

Abstract
Tissue-resident memory CD8 T cells (TRM) offer rapid and long-term protection at sites of re-infection1. Tumor-infiltrating lymphocytes (TIL) with characteristics of TRM maintain enhanced effector functions, predict responses to immunotherapy, and accompany better prognoses2,3. Thus, an improved understanding of the metabolic strategies that enable tissue residency by T cells could inform new approaches to empower immune responses in tissues and solid tumors. To systematically define the basis for the metabolic reprogramming supporting TRM differentiation, survival, and function, we leveraged in vivo functional genomics, untargeted metabolomics, and transcriptomics of virus-specific memory CD8 T cell populations. We found that memory CD8 T cells deployed a range of adaptations to tissue residency, including reliance on non-steroidal products of the mevalonate/cholesterol pathway, such as Coenzyme Q (CoQ), driven by increased activity of the transcription factor Srebp2. This metabolic adaptation was most pronounced in the small intestine (SI), where TRM interface with dietary cholesterol and maintain a heightened state of activation4, and was shared by functional TIL in diverse tumor types in mice and humans. Enforcing CoQ synthesis through Fdft1 deletion or Pdss2 overexpression promoted mitochondrial respiration, memory formation upon viral infection, and enhanced antitumor immunity. In sum, through a systematic exploration of TRM metabolism, we reveal how these programs can be leveraged to fuel CD8 T cell memory formation in the context of acute infections and enhance antitumor immunity.
To maximize immune protection, memory CD8 T cells differentiate into functionally distinct populations with different surveillance capacities. These include effector memory T cells (TEM) that mostly recirculate through tissues, central memory T cells (TCM) that act as sentinels in lymphoid organs, and TRM, which become a permanent asset to tissues throughout the body1. TRM constitute the majority of the CD8 T cell memory pool5, can retain the potential for supernumerary division and longevity6,7, and offer a rapid and potent first line of defense due to their proximity to sites of reinfection and enhanced cytotoxic potential8–13, mediating protection against viral and bacterial infections8–11,14–17. Similarly in tumors, pre-existing TRM as well as TIL with TRM features (TRM-like) play a crucial role in suppressing the emergence of malignancies and mediating tumor control upon immune checkpoint blockade due to their superior intrinsic functional activity3,17–21. The different environments in which these memory CD8 T cell subsets reside condition their function and differentiation22, potentially imposing distinct metabolic adaptations that support cell fitness. In addition, cellular metabolism can steer the course of CD8 T cell differentiation by linking specific enzymes and metabolites to cell fates and functions22,23. Thus, an understanding of such mechanisms offers actionable strategies to program T cell subtypes that augment protection against infection and enhance current immunotherapies. While the major transcriptional programs that guide TRM differentiation are established4,17,24–27, a systematic investigation of the metabolic changes that allow TRM formation and function is lacking.
A systematic profiling of TRM metabolism
The transcriptional and functional inter-tissue heterogeneity of TRM24,27 indicate that metabolic adaptions to diverse tissue environments accompany their differentiation. Indeed, we observe activation-dependent and tissue-specific gene-expression changes of metabolic signatures by different lymphocytic choriomeningitis virus (LCMV)-specific CD8 T cells subsets (Extended Data Fig. 1a). To uncover the functional metabolic regulators of TRM formation, we performed a pooled CRISPR/Cas9-mediated loss-of-function screen targeting 3017 metabolic genes in the context of LCMV infection in vivo (Fig. 1a). We utilized P14 T cell receptor transgenic CD8 T cells, specific for the LCMV glycoprotein 33–41 peptide (GP33–41) presented by H2-Db, that constitutively expressed Cas9, allowing gene-editing by CRISPR/Cas9 (P14 Cas9eGFP). P14 Cas9eGFP cells transduced with the metabolic regulator targeting sgRNA library were adoptively transferred into WT recipients that were then infected with LCMV-Armstrong. Two weeks after infection, sgRNA-transduced P14 Cas9eGFP T cells were recovered, including circulating memory populations TEM, TCM, and t-TEM from the spleen; small intestine (SI) TRM, and liver TRM and TEM, allowing comparison of circulating to tissue-resident populations as well as comparison of two different tissue-resident populations. At this time point, P14 Cas9eGFP cells have seeded peripheral tissues and show a TRM phenotype while allowing sufficient cell recovery17 (Fig. 1a). Intravenous (IV) labeling of CD8 T cells before cell isolation allowed discrimination of tissue-localized TRM (IV−) from cells in circulation5. Consistent with a process of in vivo selection, sgRNA frequencies were more heterogenous in the recovered memory populations than in the input populations (Extended Data Fig. 1b). To assess the impact of each gene on memory T cell subsets, individual sgRNA abundance for each sample was compared to the initial input to calculate Log2 Fold Change (Log2FC) enrichment and pooled to obtain gene-level scores. We focused first on the 155 metabolic genes that were statistically significant in their impact on SI TRM formation (Fig. 1b and Supplementary Table 1). Most gene disruptions showed detrimental effects on memory CD8 T cell formation irrespective of subset or tissue (Fig. 1b). Unsupervised clustering grouped SI and liver TRM together, suggesting that, despite clear differences between these two tissues, TRM were metabolically different from circulatory populations (Fig. 1b). Further, a subset of genes was uniquely depleted in SI TRM, including Cardiolipin synthase 1 (Crls1), a metabolic regulator known to support the mitochondrial function of SI TRM4 (Fig. 1b). Finally, 6 genes led to increased SI TRM formation when disrupted (Fig. 1b and Extended Data Fig. 1c). These genes included known negative regulators of CD8 T cell function and proliferation, such as Regnase-1 (Zc3h12a)28, Pten29, and Inpp5d30. To gain a higher-level view of the processes represented by these individual gene scores, we classified each target by metabolic pathway (Supplementary Table 2). Among the positive metabolic regulators of SI TRM, the “Oxidative phosphorylation complex III (OXPHOS Complex III)” and the “mevalonate/cholesterol synthesis” pathways were highly enriched in SI TRM (Fig. 1c and Supplementary Table 3).
Figure 1. Functional genetics in vivo, metabolomics and transcriptional analysis of ex vivo populations of memory CD8 T cells identify a graded upregulation of a Srebp2-dependent metabolic programming across TRM.
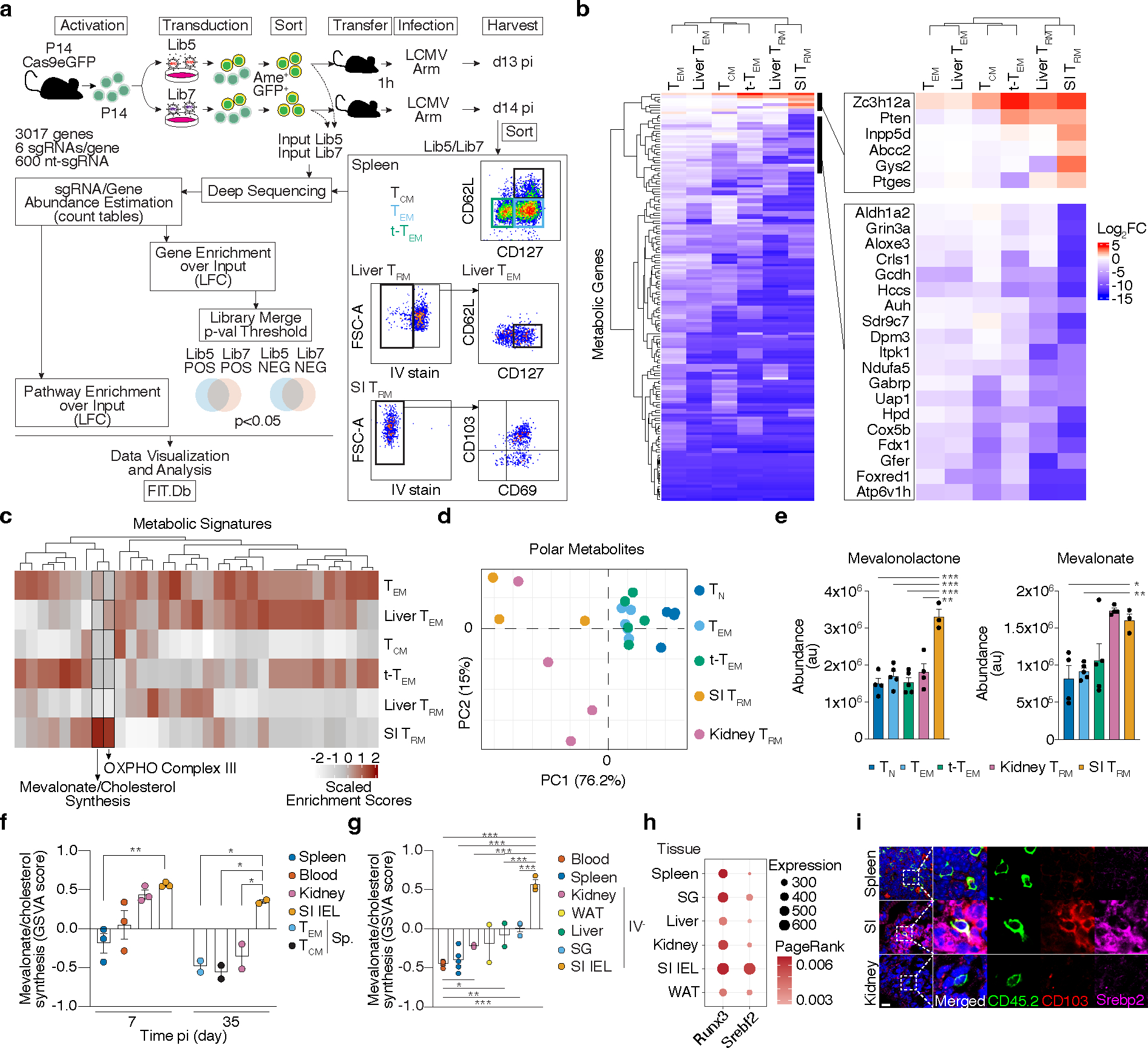
a, Experimental design and data analysis approach for an in vivo CRISPR/Cas9-mediated loss-of-function screen of metabolic regulators of memory CD8 T cell differentiation in LCMV infection. b, Unsupervised hierarchical clustering heatmap of averaged gene essentiality scores for significant genes in SI TRM in both libraries. c, Unsupervised hierarchical clustering of scaled averaged enrichment scores of significant metabolic signatures in SI TRM in both libraries. d, PCA of relative metabolite abundances of indicated P14 CD8 T cells 13 days after LCMV infection. e, Abundance of selected identified annotated metabolites of mevalonate/cholesterol synthesis pathway on indicated P14 CD8 T cells. TN, Naïve. f, Mevalonate/cholesterol synthesis pathway GSVA scores from RNAseq analysis of indicated P14 CD8 T cells, GSE107278. g, Mevalonate/cholesterol synthesis pathway GSVA scores from RNAseq analysis of indicated memory P14 CD8 T cells (>30 days pi), GSE182276. SG, salivary gland. WAT, white adipose tissue. h, PageRank scores and gene expression of Srebp2 and Runx3 in memory P14 CD8 T cells, GSE182276. i, Representative detection of Srebp2 and CD103 by immunofluorescence of congenically labeled memory P14 CD8 T cells. Scale bar, 20μm. P14 CD8 T cells recovered from tissues other than the spleen were pregated on IV− populations. Images are representative of 2 independent experiments (i). Data are mean +/− s.e.m., and representative of at least two independent experiments (f, g) with a total of n=2 (day 35) and n=3 (day 7) (f), n=2 (liver and WAT), n=3 (SG, kidney, and SI), n=4 (blood), and n=5 (spleen) (g) mice, or one experiment (a, b, c, d and e), with a total of n=7 (Lib7) and n=9 (Lib5) mice pooled per library (a, b, and c), and n=3 (SI), n=4 (TN and kidney), and n=5 (rest) (d and e) samples, where each sample contains cells pooled cells from 2 to 5 mice. Two-sided un-paired t-Test (e, f, and g). *P<0.05, **P<0.01, ***P< 0.005.
