

ABSTRACT
Detection of bacterial RNA by nucleic acid amplification tests (NAATs), such as reverse transcription PCR (RT-PCR) and reverse transcription loop-mediated isothermal amplification (RT-LAMP), offers distinct advantages over DNA-based methods. However, such assays also present challenges in ascertaining positive and internal control material that can reliably monitor success over all phases of testing (bacterial lysis, nucleic acid recovery, reverse transcription, amplification, and signal detection): since they are unable to distinguish between amplification of bacterial RNA transcripts and the DNA templates that encode them, using intact organisms as controls can inform cell lysis but not successful detection of RNA. We developed a control strategy for RNA-based bacterial NAATs that allows ready discrimination of RNA from DNA templates using self-splicing bacterial introns, such that those nucleic acids ultimately encode different sequences. We engineered two vectors encoding synthetic transgenes based on this principle, one that is active in the Gram-negative bacterium Escherichia coli and one that functions in both E. coli and the Gram-positive organism Staphylococcus aureus. We subsequently designed RT-LAMP assays that either target RNA and DNA from transgenic organisms or target RNA exclusively and demonstrated the specificity of amplification using purified nucleic acids. Using multiplex fluorescent RT-LAMP of heat-lysed specimens, we showed the practicality of deploying such transgenic organisms as an internal control to ascertain sample integrity and assay performance during clinical diagnostic testing. Our approach has broad utility for RNA-based bacterial NAATs, especially point-of-care assays and other applications where nucleic acids are nonspecifically liberated for testing.
KEYWORDS: molecular methods, diagnostics
INTRODUCTION
Although DNA-based nucleic acid amplification tests (NAATs) presently comprise the dominant molecular detection methods for bacteria, RNA-based NAATs are beginning to be developed and adopted (1–3). RNA-based microbial NAATs have demonstrated advantages over DNA-based methods in their specificity for live bacteria (1, 4–8) and enhanced sensitivity resulting from signal amplification of highly transcribed bacterial genes (1, 2, 4, 5, 9, 10). Few such assays are able to directly recognize and amplify RNA templates using transcription-mediated amplification (TMA) (2, 3, 11, 12), while most depend on the action of reverse transcriptases to convert RNA to DNA templates prior to their amplification by DNA polymerases, as in reverse transcription PCR (RT-PCR) (10, 13, 14) and reverse transcription loop-mediated isothermal amplification (RT-LAMP) (5). Of particular appeal are RT-LAMP assays, which integrate reverse transcription and isothermal template amplification into a single, one-step reaction (15). RT-LAMP provides a low-cost, reliable testing approach that can be performed without specialized instrumentation and can be made compatible with point-of-care or at-home testing (16–20). Already widely used for the detection of RNA viruses (15, 21), RT-LAMP assays for pathogenetic bacteria are rapidly emerging (4, 5, 22–27).
When performing NAATs for clinical molecular diagnosis, it is critical to include an appropriate combination of negative, positive, and internal controls to assess successful assay performance (28–30). Negative controls allow the exclusion of false-positive results from contamination or other artifacts, while positive controls ensure that false-negative results from assay technical failures can be recognized. In contrast, internal controls allow verification of assay performance for each individual sample that is subjected to testing, permitting the recognition of inhibitors from specimen matrices that may impair testing and yield false-negative outcomes. These well-accepted paradigms for assay controls, however, pose a challenge for most RNA-based NAATs. A comprehensive positive or internal control for such assays must allow the user to ascertain success through all phases of testing, from bacterial lysis and recovery of intact cellular RNA through reverse transcription, amplification, and the ultimate detection of RNA templates. Yet, in RT-PCR and RT-LAMP, one cannot distinguish between amplification of bacterial RNA transcripts and the DNA that encodes them because DNA is recognized by the polymerases used in such assays and because RNA bears the same sequence as the DNA from which it is derived. Consequently, using intact bacteria as controls can identify successful release of nucleic acids but does not have specificity for the recovery or detection of RNA itself. This limitation is especially problematic for qualitative tests where the degree of amplification cannot be accurately measured and for applications where nucleic acid recovery is achieved via nonselective approaches like thermal bacterial lysis, where both DNA and RNA templates are present in the testing matrix.
Here, we have developed an approach that enables users to readily distinguish an expressed RNA transcript from its template DNA sequence in bacteria and have used it to construct transgenic bacteria with value as positive and internal control material for RNA-based molecular diagnostic assays. This strategy (Fig. 1) takes advantage of autocatalytic, self-splicing bacterial introns (31, 32), which result in RNA transcripts that differ in sequence from the DNA that encodes them. An intron with activity in the target organism, including its catalytic exonic components, is inserted into a marker gene and expressed. Primers that specifically amplify the spliced RNA transcript can be designed, either by virtue of their binding across exon junctions that are absent from template DNA or because the target length is prohibitively large prior to excision of the intron. We demonstrate the feasibility of this approach in two different organisms (Escherichia coli and Staphylococcus aureus) and show that transgenic strains can be used to provide comprehensive positive and internal controls in RT-LAMP assays.
Fig 1.

The engineered control material allows differential amplification of RNA and DNA from transgenic organisms. The bacterium of interest is transformed with a plasmid vector containing a target gene construct, wherein the target gene is disrupted by an autocatalytic, self-splicing intron that has activity in the chosen organism. Primers are designed that specifically amplify the spliced RNA transcript, rather than the template DNA, either by binding across the exon–exon junction (top primer) or because the amplicon length is prohibitively large, unless the intron has first been successfully excised (bottom primer). This schema allows selective amplification of RNA from the transgenic organism, which can be used to monitor successful assay performance through all phases of testing (bacterial lysis, nucleic acid recovery, reverse transcription, template amplification, and signal detection).
MATERIALS AND METHODS
Bacterial strains and growth conditions
S. aureus strain JE2 and E. coli IM08B were obtained from the Biodefense and Emerging Infections Research Resources Repository (BEI Resources). E. coli DH5 alpha was obtained from New England Biolabs. Pseudomonas aeruginosa PAO1 was generously gifted by Dr. Colin Manoil (University of Washington). All bacteria were cultured at 37°C in Luria–Bertani (LB) medium containing antibiotics if appropriate to select for plasmid vector maintenance (100 µg/mL ampicillin for E. coli and 10 µg/mL chloramphenicol for S. aureus).
