

Abstract
Regulatory authorities must balance ensuring evidence of efficacy and safety of new drugs. Various regulatory pathways, such as the accelerated approval program in the United States (US), allow authorities to quickly approve drugs for severely ill patients by granting market authorization based on surrogate end points and pending confirmatory trials. In this cross‐sectional study, we considered 23 indications of cancer drugs that received accelerated approval by the US Food and Drug Administration (FDA) but were subsequently withdrawn as of April 2023. Our investigation extended to assessing the regulatory status of these accelerated approvals in the European Union (EU) and Japan, examining relevant regulatory documents and identifying factors contributing to the withdrawal in the United States. Comparing regions, we found that for 52% (12/23) and 30% (7/23) of withdrawn accelerated approvals in the United States, sponsors had also sought marketing authorization from the European Medicines Agency (EMA) and Japan's Pharmaceuticals and Medical Devices Agency (PMDA), respectively. As of the April 30, 2023 study cutoff date, 83% (10/12) of drug–indication pairs remained approved by the EMA, while the PMDA retained 100% (7/7). For these indications, the time from FDA withdrawal until the study cutoff date ranged from 0.23 years to 11.45 years for EMA approvals (median: 1.28 years) and 1.10 years to 11.45 years for PMDA approvals (median: 3.22 years). These findings highlight substantial regulatory discrepancies concerning cancer drugs with unconfirmed benefits. Addressing these discrepancies may involve requiring pharmaceutical companies to confirm clinical benefits using more robust end points and fostering international harmonization in regulators' assessment.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Various regulatory pathways, such as the accelerated approval program in the United States (US), allow authorities to quickly approve drugs for severely ill patients by granting market authorization based on surrogate end points. Nonetheless, this approach has a notable downside: significant uncertainty regarding the clinical benefit of many novel cancer drugs at the time of market launch.
WHAT QUESTION DID THIS STUDY ADDRESS?
Are there any regulatory discrepancies in expedited cancer drug approvals across the United States, European Union (EU), and Japan—three globally leading pharmaceutical markets that have approved numerous drugs through expedited programs?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Our findings highlight significant regulatory discrepancies across regions for cancer drugs initially granted accelerated approval by the FDA but later withdrawn from the US market. Despite the absence of confirmed improvements in critical end points, such as overall survival (OS) in post‐approval confirmatory trials, a considerable number of indications—100% (7/7) in Japan and 83% (10/12) in the EU—maintained authorization even after withdrawal in the United States.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
This implies that regulatory authorities in the EU and Japan should undertake a re‐evaluation of these oncology drugs and consider their withdrawal to protect patients from dubious treatments. At the very least, our results emphasize the need for regulatory authorities to better explain and justify the rationale behind different regulatory conclusions.
INTRODUCTION
Swiftly providing effective therapeutic medications is critical, yet the prolonged duration of clinical trials assessing overall survival (OS) in cancer treatment presents significant challenges. 1 In response to the pressing needs of patients with serious conditions and with unmet medical needs, the accelerated approval pathway was established in the United States (US) in 1992. 2 This pathway, predominantly used for cancer drugs, 3 speeds up drug approvals by the US Food and Drug Administration (FDA) by majorly relying on surrogate or intermediate clinical end points, such as objective response rate (ORR) and sometimes progression‐free survival (PFS), although many accelerated approvals are based on data from single non‐randomized trials that require confirmation. Additionally, there are also cases where such approvals are granted due to the lack of evidence of safety or studies in certain patient sub‐populations.
Nonetheless, this approach introduces a notable downside: uncertainty regarding the clinical benefit of many novel cancer drugs at the time of market launch. Consequently, sponsors are obligated to conduct post‐marketing confirmatory trials within a specified timeframe to confirm anticipated benefits and subsequently transition drugs to traditional approval status. However, in the event of confirmatory trial failure or undue sponsor delay, the FDA retains the authority to withdraw the drug's approval 4 , 5 , 6 , 7 with the median time from accelerated approval to withdrawal reported to be 3.8 years. 8
Similar regulatory pathways for expediting drug approvals have been established in other regions. The European Medicines Agency (EMA) introduced Conditional Marketing Authorization (CMA) in 2006, 9 requiring sponsors to perform confirmatory trials with more robust end points after CMA. Similarly, Japan's Pharmaceuticals and Medical Devices Agency (PMDA) implemented Conditional Early Approval in 2017, 10 , 11 albeit without the requirement for post‐marketing confirmatory trials that exist in the United States and the European Union (EU). Instead, the PMDA relies on post‐marketing observational studies with “real‐world data” to substantiate the anticipated benefit. 12 , 13 , 14 All these expedited approvals (accelerated approval in the United States, CMA in the EU, and conditional early approval in Japan) are based on a drug matched to a single indication, but a drug can have multiple expedited approvals in different indications.