Next, we determined the relative abundance of intracellular metabolites in ex vivo populations of TRM from the SI and kidney, and TEM, TCM and t-TEM from the spleen by untargeted metabolomics (Extended Data Fig. 1d). Principal component analysis (PCA) of metabolite composition of these populations revealed a separation among TRM and circulatory memory CD8 T cell fractions along PC1, and separation between kidney and SI TRM along PC2 (Fig. 1d). In agreement with our transcriptional and functional data, these data suggested that TRM in general have a distinct metabolic profile compared to their circulatory counterparts, as well as unique profiles associated with specific tissues. Unsupervised clustering based on identified, annotated metabolites revealed a similar sample aggregation and highlighted key intermediaries of the mevalonate/cholesterol synthesis pathway, mevalonate and mevalonolactone (loss-of-water adduct of mevalonate), as elevated in TRM, and SI TRM, respectively (Fig. 1e, Extended Data Fig. 1e, f, and Supplementary Table 4). Thus, data from our functional screen as well as gene expression and metabolomics profiling each revealed increased mevalonate/cholesterol synthesis as a potential metabolic adaptation of SI TRM.
Rapid generation of mevalonate-derived products, including cholesterol, is required for initial rapid CD8 T cell proliferation after TCR activation31. Consistent with this, the transcriptional upregulation of enzymes of this pathway peaked early after LCMV infection, followed by a decline as CD8 T cells differentiate into memory CD8 T cell populations, with the notable exception of SI TRM, where expression remained elevated well after resolution of infection (Fig. 1f and Extended Data Fig. 2a, b). Sustained expression of the mevalonate/cholesterol synthesis pathway was not unique to SI TRM, as TRM from other tissues maintained enrichment of this pathway compared to circulating memory CD8 T cell populations in the spleen and blood, however to a lesser degree (Fig. 1g and Extended Data Fig. 2c,d). Multiple data sets of mouse and human CD8 T cells showed similar enrichment patterns for expression of genes in the mevalonate/cholesterol synthesis pathway (Extended Data Fig. 2a–f).
Expression of the enzymes of the mevalonate/cholesterol synthesis pathway are collectively enhanced by the transcription factor (TF) sterol regulatory element binding protein 2 (Srebp2, encoded by Srebf2). Bioinformatics analysis by Taiji32, which leverages chromatin accessibility data combined with RNA expression, predicted Srebp2 as an upstream regulator of gene expression in SI TRM24 (Fig. 1h). Srebp2 protein was more abundant in TRM of the SI compared to those of the kidney, and CD8 T cells in the spleen (Fig. 1i). Similarly, protein levels of Hmgcr, an Srebp2 target, and the rate-limiting enzyme of the mevalonate/cholesterol synthesis pathway, were also higher in human SI and colonic CD8 T cells compared to those in the spleen (Extended Data Fig. 2g). Runx3, necessary for the programming of CD8 T cell tissue residency17, also regulated expression of mevalonate/cholesterol pathway enzymes by CD8 T cells (Extended Data Fig. 2h). Thus, TRM possessed a distinct metabolic program compared to their circulatory counterparts, including adaptations more prevalent in certain tissues, such as enrichment of the mevalonate/cholesterol synthesis pathway driven by Srebp2, which was most active in SI TRM.
Srebp2 promotes TRM formation
To functionally assess the contribution of Srebp2 to TRM formation, we adoptively transferred a 1:1 mix of P14 T cells transduced with control Cd19 shRNAmir- or Srebf2 shRNAmir-encoding retroviruses into mice that were subsequently infected with LCMV (Fig. 2a). Srebf2 shRNAmir suppressed Srebf2 expression as well as its downstream target Hmgcr by 5–10-fold (Extended Data Fig. 3a), and impaired TRM formation in the SI with minimal impact on T cell accumulation in kidney, WAT, or liver (Fig. 2a). CD8 T cell populations in the spleen were unaffected by Srebf2 KD at both effector and memory time points (Extended Data Fig. 3b,c). However, full deletion of Srebp2 (P14 CD8 T cells transduced with control (sgCd19) or Srebf2 sgRNA-encoding retroviruses (sgSrebf2)), impaired TRM formation in SI, kidney and WAT, but spared circulatory memory CD8 T cell populations despite decreased effector T cell accumulation at day 7 (Fig. 2b and Extended Data Fig. 3d–f). Liver TRM showed less dependence on Srebp2; IV− P14 CD8 T cells were modestly impaired and when defined as CD69+CD62L−, an alternative gating strategy33, liver TRM were unaffected by loss of Srebp2 (Fig. 2b). SI TRM populations induced by Listeria monocytogenes (LM) infection also required Srebp2, suggesting the reliance on this pathway in infection was generalizable (Fig. 2c). Together, these results showed that sustained Srebf2 expression was functionally relevant for the formation of TRM but not for circulating memory T cell populations.
Figure 2. An Srebp2-dependent metabolic program controls SI TRM formation.