Vector design and construction
The general design of expression cassettes used in this study was the same in both engineered vectors. Cassettes incorporated a T7 promoter sequence for possible in vitro transcription of the cloned transgenes, followed by a broad host range Pcap promoter and ribosome-binding site (33) to drive in vivo transcription. Downstream of the transgene, cassettes incorporated the T7hyb10 terminator sequence, which has strong activity against both T7 RNA polymerase and native bacterial RNA polymerases (34).
The E. coli splicing vector (pKTM_IAC) was synthesized by IDT into the pUC19-derived pUCIDT (Amp) Golden Gate vector. The KTM_IAC transgene (Fig. S1) contained the Tet-119-derivative of the Tetrahymena group I intron (wild-type sequence from Genbank JN547815.1) inserted into a version of KTM (Genbank HM151400.1), as previously described (35), except that KTM was re-encoded using E. coli codon optimization (Fig. S2) through the IDT codon optimization tool (https://www.idtdna.com/pages/tools/codon-optimization-tool), while preserving exonic residues deemed critical to splicing.
The S. aureus splicing cassette (Fig. S3) was initially synthesized by GenScript into pUC19. The transgene comprised the first exon, intron, and initial 22 bp of the second exon from bacteriophage Twort orf142 (Genbank AF132670.1) with a short spacer sequence added at the 5′ end to improve primer design characteristics and followed by an in-frame superfolder green fluorescent protein (GFP) (36) codon optimized for S. aureus as above and from which the start codon was removed. We generated a temperature-stable derivative of the E. coli–S. aureus shuttle vector pCN50-BSAI (37) by replacing the temperature-sensitive pT181 cop–634ts repC4 replicon with the wild-type pT181 cop-wt repC replicon from pCN33 (38), yielding pCN50wt. The expression cassette was subsequently cloned into pCN50wt to yield the final S. aureus splicing vector (pOrf142+gfp_mod). Vectors were transformed into S. aureus JE2 after plasmid conditioning in IM08B, as previously described (39).
Nucleic acid extraction
Total RNA was extracted from E. coli after lysis with lysozyme (Sigma) using the Monarch Total RNA Miniprep Kit (NEB), with on-column DNase I (NEB) treatment. S. aureus total RNA was similarly extracted after lysis with lysostaphin (Sigma) using the RNeasy Kit (Qiagen), followed by an in-solution DNase I digestion and secondary cleanup with the RNeasy Kit. Total DNA was extracted from both organisms using the DNeasy UltraClean Microbial Kit (Qiagen), followed by removal of residual RNA from the eluate by RNase A digestion (Thermo Fisher) and secondary purification using AMPure XP beads (Beckman Coulter).
RT-LAMP assays
Primers, probes, and quenchers were synthesized by IDT (Table S1). LAMP primers were designed using NEB LAMP Primer Design tool v 1.4.1 (https://lamp.neb.com/), with LB primers subsequently modified to carry 5′ adapter sequences for interaction with primer-specific fluorescent probes and quenchers (21). Fluorescent probes and quencher sequences were synthesized as previously described (21).
Cell equivalents of DNA used as the template for RT-LAMP were calculated, as elsewhere (40), based on the genomic sizes of E. coli DH5alpha and S. aureus JE2. Cell equivalents of RNA used were based on an estimate of 100 fg total RNA per bacterial cell (41).
All RT-LAMP reactions were conducted at 65°C using the WarmStart LAMP Kit (DNA & RNA) (NEB) following the manufacturer’s instructions. Primer and probe concentrations were as specified elsewhere (21). Reaction fluorescence was measured using QuantStudio 3 (Applied Biosystems) in 30-second intervals over the course of a 1-hr incubation.
Bacterial thermal lysis experiments used aliquots of 10,000 cells per RT-LAMP reaction, based on spectrophotometric readings of overnight cultures (42, 43). Cells were diluted in TE buffer (pH 8.0), heated to 65°C for 10 m, followed by cooling on ice. Varying concentrations of RNase A were then added to lysed cell aliquots prior to RT-LAMP.
Fully deidentified residual stool specimens (~20–100 mg) were resuspended in 500 µL molecular-grade water by vortexing. Serial dilutions were prepared using molecular-grade water. E. coli carrying pKTM_IAC was added to each specimen at a final concentration of 1,140 cells per microliter. Aliquots were spiked with P. aeruginosa to a final concentration of 57 cells per microliter. Bacterial thermal lysis followed by RT-LAMP was conducted as before, using 8.75 µL specimen volume per reaction.
RESULTS
Design of a self-splicing marker gene in E. coli and RNA-specific RT-LAMP
We initially developed a system for enabling specific detection of RNA in the canonical Gram-negative bacterium E. coli. We repurposed a previously described, self-splicing construct comprising a bacterial antibiotic resistance gene (kanamycin nucleotidyltransferase, KNT) into which was inserted a 413-bp self-splicing intron derived from Tetrahymena (35) (Fig. S1). It is theoretically possible that a native form of the KNT gene could be present in test specimens, for example, if carried by other organisms in a patient microbiome or contaminating organisms from the environment and that this could lead to false-positive amplification from exogenous DNA sequences. To ensure specificity for the synthetic, spliced RNA target, we, therefore, re-encoded the sequence of this resistance gene to a form that is not found in nature (Fig. S2).
E. coli transformed with the transgene-containing vector (pKTM_IAC), but not the parental vector backbone (pUC19), proved resistant to kanamycin at a level of 50 µg/mL, indicating successful splicing of the synthetic construct into an active form of the RNA transcript.
We designed RNA-specific RT-LAMP assay primers that spanned the target gene’s intron such that it fell between both the FIP and BIP primers and also between the F1 and F2 domains targeted by the FIP primer itself (8, 20). This configuration disrupts the binding of FIP primer to DNA templates and simultaneously makes the target region too large (621 bp from the ends of the outermost F3 and B3 primers) to be effectively amplified by RT-LAMP (44), unless splicing has successfully occurred (208 bp from the ends of F3 and B3 primers). RT-LAMP primers were additionally designed to leverage sites of sequence divergence between the native and re-encoded forms of the gene, with discrimination for the synthetic construct further enhanced by incorporating locked nucleic acid (LNA) bases at mismatched sites overlying the 3′ termini of assay primers (45).
