

Abstract
We have recently generated a pool of murine cytomegalovirus (MCMV) mutants by using a Tn3-based transposon mutagenesis approach. In this study, one of the MCMV mutants, RvM43, which contained the transposon inserted in open reading frame M43, was characterized. Our results provide the first direct evidence to suggest that M43 is not essential for viral replication in vitro in NIH 3T3 cells. Moreover, RvM43 exhibited a titer similar to that of the wild-type virus in the lungs, livers, spleens, and kidneys of both BALB/c and SCID mice and was as virulent as the wild-type virus in killing SCID mice that had been intraperitoneally infected with the viruses. In contrast, titers of the mutant virus in the salivary glands of the infected animals at 21 days postinfection were significantly (100 to 1,000-fold) lower than those of the wild-type virus and a rescued virus that restored the M43 region and its expression. Thus, M43 appears to be not essential for viral growth in vivo in the lungs, livers, spleens, and kidneys of infected animals and is also dispensable for virulence in killing SCID mice. Moreover, our results suggest that M43 is an MCMV determinant for growth in the salivary glands. Studies of viral genes required for replication in the salivary glands are important in understanding the mechanism of viral tropism for the salivary glands and shedding in saliva, which is believed to be one of the major routes of CMV transmission among healthy human populations.
Human cytomegalovirus (HCMV) is a serious opportunistic pathogen for both newborn and immunocompromised individuals (2, 26). It is the leading viral cause of birth defects, affecting about 7,000 infants yearly in the United States alone (13). Moreover, nearly 10% of the deaths of AIDS patients in 1992 were attributed to HCMV diseases (38, 39). Meanwhile, HCMV infection continues to be a major cause of morbidity and mortality in bone marrow and solid organ transplant recipients (2, 26). Understanding the mechanism of HCMV pathogenesis and the function of viral genes in HCMV-related diseases is necessary for developing new drugs and novel strategies for the treatment and prevention of HCMV infections.
HCMV is a betaherpesvirus with a genome of about 230 kb and a coding capacity of more than 220 open reading frames (6, 26). HCMV propagates only in human cells and grows slowly due to a long lytic replication cycle. Strict species specificity has prevented the study of HCMV in any animal model. Related viruses, such as murine CMV (MCMV), must be used to provide insight into the tissue tropism, virulence, and latency of HCMV.
MCMV provides an excellent model for studying the biology of CMV infection. MCMV offers several advantages, including rapid growth, the availability of a reliable animal model, and the fact that it has sequence homology with HCMV (2, 17, 19, 26). Infection of mice by MCMV resembles in many ways its human counterpart with respect to transmission, pathogenesis during acute infection, establishment of latency, and reactivation after immunosuppression, transfusion, or transplantation (2, 17, 19, 26). For example, tropism for the salivary glands is believed to be important in the infection of both HCMV and MCMV (2, 26). Persistent and recurrent shedding of viral particles from the salivary glands appears to be the principal means by which these viruses spread in the population. The MCMV genome is also 230 kb long and is predicted to encode more than 170 open reading frames, 78 of which have extensive homology to those of HCMV (6, 34). A complete understanding of the biology of MCMV and the function of its genes may provide insight into the pathogenesis of HCMV.
One of the most powerful approaches for studying the function of virus-encoded genes is to introduce mutations into the viral genome and to screen viral mutants in both tissue culture and animals for possible growth defects in vitro and in vivo. The construction of herpesvirus mutants by using site-directed homologous recombination and transposon-mediated insertional mutagenesis has been reported (18, 27, 28, 33, 36, 47), as have methods using overlapping cosmid DNA fragments to generate mutants of HCMV and other herpesviruses (8, 9, 20, 43, 44). More recently, the MCMV genome as well as the genomes of other herpesviruses have been cloned into a bacterial artificial chromosome (BAC), and viral mutants were successfully generated from the BAC-based viral genome by a bacterial mutagenesis procedure (3, 12, 25, 37, 40–42, 46). It has been shown that the BAC sequence in a BAC-based pseudorabies virus does not affect viral pathogenesis in vivo in animals (40, 41). Moreover, two different approaches to excise the BAC sequence from the viral genome have been described (41, 46). These studies have greatly facilitated the identification of the functions of viral genes in tissue culture and in animals.
Many of the CMV genes have been found to be dispensable for growth in cultured cells. Their presence in the viral genome indicates that they are likely needed to perform functions involved only in modulating the interactions between the virus and its human or animal hosts. For example, HCMV US11, a nonessential protein, functions to downregulate the expression and presentation of major histocompatibility complex class I molecules (48). Meanwhile, MCMV open reading frame m133, which is also called salivary gland gene 1 (sgg1) and is dispensable for viral replication, is a determinant for MCMV replication in salivary glands in vivo (21, 24). Thus, studies of viral mutants carrying mutations in genes found to be dispensable in tissue culture are valuable for an understanding of gene function in viral pathogenesis and virus-host interactions, including tissue tropism and virulence.
We have previously generated a pool of MCMV mutants using a Tn3 transposon-mediated shuttle mutagenesis system (49, 50). In this approach, the transposon is randomly inserted into the MCMV genomic DNA fragments in a plasmid library in Escherichia coli. Regions bearing an insertion mutation are then transferred to the MCMV genome by cotransfecting the plasmid library and purified MCMV genomic DNA into NIH 3T3 cells. Characterization of one of the viral mutants (e.g., Rvm09) in NIH 3T3 cells and in both BALB/c and SCID mice indicated that the presence of the transposon sequence per se in the viral genome does not significantly affect viral replication and growth in vitro and in vivo (50). In the present study, we have characterized an MCMV mutant, RvM43, which contains a transposon insertion in open reading frame M43. The function of M43 in viral replication and pathogenesis is unknown. Indeed, this open reading frame has not been extensively characterized transcriptionally or translationally. Our results provide the first direct evidence to suggest that M43 is not essential for viral replication in vitro in tissue culture. Moreover, RvM43 replicated as well as the wild-type virus in the lungs, livers, spleens, and kidneys of both the BALB/c and SCID mice and was as virulent as the wild-type virus in killing SCID mice that had been intraperitoneally infected with the viruses. In contrast, at 21 days postinfection, titers of the mutant virus were at least 100- and 1,000-fold lower than those of the wild-type virus in the salivary glands of BALB/c and SCID mice, respectively. These results suggest that M43 functions as a viral determinant for replication in the salivary glands.
MATERIALS AND METHODS
Cells and viruses.