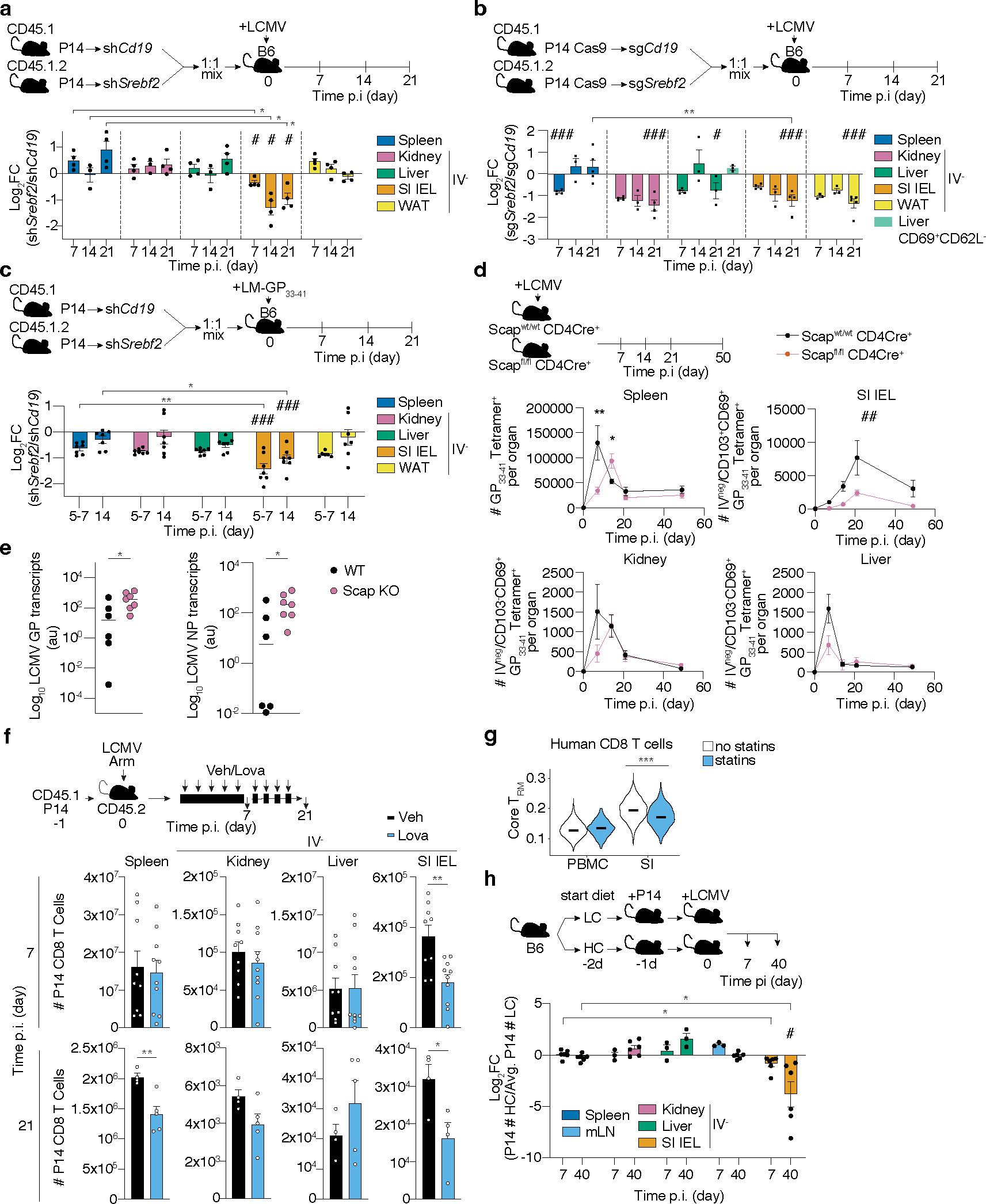
a-c, Ratio of transduced transferred P14 CD8 T cells (a and b), or P14 Cas9eGFP CD8 T cells (b), harvested from indicated tissues at indicated times after LCMV (a and b), or LM-GP33–41 (c) infection, evaluated by flow cytometry. d, Number of total Tet+ cells in the spleen, and total Tet+ TRM cells in the SI, kidney, and liver of Scapfl/fl Cd4-Cre+ and Scapwt Cd4-Cre+ at indicated times after LCMV infection. e, LCMV titers measured by qPCR in the SI of WT and Scap KO mice previously infected with LM-GP33–41 3days after LCMV rechallenge. NP, nucleoprotein. GP, glycoprotein. f, Number of total P14 CD8 T cells isolated from indicated tissues at days 7 and 21 after LCMV infection in mice treated with vehicle or Lovastatin (Lova). g, TRM scores of human CD8 T cells of the blood and SI profiled by scRNAseq grouped by statin use; no statins = 8 donors, statins = 5 donors. h, Ratio of total P14 CD8 T cells from indicated tissues in mice fed high versus low-cholesterol-containing diet. Data are geometrical distributions of single-cell gene expression values with median values (g) or mean +/− s.e.m. and representative of two independent experiments (a, b, d, e and f), or pooled from two independent experiments (c, e, f, and h), with a total of n=7 (a), n=7 (b), n=7 (c), n=8 (d), n=6 in WT and n=7 in Scap KO (e), n=9 (day 7) and n=4 (day 21) (f), and n=3 (mLN, kidney, and liver day 7, and liver day 40), and n=6 (rest) (h) mice. Two-sided unpaired (f), and paired t-Test, (a, b, c, d, f, g, and h), two-sided Mann-Whitney U Test (e). *P<0.05, **P<0.01, ***P< 0.005. Two-sided one-sample t-Test (a-c, and h) #P<0.05, ##P<0.01, ###P< 0.005. Two-way ANOVA (d) ##P<0.01.
The inactive form of Srebp2 in the endoplasmic reticulum (ER) becomes activated under conditions of low ER cholesterol by its translocation to the Golgi by Srebp activating-cleavage protein (Scap)34, and thus loss of Scap impairs Srebp2 activation34. To understand the implications of mevalonate/cholesterol synthesis inhibition in polyclonal endogenous CD8 T cell responses, we infected mice with a T cell-specific deletion of Scap (Scapfl/fl Cd4-Cre+, Scap KO) and followed the LCMV-specific CD8 T cell response of H-2Db-GP33–41 tetramer (Tet+) compared to WT (Scapwt Cd4-Cre+) mice. As previously shown33, Tet+ Scap KO cells displayed reduced accumulation during the effector phase compared to WT Tet+ cells31 (Fig. 2d and Extended Data Fig. 3g). Scap KO showed impaired differentiation of Tet+ CD103+CD69+ SI TRM, but not kidney or liver TRM (Fig. 2d, Extended Data Fig. 3h). Concomitant with impaired TRM accumulation, Scap KO mice that had been infected with LM-GP33–41 had higher viral titers in the SI when rechallenged with LCMV compared to WT mice (Fig. 2e). While loss of Scap reduces fatty acid synthesis, as it also controls Srebp1 (Srebf1) activity, neither Srebf1 nor Fatty Acid Synthase (Fasn) impacted memory T cell differentiation (Supplementary Table 1).
Hmgcr inhibitors (statins) are widely consumed lipid-lowering drugs with known immunomodulatory effects35. We found that lovastatin treatment impaired the accumulation of P14 CD8 T cells responding to LCMV in the SI compared to the control group on day 7 of infection; by day 21 there was a sustained loss of SI TRM and impaired accumulation of memory T cells in the spleen as well (Fig. 2f). These observations are of potential human significance as we observed lower expression of the TRM gene-expression signature17 in SI CD8 T cells from statin users than non-statin users, revealing a potentially underappreciated effect of statins on TRM populations of the SI (Fig. 2g and Supplementary Table 5).
Each of these conditions inhibit the mevalonate/cholesterol synthesis pathway to varying degrees. Combined, these data suggested that enhanced activity downstream of Hmgcr, Scap, and Srebp2 is a metabolic requirement of CD8 T cells to become SI TRM. We observed graded reliance on Srebp2 among the different tissues; where SI TRM showed a greater dependence on the pathway than kidney, WAT, and liver resident cells, reflecting the pattern of expression for genes involved in this pathway (Fig. 1g).
Interestingly, enforcing Srebp2 activity by ectopic overexpression of a constitutively active nuclear isoform actually prevented P14 CD8 T cell accumulation in the SI after LCMV infection, despite successfully increasing total cellular cholesterol and its downstream target, LDLR (Extended Data Fig. 3i, j). This additional observation is consistent with the idea that generating high cholesterol levels may not be the rate-limiting step for TRM populations. To further investigate the impact of Srebp2 loss, we compared gene expression by shCd19- or shSrebf2-transduced P14 CD8 T cells after LCMV infection. Srebf2 and the mevalonate/cholesterol synthesis pathway-associated enzymes were most highly expressed in SI TRM and reduced in the shSrebf2-transduced P14 CD8 T cells (Extended Data Fig. 4a–c), and a core TRM gene-expression signature17 was downregulated in shSrebf2 P14 CD8 T cells in the SI (Extended Data Fig. 4d–e). Strikingly, few genes were differentially expressed beyond Srebp2 and Srebp2-dependent genes (Extended Data Fig. 4f), suggesting that impaired SI TRM accumulation upon Srebp2 knockdown was not likely the result of transcriptional changes beyond the mevalonate/cholesterol synthesis pathway.
SI TRM are unique in their close contact with food-derived nutrients in the absorptive lining of the intestine36. Given that Srebp2 activity is regulated by the strong inhibitory feedback exerted by cholesterol (present in the diet), we sought to understand how the intestinal environment affected Srebp2-dependent metabolic programs on SI TRM. Notably, memory P14 CD8 T cells in the SI, but not the spleen, showed significant uptake of orally administered cholesterol (Extended Data Fig. 5a). On the other hand, when administered IV, none of the memory CD8 T cell populations profiled, including SI TRM, showed significant in vivo uptake of cholesterol-containing low-density lipoproteins (LDL), the main form of systemic delivery of cholesterol to cells (Extended Data Fig. 5b). Using imaging flow cytometry, we found that SI TRM of mice fed regular chow had the lowest amount of intracellular cholesterol compared to the kidney, liver, and splenic population (Extended Data Fig. 5c), consistent with maintained Srebp2 activity. Feeding a high cholesterol-containing diet partially reduced Srebp2-dependent genes, Hmgcr, Hmgcs1, and Ldlr in CD8 T cells of the SI, with minimal effects in splenic CD8 T cells (Extended Data Fig. 5d) and impaired SI TRM, but not kidney or liver TRM, formation and maintenance (Fig. 2h and Extended Data Fig. 5e,f). Of note, the diet high in cholesterol did not impact food intake or mouse weight, and only transiently raised circulating cholesterol levels compared to the low cholesterol diet control (Extended Data Fig. 5g–i). Ultra-low input (ULI)-RNAseq analysis of SI TRM from mice fed a high cholesterol diet showed marked suppression of Srebp2 targets, as well as induction of programmed cell death-related genes and cholesterol efflux transporters, compared SI TRM from mice fed a low cholesterol diet (Extended Data Fig. 5j–l and Supplementary Table 6). Thus, SI TRM take up and adapt to differences in dietary cholesterol, but when in excess, either by enforcing Srebp2 OE (Extended Data Fig. 3i) or over-feeding (Fig. 2h), cholesterol was detrimental to SI TRM accumulation. While increasing cholesterol in the diet attenuated Srebp2 activity, it was insufficient to reduce it to levels observed in the spleen (Extended Data Fig. 5d) suggesting additional cues, beyond environmental cholesterol, likely maintain Srebp2 activity in this environment, such as Runx3 (Extended Data Fig. 2g). Further, these data suggested that, rather than cholesterol, alternative products of the mevalonate/cholesterol synthesis pathway are the relevant mediators of TRM formation downstream of Srepb2.