We separately developed an RT-LAMP primer set for the ampicillin resistance gene (ampR) encoded by the pUC19 plasmid vector backbone itself, which is expected to amplify both the DNA that encodes the gene and the RNA that is constitutively expressed from it.
RT-LAMP assays can specifically identify RNA templates in E. coli
To test our strategy for differential detection of nucleic acids in RNA-based NAATs, we selectively purified RNA and DNA from transgenic bacteria and conducted RT-LAMP reactions with primer sets targeting either plasmid DNA and RNA (ampR primers) or those designed to be specific for spliced RNA from the plasmid-encoded target gene (transgene primers). RT-LAMP was performed using a commercially available cocktail of Bst polymerase and reverse transcriptase. We tested tenfold serial dilutions of purified RNA and DNA templates spanning 10,000 to 100 cell equivalents of E. coli that carried either transgene-expressing pKTM_IAC or the precursor pUC19 vector (Fig. 2A).
Fig 2.
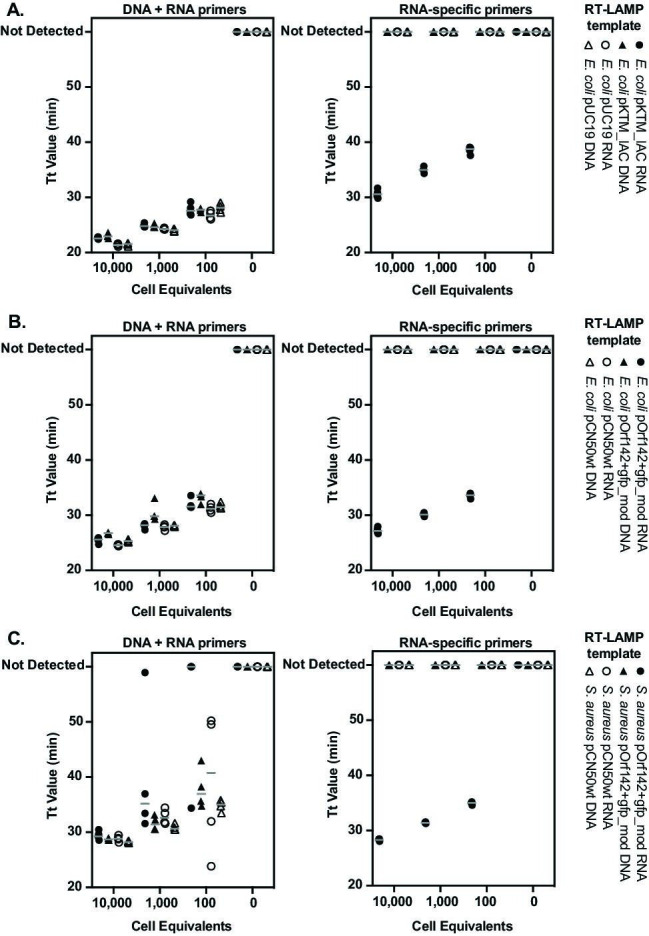
Selective amplification of RNA from self-splicing transgenes by RT-LAMP. Time to threshold (Tt) values are shown for RT-LAMP reactions targeting both RNA and DNA or spliced, transgenic RNA. Organisms carrying transgenic expression vectors and precursor vectors lacking the transgene were tested:(A) E. coli carrying pKTM_IAC or the pUC19 precursor vector,(B) E. coli carrying pOrf142+gfp_mod or the pCN50wt precursor vector, and(C) S. aureus carrying pOrf142+gfp_mod or the pCN50wt precursor vector. Various cell equivalents of purified RNA and DNA templates were tested in triplicate, with results of four technical replicates from each test superimposed. Gray horizontal bars represent the mean for replicates.
As expected, RT-LAMP primers targeting pUC19-encoded ampR amplified both DNA and RNA fractions purified from bacteria proportionately to the amount of those templates provided. Amplification was achieved from E. coli carrying either pUC19 or pKTM_IAC, indicating the presence of intact nucleic acids for each condition tested. In contrast, amplification using the RNA-specific transgene primers occurred only for RNA purified from E. coli expressing the pKTM_IAC construct, with no measurable signal generated from the DNA fraction of those cells. RNA-specific RT-LAMP similarly failed to amplify either RNA or DNA of E. coli transformed with pUC19, which lacks the spliced transgene.
We conclude that RT-LAMP targeted against the spliced KTM target is highly specific for RNA templates and does not amplify the DNA template that encodes it.
Design of a self-splicing transcript with activity in both E. coli and S. aureus
To expand our strategy to a harder-to-lyse, Gram-positive organism, we next designed an analogous system that would function in S. aureus. We constructed a second vector (pOrf142+gfp_mod) housing a synthetic fusion of the self-splicing orf142 gene from bacteriophage Twort (46, 47) and a start-loss mutated GFP (36) that was codon-optimized for S. aureus and cloned in-frame with the second exon of orf142 (Fig. S3). Since in this implementation the GFP target gene is both synthetic and exogenous to bacteria, the design does not pose the risk of contamination from organisms in the environment or in tested specimens. The construct was built into an E. coli-S. aureus shuttle vector (pCN50wt) that carried pUC19- and pT181-derived replicons (38) in order to facilitate its construction and ultimate transformation into S. aureus (39).
The pOrf142+gfp_mod transcript underwent successful splicing in S. aureus, as evidenced by visible GFP fluorescence in transgenic bacteria, but this phenotype was unexpectedly also observed for E. coli carrying the vector. This result indicated that autocatalytic activity in the intron was conserved across these two distantly related bacteria and that the vector construct could be used in both organisms.
We therefore tested the ability of pOrf142+gfp_mod to act as a control in both E. coli and S. aureus. We designed primers for RT-LAMP of the spliced target gene using the same schema as in our earlier construct and again performed serial dilutions of purified nucleic acids from transgenic E. coli (Fig. 2B) and S. aureus (Fig. 2C) that harbored either pOrf142+gfp_mod or the precursor pCN50wt vector, which lacks the transgene construct.