Comparing the regulatory landscape of expedited cancer drug approvals in the United States, EU, and Japan—three globally leading pharmaceutical markets 15 that have approved numerous drugs through expedited pathways 16 , 17 , 18 —is of major interest due to some of these drugs' unconfirmed advantage for both patient health and health systems. Furthermore, despite successful collaborations between regulators and the industry in these three regions to harmonize global drug development, including in oncology, there remain important divergences in managing failed post‐marketing commitments and in withdrawing market authorizations. 16 , 19
This cross‐sectional study focuses on such divergences by examining the regulatory histories of cancer drugs in the EU and Japan that received accelerated approval for one or more indications by the FDA but were subsequently withdrawn from the US market. 20 , 21 While the regulatory status of such drugs in the EU has been examined, 20 the situation in Japan remains unexplored. Specifically, our focus is on cancer accelerated approvals that were withdrawn from the US market, with April 30, 2023, as the study cutoff date.
MATERIALS AND METHODS
Assessment of the regulatory status of cancer accelerated approvals in the United States
We used publicly available data from the FDA database of accelerated approvals 21 to identify oncology drugs that were granted accelerated approval but were later withdrawn from the US market between December 11, 1992, and April 30, 2023. We recorded the date of the accelerated approval, date of withdrawal, the surrogate end points used in pre‐marketing trials, and the FDA's rationale for the withdrawal. For comprehensive insights into the results of post‐marketing confirmatory trials, we conducted searches across multiple sources. This involved searching the FDA database of Postmarketing Requirements and Commitments, 22 FDA news, ClinicalTrials.gov, 23 press releases issued by pharmaceutical companies, as well as scholarly articles accessible on PubMed and Google Scholar. Our search strategy for scholarly articles utilized relevant terms, such as the name of drugs, trial names, and other identifiers extracted from the FDA database of accelerated approvals. 21 Searches were initially performed in March 2023 and subsequently updated on April 30, 2023, to ensure current and relevant information.
Assessment of the regulatory status of withdrawn cancer accelerated approvals in the EU and Japan
For each drug–indication pair that underwent FDA withdrawal, we conducted a comprehensive assessment of the corresponding regulatory statuses in the EU and Japan (whether the indication had been applied for, and whether the application had been rejected, approved, market‐authorized, and/or withdrawn by the regulators). If the indication had been approved, the end point in the trials leading to the approval was also extracted. We also recorded the date of approval and, when relevant, withdrawal in each region to assess the time between key regulatory events across regions for similar indications. In instances where the approved indication slightly varied between regions, we defined “similar indication” as an indication in which the patient population covered in the EMA or PMDA approval was a subgroup of those included in the withdrawn accelerated approval in the United States, or vice versa. Two authors independently made this decision for each such case, resolving any discrepancies through discussion and consensus.
This involved using databases hosted by the regulatory authorities and examining the regulatory review reports and PDF version of package inserts from each region. In the case of the EU, we systematically searched European Public Assessment Reports (EPARs), EPAR variation assessment reports, and withdrawn applications to identify approved indications similar to the withdrawn US‐accelerated approval indications. In the case of Japan, we utilized product information databases from the Kyoto Encyclopedia of Genes and Genomes (KEGG) and the PMDA to obtain drug approval information. 24 , 25 If no EPAR or withdrawn applications were found on the EMA website or no review reports were found in the PMDA database, respectively, with a similar indication to the withdrawn accelerated approval, we considered that the EMA or PMDA did not receive an application for marketing authorization.
Comparison of regulatory histories of withdrawn cancer accelerated approvals between regions
When marketing authorization was applied for either in the EU or Japan, or in both regions, for an FDA‐withdrawn indication, we compared regulatory histories of the drugs between the regions. This involved plotting a timeline with dates of key regulatory events for each drug–indication pair, including the accelerated approval and withdrawal in the United States, and approvals and potential withdrawals in EU and Japan. For those withdrawn accelerated approvals in the United States that remained approved by the EMA and/or PMDA, the time intervals between FDA accelerated approval and EMA and PMDA approvals, FDA withdrawal and EMA and PMDA approvals, and FDA withdrawal and the study cutoff date were calculated in years. Data were analyzed in Microsoft® Excel.
RESULTS
As of April 30, 2023, we identified 23 accelerated approvals, across 18 oncology drugs, that were withdrawn from the US market after being granted accelerated approval. Table 1 summarizes the characteristics of these 23 accelerated approvals, including generic names, indications, and their respective regulatory status in the EU and Japan, as of April 30, 2023.
