

Abstract
Thioredoxin reductase (TrxR) is an essential antioxidant in most cells; it reduces thioredoxin (Trx) and several more substrates, utilizing NADPH. However, the enzyme’s internal active site is too small to accommodate the Trx substrate. Thus, TrxR evolved a disulfide shuttle that can carry reducing equivalents from the active site to the docking site of thioredoxin on the enzyme surface. Yet, in all available atomic structures of TrxR, access to the active site by the shuttle is sterically blocked. We find with computational dynamics that thermal motion at 37 °C allows the oxidized shuttle x to transiently access the active site. Once the shuttle is reduced, it becomes polar. Again, with molecular dynamics, we show that the polar shuttle will move outward toward the solution interface, whereas the oxidized, neutral shuttle will not. This work provides physical evidence for crucial steps in the enzyme mechanism that thus far were just conjectures. The total shuttle motion, from the active site toward the surface, is over 20 Å. TrxR may thus also be termed a molecular machine.
Introduction
Thioredoxin reductase-1, TrxR, (EC 1.6.4.5), catalyzes the reduction of thioredoxin (Trx) using disulfide exchange:
| 1 |
Reduced thioredoxin subsequently reduces a wide variety of proteins including transcription factors and ribonucleotide reductase1 and plays a major role in the regulation of reactive oxygen species.2 In addition to the canonical substrate thioredoxin, TrxR reduces many other redox-active proteins, such as protein disulfide isomerase and NK-lysin, and smaller compounds such as lipid hydroperoxides and lipoic acid.3
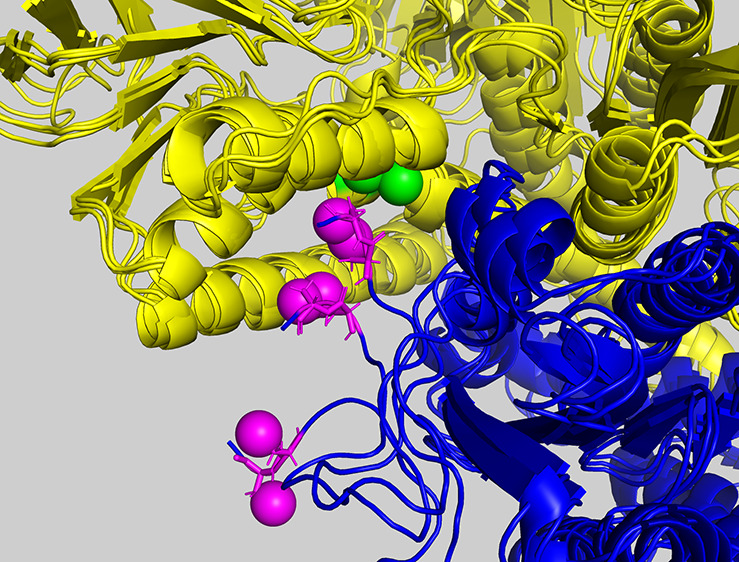
A crystal structure of human TrxR is shown in Figure 1.4 TrxR is an obligate crossover dimer, where the active site of each monomer contains participants from both polypeptide chains. We follow here sequence numbers as in the available crystal structures; the 499-residue monomers are N-terminally truncated compared to the 649-residue protein described in the UniProtKB entry Q16881. The 499-residue monomers contain the coenzyme flavin adenine dinucleotide (FAD(H)) and a Cys59/Cys64 redox-active disulfide from one monomer and a Cys497/SeCys498 disulfide from the other monomer. The Cys59/Cys64 disulfide is referred to as the distal/proximal pair, where Cys 64 is the closest to the FAD. All mammalian TrxR are seleno proteins containing the “21st amino acid” SeCys.5 Ten residues comprising the C-termini of each monomer are loosely structured and contain the penultimate Cys497/SeCys498 pair, which we will refer to as the “shuttle”.
Figure 1.
Crystal structure of the catalytically active human thioredoxin reductase dimer. The figure shows the central dimer (chains C and D) of 2J3N.pdb. The yellow (chain C) and blue monomers (chain D) are related by a 2-fold axis, running through the center of the plane of the view. The FAD molecules are shown in orange. The C-terminal Cysteines 497 and 498 (magenta) of one monomer form a redox pair with the proximal-distal disulfide 64–59 (green) of the other monomer. Other colored residues are identified in Figure 3.
As the catalyst of the reaction 1, TrxR carries out the following steps (Williams, 1992):
-
1.
NADPH docks to TrxR.
-
2.
NADPH reduces FAD of TrxR.
-
3.
FADH reduces the TrxR Cys59/Cys64 S2 disulfide.
-
4.
The TrxR Cys497/SeCys498 disulfide shuttle moves to the vicinity of Cys59/Cys64.
-
5.
Cys59/Cys64 reduces the TrxR Cys497/SeCys498 disulfide shuttle.
-
6.
The Cys497/SeCys498 SH-Se– shuttle seeks and binds to the external substrate Trx-S2.
-
7.
The TrxR Cys497/SeCys498 SH-Se– shuttle reduces Trx Cys32/Cys35 disulfide.
Steps (1), (2), and (3) have been extensively described by Williams.6 In Figure 2, we show the three crucial participants in the reductive half reaction of TrxR, NADPH, flavin, and the redox active disulfide.7,4
Figure 2.