As in previous experiments, amplification of DNA and RNA fractions using ampR-targeted primers confirmed the presence of both nucleic acids in specimens derived from E. coli and S. aureus. We noted that RT-LAMP of ampR was less robust from S. aureus RNA fractions at 1,000 and 100 cell equivalents than from corresponding cell equivalents of S. aureus DNA, E. coli DNA, and E. coli RNA, evidenced both by increased time to threshold (Tt) values and greater variability between replicates. These outcomes (48) are consistent with less efficient transcription being driven by the Gram-negative ampR promoter when transferred from E. coli to S. aureus, resulting in fewer RNA template molecules per organism. Regardless of this, we again observed that amplification with transgene-specific primers was specific for purified RNA from both E. coli and S. aureus expressing the Orf142+gfp_mod cassette. No product was detected from the DNA of those organisms or from RNA or DNA of bacteria carrying the empty pCN50wt vector backbone. These findings indicate that selective amplification of the spliced RNA transcript was achieved in both E. coli and S. aureus, without measurable amplification from plasmid DNA that encodes the expression cassette.
Multiplex RT-LAMP provides an internal control for monitoring assay performance
We next determined whether our approach could be used to provide an internal control for monitoring RT-LAMP assay performance when deployed alongside testing for a diagnostic target. To allow for simultaneous detection of the signals from separate primer sets within the same reaction, we adopted a strategy for multiplex RT-LAMP employing fluorescently labeled probes and quencher complexes that differentially bind to nucleotide tails on assay loop primers (21), allowing distinct amplification products to be fluorometrically distinguished.
We initially ascertained the viability of this multiplex assay strategy by ascertaining nucleic acid recovery and detection after thermal lysis of E. coli carrying pKTM_IAC, a crude method for total nucleic acid extraction that is compatible with point-of-care testing (49–51). In order to test a range of conditions under which RNA would be variably degraded, aliquots of the cell lysate were spiked with varying amounts of RNase A prior to multiplex RT-LAMP containing primers targeting pUC19 ampR (serving as a diagnostic target) and the spliced KTM transgene (acting as an internal control) (Fig. 3).
Fig 3.
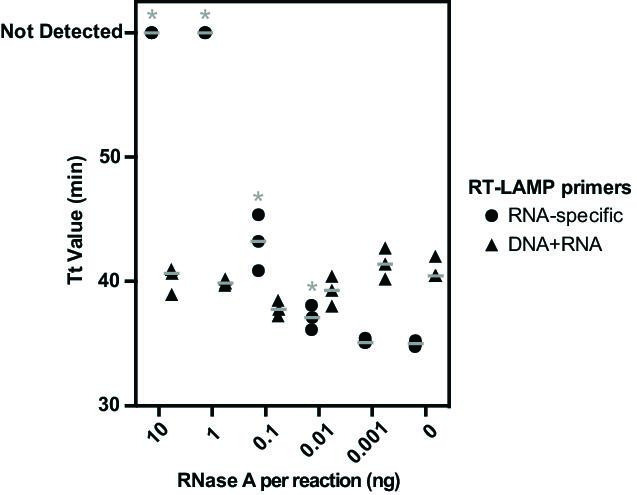
Self-splicing transgenes serve as internal assay controls. Time to threshold (Tt) values are shown for multiplex RT-LAMP reactions incorporating primer sets that target both RNA and DNA and those that are specific for spliced RNA from transgenic expression vectors. Heat-lysed specimens of E. coli carrying pKTM_IAC were combined with the indicated quantity of RNase A prior to RT-LAMP. Results from three technical replicates are superimposed, with Tt values significantly (P ≤ 0.05, two-tailed T test) higher than controls lacking RNase A, indicated with an asterisk (*). Gray horizontal bars express the mean for replicates.
Amplification of ampR and the spliced KTM transgene was readily achieved by multiplex RT-LAMP after heat lysis, demonstrating robust signal detection from both targets simultaneously. Signals from the spliced KTM internal control decreased inversely with the amount of RNase A added to reactions, indicating that the assay’s ability to detect RNA had been compromised. Significantly (P ≤ 0.05, two-tailed T test) delayed Tt values relative to a lysed cell control were observed when using ≥0.01 ng RNase A per reaction, and complete signal loss from RNA-specific primers occurred with inclusion of 1 ng or more of RNase A. In contrast, the signal from the ampR target was not measurably decreased under any test condition, consistent both with amplification occurring off the DNA present in those specimens and with the insensitivity of the pan-nucleic acid primer set to identify reduced RT-LAMP assay performance for RNA templates.
Finally, we explored the feasibility of using transgenic organisms to monitor assay performance in a clinical context. To this end, we tested an independent diagnostic target within clinical matrices that could contain additional microorganisms and inhibitory substances capable of confounding testing. We designed organism-specific RT-LAMP primers targeting a gene that identifies Pseudomonas aeruginosa, (52) an opportunistic pathogen able to seed hematogenous and respiratory infections after colonizing the gut of critically ill patients (53), and multiplexed this diagnostic assay with RNA-specific primers for the pKTM_IAC control. For these studies, we evaluated stool specimens as a relevant and challenging clinical matrix containing a high amount of molecular inhibitors (54, 55) and resident microbiota (56). E. coli carrying pKTM_IAC was added to stool specimens as an internal control, and aliquots were spiked with exogenous, cultured P. aeruginosa to generate positive reference material. Thermal bacterial lysis and multiplex RT-LAMP testing were then performed (Fig. 4).
Fig 4.