Mouse NIH 3T3 cells and STO cells and the wild-type Smith strain of MCMV were obtained from the American Type Culture Collection, Manassas, Va. Cells were maintained in complete growth medium consisting of Dulbecco's modified Eagle medium (DMEM) supplemented with 10% NuSerum (Becton Dickenson, Mountain View, Calif.), amino acids, and antibiotics (each from a stock solution purchased from Life Technologies Inc. [Gibco BRL, Grand Island, N.Y.]) (49). The wild-type Smith strain, mutant RvM43, and the rescued virus RqM43 were propagated in NIH 3T3 cells as described previously (49).
Construction of an MCMV DNA subclone pool, transposon shuttle mutagenesis, and generation of MCMV recombinant mutants.
Isolation of viral genomic DNA, construction of an MCMV genomic subclone pool, and transposon-based shuttle mutagenesis to generate a pool of MCMV DNA fragments containing a transposon insertion were performed as described by Zhan et al. (49). To generate a pool of MCMV mutants that contained the transposon sequence, intact MCMV genomic DNAs and plasmid DNAs containing the Tn3-guanine phosphorlbosyltransferase gene (gpt)-MCMV fragments were cotransfected into NIH 3T3 cells using a calcium phosphate precipitation protocol (Gibco BRL). The recombinant MCMV mutants were selected in the presence of mycophenolic acid (25 μg/ml; Gibco BRL) and xanthine (50 μg/ml; Sigma, St. Louis, Mo.) and then plaque purified six times as described previously (49). To confirm the integration of the transposon in the viral genome and to identify the genes that were disrupted by the transposon insertion, viral DNA was purified and directly sequenced using the primer FL110PRIM (5′-GCAGGATCCTATCCATATGAC-3′) and an fmol cycle sequencing kit (Promega, Inc., Madison, Wis.).
To construct the rescued virus RqM43, the full-length genomic DNA of RvM43 was isolated from infected cells as described previously (49). The DNA sequence that contained the coding sequence of M43 was generated by PCR using the viral genomic DNA as the template, the 5′ primer M43-sense (5′-CATGCCTGGCGGACTGGAAA-3′), and the 3′ primer M43-antisense (5′-TCATAGTTCCGGTTCGGATG-3′). The PCR product that contained the M43 coding sequence (1 to 3 μg) and the full-length intact RvM43 viral genomic DNA (8 to 12 μg) were subsequently cotransfected into mouse STO cells using a calcium phosphate precipitation protocol (Gibco BRL). The recombinant virus was selected in STO cells in the presence of 6-thioguanine (25 μg/ml; Sigma) and purified by six rounds of amplification and plaque purification as described previously (14). For each cotransfection, several viral plaques were picked and expanded. Viral stocks were prepared by growing the viruses in T-150 flasks of NIH 3T3 cells.
Southern and Northern analyses of recombinant viruses.
Viral genomic DNA was purified from NIH 3T3 cells infected with the viruses as described previously (45, 49). Briefly, cells that exhibited 100% cytopathic effect were washed with phosphate-buffered saline and then subjected to proteolysis with a solution containing sodium dodecyl sulfate and proteinase K. The genomic DNA was purified by extraction with phenol-chloroform followed by precipitation with 2-propanol. The DNA was then digested with HindIII or NotI, separated on agarose gels (0.8%), transferred to Zeta-Probe nylon membranes (Bio-Rad, Hercules, Ca), and hybridized with 32P-labeled DNA probes specific for both the transposon and the MCMV sequences. A STORM 840 PhosphorImager was used to analyze the blots.
For Northern blot analysis, cells were infected with viruses at a multiplicity of infection (MOI) of 5 and harvested at different time points postinfection. In the experiments to assay the expression of immediate-early (IE) transcripts, cells were pretreated with cycloheximide (100 μg/ml; Sigma), then infected with viruses, and harvested at 6 h postinfection. Total cytoplasmic RNA was isolated from NIH 3T3 cells infected with the viruses as described previously (22). Viral RNAs were separated in a 1% agarose gel that contained formaldehyde, transferred to a nitrocellulose membrane, hybridized with the 32P-radiolabeled DNA probes that contained specific MCMV sequences, and finally analyzed with a STORM 840 PhosphorImager. The DNA probes used for Northern analyses were generated by PCR using viral DNA as the template and radiolabeled with a random primer synthesis kit in the presence of [32P]dCTP (Boehringer Mannheim, Indianapolis, Ind.). The 5′ and 3′ PCR primers used for construction of the DNA probe for Northern analysis were M43/1 (5′-TACACGTTGGCTGTTACCGCTAGGT-3′) and M43/2 (5′-TCACCGTCGGTTCGACGTTCTCGTA-3′), respectively.
Analysis of growth of the viruses in vitro.
The growth kinetics of the viruses was determined as described previously (50). Briefly, NIH 3T3 cells grown to 50 to 60% confluence were inoculated with virus at an MOI of either 0.5 or 5. At 0, 1, 2, 4, and 7 days postinfection, the infected cells together with medium were harvested, and an equal volume of 10% skim milk was added before sonication. Virus titer was determined by plaque assays in NIH 3T3 cells. Viral titers reported were averages from triplicate experiments.
Analysis of growth of the viruses in animals.
Four-week-old male BALB/c-Byj mice (Jackson Laboratory, Bar Harbor, Maine) or 6-week-old CB17 SCID mice (National Cancer Institute, Bethesda, Md.) were infected intraperitoneally with 104 PFU of each virus. The animals were sacrificed at 1, 3, 7, 10, 14, and 21 days postinoculation. For each time point, at least three animals were used as a group and infected with the same virus. The salivary glands, lungs, spleens, livers, and kidneys were harvested and sonicated as a 10% (wt/vol) suspension in a 1:1 mixture of DMEM and skim milk. The sonicates were stored at −80°C until plaque assays were performed.
Viruses harvested from the mice were titered on NIH 3T3 cells in six-well tissue culture plates (Corning Inc., Corning, N.Y.). Briefly, cells were first infected with the viruses at 10-fold serial dilutions. After 2 h of incubation with the homogenates diluted in 1 ml of complete medium at 37°C with 5% CO2, the cells were overlaid with fresh complete medium containing 1% agarose and cultured for 3 to 4 days before the plaques were counted under an inverted microscope. Viral titers were recorded as PFU per milliliter of organ homogenates. Each sample was titered in triplicate. The limit of virus detection in the organ homogenates was 10 PFU/ml of the sonicated mixture. Those samples that were negative at a 10−1 dilution were assigned a titer of 10 (101) PFU/ml.
Virulence assays.
Virulence of the viruses was studied by determining the mortality of the animals infected with the Smith strain, RvM43, or RqM43. The CB17 SCID mice (five animals per group) were infected intraperitoneally with 104 PFU of each virus. The animals were observed twice daily; mortality of the infected animals was monitored for at least 28 days postinfection, and the survival rates were determined.
RESULTS
Isolation of an MCMV mutant containing the transposon insertion at open reading frame M43.
