

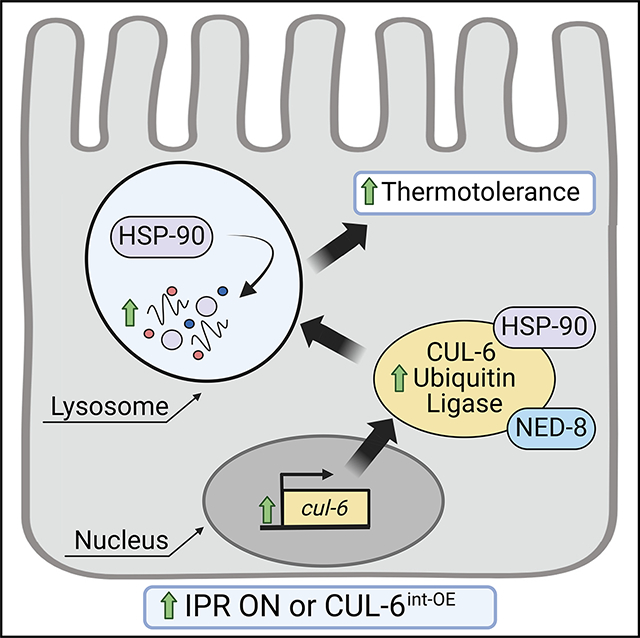
SUMMARY
Heat shock can be a lethal stressor. Previously, we described a CUL-6/cullin-ring ubiquitin ligase complex in the nematode Caenorhabditis elegans that is induced by intracellular intestinal infection and proteotoxic stress and that promotes improved survival upon heat shock (thermotolerance). Here, we show that CUL-6 promotes thermotolerance by targeting the heat shock protein HSP-90 for degradation. We show that CUL-6-mediated lowering of HSP-90 protein levels, specifically in the intestine, improves thermotolerance. Furthermore, we show that lysosomal function is required for CUL-6-mediated promotion of thermotolerance and that CUL-6 directs HSP-90 to lysosome-related organelles upon heat shock. Altogether, these results indicate that a CUL-6 ubiquitin ligase promotes organismal survival upon heat shock by promoting HSP-90 degradation in intestinal lysosomes. Thus, HSP-90, a protein commonly associated with protection against heat shock and promoting degradation of other proteins, is itself degraded to protect against heat shock.
In brief
Bardan Sarmiento et al. demonstrate in C. elegans that heat shock protein HSP-90 levels are lowered by CUL-6 ubiquitin ligase function in the intestine, which promotes thermotolerance. These degradative effects are mediated by the lysosome, and heat shock exposure directs HSP-90 to lysosomes and lysosome-related organelles, in a CUL-6-dependent manner.
Graphical Abstract
INTRODUCTION
Exposure to dangerously high temperatures, i.e., heat shock, can lead to organismal sickness and death. These negative impacts are due, at least in part, to heat-induced denaturation and aggregation of proteins, which impair normal cellular function. To combat these negative impacts, organisms have evolved dedicated stress resistance pathways such as the heat shock response, which upregulates and deploys heat shock proteins (chaperones) to refold denatured proteins and restore protein homeostasis (proteostasis) in response to acute increases in temperature.1–3 A central regulator of the heat shock response and proteostasis is the transcription factor heat shock factor 1 (HSF-1), which mediates the transcriptional upregulation of chaperones upon heat shock to promote thermotolerance.4–6 HSF-1 is conserved across organisms from yeast and the nematode C. elegans to humans.
In C. elegans, we recently described the intracellular pathogen response (IPR) as a stress resistance pathway that appears to promote thermotolerance in a manner that is separate from the upregulation of chaperones by HSF-1.7–10 The IPR comprises a common set of genes induced by natural intracellular pathogens of the intestine, including microsporidia and the Orsay virus, as well as by abiotic stressors like proteasome blockade and chronic heat stress.11 A negative regulator of the IPR is pals-22, a protein of unknown biochemical function but that nonetheless serves a critical physiological function to repress IPR mRNA expression in the absence of infection or stress. Constitutively upregulated IPR gene expression in pals-22 mutants causes slowed development but increased resistance to infection and heat shock.7,11,12
IPR genes are not enriched for chaperones and instead are enriched for transcriptionally upregulated E3 cullin-ring ubiquitin ligase components, which we found are required for the increased thermotolerance of pals-22 mutants.8,11 E3 ubiquitin ligases are enzymes that conjugate the small protein ubiquitin onto lysine residues of substrate proteins, which then alters the fate of these proteins.13 Skp-Cullin-F-box (SCF) ubiquitin ligases are a large class of cullin-ring ubiquitin ligases and are multi-subunit enzymes composed of three core components: (1) Cullins, (2) Skp proteins, and (3) RING proteins.14 These core components assemble with F-box proteins that serve as adaptors to recognize substrate proteins for ubiquitylation. IPR-induced SCF genes include cul-6/cullin, as well as a previously uncharacterized RING protein (rcs-1), three Skp-related proteins (skr-3, skr-4, skr-5), and two previously uncharacterized F-box proteins (fbxa-75, fbxa-158). Using genetics and biochemistry, we demonstrated that the CUL-6 protein assembles with these other SCF protein components into a multi-subunit ubiquitin ligase complex that promotes thermotolerance in pals-22 mutants.15 Both pals-22 and cul-6 are normally expressed in the intestine, among other tissues, and expression of pals-22 or cul-6 only in the intestine can regulate thermotolerance.7,15
The ubiquitin ligase activity of cullins can be increased by post-translational modification of a conserved lysine by the ubiquitin-like protein NEDD8 in a process called neddylation.16 We found that the ability of CUL-6 to promote thermotolerance in C. elegans depends on a conserved neddylation site,15 indicating that the CUL-6 ubiquitin ligase complex likely promotes thermotolerance through its ability to conjugate ubiquitin onto substrates, but it was unclear what these substrates were. One hypothesis was that a CUL-6 ubiquitin ligase could target misfolded proteins for destruction, including pathogen proteins delivered into host cells in the context of infection, and/or that it might target misfolded cytosolic host proteins in the context of heat shock.8 The ultimate fate of CUL-6 target proteins was also not clear. The most common fate for ubiquitylated proteins is degradation by the proteasome, but the lysosome can also degrade them,17 or ubiquitylation can result in non-degradative effects such as altered trafficking, subcellular localization, or biochemical function.18
Here, we provide evidence that the highly abundant heat shock protein HSP-90 is a target for degradation by the CUL-6 ubiquitin ligase complex. Unlike other heat shock proteins, HSP-90 is highly expressed in the absence of heat shock and has many functions, including facilitating the proper folding of hundreds of proteins under unstressed conditions.19–21 We find that CUL-6 activity reduces HSP-90 protein levels in the absence of heat shock and that decreased expression of HSP-90, specifically in the intestine, leads to higher thermotolerance. In contrast, overexpression of HSP-90 in the intestine leads to lower thermotolerance. The current paradigm in the field indicates that loss of HSP-90 activates HSF-1,22,23 but here, we find that CUL-6-mediated effects of lowering HSP-90 levels on thermotolerance may be independent of HSF-1, as assessed by RNAi and a partial-loss-of-function mutant. To investigate where HSP-90 is degraded in the cell, we show that lysosomes regulate the levels of HSP-90 protein, and we show that the effects of HSP-90 and CUL-6 on thermotolerance depend on lysosomal function. Furthermore, we show that CUL-6 directs HSP-90 to lysosome-related organelles (LROs) in the intestine upon heat shock. Altogether, our results support a model that the CUL-6 ubiquitin ligase targets HSP-90 for ubiquitylation, which leads to its subsequent degradation by the lysosome and/or LROs to promote organismal survival upon heat shock.
RESULTS
The lysosome is specifically required for CUL-6-mediated thermotolerance as part of the IPR
Ubiquitylated substrates can be degraded by the proteasome or by the lysosome. To determine whether the proteasome or the lysosome might degrade the substrate(s) of the IPR-induced CUL-6 ubiquitin ligase, we blocked each pathway with pharmacological inhibitors while performing heat shock to assess the changes in CUL-6-dependent thermotolerance. First, we treated wild-type animals or pals-22 mutants (which have cul-6 upregulated as part of the IPR and have increased thermotolerance)7 with the proteasome inhibitor bortezomib. Here, we found that bortezomib treatment impaired thermotolerance in both a wild-type and a pals-22 mutant background, indicating that proteasome blockade negatively affects heat shock survival in a CUL-6-independent manner (Figures 1A and S1A). In contrast, we found that treatment with the lysosome inhibitor bafilomycin impaired thermotolerance only in a pals-22 mutant background and not in a wild-type background, suggesting that the lysosome is specifically important to promote CUL-6-mediated thermotolerance when the IPR is induced (Figure 1B).
Figure 1. The lysosome is required for CUL-6-mediated thermotolerance as part of the IPR.
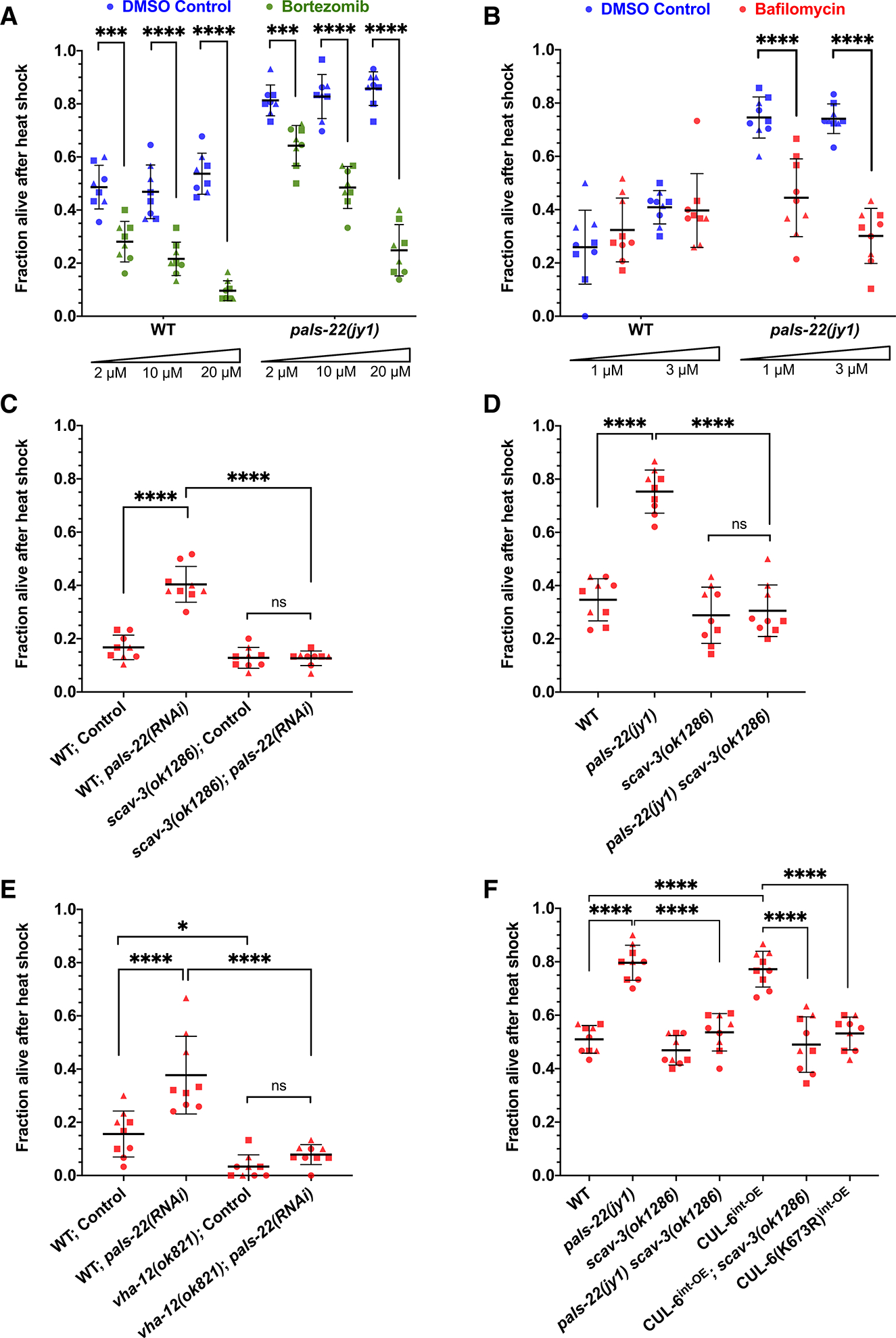
(A and B) Survival of wild-type or pals-22(jy1) mutant animals treated with bortezomib (A) or bafilomycin (B) after 2 h of 37.5°C heat shock with a 15 min gradual ramp-up, followed by a 24 h recovery period at 20°C (hereafter referred to as heat shock treatment). Bortezomib-treated animals (green dots) were tested with eight plates over three experiments with 30 animals per plate, and bafilomycin-treated animals (red dots) were tested in triplicate experiments with three plates per experiment and 30 animals per plate. DMSO-treated animals (blue dots) were tested as the vehicle control for both inhibitors. Unpaired t tests for each genotype and concentration of inhibitor were used to calculate p values.