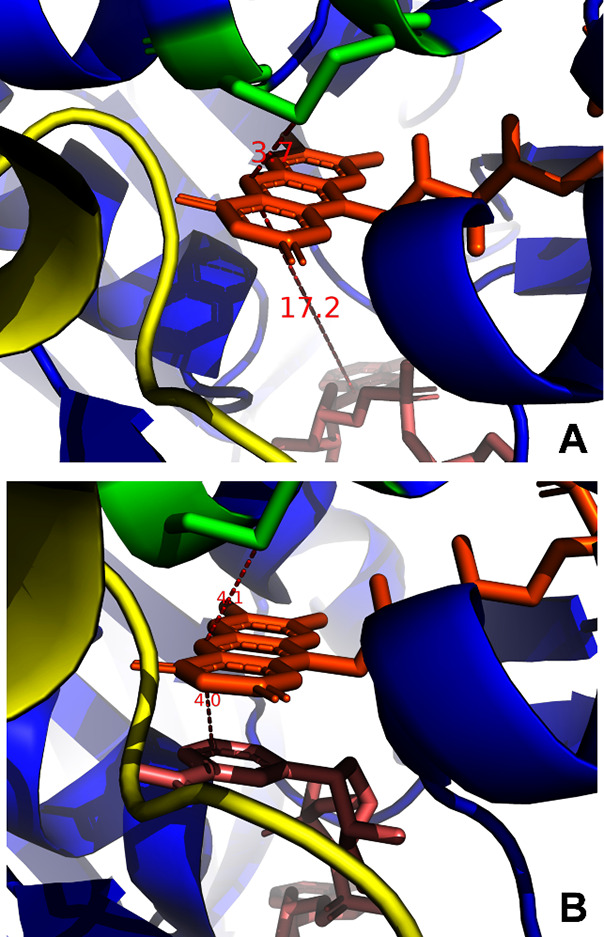
Juxtaposition of nicotinamide and flavin rings and the redox-active distal and proximal Cys. (A) Central dimer (chains C and D) of 2J3N. pdb. Color coding is as in Figure 1. The distance between Cys64-SG and FAD-N5 is 3.7 Å. The distance between the stacked rings of FAD and NAPH is about 18 Å. (B) Central dimer (chains C and D) of 2ZZC. Color coding is as in Figure 1. The distance between Cys64-SG and FAD-N5 is 4.1 Å. The distance between the stacked rings of FAD and NAPH is 4.0 Å.
The docking of NADPH (see Figure 2B) results in the stacking of its C4 ring of NADPH and the flavin ring in to favor hydride transfer to form a N5=C4a double bond.8 The N5 area of the flavin is also within bonding distance of the sulfur of the proximal Cys, which forms a disulfide with the distal Cys.7,9,10 The N5=C4a double bond electron pair attacks the proximal SG of the disulfide and resolves the disulfide. The thiolate of the distal Cys is thought to be stabilized by the His472-Glu477 ion pair (Figure 3) that is conserved in all TrxR and in the other family members GR and LipDH.
Figure 3.
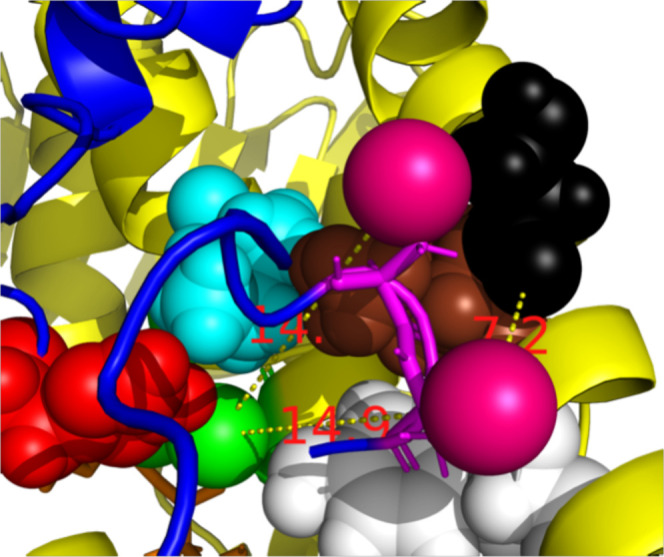
Detail of the key residues in Catalysis (Human TrxR-1). The central dimer chains C (yellow) and D (blue) of 2J3N. pdb. FAD is orange, the distal Cys-C59-SG (space fill) atom is green. The shuttle’s Cys-D489-SG (magenta space fill) is docked against Tyr-C116 (white). Cys-D488-SG is docked against Leu-C103 (brown). The shuttle is quite remote from its green redox partner Cys-C59 (>14.0 Å). Leu-C103, Tyr C116, Ile C65 (cyan), and His472 (red) hinder access from the shuttle to the redox partners. Residue Ser C-111 (black) is not involved in catalysis but indicates the (approximate) docking site of thioredoxin (Figure 5).
Step (4) of the mechanism has not been captured so far, but it has been modeled.3 In all available crystal structures, the shuttle is docked internally to Leu 112 and Tyr116 at helix 100–122 approximately 11 Å away from the Cys59/Cys64 pair. This site has been termed the “oxidized waiting position“11 (see Figure 3). In a recent cryo-EM structure, the last 6 residues of TrxR, which include the shuttle, are not detected12 and presumably disordered. Thus, the detailed chemistry of Step (5) is not fully understood either, only modeled.3 Step (6), a movement of the C-terminal Cys pair from the TrxR interior to the surface-bound Trx has been hypothesized before4,5 but has not been demonstrated by computation. Step (7) has been described before,4 but our results sheds additional light on that mechanism.
