

Abstract

Zeolites are crystalline aluminosilicates with well-defined microporous structures that have found several applications in catalysis. In recent years, great effort has been devoted to defining strategies aimed at tuning structural and acidity properties to improve the catalytic performance of zeolites. Depending on the zeolitic structure, the acid sites located inside the crystals catalyze reactions by exploiting the internal channel shape-selectivity. In contrast, strong acid sites located on the external surface do not offer the possibility to control the size of molecules involved in the reactions. This aspect generally leads to a loss of selectivity toward desired products and to the uncontrolled production of coke. Passivating surface acidity is a promising way to overcome deactivation issues and to enhance the catalytic performance of zeolites. This Mini-Review aims to provide, for the first time, a complete overview of the techniques employed in recent years to neutralize strong external acid sites. Both chemical and liquid vapor deposition of silicates have been widely employed to passivate the external surface acidity of zeolites. In recent years, the epitaxial growth of layers of aluminum-free zeolite, e.g., silicalite-1, over the surface of the acidic zeolite has been proposed as a new approach to neutralize strong external acid sites controlling diffusional phenomena. NH3-TPD, FT-IR, SEM-EDX, and other techniques have been used to provide information about the level of control of the external strong acidity of passivated zeolites. In this Mini-Review, both passivation treatments and characterization techniques are compared and advantages and disadvantages deeply discussed to elucidate the effect of passivation procedures on physical features and especially the catalytic behavior.
1. Introduction
The term “shape-selectivity” defines the capability of a catalytic structure to confine specific molecules involved in a chemical reaction, enhancing the reaction specificity (i.e., selectivity). In the field of heterogeneous catalysis, the shape-selectivity consists of matching the size and shape of interacting species, reactants, products and even transition states with the structural features of the voids of the catalytic solid. In this regard, due their well-defined microporous structures with molecular-sized pores, zeolites are widely used as catalytic materials in industry, ensuring tunable molecular shape-selectivity in several processes. As relevant examples, the fluid catalytic cracking (FCC) process adopts modified zeolite Y (FAU structure) with a few percent of ZSM-5 (MFI structure) as an additive to promote the isomerization reaction, a MOR-based catalyst is used in the isomerization of light paraffins, the MFI-type zeolite structure is also a state-of-the art catalyst in the isomerization and alkylation xylenes, BEA is usually used for ethylbenzene and cumene production, SAPOs are largely used for methanol-to-olefin processes, and TS-1 is used for propylene oxide production via direct peroxidation.1
One of the reasons for the expanding use of zeolites as catalysts at industrial scale is the possibility to synthesize these materials with specific features in terms of acidity, pore system, and crystal size. Due to the reduced mass transfer limitations, it has been demonstrated that zeolites exhibiting a hierarchical meso/micropore system or in form of nanosized crystals exhibit improved performance in terms of efficiency and coke deposition. When dealing with organic reactants, a side reaction might lead to the formation of an unwanted carbonaceous deposit, often called “coke”, that is the main cause of deactivation of zeolites. Deactivation by coke is usually accompanied by a reduction of micropore volume and surface area as a consequence of channel blockage.
In order to carefully control either the micro/mesopore structure or the size and morphology of the crystal, different synthesis and postsynthesis strategies have been developed and optimized. For instance, Gilson and co-workers studied the application of both hierarchical and nanosized H-ZSM-5 zeolites in ethanol to hydrocarbon transformation, revealing that the coke formation is positively related to the density of pore mouths on the zeolite surface and that hierarchization improves the stability, also elucidating the positive impact of nanosized zeolites on the catalyst performance.2
Similarly, the control of acidity (i.e., concentration of acid sites, Brønsted/Lewis distribution, strength, and location) plays a crucial role in improving both turnover frequency and selectivity. For example, it is well recognized that, in reactions involving hydrocarbons, the presence of strong acid sites promotes the formation of coke; in order to delay the induced deactivation, several strategies can be employed to modulate the acidity, reducing the concentration of strong acid sites. Still dealing with acid-catalyzed reactions involving hydrocarbons, Guisnet well described the actions to be adopted to prevent coke-induced deactivation, showing how the phenomenon is somehow reduced when dealing with three-dimensional zeolites without trap cavities and modulating the density and strength of the acid sites to improve the catalyst selectivity.3 In some cases, the formation of secondary carbonaceous species is a fundamental condition to promote the formation of the desired species. This is the case of the methanol-to-olefin process, where the formation of coke precursors (mainly aromatic methyl-substituted cations) is a key step to activate the reaction mechanism known as “hydrocarbon pool” to achieve a high yield of olefins. Also, in that case, the control of shape-selectivity is a crucial aspect to control the mechanism and thus the process performance.
“Shape-selectivity” is a concept strictly related to the inner surface of the crystal of zeolites, although both the pore mouth and the external surface have a role in catalysis. Wei et al. reported a study on the formation mechanism of olefins during methanol conversion of TON and MTT zeolites, both structures not suitable for the hydrocarbon pool mechanism, due to steric constraints.4 The authors demonstrated that olefins were still generated through a “hydrocarbon pool” mechanism, which was catalyzed by external and/or pore mouth acid sites. In another work, Xu et al. highlighted the importance of controlling the acidity in a SSZ-13 zeolite for the MTO process, indicating that low acid site density can suppress both the hydrogen transfer reaction and heavy coke formation, leading to the preferential formation of polymethylbenzenes as primary hydrocarbon pool species.5
Several works reported data about the important role played by the acid sites not located in the channels, such as in the case of alkane hydro-isomerization, the synthesis of iso-paraffins from syngas, skeletal isomerization reactions, and so on. Several strategies are proposed for controlling the location of acid sites on the external surface. Chemical vapor/liquid deposition of silicates is the most common route adopted to neutralize the external surface acidity. In recent years, leading to the formation of a so-called “core-shell” zeolite, the epitaxial growth of a nonacidic zeolitic shell over an acidic zeolitic core has received a great attention because of the several benefits offered in terms of control of textural properties.
On the basis of the important role attributed to the external surface when zeolites are used in catalysis, this Mini-Review focuses on the strategies adopted to obtain zeolites with reduced external strong surface acidity, with correlations to characterization techniques used to assess the quality of external passivation. Both advantages and disadvantages of routes commonly adopted are deeply discussed and compared, especially in terms of the impact on catalysis.
2. Zeolite Surface Passivation and Catalysis
Techniques proposed in the recent literature to passivate external surfaces of zeolites are summarized in Table 1; among them, neutralization through the epitaxial growth of a layer of silicalite-1 was also included. Moreover, catalytic applications of passivated catalysts are also summarized in Table 1, underlining the advantages and drawbacks related to their usage.
Table 1. Summary of the Main Surface Passivation Techniques Recently Employed, the Catalytic Applications of Passivated Catalysts, and the Main Advantages and Disadvantages Related to Their Use.
| catalytic
application |
|||||
|---|---|---|---|---|---|
| ref | passivation technique employed | parent zeolite | reaction | advantages | disadvantages |
| (9) | epitaxial growth of Sil-1 via hydrothermal synthesis | ZSM-5 (Si/Al = 150) | toluene methylation with methanol | longer diffusion time of species | none |
| higher catalyst stability | |||||
| higher para-xylene selectivity | |||||
| lower coke formation | |||||
| (8) | epitaxial growth of Sil-1 via hydrothermal synthesis | ZSM-5 (Si/Al = 75) | toluene methylation with methyl bromide | higher catalyst stability | lower initial conversion value |
| higher para-xylene selectivity | |||||
| (10) | Al incorporation by organotemplate-free synthesis and sequential crystallization of residual silica species by organotemplate-oriented synthesis | direct synthesis of core–shell material | catalytic cracking of n-hexane | higher catalyst stability and higher para-xylene selectivity compared to a conventional ZSM-5 with the same acidity | lower initial n-hexane conversion value |
| (11) | epitaxial growth of Sil-1 via hydrothermal synthesis (different shell thickness) | ZSM-5 (Si/Al = 25) | catalytic cracking of n-hexane | higher light olefin selectivity | lower drop in total acidity with the increase of Sil-1 thickness |
| lower initial n-hexane conversion value | |||||
| (12) | epitaxial growth of Sil-1 via hydrothermal synthesis (one and two layers of Sil-1) | commercial ZSM-5 (Si/Al = 38) | catalytic fast pyrolysis of biomass | slightly higher BTX selectivity | nonhomogeneous Sil-1 coating and low effect of passivation on catalytic performances |
| (13) | steam-assisted crystallization of Sil-1 layer (SAC) | mesoporous ZSM-5 (Si/Al≈140) | methanol to hydrocarbons | higher catalyst stability | none |
| (14) | epitaxial growth of Si-FER via hydrothermal synthesis | FER (Si/Al = 10) | benchmark reaction of Friedel–Crafts alkylation of mesitylene and benzyl alcohol | higher benzyl alcohol conversion (demonstration of passivation) | none |
| (16) | chemical liquid deposition (CLD) of tetraethyl orthosilicate | Nano-ZSM-5 | methanol to olefins | higher light olefin selectivity | lower surface area and micropore volume |
| lower stability | |||||
| (17) | chemical liquid deposition (CLD) of tetraethyl orthosilicate | SAPO-34 | methanol to olefins | none | lower stability due to higher surface barrier |
| (18) | chemical liquid deposition (CLD) of tetraethyl orthosilicate | ZSM-22 | 1-butene skeletal isomerization | higher isobutene yield and selectivity | none |
| (19) | chemical liquid deposition (CLD) of tetraethyl orthosilicate and 3,5-dimethylphenylmagnesium bromide (DPB) | MOR | isomerization of o-ethyltoluene | higher selectivity toward desired products for sample passivated with DPB | lower surface area and micropore volume for the sample treated with TEOS |
| (20) | chemical liquid deposition (CLD) of tetraethyl orthosilicate | Ga/HZSM-5 | propane dehydroaromatization | higher BTX selectivity for the sample with a SiO2 content of 4% | lower surface area for the passivated samples with up to 4 wt % SiO2 |
| (21) | external surface charge reversal with hexadecyltrimethylammonium (HDTMA) followed by impregnation of phosphoric acid | ZSM-5 (Si/Al = 70) | toluene methylation with methanol | higher para-xylene selectivity and yield | none |
| (22) | chemical liquid deposition (CLD) of tetraethyl orthosilicate | BEA | cyclohexanol dehydration | extended catalyst lifetime | none |
| (23) | surface modification with organodisilane compound | ZSM-5 | dimethyl ether-to-olefin (DTO) | higher propylene yield for sample passivated with 10 wt % disilane | loss of catalytic performances for higher silylation levels |
2.1. Core–Shell Zeolites
The “core-shell concept” involves materials composed of two distinct parts: an outer shell and an inner core. In recent years, core–shells have attracted the attention of scientists, as this configuration allows the coupling of materials with different properties and makes them suitable for numerous applications. While largely preserving the original properties of each phase, the different properties of core (or hollow region) and shell allow the generation of new characteristics, resulting in enhanced features compared to any pure phase or their physical blends. Different core–shell configurations have already been explored in the literature, where zeolites could represent the shell or the core. Moreover, depending on the core material, core–shells could be separated into two main categories: zeolite@zeolite and metal-oxide@zeolite core–shells. While the production of metal-oxide@zeolite core–shells is generally exploited when acid–base function is required to avoid the sintering of the metal catalyst and its deactivation, in recent years the synthesis of composite materials characterized by both a zeolitic core and a zeolitic shell has been widely employed to neutralize external strong acid sites or to achieve a gradient of distribution of zeolite acid sites. In particular, growing a nonacidic zeolite shell, such as silicalite-1, on the surface of the starting core allows the passivation of the external strong acidity and meanwhile preserves the catalytic activity without limiting the accessibility of internal acid sites.6 ZSM-5@silicalite-1 composites have been synthesized using different techniques and employed for various applications. This approach usually consists of two steps: first the core crystals, eventually pretreated, are placed in contact with the silicalite-1 precursor solution, and the hydrothermal crystallization of the shell is carried out during the second step. The precursor solution usually employed for the synthesis of the silicalite-1 layer includes water, an organic template (generally tetrapropylammonium), a silica source, e.g., tetraethyl orthosilicate (TEOS) or fumed silica, and, eventually, ethanol used as solvent to enhance the crystallization of the zeolite shell.7 The growth of the layer of silicalite-1 is usually performed at 180 °C for 24 h. High molar H2O/SiO2 ratios are always required to prevent the segregated nucleation of silicalite-1 crystals and to promote the epitaxial growth around the core.
The analysis of catalytic applications in which core–shells have been used provides a tool to understand the true effectiveness of this technique. Coating of the external surface of ZSM-5 zeolites has widely been employed to enhance the performance of catalysts in the production of para-xylene from toluene methylation.8,9para-Xylene is essential to produce polyesters, but it also represents a key intermediate for pharmaceutical or pesticide industries. Traditionally, it is produced from the reforming of fossil naphtha. Exploiting the shape-selectivity of zeolites in this reaction is a promising alternative route to produce para-xylene. In more detail, para-xylene can diffuse into the 10-membered rings of ZSM-5 more easily than its isomers (ortho- and meta-xylene), and this aspect enhances p-selectivity. Nevertheless, the external strong acidity of ZSM-5 leads to a rapid isomerization of para-xylene to ortho-xylene and meta-xylene, causing a loss of selectivity. The growth of a layer of silicalite-1 over the surface of a ZSM-5 represents a way to neutralize external strong acid sites and to increase the diffusion path length for xylenes, forcing the production of para-xylene at the same time. Pan et al. prepared H-ZSM5@silicalite-1 core–shells at different levels of silicalite-1 coating, employing the hydrothermal crystallization method, and tested them in a toluene alkylation reaction with methanol in a fixed-bed tubular quartz reactor operated at atmospheric pressure and 470 °C (WHSV = 8 h–1, toluene/methanol molar ratio equal to 6).9 SEM micrographs provided direct proof of the success of the silicalite-1 coating, showing evident differences between the parent zeolite crystals and the core–shells crystals. After being coated with silicalite-1, the crystals maintained the same hexagonal prism shape, but the external surfaces appeared roughened. Moreover, total acidity was studied by NH3-TPD measurements, while Brønsted external strong acidity was analyzed using in situ FT-IR with di-tert-butyl-pyridine (DTBPy) as the probe molecule. Even if the number of acid sites was not quantified, a decreasing trend of total acidity and external Brønsted acid sites was found with the increase in the amount of silicalite-1 coated on the surface of the ZSM-5 zeolite (Figure 1).
Figure 1.