(C) Heat shock treatment of wild-type or scav-3(ok1286) mutant animals fed E. coli OP50 expressing control or pals-22 double-stranded RNA (dsRNA) to induce RNAi (see STAR Methods).
(D) Survival of wild-type, pals-22(jy1), scav-3(ok1286), or pals-22(jy1) scav-3(ok1286) mutant animals after heat shock treatment.
(E) Heat shock treatment of wild-type or vha-12(ok821) mutant animals fed OP50 expressing control or pals-22 dsRNA to induce RNAi.
(F) Heat shock treatment of animals from (D) but also including strains with transgenes overexpressing CUL-6 in the intestine (superscript int-OE). CUL-6(K673R) indicates a lysine-to-arginine mutation at CUL-6’s neddylation site with reduced ubiquitylation activity, which controls for CUL-6 overexpression in the intestine.
For (C)–(F), animals were tested in triplicate experiments with three plates per experiment and 30 animals per plate. A one-way ANOVA with Tukey’s multiple comparisons test was used to determine p values. For (A)–(F), the mean fraction of animals alive for the pooled replicates is indicated by the black bar with error bars as the standard deviation (SD). Each dot represents a plate, and different shapes represent the experimental replicates performed on different days. *p < 0.05, ***p < 0.001, and ****p < 0.0001.
Next, we tested the role of the lysosome in promoting thermotolerance using genetic approaches. Here, we performed RNAi against pals-22 in animals with a mutation in scav-3, a lysosomal membrane protein important for lysosomal integrity.24 We found that pals-22 RNAi, which increases thermotolerance in wild-type animals, no longer increased thermotolerance in scav-3 mutants (Figure 1C). Similarly, pals-22 scav-3 double mutants have a thermotolerance phenotype similar to that of wild-type animals (Figure 1D). Furthermore, pals-22 RNAi no longer increased thermotolerance in mutants defective in vha-12, which encodes a vacuolar-ATPase subunit important for lysosomal function25 (Figure 1E), again indicating that the lysosome is required for the increased thermotolerance in pals-22-defective animals.
The increased thermotolerance of pals-22 mutants depends on cul-6 expression in the intestine, and overexpression of cul-6 specifically in the intestine, even in a wild-type background, promotes thermotolerance.15 Investigating the role of the lysosome in this context, we saw that the increased thermotolerance due to cul-6 overexpression depended on scav-3 (Figure 1F). As a control, we show that overexpression of a neddylation-deficient CUL-6(K673R) that lacks the lysine used for neddylation does not promote thermotolerance (Figure 1F), consistent with prior results.15 Taken together, these findings indicate that the lysosome is required for the increased thermotolerance associated with transcriptional upregulation of cul-6.
RNAi knockdown of hsp-90 specifically in the intestine increases thermotolerance
Given the results above indicating that lysosomal degradation of a CUL-6 ubiquitin ligase substrate promotes thermotolerance, we searched for candidate proteins whose reduction might promote thermotolerance. Previously published studies have shown that, in some cases, levels of the heat shock protein HSP-90 paradoxically appear to be negatively associated with thermotolerance.23,26,27 Therefore, we investigated the possible role of HSP-90 in our heat shock paradigm and indeed found that feeding animals hsp-90 double-stranded RNA (dsRNA) significantly increased their thermotolerance (Figure 2A).
Figure 2. Knockdown of hsp-90 in the intestine increases thermotolerance.
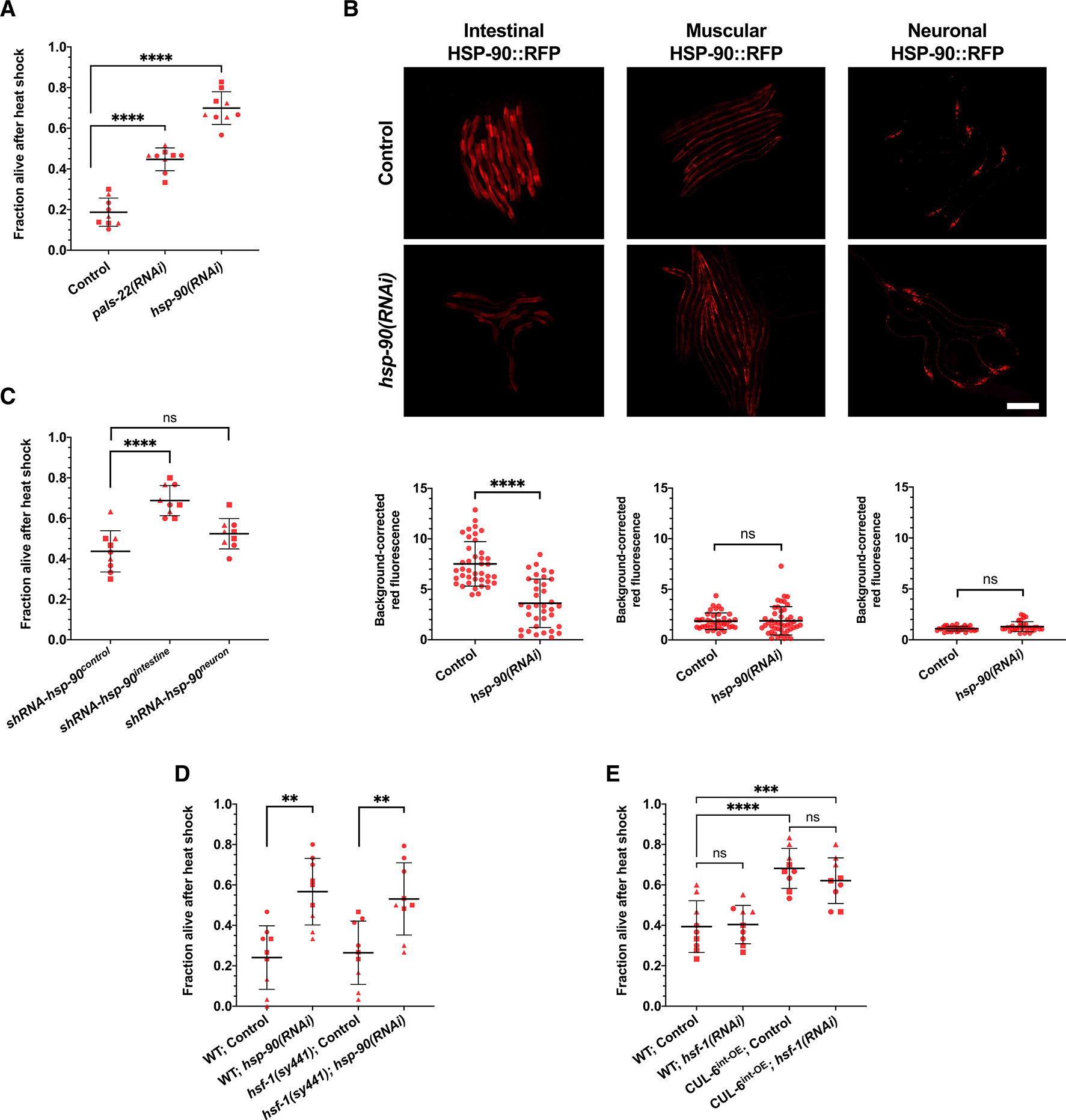
(A) Heat shock treatment of wild-type animals fed OP50 expressing control, pals-22, or hsp-90 dsRNA to induce RNAi.
(B) Top: representative fluorescent images of L4 animals with HSP-90::RFP overexpression under different tissue-specific promoters fed OP50 expressing control or hsp-90 dsRNA to induce RNAi. Scale bar: 200 mm. Bottom: quantification of the RFP fluorescent signal for each condition. Each dot represents one animal measured, and the data shown are the results of three experimental replicates. n = 36–47 worms quantified. A Mann-Whitney test was used to calculate p values for all strains.
(C) Heat shock treatment of animals expressing tissue-specific short hairpin RNA (shRNA) against hsp-90 in the intestine and neurons relative to a control strain.
(D) Heat shock treatment of wild-type or hsf-1(sy441) mutant animals fed OP50 expressing control or hsp-90 dsRNA to induce RNAi.
(E) Heat shock treatment of wild-type and CUL-6int—OE animals fed OP50 expressing control or hsf-1 dsRNA to induce RNAi.
For (A), (C), (D), and (E), animals were tested in triplicate experiments with three plates per experiment and 30 animals per plate. The mean fraction of animals alive for the pooled replicates is indicated by the black bar with error bars as the SD. Each dot represents a plate, and different shapes represent the experimental replicates performed on different days. A one-way ANOVA with Tukey’s multiple comparisons test was used to calculate p values. For (A)–(E), **p < 0.01, ***p < 0.001, and ****p < 0.0001.
To examine the efficacy of hsp-90 RNAi in various tissues, we next performed whole-animal hsp-90 RNAi by feeding dsRNA to transgenic strains that express HSP-90::RFP specifically in the intestine, the body wall muscle, or in neurons.26 Here, we only observed a significant reduction of HSP-90::RFP levels in the intestine (Figure 2B). In C. elegans, the neurons are often refractory to RNAi delivered by feeding, so the lack of knockdown there is not surprising.28 However, the muscle is generally susceptible to feeding RNAi. One potential explanation for this lack of effect is the compensatory mechanisms that have been reported to upregulate hsp-90 expression after hsp-90 RNAi in C. elegans26 or after deleting 3 of the 4 hsp-90 alleles found in mice.29 These compensatory mechanisms may be responsible for maintaining stable HSP-90::RFP protein levels in muscle after whole-animal RNAi in C. elegans (Figure 2B).