We have constructed a pool of MCMV mutants using an E. coli Tn3-based transposon mutagenesis system (49). Figure 1A shows the structure of the transposon used to generate MCMV mutants. The transposon contains the gpt expression cassette (which includes the gpt coding sequence driven by a promoter and a transcription termination signal) and an additional transcription termination site to allow selection of mutant MCMV in mammalian cells and transcription truncation of the disrupted gene (49). Since the gpt expression cassette and the additional poly(A) signal in the transposon are in opposite orientations, the expression of nearby genes that may share a common poly(A) signal with the target gene would not be disrupted (Fig. 1A). In our mutagenesis procedure, an MCMV genomic library containing a randomly inserted transposon in each viral DNA fragment was first generated using a shuttle mutagenesis method as described previously (49). Such a pool of MCMV genomic fragments containing randomly inserted Tn3-gpt sequence was then cotransfected with the full-length genomic DNA of the wild-type virus (Smith strain) into mouse NIH 3T3 cells in which homologous recombination occurred. Cells that harbored the progeny viruses expressing the gpt gene were selected for growth in the presence of mycophenolic acid and xanthine (14, 30, 45). After multiple rounds of selection and plaque purification, we isolated individual recombinant viruses and determined the location of the inserted transposon by directly sequencing the genomic DNA of the recombinants. One of the recombinant viruses was further characterized and is reported here. This viral mutant, designated RvM43, contains the transposon sequence inserted within open reading frame M43 (Fig. 1B). Sequencing analyses of the junction between the transposon and the viral sequence revealed that the transposon in RvM43 is located at nucleotide position 56204 (the codon for amino acid 313 of M43), with reference to the genome sequence of the wild-type Smith strain (34) (Fig. 1B and data not shown).
FIG. 1.

(A) Schematic representation of the structure of the transposon construct used for mutagenesis. The gpt expression cassette includes the gpt coding sequence driven by a promoter and a transcription termination signal. TR, terminal repeat; Tet, tetracycline resistance gene; poly(A), transcription termination signal. (B) Location of the transposon insertion in the recombinant virus. The transposon sequence is shown as a filled bar; the coding sequence of open reading frame M43 is represented by an open arrow. The orientation of the arrow represents the direction of the translation and transcription predicted based on the nucleotide sequence (34). The numbers represent sizes (in kilobases) of the DNA fragments of the viruses that were generated by digestion with HindIII (H) or NotI (N). (C) Southern analyses of the recombinant viruses. The DNA fractions were isolated from cells infected with the wild-type virus (WT), RvM43, or RqM43. The DNA samples (20 μg) were digested with either HindIII (H) or NotI (N), separated on 0.8% agarose gels, transferred to a Zeta-Probe membrane, and hybridized to a DNA probe. The probe used for the analyses was the plasmid that contained the MCMV DNA fragment inserted with the transposon sequence.
Southern analyses of RvM43 and rescued virus RqM43.
The genomic structure of RvM43 and the location of the transposon insertion in the viral genome were examined by Southern blot hybridization with a DNA probe containing both the transposon and the viral sequence (Fig. 1B and C). When the viral DNA samples were digested with HindIII and subjected to Southern analyses, a small fragment of 1.8 kb representing the gpt gene was detected, indicating the presence of the transposon sequence within the viral genome (Fig. 1C, lane 1). This finding was further supported by the results of Southern analyses of the RvM43 DNA samples digested with another restriction enzyme, NotI (Fig. 1C, lanes 4). In these experiments, the genomic fragments containing the transposon were found to be larger than those of the wild-type virus by 3.6 kb, which is the size of the transposon (Fig. 1B and C).
The Southern blots also showed that the stocks of the mutant virus were pure and free of the wild-type strain since the hybridizing DNA fragments from the mutant did not comigrate with those of the wild-type Smith strain (Fig. 1C, lanes 1, 3, 4, and 6). For example, the hybridization patterns of the RvM43 and Smith strain DNAs digested with HindIII gave rise to three (20, 8.2, and 1.8 kb) and one (26.4 kb) DNA band(s), respectively (Fig. 1C, lanes 1 and 3). Meanwhile, the hybridized species (12.8 kb) of the NotI-digested RvM43 DNA migrated differently from that (9.2 kb) of the wild-type viral DNA digested with the same enzyme (Fig. 1C, lanes 4 and 6). The sizes of the hybridized DNA fragments (Fig. 1C) were consistent with the predicted digestion patterns of the recombinant virus, based on the MCMV genomic sequence (34) and the location of the transposon insertion in the viral genome as determined by sequence analysis (Fig. 1B). The restriction enzyme digestion patterns of the regions of the mutant genomic DNA other than the transposon insertional site appeared to be identical to those of the parental Smith strain, as indicated by ethidium bromide staining of the digested DNAs (data not shown). This observation suggests that regions of the viral genome other than that containing the transposon insertion remained intact in this MCMV mutant.
To restore the M43 open reading frame, a rescued virus, designated RqM43, was derived from RvM43, using a protocol similar to the procedure used for construction of the viral mutant. A DNA fragment that contained the M43 coding region was cotransfected with the full-length RvM43 genomic DNA into mouse STO cells to allow homologous recombination to occur. The cells that harbored the progeny viruses were allowed to grow in the presence of 6-thioguanine, which selects against gpt expression (14, 30). The rescued virus, RqM43, which did not express the gpt protein and no longer contained the transposon, was isolated after multiple rounds of selection and plaque purification.
Southern hybridization analyses with DNA probes containing the transposon and the viral sequence were used to examine the genomic structure of RqM43 and determine if the M43 region was restored (Fig. 1B and C). Analysis of the RqM43 DNA samples digested with HindIII and NotI showed that the hybridized DNA fragments for RqM43 were the same size as the hybridized fragments for the Smith strain but not those for RvM43. These results indicated that RqM43 did not contain the transposon sequence and the M43 region was restored (Fig. 1C, lanes 2 and 5). Moreover, the restriction enzyme digestion patterns of the regions of the rescued RqM43 genomic DNA samples other than the M43 region appeared to be identical to those of the parental RvM43, as indicated by ethidium bromide staining of the digested DNAs (data not shown). This observation suggests that the regions of the RqM43 genome other than the M43 region remained intact and were identical to those of RvM43. Thus, RqM43 may represent a rescued virus for RvM43.
Expression of the transcripts from the viral M43 region in tissue culture.