2,6-DTBPy FT-IR spectra of parent zeolite and core–shell samples. Reprinted with permission from ref (9). Copyright 2022 MDPI.9
Carried out at 220 °C to avoid para-xylene isomerization, pulsed chromatography experiments have been further employed to study the diffusion of xylene isomers into ZSM-5 channels. ortho- and meta-xylene exhibited the shortest retention times in all the experiments because ZSM-5 channels hinder their diffusion. On the contrary, for para-xylene, two peaks were always detected: a small peak at a low retention time related to a small quantity of para-xylene that did not diffuse inside zeolite pores and a broader peak at a high retention time associated with para-xylene that entered ZSM-5 channels. A translation of this second peak to higher retention times was found when experiments were carried out using HZSM5@silicalite-1 core–shells, and it was found that the peak elution time increased with the increase of the amount of silicalite-1 deposited over ZSM-5 parent zeolites. These results proved that the silicalite-1 layer increased the diffusion path, prolonging the diffusion time of species involved in the reaction and making this aspect crucial to enhance para-xylene selectivity. Although toluene conversion remained unchanged if compared with the parent zeolite, catalytic results confirmed that HZSM-5@silicalite-1 catalysts enhanced the initial para-selectivity. Moreover, the authors performed time-on-stream (TOS) tests to evaluate the effect of silicalite-1 deposition on long-term catalyst behavior, as well as coke formation and deactivation. Unlike the parent zeolite, which showed a decrease in toluene conversion from 11% to 9% after about 50 h of reaction, HZSM5@silicalite-1 samples were more stable and exhibited an initial conversion value equal to the parent zeolite, and the same drop in conversion was found after about 160 h as shown in Figure 2.
Figure 2.
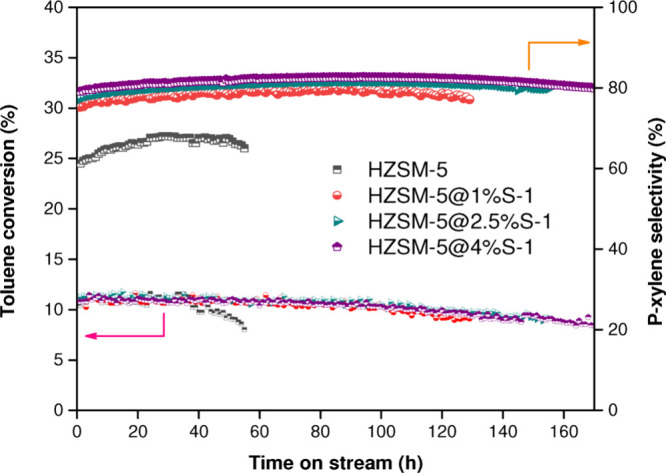
Toluene conversion and p-xylene selectivity as a function of time (TOS) in a toluene alkylation reaction over an untreated H-ZSM-5 sample and HZSM5@X@S-1 core–shells. Reprinted with permission from ref (9). Copyright 2022 MDPI.9
Furthermore, passivated samples showed a higher para-xylene selectivity that reached a maximum value of about 80%, higher than that exhibited by parent zeolite (67.9%). TGA analyses of spent catalysts also revealed the positive effect of silicalite-1 deposition over coke formation; weight loss detected for the passivated sample coated with the highest amount of silicalite-1 was six times lower than that found for the uncoated ZSM-5 zeolite, demonstrating that the reduction of external crystal surface acidity delays coke deposition. The passivation of external strong acidity through the epitaxial growth of a layer of silicalite-1 was also studied by Deng et al. through the catalytic cracking of 1,3,5-triisopropylbenzene (TIPB) to verify the role of external surface acid sites of the crystals. It happened because TIPB, with a critical diameter of 9.5 Å, cannot diffuse into ZSM-5 channels and can then be converted by external acid sites only. A TIPB conversion of only 1.2% was found when the HZSM5@silicalite-1 catalyst was employed for the cracking reaction, significantly lower than that detected for the parent zeolite (16.9%), thus demonstrating that a large amount of external strong acid sites were effectively neutralized. The same paper also investigated a core–shell sample in the reaction of toluene methylation with CH3Br (produced from methane in the presence of HBr).8 Results obtained from TOS tests at 400 °C for 360 min (WHSV = 2 h–1, toluene/CH3Br molar ratio equal to 0.5) revealed that the coated sample was more stable than the parent zeolite. In fact, although ZSM-5 catalysts exhibited an initial conversion value of about 70%, they were rapidly deactivated and toluene conversion dropped to a value of about 20% after 360 min of reaction. Despite the fact that HZSM@silicalite-1 showed an initial conversion of 50% (likely due to its reduced acidity), the toluene conversion decreased to 40% after 360 min, showing a positive effect on stability.
Recently, the synthesis of ZSM5@silicalite-1 core–shells was achieved using a novel tandem synthesis approach.10 This innovative strategy involved two sequential steps: the incorporation of nearly all aluminum atoms through organic-template-free (or organotemplate-free) synthesis, followed by the crystallization of the remaining silicate precursor using organic template synthesis. Referring to the organic-template-free synthesis, chemicals including colloidal silica, Al2(SO4)3·18H2O, NaOH, H2O, and seeds were mixed and stirred at room temperature and then transferred to a stainless-steel autoclave for dynamic crystallization for 8 h at 170 °C. Subsequently, in the organic-template-oriented synthesis phase, a specific quantity of the template agent was added as TPABr, and this mixture again underwent dynamic crystallization at 170 °C for 72 h. The so-obtained core–shell sample was tested in an n-hexane catalytic cracking reaction carried out at 550 °C (WHSV = 4 h–1) to evaluate the effect of surface passivation on coke deposition and product distribution in respect to a conventional ZSM-5 catalyst, which was characterized by the same total acidity and a different aluminum distribution along the crystals. The authors employed TEMEDS and XPS analysis to assess the aluminum distribution along the crystal synthesized with this novel strategy. In particular, these results revealed that no aluminum was present on the external surface of the “Si-rich-ZSM-5” crystals. Only total acidity was measured by NH3TPD analysis, while external strong acidity was evaluated via the catalytic cracking of TIPB. Although “Si-rich-ZSM-5” showed an initial TIPB conversion lower than that of the conventional ZSM-5 catalyst (about 24% and 40%, respectively), these results probably suggested that external strong acidity was not totally “neutralized”. Despite this evidence, results of n-hexane catalytic cracking revealed the better performance of the core–shell sample in terms of stability. While the passivated catalyst showed a lower initial conversion compared to conventional ZSM-5 (74.6% versus 99.5%, respectively), its rate of deactivation was significantly slower. After 100 h of reaction, the core–shell catalyst maintained a conversion of 57.7%, substantially higher than the 24.6% achieved using the ZSM-5 zeolite.
The effect of surface passivation on the performance of catalysts for the reaction of n-hexane cracking has also been evaluated by Zhu et al.11 The authors employed the common hydrothermal coating of silicalite-1 over the external surface of a ZSM-5 zeolite to produce core–shells with different shell thicknesses. The synthesis procedure consists of two steps: ZSM-5 crystals were first stirred for 2 h with a solution of TPAOH and water, then a proper amount of TEOS was added and the stirring prolonged for other few hours. Shell thickness was controlled by varying the mass ratio between the ZSM-5 and the SiO2 in the synthesis gel with TEOS (from 0.5 to 2.5 at 180 °C for 72 h). The growth of the silicalite-1 layer was demonstrated using different techniques. EDS mapping images collected for the ZSM-5 zeolite showed a uniform distribution of Si and Al, while those obtained for the passivated sample revealed the presence of a core in which the Al distribution was unchanged and a shell where only Si was detected (Figure 3).
Figure 3.