SI TRM rely on CoQ-producing enzymes
Intermediate products of the mevalonate/cholesterol synthesis pathway supply substrates to produce important nonsterol isoprenoids, such as CoQ, through branched reactions. To explore the mechanistic basis for TRM dependence encompassing these alternative fates of mevalonate, we focused on additional enzymes that regulate diversion of intermediates to synthesis of non-steroidal metabolites from the in vivo CRISPR/Cas9 (extended mevalonate/cholesterol synthesis pathway) (Fig. 3a and Extended Data Fig. 6a). Clustering of these genes by their relative impact on CD8 T cell memory populations revealed a group that were more specific to SI TRM (Fig. 3a). Among these genes, Pdss2 and Hpd, two enzymes that divert metabolites to the synthesis of CoQ, were both particularly required for SI TRM, and targeting led to similar loss of TRM phenotypes as disruption of Srebf2 and Scap (Fig. 3a). Notably, our metabolomics analysis found 4-hydroxybenzaldehyde (4-Hb), the product of Hpd enzyme, to be especially elevated in SI TRM compared to other CD8 T cell populations, indicating this pathway might be increased in SI TRM (Extended Data Fig. 6b,c). Validation of the CRISPR/Cas9 screen by targeting Hpd revealed impaired formation of SI TRM, but not P14 memory T cells in the spleen, liver, or kidney (Fig. 3b and Extended Data Fig. 6d). These results suggested SI TRM have a heightened requirement for CoQ synthesis compared to other memory CD8 T cell populations. Further, deletion of squalene synthase (Fdft1), which catalyzes the first step of the isoprenoid branch committed specifically to cholesterol production, appeared to promote several populations of memory CD8 T cells (Fig. 3a). Consistent with the CRISPR/Cas9 screen, targeting Fdft1 resulted in increased accumulation of liver TRM, as well as circulating memory subsets, but it was not sufficient to further enhance SI TRM (Fig. 3c and Extended Data Fig. 6e). These data reinforce the notion that the production of cholesterol by the mevalonate pathway is not required and revealed a metabolic enzyme, Fdft1, to be a potentially actionable target to improve memory CD8 T cell formation.
Figure 3. Non-steroidal products of the mevalonate/cholesterol synthesis pathway mediate TRM adaptations.
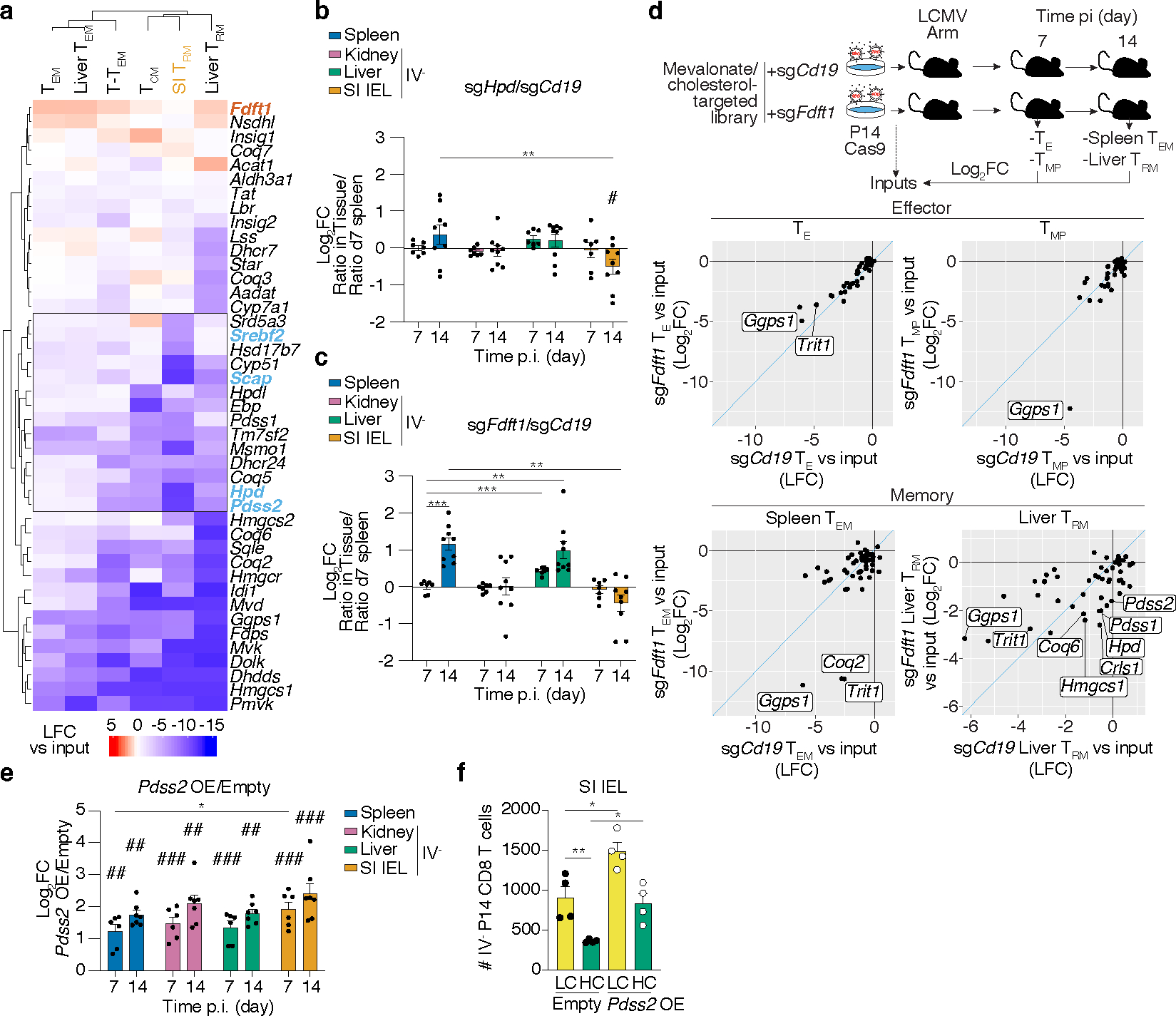
a, Unsupervised hierarchical clustered heatmap of gene essentiality scores for genes related to the mevalonate/cholesterol synthesis pathway in the cellular populations profiled in our in vivo CRISPR/Cas9-mediated loss-of-function screen. Blue and red denote positive and negative regulators of memory CD8 T cell formation, respectively. b,c, Ratio of indicated transduced transferred P14 Cas9eGFP CD8 T cells, sgHpd (b) and sgFdft1 (c), harvested from indicated tissues at indicated times after LCMV infection, evaluated by flow cytometry. d, Targeted in vivo CRISPR/Cas9-mediated loss-of-function screen of mediators of the effect of Fdft1 deletion on CD8 T cell memory formation. Data visualization represents the enrichment as Log2FC of sgRNA frequencies in sgCD19 outputs versus sgCd19 input (x axis), and sgFdft1 outputs versus sgFdft1 input (y axis), for each effector and memory CD8 T cell subset. e, Ratio of transduced transferred P14 CD8 T cells harvested from indicated tissues at indicated times after LCMV infection, evaluated by flow cytometry. f, Total cell numbers of Empty and Pdss2 OE SI IEL P14 CD8 T cells in mice fed low or high cholesterol-containing diets. Data are mean +/− s.e.m.. and representative or pooled from at least two independent experiments (b, c, e, and f), or one experiment (d), with a total of n=7 (day 7) and n=9 (day 14) (b, c), n=9 (c), n=6 (d), n=6 (day 7) and n=7 (day 14) (e), n=4 (f) mice. Two-sided unpaired t-Test (b, c, e, and f) *P<0.05, **P<0.01, ***P< 0.005. Two-sided one-sample t-Test (b, c, and e) #P<0.05.
To gain greater insight into the identity and mechanism of action of the relevant non-steroidal products mediating greater memory potential to Fdft1 KO CD8 T cells, we conducted a secondary targeted in vivo CRISPR/Cas9 screen of potential non-steroidal mediators of Fdft1 impact on T cell accumulation. This library, composed of 153 sgRNAs targeting 50 genes of the extended mevalonate/cholesterol synthesis pathway (Extended Data Fig. 6a and Supplementary Table 7), was transduced into either control (sgCd19) or sgFdft1 P14 Cas9eGFP T cells to test their function in mediating Fdft1-enhanced TEM and liver TRM populations (Fig. 3c,d). By plotting their relative impact, we looked for genes that had a significant effect in reducing the accumulation of sgFdft1 while sparing control sgCd19 P14 T cells in memory populations, as these likely mediate the enhanced accumulation observed (Fig. 3d, Extended Data Fig. 6f, and Supplementary Table 8). Comparison to TE and TMP cells from day 7 of infection provided a reference for identifying genes that were required only at the memory timepoint rather than at the effector stage, such as Ggps1 or Trit1. Genes that affected both populations equally, and thus fall in the plot’s diagonal, do not likely mediate the specific advantage observed upon Fdft1 deletion (Fig. 3d). This analysis revealed that several enzymes needed for CoQ synthesis (Coq2), geranylgeranylation (Ggps1) and tRNA modification (Trit1) showed a larger effect on Fdft1 KO CD8 T cells than control CD8 T cells in spleen TEM, of which only Coq2 was specific to memory cells (Fig. 3d). Further, the dependence on CoQ synthesis was more striking for the TRM population, as it included additional enzymes, such as Hpd, Pdss1, and Pdss2 (Fig. 3d). Combined, these data revealed that the advantage endowed by Fdft1 loss is supported by the production of non-steroidal metabolites, including CoQ (Extended Data Fig. 6g).
Given the critical role of Pdss2 in our functional screens and its role in the first reaction producing CoQ from mevalonate products, we tested whether Pdss2 overexpression could mediate enhanced TRM formation. Indeed, enforced Pdss2 expression led to a 3- to 8-fold increase in T cell accumulation compared to control vector across all tissues examined (Fig. 3e and Extended Data Fig. 6h). Of note, Pdss2 OE restored SI TRM formation to WT levels in mice fed a high cholesterol diet (Fig. 3f). Combined, these data suggest that TRM increase Srebp2 activity to meet a heightened requirement, particularly elevated in the SI, for non-steroidal products, such as CoQ. As such, promoting the accumulation of metabolic intermediates that can be diverted to CoQ production by blocking Fdft1, or boosting the capacity to shunt intermediates into the pathway by enforcing Pdss2 overexpression, enhances TRM formation and can offset some of the toxicities induced by an excess of dietary cholesterol. Of note, circulating memory CD8 T cell populations similarly benefitted from Pdss2 OE (Fig. 3e), suggesting this pathway might generally increase T cell-mediated immunological memory.