In Figure 4, we show a superposition of Human TrxR and human glutathione reductase with bound glutathione.9 Glutathione reductase (GR) is sequentially and functionally homologous to TrxR. In this structure, the glutathione substrate has entered the active site and is S–S linked to the distal Cys of GR (yellow). In contrast, in TrxR, the Trx substrate is much too large to approach the corresponding proximal Cys. Instead, the covalently bound disulfide shuttle evolved, which, in analogy with GR, is thought to form a mixed disulfide with the distal cysteine, becoming reduced in the following resolution Step (3). However, as mentioned above, all available crystal structures, this shuttle is docked at a distance of >11 Å of the TrxR active site.
Figure 4.
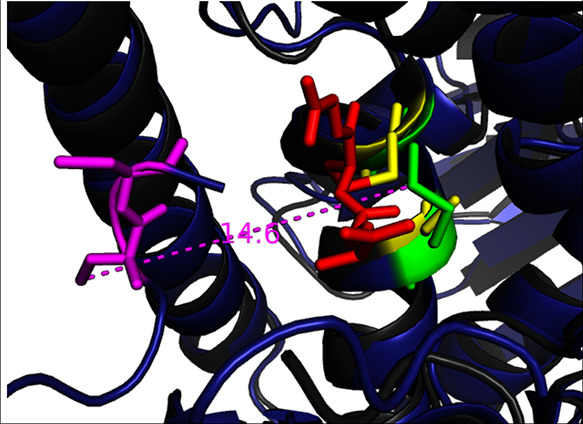
Superposition of human TrxR (dark blue) and human glutathione reductase (GR; 1GRE.pdb dark gray). The glutathione (red) is S–S linked to the distal Cys in the active site of GR (yellow). In TrxR, the Trx substrate is too large to approach the corresponding proximal Cys. Instead, a covalently bound disulfide shuttle evolved (Cys 497 and 498, in magenta) that seems to serve as a transient analogue of the Trx substrate. However, in all available crystal structures of TrxR, this shuttle is docked at a distance of >14 Å of the TrxR active site (green).
Figure 3 shows with CPK rendering that Leu-C103, Tyr-C116, Ile-C65, and His-C472 hinder access from the shuttle to the redox partners to distal Cys-D64. This suggests that major (transient) rearrangement of the active site is required to grant the Cys497/SeCys 498 shuttle access for Steps (4) and (5) of the mechanism.
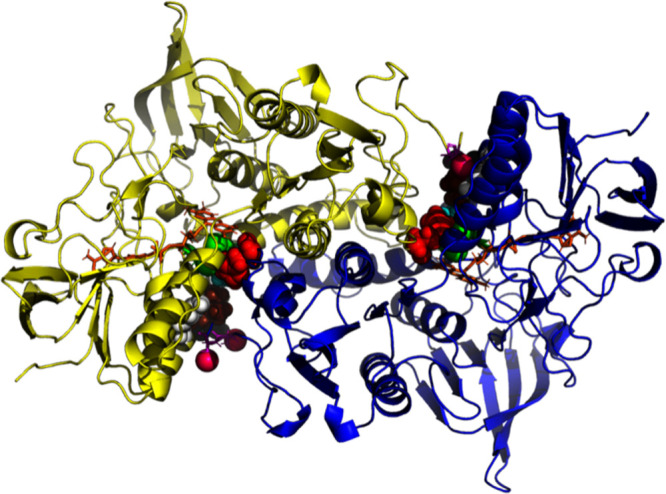
Significantly, the Cys497/SeCys 498 pair is also found on the surface of TrxR in the crystal structure of a TrxR-Trx complex (Figure 5) where it forms a mixed disulfide with residue 35 of the target disulfide on thioredoxin (Trx). The target substrate, thioredoxin, Trx, is located at a conserved interface of each TrxR monomer.11,5 The displacement of the Cys 497/SeCys 498 shuttle between the helix and surface locations is more than 10 Å. It was hypothesized that the C-terminal ten or so residues in TrxR must be disordered and mobile4,5 to allow such travel. However, the path of that motion has not been established.
Figure 5.
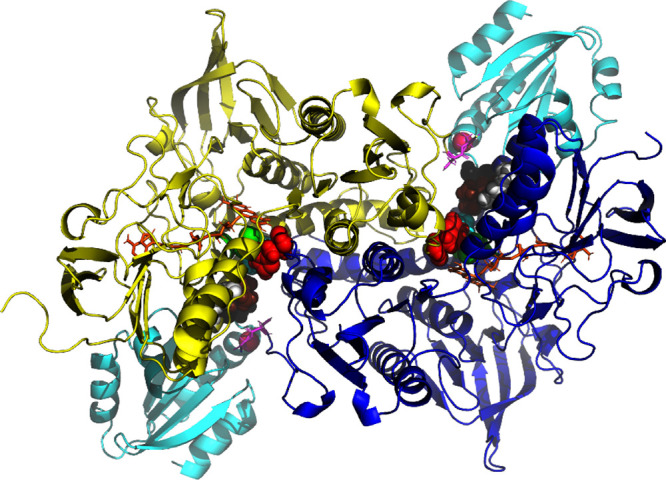
Complex between human thioredoxin reductase and thioredoxin (3QFB.pdb). The TrxR monomers are colored yellow and blue. Thioredoxin (Trx) is shown in cyan. The FAD molecules are shown in orange. The C-terminal Cys 497 and SeCys 498 are in pink, and the proximal-distal disulfides 64–59 are in green. Tyr-116 is white, Leu-103 is brown, Ile-65 is cyan, and His-472 is red. Residue Ser C-111 (black) is not involved in catalysis but indicates the (approximate) docking site of thioredoxin.