To knock down HSP-90 in specific tissues, and to circumvent the issues with neurons being refractory to feeding RNAi, we next used strains that genetically encode short hairpin RNA (shRNA) against hsp-90 specifically expressed in different tissues.27 Here, we found that by targeting hsp-90 for RNAi specifically in the intestine, there was increased thermotolerance (Figure 2C). In contrast, a strain with hsp-90 shRNA expressed specifically in neurons did not exhibit significantly increased thermotolerance (Figure 2C). Altogether, these results demonstrate that reducing HSP-90 levels specifically in the intestine leads to increased thermotolerance.
Prior studies have indicated that HSP-90 binds to the transcription factor HSF-1 and holds it in an inactive state until heat shock, at which point HSP-90 binds to misfolded proteins and releases HSF-1 to activate the transcription of chaperones.22 Therefore, we examined whether HSF-1 might be involved in the increased thermotolerance either through the reduction of HSP-90 levels or the overexpression of CUL-6. First, we performed hsp-90 RNAi in an hsf-1(sy441) mutant background and saw increased thermotolerance similar to hsp-90 RNAi in a wild-type background (Figure 2D). Of note, a complete loss of hsf-1 is lethal, and hsf-1(sy441) is a non-null allele that still retains basal activity of hsf-1, although it is defective in HSP induction.30,31 Therefore, we cannot rule out a role for hsf-1 based on these results. Also important to note is that while the overexpression of hsf-1 in C. elegans consistently promotes thermotolerance, there are varying results in the loss of hsf-1 function; some studies show reduced thermotolerance, and others show no reduction in thermotolerance, similar to our results here.32–35 Next, we performed hsf-1 RNAi in a cul-6 intestinal overexpression strain and saw no significant impairment on thermotolerance (Figure 2E). As a control, we ensured that hsf-1 RNAi significantly reduced hsp-16.2p::GFP induction, indicating that the RNAi clone was effective (Figures S2A and S2B). Altogether, these results suggest that the increased thermotolerance due to the effects of CUL-6 overexpression and loss of HSP-90 may be independent of the heat shock-inducible functions of HSF-1 and the canonical heat shock response.
Overexpression of HSP-90 in the intestine decreases thermotolerance
Next, we investigated whether elevating HSP-90 levels in the intestine would impair thermotolerance. We performed heat shock assays on transgenic strains where HSP-90::RFP is overexpressed specifically in the intestine, body wall muscle, or neurons.26 Here, we found that HSP-90::RFP overexpression in the intestine decreased thermotolerance, while overexpression in body wall muscle and neurons did not negatively affect thermotolerance (Figure 3A). These results are consistent with a prior study that found HSP-90 overexpression in the intestine impaired thermotolerance.26 However, that study found that overexpression of HSP-90 in muscle and neurons also impaired thermotolerance. Several differences may account for this discrepancy, including the different copy numbers and fluorophores of the strains used in that study. In addition, there were distinct thermotolerance assay conditions used in that study, which notably did not include a 24 h recovery step, whereas our assay did (Figure S1). Overall, because of our findings with the intestine, and because CUL-6 is expressed in that tissue, but not in neurons and muscle,15 we focused our efforts on the intestine.
Figure 3. Overexpression of HSP-90 in the intestine decreases thermotolerance, which is reversed by CUL-6 expression and lysosome function.
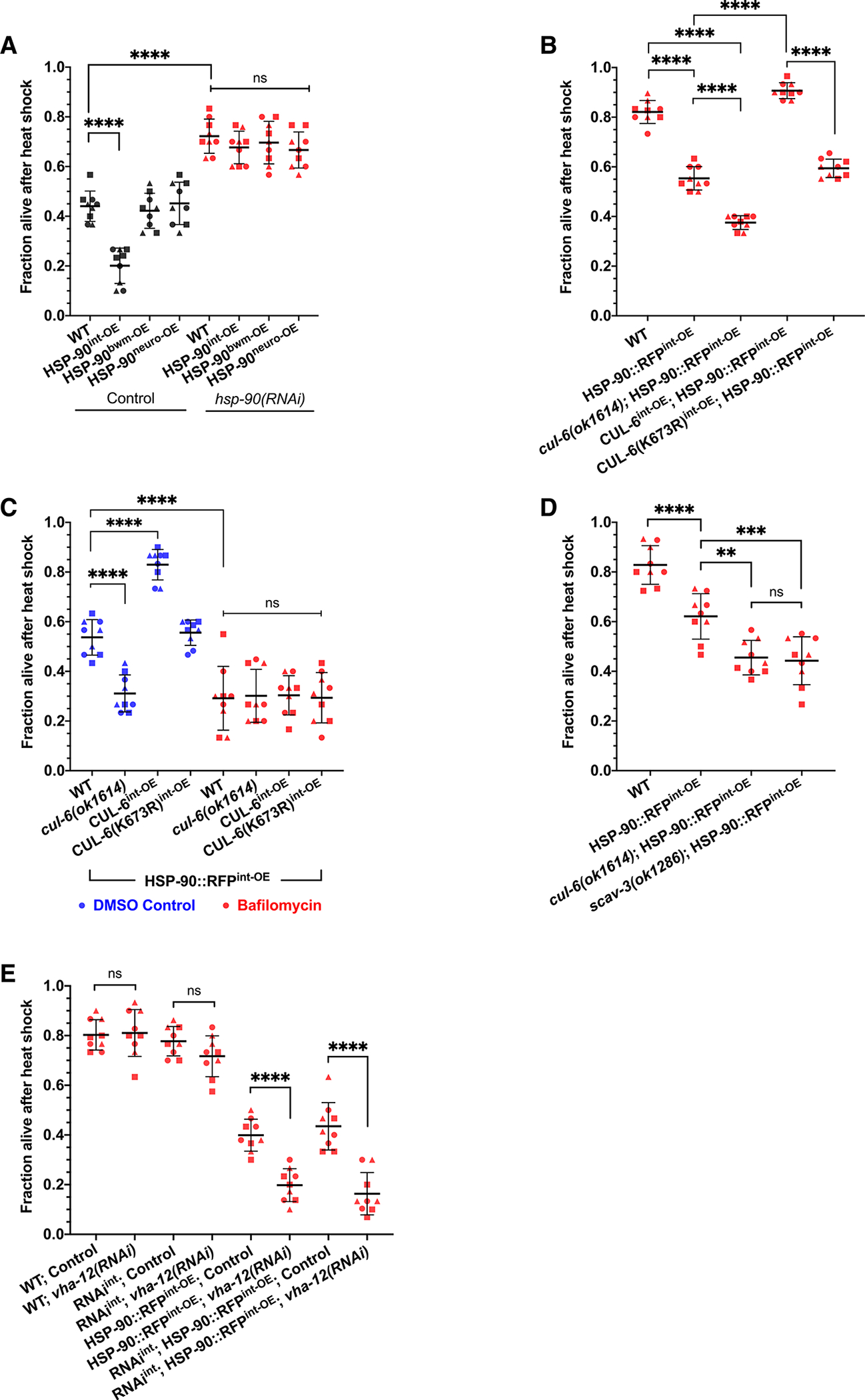
(A) Heat shock treatment of animals with HSP-90::RFP (hereafter HSP-90 for brevity) overexpression under the control of intestinal (superscript int-OE), body wall muscular (superscript bwm-OE), or neuronal (superscript neuro-OE) tissue-specific promoters fed OP50 expressing control (black) or hsp-90 (red) dsRNA to induce RNAi.
(B) Survival of wild-type, HSP-90int-OE, and HSP-90int-OE animals with cul-6(ok1614) mutation or CUL-6int-OE after reduced heat shock treatment (see STAR Methods).
(C) Heat shock treatment of wild-type, cul-6(ok1614), or CUL-6int-OE animals in an HSP-90int-OE background treated with bafilomycin (red). DMSO-treated animals (blue) were tested as a vehicle control.
(D) Survival of wild-type, HSP-90int-OE, and HSP-90int-OE animals in a cul-6(ok1614) or scav-3(ok1286) mutant background after reduced heat shock treatment.
(E) Heat shock treatment after whole-animal or intestinal-specific RNAi (RNAiint) against vha-12 in either a wild-type or HSP-90::RFPint-OE background. For (A)–(E), animals were tested in triplicate experiments with three plates per experiment and 30 animals per plate. The mean fraction of animals alive for the pooled replicates is indicated by the black bar with error bars as the SD. Each dot represents a plate, and different shapes represent the experimental replicates performed on different days. A one-way ANOVA with Tukey’s multiple comparisons test was used to calculate p values. **p < 0.01, ***p < 0.001, and ****p < 0.0001.
To confirm that this intestinal-specific effect on thermotolerance was due to HSP-90 levels, we treated these tissue-specific expression strains with hsp-90 RNAi that should knock down hsp-90 systemically and found that all of them had thermotolerance levels increased to the same level (Figure 3A). In fact, all of the overexpression strains had greatly increased thermotolerance after hsp-90 RNAi, comparable with the survival rates after hsp-90 RNAi in wild-type animals mentioned above (Figure 2A). Furthermore, intestinal HSP-90::RFP impairment of thermotolerance was not exacerbated by an hsf-1(sy441) mutation, again suggesting that these effects are independent of the heat shock-inducible functions of HSF-1 (Figure S2C). To ensure that the effects on thermotolerance were specific to overexpression of HSP-90 in the intestine and not overexpression of the RFP tag, we tested thermotolerance of two other strains that specifically express cytoplasmic RFP in the intestine. We did not observe decreased thermotolerance in either of these strains (Figure S2D). Therefore, we conclude that overexpression of HSP-90 specifically in the intestine impairs thermotolerance.
CUL-6 ubiquitin ligase and lysosomal activity promote thermotolerance when HSP-90 is overexpressed in the intestine
Next, we investigated the role of CUL-6 in promoting thermotolerance in the context of HSP-90 overexpression. Our previous studies found that loss of cul-6 in a pals-22 mutant background impaired thermotolerance, but the loss of cul-6 in a wild-type background did not, suggesting that only when cul-6 is upregulated does it have a role in thermotolerance.7,15 However, we reasoned that if a CUL-6 ubiquitin ligase targets the HSP-90 protein, then there would be increased potential for an interaction between the HSP-90 protein with even low levels of the CUL-6 ubiquitin ligase complexes (i.e., in a wild-type background) when HSP-90 is overexpressed. Therefore, we predicted we might see a decrease in thermotolerance upon the loss of cul-6 in wild-type animals when HSP-90 is overexpressed. Indeed, we found that when HSP-90 is overexpressed in the intestine, the loss of cul-6 caused a decrease in thermotolerance to a level below that caused by HSP-90 overexpression alone (Figures 3B and S1B). Furthermore, we found that CUL-6 overexpression in the intestine increases thermotolerance in an HSP-90 overexpression background (Figures 3B and 3C). Importantly, overexpression of CUL-6(K73R) did not have an impact on thermotolerance when HSP-90 was also overexpressed, indicating that the effect of CUL-6 on thermotolerance is dependent on the neddylation site of CUL-6, as would be expected if thermotolerance were promoted by the activity of a CUL-6 ubiquitin ligase (Figures 3B and 3C).