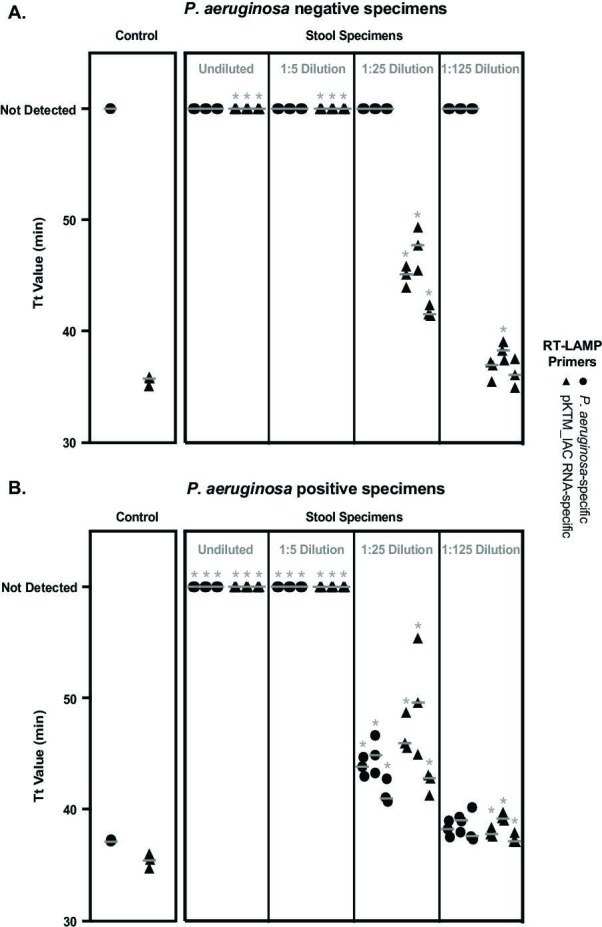
Diagnostic RT-LAMP assay for P. aeruginosa using transgenic E. coli as an internal assay control. Time to threshold (Tt) values are shown for multiplex RT-LAMP reactions incorporating diagnostic primer sets for P. aeruginosa and RNA-specific primers for the transgenic expression vector pKTM_IAC to serve as an internal assay control. Results are shown for control reactions lacking the clinical specimen matrix and for a series of three separate stool specimens. All specimens were tested in the presence of E. coli carrying pKTM_IAC, (A) without or (B) with the introduction of exogenous P. aeruginosa. Dilutions of stool specimens are indicated. Results from three technical replicates for each specimen are superimposed, with Tt values significantly (P ≤ 0.05, two-tailed T test) higher than those of controls lacking clinical matrices indicated with an asterisk (*). Gray horizontal bars express the mean for replicates.
In control specimens tested without clinical matrices, multiplex amplification from P. aeruginosa and transgenic control targets yielded similar Tt when both templates were present. Stool specimens showed complete inhibition of amplification from the internal control target, indicating that assay performance had been compromised by using that sample type. We therefore evaluated serial dilutions of the stool specimens that had been similarly spiked with control and target organisms to identify conditions under which valid testing results could be achieved. Amplification from the internal control and spiked P. aeruginosa, if present, could be detected using 1:25 dilutions of specimens, although the Tt from both targets was significantly delayed, relative to controls (P ≤ 0.05, two-tailed T test). Assay performance was more fully restored with 1:125 dilution, although some specimens continued to show inhibition, as indicated by nominally higher Tt for internal controls (P ≤ 0.05, two-tailed T test).
These experiments demonstrate the feasibility of using autocatalytic splicing constructs as an internal control for monitoring the RNA-dependent functions and diagnostic performance of RT-LAMP reactions, as well as the compatibility of such an approach with point-of-care testing procedures used in clinical diagnostic assays.
DISCUSSION
We have developed a robust strategy that enables users to specifically detect a targeted RNA transcript in bacteria using synthetic genes containing autocatalytic introns (Fig. 1). This system can be leveraged to design NAATs that are able to amplify templates only after self-splicing has occurred, achieving specificity for successfully reverse-transcribed RNA (Fig. 2). Although this concept has long been applied to the design of RT-qPCR assays for monitoring human gene expression (57), its application in prokaryotes represents a novel means to provide control material suitable for RNA-based bacterial NAATs, including RT-PCR and RT-LAMP. Because the system is self-contained within transgenic bacteria, the intact organisms themselves can be used as a control for all phases of testing: cell lysis, nucleic acid extraction, reverse transcription, template amplification, and detection. We have shown that control-encoded RNA transcripts and an experimental target can be simultaneously detected in multiplex RT-LAMP assays, which can be leveraged in clinical molecular diagnostic assays to provide an internal control for assessing test performance (21) (Fig. 3 and 4). This internal control schema is expected to provide an especially high value to applications such as point-of-care testing, where nucleic acid release is typically achieved by nonspecific methods and inhibitors present in the testing matrix are not removed prior to testing. Indeed, several P. aeruginosa-containing clinical matrices subjected to diagnostic RT-LAMP in this study would have been falsely reported as negative for that organism, rather than indeterminate due to assay inhibition, without the interpretive framework enabled by the internal control. Inhibited tests could be validly interpreted after dilution of the specimen matrix was sufficient to allow amplification from the internal control (Fig. 4), illustrating how this schema could be used to assess assay performance in clinical practice.
We generated two versions of a transgenic control vector in order to facilitate its use in bacteria having different physical and genetic properties: One encodes an antibiotic resistance gene and is active in E. coli, while the other contains a synthetic GFP gene and functions in both E. coli and S. aureus. The functionality of the latter construct in Gram-positive and Gram-negative species alike was surprising, as prior work has suggested that the autocatalytic activity of such sequences requires species-specific optimization (35). This finding instead suggests that the synthetic target gene developed here may have even broader range generalizability outside of the bacteria tested in this study and could potentially be utilized in other species after integration with vector backbones and/or promoters that are compatible with those organisms.
Future improvements to this system can be envisioned. Transgenic bacteria expressing the target genes could be lyophilized or otherwise preserved to be rendered as stable control reagents for use in molecular assays (58). Inducible or repressible promoters could be introduced to tailor the expression of the target gene to specific levels. Separately, although the phenotypes encoded by our target genes were convenient for confirming proper splicing in vivo, they are not relevant to the control schema during molecular testing. Transgenes could consequently be altered to exhibit a particular GC content or amplicon length, making control assays targeting them better-matched to the primary diagnostic targets they are intended to be used with. Relatedly, the fluorescence of the GFP does not interfere with our molecular detection assays, but the function of that gene could be ablated through inactivating mutations if deemed necessary for some applications.