In the current work, we have employed molecular dynamics (MD) simulations13 of a complete dimer of TrxR, in explicit water. The simulations covered hundreds of nanoseconds and showed that
-
I.
the internal active site is sufficiently flexible to allow a (transient) approach between the reduced distal Cys SH and the C-terminal Cys-S-Se-Cys disulfide shuttle.
-
II.
the C-terminal Cys-S-Se-Cys disulfide can also reach the Trx docking site on the protein surface.
-
III.
the oxidized shuttle does not escape from the protein interior in 500 ns of simulation.
-
IV.
When reduced, the shuttle carries a negative charge on the Se atom. In this oxidation state, the shuttle can also reach the surface docking site, but it can also reach out into solution within 200 ns.
Materials and Methods
We utilized the computational molecular dynamics simulation package Amber 19,14 with the ff19SB force field15 running on a NVIDIA cluster at Rutgers University. The following protocol was used.
MD Calculation for TrxR with the Oxidized Shuttle
From the crystal structure 2J3N.pdb the “middle dimer”, chains C (residues 10–499) and D (residues 9–499), were copied. This pdb file reports the coordinates for human TrxR, fully reduced and with FAD bound, with a resolution of 2.8 Å, and a coordinate precision of 0.43 Å. The shuttle residues Cys-C497, Cys-C498, Cys-D497, and Cys-D498 were changed to CYX, representing an oxidized disulfide for the Amber package.
Force constants for FAD were created through antechamber (a part of the AmberTools suite) using the GAFF2 force field.16 Using the program tleap (also from AmberTools), the disulfide bonds Cys-C497/Cys-C498 and Cys-D497/Cys-D498 were created. The partially oxidized protein was briefly minimized with all CA restrained at 4 kCal/M/Å2, in order to obtain the proper disulfide bond lengths. This oxidized protein was then solvated in an octahedral OPC17 water box, using a layer of 14 Å, and 7 sodium ions were added to neutralize the protein. The ff19SB force field15 was used for the protein. This starting structure contained 981 protein residues, 2 FAD residues, 7 Na+ ions, and 40881 water molecules for a grand total of 178,825 atoms. A 9 Å cutoff was used for the computation for pairwise energies. After heating and equilibrating with protein heavy atom restraints decreasing from 5 to 0.01 kCal/M/Å2, all restraints were turned off during the production run. The production run was 500 ns, using 4 fs integration steps. The temperature was kept at 310 K using a Langevin thermostat with a 5.0 collisions per picosecond, at constant pressure using a Monte Carlo barostat, and with periodic boundaries. The SHAKE procedure was used to constrain the C–H bond lengths.
MD Calculation for TrxR with the Reduced Shuttle
We selected from the MD simulation from the oxidized shuttle a frame occurring at 208 ns in which the Cys-D488/Cys D489 disulfide approached the distal Cys-C59 to within 4.8 Å. The water molecules were removed. The disulfides Cyx-C497/Cyx-C498 and Cyx-D497/Cyx-D498 were “reduced” to Cys-C497, Cym-C498, Cys-D497, and Cym-D498 where Cym represents a reduced and ionized Cys residue in the Amber package. We used the Cym residues as stand-ins for ionized SeCys. SeCys has a pKa of 5.8 and is thus negatively charged at neutral condition. However, the physical properties of S and Se are not very different: the van der Waals radius of S (1.89 Å) is very similar to that of Se (1.81 Å).18 DFT computations19 have shown that the side-chain charge changes (CYS → SE CYS) are just 0.026 and 0.061 units for CA+HA and CB+HB2,3, respectively. Hence, for MD simulations, Cys is a good stand in for SeCys.
Using a tleap, the disulfide bonds for Cys-C59/Cys-C64 and Cys-D59/Cys-D64 were formed. The protein was briefly minimized with all CA restraints restraints at 4 kCal/M/Å2, in order to obtain the proper disulfide bond lengths. This protein was then solvated in an octahedral OPC water box using a layer of 14 Å, and 9 sodium ions were added to neutralize the protein. A 9 Å cutoff was used for the computation for pairwise energies. After heating and equilibrating with protein heavy atom restraints decreasing from 5 to 0.01 kCal/M/Å2, we obtained a “starting” structure for three production runs. The three independent MD simulations were performed at 310 K, each 500 ns long with 4 fs integration steps with all restraints turned off. Each of the three runs started with a new random velocity distribution at 310 K.
Results and Discussion
The mechanism comprises many steps, both chemical (steps 2, 3, 5, and 7) and physical (steps 1, 4, and 6). In the crystal structure of human TrxR, 2J3N, which contains 3 dimers in the unit cell, the C-terminal shuttle residues occupy significantly different positions. In the three monomers, there is no electron density for residues 496–499 containing the shuttle Cys pair. In the three other monomers, residues 496–499 are tucked in and docked against Tyr-116 and Leu-103 of the other chain, showing full density for residues 496–499. The minimum distances between either of the C-terminal-Cys sulfur atoms and the sulfur atom of the distal Cys 59 are 10.7, 14.5, and 11.1 Å (not shown). At these distances, formation of disulfides as required by the mechanism (Step (5)), is obviously impossible. One must postulate that the C-terminal cysteine shuttle can transiently approach the distal cysteine deep in the protein interior.3 The variation in positions for the shuttle, as seen in the crystal structure, also strongly suggests that shuttle motion is possible. However, the static structure does not provide a clue how the shuttle can approach the distal cysteine since the path is almost completely occluded by Tyr-116, Ile-65, Leu-103, and His-472 (see Figure 3).
