

Abstract
Multiple myeloma (MM) is a genetically heterogeneous disease and the management of relapses is one of the biggest clinical challenges. TP53 alterations are established high‐risk markers and are included in the current disease staging criteria. KRAS is the most frequently mutated gene affecting around 20% of MM patients. Applying Clonal Competition Assays (CCA) by co‐culturing color‐labeled genetically modified cell models, we recently showed that mono‐ and biallelic alterations in TP53 transmit a fitness advantage to the cells. Here, we report a similar dynamic for two mutations in KRAS (G12A and A146T), providing a biological rationale for the high frequency of KRAS and TP53 alterations at MM relapse. Resistance mutations, on the other hand, did not endow MM cells with a general fitness advantage but rather presented a disadvantage compared to the wild‐type. CUL4B KO and IKZF1 A152T transmit resistance against immunomodulatory agents, PSMB5 A20T to proteasome inhibition. However, MM cells harboring such lesions only outcompete the culture in the presence of the respective drug. To better prevent the selection of clones with the potential of inducing relapse, these results argue in favor of treatment‐free breaks or a switch of the drug class given as maintenance therapy. In summary, the fitness benefit of TP53 and KRAS mutations was not treatment‐related, unlike patient‐derived drug resistance alterations that may only induce an advantage under treatment. CCAs are suitable models for the study of clonal evolution and competitive (dis)advantages conveyed by a specific genetic lesion of interest, and their dependence on external factors such as the treatment.
INTRODUCTION
Intra‐tumor genetic heterogeneity is one hallmark of multiple myeloma (MM) and an important factor in tumor evolution and disease progression. 1 , 2 , 3 , 4 , 5 The typical disease course for patients with MM is frequent relapses upon different therapies and the development of multidrug resistance. 6 In MM, a plethora of different treatments are available. Notably, recent studies show that novel therapies, such as bispecific antibodies or CAR T cells, may induce deep and ongoing remission in multidrug‐resistant patients. Regrettably, no cure is achieved and primary or secondary resistance is observed, 7 , 8 known already from standard drugs like immunomodulators (IMiDs) or proteasome inhibitors (PIs). To decipher the underlying evolutionary trajectories and to better understand the clonal variation from disease diagnosis to the relapsed‐refractory state, intensive research has been conducted on the genomic landscape of MM, mostly using bulk sequencing approaches like whole exome and whole genome sequencing (WES/WGS). 4 , 9 , 10 A number of genetic alterations have been identified and were associated with increased clonal fitness or disease relapse. 2 , 7 , 11 , 12 , 13 There is growing evidence for clonal diversity as the major driver of treatment failure and drug resistance. Still, the specific impact on clinics and biology remains unclear for the vast majority of identified genetic alterations. Many studies remain largely descriptive, lacking functional characterization, especially scalable methods for the analysis of interaction dynamics between different clones and cell competition. Moreover, cell competition occurs against healthy tissue when the space is limited, 14 and MM cells are located in the bone marrow niche, which is a confined space. 15 It also plays a role in mammalian development, organ homeostasis, and aging, and critically influences the initiation and development of all cancer types. 14 , 16 , 17
The concept of cell fitness describes both, the cell's ability to remain in a cell population and the capacity of a clone to expand under the given environmental conditions. Less fit cells get replaced by fitter congeners in a continuous process that ensures the highest fitness of a tissue. The replacement can happen actively by the removal of less fit cells (e.g., clone extinction via apoptosis, phagocytosis, or entosis) or passively by outcompeting over time through enhanced cell survival, proliferation, or differentiation. 17 The vast majority of healthy cells show a neutral competition fitness, meaning they are overall equally fit and the replacement is stochastic though maintaining the number of cells within a tissue. 14 , 18 Nonneutral competition fitness, on the other hand, is associated with development, aging, or oncogenic processes. 14 , 17 In cancer, clonal expansion can be explained in two ways: cancer cells die less than their competitors and/or they proliferate more. When mutations arise, they confer a neutral or selective fitness advantage or disadvantage to the cell. 19 The cancer cell is in constant competition against normal tissue, microenvironment, and other tumor cells. Diverse intrinsic and extrinsic pressures suffered by the mutant cells promote the selection and expansion of distinct subclones. 14
Here we apply Clonal Competition Assays (CCAs) based on the coculturing of fluorescence‐marked isogenic MM cells with or without a genetic alteration under study, for example, point mutations or deletions. This experimental setting allows us to study the implications of specific genetic lesions on cellular fitness and their impact under different environmental conditions such as drug exposure (Figure 1A). In this study, our focus was on KRAS as the activation of RAS‐mediated oncogenic signaling pathways is a hallmark of various cancers 22 , 23 and it is frequently mutated in relapsed MM. 24 , 25 Other emblematic cancers prone to KRAS mutations are lung, colorectal, and pancreatic adenocarcinomas and urogenital cancers. 23 In MM, mutations in KRAS and NRAS are the most recurring somatic variants, with a mutational frequency at diagnosis of around 20%–25% each. 4 , 25 , 26 , 27 , 28 Persistence or de novo occurrence of KRAS alterations at disease relapse was also described. 24 , 29 Mutational hotspots for KRAS are regions in exon 2, for example, p.G12 in the P‐loop (representing up to 40% of KRAS mutations in MM) or in exon 3, for example, p.Q61 in switch 1 (35%) (Figure 1B). However, several other KRAS mutations also occur. 25 In our CCAs we investigated two different KRAS point mutations, p.G12A within the exon‐2 hotspot and a second less frequent mutation p.A146T in exon 4 (switch 2) as these two mutations represent two different mechanisms for RAS activation. Furthermore, we compare the fitness signatures of KRAS and TP53 mutations to confirmed resistance mutations in CUL4B, IKZF1, and PSMB5.
Figure 1.