TEM images of (a) ZSM-5 and (b) Z5@S(1.0). (c) TEM image of ZSM-5 and the corresponding EDS mapping images representing (d) Si and (e) Al. (f) TEM image of Z5@S(1.0) and the corresponding EDS mapping images representing (g) Si and (h) Al. Reprinted with the permission from ref (11). Copyright 2023 John Wiley and Sons.11
Moreover, XPS measurements proved that with the increase of the silicalite-1 shell thickness, i.e., with the increase of TEOS used in the synthesis procedure, the peak attributed to Si 2p in SiO2 increased while the peak related to Al 2p decreased. Total acidity was measured and quantitatively estimated via NH3-TPD measurements and FT-IR analyses with pyridine as the probe molecule. Comparing the ZSM-5 parent zeolite with the composite sample with the thickest silicalite-1 layer, a drop in total specific acidity was found after the passivation treatment. The core–shell sample with the thickest layer of silicalite-1 exhibited a total amount of Brønsted and Lewis acid sites of 5.5 μmol/g while the nonpassivated ZSM-5 zeolite showed a value 5× higher. FT-IR spectra collected after the adsorption of 2,4,6-collidine were collected to assess the external strong acidity, which was not quantified. Results demonstrated that the external strong acidity decreased as the thickness of the shell increased, even though quantification of the external acidity would have been useful to understand whether the drop detected in the total acidity was related to the passivation treatment or, even partially, to the segregated crystal growth of silicalite-1 crystals. Parent zeolite and core–shell catalysts were tested for n-hexane cracking in a fixed bed reactor at 550 °C (WHSV = 3 h–1). The authors found that the H-ZSM5 zeolite showed the highest initial conversion of hexane (99.8%), as well as the highest selectivity toward light alkanes and BTX (62.9% and 12.5%, respectively). On the contrary, core–shell catalysts exhibited the highest selectivity toward light olefins, with a slight decrease in hexane conversion. A passivated sample with an estimated 150 nm silicalite-1 shell (ZS@S(2.0)) exhibited a conversion value of 91.6% and a light olefin selectivity of 58.2%, significantly overcoming the performance of the parent zeolite (20.3%). External strong acid sites of the H-ZSM5 zeolite led to the conversion of hexane to aromatics and the aromatization of light olefins and alkanes through oligomerization, cyclization, aromatization, and hydrogen transfer reactions. On the contrary, the decrease in external strong acid sites led to a suppression of the aromatization reaction of hexane, resulting in decreased BTX selectivity and consequently increased selectivity towards olefins. Additionally, the passivation process prevented the formed olefins from undergoing consecutive and undesired reactions. Moreover, to prove the effectiveness of the deposition of silicalite-1 over the external surface of the parent zeolite, the authors tested a mechanical mixture of silicalite-1 and ZSM-5 (Z5/S) with similar total acidity. FT-IR spectra collected after the adsorption of 2,4,6-collidine demonstrated that although a similar acidity was measured by NH3-TPD and pyridine FT-IR analysis, peaks related to external Brønsted and Lewis acid sites were higher in the case of the mechanical mixture. Furthermore, the mechanical mixture was employed in hexane cracking to obtain a conversion of 91.6%, which was equal to that exhibited by the core–shell sample with the same total acidity (ZS@S(2.0)). On the contrary, using the Z5/S sample, a light olefin selectivity of 43.1% was reached, lower than that obtained when the core–shell catalyst was tested (48.2%). Although the mechanical mixture made it possible to lower the selectivity toward BTX compared to solely ZSM-5 (4.3% and 12.5% respectively), the lowest value of selectivity is always obtained using the passivated sample (2.4%). Comparisons among catalytic results of the parent zeolite, the mechanical mixture (Z5/S), and core–shell samples are summarized in Figures 4 and 5.
Figure 4.

n-Hexane conversion and product selectivity for parent zeolite and passivated samples. Reprinted with the permission from ref (11). Copyright 2023 John Wiley and Sons.11
Figure 5.

n-Hexane conversion and product selectivity for core–shell Z5@S(2.0) and a mechanical mixture (Z5/S). Reprinted with the permission from ref (11). Copyright 2023 John Wiley and Sons.11
The authors employed a TOS test of about 80 h to study the stability of the parent zeolite and one of the synthesized core–shell samples. Employing the parent zeolite, hexane conversion dropped from a value of 99% to a value of about 90%. The core–shell sample exhibited a lower conversion value, because of its lower acidity, but a more stable trend with an initial conversion value of 91.5% and a decrease, after 80 h of reaction, of 2.8%. An analysis of the stability of the mechanical mixture during the TOS test would have been useful to fully understand whether the lower deactivation found with the passivated catalyst was linked only to the success of the passivation treatment or if there was an effect from the lower acidity.
Catalytic fast pyrolysis (CFP) is a promising method for converting renewable biomass into liquid fuels and chemicals, and H-ZSM-5 is usually investigated as catalyst to produce BTX. While zeolites, especially H-ZSM-5, are efficient for the deoxygenation of fast pyrolysis vapors, facilitating hydrocarbon production, the formation of coke, particularly on the external surface, quickly deactivates the catalysts. To address this issue, Hu et al. have explored core–shell properties in catalytic pyrolysis to improve catalyst stability.12 The authors synthesized two core–shell catalysts using the hydrothermal synthesis method, coating a commercial H-ZSM-5 zeolite with one layer (sample named CS1) or two layers of silicalite-1 (sample called CS2). SEM micrographs of the parent zeolite and the CS2 sample (Figure 6) showed an incomplete coating of silicalite-1.
Figure 6.