The systems presented in this work enable a novel form of internal and positive control material that is useful for RT-LAMP, RT-PCR, and other RNA-based bacterial NAATs. The strategy enables testing to simultaneously control for the full complement of pre-analytic and analytic points of failure possible with such assays. We project that this approach will be applicable for both laboratory-based and point-of-care molecular diagnostics and may be generalizable or adaptable to other bacterial species that are targeted for testing or are selected to emulate key properties of such targeted organisms.
ACKNOWLEDGMENTS
This study was funded in part by the Centers for Disease Control and Prevention (75D30122C15478).
S.J.S. discloses a patent application based on the approach described in this work.
Contributor Information
Stephen J. Salipante, Email: stevesal@uw.edu.
Nathan A. Ledeboer, Medical College of Wisconsin, Milwaukee, Wisconsin, USA
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/jcm.00243-24.
Fig. S1 to S3 and Table S1.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Ma Y, Jiang J, Han Y, Chen Y, Diao Z, Huang T, Feng L, Chang L, Wang D, Zhang Y, Li J, Zhang R. 2023. Comparison of analytical sensitivity of DNA-based and RNA-based nucleic acid amplification tests for reproductive tract infection pathogens: implications for clinical applications. Microbiol Spectr 11:e0149723. doi: 10.1128/spectrum.01497-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gaydos CA, Quinn TC, Willis D, Weissfeld A, Hook EW, Martin DH, Ferrero DV, Schachter J. 2003. Performance of the APTIMA combo 2 assay for detection of Chlamydia trachomatis and Neisseria gonorrhoeae in female urine and endocervical swab specimens. J Clin Microbiol 41:304–309. doi: 10.1128/JCM.41.1.304-309.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boyadzhyan B, Yashina T, Yatabe JH, Patnaik M, Hill CS. 2004. Comparison of the APTIMA CT and GC assays with the APTIMA combo 2 assay, the Abbott LCx assay, and direct fluorescent-antibody and culture assays for detection of Chlamydia trachomatis and Neisseria gonorrhoeae. J Clin Microbiol 42:3089–3093. doi: 10.1128/JCM.42.7.3089-3093.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen D, Tian F, Liu W, Yu J, Song D. 2022. Reverse transcription loop-mediated isothermal amplification assay with high sensitivity to rapid detection of viable Salmonella in foods. Jundishapur J Microbiol 14. doi: 10.5812/jjm.117938 [DOI] [Google Scholar]
- 5. Wu D, Kang J, Li B, Sun D. 2018. Evaluation of the RT-LAMP and LAMP methods for detection of Mycobacterium tuberculosis. J Clin Lab Anal 32:e22326. doi: 10.1002/jcla.22326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burton MJ, Holland MJ, Jeffries D, Mabey DCW, Bailey RL. 2006. Conjunctival chlamydial 16S ribosomal RNA expression in trachoma: is chlamydial metabolic activity required for disease to develop? Clin Infect Dis 42:463–470. doi: 10.1086/499814 [DOI] [PubMed] [Google Scholar]
- 7. Fang X-Y, Li W-B, Zhang C-F, Huang Z, Zeng H-Y, Dong Z, Zhang W-M. 2018. Detecting the presence of bacterial DNA and RNA by polymerase chain reaction to diagnose suspected periprosthetic joint infection after antibiotic therapy. Orthop Surg 10:40–46. doi: 10.1111/os.12359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li R, Tun HM, Jahan M, Zhang Z, Kumar A, Dilantha Fernando WG, Farenhorst A, Khafipour E. 2017. Comparison of DNA-, PMA-, and RNA-based 16S rRNA Illumina sequencing for detection of live bacteria in water. Sci Rep 7:5752. doi: 10.1038/s41598-017-02516-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mei X, Zhai X, Lei C, Ye X, Kang Z, Wu X, Xiang R, Wang Y, Wang H. 2019. Development and application of a visual loop-mediated isothermal amplification combined with lateral flow dipstick (LAMP-LFD) method for rapid detection of Salmonella strains in food samples. Food Control 104:9–19. doi: 10.1016/j.foodcont.2019.04.014 [DOI] [Google Scholar]
- 10. Wang X, Li X, Liu S, Ren H, Yang M, Ke Y, Huang L, Liu C, Liu B, Chen Z. 2016. Ultrasensitive detection of bacteria by targeting abundant transcripts. Sci Rep 6:20393. doi: 10.1038/srep20393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yasukawa K, Agata N, Inouye K. 2010. Detection of cesA mRNA from Bacillus cereus by RNA-specific amplification. Enzyme Microb Technol 46:391–396. doi: 10.1016/j.enzmictec.2009.12.009 [DOI] [Google Scholar]
- 12. Getman D, Lin M, Barakat N, Skvoretz R, Godornes C, Swenson P, Nenninger A, Golden MR, Lukehart SA. 2021. Analytical performance characteristics of a new transcription-mediated amplification assay for Treponema pallidum. J Clin Microbiol 59:e0051121. doi: 10.1128/JCM.00511-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Merold V, Silberreis K, Stoecker K. 2021. Development and validation of ribosomal RNA-targeted reverse transcription real-time PCR assays for the sensitive and rapid diagnostics of high consequence pathogens. Front Microbiol 12:738868. doi: 10.3389/fmicb.2021.738868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aarthi P, Bagyalakshmi R, Therese KL, Madhavan HN. 2013. Development of a novel reverse transcriptase polymerase chain reaction to determine the Gram reaction and viability of bacteria in clinical specimens. Microbiol Res 168:497–503. doi: 10.1016/j.micres.2013.03.005 [DOI] [PubMed] [Google Scholar]