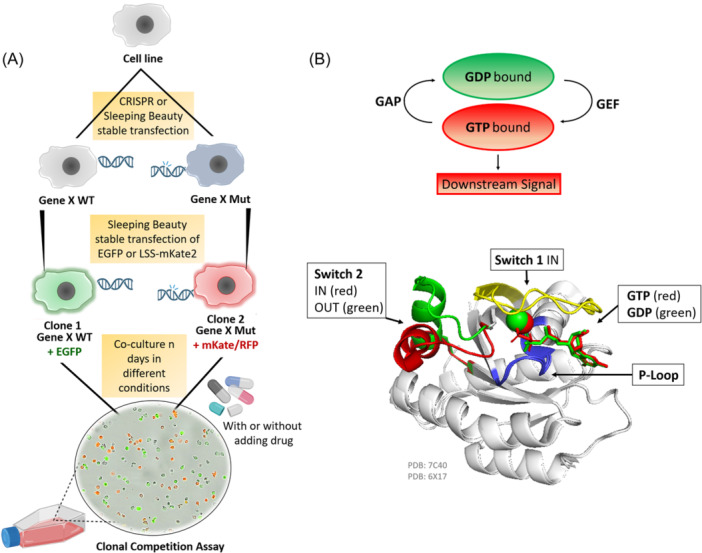
Clonal competition assay (CCA) for the study of the long‐term impact of genomic alterations on the overall cell fitness, and KRAS in its active and inactive conformational shape. (A) Schematic illustration of the general principle of the CCA. Initially, the alteration of interest (e.g., point mutation, deletion, etc.) is introduced into a cell line by gene editing approaches like CRISPR Cas9 or Sleeping Beauty (SB). Then, SB vectors for the stable expression of fluorescent proteins (e.g., EGFP or mKate/RFP) are integrated. Different colors are used to differentiate between the WT and the mutant or between different alterations of interest. Co‐cultures are set up by mixing cells of different colors. The culture is regularly split and drugs can be added. Every 4–5 days a sample is taken from the culture and the ratio of differently colored cells is measured by flow cytometry. (B) Schematic representation of RAS superfamily proteins activation and inactivation cycle by hydrolyzing GTP into GDP via the GTPase activating proteins (GAPs) and the Guanine exchange factor (GEF) proteins triggering or stopping downstream signaling. Superimposition of the cartoon diagram of KRAS displaying the conformational change in switch 2 induced by the hydrolysis of GTP changing from the signaling conformation (red, PDB: 6XI7, 20 ) to the nonsignaling conformation (green, PDB: 7C40, 21 ). The backbone is shown in white, the P‐loop Walker Motif A in blue, and switch 1 in yellow as it is in the IN conformation in both PDBs.
METHODS
Generation of the mutant cell lines
The OPM2 MM cell line was chosen for the generation of monoallelic KRAS‐altered variants (A146T and G12A) because it originally has an intact RAS pathway. Notably, many other commonly used MM cell lines are not suitable models as they already harbor mutations in the RAS genes, for example, KRAS A146T in AMO1, NRAS Q61L in L363, or KRAS G12A in MM1S. The two OPM2 KRAS mutants were generated as previously described by stable transfection with Sleeping Beauty (SB) transposon plasmids. 25 AMO1, on the other hand, is one of the few MM cell lines harboring wild‐type (WT) TP53 and was therefore chosen to generate mono‐ and/or bi‐allelic TP53 lesions as previously published. 30 AMO1 and L363 cell lines were used to study mutations associated with drug resistance as they are sensitive to immunomodulatory agents (IMiDs) or proteasome inhibitors (PIs), respectively. The inserted mutations are either located in the drug binding site, for example, for PSMB5 in the S1 pocket essential for the proteasomal chymotrypsin‐like activity (CCA: PSMB5 A20T vs. WT) 31 or they affect associated genes or downstream transcription factors like IKZF1 (CCA: IKZF1 A152T vs. WT) 2 or CUL4B (CCA: CUL4B KO vs. WT) that forms part of the CRBN‐CRL4 E3 ubiquitin ligase network. 2 All investigated mutations were derived from drug‐resistant MM patients and have previously been functionally confirmed to confer drug resistance. 2 , 31 For choosing a suitable cell line, an intact wild‐type pathway of interest is important. When designing a CCA, the Cancer Cell Line Encyclopedia (CCLE), the MM cell line characterization dataset created by Keat's Lab (https://myelomagenomics.tgen.org/), and published datasets, for example, Vikova et al. 32 Theranostics might be considered to pick the best cell lines. 32
Generation of fluorescently labeled cells (stable transfection)
The cells of interest for the CCA were stably transfected with SB expression plasmids for the fluorescent proteins EGFP, 33 LSS‐mKate2, 34 and a transposase vector by electroporation using a Gene Pulser XCell (Bio‐Rad Laboratories) with a single exponential decay pulse of 280–300 V. After overnight recovery, geneticin (G418) was added to the electroporated cell culture to extinguish untransfected and only transiently transfected cells and select the stably transfected cells. It took 10–14 days for the selection process to be completed. Cultures were routinely checked for mycoplasma negativity.
Setup of the CCA and flow cytometry
CCAs were set up by mixing WT and mutant cells in different ratios, for example, 3:1 for the KRAS WT and the KRAS mutants. To confirm reproducibility, the CCAs were performed at least in two identical technical replicates. Additionally, the fluorescent markers were switched between WT and mutants in independent experiments. For each CAA experiment, two million cells were seeded in a T75 culture flask. Cells were cultured in RPMI‐1640 medium supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 2 mM glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin and kept at 5% CO2 and 37°C. Cultures were regularly split (every 3–4 days). OPM2 cells are semi‐adherent and were scratched with a cell scraper before each splitting, the other cell lines were grown as suspension cultures. Every 4–5 days, a sample of the CCA co‐culture was taken, washed, and resuspended in phosphate‐buffered saline for flow cytometry using a FACS Calibur (BD Biosciences). EGFP and mKate/RFP are both excitable with a 488 nm laser. A total of 10,000 cells was counted per sample for each time point. The relative number/ratio of color‐coded cells in the cultures under study was analyzed with FlowJo version 8.8.7 (BD Biosciences). To generate the plots, PRISM GraphPad was used with the following settings: standard curve to interpolate, sigmoidal, 4PL (parameter logistic), X is log, and no special handling of outliers.
RNA extraction, library preparation, and sequencing
For RNA‐Seq analysis, the OPM2 KRAS WT and the OPM2‐derived KRAS A146T and G12A cells were seeded in triplicates at a density of 100,000 cells/mL. Three days after seeding, two million cells were pelleted and stored at −80°C. The RNeasy Mini kit (Qiagen) was used to extract the RNA and the quality was checked with a 2100 Bioanalyzer and the RNA 6000 Nano kit (Agilent). The RNA integrity number (RIN) was ≥9.3 for all samples. Libraries were prepared from 1 µg of total RNA using the TruSeq stranded mRNA kit (Illumina). Quantification was conducted with a Qubit 3.0 Fluorometer (Thermo Fisher) and a 2100 Bioanalyzer with the DNA 1000 kit (Agilent). Pooled libraries were sequenced (single‐end) on the NextSeq 500 (Illumina) using the High Output Kit v2.5 (75 cycles).
Bioinformatic analysis
Sequencing quality was evaluated via FastQC (v. 0.11.7) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). RNA‐sequencing reads were quality‐ and adapter‐trimmed using Cutadapt (v. 2.5) with a cutoff Phred score of 20 in NextSeq mode, and reads without any remaining bases were discarded (parameters: ‐‐nextseq‐trim=20 ‐m 1 ‐a ACTGTCTCTTATACACATCT). Trimmed reads were mapped to the human genome (RefSeq GRCh38.p13 primary assembly and mitochondrion) using STAR (v. 2.7.2b) 35 with default parameters except for including transcript annotations from RefSeq annotation version 109.20190905 for GRCh38.p13. RNA‐Seq quality data per sample is given in Supporting Information S1: Table 1. Mapped reads were quantified at the exon level and summarized for each gene using featureCounts (v. 1.6.4) from the Subread package 36 based on the annotation above. Multimapping and multioverlapping reads were counted unstranded with a fractional count for each alignment and overlapping feature (parameters: ‐s 0 ‐t exon ‐M ‐O ‐‐fraction).