TABLE 1.
Cancer accelerated approvals withdrawn by the Food and Drug Administration (FDA) and their regulatory status in the European Union (EU) and Japan.
| Drug | Indication | Regulatory status in EUb | Regulatory status in Japanb |
|---|---|---|---|
| Umbralisib | MZL | Not applied | Not applied |
| Umbralisib | FL | Not applied | Not applied |
| Belantamab | R/R MM | CMA | Not applied |
| Pembrolizumab | SCLC | Not applied | Not applied |
| Atezolizumab | PDL1(+) TNBC | Approved | Approved |
| Duvelisib | FL | Approved | Not applied |
| Nivolumab | SCLC | Not applied | Not applied |
| Nivolumab | Hepatocellular carcinoma | Applied but application rejected | Not applied |
| Pembrolizumab | PDL1(+) gastric or gastroesophageal Cancer | Approved | Not applied |
| Durvalumab | UC | Not applied | Not applied |
| Atezolizumab | UC | Approved | Not applied |
| Olaratumab | Soft tissue sarcoma | CMA → withdrawn | Not applied |
| Atezolizumab | UC | Approved | Not applied |
| Panobinostat | MM | Approved | Approved |
| Idelalisib | FL | Approved | Not applied |
| Vincristine sulfate liposomal | Philadelphia chromosome (−) leukemia | Not applied | Not applied |
| Romidepsin | PTCL | Not applied | Approved |
| Fludarabine phosphate | B‐cell CLL | Not applieda | Approved |
| Bevacizumab | HER2(−) breast cancer | Approved | Approved |
| Tositumomab | Non‐Hodgkin's lymphoma | Not applied | Not applied |
| Gefitinib | NSCLC | Approved | Approved |
| Gemtuzumab ozogamicin | AML | Approved | Approved |
| Celecoxib | FAP | Regular approval → withdrawn | Not applied |
Abbreviations: AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; CMA, conditional marketing authorization; FAP, familial adenomatous polyposis; FL, follicular lymphoma; HER2, human epidermal growth factor receptor 2; MM, multiple myeloma; MZL, marginal zone lymphoma; NSCLC, non‐small cell lung cancer; PDL1, programmed death‐ligand 1; PTCL, peripheral T‐cell lymphoma; R/R, relapsed or refractory; SCLC, small cell lung cancer; TNBC, triple‐negative breast cancer; UC, urothelial carcinoma.
Note that Fludarabine phosphate is not authorized by the EMA, although oral products containing this drug have been authorized by national competent authorities in individual European countries.
Represented data is as of April 30, 2023.
Among these 23 accelerated approvals, sponsors sought marketing authorization in the EU and/or Japan for 15: only EU (n = 8), only Japan (n = 2), both regions (n = 5) (Figure 1). While the PMDA approved all seven submissions in Japan, one of the 13 submissions to the EMA was rejected (nivolumab for hepatocellular carcinoma) due to uncertainties about the main study's design, resulting in a total of 12 approvals in the EU (Figure 1). The regulatory histories of these approvals in each region are depicted in Figure 2. At the time of this study, the EMA had maintained marketing authorization for 83% (10/12) and the PMDA 100% (7/7) (Figure 2). Among the 10 withdrawn cancer accelerated approvals in the United States still authorized by the EMA (Figure 1), two received re‐approval by the FDA but for different populations (gemutuzumab ozogamicin for acute myeloid leukemia 26 , 27 and gefitinib for non‐small cell lung cancer 28 , 29 , 30 ) (Figure 2). In both cases, the FDA re‐approval was for a similar indication as the EMA approval (Table S1).
FIGURE 1.

Flowchart of the regulatory status of cancer accelerated approvals in the European Union (EU) and Japan that were approved via the Food and Drug Administration (FDA's) accelerated approval pathway but subsequently withdrawn by the FDA. * Among the eight indications applied for two were through CMA pathway. One of the approved indications through CMA were withdrawn subsequently based on the result of the confirmatory trial (olaratumab for soft tissue sarcoma). †“Rejected” means that the sponsor's application was rejected by the regulatory authority. ‡“Withdrawn” means that after the initial CMA, the market authorization was withdrawn by the regulatory authority.
FIGURE 2.