Transcription from the target M43 region is expected to be disrupted due to the presence of the two transcription termination signals within the transposon (Fig. 1A). In particular, the region of the M43 open reading frame downstream from the transposon insertion site is not expected to be expressed. To determine whether this is the case, cytoplasmic RNAs were isolated from cells infected with the mutant virus at different time points (e.g., 4, 12, and 24 h) postinfection. Northern analysis was carried out to examine the expression of the transcripts from the M43 open reading frame downstream from the transposon insertion site (Fig. 2). The probe used in the Northern analyses contained the DNA sequence complementary to the M43 coding region about 50 nucleotides downstream from the site of the transposon insertion. An abundant RNA species was detected in RNA fractions isolated from cells that were infected with the wild-type Smith strain (Fig. 2, lane 3) but not in RNA fractions isolated from cells infected with RvM43 when the same probe was used (lane 2). Indeed, no truncated transcripts or bands were detected from cells infected with RvM43 (Fig. 2 and data not shown), indicating that the region downstream from the transposon insertion site was not expressed. Meanwhile, expression of the transcript was found in RNA fractions of cells infected with the rescued virus RqM43 (Fig. 2, lane 4) and was at a level similar to that of the RNA fractions from cells infected with the Smith strain (compare lanes 3 and 4). The level of MCMV M25 transcript (10, 49) was used as the internal control for the expression of the M43 transcript. As shown in Fig. 2 (lanes 5 to 8), the levels of the M25 transcript detected in cells that were infected with RvM43 and RqM43 were found to be similar to that of M25 transcript in cells infected with the Smith strain. Thus, the transposon insertion in RvM43 truncated or disrupted the transcript expressed from the M43 open reading frame, whereas expression of the transcript was restored in RqM43.
FIG. 2.

Northern analyses of RNA fractions isolated from cells that were mock infected (lanes 1 and 5) or infected with the wild-type virus (WT; lanes 3 and 7), RvM43 (lanes 2 and 6), and RqM43 (lanes 4 and 8); 107 NIH 3T3 cells were infected with each virus at an MOI of 5 PFU per cell, and cells were harvested at 24 h postinfection. Equal amounts of RNA samples (30 μg) were separated on agarose gels that contained formaldehyde, transferred to a nitrocellulose membrane, and hybridized to a 32P-radiolabeled probe that contained the sequence of M43 (lanes 1 to 4) or M25 (lanes 5 to 8).
Replication of RvM43 and RqM43 in vitro in tissue culture.
Growth rates of the recombinant viruses in NIH 3T3 cells were studied in order to determine whether these viruses had any growth defects in vitro. Cells were infected with the viruses at both low and high MOIs, and their growth rates were assayed by triplicate experiments. No significant difference was found in growth rates among RvM43, RqM43, and the Smith strain. For example, the peak titers of RvM43 and RqM43 were similar to those of the Smith strain after both high- and low-MOI infections (Fig. 3). Combined with the results from the Southern and Northern analyses, these observations indicate that M43 is not essential for viral growth in vitro.
FIG. 3.

In vitro growth of MCMV mutants in tissue culture. Mouse NIH 3T3 cells were infected with each virus at an MOI of either 0.5 (A) or 5 (B) PFU per cell. At 0, 1, 2, 4, and 7 days postinfection, cells and culture media were harvested and sonicated. The viral titers were determined by plaque assays on NIH 3T3 cells. The virus titers represent averages from triplicate experiments. Standard deviations are indicated by error bars. WT, wild type.
Growth of recombinant viruses RvM43 and RqM43 in immunocompetent animals.
To determine whether disruption of M43 significantly affects viral replication in vivo, BALB/c-Byj mice were injected intraperitoneally with 104 PFU of RvM43, RqM43, or the wild-type Smith strain. At 1, 3, 7, 10, 14, and 21 days postinfection, salivary glands, lungs, spleens, livers, and kidneys were harvested and virus titers in these five organs were determined on NIH 3T3 cells; titers in the salivary glands at 28 and 35 days postinfection were also determined. These organs are among the major targets for MCMV infection (2, 17, 19, 26). RvM43 exhibited similar titers as the Smith strain and RqM43 in the lungs, spleens, livers, and kidneys of the infected animals (Fig. 4B to E). These results suggest that open reading frame M43 is dispensable for MCMV replication in these organs in vivo. However, the titers of mutant RvM43 in the salivary glands at 14 and 21 days postinfection were found to be at least 100-fold less than those of the Smith strain and RqM43 (Fig. 4). Previous studies have shown that the presence of the transposon sequence per se within the viral genome does not significantly affect viral growth in mice in vivo (50). Thus, these results suggest that the attenuated growth of RvM43 in the salivary glands is due to the disruption of M43 and that open reading frame M43 is important for viral growth in the salivary glands in vivo.
FIG. 4.

Titers of MCMV mutants in the salivary glands (A), lungs (B), spleens (C), livers (D), and kidneys (E) of infected BALB/c mice. BALB/c-Byj mice were infected intraperitoneally with 104 PFU of each virus. At 1, 3, 7, 10, 14, and 21 days postinfection, the animals (three mice per group) were sacrificed. The salivary glands, lungs, spleens, livers, and kidneys were collected and sonicated; salivary glands were also collected from animals at 28 and 35 days postinfection. Viral titers in the tissue homogenates were determined by standard plaque assays in NIH 3T3 cells. The limit of detection was 10 PFU/ml of the tissue homogenate. The virus titers represent averages from triplicate experiments. Error bars indicate standard deviations; error bars that are not evident indicate that the standard deviation was less than or equal to the height of the symbols. WT, wild type.
Virulence and growth of viral mutants in SCID mice.
It has been shown that immunodeficient animals are extremely susceptible to MCMV infection (15, 31, 32, 35). For example, the CB17 SCID mice, which lack functional T and B lymphocytes, are extremely sensitive to viral infection (as few as 10 PFU of MCMV cause serious infection) (31, 32). Analysis of viral replication in these mice therefore serves as an excellent means for determining the virulence of different MCMV strains and mutants and for studying the mechanism whereby they cause opportunistic infections in immunocompromised hosts. To determine whether the M43 open reading frame plays a significant role in CMV virulence, we compared the survival rates of animals infected with RvM43 and those of mice infected with RqM43 and the wild-type Smith strain. For each virus, five SCID mice were injected intraperitoneally with 104 PFU of RvM43, RqM43, or the Smith strain. We found that 50% of the infected mice died within 24 to 26 days postinfection, indicating that disruption of the M43 open reading frame does not significantly affect viral virulence (Fig. 5). Moreover, these results are consistent with our previous observations that the presence of the transposon sequence per se in the viral genome does not significantly affect the virulence of MCMV in SCID mice (50).
FIG. 5.

Mortality of SCID mice infected with the Smith strain (wild type [WT]), RvM43, and RqM43. CB17 SCID mice (five animals per group) were infected intraperitoneally with 104 PFU of each virus. Mortality of mice was monitored for at least 28 days postinfection, and survival rates were determined.