Differential gene expression analysis was performed in R (v. 4.3.1) with DESeq. 2 (v. 1.22.2) 37 incorporating log fold change shrinkage using apeglm. 38 Genes with a log2FC >│1│ and adjusted p < 0.025 were considered as differentially expressed. Gene Set Enrichment Analysis (GSEA) was carried out using the gseKEGG function from clusterProfiler R package (v. 4.8.1) 39 and the KEGG pathways annotations (v. 106). Default parameters were used unless otherwise specified. Notably, the keyType parameter was set to “ncbi‐geneid” to ensure consistent gene identification across databases, and the minimum gene set size (minGSSize) was set to 15 to filter out small gene sets. Genes identified from the DESeq2 analysis were ranked based on the absolute |log2FC| between the KRAS A146T/G12A group and the KRAS WT group. Genes were excluded from the analysis if they lacked log2FC information or if they were duplicates based on HGNC symbols or gene IDs. A false discovery rate (FDR) < 0.25 was applied to select significant pathways. We conducted a streamlined analysis to pinpoint pivotal genes within three chosen proliferation pathways (JAK‐STAT, PI3K‐Akt, and MAPK), considering their significance in our biological framework. To achieve this, we focused on genes that were common to at least two of the three pathways and exhibited notable effects in the GSEA analysis, thus identifying them as pertinent in the “core enrichment” section.
Structural analysis
The structural 3D representations (Figures 1B, 2A,B and Supporting Information S1: Figure 1) were performed with PyMOL Molecular Graphics System (2002; DeLano Scientific) and data from public databases (Protein Data Bank) as listed in Supporting Information S1: Table 2. The 2D representations (interaction maps) were directly extracted from PDBe from PDB IDs 6BOF (chain B) for KRAS A146T mutant 40 and from 6MBU for KRAS WT 41 (Supporting Information S1: Figure 2).
Figure 2.
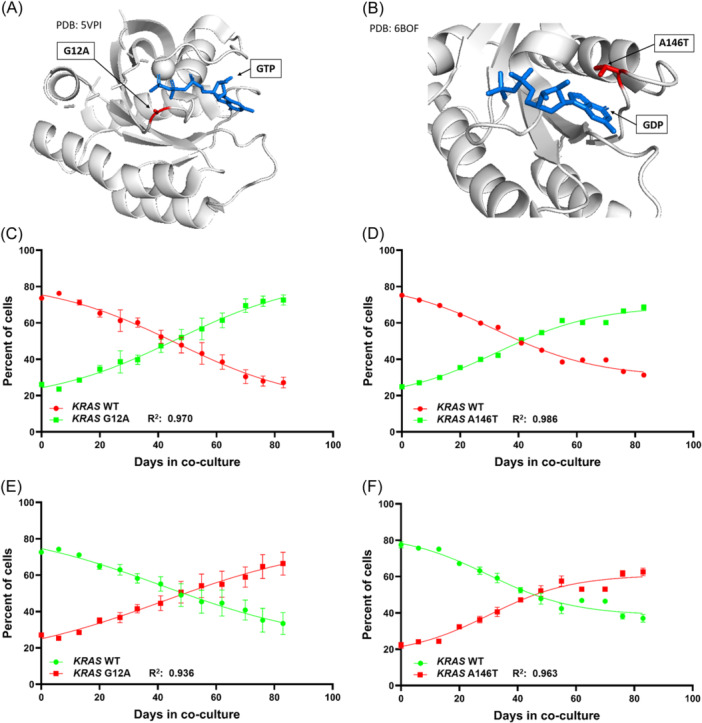
Clonal competition assays of OPM2 sublines bearing KRAS mutations G12A or A146T compared to OPM2 KRAS WT cells. (A) Structural representation of the oncogenic KRAS mutation G12A (PDB: 5VPI, 42 ) in complex with GTP and the magnesium ion cofactor. (B) KRAS A146T mutation in complex with GDP (PBD: 6BOF, 40 ), there is no magnesium ion coordination in this structure. The mutated residues are represented in red. GTP and GDP are displayed in blue; for clarity no different colors indicating charges are assigned to any atom. Clonal Competition Assays reveal a gradual increase of the mutants KRAS G12A (C, E) and KRAS A146T (D, F) outcompeting the OPM2 KRAS WT cells due to enhanced fitness. This effect was confirmed by technical replicates and by switching the color coding between WT and mutant cells. The error bars indicate the two‐paired 95% CI.
RESULTS
KRAS mutations confer a growth advantage to the mutated cells
Here we show not‐treatment‐related, increased fitness for cells harboring the KRAS mutations G12A (Figure 2, left) and A146T (Figure 2, right) in respective comparison to OPM2 KRAS WT cells. Starting with a co‐cultivation ratio of 1:3 for the KRAS mutants to the WT, both mutants expanded faster than the WT and reached 50% of the cells in the flask between co‐culturing days 41 and 48. By day 83 the starting values were reversed (Figure 2C–F, Supporting Information S1: Figure 3) and mutant to WT ratios of 3:1 to 4:3 had been obtained. The experiments were set up in two independent replicates for each setting (Supporting Information S1: Figure 3). An almost linear progression was observed for the CCA. The R 2 of the sigmoidal growth trajectory was 0.970 for KRAS G12A green (Figure 2C), 0.986 for KRAS A146T green (Figure 2D), 0.936 for KRAS G12A red (Figure 2E), and 0.963 for KRAS A146T red (Figure 2F). These results fit with other published mathematical models 13 and were confirmed by switching the red and green color‐labeling between the WT and the respective mutants. There were no significant differences in the outcome of the CCAs if G12A mutant cells were labeled in red (range: 25.0%–70.8%) or in green (range: 23.2%–74.6%) with p = 0.74 (two‐tailed MWU‐test, Figure 2C,E). Both showed similar dynamics with the mutant cells outcompeting the WT cells. The same holds true for cells harboring the A146T mutation coded either in red (range: 20.9%–64.0%) or in green (range: 24.1%–69.6%) (Figure 2D,F) with p = 0.44. We furthermore observed a similar fitness advantage for both KRAS mutants (p = 0.88) taking all replicate experiments into account.