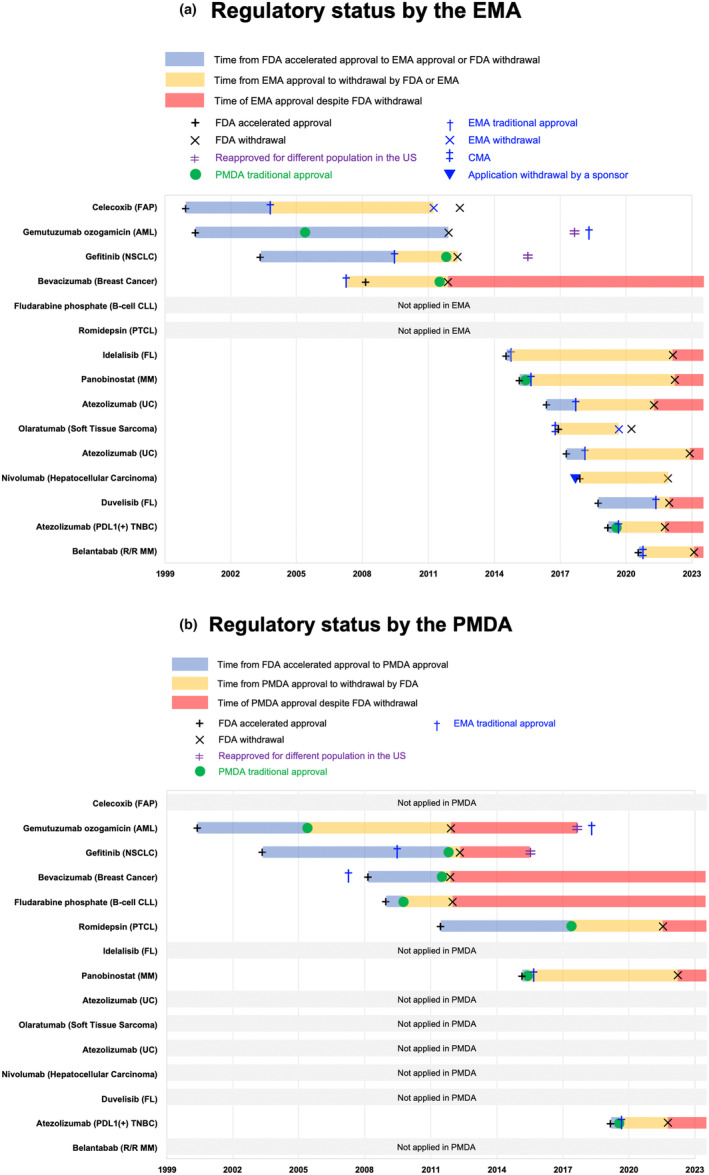
Timeline of cancer accelerated approvals and subsequent withdrawals from the US market, and key regulatory events in the European Union (EU) and Japan. Fifteen cancer accelerated approvals that were withdrawn by the FDA and for which marketing authorization was sought either in the EU or Japan, or both regions. (a) EU. (b) Japan. AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; FAP, familial adenomatous polyposis; FL, follicular lymphoma; MM, multiple myeloma; NSCLC, non‐small cell lung cancer; PDL1, programmed death‐ligand 1; PTCL, peripheral T‐cell lymphoma; R/R, relapsed or refractory; TNBC, triple‐negative breast cancer; UC, urothelial carcinoma.
Two submissions—celecoxib in familial adenomatous polyposis and olaratumab in soft tissue sarcoma 31 —were initially approved but later withdrawn also by the EMA. Table S2 compares the regulatory history of the two approvals withdrawn by both the FDA and EMA. Celecoxib in familial adenomatous polyposis was conditionally approved by the EMA in 1999, albeit before the implementation of the CMA pathway in 2006. It was withdrawn from the EU market in March 2011 32 and from the US market in June 2012 due to the sponsor's inability to provide the required efficacy and safety data to the regulators. 32 , 33 For olaratumab in soft tissue sarcoma, accelerated approval in the United States and CMA in the EU was granted on the basis of the same study, but both regulators subsequently withdrew the authorization due to the confirmatory trial failing to demonstrate improved OS. 31 The EMA's period from initial approval to withdrawal was shorter in both cases compared with the FDA (celecoxib; 12.5 years for FDA vs. 7.5 years for EMA; olaratumab; 3.4 years for FDA vs. 2.9 years for EMA) (Table S2).
All the EMA approvals were later granted traditional marketing authorization, except for belantamab for relapsed or refractory multiple myeloma 34 which received CMA and was under evaluation at the time of our analysis (Figure 2). In all cases, except for the two that received re‐approval by the FDA, drugs were initially approved by the EMA on the basis of PFS or the surrogate end‐point ORR in the exploratory trials that also led to the accelerated approvals by the FDA (Table S3). Similarly, the PMDA granted traditional marketing authorization for all its seven approvals based on PFS or ORR (Table S4).