CoQ supports mitochondrial respiration
CoQ is a highly hydrophobic redox lipid in the inner mitochondrial membrane that transports electrons from complexes I and II to complex III in the electron transport chain (ETC). We next tested whether CoQ production driven by the mevalonate/cholesterol synthesis pathway impacted mitochondrial respiration. Srebf2 KD CD8 T cells had lower rates of basal oxygen consumption (OCR) and reduced maximal respiration upon FCCP treatment (spare respiratory capacity, SRC) (Fig. 4a). Similarly, statin treatment reduced mitochondrial respiration of CD8 T cells in a mevalonate-dependent, but cholesterol-independent manner (Fig. 4b). A comparable loss of mitochondrial respiration was also observed in Pdss2-deficient CD8 T cells (Fig. 4c). Conversely, Fdft1 deletion increased basal OCR and SRC (Fig. 4d), in a Pdss2-dependent manner (Fig. 4e). Of note, Pdss2 OE was sufficient to promote higher basal and maximal mitochondrial respiration (Fig. 4f). In line with these data, deletion of Fdft1 or enforced expression of Pdss2, lead to higher production of CoQ species, including the most abundant form in mice, CoQ9, and its precursor DMQ9 (Fig. 4f,g). Taken together, these data suggest that elevated rates of mevalonate/cholesterol synthesis intermediaries supply the CoQ pool to potentiate mitochondrial respiration in CD8 T cells.
Figure 4. Pdss2 expression is required and sufficient to promote mitochondrial respiration and CoQ synthesis in CD8 T cells.
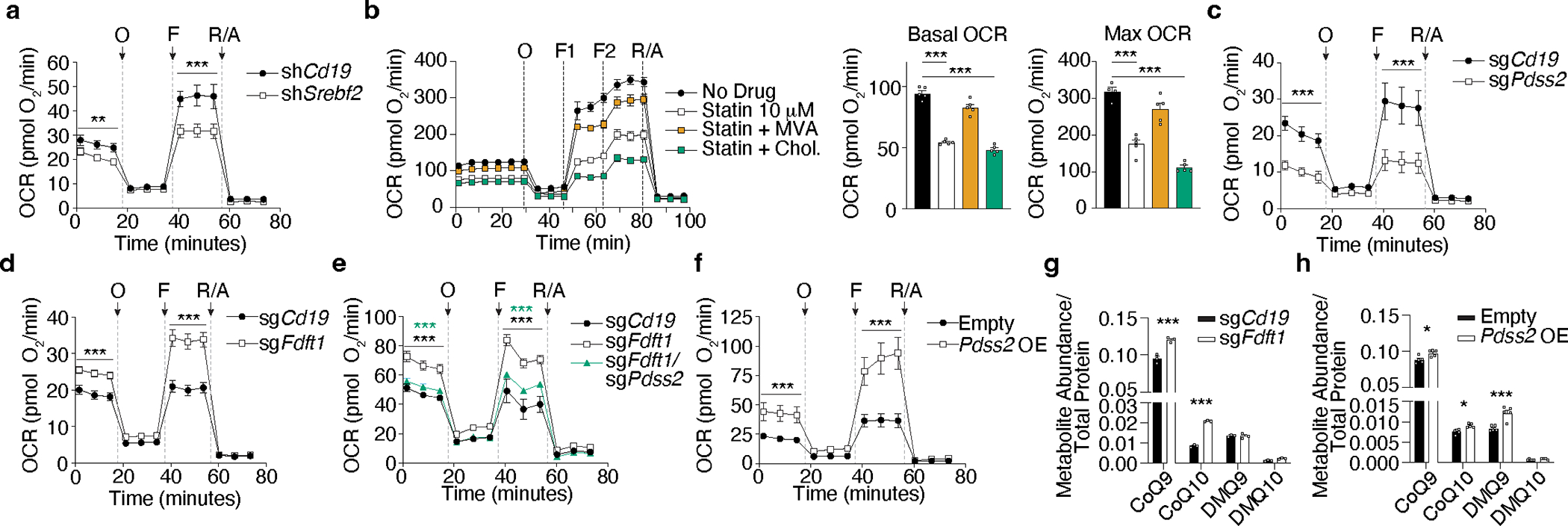
a, Oxygen consumption rate (OCR) of in vitro activated shCd19 and shSrebf2 CD8 T cells subjected to the MitoStress test (Seahorse). Oligomycin (O), FCCP (F), Rotenone/Antimycin A (R/A). b, Oxygen consumption rate (OCR) of in vitro activated CD8 T cells treated with statin (simvastatin) in combination with mevalonolactone (MVA) or cholesterol (Chol.) and subjected to the MitoStress test (Seahorse). c, OCR of in vitro-activated sgCd19 and sgPdss2 Cas9eGFP CD8 T cells subjected to the MitoStress test (Seahorse). d, OCR of in vitro activated sgCd19 and sgFdft1 Cas9eGFP CD8 T cells subjected to the MitoStress test (Seahorse). e, OCR of in vitro activated sgCd19, sgFdft1, and sgFdft1/Pdss2 Cas9eGFP CD8 T cells subjected to the MitoStress test (Seahorse). f, OCR of in vitro activated empty vector control (Empty) and Pdss2 OE CD8 T cells subjected to the MitoStress test (Seahorse). g, Quantification of CoQ and demethoxyubiquinone (DMQ) species in in vitro activated sgCd19 and sgFdft1 Cas9eGFP CD8 T cells normalized to total protein content. h, Quantification of CoQ and DMQ species in in vitro activated Empty and Pdss2 OE CD8 T cells normalized to total protein content. Data are mean +/− s.e.m. and representative of at least two independent experiments (a-h), with a total of n=4 (a), n=5 (b), n=3 (c), n=2 (e), n=3 (f), n=3 (g), n=5 (h) cell replicates. Two-way ANOVA (a, c, d, e, and f). Two-sided unpaired t-Test (b, g, and h) *P<0.05, **P<0.01, ***P< 0.005.
Given the relationship between TRM and TRM-like TIL, including the importance of mitochondrial respiration and transcriptional programming17,37,38, we explored if common metabolic requirements might be shared between SI TRM and functional TIL. To put our previous findings into the context of anti-tumor CD8 T cell responses, we performed scRNAseq profiling of P14 CD8 T cells responding to a murine tumor model (MC38-GP33–41). The mevalonate/cholesterol synthesis pathway was elevated in P14 CD8 TIL versus those in the spleen (Extended Data Fig. 7a,b). Examining the different subpopulations revealed that the proliferating (C4), terminally exhausted (C3), and TRM-like TIL (C7) expressed the highest levels of Srebp2-dependent targets, enzymes Hmgcs1 and Hmgcr (Extended Data Fig. 7c). Similarly, reanalysis of a human pan-tumor dataset revealed that the mevalonate/cholesterol pathway was most upregulated in proliferating, exhausted, effector, and TRM-like populations (found mostly within the tumor) and low in memory and naive CD8 T cell populations (found mostly in circulation) across 21 different cancer types39 (Extended Data Fig. 7d). Thus, in both human and mouse, increased expression of genes involved in mevalonate/cholesterol synthesis in TIL implicate this pathway in intratumoral T cell accumulation.
An Srebp2-CoQ axis boosts tumor immunity
To functionally test whether TRM and TIL shared common metabolic adaptations related to the mevalonate/cholesterol synthesis pathway and to pin down which of its components were most relevant, we performed two in vivo CRISPR/Cas9 screens in P14 CD8 T cells in parallel using the 50-gene library targeting key elements of this pathway (Supplementary Table 5): one in the context of LCMV infection for the formation of SI TRM, and the second in the context of MC38-GP33–41 tumors for the accumulation of TIL (Fig. 5a). This experiment revealed a significant correlation between the impact of these genes on TIL and SI TRM accumulation, with 19 significant genes commonly required for both cellular populations (67% overlap) (Fig. 5b,c, Extended Data Fig. 7e and Supplementary Table 9). Targeted genes were essential for T cells in the tumor and for TRM but were largely dispensable for T cells in the draining lymph node (dLN) (Fig. 5b). Most of the 19 genes with a shared requirement by TRM and TIL mediate the production of non-steroidal metabolites, including Hpd and Coq2 that lead to CoQ production (Fig. 5c,d). As we observed for SI TRM, these data showed that synthesis of cholesterol was not required for TIL accumulation (Dhcr7 and Dhcr24 were not essential) (Fig. 3a and Fig. 5c). Consistent with our studies in the context of infection, Srebf2 deficiency impaired accumulation of P14 CD8 T cells in the tumor (Fig. 5e), loss of Pdss2 reversed the increased accumulation of Fdft1 KO P14 CD8 T cells in the tumor (Fig. 5f), and Pdss2 OE promoted an even higher accumulation of P14 CD8 T cells in the tumor than in the spleen (Fig. 5g). Taken together, these data showed that both TRM and TIL populations rely on a sustained and elevated Srebp2-driven program to synthesize non-steroidal metabolites, such as CoQ, to support their accumulation.
Figure 5. Increased production of non-steroidal products of the mevalonate/cholesterol synthesis pathway is a common requirement for SI TRM and TIL.