Global transcriptomics analysis reveals increased proliferation signaling as key dysregulated pathways
To define the gene expression profile associated with KRAS mutations, we performed differential gene expression analysis comparing KRAS G12A/A146T mutants with the WT (Figure 3). The Principal‐Component Analysis (PCA) with PC1 (57% variance) and PC2 (34% variance) revealed a clustering according to the mutational alteration (Figure 3A and Supporting Information S1: Figure 4). The KRAS expression was significantly lower in both the KRAS A146T (p = 0.017) and KRAS G12A (p = 0.001) mutant groups compared to the WT group (Figure 3B). We identified a total of 186 differentially expressed genes (DEGs) in the comparison between KRAS A146T and the WT group with the cut‐off criterion log2FC > |1| and adjusted p < 0.05 (Figure 3C and Supporting Information S1: Table 3). Similarly, while comparing the KRAS G12A mutant and the WT group, 130 DEGs were found (Figure 3C and Supporting Information S1: Table 4). The gene expression analysis revealed distinctive gene regulation patterns. The KRAS A146T mutants showed a predominant upregulation pattern, with 132 genes upregulated and 54 genes downregulated. The KRAS G12A mutants exhibited a more balanced pattern, with 69 upregulated and 61 downregulated genes. Remarkably, 56 DEGs overlapped between both mutant groups, with 43 upregulated and 13 downregulated genes (Figure 3D and Supporting Information S1: Table 5). The SHISAL1 gene exhibited differential regulation, being upregulated in the KRAS G12A mutant and downregulated in the KRAS A146T mutant (Figure 3D). Gene Set Enrichment Analysis (GSEA) was performed on KEGG pathways to delve into the molecular intricacies of KRAS mutations. The analysis identified a total of 46 and 133 significantly enriched pathways in KRAS A146T and KRAS G12A, respectively (FDR < 0.25) (Supporting Information S1: Tables 6 and 7). Within the significantly enriched pathways in G12A, we observed an accumulation of pathways linked to pivotal cellular processes, such as cell proliferation, cell growth, and cell death. Those pathways are listed in the barplots in Figure 4A and Supporting Information S1: Figure 5. The enrichment of the MAPK signaling pathway (NES = 1.30, FDR = 0.09), the PI3K‐Akt signaling pathway (NES = 1.34, FDR = 0.06), and the JAK‐STAT signaling pathway (NES = 1.57, FDR = 0.02) were emphasized for the KRAS G12A versus WT comparison (Figure 4B), as alterations in KRAS have been described to primarily activate the MAPK pathway. 43 , 44 In Figure 4C and Supporting Information S1: Figure 5C, we have simplified the “Leading Edge Analysis” to identify genes with the most significant impact on the studied biological process. In Figure 4C, we showed the importance of these genes as DEGs, highlighting AREG, BCL2, and CDKN1A. For KRAS A146T, the MAPK and the PI3K‐Akt signaling pathways were also enriched in cells carrying the mutation with an NES of 1.02 and 1.11, respectively; however, the enrichment did not reach statistical significance. The JAK‐STAT signaling pathway, on the other hand, was found significantly enriched (NES = 1.72, FDR = 0.02) (Supporting Information S1: Figure 5A,B). For KRAS A146T, the important genes among these pathways were AREG, IL2RB, and CSF1R (Supporting Information S1: Figure 5C). Of note, the drug metabolism‐cytochrome P450 pathway was downregulated in both KRAS mutants compared to the WT with NES = −1.28, FDR = 0.24, for KRAS G12A, and NES = −1.63, FDR = 0.11, for KRAS A146T. Notably, the total number of significantly shared pathways in KRAS A146T and KRAS G12A is 30.
Figure 3.
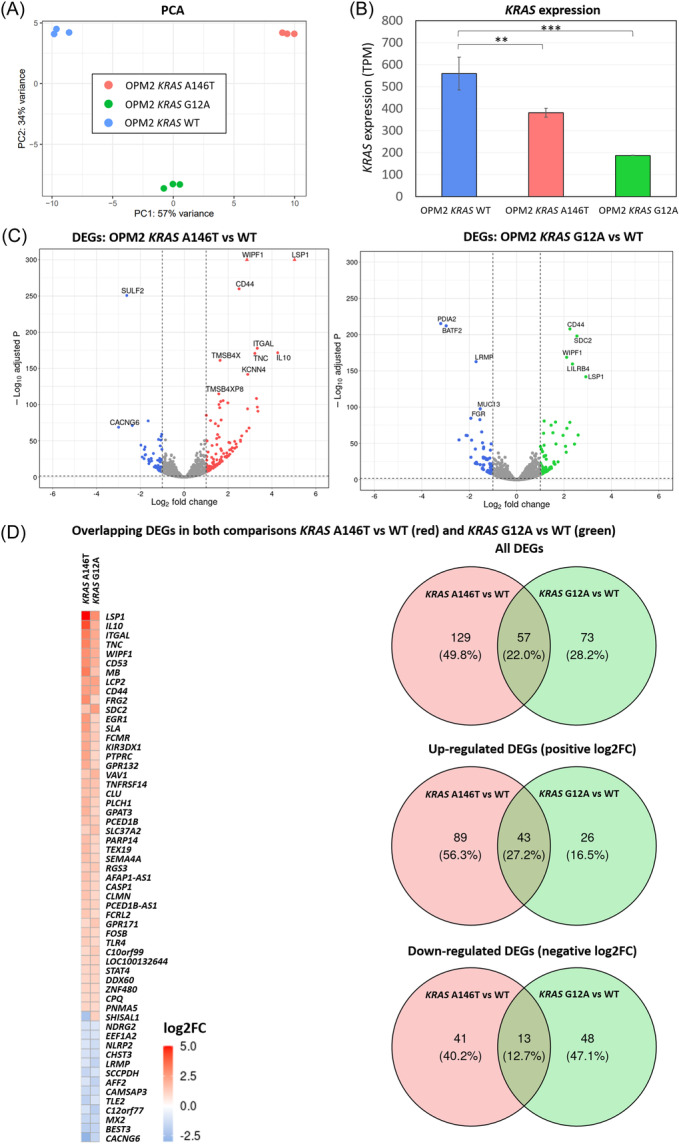
RNA‐seq transcriptome analysis and differentially expressed genes (DEGs) in the KRAS A146T and G12A mutation‐bearing cells. (A) Principal‐component analysis (PCA) results. The figure shows a scatter plot of the first two principal components (PC1 and PC2) depicting the distribution of the gene expression profile according to the mutational status. Percentages on each axis represent the percentages of variation explained by the principal components. (B) Bar plots showing expression levels in transcripts per million (TPM) in KRAS mutants and KRAS WT. (C) Volcano plots indicating gene expression differences between KRAS mutants and KRAS WT. DEGs with an adjusted p < 0.05 and a log2FC >│1│ are depicted in colors. Triangles indicate genes with a –Log10 adjusted p‐value higher than 300. (D) Heatmap of the 57 overlapping DEGs in both comparisons. Log2FC > 0 is indicated in red and log2FC < 0 in blue. Venn diagrams of all DEGs (upper panel), upregulated DEGs (middle panel), and downregulated DEGs (lower panel) found in the two comparisons.
Figure 4.