Finally, for the cancer accelerated approvals that were still on the market in either the EU or Japan, but remained withdrawn in the United States (i.e., excluding those re‐approved), we measured the time from FDA withdrawal to the study's cutoff date on April 30, 2023 (highlighted in red in Figure 2 ). In the EU, this time ranged from 0.23 years to 11.45 years, with a median of 1.28 years, while in Japan, it ranged from 1.10 years to 11.45 years, with a median of 3.22 years.
DISCUSSION
Our findings highlight significant regulatory disparities across regions for cancer drugs initially granted accelerated approval by the FDA but later withdrawn from the US market, mirroring previous observations regarding the distinctions between the EMA and FDA. 20 , 35 , 36 Despite the absence of confirmed improvements in critical end points, such as OS in post‐approval confirmatory trials, a considerable number of indications—100% (7/7) in Japan and 83% (10/12) in the EU—maintained authorization and were still on the market as of the study's cutoff date even after withdrawal in the United States. Notably, the PMDA endorsed FDA‐withdrawn indications for a more prolonged period than the EMA, and interestingly, no market withdrawal by the PMDA was observed for the indications once granted approval, possibly affected by the FDA's initial accelerated approval. 37 The underlying reasons for these regulatory discrepancies are likely multifaceted. One possible factor could be a lower threshold in the United States for both marketing authorization 19 , 35 , 38 and withdrawal, 20 especially when compared to Japan, where fewer accelerated approvals were granted, and none were withdrawn.
These findings suggest a critical issue: either cancer patients in the United States may no longer have access to numerous beneficial medications, as judged by European or Japanese regulatory standards, or patients in the EU or Japan might be receiving treatments with medications that lack clinical benefits, as judged by the US regulatory standards. The latter scenario appears more plausible, considering that FDA's withdrawal decisions were primarily based on a failure to meet primary end points in confirmatory trials (refer to Tables S3 and S4).
This implies that regulatory authorities in the EU and Japan should undertake a re‐evaluation 39 of such oncology drugs and consider their withdrawal to protect patients from dubious treatments. At the very least, our results emphasize the need for regulatory authorities to better explain and justify the rationale behind different regulatory conclusions. 40 This is particularly crucial in light of reported incomplete or delayed post‐marketing studies in the United States and the EU, 41 , 42 , 43 , 44 along with potential safety risks associated with drugs approved through expedited programs. 45 , 46 Investigating clinical guidelines, such as from the National Comprehensive Cancer Network in the United States, for these drugs may be warranted to further illustrate the potential clinical effects of these regulatory discrepancies.
More broadly, recognizing the inherent challenge faced by regulatory authorities in evaluating clinical benefits of many novel cancer drugs, 47 , 48 our results underscore the importance for pharmaceutical sponsors and regulators to intensify efforts in verifying clinical benefits earlier using more robust end points in order to withdraw ineffective or unsafe drugs from the market quickly. Indeed, the FDA recently released a guideline, suggesting it may require that studies intended to confirm clinical benefit be underway prior to approval, or within a specified time period subsequent to approval, with the aim of more quickly strengthening evidence on benefits and harms and withdraw drugs that fail to meet acceptable therapeutic standards. 49 , 50 However, the significant regulatory disparities across regions we have identified suggests that this may require a global regulatory harmonization of expedited programs for cancer drugs, mirroring, for example, the existing practices for rare disease drugs in the United States, Europe, and Japan. 51 , 52 , 53
Limitations of this study
The current study has several limitations. Firstly, it relied exclusively on the authors' assessment of publicly available information, lacking access to internally held data within companies or regulatory agencies. This constraint may impact the comprehensiveness and accuracy of the data analysis. Secondly, the study's exclusive focus on oncology drugs within the accelerated approval program might constrain its generalizability to other therapeutic areas, limiting the broader applicability of the findings. Thirdly, the regulatory statuses in the EU and Japan were determined based on data as of April 2023, and there is a possibility that these statuses could undergo changes in the future, especially CMAs still under evaluation.
AUTHOR CONTRIBUTIONS
H.H., A.O., and T.T. conceptualized and designed the research. H.H. and F.M. majorly acquired data and performed research with help from S.M., A.O., and T.T. H.H., F.M., and S.M. analyzed the data. H.H. completed the original manuscript and all listed authors contributed to this manuscript with a critical revision.
FUNDING INFORMATION
No funding was received for this work.