EM images of (a, b) HZSM-5 and (c, d) CS2 samples, where the sporadic silicalite-1 crystals (C), the incomplete silicalite-1 layer (B), and the layer incompletely covered by layer B (A) are indicated. Reprinted with the permission from ref (12). Copyright 2018 Elsevier.12
Further passivation cycles could likely lead to the generation of a homogeneous layer of silicalite-1. To evaluate the effect silicalite-1 on both external and internal acidity, two reactions were studied: cracking of 1,3,5-triisopropylbenzene (TIPB) and the ketonization reaction of acetic acid, with this molecule being small enough to enter the pore mouth of the MFI-type zeolite.
Results are shown in Figure 7 and reveal that TIPB conversion decreased when core–shell catalysts were tested: while the H-ZSM-5 parent zeolite exhibited a conversion of 67.1%, employing CS1 and CS2 led to conversions of 38.7% and 14.3%, respectively. These results confirm that while the growth of silicalite-1 effectively reduces external strong acidity, homogeneous and complete coverage was not attained with two cycles of passivation. Moreover, H-ZSM-5, CS1, and CS2 samples showed almost unchanged conversion during the acetic acid ketonization reaction, confirming that the internal acidic properties of the catalyst were preserved. To perform catalytic fast pyrolysis of biomass, the authors used a bench-scale continuous feeding two-stage fluidized-bed reactor, where the fast pyrolysis of biomass was carried out, and a fixed-bed reactor for the catalytic conversion of pyrolysis vapors with a WHSV of 2 h –1. No significant effect of the passivation on the investigated reaction was found, and the results probably do not justify the effort made to synthesize the samples. Using passivated samples, the results demonstrated only a slight increase in BTX carbon yield as the level of silicalite-1 coating increased. In particular, the CS2 sample exhibited a BTX carbon yield of 9.9% while the parent zeolite showed a value of 8.5%. The small increase in BTX carbon yield was accompanied by a decrease in coke carbon yield of 22.1% when the passivated sample was adopted. In order to study the long-term catalytic performance of catalysts, the authors performed six cyclic experiments using spent samples without regeneration, and results are reported in Figure 8.
Figure 7.

1,3,5-TIPB and acetic acid conversion over untreated zeolite and passivated samples. Reprinted with the permission from ref (12). Copyright 2018 Elsevier.12
Figure 8.

Product distributions over untreated HZSM-5 and CS2 as a function of catalyst cycle number: carbon yields of BTX, olefins, and BTX + olefins. Reprinted with the permission from ref (12). Copyright 2018 Elsevier.12
Although the CS2 sample showed a carbon yield of BTX and olefins higher than that of the parent zeolite, a decrease in this value after each testing cycle was also detected. This phenomenon might be attributed in part to the inhomogeneous coverage of the silicalite-1 layer, which may result in the presence of some unpassivated external strong acid sites.
Goodarzi et al.13 proposed a straightforward solid-state steam-assisted technique aimed at producing a nonacidic ultrathin layer of silicalite-1 over mesoporous ZSM-5 crystals. The technique consists of a pulverizing step of the as-synthesized mesoporous ZSM5 in a mortar, followed by drying overnight. Then, sequential impregnation with TPAOH and TEOS was performed in a Teflon beaker, followed by drying and heating in a Teflon-lined stainless-steel autoclave with distilled water to generate steam at 180 °C for 72 h. The authors used different characterization techniques to investigate the effect of silicalite-1 coating on the properties of the starting mesoporous ZSM-5. Even if the external strong acidity was not even quantified, XPS analysis results confirmed the growth of the layer of silicalite-1 over the ZSM-5 core, as a drop in the peak at 76.7 eV, related to the presence of Al on the surface of the sample, was detected. Conventional ZSM-5, mesoporous ZSM-5, and core–shell samples with a mesoporous core were tested in the methanol-to-hydrocarbons (MTH) reaction at 400 °C and 4 bar with a WHSV of 4.7 gMeOH/(gcat·h). Results in terms of methanol conversion during TOS tests are shown in Figure 9.
Figure 9.

Methanol conversion over Conv-ZSM-5, Meso-ZSM-5, and Shell-Meso-ZSM-5 catalysts as a function of time. Reprinted with the permission from ref (13). Copyright 2020 Elsevier.13
It appeared clear that passivation treatment improved catalyst stability, as conventional microporous ZSM-5 deactivated after 10 h of reaction, while the core–shell sample (Shell-Meso-ZSM-5) deactivated much more slowly and significant methanol conversion was still observed after 70 h of reaction. TGA analysis was employed to quantify the coke deposited over the surface of spent catalysts, and GC-MS analysis was used to analyze soluble coke species. The quantity of deposited coke was about 5% for conventional ZSM-5, 8.2% for mesoporous ZSM-5, and 7% for the core–shell sample, although the amount should be assessed on the basis of total converted methanol during the test. The parent zeolite (Conv-ZSM-5) displayed the lowest coke deposition but was deactivated after only 10 h, likely due to rapid pore blockage by coke formed on its external surface. This hypothesis was confirmed by GC-MS analyses (Figure 10).
Figure 10.