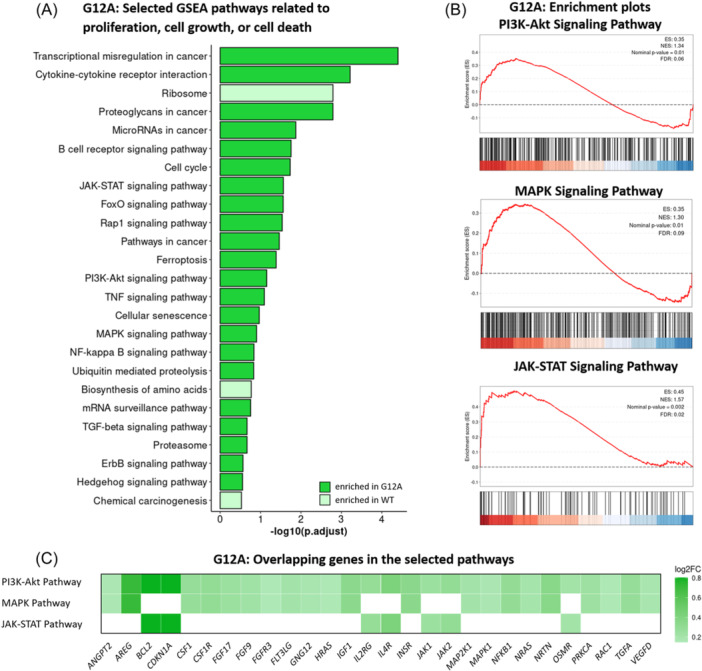
Gene Set Enrichment Analysis (GSEA) results of the G12A mutation‐bearing cells. (A) Barplot with GSEA results depicting positively and negatively enriched gene sets in KRAS G12A altered cells related to proliferation, cell division, growth, or cell death. A false discovery rate (FDR) of <0.25 was chosen as the cut‐off. (B) Enrichment plots of the PI3K‐Akt, MAPK, and JAK‐STAT signaling pathways. The enrichment profile is indicated by the red line. (C) Heatmap of log2FC on overlapping genes shared by the MAPK, JAK‐STAT, and PI3K‐Akt pathways in the GSEA analysis. Each row represents one of the three evaluated pathways and the columns represent those 28 genes that were shared at least by two of the three pathways and had a substantial impact on the GSEA analysis.
Implications of drug resistance‐related mutations on cell fitness
The described KRAS CCA was compared with other published CCAs investigating different genetic alterations from previous studies in our laboratory. 2 , 13 Both, the L363 IKZF1 A152T and the L363 CUL4B KO mutants outcompeted WT cells when Lenalidomide (LEN) was added to the culture (Figure 5A). 2 The same effect was observed for the L363 PSMB5 A20T mutant in the presence of Bortezomib (BOR). In the absence of the drug, this selection or survival fitness was not observed, even reversed, as the mutants were overgrown by the WT cells as long as the drug was not added to the co‐culture. Biallelic TP53 lesions in AMO1 cells, on the other hand, transmitted a fitness advantage in the absence of a drug, similar to the one described for the two KRAS‐altered OPM2 cell lines.
Figure 5.
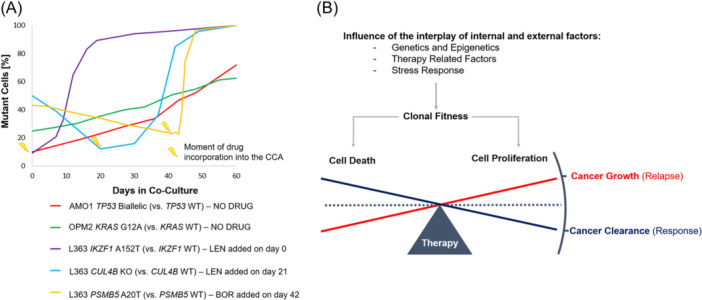
Clonal competition assays (CCA) examples of different genetic alterations and models of the cell death/cell growth equilibrium with regard to clonal fitness and therapy. (A) Alterations in drug resistance‐associated genes (CUL4B and PSMB5) only confer a proliferative fitness in the presence of the respective drug (Lenalidomide or Bortezomib). In the absence of selection pressure, the WT cells outcompete the mutant‐bearing sublines in the CCAs. On the contrary, KRAS and TP53 alterations intrinsically confer a growth advantage to the affected cells. Shown here is a meta‐analysis of CCAs made for previously published studies 2 , 13 and the OPM2 KRAS WT vs KRAS G12A comparison. (B) The complex interplay between clonal fitness and antitumor therapy determines if the cancer gets cleared as a response to the treatment or if it grows, provoking a relapse. The CCA is a suitable tool to study the interaction and outcome by modulating internal and external factors of the system.
DISCUSSION
Our data show that lesions in cancer driver genes like the here described KRAS G12A and A146T or mono‐ and biallelic TP53 alterations, 45 intrinsically confer a fitness advantage to the cells. This provides a biological rationale for the observation that such alterations are frequently found in patients at diagnosis and become even more prominent during the course of the disease, 24 whereas mutations in drug resistance‐associated genes are rare at diagnosis and generally found in low allele frequencies 2 , 12 , 46 at relapse. Alternative mechanisms such as epigenetic dysregulation in regulatory regions for gene expression also contribute to drug resistance. 46 , 47 However, concerning single point mutations and small gene deletions, our recently performed meta‐analysis of 1838 MM cases reported mutation frequencies of not higher than 0.44% at diagnosis (N = 1373) and 2.15% in pretreated MM patients (N = 465) for IKZF1, CUL4B, CRBN, 2 the direct binding site of all IMiDs. Equally low percentages were reported with regard to mutations affecting genes encoding proteasomal subunits. Even PSMD1, the highest‐scoring out of 46 genes, exhibited a mutation frequency of only 0.44% at diagnosis (N = 1137) and of 1.47% in pretreated patients (N = 447). 46 Mutations in the primary binding target of all PIs, PSMB5, were even rarer with 0.3% and 0.5%. 46 These numbers are surprisingly low considering the high number of clinically resistant cases. 6 In treatment‐free intervals, when the cancer is not under the selective pressure of a drug, such resistance mutations might get lost or be drastically reduced to small subclones, even to the single‐cell level, making their detection challenging. In this way, they can stay hidden and still expand later. A recent longitudinal sequencing study describes three different patterns of subclonal evolution for the rise of drug‐resistant disease: first coexistence of competing and expanding subclones, second spatial dominance, and third mono‐clonal single‐cell expansion. 48 Focusing on TP53, KRAS, and NRAS, key drivers of relapse in MM, parallel evolution was reported in this study, referring to subclones that accumulate new mutations during their evolution. 48 Monoallelic point mutations in TP53 and del17p are considered high‐risk alterations for an adverse disease course in MM as defined in the International Staging System (ISS). 45 , 49 Around 8% of newly diagnosed MM patients have a del17p, around 6% harbor monoallelic mutations, and around 4% show biallelic TP53 lesions. 50 Using CCAs, we previously unraveled a proliferative fitness advantage for mono‐ and biallelic TP53 alterations. 45 Unlike for TP53 somatic variants, there is no general consent on whether point mutations in KRAS impact the survival of MM patients. 4 In the current study, we report competitive advantages for two KRAS alterations, one affecting the common p.G12 exon‐2 hotspot that comprises about 40% of all KRAS mutations in MM, and the second, a less frequently affected downstream exon‐4 mutation at location p.A146, which is present in 2.3% of the KRAS mutated cases in CoMMpass. 24 The Ras superfamily of proteins is a group of enzymes with GTPase activity that rule cell signaling functions and trigger cell division, proliferation, migration, survival, and growth. 4 , 22 , 51 Oncogenic mutation of the RAS genes can cause incessant RAS activation by impairing the intrinsic GTPase activity, which for its part, allosterically activates downstream effectors and impacts a plethora of important cellular activities by activating the MEK (mitogen‐activated protein kinase)‐ERK (extracellular signal‐regulated kinase) module, the PI3K (phosphoinositide‐3 kinase)‐AKT pathway, and other signaling cascades. 