CONFLICT OF INTEREST STATEMENT
T.T. receives personal fees from Medical Network Systems MNES Inc., and Bionics Inc., outside the submitted work. A.O. receives personal fees from MNES Inc., Becton, Dickinson and Company, Taiho Pharmaceutical Co., Ltd., and Kyowa Kirin Inc., outside the submitted work. H.S. received personal fees from TAIHO pharmaceutical company outside the submitted work. S.M's partner is employed by ICON, a global contract research organization whose customers include many pharmaceutical companies. Other authors (H.H. and F.M.) have no relevant financial or non‐financial interests to disclose.
Supporting information
Table S1.–S4.
ACKNOWLEDGMENTS
The authors thank Dr. M. Kami and Dr. H. Maeda for the fruitful discussion. H.H. is supported by TOYOBO Biotechnology Foundation Fellowship and JSPS Overseas Research Fellowships, outside the submitted work.
Hakariya H, Moriarty F, Ozaki A, Mulinari S, Saito H, Tanimoto T. Continued cancer drug approvals in Japan and Europe after market withdrawal in the United States: A comparative study of accelerated approvals. Clin Transl Sci. 2024;17:e13879. doi: 10.1111/cts.13879
REFERENCES
- 1. Chen EY, Joshi SK, Tran A, Prasad V. Estimation of study time reduction using surrogate end points rather than overall survival in oncology clinical trials. JAMA Intern Med. 2019;179:642‐647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. FDA . Accelerated approval program. Accessed June 30, 2023. https://www.fda.gov/drugs/nda‐and‐bla‐approvals/accelerated‐approval‐program?fbclid=IwAR213pV8uj9CSmdXvmTBF92X_LXiFXQW8tvsAKGq7ZdhkNLfJYpAF0LsEOg
- 3. Beaver JA, Pazdur R. “Dangling” accelerated approvals in oncology. N Engl J Med. 2021;384:e68. [DOI] [PubMed] [Google Scholar]
- 4. Sachs RE, Donohue JM, Dusetzina SB. Accelerated approval — taking the FDA's concerns seriously. N Engl J Med. 2022;387:199‐201. [DOI] [PubMed] [Google Scholar]
- 5. Gyawali B, Rome BN, Kesselheim AS. Regulatory and clinical consequences of negative confirmatory trials of accelerated approval cancer drugs: retrospective observational study. BMJ. 2021;374:n1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cliff ERS, Rome RS, Kesselheim AS, Rome BN. Association of accelerated approval (AA) status with National Comprehensive Cancer Network (NCCN) guideline recommendation for cancer drug indications: a cross‐sectional study. J Clin Oncol. 2023;41:1599. [Google Scholar]
- 7. Mooghali M, Wallach JD, Mitchell AP, Skydel JJ, Ross JS, Ramachandran R. Clinical practice guideline descriptions of oncologic drugs granted FDA accelerated approval: a cross‐sectional study. J Clin Oncol. 2023;41:1590.36599114 [Google Scholar]
- 8. Fashoyin‐Aje LA, Mehta GU, Beaver JA, Pazdur R. The on‐ and off‐ramps of oncology accelerated approval. N Engl J Med. 2022;387:1439‐1442. [DOI] [PubMed] [Google Scholar]
- 9. European Medicines Agency . Human regulatory: conditional marketing authorization. Accessed January 9, 2024. https://www.ema.europa.eu/en/human‐regulatory/marketing‐authorisation/conditional‐marketing‐authorisation
- 10. Japan Pharmaceuticals and Medical Devices Agency . Implementation of a conditional early approval system for pharmaceutical products, October 20, 2017. Accessed December 7, 2023. https://www.pmda.go.jp/files/000227089.pdf
- 11. Tanaka M, Miyazawa H, Terashima R, Ikuma M. Conditional early approval for new drug applications in Japan: current and emerging issues. Clin Transl Sci. 2023;16:1289‐1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murayama A, Ueda M, Shrestha S, Tanimoto T, Ozaki A. Japan's conditional early approval program for innovative cancer drugs: comparison of the regulatory processes with the US FDA and the EMA. Cancer Cell. 2021;39:1165‐1166. [DOI] [PubMed] [Google Scholar]
- 13. Matsushita S, Tachibana K, Kusakabe T, Hirayama R, Tsutsumi Y, Kondoh M. The roadmap to approval under Japan's two‐track regulatory system: comparing six regenerative medical products. Cell Stem Cell. 2020;27:515‐518. [DOI] [PubMed] [Google Scholar]
- 14. Ozaki A, Senoo Y, Saito H, et al. Japan's drug regulation framework: aiming for better health or bigger profits? Int J Health Policy Manag. 2021;10:47‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shibata S, Uemura R, Suzuki T. Comparative analysis between the top‐selling drugs in the Japanese pharmaceutical market and those in the United States, the United Kingdom, France, and Germany. Ther Innov Regul Sci. 2016;50:221‐227. [DOI] [PubMed] [Google Scholar]
