- Malaria
a disease caused by the pathogen Plasmodium that is characterized by intense cyclic fevers
- Merozoite
the invasive, asexual form of Plasmodium in the blood stage that infects erythrocytes
- Micrograph
an image taken using a microscope
- Recombinant or heterologous expression
expression of protein(s) in model organisms like E. coli, S. cerevisiae, baculovirus, insect, or mammalian cell lines rather than in the source organism, often at much higher expression levels (known as overexpression)
- Sporozoites
develop from sexual reproduction in the mosquito midgut and are injected into a human host during a mosquito blood-feed
- Structural proteomics
uses structure determination to identify specific proteins in a complex mixture of proteins
- Tilt series
a series of two-dimensional images taken of an object or area of interest from a range of angles during cryoET data collection
- Tomogram
a three-dimensional reconstruction of a slice or section through a three-dimensional object, reconstructed from a tilt series
Footnotes
Declaration of Interests
No interests declared.
Resources
Protein Data Bank (PBD): https://www.rcsb.org/
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.WHO, World malaria report 2018, 2018, p. 210. [Google Scholar]
- 2.Uwimana A. et al. (2020) Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathieu LC et al. (2020) Local emergence in Amazonia of Plasmodium falciparum k13 C580Y mutants associated with in vitro artemisinin resistance. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conrad MD and Rosenthal PJ (2019) Antimalarial drug resistance in Africa: the calm before the storm? Lancet Infect Dis 19 (10), e338–e351. [DOI] [PubMed] [Google Scholar]
- 5.Menard D. et al. (2018) Multidrug-resistant Plasmodium falciparum malaria in the Greater Mekong subregion. Lancet Infect Dis 18 (3), 238–239. [DOI] [PubMed] [Google Scholar]
- 6.Thu AM et al. (2017) Combating multidrug-resistant Plasmodium falciparum malaria. FEBS J 284 (16), 2569–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menard D. and Fidock DA (2019) Accelerated evolution and spread of multidrug-resistant Plasmodium falciparum takes down the latest first-line antimalarial drug in southeast Asia. Lancet Infect Dis 19 (9), 916–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.EMBL-EBI (2018) EMDB statistics. https://www.ebi.ac.uk/pdbe/emdb/statistics_main.html/, (accessed 12/9/2018 2018). [Google Scholar]
- 9.PDB R. (2018) PDB Statistics: Overall Growth of Released Structures Per Year. https://www.rcsb.org/stats/growth/overall, (accessed 2018). [Google Scholar]
- 10.Carlton JM et al. (2008) Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature 455 (7214), 757–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weber JL (1987) Analysis of sequences from the extremely A + T-rich genome of Plasmodium falciparum. Gene 52 (1), 103–9. [DOI] [PubMed] [Google Scholar]
- 12.Burgess-Brown NA et al. (2008) Codon optimization can improve expression of human genes in Escherichia coli: A multi-gene study. Protein Expr Purif 59 (1), 94–102. [DOI] [PubMed] [Google Scholar]
- 13.Aravind L. et al. (2003) Plasmodium biology: genomic gleanings. Cell 115 (7), 771–85. [DOI] [PubMed] [Google Scholar]
- 14.Muralidharan V. and Goldberg DE (2013) Asparagine repeats in Plasmodium falciparum proteins: good for nothing? PLoS Pathog 9 (8), e1003488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Derewenda ZS (2004) The use of recombinant methods and molecular engineering in protein crystallization. Methods 34 (3), 354–63. [DOI] [PubMed] [Google Scholar]
- 16.Hopp TP et al. (1988) A Short Polypeptide Marker Sequence Useful for Recombinant Protein Identification and Purification. Bio-Technology 6 (10), 1204–1210. [Google Scholar]
- 17.Porath J. et al. (1975) Metal Chelate Affinity Chromatography, a New Approach to Protein Fractionation. Nature 258 (5536), 598–599. [DOI] [PubMed] [Google Scholar]
- 18.Rosenberg AH et al. (1987) Vectors for selective expression of cloned DNAs by T7 RNA polymerase. Gene 56 (1), 125–35. [DOI] [PubMed] [Google Scholar]
- 19.Cheng Y. (2015) Single-Particle Cryo-EM at Crystallographic Resolution. Cell 161 (3), 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X. et al. (2013) Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods 10 (6), 584–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu H. et al. (2010) Atomic structure of human adenovirus by cryo-EM reveals interactions among protein networks. Science 329 (5995), 1038–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McMullan G. et al. (2009) Detective quantum efficiency of electron area detectors in electron microscopy. Ultramicroscopy 109 (9), 1126–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMullan G. et al. (2009) Enhanced imaging in low dose electron microscopy using electron counting. Ultramicroscopy 109 (12), 1411–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X. et al. (2010) 3.3 A cryo-EM structure of a nonenveloped virus reveals a priming mechanism for cell entry. Cell 141 (3), 472–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bai XC et al. (2015) An atomic structure of human gamma-secretase. Nature 525 (7568), 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Punjani A. et al. (2017) cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nature Methods 14 (3), 290-+. [DOI] [PubMed] [Google Scholar]