To further study the pathogenesis of the mutant virus in these immunodeficient animals, the replication of RvM43 in different organs of the animals was studied during a 21-day infection period before the mortality of the infected animals became apparent. In these experiments, SCID mice were injected intraperitoneally with 104 PFU of each virus (RvM43, RqM43, or the wild-type Smith strain). At 1, 3, 7, 10, 14, and 21 days postinfection, three mice from each virus group were sacrificed and the salivary glands, lungs, spleens, livers, and kidneys were harvested. The viral titers in these five organs were determined. The titers of RvM43 in the lungs, spleens, livers, and kidneys were similar to those of the Smith strain and the rescued RqM43 (Fig. 6), indicating that disruption of M43 does not significantly affect MCMV growth in these four organs. In contrast, titers of the mutant virus in the salivary glands at 21 days postinfection were at least 1,000-fold lower than those of the wild-type virus and RqM43 (Fig. 6). Thus, RvM43 is attenuated in growth in the salivary glands. These results suggest that open reading frame M43 may be essential for efficient replication of MCMV in the salivary glands.
FIG. 6.

Titers of MCMV mutants in the salivary glands (A), lungs (B), spleens (C), livers (D), and kidneys (E) of infected SCID mice. CB17 SCID mice were infected intraperitoneally with 104 PFU of each virus. At 1, 3, 7, 10, 14, and 21 days postinfection, the animals (three mice per group) were sacrificed. The salivary glands, lungs, spleens, livers, and kidneys were collected and sonicated. Viral titers in the tissue homogenates were determined by standard plaque assays in NIH 3T3 cells. The limit of detection was 10 PFU/ml of the tissue homogenate. The virus titers represent averages obtained from triplicate experiments. Error bars indicate standard deviations; error bars that are not evident indicate that the standard deviation was less than or equal to the height of the symbols. WT, wild type.
The transposon insertion at the M43 open reading frame is stable in the viral genome.
Our previous studies indicated that a transposon sequence inserted at several regions (e.g., m09 and M83) of the MCMV genome is stable during viral replication in NIH 3T3 cells and in both BALB/c and SCID mice (50). However, it has been reported that viral mutants with an additional inserted sequence were not stable and generated spontaneous mutations during replication in vitro and in vivo (1, 24). It is possible that the transposon sequence in RvM43 is not stable during viral replication in vivo and that introduction of the second mutation may be responsible for the observed phenotypes of the virus in animals. To address this issue, viruses were recovered from the lungs and salivary glands of the BALB/c and SCID mice at 21 days postinfection and subsequently used to infect NIH 3T3 cells. Viral DNAs were purified from the infected cells, and their restriction digestion patterns were analyzed in agarose gels. Figure 7 shows a Southern analysis of the RvM43 viral DNAs with a DNA probe that contained the transposon and the M43 sequence. These results indicated that no change in the hybridization patterns of RvM43 occurred as a result of viral growth in animals for 21 days (lanes 1 to 4). Moreover, the overall HindIII digestion patterns of RvM43 DNA isolated from either infected cultured cells or animals were identical to those of the original recombinant virus RvM43, as visualized by ethidium bromide staining of the viral DNAs (data not shown). Thus, the transposon insertion in RvM43 appeared to be stable and the genome of RvM43 remained intact during replication in both BALB/c and SCID mice.
FIG. 7.
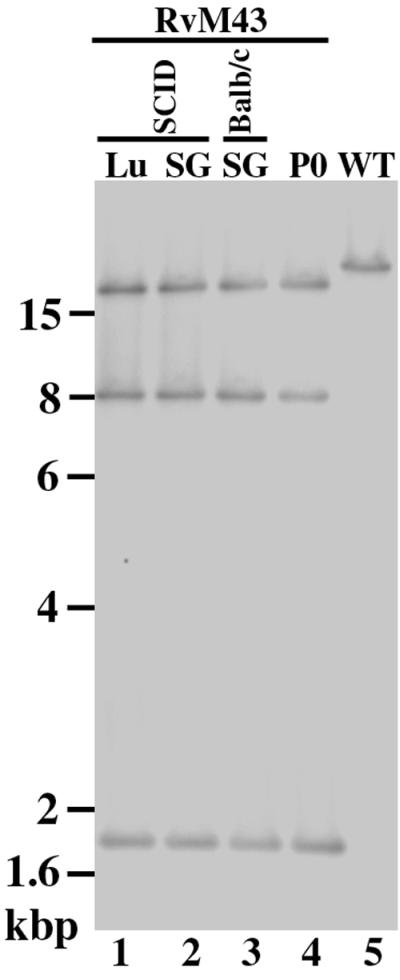
The stability of the transposon mutation of RvM43 in BALB/c and SCID mice. Viral DNAs were isolated either from cells that were infected with RvM43 (MOI, <0.01) and allowed to grow in culture for 5 days (P0; lane 4) or from cells that were infected with the virus collected from the salivary glands (SG; lanes 2 and 3) and lungs (Lu; lane 1) of either BALB/c (lane 3) or SCID (lanes 1 and 2) mice 21 days after intraperitoneal inoculation with 104 PFU of RvM43. Southern analyses of the viral DNA fractions digested with HindIII are shown. The DNA of the wild-type virus (WT) is shown in lane 5. The 32P-radiolabeled probe was derived from the same plasmid as was used for Southern analyses of RvM43 in Fig. 1 and contained the transposon and the M43 open reading frame sequence.
DISCUSSION
In this study, a recombinant virus that contained the insertional mutation at open reading frame M43 was characterized. Our results provide the first direct evidence to suggest that M43 is not essential for viral replication in NIH 3T3 cells in vitro. Moreover, M43 appears to function as a viral determinant for MCMV growth in the salivary glands.
While it is possible that the functional protein product is synthesized from the transposon-disrupted region, several lines of evidence strongly suggest that this is not the case. First, the transposon sequence was inserted into the coding sequence of the M43 open reading frame (Fig. 1B). Second, transcription from the region downstream from the transposon insertion site was not detected in cells infected with the mutant virus (Fig. 2). Thus, the region of the target open reading frame downstream from the transposon insertion site was not expressed, and the transcript expressed from the disrupted open reading frame was truncated.
The function of M43 is unknown. Indeed, to our knowledge, neither the transcript nor the protein product coded by this open reading frame has been reported. Our results indicate that a transcript of about 5,000 nucleotides is expressed from the M43 open reading frame. The growth rate of RvM43 in NIH 3T3 cells was not significantly different from that of the Smith strain. Since the transposon insertion in RvM43 is within the M43 coding region and the coding sequence 3′ from the insertion site was not transcribed, it is likely that no functional M43 protein was expressed from the viral mutant. Thus, our results suggest that M43 is not essential for viral replication in NIH 3T3 cells.