- 16. Wang S, Yang Q, Deng L, et al. An overview of cancer drugs approved through expedited approval programs and orphan medicine designation globally between 2011 and 2020. Drug Discov Today. 2022;27:1236‐1250. [DOI] [PubMed] [Google Scholar]
- 17. Hwang TJ, Kesselheim AS, Tibau A, Lee CWC, Vokinger KN. Clinical benefit and expedited approval of cancer drugs in the United States, European Union, Switzerland, Japan, Canada, and Australia. JCO Oncol Pract. 2022;18:e1522‐e1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Muensterman ET, Luo Y, Parker JM. Breakthrough therapy, PRIME and Sakigake: a comparison between neuroscience and oncology in obtaining preferred regulatory status. Ther Innov Regul Sci. 2020;54:658‐666. [DOI] [PubMed] [Google Scholar]
- 19. Alqahtani S, Seoane‐Vazquez E, Rodriguez‐Monguio R, Eguale T. Priority review drugs approved by the FDA and the EMA: time for international regulatory harmonization of pharmaceuticals? Pharmacoepidemiol Drug Saf. 2015;24:709‐715. [DOI] [PubMed] [Google Scholar]
- 20. Cramer A, Sørup FKH, Christensen HR, Petersen TS, Karstoft K. Withdrawn accelerated approvals for cancer indications in the USA: what is the marketing authorisation status in the EU? Lancet Oncol. 2023;24:e385‐e394. [DOI] [PubMed] [Google Scholar]
- 21. US Food and Drug Administration . Withdrawn: cancer accelerated approvals. Accessed April 30, 2023. https://www.fda.gov/drugs/resources‐information‐approveddrugs/withdrawn‐cancer‐accelerated‐approvals
- 22. FDA database of postmarketing requirements and commitments. Accessed January 9, 2024. https://www.accessdata.fda.gov/scripts/cder/pmc/index.cfm
- 23. ClinicalTrials.gov. Accessed June 30, 2023. https://clinicaltrials.gov/
- 24. KEGG, new drug approvals in the USA, Europe and Japan. Accessed June 30, 2023. https://www.kegg.jp/kegg/drug/br08328.html
- 25. Japan Pharmaceuticals and Medical Devices Agency . Ethical drug information research. Accessed June 30, 2023. https://www.pmda.go.jp/PmdaSearch/iyakuSearch/
- 26. Tanimoto T, Tsubokura M, Mori J, Pietrek M, Ono S, Kami M. Differences in drug approval processes of 3 regulatory agencies: a case study of gemtuzumab ozogamicin. Investig New Drugs. 2013;31:473‐478. [DOI] [PubMed] [Google Scholar]
- 27. Selby C, Yacko LR, Glode AE. Gemtuzumab ozogamicin: back again. J Adv Pract Oncol. 2019;10:68‐82. [PMC free article] [PubMed] [Google Scholar]
- 28. Fukuoka M, Yano S, Giaccone G, et al. Multi‐institutional randomized phase II trial of gefitinib for previously treated patients with advanced non–small‐cell lung cancer. J Clin Oncol. 2003;21:2237‐2246. [DOI] [PubMed] [Google Scholar]
- 29. Mok TS, Yi‐Long W, Sumitra T, et al. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947‐957. [DOI] [PubMed] [Google Scholar]
- 30. Douillard J‐Y, Ostoros G, Cobo M, et al. First‐line gefitinib in Caucasian EGFR mutation‐positive NSCLC patients: a phase‐IV, open‐label, single‐arm study. Br J Cancer. 2014;110:55‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tap WD, Wagner AJ, Schöffski P, et al. Effect of doxorubicin plus olaratumab vs doxorubicin plus placebo on survival in patients with advanced soft tissue sarcomas. JAMA. 2020;323:1266‐1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. European Medicines Agency . Onsenal (celecoxib) ‐ withdrawal of the marketing authorization in the European Union. Accessed January 9, 2024. https://www.ema.europa.eu/en/documents/public‐statement/public‐statement‐onsenal‐celecoxib‐withdrawal‐marketing‐authorisation‐european‐union_en.pdf
- 33. Federal Register. Pfizer, Inc . Withdrawal of approval of familial adenomatous polyposis indication for CELEBREX. 8 June 2012. Accessed January 9, 2024. https://www.federalregister.gov/documents/2012/06/08/2012‐13900/pfizer‐inc‐withdrawal‐of‐approval‐of‐familial‐adenomatous‐polyposis‐indication‐for‐celebrex#:~:text=On%20February%202%2C%202011%2C%20FDA,subpart%20H%20was%20never%20completed
- 34. Tzogani K, Penttilä K, Lähteenvuo J, et al. EMA review of Belantamab Mafodotin (Blenrep) for the treatment of adult patients with relapsed/refractory multiple myeloma. Oncologia. 2020;26:70‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Michaeli DT, Mills M, Michaeli T, Miracolo A, Kanavos P. Initial and supplementary indication approval of new targeted cancer drugs by the FDA, EMA, Health Canada, and TGA. Investig N Drugs. 2022;40:798‐809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vokinger KN, Kesselheim AS, Glaus CEG, Hwang TJ. Therapeutic value of drugs granted accelerated approval or conditional marketing authorization in the US and Europe from 2007 to 2021. JAMA Health Forum. 2022;3:e222685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Madhusoodanan J. How a controversial US drug policy could be harming cancer patients worldwide. Nature. 2023;620:264‐267.37558845 [Google Scholar]