- 27.Rohou A. and Grigorieff N. (2015) CTFFIND4: Fast and accurate defocus estimation from electron micrographs. Journal of Structural Biology 192 (2), 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheres SHW (2012) RELION: Implementation of a Bayesian approach to cryo-EM structure determination. Journal of Structural Biology 180 (3), 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tegunov D. and Cramer P. (2019) Real-time cryo-electron microscopy data preprocessing with Warp. Nat Methods 16 (11), 1146–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tegunov D. et al. (2021) Multi-particle cryo-EM refinement with M visualizes ribosome-antibiotic complex at 3.5 A in cells. Nat Methods 18 (2), 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang K. (2016) Gctf: Real-time CTF determination and correction. Journal of Structural Biology 193 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang K, Gautomatch: a GPU-accelerated program for accurate, fast, flexible and fully automatic particle picking from cryo-EM micrographs with or without templates, 2016. [Google Scholar]
- 33.Zheng SQ et al. (2017) MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Methods 14 (4), 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grant T. et al. (2018) cisTEM, user-friendly software for single-particle image processing. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liao M. et al. (2013) Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504 (7478), 107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Punjani A. and Fleet DJ (2021) 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J Struct Biol 213 (2), 107702. [DOI] [PubMed] [Google Scholar]
- 37.Punjani A. et al. (2020) Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat Methods 17 (12), 1214–1221. [DOI] [PubMed] [Google Scholar]
- 38.Nakane T. and Scheres SHW (2021) Multi-body Refinement of Cryo-EM Images in RELION. Methods Mol Biol 2215, 145–160. [DOI] [PubMed] [Google Scholar]
- 39.Zhong ED et al. (2021) CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks. Nat Methods 18 (2), 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen M. and Ludtke SJ (2021) Deep learning-based mixed-dimensional Gaussian mixture model for characterizing variability in cryo-EM. Nat Methods 18 (8), 930–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Böhme U. et al. (2019) Progression of the canonical reference malaria parasite genome from 2002?2019 [version 2; peer review: 3 approved]. Wellcome Open Research 4 (58). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nogales E. (2016) The development of cryo-EM into a mainstream structural biology technique. Nat Methods 13 (1), 24–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin RE and Kirk K. (2004) The malaria parasite’s chloroquine resistance transporter is a member of the drug/metabolite transporter superfamily. Mol Biol Evol 21 (10), 1938–49. [DOI] [PubMed] [Google Scholar]
- 44.Dhingra SK et al. (2017) A Variant PfCRT Isoform Can Contribute to Plasmodium falciparum Resistance to the First-Line Partner Drug Piperaquine. mBio 8 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fidock DA et al. (2000) Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell 6 (4), 861–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J. et al. (2019) Structure and drug resistance of the Plasmodium falciparum transporter PfCRT. Nature 576 (7786), 315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dijkman PM et al. (2021) Structure of the merozoite surface protein 1 from Plasmodium falciparum. Sci Adv 7 (23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wong W. et al. (2019) Structure of Plasmodium falciparum Rh5-CyRPA-Ripr invasion complex. Nature 565 (7737), 118–121. [DOI] [PubMed] [Google Scholar]
- 49.Cowman AF et al. (2017) The Molecular Basis of Erythrocyte Invasion by Malaria Parasites. Cell Host Microbe 22 (2), 232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baldwin MR et al. (2015) Merozoite surface protein 1 recognition of host glycophorin A mediates malaria parasite invasion of red blood cells. Blood 125 (17), 2704–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boyle MJ et al. (2010) Interactions with heparin-like molecules during erythrocyte invasion by Plasmodium falciparum merozoites. Blood 115 (22), 4559–68. [DOI] [PubMed] [Google Scholar]