22 , 52 Our RNA sequencing analysis comparing the whole transcriptomes of the KRAS WT OPM2 cells with the two mutation‐bearing sublines revealed an enrichment of several proliferation‐associated pathways including the above‐mentioned ones (Figure 4B,C and Supporting Information S1: Figure 5). To regulate activity, RAS proteins hydrolyze GTP gamma phosphate to GDP switching from an active state (GTP‐bound) to an inactive state (GDP‐bound) (Figure 1B and Supporting Information S1: Figure 1). The reaction kinetic is rather slow but gets accelerated by GAPs. The dissociation of the GDP and reloading of the pocket with a new GTP is mediated by GEFs. 53 The phosphates of the GTP are in contact with three loops of KRAS (Figure 1B), first, the so‐called P‐loop or glycine‐rich loop (residues 10–17), where glycine nitrogens surround the phosphates, a lysine causes nucleophilic attack and the last serine/threonine binds to a magnesium ion and stabilizes the beta and gamma phosphates in the right position. 54 The p.G12 hotspot mutations are located within this glycine‐rich P‐loop; they disrupt the hydrolysis of the GTP and therefore provoke a constitutively active state and downstream signaling of the protein (Supporting Information S1: Figure 1). 53 The p.A146T mutation is located within another loop (residues 30 to 40), which forms part of switch I and closes the GDP/GTP pocket from the top (Figure 1B and Supporting Information S1: Figure 1). GEFs take this loop into the OUT conformation allowing the release of the GDP. The p.A146T mutation promotes the switch I OUT conformation and diminishes the affinity for the GDP, thus it fosters faster exchange and reloading (Supporting Information S1: Figure 2). 40 , 55 The next loop in the sequence forms part of switch II (residues 60–76), and is catalytically relevant and necessary for the hydrolysis of GTP. Once the GTP is hydrolyzed, switch II changes its conformation by separating from the pocket and pointing the alpha helix upside down (Supporting Information S1: Figure 1). Because of the inability to bind to effector proteins, this is considered the inactive OUT conformation. Mutations within this loop provoke the IN conformation. 54 Thus, the two functionally investigated mutations G12A and A146T are representative of the impact of all alterations found in these two regulatory sites and, by different modes of action, induce proto‐oncogenic activation.
In patients, the emergence of unique subclones after years of treatment and multiple relapses is a clear indicator of the clonal competition hypothesis. It is only a matter of time until clones with the highest proliferation rate and potential for adaptation will become dominant. 48 Comparing the dynamics of different genetic alterations in a clonal competition setting, resistance‐associated mutations only provided a fitness advantage under drug pressure, unlike TP53 13 and KRAS alterations, which transmitted a fitness benefit per se (Figure 5A). However, our view on the complex subclonal architecture, on clonal evolution, and fitness signatures is still incomplete and requires further investigation. Most functional studies aiming to understand the impact of an alteration of interest are currently performed individually, not in a competitive setting, which better describes the real situation in a patient. A CCA therefore represents a useful approach to mimic the real‐life dynamics in a laboratory‐controlled environment, and moreover, the opportunity to variegate external factors such as treatments (Figure 5B). In this work, we have used stable transposition of cDNA genes for the fluorescent proteins EGFP 33 and LSS‐mKate2, 34 to color‐code genetically altered cells and their wild‐type parental cell line and make them visually distinguishable in a co‐culture. However, to compare more than just two different cell varieties, for example, to study genetic alterations in different genes, multiple alterations in the same gene, or to compare mono‐ with biallelic hits, it is necessary to expand the portfolio of available colors. The preparation of expression plasmids for different fluorescent proteins is a possibility. Noncolored cells also constitute a defined population (“black” in flow cytometry) and can thus also be added as another component in CCAs. Importantly, expression of the fluorescent protein alone did not affect the fitness of the cells employed in this study, although this should initially be tested for each new expression plasmid.
Altogether, our results show that it is possible to perform analytical, even quantitative genetics with CCA. This approach can be applied to characterize fitness advantages transmitted through patient‐specific lesions and to select promising treatments.
AUTHOR CONTRIBUTIONS
Santiago Barrio, Larissa Haertle, Umair Munawar, Thorsten Stühmer, K. Martin Kortüm, Tobias Heckel: Conceptualization. Larissa Haertle, Umair Munawar, Hipólito N. C. Hernández, Nicole Müller, Cornelia Vogt, Tobias Heckel, Seungbin Han: Data curation. Larissa Haertle, Santiago Barrio, K. Martin Kortüm, Hipólito N. C. Hernández, Andres Arroyo‐Barea, Thorsten Bischler, Natalia Buenache, Paula L. del Campo, Xiang Zhou, Florian Bassermann, Johannes Waldschmidt, Umair Munawar, Torsten Steinbrunn, Thorsten Stühmer, Joaquin Martinez‐Lopez: Formal analysis. Andres Arroyo‐Barea, Tobias Heckel, Thorsten Bischler, Larissa Haertle: RNA seq analysis and bioinformatic support. Larissa Haertle, Isabel Cuenca, Lucia Martin, Carlotta Höschle, Nicole Müller, Cornelia Vogt, Seungbin Han, Umair Munawar: Validation. Larissa Haertle, Nicole Müller, Umair Munawar, Santiago Barrio: Investigation. Hipólito N. C. Hernández, Larissa Haertle: Visualization 3D structural projections and help with the interpretation. Larissa Haertle, Santiago Barrio, Hipólito N. C. Hernández, Andres Arroyo‐Barea, K. Martin Kortüm: Writing—original draft. Santiago Barrio, K. Martin Kortüm, Larissa Haertle, Leo Rasche: Project administration. Umair Munawar, Hipólito N. C. Hernández, Andres Arroyo‐Barea, Santiago Barrio, Nicole Müller, Torsten Steinbrunn, Thorsten Stühmer: Methodology. K. Martin Kortüm, Joaquin Martinez‐Lopez, Leo Rasche, Thorsten Stühmer: Resources. K. Martin Kortüm, Joaquin Martinez‐Lopez, Florian Bassermann, Leo Rasche, Larissa Haertle, Santiago Barrio: Funding acquisition. Larissa Haertle, Santiago Barrio, Thorsten Stühmer, Tobias Heckel, Hipólito N. C. Hernández, Andres Arroyo‐Barea, Umair Munawar, Thorsten Bischler, Natalia Buenache, Florian Bassermann, Torsten Steinbrunn, Leo Rasche, Joaquin Martinez‐Lopez, K. Martin Kortüm: Writing—review and editing.