GC-MS spectra of spent catalysts. Reprinted with the permission from ref (13). Copyright 2020 Elsevier.13
In fact, among soluble coke species, heavier compounds have also been detected for Conv-ZSM-5, such as naphthalene and anthracene derivatives, while only polymethylbenzenes were found for the other spent samples. Conversely, GC-MS analysis of both the mesoporous catalyst and the core–shell variant revealed identical compounds and comparable total areas. This means that the mesoporous sample tends to form heavier, nonsoluble coke species in a higher amount than the core–shell sample, also deactivating much more quickly.
Besides silicalite-1, other aluminum-free zeolitic shells have been employed to passivate zeolites other than ZSM-5. In a recently published work, Rimer and co-workers14 proposed a new method to synthesize a Si-rich FER shell over the surface of an Al-FER core with the aim of passivating external strong acid sites. In particular, a FER core with a Si/Al = 10 was prepared through a hydrothermal synthesis at 165 °C for 2 days by using cyclohexylamine as an organic structure directing agent. After washing, core crystals were stored in distilled water to prevent them from drying out. Then, the solution of Si–FER–shell was prepared and aged for 1 day at room temperature before adding FER-core crystals; the hydrothermal synthesis was then conducted at 120 °C for 2 days. Characterizations proved the growth of a Si-rich shell; XRD patterns of the core–shell sample revealed the presence of a well-defined peak at 2θ = 9.7°, corresponding to the (200) plane of FER crystals that represents the nonporous basal plane of FER crystals. This result suggests that the shell growth is preferentially oriented along the (200) plane. XPS analysis indicated the increase of the external Si/Al ratio for the passivated sample, confirming the growth of a Si-rich shell. Moreover, the benchmark reaction of Friedel–Crafts alkylation of mesitylene and benzyl alcohol has been performed in liquid phase at 100 °C and used to assess the quality of the passivation procedure.
Results, as shown in Figure 11, highlighted distinctions between the original FER zeolite and the core–shell structure. Passivated catalysts showed a 90% decrease in benzyl alcohol conversion compared to the core sample.
Figure 11.
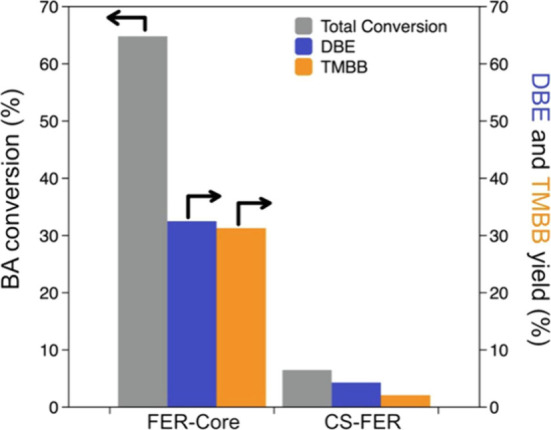
Benzyl alcohol conversion and yields of 1,3,5-trimethyl-2-benzylbenzene (TMBB) and dibenzyl ether (DBE) obtained testing parent zeolite (FER-Core) and core–shell sample (CS-FER): the BA conversion (gray) and yields of products DBE (blue) and TMBB (orange) from H-form FER-Core and CS-FER catalysts. Reprinted with the permission from ref (14). Copyright 2023 Elsevier.14
It should be clarified that the formation of an aluminum-free zeolitic layer on the external surface of an initial zeolite is a complex procedure. Indeed, achieving zeolitic structures with an infinite Si/Al ratio is not universally feasible, and coupling different zeolitic frameworks can present challenges. Regarding this topic, an interesting work has been developed by Valtchev and co-workers, who conducted research on core–shell zeolite–zeolite composites through a two-step method.15 Initially, presynthesized seeds of the zeolitic phase to be formed as the shell were deposited onto the surface of cores pretreated with a polycation agent. Then, the hydrothermal synthesis of the shell was performed. Different zeolitic structures were employed to investigate the influence of framework composition and synthesis conditions on the production of these microcomposites. The study included different combinations: pure-silica sodalite@zeolite A, β@zeolite A, zeolite X@silicalite-1, ZSM-5@zeolite-β, and ZSM-5@silicalite-1. The authors found that successful formation of core–shell structures occurred when materials had a compatible zeolite framework and closely aligned crystallization conditions. A wider overlap between crystallization regions promoted composite formation, while less overlap indicated difficulty in achieving core–shell structures. The formation of a FAU@MFI core–shell structure failed due to insufficient overlap between compositions and synthesis conditions, e.g., overlapping of crystallizing fields on ternary phase diagrams of mentioned zeolites. Conversely, close similarity in framework compositions and synthesis conditions of zeolite-β and ZSM-5 facilitated easier formation of core–shell materials.
2.2. Chemical Neutralization
As reported in the previous paragraph, epitaxial growth of pure siliceous zeolite layers over an acidic material is an effective method to reduce the number of strong acid sites located on the external surface of the crystals, with an effect on the catalysis. Nevertheless, the synthesis of core–shell systems requires effort for the optimization of the synthesis procedure in order to obtain an acidic core effectively and homogeneously covered by a nonacidic shell, as described above. The inactivation of external strong acid sites by neutralization reactions with specific chemicals is also usually adopted as a postsynthesis strategy to passivate the external surfaces of crystals.
Valtchev and co-workers16 passivated the surface of nanosized ZSM-5 by the chemical liquid deposition (CLD) method using tetraethylortosilicate (TEOS) to assess the impact of surface modification by silylation on the light olefin production via methanol conversion. In order to control the concentration of acid sites on the external surface, the authors carried out three cycles of passivation. The surface passivation influenced the Si/Al ratio measured by XRF analysis, as an increase in the Si/Al ratio after passivation was observed, and the amount of deposited silica coating was estimated to be 0.33 and 0.85 wt % after the first and the third passivation step, respectively. Although the adopted passivation procedure has a negative effect on both surface area and micropore volume, a significant decrease in the external strong acidity was measured by FT-IR after collidine titration. In particular, the collidine uptake drops from 8 to 0.006 μmol/g after three consecutive CLD cycles. At the same time, for up to two consecutive passivation steps, no significant change in the concentration of Brønsted and Lewis acid sites was observed (estimated by pyridine titration), while both Brønsted acid sites and the pyridine accessibility index noticeably decrease after a third passivation cycle. The impact of passivation on catalysis was well-described by the authors, revealing a trade-off between the reduction of external acid sites and textural modification. In fact, the selectivity for light olefins was significantly improved by reducing the external acid sites, while the passivation negatively affects the stability due to coke deposition that preferably occurs inside the zeolite pores in the case of passivated zeolites.
The chemical liquid deposition of TEOS not only impacts on the external strong acidity, and in this regard Liu and co-workers17 evidenced the importance of controlling the surface barriers in mass transfer in SAPO-34 zeolites to modulate the methanol-to-olefin reaction. The authors modified the external surface of homemade SAPO-34 by CLD of TEOS, claiming that the decoration with silica mainly occurs on the external surface, as TEOS hardly diffuses inside the channels of the investigated material, allowing the production of a material with a more pronounced surface barrier. Furthermore, the authors also treated the starting material with an acetic acid solution that removed the structural defects reducing the surface barrier. XRD, SEM, N2 adsorption–desorption isotherms, and reflectance FT-IR measurements revealed that the treatments do not affect the crystallinity, morphology, or textural and acidic properties of the SAPO-34 crystals. Therefore, the authors focused on the effect of the surface modification to lower surface barrier and increased intracrystalline diffusion of propane and methanol by a dual resistance model. For both methanol and propane, a similar intracrystalline diffusivity over the investigated samples was calculated, while an evident effect of treatment on apparent surface permeability was observed. In particular, the surface permeability was the lowest for the CLD-treated sample and the highest for the acid acid-treatment sample, while the untreated sample shows an intermediate behavior. The authors used transport phenomena information to explain the effect of surface barrier modification on catalytic performances during the methanol-to-olefin (MTO) reaction.
As reported in Figure 12, the control of surface barriers is a paramount aspect in catalysis with zeolites. In fact, when treated with acetic acid, the SAPO-34 sample kept a methanol conversion higher than 95% for 69 min, while in the case of TEOS CLD the methanol conversion dropped after 19 min only. On the basis this evidence, the differences in terms of stability were mainly related to a change on the surface barrier, as the interior structure of the SAPO-34 material was almost unaffected by the treatment. The authors positively associated the stability with the surface permeability even with a linear trend. The increase in surface barriers induced by CLD of TEOS hinders the diffusion of light olefins, which are further converted into heavier hydrocarbons with consequent selectivity loss. In contrast, in the case of reduced surface barriers, light olefins (ethene and propene) can diffuse out of the crystals with no consecutive transformation, reaching a selectivity higher than 80% in the case of SAPO-34 treated with acetic acid, indicating that the surface barriers can be affected by chemical treatment with a significant effect on catalysis.
Figure 12.

(a) Methanol conversion and (b) ethylene/propene selectivity as a function of time on stream (TOD) in a MTO reaction over untreated SAPO-34 samples (SAPO-34-B) and samples treated with TEOS CLD (SAPO-34-L) and acetic acid etching (SAPO-34-H). Reaction condition: 450 °C, methanol WHSV = 5.0 h–1. Reprinted with the permission from ref (17). Copyright 2020 Jhonson Wiley and Sons.17
In a recent study, Wu et al.18 investigated the effect of chemical treatment of ZSM-22 zeolite on the external acidity and then on catalysis of the 1-butene skeletal isomerization reaction. The authors combined alkali treatment (NaOH solution at different molar concentrations), acidic treatment (HCl solution), and CLD of TEOS in order to tune the external acidity and to study its impact on catalysis. The treatment with a strong NaOH solution causes crystallinity loss and modifications of the crystal morphology, indicating that desilication occurs to generate mesopores. Furthermore, alkali treatment promotes the migration of aluminum atoms from the framework of zeolite, causing a decrease in the concentration of Brønsted acid sites and the growth of Lewis acid sites due to the extra-framework aluminum species. In order to assess the role of the latter type of acid sites, the alkali-treated ZSM-22 samples were washed with a HCl solution to promote dealumination and the elimination of extra-framework species, like Al(OH)3 or AlOOH. Surprisingly, the authors observed that the crystallinity of samples subjected to a severe alkali treatment was recovered after acid washing, along with a partial restoration of both micropore volume and Brønsted acidity. The modification of external acidity has a direct impact on catalysis, as alkali-treated samples show a higher isobutene yield and selectivity with respect to acid-washed sample. Furthermore, in the case of samples with high external aluminum concentrations, coke deposits were formed mainly on the external surface. However, the best results in terms of isobutene yield and selectivity were obtained when the surface acidity was properly passivated by CLD, indicating that the enhancement of the catalytic properties is related to the monomolecular reaction of 1-butene over external diluted acid sites.
Cao et al.19 emphasized the effect of CLD of TEOS on the textural properties of MOR, indicating that this method could shrink and block the microporous channels of the zeolite. For comparison, the authors also used 3,5-dimethylphenylmagnesium bromide (DPB) as a bulk precursor to passivate the external surface of the crystals with MgO, claiming that both the passivating molecules, i.e., TEOS and DPB, are not able to access the 12 MR channels of MOR. XRD measurements indicated that the passivation with either TEOS or DPB does not impact on the crystallinity, and no further phases were detected after the treatment. On the contrary, N2 adsorption isotherms at −196 °C clearly demonstrated the negative impact of TEOS CLD on both the micropore volume and the BET surface area, which drop from 0.180 to 0.162 cm3/g and from 482 to 458 m2/g, respectively, with respect to parent material when 1% of SiO2 is deposited. No loss of textural features was observed when DPB was used for passivation with 4% MgO instead. When the acidity of treated samples is investigated, the impact of passivation on the total and external acidity is clearly observed by FT-IR study of pyridine and 2,6-di-tert-butylpyridine (DTBPy) desorption, respectively. Despite the fact that both molecules are effective toward the reduction of external strong acid sites, pyridine FT-IR revealed that TEOS deposition causes Brønsted acidity loss, while DPB does not affect the distribution of acid sites of the zeolite. The authors also adopted micro/mesohierarchical mordenite to obtain insights about the role of mesopores on the passivation impact, observing that a higher amount of Brønsted acid sites are deactivated as the diffusion of passivating molecules in the crystals is enhanced. The investigated materials were tested as catalysts in the isomerization of o-ethyltoluene to produce m- and p-ethyltoluene, and o-ethyltoluene conversion and m- and p-ethyltoluene selectivity as a function of time on stream are reported in Figure 13.
Figure 13.