Our results indicate that RvM43 replicated as well as the wild-type Smith strain in the lungs, livers, spleens, and kidneys of both BALB/c and SCID mice that were infected intraperitoneally with these viruses. These results suggest that RvM43 is not essential for viral growth in these organs in vivo. Moreover, RvM43 was as virulent as the Smith strain in killing the SCID mice, suggesting that the M43 open reading frame does not play a significant role in viral virulence in SCID mice. In contrast, RvM43 was attenuated in replication in the salivary glands of the infected animals. The titers of RvM43 in the salivary glands of SCID mice at 21 days postinfection were at least 1,000-fold lower than those of the wild-type virus, whereas the rescued virus RqM43 exhibited similar titers as the Smith strain. These results strongly suggest that M43 is a viral determinant for replication in the salivary glands. Furthermore, our data suggest that viral replication in the salivary glands does not significantly contribute to MCMV virulence in killing the SCID mice.
It is possible that the observed change in the levels of replication of the mutant is due to other adventitious mutations accumulated during the construction and growth of the recombinant virus in cultured cells or in animals. However, several lines of evidence strongly suggest that this is unlikely. First, rescued virus RqM43 replicated as well as the wild-type virus in the salivary glands (Fig. 4 and 6). The genomic sequence of M43 and its expression in the rescued virus were restored (Fig. 1 and 2). These observations suggest that the transposon insertion, rather than a second mutation, is responsible for the observed attenuation of RvM43 replication in the salivary glands. Second, our previous studies indicated that a virus with a transposon insertion at the m09 open reading frame replicated as well as the wild-type virus in the salivary glands, indicating that the transposon sequence per se in the viral genome does not significantly affect viral replication in the organs (50). Third, the transposon insertion was stable during replication in animals. There was no change in the hybridization patterns of the DNAs from the mutant virus recovered from the salivary glands and lungs of the infected animals at 21 days postinfection (Fig. 7). Moreover, the HindIII digestion patterns of the RvM43 mutant DNAs, other than the region with the inserted transposon, appeared to be identical to those of the wild-type virus DNA (data not shown). Thus, the observed change in the RvM43 titer in the salivary glands is probably due to the insertional mutation at M43 introduced by the transposon.
Our results suggest that M43 functions as a viral determinant for growth in salivary glands and that the lack of expression of the functional full-length M43 product is responsible for the observed low level of growth of RvM43 in the salivary glands. However, it is possible that expression of the N-terminal truncated portion of the M43 open reading frame rather than the lack of expression of the full-length M43 product may be responsible for the observed phenotype of the mutant in vivo. This is because the truncated protein, which is possibly expressed under the control of the wild-type viral M43 promoter and translation initiation site, may interfere with viral growth in the salivary glands. To address this issue, another viral mutant with a transposon insertion at M43 (at the codon for amino acid 116) was isolated independently from RvM43. The titers of this second mutant in the salivary glands of the infected animals were similar to those of RvM43 and lower than those of the Smith strain and RqM43 (J. Xiao, X. Zhan, E. Haghjoo, and F. Liu, unpublished results). Thus, it is unlikely that expression of the truncated N-terminal product rather the lack of expression of the full-length M43 product is responsible for the low level of growth of the viral mutant in the salivary glands, although we cannot completely rule out this possibility.
Open reading frame M43 and its HCMV counterpart, UL43, belong to the MCMV M23 and HCMV US22 gene families, respectively (6, 34). The MCMV M23 gene family, consisting of 12 open reading frames, includes M23, M24, M36, M43, m25.1, m25.2, m128 (ie2), m139, m140, 141, m142, and m143. The HCMV US22 gene family includes UL23, UL24, UL28, UL29, UL36, UL43, US22, US23, US24, US26, IRS1, and TRS1. A spontaneous deletion at the UL42 and UL43 region during HCMV growth in vitro has been observed, and UL43 appeared to be dispensable for HCMV replication in vitro (11, 29). Previous studies have also shown that several members of the MCMV M23 gene family (e.g., m128 [ie2] and m140) are dispensable for MCMV replication (4, 5, 16, 23). Indeed, the replication of a virus with a deletion in m139, m140, and m141 was significantly attenuated in growth in animals (5, 16), while a virus carrying a deletion in m128 (ie2) appeared to replicate as well as the wild-type virus (4, 23). Moreover, some members (e.g., m140 and m141) of the M23 gene family have been implicated as important for cell and tissue tropism of MCMV infection (5, 16). Our results suggest that M43 plays a significant role in MCMV infection in the salivary glands. It will be interesting to determine whether UL43 is also a viral determinant for HCMV replication in the same organ.
Specific tropism for host tissues, particularly the salivary glands, is an important determinant of CMV biology. Tropism for the salivary glands and persistent and recurrent viral shedding from this organ are believed to be among the main routes for HCMV transmission in normally healthy individuals (2). Previous studies have suggested that open reading frame m133 (sgg1) is a viral determinant for MCMV replication in the salivary glands (21, 24). A viral mutant with a deletion in sgg1 was defective in growth in the salivary glands of BALB/c mice (24). Meanwhile, this mutant did not exhibit any growth defects in other organs examined and was as virulent as the wild-type virus in killing BALB/c mice (21, 24). Although there is little sequence homology between M43 and sgg1, it is conceivable that the function of the M43 gene is related to that of sgg1. There are other examples in the herpesvirus family that some viral genes are necessary for viral replication in specific tissues or organs. For example, herpes simplex virus type 1 γ1-34.5, which is dispensable for viral replication in cultured cells, is essential for viral replication in neuronal cells and has been suggested to play a significant role in viral neuronal tropism and neuroinvasiveness (7). It is intriguing that more than one gene is needed for MCMV tissue tropism in a specific organ. It would be interesting to determine how these determinants function in concert with the replication cycle of MCMV in vivo. These studies will further provide insight into the biology of CMVs and functions of the viral genes in CMV pathogenesis.
ACKNOWLEDGMENTS
We thank Edward Mocarski, Stanford University, for insightful advice and Michael Snyder, Yale University, for providing the Tn3 constructs and the E. coli strains for transposon shuttle mutagenesis. Gratitude also goes to Gerry Abenes and Manfred Lee for sharing unpublished results and helpful discussions and to Ilse Von Reis and John Kim for technical assistance.
F.L. is a Pew Scholar in Biomedical Sciences and a recipient of a Hellman Family Faculty Award, a Basil O'Connor Starter Scholar Research Award (March of Dimes National Birth Defects Foundation), and a Regents Junior Faculty Fellowship (University of California). This research was supported in part by a Chancellor's Special Initiative Grant Award (UC-Berkeley) and a grant from the State of California AIDS research program.