- 15. Garg N, Ahmad FJ, Kar S. 2022. Recent advances in loop-mediated isothermal amplification (LAMP) for rapid and efficient detection of pathogens. Curr Res Microb Sci 3:100120. doi: 10.1016/j.crmicr.2022.100120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Song X, Coulter FJ, Yang M, Smith JL, Tafesse FG, Messer WB, Reif JH. 2022. A lyophilized colorimetric RT-LAMP test kit for rapid, low-cost, at-home molecular testing of SARS-CoV-2 and other pathogens. Sci Rep 12:7043. doi: 10.1038/s41598-022-11144-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Agarwal S, Warmt C, Henkel J, Schrick L, Nitsche A, Bier FF. 2022. Lateral flow-based nucleic acid detection of SARS-CoV-2 using enzymatic incorporation of biotin-labeled dUTP for POCT use. Anal Bioanal Chem 414:3177–3186. doi: 10.1007/s00216-022-03880-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hambalek JA, Kong JE, Brown C, Munoz HE, Horn T, Bogumil M, Quick E, Ozcan A, Di Carlo D. 2021. Methylation-sensitive loop-mediated isothermal amplification (LAMP): nucleic acid methylation detection through LAMP with mobile fluorescence readout. ACS Sens 6:3242–3252. doi: 10.1021/acssensors.1c00902 [DOI] [PubMed] [Google Scholar]
- 19. Das D, Lin C-W, Chuang H-S. 2022. LAMP-based point-of-care biosensors for rapid pathogen detection. Biosensors (Basel) 12:1068. doi: 10.3390/bios12121068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wong Y-P, Othman S, Lau Y-L, Radu S, Chee H-Y. 2018. Loop-mediated isothermal amplification (LAMP): a versatile technique for detection of micro-organisms. J Appl Microbiol 124:626–643. doi: 10.1111/jam.13647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Panpradist N, Kline EC, Atkinson RG, Roller M, Wang Q, Hull IT, Kotnik JH, Oreskovic AK, Bennett C, Leon D, Lyon V, Gilligan-Steinberg SD, Han PD, Drain PK, Starita LM, Thompson MJ, Lutz BR. 2021. Harmony COVID-19: a ready-to-use kit, low-cost detector, and smartphone app for point-of-care SARS-CoV-2 RNA detection. Sci Adv 7:eabj1281. doi: 10.1126/sciadv.abj1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li N, Shen M, Liu J, Zhang L, Wang H, Xu Y, Cheng J. 2021. Multiplexed detection of respiratory pathogens with a portable analyzer in a “raw-sample-in and answer-out” manner. Microsyst Nanoeng 7:94. doi: 10.1038/s41378-021-00321-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee M-F, Chen Y-H, Peng C-F. 2009. Evaluation of reverse transcription loop-mediated isothermal amplification in conjunction with ELISA-hybridization assay for molecular detection of Mycobacterium tuberculosis. J Microbiol Methods 76:174–180. doi: 10.1016/j.mimet.2008.10.005 [DOI] [PubMed] [Google Scholar]
- 24. Techathuvanan C, D’Souza DH. 2012. Reverse-transcriptase loop-mediated isothermal amplification as a rapid screening/monitoring tool for Salmonella enterica detection in liquid whole eggs. J Food Sci 77:M200–M205. doi: 10.1111/j.1750-3841.2011.02601.x [DOI] [PubMed] [Google Scholar]
- 25. Techathuvanan C, Draughon FA, D’Souza DH. 2010. Loop-mediated isothermal amplification (LAMP) for the rapid and sensitive detection of Salmonella Typhimurium from pork. J Food Sci 75:M165–M172. doi: 10.1111/j.1750-3841.2010.01554.x [DOI] [PubMed] [Google Scholar]
- 26. Techathuvanan C, Draughon FA, D’Souza DH. 2011. Comparison of reverse transcriptase PCR, reverse transcriptase loop-mediated isothermal amplification, and culture-based assays for Salmonella detection from pork processing environments. J Food Prot 74:294–301. doi: 10.4315/0362-028X.JFP-10-306 [DOI] [PubMed] [Google Scholar]
- 27. Zhan L-Z, Song D-F, Gu Q, Yan T-T, Ma C-C. 2019. Reverse transcription - loop-mediated isothermal amplification assay for the rapid detection of pathogenic Listeria monocytogenes in meat products. Can J Microbiol 65:913–921. doi: 10.1139/cjm-2019-0114 [DOI] [PubMed] [Google Scholar]
- 28. Burd EM. 2010. Validation of laboratory-developed molecular assays for infectious diseases. Clin Microbiol Rev 23:550–576. doi: 10.1128/CMR.00074-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Whiley DM, Tapsall JW, Sloots TP. 2006. Nucleic acid amplification testing for Neisseria gonorrhoeae: an ongoing challenge. J Mol Diagn 8:3–15. doi: 10.2353/jmoldx.2006.050045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hoorfar J, Cook N, Malorny B, Wagner M, De Medici D, Abdulmawjood A, Fach P. 2003. Making internal amplification control mandatory for diagnostic PCR. J Clin Microbiol 41:5835. doi: 10.1128/JCM.41.12.5835.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martínez-Abarca F, Toro N. 2000. Group II introns in the bacterial world. Mol Microbiol 38:917–926. doi: 10.1046/j.1365-2958.2000.02197.x [DOI] [PubMed] [Google Scholar]
- 32. Tourasse NJ, Kolstø A-B. 2008. Survey of group I and group II introns in 29 sequenced genomes of the Bacillus cereus group: insights into their spread and evolution. Nucleic Acids Res 36:4529–4548. doi: 10.1093/nar/gkn372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schwendener S, Perreten V. 2015. New shuttle vector-based expression system to generate polyhistidine-tagged fusion proteins in Staphylococcus aureus and Escherichia coli. Appl Environ Microbiol 81:3243–3254. doi: 10.1128/AEM.03803-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Calvopina-Chavez DG, Gardner MA, Griffitts JS. 2022. Engineering efficient termination of bacteriophage T7 RNA polymerase transcription. G3 (Bethesda) 12:jkac070. doi: 10.1093/g3journal/jkac070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guo F, Cech TR. 2002. In vivo selection of better self-splicing introns in Escherichia coli: the role of the P1 extension helix of the tetrahymena intron. RNA 8:647–658. doi: 10.1017/s1355838202029011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pédelacq J-D, Cabantous S, Tran T, Terwilliger TC, Waldo GS. 2006. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol 24:79–88. doi: 10.1038/nbt1172 [DOI] [PubMed] [Google Scholar]