The findings in Figures 3A and 3B suggest that thermotolerance is promoted by CUL-6-mediated degradation of HSP-90, which the findings shown in Figure 1 suggested would occur in the lysosome. Indeed, we found that treatment with the lysosome inhibitor bafilomycin in an HSP-90 overexpression background reduced thermotolerance even further, suggesting that normally, lysosomal-mediated degradation of HSP-90 promotes thermotolerance (Figure 3C). Furthermore, treatment with bafilomycin suppressed the loss or overexpression of CUL-6, as would be expected if the lysosome were downstream of CUL-6 (Figure 3C). To further test the model that the lysosome degrades HSP-90 to promote thermotolerance, we crossed a scav-3 mutation into the HSP-90 overexpression strain. Here, we found that a scav-3 mutation causes reduced thermotolerance in an HSP-90 overexpression background (Figure 3D), while it does not affect thermotolerance in a wild-type background (Figure 1D). Furthermore, SCAV-3::GFP leads to increased thermotolerance in an HSP-90 overexpression background (Figure S2E), perhaps due to increased lysosomal function caused by the overexpression of SCAV-3. While SCAV-3::GFP is not prominently visible in the intestine, possibly due to autofluorescence in this tissue, we do note that scav-3 transcripts have been found in the intestine, indicating that it is expressed there.24,36
Next, we examined whether lysosomal function was required specifically in the intestine to regulate thermotolerance. Because a scav-3 RNAi clone was not available and a vha-12 RNAi clone was, we used this clone to investigate whether the lysosome was required in the intestine for HSP-90 overexpression effects on thermotolerance. First, we used this vha-12 RNAi clone in a systemic RNAi strain, which confirmed our findings with vha-12 mutants (Figure 1E) showing that this lysosomal component is required for thermotolerance specifically in an HSP-90 overexpression background (Figure 3E). Next, we performed RNAi against vha-12 in an intestinal-specific RNAi strain and again found the suppression of thermotolerance specifically in an HSP-90 overexpression background, indicating that the lysosome is required in the intestine for these effects (Figure 3E). Altogether, these results support the model that a CUL-6 ubiquitin ligase promotes the degradation of HSP-90 in the intestine by the lysosomes to promote thermotolerance.
CUL-6 and lysosomal function regulate HSP-90 protein levels in the intestine
If CUL-6 ubiquitin ligases target HSP-90 for ubiquitylation and subsequent degradation by the lysosome, then the levels of HSP-90 protein should be regulated by CUL-6 and lysosomal function. To investigate this possibility, we quantified HSP-90::RFP levels in strains with loss or overexpression of CUL-6. Because endogenous CUL-6 is expressed more highly in the anterior intestine compared to the rest of the intestine,15 we focused our quantification on the anterior half of the intestine. Here, we found that HSP-90::RFP levels varied inversely with CUL-6 activity. In particular, we saw significantly higher HSP-90::RFP levels in cul-6 mutants compared to wild-type animals and significantly lower HSP-90::RFP levels in animals overexpressing wild-type CUL-6 specifically in the intestine but not in those expressing neddylation-deficient CUL-6 (Figures 4A–4C).
Figure 4. CUL-6 expression and lysosomal function reduce HSP-90 protein levels in the intestine.
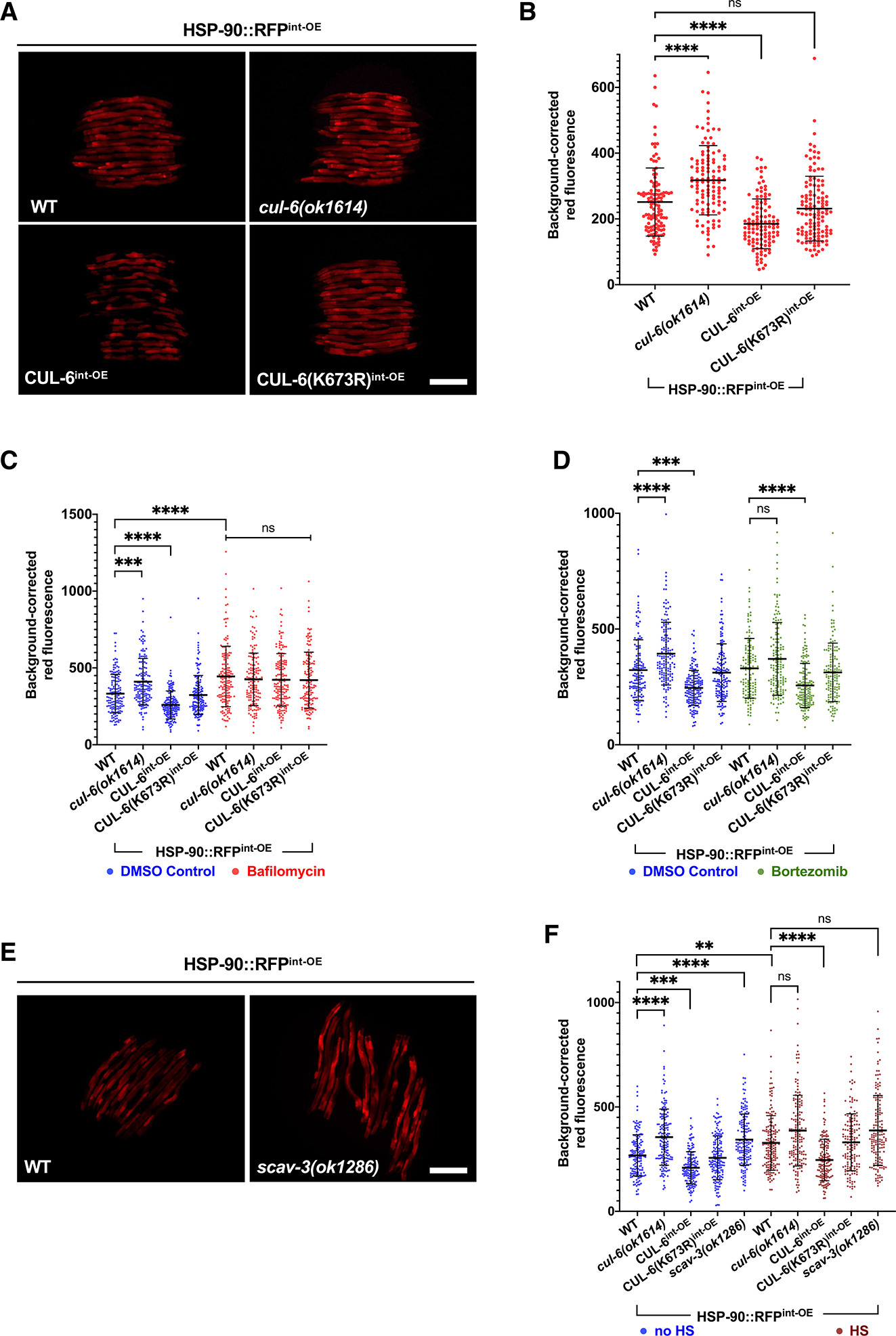
(A) Fluorescent images of L4 HSP-90int-OE L4 animals with mutation or overexpression of cul-6. Scale bar: 200 μm.
(B) Quantification of RFP signal in the anterior intestine of strains shown in (A). n = 113–125 worms quantified.
(C) Quantification of RFP fluorescent signal in the anterior intestine of L4 HSP-90int-OE animals with mutation or overexpression of cul-6 and treated with bafilomycin (red) or DMSO (blue) as a vehicle control. n = 141–166 worms quantified.
(D) Quantification of RFP fluorescent signal in the anterior intestine of L4 HSP-90int-OE animals with mutation or overexpression of cul-6 and treated with bortezomib (green) or DMSO (blue) as a vehicle control. n = 135–160 worms quantified.
(E) Fluorescent images of wild-type or scav-3(ok1286) mutant L4 animals expressing HSP-90int-OE. Scale bar: 200 μm.
(F) Quantification of RFP signal in the anterior intestine of HSP-90int-OE animals with mutation or overexpression of cul-6 in the absence of heat shock (blue) or 1 h after reduced heat shock treatment (maroon). n = 145–162 worms quantified.
For (B)–(D) and (F), each dot on the graph represents one animal. The black bar indicates the mean fluorescence intensity with error bars as SD. A Kruskal-Wallis test with Dunn’s multiple comparisons test was used to calculate p values. ****p < 0.0001, ***p < 0.001, and **p < 0.01.
To investigate the role of the lysosome in regulating HSP-90::RFP levels in the intestine, we treated HSP-90 overexpression animals with bafilomycin and then quantified HSP-90::RFP in the anterior intestine. Here, we saw higher levels of HSP-90 in animals treated with bafilomycin (Figure 4C). Furthermore, we found that bafilomycin treatment suppressed the effects of cul-6 mutation or overexpression on HSP-90::RFP levels, consistent with the model that the lysosome acts downstream of a CUL-6 ubiquitin ligase to regulate HSP-90::RFP protein levels (Figure 4C). As a control for specificity, when we treated HSP-90 overexpression animals with the proteasome inhibitor bortezomib, we saw that cul-6 mutation or overexpression still had an effect on HSP-90 levels (Figure 4D). We also investigated the role of the lysosome in regulating HSP-90::RFP levels by analyzing the effects of a scav-3 mutation. Again, we saw that loss of lysosomal function in scav-3 mutants led to increased levels of HSP-90 (Figures 4E and 4F), comparable to the effect of loss of cul-6 (Figure 4E). Furthermore, we found that cul-6 and scav-3 regulated the levels of HSP-90 both before and after heat shock (Figure 4F). These results suggest that CUL-6 and lysosomes degrade HSP-90 at normal growth temperatures, and the effects are still apparent after heat shock.
If HSP-90 proteins were targeted for ubiquitylation and degradation, then overexpression of ubiquitin might lower HSP-90 levels. Indeed, we found that overexpressing ubiquitin-GFP in intestinal cells led to lower HSP-90::RFP levels in the intestine (Figure S3). Ubiquitylation is the process of directly conjugating ubiquitin onto substrate proteins.13 Therefore, as a control for non-specific effects of ubiquitin-GFP overexpression that are unrelated to ubiquitylation, we overexpressed a conjugation-deficient ubiquitin-GFP, and here we saw no effect on HSP-90::RFP levels (Figure S3). Altogether, these findings support a model whereby wild-type cul-6 promotes ubiquitylation of HSP-90, which leads to its degradation in the lysosome to promote thermotolerance.
CUL-6 promotes HSP-90::RFP co-localization with LROs upon heat shock
To investigate if HSP-90 is targeted to the lysosome in a manner dependent on CUL-6, we imaged the subcellular localization of HSP-90::RFP before and after heat shock. Here, we saw that after heat shock, HSP-90::RFP localized to spherical structures in the intestine in a manner dependent on CUL-6; there were fewer spherical structures in cul-6 mutants and more structures upon wild-type CUL-6 intestinal overexpression but not upon neddylation-deficient CUL-6 intestinal overexpression (Figures 5A–5C and S4A). To determine whether this phenotype was specific to the overexpression of HSP-90 in the intestine and not a consequence of the RFP tag, we also looked at transgenic strains carrying an extrachromosomal array expressing intestinal HSP-90::GFP and found a similar CUL-6-dependent formation of ring-like structures after heat shock (Figures S4B and S4C).
Figure 5. CUL-6 promotes HSP-90::RFP co-localization with LROs upon heat shock.