We decided to carry out computational molecular dynamics (MD) simulations for the shuttle motion.20 MD is a computer simulation of the thermal motion of the protein and solvent atoms restrained by a mechanical force field, given a starting structure and a Maxwellian distribution of initial velocities for all atoms at the required temperature. As described in the Materials and Methods section, we simulated the solution dynamics of the central dimer of the TrxR crystal structure 2J3N.pdb, embedded in explicit water and ions. The calculations simulate the dynamics at 37 °C, without any restraints or steering, and were carried out for different oxidation/ionization states of the shuttle pairs and the active site pairs. The longest calculations simulated the dynamics for up to 500 ns. We computed three such 500 ns simulations in each oxidation state, and each simulation included two independent chains. Thus, we have a total of 6 μs of sampling of the shuttle motion.
We carried out two series of calculations. The first series started from the crystal structure coordinates, with the shuttle docked against the 100s helix (especially Tyr-116) and >14 Å remote from the distal Cys-59. The shuttle was modeled as an oxidized disulfide, and the distal-proximal pairs were modeled with reduced SH groups. This calculation simulates the dynamics of the shuttle in Step (4) of the mechanism. The second simulation starts with the shuttle close to the active site and mimics the dynamics at Step (6), after the shuttle has just been reduced.
Figure 6A shows the active site after 208 ns of unrestrained molecular dynamics simulation at 37 °C, in which the shuttle disulfide has “struggled” past the blockage and approached Cys-59-SH to 4.8 Å (see the “struggle” in Movie A of the Supporting Information). At this distance, the two sulfurs virtually touch each other, which should make mixed disulfide formation possible. We reiterate that this event is observed in a simulation carried out with a kinetic energy corresponding to 37 °C, without any restraints or steering. The close approach did not last very long; a few nanoseconds later, the relevant distances were back to 10 Å (Figure 7A). We also note that we observed ten more close approaches over the duration of the 500 ns simulation, all with short lifetimes (see Figure 7A). The oxidized shuttle did not move past the Tyr-116 docking site toward solution during this simulation (see Figure 7B and Movie B in the Supporting Information)
Figure 6.
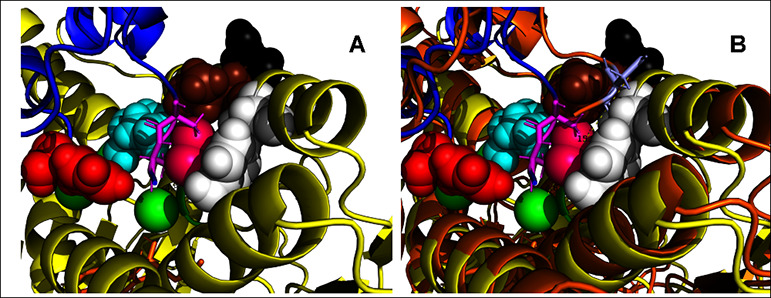
Snapshots of the simulated motion of the oxidized shuttle. (A) Detail of the close approach of the Cys D488-D489 disulfide to the distal Cys-C59, as occurring at 208 ns in the unrestrained, unguided, molecular dynamics simulation of the full dimer in explicit water, 37 °C. The distance between Cys-D489-SG and Cys-C59-SG is 4.8 Å. The starting structure was the central dimer (chains C and D) from 2J3N.pdb. Color coding is as in Figure 5. See also Figure 7 and the movie in the Supporting Information. (B) Detail of the close approach of the Cys–C488-C489 disulfide to Ser 111, which represents the Trx docking site, as occurring at 348 ns. The distance between Cys-D489-SG and Cys-C59-SG is 4.8 Å. Color coding is as in Figure 5. See also Figure 7, and the movie in the Supporting Information.
Figure 7.

Selected distances in the MD simulation of TrxR with the shuttle in the disulfide state. The simulation comprised the complete dimer in explicit water at 37 °C. (A) Trajectories for the distances Cys-D498-SG–Cys-C64-SG, Cys-D498-SG–Cys-C59-SG, Cys-D497-SG–Cys-C64-SG, and Cys-D497-SG–Cys-C59-SG are light green. The trajectories for the distances Cys-C498-SG–Cys-D64-SG, Cys-C498-SG–Cys-D59-SG, Cys-C497-SG–Cys-D64-SG, and Cys-C497-SG–Cys-D59-SG are dark green. The trajectories for the distances Cys-D498-SG–Tyr-C116-OZ and Cys-D497-SG–Tyr-C116-OZ, are light blue. The trajectories for the distances Cys-C498-SG–Tyr-D116-OZ and Cys-C497-SG–Tyr-D116-OZ are dark blue. The trajectories for the distances Cys-D498-SG–Ser-C111-OG and Cys-D497-SG–Ser-C111-OG are orange. The trajectories for the distances Cys-C498-SG–Ser-D111-OG and Cys-C497-SG–Ser-D111-OG are red. Note the proximity of C498 to D111 (RED) at 345 ns simulation. (B) Trajectories for the distances Cys-D498-SG–Cys-C64-SG and Cys-D497-SG–Cys-C64-SG are light green. The trajectories for the distances Cys-C498-SG–Cys-D64-SG and Cys-C497-SG–Cys-D64-SG are dark green. The trajectories for the distances Cys-D498-SG–Cys-C59-SG and Cys-D497-SG–Cys-C59-SG are purple. The trajectories for the distances Cys-C498-SG–Cys-D59-SG and Cys-C497-SG–Cys-D59-SG are black. Note the multiple close approaches D498-C59 (purple).