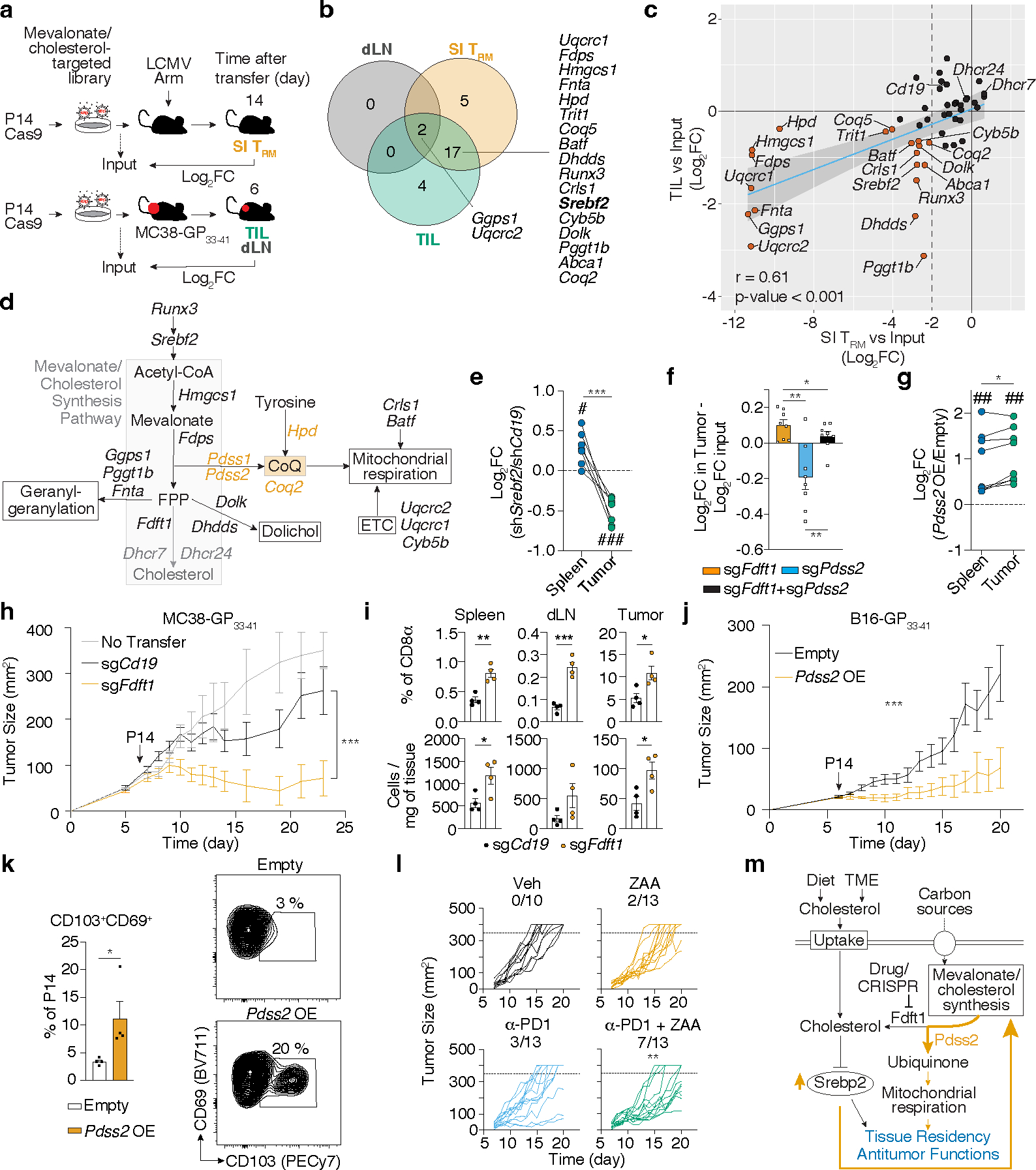
a, Targeted in vivo screen of mevalonate/cholesterol synthesis pathway genes for SI TRM formation in LCMV infection, and CD8 T cell accumulation in dLN and MC38-GP33–41 tumors. b, Venn diagram of significant genes (fdr < 0.01; dLN and TIL, fdr < 0.01 and Log2FC < −2, SI TRM) from (a). c, Log2FC of sgRNA frequencies for each screen from a. d, Metabolic pathway with relevant genes from a. e-g, Ratio of indicated transduced P14 CD8 T cells (e,g) or P14 Cas9eGFP CD8 T cells (f) in indicated tissues 5–7 days after adoptive cell transfer (ACT). h, MC38-GP33–41 tumor growth curves of mice receiving indicated transduced P14 Cas9eGFP CD8 T cells. i, Cell frequencies and total numbers of transduced P14 Cas9eGFP CD8 T cells in MC38-GP33–41 tumors, dLN, and spleens 4 days after ACT. j, B16-GP33–41 tumor growth curves in mice adoptively transferred with indicated transduced P14 CD8 T cells. k, Cell frequencies in Empty and Pdss2 OE P14 CD8 T cells in MC38-GP33–41 tumors. l, B16-GP33–41 tumor growth curves of mice receiving indicated treatments. m, Proposed model of the Srebp2-dependent metabolic programming of SI TRM and TIL. Data are mean +/− s.e.m. and representative or pooled from of at least two independent experiments (e-l), with a total of n=4 (SI TRM) and n=5 (tumor) (a-c), n=9 (e), n=8 (f), n=8 (g), n=3 (no transfer), n=15 (sgCd19), and n=16 (sgFdft1) (h), n=8 (i), n=9 (sgCd19) and n=7 (sgPdss2) (j), n=13 (k), n=10 (veh) and n=13 (rest) (l) mice, and one experiment (a-c), with n=3–5 mice pooled in each sample (a-c). Two-sided unpaired (f and i) and paired (e, g, and k) t-Test. Two-way ANOVA (h and j). Pearson correlation (r) (c). Fisher’s exact test (l). *P<0.05, **P<0.01, ***P< 0.005. Two-sided one-sample t-Test (e and g) #P<0.05, ###P<0.005.
We next tested if it was possible to enhance T cell activity in tumors by enforcing the TRM metabolic state. Fdft1 deletion increased P14-dependent tumor control of MC38-GP33–41 tumors, accompanied by improved P14 CD8 T cell persistence in tumors, dLN, and spleens (Fig. 5h,i). Similarly, Pdss2 OE enhanced tumor control in B16-GP33–41 melanoma tumors (Fig. 5j), and promoted a higher frequency of TRM-like TIL (Fig. 5k). Of note, we did not find changes in the expression of exhaustion-associated markers or differences in cytokine production upon ex vivo restimulation (Extended Data Fig. 7g,h,i,j). Finally, Fdft1 inhibition by administration of a competitive metabolite derived from fungi, Zaragozic Acid A (ZAA), reduced tumor growth and improved the survival of mice implanted with MC38-GP33–41 tumors in a CD8-dependent manner (Extended Data Fig. 7k,l,m). These data are consistent with previous reports of the immune system-dependent effects on antitumor control of ZAA40. When combined with anti-PD1 treatment, ZAA led to greater control of B16-GP33–41 melanoma tumors, further indicating that Fdft1 may be a valid treatment target (Fig. 5l). Taken together, this study revealed that an understanding of the metabolic regulators of TRM populations can be leveraged to improve CD8 T cell immunity against infections and tumors, including avenues to increase the production of CoQ downstream of Srebp2 (Fig. 5m).
Discussion
Combining functional screens of metabolic dependencies with transcriptomic and metabolomic profiling of circulating and resident subsets of memory CD8 T cells offered a systematic approach to identifying how metabolism impacts memory CD8 T cell formation in response to viral infections. In agreement with previous findings25,27, our results support the emerging concept that TRM formation requires distinct transcriptional and metabolic adaptations to different tissue environments. These data describing multiple potentially novel regulators of CD8 T cell memory differentiation have been made publicly available in a web-based application, Functional Immunometabolism and Transcriptomics Database (FITdb, fitdb.de).
Here, we chose to focus on SI TRM because these cells display profound metabolic differences from their circulating counterparts, maintain a controlled basal state of heightened activation resembling effector cells4, and possess molecular features associated with superior CD8 T cell functionality against tumors17,26. We found the Srebp2-dependent upregulation of the mevalonate/cholesterol synthesis pathway as the most distinguishable metabolic feature of SI TRM. Mechanistically, Srebp2 sustained transcriptional activation of the mevalonate/cholesterol biosynthesis pathway to support the production of non-steroidal products, including CoQ, which promoted mitochondrial respiration in vitro and in vivo accumulation in infection and tumors. We also showed that SI TRM respond to dietary cholesterol, which when in excess, was detrimental to their formation. Together, these adaptations might explain previous studies reporting an increase of mevalonate/cholesterol enzymes by T cell populations of the intestine4,41,42, and offer new insights into how dietary interventions could be leveraged to improve cellular immunity in the gastrointestinal tract36,43. Statins impact immune responses35, including reducing lymphocyte concentrations of CoQ44. Interestingly, our results highlight the dependency of SI TRM on this pathway and implicate a potentially significant effect in humans. Overall, our study identifies both dietary and pharmacological approaches to modulate T cell responses in the intestine.
Our data further reveal a common dependency on mevalonate/cholesterol synthesis enzymes for SI TRM formation and TIL accumulation. Based on these observations, we found two actionable enzymes, Fdft1 and Pdss2, which can be manipulated to enforce CoQ production, enhance the mitochondrial respiratory capacity of CD8 T cells, and promote in vivo accumulation of CD8 T cells in the context of infection and tumors. Whether these adaptations are conserved across all tumor types and contexts remains to be further investigated. The fact that loss of most genes of the mevalonate/cholesterol synthesis pathway impaired TIL accumulation in the melanoma model B16-OVA from a previous study28 (Extended Data Fig. 8a), suggests these observations may be generalized to other tumor contexts.
TRM-like TIL, and cells genetically endowed with superior antitumor properties, such as Regnase 1 KO CD8 T cells28, upregulate the mevalonate/cholesterol synthesis pathway in the absence of increased proliferation (Extended Data Fig. 8b). Based on our data, and given that cholesterol accumulation in the tumor microenvironment leads to intracellular cholesterol buildup in TIL conducive to dysfunction45,46, we propose increased Srebp2-driven mevalonate/cholesterol pathway synthesis in TIL serves to generate non-steroidal metabolites, rather than cholesterol (Fig. 5m). Of note, deletion of genes leading to CoQ synthesis (ie Pdss2) were able to abolish the heightened antitumor potential of Regnase-1 KO T cells (Extended Data Fig. 8c)28. Certainly, the reliance on non-steroidal intermediates does not preclude the need for generating or obtaining sufficient cholesterol to support T cell functions during priming and activation in responses against pathogens and tumors. In sum, we provide conclusive evidence that TRM utilize distinct metabolic adaptations, which can be exploited to enhance the function of CD8 T cells in solid tumors.