- 38. Vokinger KN, Kesselheim AS. Application of orphan drug designation to cancer treatments (2008–2017): a comprehensive and comparative analysis of the USA and EU. BMJ Open. 2019;9:e028634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maeda H, Shingai R, Takeda K, Hara A, Murai Y, Ofuchi M. Assessment of surrogate end point trends in clinical trials to approve oncology drugs from 2001 to 2020 in Japan. JAMA Netw Open. 2023;6:e238875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mulinari S, Davis C. Why European and United States drug regulators are not speaking with one voice on anti‐influenza drugs: regulatory review methodologies and the importance of ‘deep’ product reviews. Heal Res Policy Syst. 2017;15:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Naci H, Smalley KR, Kesselheim AS. Characteristics of preapproval and Postapproval studies for drugs granted accelerated approval by the US Food and Drug Administration. JAMA. 2017;318:626‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gyawali B, Hey SP, Kesselheim AS. Assessment of the clinical benefit of cancer drugs receiving accelerated approval. JAMA Intern Med. 2019;179:906‐913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cherla A, Mossialos E, Salcher‐Konrad M, Kesselheim AS, Naci H. Post‐marketing requirements for cancer drugs approved by the European medicines agency, 2004–2014. Clin Pharmacol Ther. 2022;112:846‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Parikh RB, Hubbard RA, Wang E, et al. Exposure to US cancer drugs with lack of confirmed benefit after US Food and Drug Administration accelerated approval. JAMA Oncol. 2023;9:567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shepshelovich D, Tibau A, Goldvaser H, et al. Postmarketing modifications of drug labels for cancer drugs approved by the US Food and Drug Administration between 2006 and 2016 with and without supporting randomized controlled trials. J Clin Oncol. 2018;36:1798‐1804. [DOI] [PubMed] [Google Scholar]
- 46. Mostaghim SR, Gagne JJ, Kesselheim AS. Safety related label changes for new drugs after approval in the US through expedited regulatory pathways: retrospective cohort study. BMJ. 2017;358:j3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Olivier T, Haslam A, Prasad V. Anticancer drugs approved by the US Food and Drug Administration from 2009 to 2020 according to their mechanism of action. JAMA Netw Open. 2021;4:e2138793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mitra‐Majumdar M, Gunter SJ, Kesselheim AS, et al. Analysis of supportive evidence for US Food and Drug Administration approvals of novel drugs in 2020. JAMA Netw Open. 2022;5:e2212454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shahzad M, Naci H, Wagner AK. Association between preapproval confirmatory trial initiation and conversion to traditional approval or withdrawal in the FDA accelerated approval pathway. JAMA. 2023;329(9):760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hakariya H, Ozaki A, Mulinari S, Moriarty F, Tanimoto T. Evaluating time to withdrawal of anti‐cancer drug indications that received FDA’s accelerated approval. QJM: An International Journal of Medicine. 2024. 10.1093/qjmed/hcae115 [DOI] [PubMed] [Google Scholar]
- 51. Murakami M, Narukawa M. Matched analysis on orphan drug designations and approvals: cross regional analysis in the United States, the European Union, and Japan. Drug Discov Today. 2016;21:544‐549. [DOI] [PubMed] [Google Scholar]
- 52. Döring JH, Lampert A, Hoffmann GF, Ries M. Thirty years of orphan drug legislation and the development of drugs to treat rare seizure conditions: a cross sectional analysis. PLoS One. 2016;11:e0161660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Miller KL, Kraft S, Ipe A, Fermaglich L. Drugs and biologics receiving FDA orphan drug designation: an analysis of the most frequently designated products and their repositioning strategies. Expert Opin Orphan Drugs. 2022;9:265‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.–S4.