CONFLICT OF INTEREST STATEMENT
Joaquin Martinez‐Lopez and Santiago Barrio are equity shareholders of Altum Sequencing Co. The remaining authors declare no competing financial interests.
FUNDING
Larissa Haertle was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) project no. 493951700 and 544189139, Torsten Steinbrunn was supported by the DFG project no. 442740310. Santiago Barrio was funded by the Instituto de Salud Carlos III (FIS No. PI21/00314 and Miguel Servet CP22/00082). K. Martin Kortüm was supported by “Stiftung zur Förderung der Krebsforschung an der Universität Würzburg,” the “Stifterverband.” K. Martin Kortüm and Leo Rasche were funded by German Cancer Aid (Deutsche Krebshilfe) via the “Mildred Scheel Early Career Center” (MSNZ) program. Joaquin Martinez‐Lopez was funded by Instituto de Salud Carlos III (ISCIII) and was cofunded by the European Union and the CRIS Cancer Foundation.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
The authors would like to thank Myrto Kostopoulou from the Experimental Oncology Group at the CNIO under Mariano Barbacid for her input and impulse to write this manuscript.
Contributor Information
Larissa Haertle, Email: lara.haertle@freenet.de.
Santiago Barrio, Email: santibarrio0.3@gmail.com.
DATA AVAILABILITY STATEMENT
The RNA Seq data is available at NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under the accession number GSE247219. All requests for raw and analyzed data and materials will be promptly honored. Any data and materials that can be shared will be released via a Material Transfer Agreement. For requests, contact L.H. via e‐mail at lara.haertle@freenet.de.
REFERENCES
- 1. John L, Poos AM, Brobeil A, et al. Resolving the spatial architecture of myeloma and its microenvironment at the single‐cell level. Nat Commun. 2023;14(1):5011. 10.1038/s41467-023-40584-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barrio S, Munawar U, Zhu YX, et al. IKZF1/3 and CRL4(CRBN) E3 ubiquitin ligase mutations and resistance to immunomodulatory drugs in multiple myeloma. Haematologica. 2020;105(5):e237‐e241. 10.3324/haematol.2019.217943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12(5):335‐348. 10.1038/nrc3257 [DOI] [PubMed] [Google Scholar]
- 4. Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14(2):100‐113. 10.1038/nrclinonc.2016.122 [DOI] [PubMed] [Google Scholar]
- 5. Bolli N, Avet‐Loiseau H, Wedge DC, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997. 10.1038/ncomms3997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kastritis E, Terpos E, Dimopoulos MA. How I treat relapsed multiple myeloma. Blood. 2022;139(19):2904‐2917. 10.1182/blood.2020008734 [DOI] [PubMed] [Google Scholar]
- 7. Truger MS, Duell J, Zhou X, et al. Single‐ and double‐hit events in genes encoding immune targets before and after T cell‐engaging antibody therapy in MM. Blood Adv. 2021;5(19):3794‐3798. 10.1182/bloodadvances.2021004418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Da Vià MC, Dietrich O, Truger M, et al. Homozygous BCMA gene deletion in response to anti‐BCMA CAR T cells in a patient with multiple myeloma. Nat Med. 2021;27(4):616‐619. 10.1038/s41591-021-01245-5 [DOI] [PubMed] [Google Scholar]
- 9. Keats JJ, Chesi M, Egan JB, et al. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012;120(5):1067‐1076. 10.1182/blood-2012-01-405985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dutta AK, Alberge JB, Sklavenitis‐Pistofidis R, Lightbody ED, Getz G, Ghobrial IM. Single‐cell profiling of tumour evolution in multiple myeloma ‐ opportunities for precision medicine. Nat Rev Clin Oncol. 2022;19(4):223‐236. 10.1038/s41571-021-00593-y [DOI] [PubMed] [Google Scholar]
- 11. Da Vià MC, Solimando AG, Garitano‐Trojaola A, et al. CIC mutation as a molecular mechanism of acquired resistance to combined BRAF‐MEK inhibition in extramedullary multiple myeloma with central nervous system involvement. Oncologist. 2020;25(2):112‐118. 10.1634/theoncologist.2019-0356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haertle L, Cuesta Hernandez HN, Buenache NS, et al. Genetic alterations in members of the proteasome 26S subunit, AAA‐Atpase (PSMC) gene family transmit proteasome inhibitor resistance in multiple myeloma by impacting the ADP/ATP binding pocket. Blood. 2022;140(Suppl 1):4318‐4319. 10.1182/blood-2022-158763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Munawar U, Rasche L, Müller N, et al. Hierarchy of mono‐ and bi‐allelic TP53 alterations in multiple myeloma cell fitness. Blood. 2019;134:836‐840. 10.1182/blood.2019000080 [DOI] [PubMed] [Google Scholar]
- 14. van Neerven SM, Vermeulen L. Cell competition in development, homeostasis and cancer. Nat Rev Mol Cell Biol. 2023;24(3):221‐236. 10.1038/s41580-022-00538-y [DOI] [PubMed] [Google Scholar]
- 15. Basak G, Srivastava A, Malhotra R, Carrier E. Multiple myeloma bone marrow niche. Curr Pharm Biotechnol. 2009;10(3):335‐346. 10.2174/138920109787847493 [DOI] [PubMed] [Google Scholar]
- 16. Baker NE. Emerging mechanisms of cell competition. Nat Rev Genet. 2020;21(11):683‐697. 10.1038/s41576-020-0262-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim W, Jain R. Picking winners and losers: cell competition in tissue development and homeostasis. TIG. 2020;36(7):490‐498. 10.1016/j.tig.2020.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Simons BD, Clevers H. Strategies for homeostatic stem cell self‐renewal in adult tissues. Cell. 2011;145(6):851‐862. 10.1016/j.cell.2011.05.033 [DOI] [PubMed] [Google Scholar]
- 19. Snippert HJ, Schepers AG, van Es JH, Simons BD, Clevers H. Biased competition between Lgr5 intestinal stem cells driven by oncogenic mutation induces clonal expansion. EMBO Rep. 2014;15(1):62‐69. 10.1002/embr.201337799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tran TH, Chan AH, Young LC, et al. KRAS interaction with RAF1 RAS‐binding domain and cysteine‐rich domain provides insights into RAS‐mediated RAF activation. Nat Commun. 2021;12(1):1176. 10.1038/s41467-021-21422-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Han CW, Jeong MS, Ha SC, Jang SB. A H‐REV107 peptide inhibits tumor growth and interacts directly with oncogenic KRAS mutants. Cancers. 2020;12(6):1412. 10.3390/cancers12061412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khan AQ, Kuttikrishnan S, Siveen KS, et al. RAS‐mediated oncogenic signaling pathways in human malignancies. Sem Cancer Biol. 2019;54:1‐13. 10.1016/j.semcancer.2018.03.001 [DOI] [PubMed] [Google Scholar]