Methods
Mice
Mice were maintained in specific-pathogen-free conditions at a temperature between 18 °C and 23 °C with 40–60% humidity and a 12h-light and 12h-dark light cycle in accordance with the Institutional Animal Care and Use Committees (IACUC) of the University of California, San Diego (UCSD) and the University of California, Los Angeles (UCLA). All mice were of C57BL6/J background and bred at UCSD, UCLA or purchased from the Jackson Laboratory. P14, Cas9eGFP (stock #026179, The Jackson Laboratory), Thy1.1, and CD45.1 congenic mice were bred in house. Male and female mice were used for experiments, and were age and sex-matched, between 1.5 and 6 months old, and randomly assigned to experimental groups, except for therapeutic and dietary interventions where mice were randomly distributed into different experimental arms. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications from our laboratory and others. No blinding was performed during mouse experiments. Investigators were not blinded to group allocation during data collection and/or analysis. Low (D12102C) and high (D12104C) cholesterol diets were procured from Research Diets51. Mice were fed ad libitum for the specified amount of time. Intravascular staining of CD8 T cells was done by administration of 3 μg of anti CD8α APC-eFluor780 (eBioscience 47–0081-82) in 200 μl of PBS by retroorbital (RO) route. CD8α− (IV−) cells were considered to be localized within non-lymphoid tissues52. Isolation of IV− P14 CD8 T cells from the liver may exclude resident T cells in the vasculature. All animal studies were approved by the Institutional Animal Care and Use Committees of the University of California, San Diego (UCSD) and performed in accordance with UC guidelines.
Statistical methods
Statistical tests were performed using Prism (7.0/8.0/9.0) (Graphpad). Two-tailed paired or un-paired t-Test was used to compare groups with normally distributed values unless otherwise specified. Mann-Whitney U test was performed when value distributions did not pass normality tests. A Two-sided one-sample t-Test was used to compare data distributions against a hypothetical value of 0. Two-way ANOVA was used to test the effects of two variables on a continuous outcome. Fisher’s exact test was performed to determine the non-random association between two categorical variables. Logrank test was performed to test for differences between the populations in the probability of an event at any time point, related to survival analysis. P values of less than 0.05 were considered significant.
Cell culture
PlatE cells were acquired from Cell Biolabs (RV-101). MC38-GP33–41 cell line was generated from the commercially available C57BL/6 murine colon adenocarcinoma MC38 parental cell line (cat.# ENH204-FP, Kerafast). HEK293T cells were a gift from the Chi lab and originally sourced from the ATCC (CRL-3216). The B16-GP33–41 cell line was a gift from Dr. Alain Lamarre and is derived from C57BL/6 murine B16-F10 skin melanoma. PlatE, MC38-GP33–41, and B16-GP33–41 cells were cultured in DMEM + D-glucose supplemented with 10% bovine growth serum, 100 U/mL Penicillin, 100 ug/mL Streptomycin, 292 ug/mL L-glutamine, and 10 mM HEPES. Enriched CD8 T cells were maintained in RPMI + L-glutamine supplemented with 10% fetal bovine serum, 100 U/mL Penicillin, 100 μg/mL Streptomycin, 292 μg/mL L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate (Gibco), 1X MEM NEAA (Gibco), and 55 μM 2-Mercaptoethanol.
Naive T cell transfers and infection models
5×104 naive P14 CD8 T cells were transferred intravenously into congenically distinct sex-matched recipient mice, or female P14 cells were transferred into male mice. Recipient mice were subsequently infected intraperitoneally with 2×105 plaque-forming units (PFU) of the Armstrong strain of LCMV or 5000 colony-forming units (CFU) of L. monocytogenes expressing GP33–41 one day after cell transfer.
Single transfer and co-transfer experiments of in vitro-activated and transduced P14 CD8 T cells
For transfections using retroviral constructs, PlatE cells were seeded in 10-cm dishes at a density of 2.5×105 cells/plate 1 day before transfection in complete DMEM media (10% FBS, 2mM L-Glutamine, 1X Penicillin/Streptomycin). Transfections were performed with 25 μg plasmid DNA from each pLMPd-Amt, or LsgA clone with TransIT-LT1 (Mirus). Retroviral supernatant was harvested 48 h and 72 h after transfection. For transductions, negatively enriched naive CD8 T cells from spleen and lymph nodes were activated in 6-well plates coated with 100 μg/ml goat anti-hamster IgG (H+L; Thermo Fisher Scientific), 1 μg/ml anti-CD3 (145–2C11; eBioscience), and 1 μg/ml anti-CD28 (37.51; eBioscience). T cell culture media was removed 18 h after activation and replaced with retroviral supernatant supplemented with 50 μM β-mercaptoethanol (Gibco) and 8 μg/ml polybrene (Millipore) followed by a 1 h spinfection centrifugation at 2,000 rpm and 37°C. One day after transduction, congenically distinct ametrine+ T cells were mixed 1:1 and 5×105 total P14 cells were transferred into recipient mice subsequently infected with LCMV. Transduced CD8 T cell 1:1 ratios were validated by flow cytometry prior to adoptive cell transfer into mice.
Preparation of single-cell suspensions for flow cytometry
Isolation of CD8 T cells was performed similarly as described5. SI IEL were prepared through the removal of Peyer’s patches and the luminal contents from the entire SI. The SI was then cut longitudinally and into 1 cm pieces, then incubated at 37°C for 30 minutes in HBSS with 2.1 mg/mL sodium bicarbonate, 2.4 mg/mL HEPES, 8% bovine growth serum, and 0.154 mg/mL of dithioetheritol (EMD Millipore). The kidneys, salivary glands, fat, liver, and tumors were minced into small pieces and then incubated in RPMI with 1.2 mg/mL HEPES, 292 μ/mL L-glutamine, 1 mM MgCl2, 1 mM CaCl2, 5% fetal bovine serum, and 100 U/mL collagenase (Worthington) at 37°C for 30 min. After enzymatic incubations (kidneys, WAT, and livers), tissues were further dissociated over a 70-μm nylon cell strainer (Falcon). Lymphocytes from the small intestine, kidney, salivary gland, and liver were separated on a 44%/67% Percoll density gradient. Spleens and lymph nodes were processed with the frosted ends of microscope slides. Red blood cells were lysed with ACK buffer (140 mM NH4Cl and 17 mM Tris-base, pH 7.4). Blood samples were treated with ACK lysis buffer.
Antibodies, intracellular staining, flow cytometry and cell sorting
The following antibodies were obtained from TONBO: CD8a (53–6.7, FITC Catalog # 35–0081-U500, 1:200 dilution), Invitrogen: CD8a (53–6.7, PerCP-Cy5.5 Catalog # 1941169, PE Catalog # 2062469, PE-Cy7 Catalog # 25–0081-82, 1:200 dilution), PD1 (J43, APC Catalog # 47–9985-82, 1:200 dilution), CD127 (A7R34 PE Catalog # 12–1271-82, 1:50 dilution), Tim3 (RMT3, PE Catalog # 12–5870-82, 1:200 dilution), CD45.2 (104, PE-Cy7 Catalog # 25–0454-82, 1:200 dilution), KLRG1 (2F1 PerCP eF710 Catalog # 2011186, 1:400 dilution), and TNFα (MP6-XT22, APC Catalog # 17–7321-82, 1:100 dilution), eBioscience: CD3ε (145–2C11 PE Catalog # 12–0031-83, 1:100 dilution), CD8β (H35–17.2 FITC Catalog # 11–0083-82, 1:200 dilution), CD11b (M1/70, PE Catalog # 12–0112-82, 1:400 dilution), CD45.1 (A20–1.7, APC Catalog # 17–0453-82, 1:200 dilution), Thy1.1 (HIS51, FITC Catalog # 11–0900-85, 1:1000 dilution), CD44 (IM7, APC Catalog # 17–0441-82, 1:400 dilution), and IL-2 (JES6–5H4, PE Catalog # 12–7021-82, dilution 1:50), BioLegend: CD8a (53–6.7, PB Catalog # 100725, 1:200 dilution), CD62L (MEL-14, BV421 Catalog # 104435, BV510 Catalog # 104441, 1:400 dilution), CD103 (2E7, PE-Cy7 Catalog # 121426, 1:200 dilution), B220 (RA3–6B2, BV711 Catalog # 103255, 1:200 dilution), CD44 (IM7, BV711 Catalog # 103057, BV510 Catalog # 103043, 1:400 dilution), CD69 (H1.2F3, BV711 Catalog # 104537, 1:100 dilution), CD45.1 (A20–1.7, BV786 Catalog # 110743, BV510 Catalog # 110741, 1:200 dilution), Thy1.1 (OX-7 BV421 Catalog # 202529, AF647 Catalog # 202508, 1:1000 dilution), and IFNγ (XMG1.2, Pacific Blue Catalog 505818, 1:200 dilution), R&D Systems: LDLR (263123, FITC Catalog # 263123, PE Catalog # FAB2255P, 1:100 dilution), or BD Biosciences: CD107α (1D4B, FITC Catalog # 561069, 1:100 dilution). The H-2Db GP33–41 tetramer was obtained from the NIH Tetramer Core conjugated to APC or BV421 fluorochromes and used at 1:200 dilution. Dead cell staining was performed with Fixable Viability Dye eFluor780 or eFluor506 (eBiosciences) and used at 1:2000 dilution. For flow cytometry analysis, all events were acquired on a BD LSRFortessa X-20 or a BD LSRFortessa running FACSDiva software v9.0. Cell sorting was performed on a BD FACSAria and flow cytometry analysis was performed on FlowJo v10.