- 52.Das S. et al. (2015) Processing of Plasmodium falciparum Merozoite Surface Protein MSP1 Activates a Spectrin-Binding Function Enabling Parasite Egress from RBCs. Cell Host Microbe 18 (4), 433–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O’Donnell RA et al. (2000) Functional conservation of the malaria vaccine antigen MSP-119across distantly related Plasmodium species. Nat Med 6 (1), 91–5. [DOI] [PubMed] [Google Scholar]
- 54.Kauth CW et al. (2006) Interactions between merozoite surface proteins 1, 6, and 7 of the malaria parasite Plasmodium falciparum. J Biol Chem 281 (42), 31517–27. [DOI] [PubMed] [Google Scholar]
- 55.Lin CS et al. (2016) Multiple Plasmodium falciparum Merozoite Surface Protein 1 Complexes Mediate Merozoite Binding to Human Erythrocytes. J Biol Chem 291 (14), 7703–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wright KE et al. (2014) Structure of malaria invasion protein RH5 with erythrocyte basigin and blocking antibodies. Nature 515 (7527), 427–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paul G. et al. (2018) Protein-protein interaction studies reveal the Plasmodium falciparum merozoite surface protein-1 region involved in a complex formation that binds to human erythrocytes. Biochem J 475 (6), 1197–1209. [DOI] [PubMed] [Google Scholar]
- 58.Ranjan R. et al. (2011) Proteome analysis reveals a large merozoite surface protein-1 associated complex on the Plasmodium falciparum merozoite surface. J Proteome Res 10 (2), 680–91. [DOI] [PubMed] [Google Scholar]
- 59.Blank A. et al. (2020) Immunization with full-length Plasmodium falciparum merozoite surface protein 1 is safe and elicits functional cytophilic antibodies in a randomized first-in-human trial. NPJ Vaccines 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fowkes FJ et al. (2010) The relationship between anti-merozoite antibodies and incidence of Plasmodium falciparum malaria: A systematic review and meta-analysis. PLoS Med 7 (1), e1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Osier FH et al. (2008) Breadth and magnitude of antibody responses to multiple Plasmodium falciparum merozoite antigens are associated with protection from clinical malaria. Infect Immun 76 (5), 2240–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crosnier C. et al. (2011) Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature 480 (7378), 534–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Volz JC et al. (2016) Essential Role of the PfRh5/PfRipr/CyRPA Complex during Plasmodium falciparum Invasion of Erythrocytes. Cell Host Microbe 20 (1), 60–71. [DOI] [PubMed] [Google Scholar]
- 64.Chen L. et al. (2011) An EGF-like protein forms a complex with PfRh5 and is required for invasion of human erythrocytes by Plasmodium falciparum. PLoS Pathog 7 (9), e1002199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reddy KS et al. (2015) Multiprotein complex between the GPI-anchored CyRPA with PfRH5 and PfRipr is crucial for Plasmodium falciparum erythrocyte invasion. Proc Natl Acad Sci U S A 112 (4), 1179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weiss GE et al. (2015) Revealing the sequence and resulting cellular morphology of receptor-ligand interactions during Plasmodium falciparum invasion of erythrocytes. PLoS Pathog 11 (2), e1004670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen L. et al. (2014) Crystal structure of PfRh5, an essential P. falciparum ligand for invasion of human erythrocytes. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wong W. et al. (2014) Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H. et al. (2016) The cryo-EM structure of the Plasmodium falciparum 20S proteasome and its use in the fight against malaria. FEBS J 283 (23), 4238–4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ho CM et al. (2020) Bottom-up structural proteomics: cryoEM of protein complexes enriched from the cellular milieu. Nat Methods 17 (1), 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ho CM et al. (2018) Malaria parasite translocon structure and mechanism of effector export. Nature 561 (7721), 70-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ho CM et al. (2021) Native structure of the RhopH complex, a key determinant of malaria parasite nutrient acquisition. Proc Natl Acad Sci U S A 118 (35). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dubochet J. and McDowall AW (1981) VITRIFICATION OF PURE WATER FOR ELECTRON MICROSCOPY. Journal of Microscopy 124 (3), 3–4. [Google Scholar]
- 74.Dubochet J. et al. (1982) Electron microscopy of frozen water and aqueous solutions. Journal of Microscopy 128 (3), 219–237. [Google Scholar]
- 75.Ghorbal M. et al. (2014) Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol 32 (8), 819–21. [DOI] [PubMed] [Google Scholar]