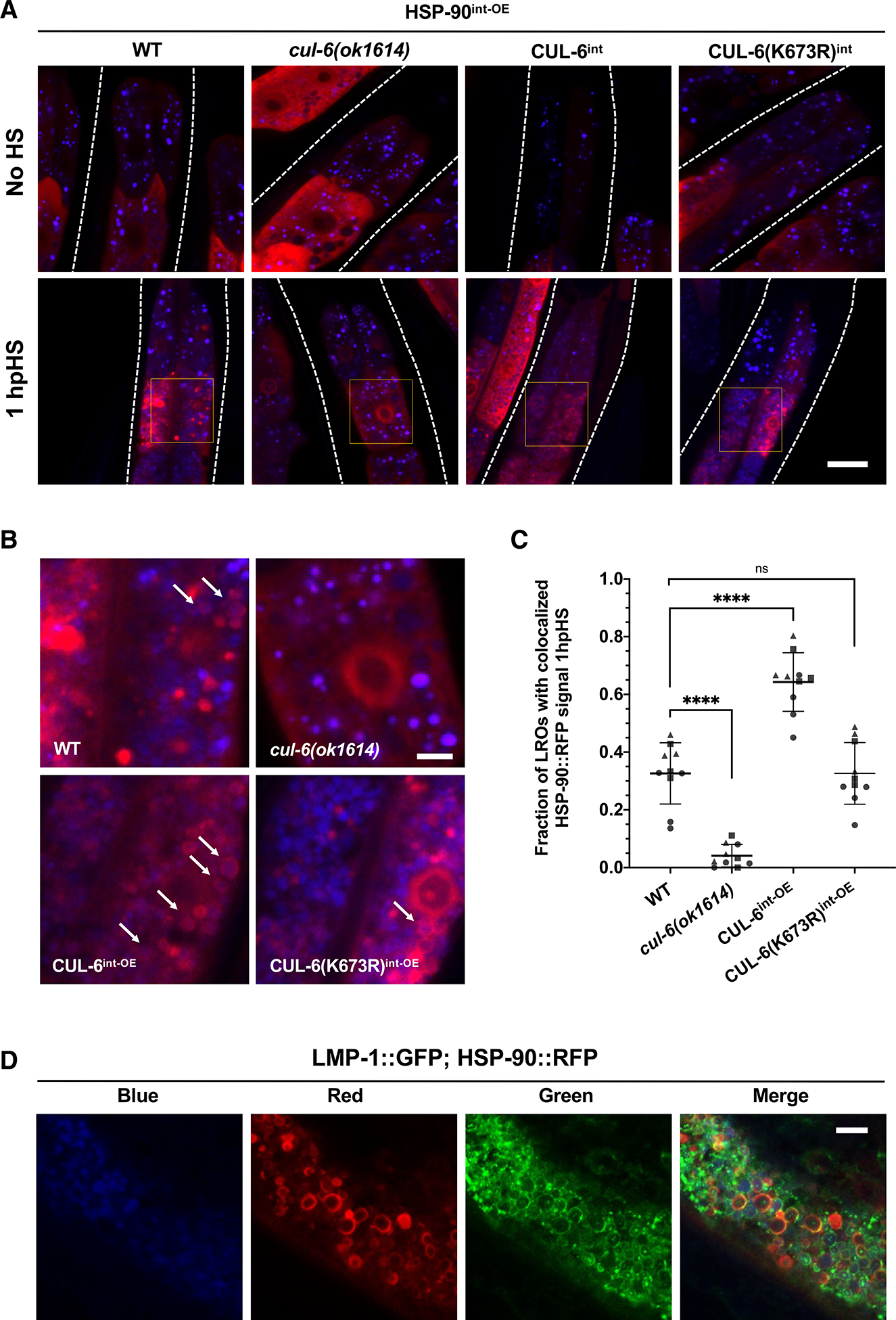
(A) Representative confocal fluorescence images of the anterior intestine in L4 animals with varying levels of CUL-6 expression in strains in an HSP-90int-OE background both 1 h after reduced heat shock and in the absence of heat shock. Lysosome-related organelles (LROs) were imaged using an autofluorescence signal in the blue channel. A region of interest showing co-localization events in the second ring of intestinal cells is highlighted in the image 1 h after reduced heat shock. See Figure S4A for individual channels of heat shocked animals. Scale bar: 20 μm.
(B) Enlarged regions of interest identified in (A) showing co-localization of HSP-90 to LROs. Arrows depict ring-like aggregation pattern of HSP-90::RFP upon heat shock around LROs. Scale bar: 5 μm.
(C) Quantification of the fraction of LROs with co-localized HSP-90 events after reduced heat shock treatment in the second ring of intestinal cells. n = 10 worms quantified per condition across three independent experiments. A one-way ANOVA with Tukey’s multiple comparisons test was used to calculate p values. ****p < 0.0001.
(D) Confocal fluorescence images of a representative L4 animal expressing both LMP-1::GFP and HSP-90::RFPint—OE 1 h after reduced heat shock. LROs were imaged using a blue autofluorescence signal. Scale bar: 5 μm.
Based on fluorescence in the blue channel, the spherical HSP-90::RFP structures are localized to LROs, which are abundant organelles in the intestine with autofluorescence prominently in this wavelength.37–40 LROs are multi-functional acidic compartments that express many canonical lysosomal markers and have degradative potential.41 To further characterize these HSP-90::RFP structures, we imaged them in animals with a GFP tag on the lysosomal protein LMP-142 and found that, indeed, LMP-1::GFP and HSP-90::RFP co-localize after heat shock (Figure 5D). Thus, our results indicate that the CUL-6 ubiquitin ligase directs HSP-90::RFP to lysosomes and/or LROs for degradation upon heat shock in the intestine.
DISCUSSION
Previously, we identified the IPR-regulated CUL-6 ubiquitin ligase as a novel proteostasis pathway in C. elegans that is upregulated by intracellular infection of the intestine and by proteotoxic stress, including chronic heat stress. In this study, we identified HSP-90 as a target of the CUL-6 ubiquitin ligase in defending the host from proteotoxic stress. In particular, we show that CUL-6 promotes the degradation of the HSP-90 protein by lysosomes and/or LROs in the intestine (Figure 6). Several other studies have shown that HSP-90 can bind to and negatively regulate HSF-1, a master regulator transcription factor of the heat shock response.22,23,43 Thus, we considered whether CUL-6-mediated degradation of HSP-90 may increase thermotolerance through the activation of HSF-1. However, we found that CUL-6-mediated degradation of HSP-90 appears to promote thermotolerance independent of HSF-1-inducible functions, a result consistent with our initial characterization of the IPR being independent of the canonical heat shock response mediated by HSF-1.7
Figure 6. A model for CUL-6-mediated degradation of HSP-90.
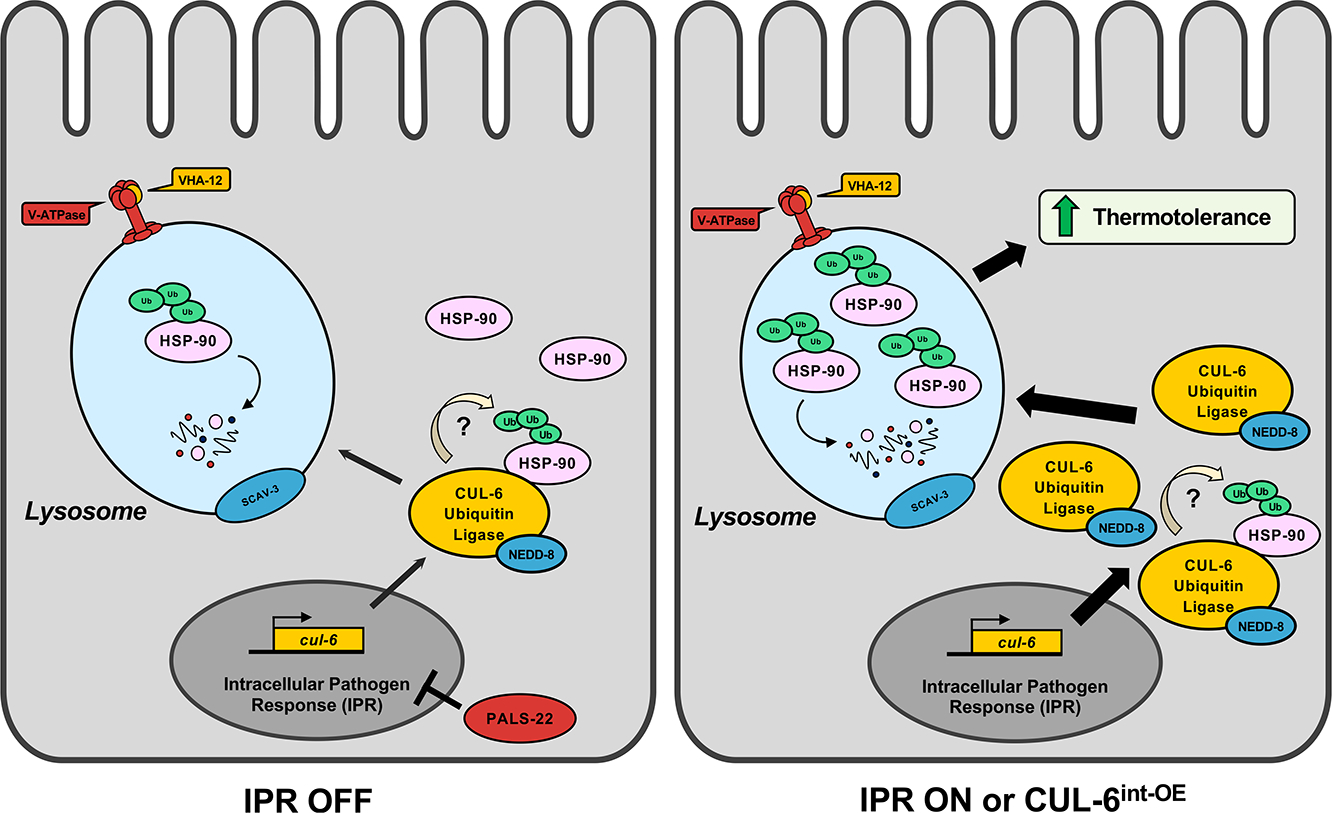
The cullin CUL-6 is upregulated as part of the IPR, a novel stress response pathway. HSP-90 is a substrate of the CUL-6 ubiquitin ligase complex and is degraded in the lysosome. The degradation of HSP-90 improves survival after heat shock stress, or thermotolerance. When the IPR is activated, upregulated expression of CUL-6 leads to increased trafficking of HSP-90 to the lysosome, resulting in higher thermotolerance relative to an inactive IPR state.
Identifying the CUL-6-mediated reduction of HSP-90 protein levels provides insights into the mechanisms by which the IPR promotes thermotolerance. Degradation of a heat shock protein to promote thermotolerance may seem counterintuitive because heat shock proteins as a protein family canonically promote thermotolerance, but it is important to note that HSP-90 is distinct from other heat shock proteins. HSP-90, together with its many co-chaperones, is a central player in protein folding in the absence of heat shock. Unlike other heat shock proteins, HSP-90 is highly expressed in the absence of heat shock, and in fact, the HSP-90 protein itself is thought to comprise 1%–2% of the entire proteome under unstressed conditions.19 Due to the high basal level of HSP-90 expression, even the modest ~25% increase or decrease of HSP-90 levels caused by the loss or overexpression of CUL-6 (Figure 4) may actually be a substantial change in the absolute amount of this highly abundant protein.