Influence of time on stream (TOS) on the catalytic yields of M-ET and P-ET over HM, HM–M, and their modified zeolites. The maximum standard deviation of each data point was <2.0%. (a) HM and modified HM samples. (b) HM–M and modified HM–M samples. Reprinted with the permission from ref (19). Copyright 2011 Royal Society of Chemistry.19
The authors commented that the passivation of external surface of microporous mordenite improves the selectivity toward the desired products, which increases from 67.27% to 77.54% in the case of DPB, also enhancing the catalyst stability. On the contrary, due to the passivation of acid sites located in the mesopores, the treatment with wither DPB or TEOS over hierarchical mordenite is not effective. Globally, the passivation with DPB led to the best results because this molecule also prevented pore blockage during the treatment, which cannot be avoided in the case of TEOS. The external surface can be effectively passivated also by using phosphoric species.
The negative impact of silylation by CLD is also reported in a study from Xu et al.20 The authors focused on controlling the external acidity of a Ga/HZSM-5 catalyst in order to inhibit the cracked byproducts formation, i.e., methane and ethylene, formed during the propane dehydroaromatization reaction. The authors studied the effect of passivation over the Ga/HZSM-5 material using a commercial HZSM-5 impregnated with 1 wt % Ga and by carrying out CLD with TEOS, obtaining catalysts with 2, 4, 6, and 8 wt % SiO2 deposited on the external surface. Also, in that case, XRD analyses reveal no effect on the crystal structure of the zeolites, although amorphous silica was deposited by CLD, which led to a higher Si/Ga molar ratio with a higher amount of deposited silica. The N2 adsorption/desorption isotherms showed a surface area loss for the passivated samples with up to 4 wt % SiO2, while new surface area can be generated with higher silica loading. The acidity assessment by NH3-TPD indicated that passivation led to a further weakening of weak acid sites (with no concentration change) and to a reduction in the number of strong acid sites with no impact of their strength, especially at low silica loading. Pyridine FT-IR also confirmed that both strong Brønsted and Lewis are selectively deactivated by silylation. At high silica loadings, a significant loss of acidity was observed as confirmation of pore mouth narrowing that suppressed the diffusion of the probe molecule.
The catalytic results (Figure 14) confirmed that the presence of gallium is of paramount importance to promote both propane conversion and BTX selectivity. In fact, the Ga-doped ZSM-5 showed a propane conversion higher than 40% and a BTX selectivity higher than 45%, while a propane conversion lower than 25% and a BTX selectivity lower than 10% were observed for the Ga-free ZSM-5 sample. The authors observed that the effect of silylation on catalysis depends on the silica loading. The sample passivated with 2 wt % silica showed a catalytic behavior similar to the nonpassivated sample, indicating that a more intensive CLD is needed to appreciate its effect in catalysis. The deposition of 4 wt % silica indeed significantly improves the selectivity that increased from 49% to about 54% after passivation due to an inhibition of the formation of cracked products whose selectivity reduced from 33% to 30%. On the other hand, higher silica loading had a negative effect on both conversion and BTX selectivity, while the formation of propene was significantly favored. Furthermore, stability was also compromised due to pore mouth narrowing that hinders aromatics diffusion, causing deactivation.
Figure 14.

(a) Propane conversion and (b) BTX selectivity as a function of time on stream for HZSM-5, Ga/HZSM-5 (Ga/Z5) and silylated samples with different TEOS amounts, i.e., 2wt % (Ga/Z5–2Si), 4 wt % (Ga/Z5–4Si), 6 wt % (Ga/Z5–6Si) and 8 wt % (Ga/Z5–8Si); The selectivity of (c) cracking products (methane and ethane) and (d) propene at 4.5 h. Reaction conditions: atmospheric pressure, 540 °C, WHSV = 6000 mL/(g·h), and N2/C3H8 = 2. Reprinted with the permission from ref (20). Copyright 2021 Elsevier.20
Yi et al.21 showed that reversing the electric charge of the external surface by impregnation with phosphoric acid to obtain a positive charged surface inhibits the formation of m-xylene during the methylation of toluene with ZSM-5. Both the crystallinity and phase purity of the modified samples were verified by XRD, and the textural properties were retained in the case of samples previously treated with HDTMA, indicating that the surface charge reversal promotes the phosphorus deposition with no pore blockage, as the pores are protected by HDTMA. Furthermore, solid-state 27Al- and 31P NMR analyses revealed that in the case of HDTMA-treated samples phosphorus exists as H4P2O7 or polyphosphates with no interaction with zeolite aluminum species. The effect of passivation on acidity was assessed by both pyridine and collidine FT-IR for their different size. The former is suitable for estimating total acidity, and the latter is suggested for quantifying external acid sites of the MFI-type zeolite. The external surface acidity of the parent sample was 38.1 μmolCollidine/g, and it decreased to 16.3, 6.5, 1.2, and 0.7 μmolCollidine/g when the surface of HDTMA-treated ZSM-5 was passivated with 0.5, 1.0, 1.5, and 2.0 wt % phosphorus, respectively. It is noteworthy that without HDTMA-based pretreatment the sample impregnated with 1.5 wt % phosphorus retains an external acid site concentration of 14.7 μmolCollidine/g, indicating that the control of surface charge is of paramount importance during impregnation or chemical neutralization to uniformly passivate the external surface, with a significant effect on catalysis by limiting the pore blocking. The toluene methylation tests (reaction temperature of 200 °C, toluene/methanol molar ratio of 2/1, WHSV = 2.0 h–1) on the investigated ZSM-5 samples showed an important impact of the passivation procedure on toluene conversion and the distribution of xylenes, as reported in Figure 15.
Figure 15.

Effects of the phosphorus content on the catalytic behavior of HZSM-5 and phosphorus-modified samples during toluene methylation: (a) HZSM-5, (b) MZS-0.5P, (c) MZS-1.0P, (d) MZS-1.5P, (e) MZS-2.0P, and (f) HZSM-5–1.5P. (A) Toluene conversion. (B) Selectivity of p-xylene. (C) Selectivity of xylene. (D) Yield of xylene and p-xylene. Reprinted with permission from ref (21). Copyright 2019 American Chemical Society.21
The untreated ZSM-5 sample exhibited the highest toluene conversion (about 30%) and the lowest p-xylene selectivity (about 30%). The passivated samples showed a significant improvement in terms of p-xylene selectivity (higher than 97.6%) in the case of the sample pretreated with HDTMA and impregnated with 1.5 wt % phosphorus (sample MZS-1.5P), with a small reduction of toluene conversion (27%). Catalytic results also confirmed the key role played by the passivation procedure, since the sample with no HDTMA treatment showed a lower p-xylene selectivity. As general comment about the proposed passivation treatment, it improved the p-xylene yield from 7–8% of untreated sample to about 25% for samples treated with 1.0–2.0 wt % phosphorus after surface charge reversal with HDTMA. Also, the samples showed a p-xylene yield higher than the P-loaded sample obtained without HDTMA, which exhibited a p-xylene yield of about 17–18%.
Surface passivation can be also employed to modify the external hydrophobicity of zeolites, where this aspect is important for catalysis. Lercher and co-workers22 obtained a surface-hydrophobized BEA zeolite by CLD with a bulky silane, octadecyltrichlorosilane (C18–SiCl3), which is not able to enter the pore of BEA crystals and remains anchored on the external surface of the zeolite. The use of this chemically passivating bulky molecule did not cause a significant loss of microporosity and significantly improved the catalytic behavior of the BEA zeolite during cyclohexanol dehydration. In particular, the silylation increased from 5 to 15 h and the apparent turnover number (TON) of silylated BEA was eight times higher than the TON measured on the parent BEA.
The use of bulky passivation species was also investigated by Fujiwara et al.23 by adopting a bulky disilane, namely 1,4-bis(hydroxydimethylsilyl)benzene, in order to enhance the catalytic performances of H-ZSM-5 during the dimethyl ether-to-olefin process. Different disilane contents up to 15 wt % were adopted. The modification did not alter the initial conversion of DME that was 100% at 1 h of TOS. In the case of the unmodified zeolite, the ethylene and propylene yields were 3.95 and 6.05 mol %, respectively, with propane as the major C3 component; a selectivity toward aromatics of about 37.5 mol % was also observed. When the H-ZSM-5 sample was modified with 10 wt % disilane, the yield toward propylene increased significantly to 13.25 mol %, also becoming the major C3 component, and significant yields of ethylene and C4 olefins were also observed after silylation. Higher silylation levels, i.e., 15 wt % disilane, caused a loss of catalytic performance in terms of propylene yield and selectivity, indicating that the modulation of the silylation degree is necessary to obtain catalytic enhancements. The authors studied the effect of crystals external surface on the stability of the catalysts, calcining H-ZSM-5 modified with disilane.
Results reported in Figure 16 indicate that both H-ZSM-5 and modified H-ZSM-5 show a similar deactivation trend, exhibiting complete DME conversion for about 6 h, with a significant reduction of the yields of aromatics when deactivation occurs. When the modified zeolite was calcined and the disilane compound decomposed to form silicon oxide species, the catalytic behavior changed completely. In particular, a rapid deactivation due to coke deposition was observed in the case of the calcined sample with an increase in p-xylene selectivity, indicating that calcination narrowed the pore mouth of H-ZSM-5, and this accelerated the coke deposition. This is a further indication that the control of the silylation procedure is a crucial aspect to obtain a catalyst suitable for a specific catalytic application.
Figure 16.