The available crystal structures of human TrxR do not give a clue about the disulfide exchange mechanism in Step (5). Nevertheless, the mechanism has been assumed to be as in GR.3 In general, formation of mixed disulfide requires that one of the thiols becomes a thiolate to carry out a nucleophilic attack on the disulfide.21 One assumes that the details of the mechanism in TrxR are similar to that in glutathione reductase (GR) (70% homologous). In GR, His-467, ion-paired with Glu-472 and within 4 Å of the distal cysteine, lowers the pKa of the latter so that it becomes a nucleophilic thiolate that can attack the glutathione disulfide.7 Indeed, the crystal structure of the glutathione–glutathione reductase complex shows a mixed disulfide with the distal cysteine9 (Figure 4). In human TrxR, the homologous residue His-472, ion-paired with Glu-477, has been proposed to play the same catalytic role. Of course, in the crystal structure of TrxR, Cys-498 and the distal Cys-59 are much too far apart to react with each other. We thus wonder whether the distances between Glu-72 and His-467, between His-467 and Cys-59 and between Cys-59 and Cys-498 are at any moment in time simultaneously short enough to allow the glutathione mechanism to take effect. In our MD calculations, this event is rare but does occur in about 5 of the 500 frames. In Figure 8, we show the best of these occurrences. However, one cannot deduce kinetic parameters from this simulation; the 500 ns is not nearly long enough as compared to kcat–1 (2,500 s–1 > 400 μs).22
Figure 8.
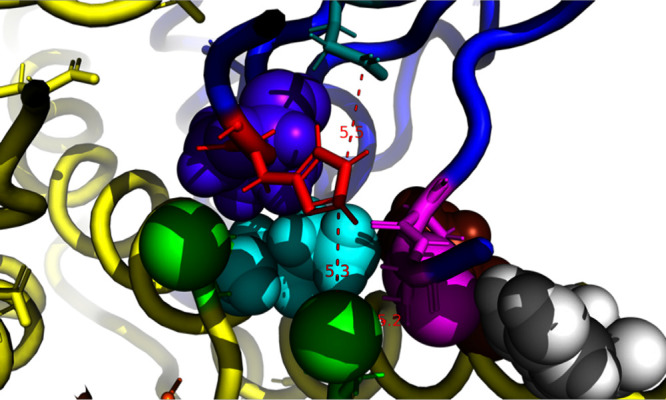
Frame at 162 ns from the MD calculation (see Figure 7) with the shuttle in the oxidized state. A rare occurrence where simultaneously, Glutamine 472 (teal) polarizes His 467 (Red), which in turn polarizes (4) the distal Cys-59 (green) so that it can attack Cys-498 (magenta). The distances are given in Å.
The simulations indicate that the oxidized shuttle cannot escape the enzyme interior in the 500 ns computation (Movie B). However, it can approach the docking site of Trx (see Figure 7B, red trace at 345 ns, and see Movie B in the Supporting Information) and return back to the protein interior (after that). This is relevant for the reverse motion of the shuttle, after it has been oxidized by the surface Trx. If one would take the 395 ns frame as the starting point for further dynamics, one would see that the oxidized shuttle has moved all the way back into the protein center at 420 ns approaching the distal Cys within 5 Å (see Figure 7A) to become reduced by the active site.
The crystal structures of the human TrxR-Trx complex11,5 illustrate the penultimate step of the mechanism, where (Se)Cys-498 in human TrxR forms a mixed disulfide with Cys-32 of Trx (see Figure 5). Just by comparing with the crystal structure of the TrxR, where the shuttle is docked against inside TrxR to Tyr-116, which in turn is 15 Å remote from where the shuttle is in the complex, one has inferred earlier that the shuttle must be mobile.5,11
In the oxidized state, the shuttle does not move into solution within 500 ns of simulation (Figure 7 and Movie B). However, after the shuttle has been reduced at the active site, it carries a negative charge: the SeH will ionize because the pKa of seleno Cys is 4.5. Such a charged system is likely to escape from the hydrophobic interior. The key questions are whether the shuttle moves along a well-defined path toward the docking site of the Trx, whether there is a conserved path or not, or if the reduced shuttles escapes the protein all together and “fishes” the Trx out of the solution and “reels” it back toward the inter molecular docking site?
We carried out several MD calculations to investigate the motion of the reduced shuttle. We ran several trajectories starting from a conformation with the shuttle close to the distal cysteine, as seen in Figure 6A (see also the description in Materials and Methods). Three 500 ns simulations were carried out at 37 °C, each starting from that situation, but with different (random) velocity distributions. Because TrxR is a dimer, we obtained information on the behavior of 6 monomers. As shown in Figure 9A, both the shuttles migrated far out in solution, a frame taken at 500 ns in Run 9 (see also Figure 10C and Movie C). In one of the computations, the shuttle approaches Ser 111 within 5 Å, indicating that it could also find a Trx when already bound to the surface (see Movie D). One of the frames showing this situation is depicted in Figure 9B. In Figure 10A–C, we show selected distances describing the motion of the shuttle in the reduced form. From the simulations, it thus appears that the dynamics of the shuttles are heterogeneous between different molecules and monomers. The shuttle can stay inside and approach the docking site of the Trx or go outside in solution.
Figure 9.