HSP-90 plays a broad role in facilitating folding and maturation of the proteome and is estimated to be required for the activity of up to 20% of all proteins and 60% of all kinases.44,45 Among its varied roles, HSP-90 promotes the active conformation of oncogenic kinases, aids the assembly of the multi-protein kineto-chore complex, and promotes ligand binding to steroid hormone receptors.19 All of these HSP-90 clients (proteins whose folding is facilitated by HSP-90) are important for cellular growth in the absence of heat shock. Therefore, one hypothesis to explain our findings is that degradation of HSP-90 in the C. elegans intestine leads to increased thermotolerance because it prevents maturation and/or degrades clients of HSP-90 that promote growth in the intestine. In this way, a CUL-6 ubiquitin ligase could promote the removal of multiple proteins at once after heat shock and provide a “factory reset” away from a growth state and toward a reparative state. However, altered levels of other HSP-90-associated proteins, as well as other distinct mechanisms, could explain our results showing that CUL-6-mediated degradation of HSP-90 promotes thermotolerance.
A simple model for our results is that degradation of HSP-90 in the intestine alone can promote organismal thermotolerance, but other tissues may also be involved. Studies of systemic signaling in stress responses have shown that RNAi knockdown of hsp-90 mRNA in either the intestine or in neurons triggers the upregulation of hsp-70 mRNA expression in muscle cells to promote thermotolerance, a phenomenon named “transcellular chaperone signaling.”20,46,47 These effects were recently found to be independent of the inducible functions of HSF-1, as are the effects we see here with CUL-6. Therefore, some of the thermotolerance benefits from CUL-6-mediated loss of the HSP-90 protein may be due to cell non-autonomous signaling to muscle. In fact, the IPR may provide a physiologically relevant stimulus to explain the activation of transcellular chaperone signaling, which has previously been studied through RNAi knockdown. Perhaps this form of systemic signaling is normally triggered by natural intracellular infection of the intestine.
While HSP-90 functions to fold its client proteins, it also aids in their degradation.48–51 To our knowledge, this study is the first to show the degradation of HSP-90 itself by the lysosome. Numerous drugs inhibit HSP-90, such as those developed to treat cancer that lead to the degradation of oncogenic kinase clients of HSP-90.52 Thus, if the pathway we have identified for degradation of HSP-90 itself in C. elegans is conserved in humans, then our findings may have relevance for the efficacy of those drugs. Several studies in C. elegans have now shown that a reduction of protein quality control factors can paradoxically promote organismal survival. In addition to the transcellular chaperone signaling described above, the repression of hsp-70 mRNA by microRNA after heat shock has been shown to aid in heat shock recovery.53 Another example is a recent study showing that inhibition of HSF-1 causes decreased protein aggregation and better functioning specifically of the pharynx.54 Interestingly, this protective benefit in the pharynx is associated with the upregulation of IPR genes and relies on the same lysosomal factors we found were important for the thermotolerance benefit of CUL-6-mediated degradation of HSP-90 in our study. In the future, it will be exciting to determine the connection among all these findings and to uncover the downstream mechanism(s) by which degradation of the central proteostasis factor HSP-90 promotes organismal survival after heat shock.
Limitations of the study
While our model posits that a CUL-6 ubiquitin ligase ubiquity-lates HSP-90 to send it to the lysosome for degradation, we have not directly shown this ubiquitylation event. Demonstrating this event will likely not be easy because of the modest relative change in HSP-90 levels (Figure 4). In addition, there are at least 7 subunits to the CUL-6 ubiquitin ligase complex(es),15 making biochemical reconstitution of this ligase together with candidate substrates difficult. Furthermore, HSP-90 has 42 lysines that are predicted ubiquitylation sites (as assessed by prediction software on this site: https://www.biocuckoo.org/), making it challenging to determine which sites are important for ubiquitylation. With these ideas in mind, a direct investigation of HSP-90 ubiquitylation by a CUL-6 ubiquitin ligase will be part of future studies. Another limitation is that we did not define the kinetics or other aspects of the degradation mechanism of HSP-90::RFP in LROs. Furthermore, all analyses were performed using HSP-90::RFP overexpression strains. While a similar C-terminally tagged HSP-90::GFP transgene rescues HSP-90 function in yeast,26 the rescue of either HSP-90::GFP or HSP-90::RFP has not been shown in C. elegans, and we did not investigate endogenous HSP-90 levels. An analysis of the functionality of HSP-90::RFP in C. elegans, as well as an analysis of endogenous HSP-90 levels, ubiquitylation status, and relocalization to LROs, can be part of future studies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further Information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Emily Troemel (etroemel@ucsd.edu).
Materials availability
C. elegans strains generated in this study are available upon request.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
The nematode Caenorhabditis elegans was used as the experimental model for this study. All experiments were performed with hermaphroditic animals, and males were used only for crosses. All experiments were carried out with L4 animals. Strains were maintained at 20°C on Nematode Growth Media (NGM) seeded with Streptomycin-resistant E. coli OP50–1 bacteria according to standard seeding methods. All strains were backcrossed a minimum of three times prior to analysis where applicable (see Table S2).
METHOD DETAILS
C. elegans maintenance and strain generation
Worms were maintained using standard methods at 20°C on Nematode Growth Media (NGM) agar plates top-plated with streptomycin-resistant Escherichia coli OP50–1 unless stated otherwise.55,56 Worm strains used in this study are listed in Table S2.
Synchronization of C. elegans
To obtain synchronized populations of C. elegans for fluorescent imaging experiments described below, gravid adult animals were collected from NGM+OP50–1 plates in M9 buffer into a 15 mL conical tube. The tubes were centrifuged, and the supernatant was removed, leaving animals in ~2 mL of M9. 800 μL of 5.65–6% sodium hypochlorite solution and 200 μL of 2M NaOH were added to the tube, and the contents were vigorously shaken for approximately 1 min and 40 s. Embryos released after bleaching were resuspended in 15 mL of M9, centrifuged, and the supernatant was discarded. The embryos were washed with M9 in this manner a total of 5 times, resuspended in a final volume of 5 mL of M9, and then placed in a 20°C incubator under continuous rotation for 16–24 h until L1s hatched.
Thermotolerance assays
Heat shock treatment: Gravid adults were picked to 6-cm NGM+OP50–1 plates and grown at 20°C. 30 F1 progeny from these adults were picked at the L4 life stage to fresh NGM+OP50–1 plates and subjected to a heat shock of 37.5°C in a dry programmable incubator for 2 h and 15 min with an initial gradual ramp-up to 37.5°C over a 15 min period (2 h stable at 37.5°C). Immediately following the completion of the 2-h and 15 min heat shock program, the plates were removed from the incubator. The animals were allowed to recover for 30 min at room temperature by placing the plates in a single layer on a benchtop, then incubated at 20°C for 24 h. Animals were then scored for survival in a blinded manner; worms not responding to touch with a worm pick, defined by a single prod to the body, were scored as dead. 3 replicate plates were scored for each condition per experiment, and the experiment was performed 3 independent times, except as noted for the DMSO and bortezomib treatment experiment performed in Figure 1A. A summary of the heat shock treatment and experiments where it was applied in this study is provided in Figure S1A.
Reduced heat shock treatment: A modified version of the assay with a shorter duration was used to accommodate the increased heat shock susceptibility of the HSP-90:RFPint—OE strain. Following the standard protocol described above, most HSP-90:RFPint—OE animals died. Therefore, to improve survival and allow for the analysis of HSP-90 intestinal overexpression on raising or lowering thermotolerance phenotypes in different contexts, the assay time was shortened to 2 h of 37.5°C heat shock with a 15 min gradual ramp-up (1 h 45 min stable at 37.5°C). Immediately following the completion of the 2-h heat shock program, the plates were removed from the incubator. Recovery, scoring, and experimental replicates were performed as described above. A summary of the reduced heat shock treatment and experiments where it was applied in this study is provided in Figure S1B.
RNAi experiments
RNAi was performed by the feeding method.57 Overnight cultures of OP50 strain (R)OP50, modified to enable RNAi58 were plated on 6-cm RNAi plates (NGM plates supplemented with 5mM β-D-1-thiogalactopyranoside [IPTG], 1 mM carbenicillin), and incubated at 20°C for 3 days. Gravid adults were transferred to these plates for growth. The F1 progeny at the L4 stage were transferred to new, matching 6-cm RNAi plates before being tested for thermotolerance as previously described. OP50 RNAi strains were generated by extracting the desired RNAi plasmid vector from HT115 E. coli in the existing Ahringer and Vidal RNAi libraries.59 The plasmids were then transformed into competent (R)OP50, and transformants were selected after ~24 h of growth based on carbenicillin resistance. Overnight cultures of transformations were then mini-prepped, and the L4440 plasmid vector was sequenced with the T7 forward primer to confirm that the insert matching the desired gene for knockdown studies was present in the vector.
Bortezomib treatment
The proteasome was inhibited with bortezomib (Selleckchem Chemicals LLC, Houston, TX) as previously described.11 For thermotolerance assays, gravid adults were plated onto 10-cm NGM+OP50–1 plates and incubated at 20°C for 72 h. Before performing heat shock treatment, a 10 mM stock solution of bortezomib in DMSO was suspended in enough M9 to cover the surface of a standard 6-cm plate and top-plated to 6-cm NGM+OP50–1 plates to reach a final concentration of 2 μM, 10 μM, or 20 μM per plate after evaporation of M9. The same volume of DMSO in M9 was added to the control plates. The plates were dried, the drug was allowed to equilibrate within plates for 1 h, and 30 L4 F1 animals were transferred onto each treatment or control plate. The plates were then subjected to the standard heat shock treatment, recovery, and scoring regimen described above.
To quantify HSP-90:RFP fluorescence, synchronized L1s were plated on 10 cm NGM+OP50–1 plates and grown for 44 h at 20°C. A 10 mM stock solution of bortezomib in DMSO was top-plated to reach a final concentration of 20 μM per plate, and the same volume of DMSO was added to control plates. Plates were dried, and worms were incubated for 4 h at 20°C. The animals were washed off the treatment plates in M9+tween 20, pelleted, anesthetized with a final concentration of 10mM levamisole, and transferred into a 96-well plate with fresh 10 mM levamisole in M9+tween 20. Imaging was performed using the ImageXpress Nano using a 4x objective (Molecular Devices, LLC) and analyzed using the FIJI program. Quantification of fluorescent signal was restricted to the anterior intestine, defined for the purposes of this assay as the beginning of the intestine to the midpoint of the vulva region, since CUL-6 is primarily expressed in the anterior intestine and we would expect that mutation of endogenous CUL-6 would have the greatest effect on HSP-90 levels in this region. The background signal was collected from three adjacent regions and the mean subtracted from the signal measured in the anterior intestines of animals.
Bafilomycin treatment
The lysosome was inhibited with bafilomycin A1 (AdipoGen Life Sciences, Liestal, Switzerland). For thermotolerance assays, a 25 mM stock solution of bafilomycin in DMSO was suspended in enough M9 to cover the surface of a standard 6-cm plate and was added to 6-cm NGM+OP50–1 plates to reach a final concentration of 1 μM or 3 μM per plate after evaporation of M9. The same volume of DMSO in M9 was added to the control plates. Plates were dried, allowed to equilibrate for 1 h, gravid adults were added, and the plates were then incubated for 72 h at 20°C. 30 L4 F1 animals from each plate were then transferred onto freshly prepared, equilibrated, and dried plates with matching treatments. The plates were then subjected to the standard heat shock treatment, recovery, and scoring regimen described above.