REFERENCES
- 1.Boname J M, Chantler J K. Characterization of a strain of murine cytomegalovirus which fails to grow in the salivary glands of mice. J Gen Virol. 1992;73:2021–2029. doi: 10.1099/0022-1317-73-8-2021. [DOI] [PubMed] [Google Scholar]
- 2.Britt W J, Alford C A. Cytomegalovirus. In: Fields B N, Knipe D M, et al., editors. Virology. 3rd ed. New York, N.Y: Raven Press; 1996. pp. 2493–2523. [Google Scholar]
- 3.Brune W, Menard C, Hobom U, Odenbreit S, Messerle M, Koszinowski U H. Rapid identification of essential and nonessential herpesvirus genes by direct transposon mutagenesis. Nat Biotechnol. 1999;17:360–364. doi: 10.1038/7914. [DOI] [PubMed] [Google Scholar]
- 4.Cardin R D, Abenes G B, Stoddart C A, Mocarski E S. Murine cytomegalovirus IE2, an activator of gene expression, is dispensable for growth and latency in mice. Virology. 1995;209:236–241. doi: 10.1006/viro.1995.1249. [DOI] [PubMed] [Google Scholar]
- 5.Cavanaugh V J, Stenberg R M, Staley T L, Virgin III H W, MacDonald M R, Paetzold S, Farrell H E, Rawlinson W D, Campbell A E. Murine cytomegalovirus with a deletion of genes spanning HindIII-J and -I displays altered cell and tissue tropism. J Virol. 1996;70:1365–1374. doi: 10.1128/jvi.70.3.1365-1374.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chee M S, Bankier A T, Beck S, Bohni R, Brown C M, Cerny R, Horsnell T, Hutchison C A, Kouzarides T, Martignetti J A. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr Top Microbiol Immunol. 1990;154:125–169. doi: 10.1007/978-3-642-74980-3_6. [DOI] [PubMed] [Google Scholar]
- 7.Chou J, Kern E R, Whitley R J, Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science. 1990;250:1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 8.Cohen J I, Seidel K E. Generation of varicella-zoster virus (VZV) and viral mutants from cosmid DNAs: VZV thymidylate synthetase is not essential for replication in vitro. Proc Natl Acad Sci USA. 1993;90:7376–7380. doi: 10.1073/pnas.90.15.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cunningham C, Davison A J. A cosmid-based system for constructing mutants of herpes simplex virus type 1. Virology. 1993;197:116–124. doi: 10.1006/viro.1993.1572. [DOI] [PubMed] [Google Scholar]
- 10.Dallas P B, Lyons P A, Hudson J B, Scalzo A A, Shellam G R. Identification and characterization of a murine cytomegalovirus gene with homology to the UL25 open reading frame of human cytomegalovirus. Virology. 1994;200:643–650. doi: 10.1006/viro.1994.1227. [DOI] [PubMed] [Google Scholar]
- 11.Dargan D J, Jamieson F E, MacLean J, Dolan A, Addison C, McGeoch D J. The published DNA sequence of human cytomegalovirus strain AD169 lacks 929 base pairs affecting genes UL42 and UL43. J Virol. 1997;71:9833–9836. doi: 10.1128/jvi.71.12.9833-9836.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delecluse H J, Hilsendegen T, Pich D, Zeidler R, Hammerschmidt W. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc Natl Acad Sci USA. 1998;95:8245–8250. doi: 10.1073/pnas.95.14.8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fowler K B, Stagno S, Pass R F, Britt W J, Boll T J, Alford C A. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N Engl J Med. 1992;326:663–667. doi: 10.1056/NEJM199203053261003. [DOI] [PubMed] [Google Scholar]
- 14.Greaves R F, Brown J M, Vieira J, Mocarski E S. Selectable insertion and deletion mutagenesis of the human cytomegalovirus genome using the Escherichia coli guanosine phosphoribosyl transferase (gpt) gene. J Gen Virol. 1995;76:2151–2160. doi: 10.1099/0022-1317-76-9-2151. [DOI] [PubMed] [Google Scholar]
- 15.Grundy J E, Melief C J. Effect of Nu/Nu gene on genetically determined resistance to murine cytomegalovirus. J Gen Virol. 1982;61:133–136. doi: 10.1099/0022-1317-61-1-133. [DOI] [PubMed] [Google Scholar]
- 16.Hanson L K, Slater J S, Karabekian Z, Virgin III H W, Biron C A, Ruzek M C, van Rooijen N, Ciavarra R P, Stenberg R M, Campbell A E. Replication of murine cytomegalovirus in differentiated macrophages as a determinant of viral pathogenesis. J Virol. 1999;73:5970–5980. doi: 10.1128/jvi.73.7.5970-5980.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hudson J B. The murine cytomegalovirus as a model for the study of viral pathogenesis and persistent infections. Arch Virol. 1979;62:1–29. doi: 10.1007/BF01314900. [DOI] [PubMed] [Google Scholar]
- 18.Jenkins F J, Casadaban M J, Roizman B. Application of the mini-Mu-phage for target-sequence-specific insertional mutagenesis of the herpes simplex virus genome. Proc Natl Acad Sci USA. 1985;82:4773–4777. doi: 10.1073/pnas.82.14.4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jordan M C. Latent infection and the elusive cytomegalovirus. Rev Infect Dis. 1983;5:205–215. doi: 10.1093/clinids/5.2.205. [DOI] [PubMed] [Google Scholar]
- 20.Kemble G, Duke G, Winter R, Spaete R. Defined large-scale alterations of the human cytomegalovirus genome constructed by cotransfection of overlapping cosmids. J Virol. 1996;70:2044–2048. doi: 10.1128/jvi.70.3.2044-2048.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lagenaur L A, Manning W C, Vieira J, Martens C L, Mocarski E S. Structure and function of the murine cytomegalovirus sgg1 gene: a determinant of viral growth in salivary gland acinar cells. J Virol. 1994;68:7717–7727. doi: 10.1128/jvi.68.12.7717-7727.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu F, Roizman B. The herpes simplex virus 1 gene encoding a protease also contains within its coding domain the gene encoding the more abundant substrate. J Virol. 1991;65:5149–5156. doi: 10.1128/jvi.65.10.5149-5156.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manning W C, Mocarski E S. Insertional mutagenesis of the murine cytomegalovirus genome: one prominent alpha gene (ie2) is dispensable for growth. Virology. 1988;167:477–484. [PubMed] [Google Scholar]
- 24.Manning W C, Stoddart C A, Lagenaur L A, Abenes G B, Mocarski E S. Cytomegalovirus determinant of replication in salivary glands. J Virol. 1992;66:3794–3802. doi: 10.1128/jvi.66.6.3794-3802.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Messerle M, Crnkovic I, Hammerschmidt W, Ziegler H, Koszinowski U H. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc Natl Acad Sci USA. 1997;94:14759–14763. doi: 10.1073/pnas.94.26.14759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mocarski E S. Cytomegaloviruses and their replication. In: Fields B N, Knipe D M, et al., editors. Virology. 3rd ed. New York, N.Y: Raven Press; 1996. pp. 2447–2492. [Google Scholar]