- 76.Wagner JC et al. (2014) Efficient CRISPR-Cas9-mediated genome editing in Plasmodium falciparum. Nat Methods 11 (9), 915–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schureck MA et al. (2021) Malaria parasites use a soluble RhopH complex for erythrocyte invasion and an integral form for nutrient uptake. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Poweleit N. et al. (2016) CryoEM structure of the Methanospirillum hungatei archaellum reveals structural features distinct from the bacterial flagellum and type IV pilus. Nat Microbiol 2, 16222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maxson ME et al. (2021) Detection and quantification of the vacuolar H+-ATPase using the Legionella effector protein SidK. bioRxiv, 2021.07.29.454369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Herbst DA et al. (2021) Structure of the human SAGA coactivator complex: The divergent architecture of human SAGA allows modular coordination of transcription activation and co-transcriptional splicing. bioRxiv, 2021.02.08.430339. [Google Scholar]
- 81.Dai DL et al. (2021) Structural Characterization of Endogenous Tuberous Sclerosis Protein Complex Revealed Potential Polymeric Assembly. Biochemistry 60 (23), 1808–1821. [DOI] [PubMed] [Google Scholar]
- 82.Puchades C. et al. (2017) Structure of the mitochondrial inner membrane AAA plus protease YME1 gives insight into substrate processing. Science 358 (6363), 609-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mashtalir N. et al. (2020) A Structural Model of the Endogenous Human BAF Complex Informs Disease Mechanisms. Cell 183 (3), 802–817 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scheres SHW (2012) A Bayesian View on Cryo-EM Structure Determination. Journal of Molecular Biology 415 (2), 406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tang G. et al. (2007) EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157 (1), 38–46. [DOI] [PubMed] [Google Scholar]
- 86.Hofmann S. et al. (2019) Conformation space of a heterodimeric ABC exporter under turnover conditions. Nature 571 (7766), 580–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhu Y. et al. (2018) Structural mechanism for nucleotide-driven remodeling of the AAA-ATPase unfoldase in the activated human 26S proteasome. Nat Commun 9 (1), 1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang J. et al. (2021) Structure of the trypanosome paraflagellar rod and insights into non-planar motility of eukaryotic cells. Cell Discov 7 (1), 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aquilini E. et al. (2021) An Alveolata secretory machinery adapted to parasite host cell invasion. Nat Microbiol 6 (4), 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lemgruber L. et al. (2013) Cryo-electron tomography reveals four-membrane architecture of the Plasmodium apicoplast. Malar J 12, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kehrer J. et al. (2018) Nuclear Pore Complex Components in the Malaria Parasite Plasmodium berghei. Sci Rep 8 (1), 11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kudryashev M. et al. (2012) Structural basis for chirality and directional motility of Plasmodium sporozoites. Cell Microbiol 14 (11), 1757–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wagner FR et al. (2020) Preparing samples from whole cells using focused-ion-beam milling for cryo-electron tomography. Nat Protoc 15 (6), 2041–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Marko M. et al. (2007) Focused-ion-beam thinning of frozen-hydrated biological specimens for cryo-electron microscopy. Nat Methods 4 (3), 215–7. [DOI] [PubMed] [Google Scholar]
- 95.Mahamid J. et al. (2016) Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science 351 (6276), 969–72. [DOI] [PubMed] [Google Scholar]
- 96.Guo Q. et al. (2018) In Situ Structure of Neuronal C9orf72 Poly-GA Aggregates Reveals Proteasome Recruitment. Cell 172 (4), 696–705 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Albert S. et al. (2017) Proteasomes tether to two distinct sites at the nuclear pore complex. Proc Natl Acad Sci U S A 114 (52), 13726–13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Albert S. et al. (2020) Direct visualization of degradation microcompartments at the ER membrane. Proc Natl Acad Sci U S A 117 (2), 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang P. (2019) Advances in cryo-electron tomography and subtomogram averaging and classification. Curr Opin Struct Biol 58, 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tuijtel MW et al. (2019) Correlative cryo super-resolution light and electron microscopy on mammalian cells using fluorescent proteins. Sci Rep 9 (1), 1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Arnold J. et al. (2016) Site-Specific Cryo-focused Ion Beam Sample Preparation Guided by 3D Correlative Microscopy. Biophys J 110 (4), 860–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hoffman DP et al. (2020) Correlative three-dimensional super-resolution and block-face electron microscopy of whole vitreously frozen cells. Science 367 (6475). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pettersen EF et al. (2021) UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci 30 (1), 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]