- 23. Timar J, Kashofer K. Molecular epidemiology and diagnostics of KRAS mutations in human cancer. Cancer Metastasis Rev. 2020;39(4):1029‐1038. 10.1007/s10555-020-09915-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kortüm KM, Mai EK, Hanafiah NH, et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood. 2016;128(9):1226‐1233. 10.1182/blood-2016-02-698092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weissbach S, Heredia‐Guerrero SC, Barnsteiner S, et al. Exon‐4 mutations in KRAS affect MEK/ERK and PI3K/AKT signaling in human multiple myeloma cell lines. Cancers (Basel). 2020;12(2):455. 10.3390/cancers12020455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mulligan G, Lichter DI, Di Bacco A, et al. Mutation of NRAS but not KRAS significantly reduces myeloma sensitivity to single‐agent bortezomib therapy. Blood. 2014;123(5):632‐639. 10.1182/blood-2013-05-504340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walker BA, Mavrommatis K, Wardell CP, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. 2018;132(6):587‐597. 10.1182/blood-2018-03-840132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steinbrunn T, Stühmer T, Gattenlöhner S, et al. Mutated RAS and constitutively activated Akt delineate distinct oncogenic pathways, which independently contribute to multiple myeloma cell survival. Blood. 2011;117(6):1998‐2004. 10.1182/blood-2010-05-284422 [DOI] [PubMed] [Google Scholar]
- 29. Sacco A, Federico C, Todoerti K, et al. Specific targeting of the KRAS mutational landscape in myeloma as a tool to unveil the elicited antitumor activity. Blood. 2021;138(18):1705‐1720. 10.1182/blood.2020010572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Munawar U, Roth M, Barrio S, et al. Assessment of TP53 lesions for p53 system functionality and drug resistance in multiple myeloma using an isogenic cell line model. Sci Rep. 2019;9(1):18062. 10.1038/s41598-019-54407-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barrio S, Stühmer T, Da‐Viá M, et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia. 2019;33(2):447‐456. 10.1038/s41375-018-0216-8 [DOI] [PubMed] [Google Scholar]
- 32. Vikova V, Jourdan M, Robert N, et al. Comprehensive characterization of the mutational landscape in multiple myeloma cell lines reveals potential drivers and pathways associated with tumor progression and drug resistance. Theranostics. 2019;9(2):540‐553. 10.7150/thno.28374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fink S, Zugelder L, Roth B, et al. A simple approach for multi‐targeted shRNA‐mediated inducible knockdowns using Sleeping Beauty vectors. PLoS One. 2018;13(10):e0205585. 10.1371/journal.pone.0205585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Piatkevich KD, Hulit J, Subach OM, et al. Monomeric red fluorescent proteins with a large Stokes shift. Proc Nat Acad Sci USA. 2010;107(12):5369‐5374. 10.1073/pnas.0914365107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29(1):15‐21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liao Y, Smyth GK, Shi W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923‐930. 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- 37. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15(12):550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhu A, Ibrahim JG, Love MI. Heavy‐tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics. 2019;35(12):2084‐2092. 10.1093/bioinformatics/bty895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu T, Hu E, Xu S, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2(3):100141. 10.1016/j.xinn.2021.100141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Poulin EJ, Bera AK, Lu J, et al. Tissue‐specific oncogenic activity of KRAS (A146T). Cancer Discovery. 2019;9(6):738‐755. 10.1158/2159-8290.CD-18-1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dharmaiah S, Tran TH, Messing S, et al. Structures of N‐terminally processed KRAS provide insight into the role of N‐acetylation. Sci Rep. 2019;9(1):10512. 10.1038/s41598-019-46846-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu S, Long BN, Boris GH, Chen A, Ni S, Kennedy MA. Structural insight into the rearrangement of the switch I region in GTP‐bound G12A K‐Ras. Acta Crystallogr D Struct Biol. 2017;73(Pt 12):970‐984. 10.1107/S2059798317015418 [DOI] [PubMed] [Google Scholar]
- 43. Drosten M, Barbacid M. Targeting the MAPK pathway in KRAS‐driven tumors. Cancer Cell. 2020;37(4):543‐550. 10.1016/j.ccell.2020.03.013 [DOI] [PubMed] [Google Scholar]
- 44. Sriskandarajah P, De Haven Brandon A, MacLeod K, et al. Combined targeting of MEK and the glucocorticoid receptor for the treatment of RAS‐mutant multiple myeloma. BMC Cancer. 2020;20(1):269. 10.1186/s12885-020-06735-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Munawar U, Rasche L, Müller N, et al. Hierarchy of mono‐ and biallelic TP53 alterations in multiple myeloma cell fitness. Blood. 2019;134(10):836‐840. 10.1182/blood.2019000080 [DOI] [PubMed] [Google Scholar]
- 46. Haertle L, Barrio S, Munawar U, et al. Single‐nucleotide variants and epimutations induce proteasome inhibitor resistance in multiple myeloma. Clin Cancer Res. 2023;29(1):279‐288. 10.1158/1078-0432.CCR-22-1161 [DOI] [PubMed] [Google Scholar]
- 47. Haertle L, Barrio S, Munawar U, et al. Cereblon enhancer methylation and IMiD resistance in multiple myeloma. Blood. 2021;138(18):1721‐1726. 10.1182/blood.2020010452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rasche L, Schinke C, Maura F, et al. The spatio‐temporal evolution of multiple myeloma from baseline to relapse‐refractory states. Nat Commun. 2022;13(1):4517. 10.1038/s41467-022-32145-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Soekojo CY, Chung TH, Furqan MS, Chng WJ. Genomic characterization of functional high‐risk multiple myeloma patients. Blood Cancer J. 2022;12(1):24. 10.1038/s41408-021-00576-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Flynt E, Bisht K, Sridharan V, Ortiz M, Towfic F, Thakurta A. Prognosis, biology, and targeting of TP53 dysregulation in multiple myeloma. Cells. 2020;9(2):287. 10.3390/cells9020287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vakiani E, Solit DB. KRAS and BRAF: drug targets and predictive biomarkers. J Pathol. 2011;223(2):220‐230. 10.1002/path.2796 [DOI] [PubMed] [Google Scholar]
- 52. Cox AD, Der CJ. Ras history: the saga continues. Small GTPases. 2010;1(1):2‐27. 10.4161/sgtp.1.1.12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170(1):17‐33. 10.1016/j.cell.2017.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kozlova MI, Shalaeva DN, Dibrova DV, Mulkidjanian AY. Common patterns of hydrolysis initiation in P‐loop fold nucleoside triphosphatases. Biomolecules. 2022;12(10):1345. 10.3390/biom12101345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moghadamchargari Z, Shirzadeh M, Liu C, et al. Molecular assemblies of the catalytic domain of SOS with KRas and oncogenic mutants. Proc Natl Acad Sci USA. 2021;118(12):e2022403118. 10.1073/pnas.2022403118 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
The RNA Seq data is available at NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under the accession number GSE247219. All requests for raw and analyzed data and materials will be promptly honored. Any data and materials that can be shared will be released via a Material Transfer Agreement. For requests, contact L.H. via e‐mail at lara.haertle@freenet.de.