- 27.Mocarski E S, Kemble G W, Lyle J M, Greaves R F. A deletion mutant in the human cytomegalovirus gene encoding IE1(491aa) is replication defective due to a failure in autoregulation. Proc Natl Acad Sci USA. 1996;93:11321–11326. doi: 10.1073/pnas.93.21.11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mocarski E S, Post L E, Roizman B. Molecular engineering of the herpes simplex virus genome: insertion of a second L-S junction into the genome causes additional genome inversions. Cell. 1980;22:243–255. doi: 10.1016/0092-8674(80)90172-5. [DOI] [PubMed] [Google Scholar]
- 29.Mocarski E S, Prichard M N, Tan C S, Brown J M. Reassessing the organization of the UL42-UL43 region of the human cytomegalovirus strain AD169 genome. Virology. 1997;239:169–175. doi: 10.1006/viro.1997.8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mulligan R C, Berg P. Selection for animal cells that express the Escherichia coli gene coding for xanthine-guanine phosphoribosyltransferase. Proc Natl Acad Sci USA. 1981;78:2072–2076. doi: 10.1073/pnas.78.4.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okada M, Minamishima Y. The efficacy of biological response modifiers against murine cytomegalovirus infection in normal and immunodeficient mice. Microbiol Immunol. 1987;31:45–57. doi: 10.1111/j.1348-0421.1987.tb03067.x. [DOI] [PubMed] [Google Scholar]
- 32.Pollock J L, Virgin H W., III Latency, without persistence, of murine cytomegalovirus in the spleen and kidney. J Virol. 1995;69:1762–1768. doi: 10.1128/jvi.69.3.1762-1768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Post L E, Roizman B. A generalized technique for deletion of specific genes in large genomes: alpha gene 22 of herpes simplex virus 1 is not essential for growth. Cell. 1981;25:227–232. doi: 10.1016/0092-8674(81)90247-6. [DOI] [PubMed] [Google Scholar]
- 34.Rawlinson W D, Farrell H E, Barrell B G. Analysis of the complete DNA sequence of murine cytomegalovirus. J Virol. 1996;70:8833–8849. doi: 10.1128/jvi.70.12.8833-8849.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reynolds R P, Rahija R J, Schenkman D I, Richter C B. Experimental murine cytomegalovirus infection in severe combined immunodeficient mice. Lab Anim Sci. 1993;43:291–295. [PubMed] [Google Scholar]
- 36.Roizman B, Jenkins F J. Genetic engineering of novel genomes of large DNA viruses. Science. 1985;229:1208–1214. doi: 10.1126/science.2994215. [DOI] [PubMed] [Google Scholar]
- 37.Saeki Y, Ichikawa T, Saeki A, Chiocca E A, Tobler K, Ackermann M, Breakefield X O, Fraefel C. Herpes simplex virus type 1 DNA amplified as bacterial artificial chromosome in Escherichia coli: rescue of replication-competent virus progeny and packaging of amplicon vectors. Hum Gene Ther. 1998;9:2787–2794. doi: 10.1089/hum.1998.9.18-2787. [DOI] [PubMed] [Google Scholar]
- 38.Selik R M, Chu S Y, Ward J W. Trends in infectious diseases and cancers among persons dying of HIV infection in the United States from 1987 to 1992. Ann Intern Med. 1995;123:933–936. doi: 10.7326/0003-4819-123-12-199512150-00006. [DOI] [PubMed] [Google Scholar]
- 39.Selik R M, Karon J M, Ward J W. Effect of the human immunodeficiency virus epidemic on mortality from opportunistic infections in the United States in 1993. J Infect Dis. 1997;176:632–636. doi: 10.1086/514083. [DOI] [PubMed] [Google Scholar]
- 40.Smith G A, Enquist L W. Construction and transposon mutagenesis in Escherichia coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J Virol. 1999;73:6405–6414. doi: 10.1128/jvi.73.8.6405-6414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith G A, Enquist L W. A self-recombining bacterial artificial chromosome and its application for analysis of herpesvirus pathogenesis. Proc Natl Acad Sci USA. 2000;97:4873–4878. doi: 10.1073/pnas.080502497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stavropoulos T A, Strathdee C A. An enhanced packaging system for helper-dependent herpes simplex virus vectors. J Virol. 1998;72:7137–7143. doi: 10.1128/jvi.72.9.7137-7143.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomkinson B, Robertson E, Yalamanchili R, Longnecker R, Kieff E. Epstein-Barr virus recombinants from overlapping cosmid fragments. J Virol. 1993;67:7298–7306. doi: 10.1128/jvi.67.12.7298-7306.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Zijl M, Quint W, Briaire J, de Rover T, Gielkens A, Berns A. Regeneration of herpesviruses from molecularly cloned subgenomic fragments. J Virol. 1988;62:2191–2195. doi: 10.1128/jvi.62.6.2191-2195.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vieira J, Farrell H E, Rawlinson W D, Mocarski E S. Genes in the HindIII J fragment of the murine cytomegalovirus genome are dispensable for growth in cultured cells: insertion mutagenesis with a lacZ/gpt cassette. J Virol. 1994;68:4837–4846. doi: 10.1128/jvi.68.8.4837-4846.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wagner M, Jonjic S, Koszinowski U H, Messerle M. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J Virol. 1999;73:7056–7060. doi: 10.1128/jvi.73.8.7056-7060.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weber P C, Levine M, Glorioso J C. Rapid identification of nonessential genes of herpes simplex virus type 1 by Tn5 mutagenesis. Science. 1987;236:576–579. doi: 10.1126/science.3033824. [DOI] [PubMed] [Google Scholar]
- 48.Wiertz E J, Jones T R, Sun L, Bogyo M, Geuze H J, Ploegh H L. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- 49.Zhan X, Abenes G, Lee M, VonReis I, Kittinunvorakoon C, Ross-Macdonald P, Snyder M, Liu F. Mutagenesis of murine cytomegalovirus using a Tn3-based transposon. Virology. 2000;266:264–274. doi: 10.1006/viro.1999.0089. [DOI] [PubMed] [Google Scholar]
- 50.Zhan X, Lee M, Xiao J, Liu F. Construction and characterization of murine cytomegaloviruses that contain a transposon insertion at open reading frames m09 and M83. J Virol. 2000;74:7411–7421. doi: 10.1128/jvi.74.16.7411-7421.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]