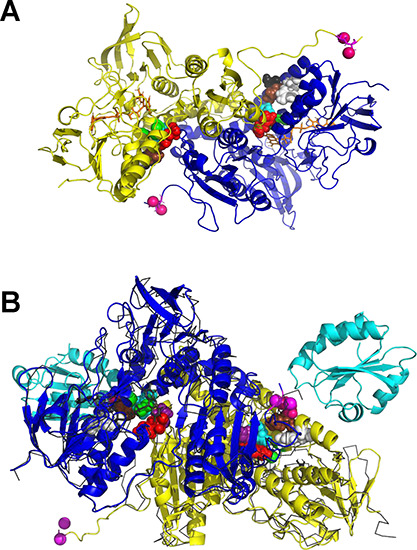
MD snapshots of the motion of the reduced shuttle. (A) MD frame at 500 ns of the motion of the reduced shuttle, starting from a configuration with the shuttle close to the distal Cys as depicted in Figure 6A. (B) MD frame at 404 ns of the motion of the reduced shuttle, starting from a configuration with the shuttle close to the distal Cys as in Figure 6A. The frame is superposed on the crystal structure of the complex (3QFB), shown with a thin black ribbon, and with the bound Trx in a cyan cartoon.
Figure 10.

Selected distances in the MD simulation of TrxR with the shuttle in the reduced state. The simulation comprised the complete dimer in explicit water at 37 °C. Selected distances describing the motion of the reduced shuttle. Three different simulations started from a configuration close to that depicted in Figure 6A, with the shuttle close to the distal Cys as shown in Figure 6A. The three simulations started with different velocity distributions. Color coding is as follows and is identical in panels (A)–(C). The trajectories for the distances Cys-D498-SG–Cys-C64-SG, Cys-D498-SG–Cys-C59-SG, Cys-D497-SG–Cys-C64-SG, and Cys-D497-SG–Cys-C59-SG are light green. The trajectories for the distances Cys-C498-SG–Cys-D64-SG, Cys-C498-SG–Cys-D59-SG, Cys-C497-SG–Cys-D64-SG, and Cys-C497-SG–Cys-D59-SG are dark green. The trajectories for the distances Cys-D498-SG–Tyr-C116-OZ and Cys-D497-SG–Tyr-C116-OZ are light blue. The trajectories for the distances Cys-C498-SG–Tyr-D116-OZ and Cys-C497-SG–Tyr-D116-OZ are dark blue. The trajectories for the distances Cys-D498-SG–Ser-C111-OG and Cys-D497-SG–Ser-C111-OG are orange. The trajectories for the distances Cys-C498-SG–Ser-D111-OG and Cys-C497-SG–Ser-D111-OG are red.
One may argue that the shuttle motion could be different in a preformed complex between TrxR and Trx. It is certainly possible that the bound Trx may help block the shuttles egress, but the fact remains that without a bound Trx, the shuttle tends to escape into solution. We argue that the latter scenario is important for the enzymatic mechanism. This suggests that it could encounter Trx while the latter is still in solution and reduce it there. This is a real possibility since the seleno-cysteine in the C-terminal residues is sufficiently nucleophilic that it needs no base activation to catalyze the disulfide interchange between TrxR and Trx.
Hence, SeCys may form a mixed disulfide with Trx in solution. The docking of Trx on TrxR may then follow after which the disulfide is resolved, requiring the participation of basic and acidic catalytic residues in TrxR, as suggested by others.5,11
As mentioned in the introduction, TrxR reduces several other redox-active proteins, such as protein disulfide isomerase and NK-lysin. These substrates are also too large to enter the TrxR active site and likely utilize the shuttle mechanism. It seems unlikely, though, that these proteins would first dock to the conserved Trx binding site on TrxR and be reduced there. However, they may be reduced in solution by an “escaped” TrxR shuttle, which, due to the Se substitution, needs no assistance from other catalytic residues.
Conclusions
In this work, we investigated the physical steps of the catalytic mechanism of human TrxR in detail. The TrxR-Trx system contains a Cys-497/SeCys-498 shuttle to carry reducing equivalents from the distal cysteine deep in the enzyme interior toward the surface-bound substrate. However, in the available crystal structures, that shuttle is very distant from the distal cysteine because access to the distal cysteine is blocked. We show here with unrestrained molecular dynamics simulations that the blockage can transiently open up and that the oxidized Cys-497/SeCys-498 shuttle can indeed reach the distal Cysteine deep into the protein interior to become reduced. This is the first time that that process has been visualized. In this equilibrium MD simulation, the oxidized shuttle oscillates between the Trx binding site and the internal active site, remaining inside the protein. Thus, as has been hypothesized previously, the shuttle is the functional equivalent of the glutathione substrate in glutathione reductase. This phenomenon is a beautiful example of adaptive evolution.
The negative charge on the C-terminal peptide increases upon reduction since the Se-Cys pKa is low. We show with molecular dynamics simulations that the shuttle can travel from being in close contact with the distal cysteine deep in the protein interior toward and ultimately into solution, sometimes approaching but mostly bypassing the Trx binding site. This is a journey of more than 20 Å, which happens spontaneously in this charged state. Once in solution, it can reduce an incoming thioredoxin or any of the other substrates for thioredoxin reductase.
Acknowledgments
This work was initiated by the late Professor Charles H. Williams Jr. He approved the virtually complete manuscript shortly before his passing. We thank Professor Bruce Palfey (University of Michigan) for critically evaluating earlier versions of this manuscript.
Glossary
Abbreviations:
- Trx
thioredoxin
- TrxR
thioredoxin reductase
- GR
glutathione reductase
- FAD
flavin adenine dinucleotide
- MD
molecular dynamics
- NADPH
nicotinamide phosphate adenine dinucleotide
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c01382.
Figure S1: Starting configuration for the MD simulations of the full TrxR dimer, embedded in explicit OPC water, with 7 Na+ counterions (PDF)
Movie A: Approach of the oxidized shuttle to the reduced Cys59 (MPG)
Movie B: Motion of the oxidized shuttle toward the active site (as in Movie A) and toward the (fictitious) Trx binding site (MPG)
Movie C:Motion of the reduced shuttle toward the (fictitious) Trx binding site and into solution (MPG)
Movie D:Detail of the motion of the reduced shuttle toward the (fictitious) Trx binding site (MPG)
Author Contributions
The MD starting structures were created by ERPZ. The MD equilibrium calculations were carried out by DAC, and the analysis was carried out by ERPZ. The figures were made by ERPZ, most the manuscript was written by ERPZ; most of the introduction was written by CHW.