- 37. Penewit K, Holmes EA, McLean K, Ren M, Waalkes A, Salipante SJ. 2018. Efficient and scalable precision genome editing in Staphylococcus aureus through conditional recombineering and CRISPR/Cas9-mediated counterselection. mBio 9:e00067-18. doi: 10.1128/mBio.00067-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Charpentier E, Anton AI, Barry P, Alfonso B, Fang Y, Novick RP. 2004. Novel cassette-based shuttle vector system for Gram-positive bacteria. Appl Environ Microbiol 70:6076–6085. doi: 10.1128/AEM.70.10.6076-6085.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Monk IR, Shah IM, Xu M, Tan M-W, Foster TJ. 2012. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio 3:e00277-11. doi: 10.1128/mBio.00277-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McLean K, Rosenthal CA, Sengupta D, Owens J, Cookson BT, Hoffman NG, Salipante SJ. 2019. Improved species-level clinical identification of Enterobacteriaceae through broad-range dnaJ PCR and sequencing. J Clin Microbiol 57:e00986-19. doi: 10.1128/JCM.00986-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu Y, Jeraldo P, Jang JS, Eckloff B, Jen J, Walther-Antonio M. 2019. Bacterial single cell whole transcriptome amplification in microfluidic platform shows putative gene expression heterogeneity. Anal Chem 91:8036–8044. doi: 10.1021/acs.analchem.8b04773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sezonov G, Joseleau-Petit D, D’Ari R. 2007. Escherichia coli physiology in Luria-Bertani broth. J Bacteriol 189:8746–8749. doi: 10.1128/JB.01368-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Armijo LM, Wawrzyniec SJ, Kopciuch M, Brandt YI, Rivera AC, Withers NJ, Cook NC, Huber DL, Monson TC, Smyth HDC, Osiński M. 2020. Antibacterial activity of iron oxide, iron nitride, and tobramycin conjugated nanoparticles against Pseudomonas aeruginosa biofilms. J Nanobiotechnol 18:35. doi: 10.1186/s12951-020-0588-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. 2000. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 28:E63. doi: 10.1093/nar/28.12.e63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. You Y, Moreira BG, Behlke MA, Owczarzy R. 2006. Design of LNA probes that improve mismatch discrimination. Nucleic Acids Res 34:e60. doi: 10.1093/nar/gkl175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Landthaler M, Shub DA. 1999. Unexpected abundance of self-splicing introns in the genome of bacteriophage Twort: introns in multiple genes, a single gene with three introns, and exon skipping by group I ribozymes. Proc Natl Acad Sci U S A 96:7005–7010. doi: 10.1073/pnas.96.12.7005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Landthaler M, Begley U, Lau NC, Shub DA. 2002. Two self-splicing group I introns in the ribonucleotide reductase large subunit gene of Staphylococcus aureus phage Twort. Nucleic Acids Res 30:1935–1943. doi: 10.1093/nar/30.9.1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Karlen Y, McNair A, Perseguers S, Mazza C, Mermod N. 2007. Statistical significance of quantitative PCR. BMC Bioinformatics 8:131. doi: 10.1186/1471-2105-8-131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Naik P, Jaitpal S, Shetty P, Paul D. 2019. An integrated one-step assay combining thermal lysis and loop-mediated isothermal DNA amplification (LAMP) in 30 min from E. coli and M. smegmatis cells on a paper substrate. Sens Actuators B Chem 291:74–80. doi: 10.1016/j.snb.2019.04.044 [DOI] [Google Scholar]
- 50. Packard MM, Wheeler EK, Alocilja EC, Shusteff M. 2013. Performance evaluation of fast microfluidic thermal lysis of bacteria for diagnostic sample preparation. Diagnostics (Basel) 3:105–116. doi: 10.3390/diagnostics3010105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heiniger EK, Buser JR, Mireles L, Zhang X, Ladd PD, Lutz BR, Yager P. 2016. Comparison of point-of-care-compatible lysis methods for bacteria and viruses. J Microbiol Methods 128:80–87. doi: 10.1016/j.mimet.2016.07.007 [DOI] [PubMed] [Google Scholar]
- 52. Li C, Shi Y, Yang G, Xia X, Mao X, Fang Y, Zhang A, Song Y. 2019. Establishment of loop-mediated isothermal amplification for rapid detection of Pseudomonas aeruginosa. Exp Ther Med 17:131–136. doi: 10.3892/etm.2018.6910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Okuda J, Hayashi N, Okamoto M, Sawada S, Minagawa S, Yano Y, Gotoh N. 2010. Translocation of Pseudomonas aeruginosa from the intestinal tract is mediated by the binding of ExoS to an Na,K-ATPase regulator, FXYD3. Infect Immun 78:4511–4522. doi: 10.1128/IAI.00428-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Buckwalter SP, Sloan LM, Cunningham SA, Espy MJ, Uhl JR, Jones MF, Vetter EA, Mandrekar J, Cockerill FR 3rd, Pritt BS, Patel R, Wengenack NL. 2014. Inhibition controls for qualitative real-time PCR assays: are they necessary for all specimen matrices? J Clin Microbiol 52:2139–2143. doi: 10.1128/JCM.03389-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nwe MK, Jangpromma N, Taemaitree L. 2024. Evaluation of molecular inhibitors of loop-mediated isothermal amplification (LAMP). Sci Rep 14:5916. doi: 10.1038/s41598-024-55241-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, Huttenhower C. 2012. Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol 8:e1002606. doi: 10.1371/journal.pcbi.1002606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Padhi BK, Pelletier G, Shwed PS. 2020. A bioinformatics workflow for the evaluation of RT-qPCR primer specificity: application for the assessment of gene expression data reliability in toxicological studies. Regul Toxicol Pharmacol 111:104575. doi: 10.1016/j.yrtph.2020.104575 [DOI] [PubMed] [Google Scholar]
- 58. Tanabe K, Kinoshita M, Nakashima M, Kariya K, Yokokawa M, Morimoto Y, Suzuki H. 2019. On‐site rapid detection of antibacterial activity of neutrophils using freeze‐dried bacteria. Med Devices Sens 2:e10030. doi: 10.1002/mds3.10030 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S3 and Table S1.