To quantify HSP-90:RFP fluorescence, a 25 mM stock solution of bafilomycin in DMSO suspended in enough M9 to cover the surface of a standard 10-cm plate was top-plated to a 10-cm NGM+OP50–1 plate to reach a final concentration of 3 μM per plate. The same volume of DMSO in M9 was added to the control plates. Once the plates were dry and equilibrated for 1 h, synchronized L1s were added, and the plates were incubated for 48 h at 20°C. The animals were washed off the treatment plates in M9+tween 20, pelleted, anesthetized with a final concentration of 10mM levamisole, and transferred into a 96-well plate with fresh 10 mM levamisole in M9+tween 20. Imaging was performed using the ImageXpress Nano using a 4x objective (Molecular Devices, LLC) and analyzed using the FIJI program. Quantification was restricted to the anterior intestine and background-corrected as described above.
Microscopy
For HSP-90:RFP-expressing animals shown in Figures 4A and 4E, and hsp-16.2p::GFP animals shown in S2A, L4 animals were picked from a mixed population grown on 6-cm NGM+OP50–1 plates incubated at 20°C, anesthetized with 10 mM levamisole in M9 buffer, and mounted on 2% agarose pads for imaging on a Zeiss Axioimager M1 compound microscope with a 10X objective. For S2A, whole animal fluorescent signal was quantified using the FIJI program, corrected by the mean background sampled from three adjacent background regions.
For images of HSP-90:RFP in the absence of and 1 h after heat shock (Figure 5), animals were synchronized by bleaching as described above, then cultured on 10 cm NGM+OP50–1 plates for 44 h at 20°C. While control (no heat shock) animals remained at 20°C, animals assigned to heat shock treatment were subjected to a reduced heat shock exposure (2 h with a 15-min ramp up to 37.5°C, 1 h and 45 min stable at 37.5°C), and allowed to recover for the standard 30 min at room temperature. Plates were then incubated at 20°C for 1 h before all conditions were anesthetized with 10 mM levamisole in M9 buffer and mounted on 2% agarose pads for imaging with a Zeiss LSM700 confocal microscope and Zen2010 software using a 63X objective. For quantification, the fraction of LROs in the second ring of intestinal cells as labeled by autofluorescence in the blue channel with positive HSP-90:RFP colocalization over total number of LROs in the region of interest was recorded for a total of four animals per condition.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analysis was performed with Prism 9 software (GraphPad). The D’Agostino & Pearson omnibus normality test was used to assess the data distribution for all experiments. Standard parametric statistical tests were performed for normally distributed data, and nonparametric tests were performed for non-normally distributed data. The corresponding figure legends describe the statistical test used for each experiment, the number of data points analyzed, replicate information, and other relevant details.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and virus strains | ||
|
| ||
| E. coli: OP50-1 | Caenorhabditis Genetics Center | N/A |
| E. coli: (R)OP50 | Lynn et al.54 | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Bortezomib | Sellecheck Chemicals | Cat#S1013 |
| Bafilomycin A1 | AdipoGen | Cat#BVT0252M001 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C. elegans: Strain N2 wild-type | Caenorhabditis Genetics Center | N2 |
| C. elegans: Strain ERT356 pals-22(jy1) III | Reddy et al.7 | ERT356 |
| C. elegans: Strain ERT571 jySi42[pET499(vha-6p::GFP::cul-6::unc-54 3’ UTR, unc-119(+))] II; unc-119(ed3) III | Panek et al.15 | ERT571 |
| C. elegans: Strain ERT740 jySi46[pET688(vha-6p::GFP::cul-6(K673R)::unc-54 3’ UTR, unc-119(+))] II; unc-119(ed3) III | Panek et al.15 | ERT740 |
| C. elegans: Strain RB938 vha-12(ok821) X | Caenorhabditis Genetics Center | WB strain: RB938 |
| C. elegans: Strain AM994 hsp-90control sid-1(pk3321); rmIs288(myo-2p::CFP;hsp-70p::mCherry) | Van Oosten-Hawle et al.26 | AM994 |
| C. elegans: Strain PVH1 sid-1(pk3321) V;rmIs288[myo-2p::CFP;hsp-70p::mCherry]; pcIs001[rgef-1p::hsp-90RNAi::unc-54 3’UTR] | Van Oosten-Hawle et al.26 | PVH1 |
| C. elegans: Strain PVH2 sid-1(pk3321) V;rmIs288;pcIs002[vha-6p::hsp-90RNAi::unc-54 3’UTR] | Van Oosten-Hawle et al.26 | PVH2 |
| C. elegans: Strain PS3551 hsf-1(sy441) I | Caenorhabditis Genetics Center | WB strain: PS3551 |
| C. elegans: Strain ERT1006 scav-3(ok1286) III | Caenorhabditis Genetics Center | WB strain: RB1227 |
| C. elegans: Strain ERT1236 rmIs346[vha-6p::HSP-90::RFP] | Van Oosten-Hawle et al.26 | ERT1236 |
| C. elegans: Strain ERT1226 rmIs345[F25B3.3p::HSP-90::RFP] | Van Oosten-Hawle et al.26 | ERT1226 |
| C. elegans: Strain ERT1227 rmIs347[unc-54p::HSP-90::RFP] | Van Oosten-Hawle et al.26 | ERT1227 |
| C. elegans: Strain ERT1166 njIs11[glr-3p::GFP + ges-1p::RFP] | Caenorhabditis Genetics Center | WB strain: IK716 |
| C. elegans: Strain ERT1167 njIs12[glr-3p::glr-1::GFP + glr-3p::RFP + ges-1p::RFP] | Caenorhabditis Genetics Center | WB strain: IK718 |
| C. elegans: Strain ERT1004 pals-22(jy1) scav-3(ok1286) III | This paper | ERT1004 |
| C. elegans: Strain ERT1152 jySi42 II; unc-119(ed3) scav-3(ok1286) III | This paper | ERT1152 |
| C. elegans: Strain ERT1046 cul-6(ok1614) IV;rmIs346 | This paper | ERT1046 |
| C. elegans: Strain ERT1104 jySi42 II; unc-119(ed3) III; rmIs346 | This paper | ERT1104 |
| C. elegans: Strain ERT1136 jySi46 II; rmIs346 | This paper | ERT1136 |
| C. elegans: Strain ERT1212 scav-3(ok1286) III; rmIs346 | This paper | ERT1212 |
| C. elegans: Strain ERT1265 frSi17[mtl-2p::rde-1 3’UTR] II; rde-1(ne300) V | Caenorhabditis Genetics Center | WB strain: IG1839 |
| C. elegans: Strain ERT1295 frSi17[mtl-2p::rde-1 3’UTR] II; rde-1(ne300) V; rmIs346[vha-6p::HSP-90::RFP] | This paper | ERT1295 |
| C. elegans: Strain ERT1297 LMP-1:GFP;HSP-90::RFP | This paper | ERT1297 |
| C. elegans: Strain CL2070 dvIs70[hsp-16-2p::GFP + pRF4 rol-6(su1006)] | Caenorhabditis Genetics Center | WB strain: CL2070 |
| C. elegans: Strain ERT1191 hsf-1(sy4410) I; rmIs346 | This paper | ERT1191 |
| C. elegans: Strain ERT1210 qxIs430 [scav-3::GFP + unc-76(+)] | Caenorhabditis Genetics Center | WB strain: XW8056 |
| C. elegans: Strain ERT1237 qxIs430;rmIs346 | This paper | ERTI 237 |
| C. elegans: Strain ERT1238 jyEx128 [vha-6p::GFP::UBQ, cb-unc-119(+)]; unc-119(ed3)III; rmIs346 | This paper | ERTI 238 |
| C. elegans: Strain ERT1239 jyEx131 [vha-6p::GFP::UBQdeltaGG, cb-unc-119(+)]; unc-119(ed3)III; rmIs346 | This paper | ERTI 239 |
| C. elegans: Strain ERT1298 rmEx315[vha-6p:: HSP-90::GFP; myo-2p::mCherry] | Van Oosten-Hawle et al.26 | ERTI 298 |
| C. elegans: Strain ERT1299 cul-6(ok1614) IV;vha-6p::HSP-90::GFP | This paper | ERTI 299 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S1 for oligonucleotide sequences used in this study | N/A | |
|
| ||
| Recombinant DNA | ||
|
| ||
| Plasmid: pL4440-RNAi control (OP50) | Ahringer RNAi Library | N/A |
| Plasmid: pL4440-unc-22 (OP50) | Ahringer RNAi Library | N/A |
| Plasmid: pL4440-pals-22 (OP50) | Ahringer RNAi Library | N/A |
| Plasmid: pL4440-hsp-90 (OP50) | Vidal RNAi Library | N/A |
| Plasmid: pL4440-hsf-1 (OP50) | Ahringer RNAi Library | N/A |
| Plasmid: pL4440-vha-12 (OP50) | Ahringer RNAi Library | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | NIH Image | RRID:SCR_003070 |
| GraphPad Prism 8 | GraphPad Software, Inc. | RRID:SCR_002798 |
| ZEISS ZEN Microscopy Software | Carl Zeiss AG | RRID:SCR_013672 |
Highlights.
CUL-6 ubiquitin ligase decreases HSP-90 in the intestine to improve thermotolerance
The lysosome decreases HSP-90 in the intestine to improve thermotolerance
Heat shock directs HSP-90 to lysosome-related organelles in a CUL-6-dependent manner
ACKNOWLEDGMENTS
We thank Michalis Barkoulas, Lakshmi Batachari, Aundrea Koger, Katie Li, Johan Panek, Deevya Raman, and Nicole Wernet for helpful comments on the manuscript. Some strains used in this study were provided by the Caenorhabditis Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This work was supported by NIH under R01 AG052622, GM114139, and AI176639 and NSF 2301657 to E.R.T., NIGMS/NIH award K12GM068524 to S.S.G., and NIH R01 AG082970 to P.v.O.-H.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114279.