None
The authors declare no competing financial interest.
Notes
∥ In memory of Professor Charles H. Williams, Jr.
Notes
UniprotKB Q16881
Supplementary Material
References
- Arner E. S. J.; Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- Gromer S.; Urig S.; Becker K. The thioredoxin system - From science to clinic. Medicinal Research Reviews 2004, 24, 40–89. 10.1002/med.10051. [DOI] [PubMed] [Google Scholar]
- Gromer S.; Wissing J.; Behne D.; Ashman K.; Schirmer R. H.; Flohé L.; Becker K. A hypothesis on the catalytic mechanism of the selenoenzyme thioredoxin reductase. Biochem. J. 1998, 591–592. 10.1042/bj3320591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz-Wolf K.; Urig S.; Becker K. The structure of human thioredoxin reductase 1 provides insights into C-terminal rearrangements during catalysis. J. Mol. Biol. 2007, 370, 116–127. 10.1016/j.jmb.2007.04.044. [DOI] [PubMed] [Google Scholar]
- Cheng Q.; Sandalova T.; Lindqvist Y.; Arnér E. S. Crystal structure and catalysis of the selenoprotein thioredoxin reductase 1. J. Biol. Chem. 2009, 284, 3998–4008. 10.1074/jbc.M807068200. [DOI] [PubMed] [Google Scholar]
- Williams C. H. (1992) Lipoamide dehydrogenase, glutathione reductase, thioredoxin reductase, and mercuric ion reductase--A family of flavoenzymes transhydrogenases., In Chemistry and Biochemistry of Flavoenzymes, pp 121–211, CRC Press, Boca Raton. [Google Scholar]
- Pai E. F.; Schulz G. E. The catalytic mechanism of glutathione reductase as derived from x-ray diffraction analyses of reaction intermediates. J. Biol. Chem. 1983, 258, 1752–1757. 10.1016/S0021-9258(18)33050-3. [DOI] [PubMed] [Google Scholar]
- Powell M. F.; Bruice T. C. Hydride vs. electron transfer in the oxidation of NADH model compounds. Prog. Clin Biol. Res. 1988, 274, 369–385. [PubMed] [Google Scholar]
- Karplus P. A.; Schulz G. E. Refined structure of glutathione reductase at 1.54 A resolution. J. Mol. Biol. 1987, 195, 701–729. 10.1016/0022-2836(87)90191-4. [DOI] [PubMed] [Google Scholar]
- Thorpe C.; Williams C. H. Jr. Spectral evidence for a flavin adduct in a monoalkylated derivative of pig heart lipoamide dehydrogenase. J. Biol. Chem. 1976, 251, 7726–7728. 10.1016/S0021-9258(17)32913-7. [DOI] [PubMed] [Google Scholar]
- Fritz-Wolf K.; Kehr S.; Stumpf M.; Rahlfs S.; Becker K. Crystal structure of the human thioredoxin reductase-thioredoxin complex. Nat. Commun. 2011, 2, 383. 10.1038/ncomms1382. [DOI] [PubMed] [Google Scholar]
- Du Z.; He Z.; Fan J.; Huo Y.; He B.; Wang Y.; Sun Q.; Niu W.; Zhao W.; Zhao L.; Cao P.; Cao K.; Xia D.; Yuan Q.; Liang X. J.; Jiang H.; Gong Y.; Gao X. Au4 cluster inhibits human thioredoxin reductase activity via specifically binding of Au to Cys189. Nano Today 2022, 47, 101686 10.1016/j.nantod.2022.101686. [DOI] [Google Scholar]
- Karplus M. Molecular dynamics of biological macromolecules: a brief history and perspective. Biopolymers 2003, 68, 350–358. 10.1002/bip.10266. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Cheatham T. E. 3rd; Darden T.; Gohlke H.; Luo R.; Merz K. M. Jr.; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C.; Kasavajhala K.; Belfon K. A. A.; Raguette L.; Huang H.; Migues A. N.; Bickel J.; Wang Y. Z.; Pincay J.; Wu Q.; Simmerling C. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. 10.1021/acs.jctc.9b00591. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Izadi S.; Anandakrishnan R.; Onufriev A. V. Building Water Models: A Different Approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. 10.1021/jz501780a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter M.. (2024) ″WebElements, https://www.webelements.com, accessed April 2024.″.
- Harki E. M. Biophysical study of selenocysteine and selenomethionine in the gas and solution phases. Comput. Theor. Chem. 2021, 1204, 113383 10.1016/j.comptc.2021.113383. [DOI] [Google Scholar]
- Karplus M.; McCammon J. A.; Peticolas W. L. The internal dynamics of globular proteins. Crit. Rev. Biochem. 1981, 9, 293–349. 10.3109/10409238109105437. [DOI] [PubMed] [Google Scholar]
- Kersteen E. A.; Raines R. T. Catalysis of protein folding by protein disulfide isomerase and small-molecule mimics. Antioxid Redox Signal 2003, 5, 413–424. 10.1089/152308603768295159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey B. M.; Eckenroth B. E.; Flemer S.; Hondal R. J. Selenium in Thioredoxin Reductase: A Mechanistic Perspective. Biochemistry 2008, 47, 12810–12821. 10.1021/bi800951f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.