Time on stream tests of DME-to-olefin reactions over H-ZSM-5 (Z0), H-ZSM-5 modified with disilane (Z10), and calcined modified H-ZSM-5 (Z10-C). Reprinted with the permission from ref (23). Copyright 2019 Elsevier.23
3. Discussion and Future Outlook
This Mini-Review aims to conduct a brief but detailed analysis of recent publications focusing on the surface passivation of zeolites. External strong acidity often represents a disadvantage in reactions where the shape-selectivity of zeolite channels is crucial. Unlike internal acid sites, external ones lack this property, resulting in undesired reactions, the formation of secondary products, and the accumulation of coke precursors on the surface. These issues stimulated researchers to develop synthesis and postsynthesis techniques aimed at adjusting the acidity of zeolites and neutralizing external surface strong acid sites. The focus of this review revolved around identifying the prevalent techniques and critically analyzing them by examining the characterization methods employed to confirm surface passivation. Moreover, the focus was on understanding the effects of neutralizing the crystal’s external surface strong acid sites on catalytic behavior. As an example, in the literature, many reviews are published on the synthesis of core–shell catalysts, but they tend to concentrate primarily on the synthesis methods used rather than exploring the catalytic applications and the advantages and disadvantages of employing such composite materials.
All the results recently reported underscored a fundamental need to quantify the external acid sites of catalyst samples prior to contemplating the adoption of any passivation methodology. This preliminary step is fundamental, as it gives invaluable information regarding the abundance and nature of acidic functional groups located on the external surface of the zeolite crystals. Quantitative data of the strength of catalyst external surface acid sites, and a comprehensive understanding of their influence on specific catalytic performance, can define the most suitable passivation procedure. The direct characterization of the catalyst external acid sites can be conducted through Fourier transform infrared (FT-IR) spectroscopy analysis utilizing probe molecules with dimensions exceeding the pore size of the zeolite. This approach enables the identification and quantification of solely the acidic sites present on the external surface of the sample. In many of the analyzed works, di-tert-butyl-pyridine and collidine adsorption have emerged as prevalent techniques for quantifying external acidic sites. Both methods and strategies for the proper characterization of acid sites of zeolites are deeply discussed in relevant papers from Guido Busca and co-workers.24
In addition to direct characterization methods, researchers often adopt indirect approaches to assess the external acidity of zeolites. One common indirect method utilizes reactions that involve reactants with dimensions exceeding those of the zeolite pores. While these reactions may not directly measure the strength of the external surface acid sites of zeolites, they provide valuable insights about their accessibility and reactivity. For example, the cracking of 1,3,5-triisopropylbenzene and the alkylation of mesitylene and benzyl alcohol have been employed to evaluate the external acidity of MFI and FER zeolites.
Recent literature revealed a large variety of methods that have been investigated to target the neutralization of external acid sites. These include the production of core–shell catalysts characterized by an aluminum-free siliceous shell, as well as techniques like chemical liquid deposition of tetraethylorthosilicate (TEOS). Additionally, various post-synthesis treatments have been proposed to further enhance catalytic performance, such as the reversing of the electric charge of the external surface with hexadecyltrimethylammonium (HDTMA), followed by impregnation of phosphoric acid or the chemical liquid deposition of various bulky silanes.
Selecting the appropriate technique for surface passivation involves considering several factors, including the influence of the neutralization treatment on catalyst properties, the degree of complexity of the technique, and its industrial applicability. For instance, in the case of chemical vapor deposition (CVD) of TEOS, a common phenomenon is the decrease of specific surface area and micropore volume. This decreases arises from the deposition of an amorphous layer onto the pore mouth during the process. The decrease in micropore volume may likewise affect the catalytic performance of the catalyst, making the effort to passivate external acid sites ineffective.
This issue has been elucidated in several studies, including that of Cao et al.,19 wherein they showed that passivating a MOR zeolite via chemical vapor deposition of TEOS resulted in pore blockage, leading to accelerated deactivation during the isomerization reaction of o-ethyltoluene. Other studies, such as the one analyzed in the review by Xu et al.,20 have demonstrated the importance of controlling the silylation level and finding a volcano-plot for the amount of SiO2 to be deposited. An excess amount of coated SiO2, causing a narrowing of the pores, leads to a worsening of the catalytic activity.
On the contrary, the epitaxial growth method with silicalite-1 successfully mitigated the reduction of the microporosity while still efficiently passivating external surface strong acidity. Moreover, some papers suggest that growing a siliceous shell on an Al-containing core with the organic template still preserves the original catalyst’s textural properties.25 Simultaneously, it is essential to address additional factors regarding the epitaxial growth of silicalite-1. In some already discussed works, the growth of the external layer may exhibit nonuniformity due to hindered diffusion of active species (different polysilicates dispersed withinthe reaction mixture) to the growing crystal’s surface. If the whole external surface of catalyst is not covered by the shell (e.g., silicalite-1), it is not passivated and acts as mechanical mixture of the original catalyst and a silicon-rich/aluminum-free one.
Several points for reflection have arisen from the analysis of the catalytic performance of passivated catalysts. In certain catalytic applications, such as toluene alkylation, strong external surface acidity plays a key role. Research conducted by Pan and co-workers9 has demonstrated that coating a ZSM-5 zeolite with a layer of silicalite-1 substantially enhances the catalyst’s performance in terms of selectivity toward the desired product and stability even with a relatively small amount of shell deposition. Notably, the coating not only neutralizes external acidity but also extends the diffusion time of the reactants involved in the reaction by enlarging the crystal size. However, passivation can sometimes yield negative effects on catalytic activity, as demonstrated in the findings reported by Liu and co-workers.17 Their study focused on the chemical vapor deposition of TEOS over a SAPO-34 zeolite tested for the MTO reaction. In this case, passivation resulted in the increase of external barriers of the initial sample, hindering the diffusion of light olefins. Consequently, these olefins were converted into heavier hydrocarbons, leading to accelerated catalyst deactivation and a decline in selectivity toward the desired products. Conversely, the epitaxial growth of silicalite-1 on ZSM-5 through steam-assisted crystallization, as investigated by Goodarzi et al.,13 has unequivocally demonstrated its efficacy in enhancing the stability of the catalyst employed for methanol-to-olefin conversion.
Alternative techniques, other than the conventional methods employed for external acidity passivation, have demonstrated effectiveness. The study conducted by Yi et al.21 explores the passivation of a ZSM-5 catalyst through a surfactant-assisted reversing of electric charge of the external surface, specifically utilizing hexadecyltrimethylammonium, followed by phosphoric acid impregnation. This approach has been effective in enhancing selectivity toward para-xylene in toluene alkylation reactions.
In conclusion, this Mini-Review comprehensively examines recent studies focusing on the external surface passivation of zeolites to mitigate the challenges induced by its strong acidity as well as the use of catalytic test to prove the effectiveness of such techniques. The review emphasizes the importance of a preliminary assessment of the strength of external surface acid sites, before selecting appropriate passivation techniques, based on a deep evaluation of demands for a specific, targeted application (catalytic reaction). Moving forward, continued research into surface passivation techniques will be crucial for advancing zeolite-based catalysis and addressing challenges in various industrial demands, as well as scientific curiosity.
Acknowledgments
The authors gratefully acknowledge the following for cofounding the work:
Next Generation EU-Italian NRRP, Mission 4, Component 2, Investment 1.5, call for the creation and strengthening of “Innovation Ecosystems”, building “Territorial R&D Leaders” (D.D. no. 2021/3277), project “Tech4You - Technologies for climate change adaptation and quality of life improvement”, Grant ECS0000009
Italian Ministry of University and Research, program PRIN 2022 PNRR founded by Next Generation EU-Italian NRRP, Mission 4, Component 2, Investment 1.1, Grant P2022KA5ZZ, CUP: H53D23008590001
Biographies

Giorgia Ferrarelli obtained her Ph.D. in “Advanced Catalytic Processes for Utilizing Renewable Energy Sources” at the University of Messina in 2024, and she is currently a postdoctoral researcher at the University of Calabria. Her research field is the synthesis and characterization of zeolitic catalysts with a focus on core–shell materials with applications in catalysis. Specifically, she is dedicated to developing innovative core–shell catalysts to neutralize external acidity and applying them in defossilization strategies and CO2 conversion processes.

Massimo Migliori is currently Full Professor in “Industrial and Technological Chemistry” at the University of Calabria. He is active in the field of chemical industrial processes, with particular attention paid to the synthesis, characterization, and application of nanostructured materials in sustainable processes oriented toward defossilization and “Green Chemistry”. He is currently the President of the Italian Zeolite Association and member of the “Synthesis Commission” of the International Zeolite Association (IZA).