REFERENCES
- 1.Lindquist S (1986). The heat-shock response. Annu. Rev. Biochem. 55, 1151–1191. 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- 2.Santiago AM, Gonç alves., and Morano KA. (2020). Mechanisms of sensing and response to proteotoxic stress. Exp. Cell Res. 395, 112240. 10.1016/j.yexcr.2020.112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fujimoto M, and Nakai A (2010). The heat shock factor family and adaptation to proteotoxic stress. FEBS J. 277, 4112–4125. 10.1111/j.1742-4658.2010.07827.x. [DOI] [PubMed] [Google Scholar]
- 4.Anckar J, and Sistonen L (2011). Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu. Rev. Biochem. 80, 1089–1115. 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- 5.Wu C (1995). Heat shock transcription factors: structure and regulation. Annu. Rev. Cell Dev. Biol. 11, 441–469. 10.1146/annurev.cb.11.110195.002301. [DOI] [PubMed] [Google Scholar]
- 6.Kyriakou E, Taouktsi E, and Syntichaki P (2022). The Thermal Stress Coping Network of the Nematode Caenorhabditis elegans. Int. J. Mol. Sci. 23, 14907. 10.3390/ijms232314907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reddy KC, Dror T, Sowa JN, Panek J, Chen K, Lim ES, Wang D, and Troemel ER (2017). An Intracellular Pathogen Response Pathway Promotes Proteostasis in C. elegans. Curr. Biol. 27, 3544–3553.e5. 10.1016/j.cub.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bakowski MA, Desjardins CA, Smelkinson MG, Dunbar TL, Lopez-Moyado IF, Rifkin SA, Cuomo CA, and Troemel ER (2014). Ubiquitin-mediated response to microsporidia and virus infection in C. elegans. PLoS Pathog. 10, e1004200. 10.1371/journal.ppat.1004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tecle E, and Troemel ER (2022). Insights from C. elegans into Micro-sporidia Biology and Host-Pathogen Relationships. Exper. Suppl. (Basel) 114, 115–136. 10.1007/978-3-030-93306-7_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lažetic V,V., Batachari LE., Russell AB., and Troemel ER. (2023). Similarities in the induction of the intracellular pathogen response in Caenorhabditis elegans and the type I interferon response in mammals. Bio-essays 45, e2300097. 10.1002/bies.202300097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reddy KC, Dror T, Underwood RS, Osman GA, Elder CR, Desjardins CA, Cuomo CA, Barkoulas M, and Troemel ER (2019). Antagonistic paralogs control a switch between growth and pathogen resistance in C. elegans. PLoS Pathog. 15, e1007528. 10.1371/jour-nal.ppat.1007528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gang SS, Grover M, Reddy KC, Raman D, Chang YT, Ekiert DC, Barkoulas M, and Troemel ER (2022). A pals-25 gain-of-function allele triggers systemic resistance against natural pathogens of C. elegans. PLoS Genet. 18, e1010314. 10.1371/journal.pgen.1010314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang S, and Weissman AM (2004). A field guide to ubiquitylation. Cell. Mol. Life Sci. 61, 1546–1561. 10.1007/s00018-004-4129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petroski MD, and Deshaies RJ (2005). Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20. 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 15.Panek J, Gang SS, Reddy KC, Luallen RJ, Fulzele A, Bennett EJ, and Troemel ER (2020). A cullin-RING ubiquitin ligase promotes thermotolerance as part of the intracellular pathogen response in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 117, 7950–7960. 10.1073/pnas.1918417117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, and Schulman BA (2008). Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell 134, 995–1006. 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuang E, Qi J, and Ronai Z (2013). Emerging roles of E3 ubiquitin ligases in autophagy. Trends Biochem. Sci. 38, 453–460. 10.1016/j.tibs.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao Y, Sumara I, and Pangou E (2022). Non-proteolytic ubiquitylation in cellular signaling and human disease. Commun. Biol. 5, 114. 10.1038/s42003-022-03060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiosis G, Digwal CS, Trepel JB, and Neckers L (2023). Structural and functional complexity of HSP90 in cellular homeostasis and disease. Nat. Rev. Mol. Cell Biol. 24, 797–815. 10.1038/s41580-023-00640-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Oosten-Hawle P (2023). Organismal Roles of Hsp90. Biomolecules 13. 10.3390/biom13020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanchez J, Carter TR, Cohen MS, and Blagg BSJ (2020). Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets 20, 253–270. 10.2174/1568009619666191202101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zou J, Guo Y, Guettouche T, Smith DF, and Voellmy R (1998). Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94, 471–480. 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- 23.Janssens GE, Lin XX, Millan-Arino L, Kavsek A, Sen I, Seinstra RI, Stroustrup N, Nollen EAA, and Riedel CG (2019). Transcriptomics-Based Screening Identifies Pharmacological Inhibition of Hsp90 as a Means to Defer Aging. Cell Rep. 27, 467–480. 10.1016/j.celrep.2019.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y, Chen B, Zou W, Wang X, Wu Y, Zhao D, Sun Y, Liu Y, Chen L, Miao L, et al. (2016). The lysosomal membrane protein SCAV-3 maintains lysosome integrity and adult longevity. J. Cell Biol. 215, 167–185. 10.1083/jcb.201602090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ernstrom GG, Weimer R, Pawar DRL, Watanabe S, Hobson RJ, Greenstein D, and Jorgensen EM (2012). V-ATPase V1 sector is required for corpse clearance and neurotransmission in Caenorhabditis elegans. Genetics 191, 461–475. 10.1534/genetics.112.139667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Oosten-Hawle P, Porter RS, and Morimoto RI (2013). Regulation of organismal proteostasis by transcellular chaperone signaling. Cell 153, 1366–1378. 10.1016/j.cell.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miles J, Townend S, Milonaitytė D, Smith W, Hodge F, Westhead DR, and van Oosten-Hawle P (2023). Transcellular chaperone signaling is an intercellular stress-response distinct from the HSF-1-mediated heat shock response. PLoS Biol. 21, e3001605. 10.1371/jour-nal.pbio.3001605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calixto A, Chelur D, Topalidou I, Chen X, and Chalfie M (2010). Enhanced neuronal RNAi in C. elegans using SID-1. Nat. Methods 7, 554–559. 10.1038/nmeth.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhattacharya K, Maiti S, Zahoran S, Weidenauer L, Hany D, Wider D, Bernasconi L, Quadroni M, Collart M, and Picard D (2022). Translational reprogramming in response to accumulating stressors ensures critical threshold levels of Hsp90 for mammalian life. Nat. Commun. 13, 6271. 10.1038/s41467-022-33916-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hajdu-Cronin YM, Chen WJ, and Sternberg PW (2004). The L-type cyclin CYL-1 and the heat-shock-factor HSF-1 are required for heat-shock-induced protein expression in Caenorhabditis elegans. Genetics 168, 1937–1949. 10.1534/genetics.104.028423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Chauve L, Phelps G, Brielmann RM, and Morimoto RI (2016). E2F coregulates an essential HSF developmental program that is distinct from the heat-shock response. Genes Dev. 30, 2062–2075. 10.1101/gad.283317.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McColl G, Rogers AN, Alavez S, Hubbard AE, Melov S, Link CD, Bush AI, Kapahi P, and Lithgow GJ (2010). Insulin-like signaling determines survival during stress via posttranscriptional mechanisms in C. elegans. Cell Metabol. 12, 260–272. 10.1016/j.cmet.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kourtis N, Nikoletopoulou V, and Tavernarakis N (2012). Small heat-shock proteins protect from heat-stroke-associated neurodegeneration. Nature 490, 213–218. 10.1038/nature11417. [DOI] [PubMed] [Google Scholar]
- 34.Hsu AL, Murphy CT, and Kenyon C (2003). Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300, 1142–1145. 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- 35.Prahlad V, Cornelius T, and Morimoto RI (2008). Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science 320, 811–814. 10.1126/science.1156093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, Qiu X, Lee C, Furlan SN, Steemers FJ, et al. (2017). Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667. 10.1126/science.aam8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schroeder LK, Kremer S, Kramer MJ, Currie E, Kwan E, Watts JL, Lawrenson AL, and Hermann GJ (2007). Function of the Caenorhabditis elegans ABC transporter PGP-2 in the biogenesis of a lysosome-related fat storage organelle. Mol. Biol. Cell 18, 995–1008. 10.1091/mbc.e06-08-0685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hermann GJ, Schroeder LK, Hieb CA, Kershner AM, Rabbitts BM, Fonarev P, Grant BD, and Priess JR (2005). Genetic analysis of lysosomal trafficking in Caenorhabditis elegans. Mol. Biol. Cell 16, 3273–3288. 10.1091/mbc.e05-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hajdú G, Somogyvari M, Csermely P, and Soti C (2023). Lysosome-related organelles promote stress and immune responses in C. elegans. Commun. Biol. 6, 936. 10.1038/s42003-023-05246-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGhee JD (2007). The C. elegans Intestine (WormBook), pp. 1–36. 10.1895/wormbook.1.133.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watts C (2022). Lysosomes and lysosome-related organelles in immune responses. FEBS Open Bio 12, 678–693. 10.1002/2211-5463.13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hadwiger G, Dour S, Arur S, Fox P, and Nonet ML (2010). A monoclonal antibody toolkit for C. elegans. PLoS One 5, e10161. 10.1371/journal.pone.0010161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eckl J, Sima S, Marcus K, Lindemann C, and Richter K (2017). Hsp90-downregulation influences the heat-shock response, innate immune response and onset of oocyte development in nematodes. PLoS One 12, e0186386. 10.1371/journal.pone.0186386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, and Lindquist S (2012). Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 150, 987–1001. 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taipale M, Jarosz DF, and Lindquist S (2010). HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 11, 515–528. 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 46.Hodge F, Bajuszova V, and van Oosten-Hawle P (2022). The Intestine as a Lifespan- and Proteostasis-Promoting Signaling Tissue. Front. Aging 3, 897741. 10.3389/fragi.2022.897741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Oosten-Hawle P (2023). Exploiting inter-tissue stress signaling mechanisms to preserve organismal proteostasis during aging. Front. Physiol. 14, 1228490. 10.3389/fphys.2023.1228490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trepel J, Mollapour M, Giaccone G, and Neckers L (2010). Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 10, 537–549. 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prince TL, Lang BJ, Okusha Y, Eguchi T, and Calderwood SK (2023). Cdc37 as a Co-chaperone to Hsp90. Subcell. Biochem. 101, 141–158. 10.1007/978-3-031-14740-1_5. [DOI] [PubMed] [Google Scholar]
- 50.Samant RS, Clarke PA, and Workman P (2014). E3 ubiquitin ligase Cullin-5 modulates multiple molecular and cellular responses to heat shock protein 90 inhibition in human cancer cells. Proc. Natl. Acad. Sci. USA 111, 6834–6839. 10.1073/pnas.1322412111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Z, Zhou L, Prodromou C, Savic V, and Pearl LH (2017). HECTD3 Mediates an HSP90-Dependent Degradation Pathway for Protein Kinase Clients. Cell Rep. 19, 2515–2528. 10.1016/j.celrep.2017.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Magwenyane AM, Ugbaja SC, Amoako DG, Somboro AM, Khan RB, and Kumalo HM (2022). Heat Shock Protein 90 (HSP90) Inhibitors as Anticancer Medicines: A Review on the Computer-Aided Drug Discovery Approaches over the Past Five Years. Comput. Math. Methods Med. 2022, 2147763. 10.1155/2022/2147763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pagliuso DC, Bodas DM, and Pasquinelli AE (2021). Recovery from heat shock requires the microRNA pathway in Caenorhabditis elegans. PLoS Genet. 17, e1009734. 10.1371/journal.pgen.1009734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jung R, Lechler MC, Fernandez-Villegas A, Chung CW, Jones HC, Choi YH, Thompson MA, Rödelsperger C, Röseler W, Kaminski Schierle GS, et al. (2023). A safety mechanism enables tissue-specific resistance to protein aggregation during aging in C. elegans. PLoS Biol. 21, e3002284. 10.1371/journal.pbio.3002284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stiernagle T (2006). Maintenance of C. elegans (WormBook), pp. 1–11. 10.1895/wormbook.1.101.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, and Ahringer J (2001). Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2, RESEARCH0002. 10.1186/gb-2000-2-1-research0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lynn DA, Dalton HM, Sowa JN, Wang MC, Soukas AA, and Curran SP (2015). Omega-3 and −6 fatty acids allocate somatic and germline lipids to ensure fitness during nutrient and oxidative stress in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 112, 15378–15383. 10.1073/pnas.1514012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jagadeesan S, and Hakkim A (2018). RNAi Screening: Automated High-Throughput Liquid RNAi Screening in Caenorhabditis elegans. Curr. Protoc. Mol. Biol. 124, e65. 10.1002/cpmb.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.