Enrico Catizzone is currently Assistant Professor in Chemical Plants at the University of Calabria. In 2013 he graduated with a degree in Chemical Engineering, and in 2017 he obtained a Ph.D. in ‘‘Science and Engineering of Environment, Buildings and Energy” at the University of Calabria. His scientific activity is mainly devoted to heterogeneous catalysis, with a main focus on the development of novel catalytic nanostructured systems with applications in the conversion of biomass/waste-derived CO2-rich gas for the production of chemicals and liquid fuels.
The authors declare no competing financial interest.
Dedication
This work was inspired by Prof. Girolamo Giordano and is dedicated to him for his tireless scientific support to his research group and to the zeolite science and technology community.
References
- Bellussi G.; Pollesel P. Industrial Applications of Zeolite Catalysis: Production and Uses of Light Olefins. Stud. Surf. Sci. Catal. 2005, 158, 1201–1212. 10.1016/S0167-2991(05)80466-5. [DOI] [Google Scholar]
- Lakiss L.; Ngoye F.; Canaff C.; Laforge S.; Pouilloux Y.; Qin Z.; Tarighi M.; Thomas K.; Valtchev V.; Vicente A.; Pinard L.; Gilson J.-P.; Fernandez C. On the Remarkable Resistance to Coke Formation of Nanometer-Sized and Hierarchical MFI Zeolites during Ethanol to Hydrocarbons Transformation. J. Catal. 2015, 328, 165–172. 10.1016/j.jcat.2014.12.030. [DOI] [Google Scholar]
- Guisnet M.; Costa L.; Ribeiro F. R. Prevention of Zeolite Deactivation by Coking. J. Mol. Catal. A Chem. 2009, 305 (1–2), 69–83. 10.1016/j.molcata.2008.11.012. [DOI] [Google Scholar]
- Wei F.-F.; Cui Z.-M.; Meng X.-J.; Cao C.-Y.; Xiao F.-S.; Song W.-G. Origin of the Low Olefin Production over HZSM-22 and HZSM-23 Zeolites: External Acid Sites and Pore Mouth Catalysis. ACS Catal. 2014, 4 (2), 529–534. 10.1021/cs400855p. [DOI] [Google Scholar]
- Xu Z.; Ma H.; Huang Y.; Qian W.; Zhang H.; Ying W. Synthesis of Submicron SSZ-13 with Tunable Acidity by the Seed-Assisted Method and Its Performance and Coking Behavior in the MTO Reaction. ACS Omega 2020, 5 (38), 24574–24583. 10.1021/acsomega.0c03075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano G.; Migliori M.; Ferrarelli G.; Giorgianni G.; Dalena F.; Peng P.; Debost M.; Boullay P.; Liu Z.; Guo H.; Yan Z.-F.; Mintova S. Passivated Surface of High Aluminum Containing ZSM-5 by Silicalite-1: Synthesis and Application in Dehydration Reaction. ACS Sustain Chem. Eng. 2022, 10 (15), 4839–4848. 10.1021/acssuschemeng.1c07198. [DOI] [Google Scholar]
- Zhang Z.; Sun Y.; Zhang H.; Wei L. Synthesis, Types, and Applications of Zeolite Capsule Catalysts. Crystal Research and Technology 2023, 58 (9), 1–19. 10.1002/crat.202200287. [DOI] [Google Scholar]
- Deng Y.-Q.; Zhou W.-F.; Lv H.-M.; Zhang Y.-Y.; Au C.-T.; Yin S.-F. Synthesis of HZSM-5@silicalite-1 Core–Shell Composite and Its Catalytic Application in the Generation of p-Xylene by Methylation of Toluene with Methyl Bromide. RSC Adv. 2014, 4 (70), 37296–37301. 10.1039/C4RA04126G. [DOI] [Google Scholar]
- Pan X.; Huang X.; Wang R.; Zhang H.; Wei H.; Chen J.; Liu S.; Sun L.; Xu D.; Liu Y. Effects of Silicalite-1 Coating on the p-Xylene Selectivity and Catalytic Stability of HZSM-5 in Toluene Methylation with Methanol. Catalysts 2022, 12 (12), 1538. 10.3390/catal12121538. [DOI] [Google Scholar]
- Shen H.; Liu M.; Li J.; Li X.; Xie S.; Chen F.; Xu L.; Guo X.; Song C.; Zhu X. Promising Strategy to Synthesize ZSM-5@Silicalite-1 with Superior Catalytic Performance for Catalytic Cracking Reactions. Ind. Eng. Chem. Res. 2021, 60 (25), 9098–9106. 10.1021/acs.iecr.1c01043. [DOI] [Google Scholar]
- Zhu J.; Yang F.; Yan S.; Wu H.; Zhu X. Fabrication and Characterization of ZSM-5@Silicalite-1 Core-shell Composites and Their Application in Hexane Catalytic Cracking. ChemistrySelect 2023, 8 (38), e202302769 10.1002/slct.202302769. [DOI] [Google Scholar]
- Hu C.; Zhang H.; Xiao R. Catalytic Fast Pyrolysis of Biomass over Core-Shell HZSM-5@silicalite-1 in a Bench-Scale Two-Stage Fluidized-Bed/Fixed-Bed Reactor. J. Anal Appl. Pyrolysis 2018, 136, 27–34. 10.1016/j.jaap.2018.11.005. [DOI] [Google Scholar]
- Goodarzi F.; Herrero I. P.; Kalantzopoulos G. N.; Svelle S.; Lazzarini A.; Beato P.; Olsbye U.; Kegnæs S. Synthesis of Mesoporous ZSM-5 Zeolite Encapsulated in an Ultrathin Protective Shell of Silicalite-1 for MTH Conversion. Microporous Mesoporous Mater. 2020, 292, 109730 10.1016/j.micromeso.2019.109730. [DOI] [Google Scholar]
- Han S.; Shilpa K.; Mallette A. J.; Li Y.; Hoke J. B.; Rimer J. D. Epitaxial Growth of Surface-Passivated Core-Shell Ferrierite. J. Cryst. Growth 2023, 603, 126992 10.1016/j.jcrysgro.2022.126992. [DOI] [Google Scholar]
- Bouizi Y.; Rouleau L.; Valtchev V. P. Factors Controlling the Formation of Core–Shell Zeolite–Zeolite Composites. Chem. Mater. 2006, 18 (20), 4959–4966. 10.1021/cm0611744. [DOI] [Google Scholar]
- Losch P.; Boltz M.; Bernardon C.; Louis B.; Palčić A.; Valtchev V. Impact of External Surface Passivation of Nano-ZSM-5 Zeolites in the Methanol-to-Olefins Reaction. Appl. Catal. A Gen 2016, 509, 30–37. 10.1016/j.apcata.2015.09.037. [DOI] [Google Scholar]
- Peng S.; Gao M.; Li H.; Yang M.; Ye M.; Liu Z. Control of Surface Barriers in Mass Transfer to Modulate Methanol-to-Olefins Reaction over SAPO-34 Zeolites. Angew. Chem., Int. Ed. 2020, 59 (49), 21945–21948. 10.1002/anie.202009230. [DOI] [PubMed] [Google Scholar]
- Wu G.; Hu Y.; Bao Q.; Zhang J.; Ge J. Improved Catalytic Performances of the NaOH-Treated ZSM-22 Zeolite in the 1-Butene Skeletal Isomerization Reaction: Effect of External Acid Sites. ACS Omega 2023, 8 (16), 14349–14364. 10.1021/acsomega.2c05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X.; Wang K.; Kong L.; Gu Z.; Wang F. Comparison of Surface Passivation Modification of Two Mordenite Zeolites and Their Application on the Isomerisation of o -Ethyltoluene. RSC Adv. 2022, 12 (48), 31326–31337. 10.1039/D2RA05390J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B.; Tan M.; Wu X.; Geng H.; Song F.; Ma Q.; Luan C.; Yang G.; Tan Y. Effects of Silylation on Ga/HZSM-5 for Improved Propane Dehydroaromatization. Fuel 2021, 283, 118889 10.1016/j.fuel.2020.118889. [DOI] [Google Scholar]
- Yi D.; Meng X.; Xu X.; Liu N.; Shi L. Catalytic Performance of Modified ZSM-5 Designed with Selectively Passivated External Surface Acidity by Phosphorus. Ind. Eng. Chem. Res. 2019, 58 (24), 10154–10163. 10.1021/acs.iecr.9b00629. [DOI] [Google Scholar]
- Prodinger S.; Shi H.; Wang H.; Derewinski M. A.; Lercher J. A. Impact of Structural Defects and Hydronium Ion Concentration on the Stability of Zeolite BEA in Aqueous Phase. Appl. Catal., B 2018, 237, 996–1002. 10.1016/j.apcatb.2018.06.065. [DOI] [Google Scholar]
- Fujiwara M.; Mimura N.; Sato O.; Yamaguchi A. Surface Modification of H-ZSM-5 with Organo-Disilane Compound for Propylene Production from Dimethyl Ether. Microporous Mesoporous Mater. 2019, 280, 219–226. 10.1016/j.micromeso.2019.02.005. [DOI] [Google Scholar]
- Busca G. Acidity and Basicity of Zeolites: A Fundamental Approach. Microporous Mesoporous Mater. 2017, 254, 3–16. 10.1016/j.micromeso.2017.04.007. [DOI] [Google Scholar]
- Ferrarelli G.; Giordano G.; Migliori M. ZSM-5@Sil-1 Core Shell: Effect of Synthesis Method over Textural and Catalytic Properties. Catal. Today 2022, 390–391, 176–184. 10.1016/j.cattod.2021.11.036. [DOI] [Google Scholar]