Imaging flow
Imaging flow cytometry data were acquired with a two-camera Amnis ImageStreamX MkII with 60X objective at Low speed using INSPIRE v6.2.187.0 acquisition software. 15,000 single, in-focus cells were collected per data file. FITC, PE, and PerCP were excited with a 488 nm laser (200 mW) and recorded in camera one. Filipin and BV510 were excited by a 405 nm laser (120 mW) and recorded in camera two. APC and APC-Cy7 were excited by 642 nm laser (150 mW) and recorded in camera two. SSC was recorded in channel 06 (camera one) and the 785 nm laser was set to 1.25 mW. Compensation was calculated with the IDEAS compensation wizard and then manually fine-tuned. Internal filipin intensities were calculated as a new feature on the Filipin channel using a mask based on M01 using Eroded 7 pixels with the IDEAS software. Feature intensities were exported as FCS files and further analyzed on FlowJo.
LCMV rechallenge and quantification of LCMV titers by qPCR
LCMV rechallenge and quantification of LCMV titers by qPCR to measure protection was done as previously published24,53. In short, mice were infected IV with 5000 CFU of L. monocytogenes expressing GP33–41. 23 days after the primary challenge, mice were infected intraperitoneally with 2×105 PFU of the Armstrong strain of LCMV. 3 days later, spleens and duodenums from the SI were harvested and flash-frozen in dry ice. RNA extraction from mice tissues was done following homogenization with mortar and pestle in liquid nitrogen, followed by homogenization with the QIAShredder (Qiagen), followed by RNA purification with the RNAeasy kit (Qiagen). cDNA was generated by retrotranscribing 1 μg of total RNA with random hexamer primers (High-Capacity cDNA Reverse Transcription Kit, Thermo Scientific). Quantification of LCMV genomes was performed by qPCR with 2x SYBR green (Agilent) and the following primers: m18S F (5’–3’): GTAACCCGTTGAACCCCAT, m18S R (5’–3’): CCATCCAATCGGTAGTAGCG, LCMV-GP F (5’–3’): CATTCACCTGGACTTTGTCAGACTC, LCMV-GP R (5’–3’): GCAACTGCTGTGTTCCCGAAA, and LCMV-NP F (5’–3’): CAGAAATGTTGATGCTGGACTGC, LCMV-NP R (5’–3’): CAGACCTTGGCTTGCTTTACACAG. The 2^-DeltaDeltaCt method, using m18S as housekeeping and WT samples as the reference group, was used to quantify the abundance of LCMV transcripts.
Mitochondrial respiration analysis of CD8 T cells
Mouse CD8 T cells from the spleen were treated with vehicle or 10 μM simvastatin (Sigma-Aldrich) at the time of activation in combination with 20 μM of Methyl-beta-cyclodextrin (MBCD)-Cholesterol (Sigma) or 10 μM mevalonolactone (Sigma) for 24 hours in complete T cell media. Analysis of in vitro activated and transduced CD8 T cells was performed 24 to 48 hours after transduction following FACS sorting of transduced cells. The Seahorse XF HS and XF96 Analyzers (Agilent) were used to measure the mitochondrial respiration capacity of CD8 T cells. Cells were seeded at a density of 20,000 cells per well in Seahorse XFp Cell Culture Miniplates with inserts without coating (Agilent Technologies) or 300,000 cells/well in CellTak-coated XF96 cell culture microplates (Agilent Technologies). Before seeding, cells were washed once with XF RPMI (Agilent) complete media. Complete media contains freshly added 2 mM glutamine (Gibco), 1 mM Sodium Pyruvate (Gibco), and 10 mM glucose. For the statin experiment, DMEM (Sigma #D5030) supplemented with 8 mM glucose, 2 mM glutamine, 2 mM pyruvate, and 5 mM HEPES was used. Cells were plated in 160 μl of volume, and plates were centrifuged at 300rcf for 4 minutes, followed by incubation at 37°C without CO2 for 30 minutes to allow for cell attachment and equilibration. Next, the chemicals for the mitochondrial stress test were prepared in assay media and loaded into the cartridge. For the mitochondrial stress test, the following compounds were sequentially injected into each well: 1 μM oligomycin, 2 μM carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), or two rounds of 1 μM FCCP injections, 0.5 μM rotenone, and 0.5 μM antimycin A (all from Agilent Technologies) and measurements were performed at 5-minute intervals. During the experiment, cells were maintained at 37°C without CO2. Three to six baseline measurements and three measurements following each injection were taken. Data were analyzed using the Seahorse XF Wave software (Agilent Technologies). Statistical analysis was performed using GraphPad Prism version 9.0 (GraphPad Software). Data were analyzed using a two-tailed paired t-test or a two-way ANOVA comparing OCR values by sample type and time.
Histology and Immunofluorescence staining of fresh frozen mouse tissues and FFPE human tissue
Mouse organs were harvested and rapidly snap-frozen in OCT in plastic molds for cryosection (Tissue-Tek Cryomold). 10 μm slices were obtained using a cryostat and mounted on glass slides, dried for 20 minutes at −20°C, submerged in cold PBS for 5 minutes and fixed in ice cold acetone at −20°C for 20 minutes. After fixation, slides were dried briefly at room temperature and stored at −80°C until stained. For staining, slides were equilibrated at room temperature, washed in PBS twice for five minutes, blocked in serum-free blocking reagent (Dako) ON at 4C, followed by staining with Srebp2 (ab30682, Abcam), CD103 PE (2E7, Invitrogen), CD45.2 FITC (104, Invitrogen) diluted in Antibody diluent solution (Dako) ON at 4 C, followed by a 2h incubation with Donkey Anti-Rabbit AF647 (Thermo Fisher), stained with DAPI, and mounted with coverslips using Vectashield hardset mounting media. Images were acquired on a Zeiss LSM700 confocal microscope and Aperio Scansope at 20X and 63X resolution. Deidentified human FFPE samples from healthy subjects were acquired from NovusBio. Slides were treated according to standard immunohistochemistry procedures and stained with anti-HMGCR (LS-B16059, LSBio), anti-CD3e (ab11089, Abcam), and anti-CD8α (66868, Thermo Fisher), followed by secondaries anti-rabbit AF647, anti-mouse AF488, and anti-rat AF594. Images were acquired at 20X with an Aperio FL fluorescence scanner system. Cell segmentation and quantification of HMGCR expression was done using Qupath54. In short, DAPI stains were used for cell detection with the following parameters; requestedPixelSizeMicrons 0.2, backgroundRadiusMicrons 2.5, sigmaMicrons 0.5, minAreaMicrons 5.0, maxAreaMicrons 50, threshold 300, waterShedPostProcess True, cellExpansionMicrons 1.5. CD8a cells were identified by training an object classifier based on “Random trees” using CD8α, and CD3ε staining, as well as cell morphology features. HMGCR expression was obtained using compute intensity features on the ROI CD8 mask with tileSizeMicrons 10.0.
Ultra-low input (ULI) RNA-sequencing
For studies pertaining to the profiling of shCd19 or shSrebf2 P14 CD8 T cells:
for each replicate, cells from 2 mice were pooled and then sorted for ULI RNA-seq following Immgen guidelines. Seven, and 14 days after initial infection with LCMV, 5 × 103 Ametrine+ P14 cells from each congenic were sorted from the spleen, and 5 × 103 iv− Ametrine+ P14 cells were sorted from the IEL, kidney, and liver into PBS + 5% BSA + 2% FBS + 0.5 M EDTA. 1 × 103 cells were then resorted into 5 μl of 1x TCL lysis buffer + 1% 2-mercaptoethanol.
For studies pertaining to the profiling of SI TRM from mice fed low or high cholesterol-containing diets:
Mouse previously infected (> 30 days) with LCMV and transferred with P14 CD8 T cells were fed a low or high cholesterol diet for 7 days. After preparation of single-cell suspensions, 5 × 103 iv− P14 CD8 T cells were sorted from the IEL, into PBS + 5% BSA + 2% FBS + 0.5 M EDTA. 1 × 103 cells were then resorted into 5 μl of 1x TCL lysis buffer + 1% 2-mercaptoethanol. Library preparation for ULI RNA-seq was performed as described online at Immgen (“ImmGenULI_RNAseq_methods.pdf”). Trimmomatic was used to remove adapters and trim low-quality reads (NexteraPE-PE.fa:2:30:10:1:TRUE LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:25)55. Trimmed reads were then aligned to the gencode M25 annotation of the mm10 genome using STAR with the default conditions. Aligned reads were then quantified with featureCounts (-t exon -g gene_id -p -B), and differentially expressed genes were identified using DEseq256. GSVA was performed using the GSVA package in R57. When raw expression counts were used as input, kcdf was set to Poisson, otherwise kcdf was set to Gaussian.
CRISPR-Cas9 mutagenesis screening using the lentiviral metabolic library
Lentiviral sgRNA metabolic library has been previously published23,28. In short, this custom mouse metabolic library contains 3,017 genes synthesized based on the gene list from reported human metabolic genes. A total of 6 gRNAs were designed for each gene according to previously published criteria. Two sub-libraries (Lib5 and Lib7) were made, with each containing 3 gRNAs targeting one gene and 500 non-targeting controls. The synthesis, purification, and quality control of the library was described previously. The in vivo screening approach was modified from previous studies. In short, lentivirus was produced by co-transfecting the lentiviral metabolic library plasmids, psPAX2, and pCAG4-Eco in HEK293T cells. At 48 h after transfection, the supernatant was harvested and frozen at −80°C. Naive P14 Cas9eGFP+ were isolated and pooled from lymph nodes and spleens from 8 P14 Cas9eGFP mice followed by in vitro activation with plate-bound anti-CD3 (1 μg/ml; 145–2C11; eBioscience) and anti-CD28 (1 μg/ml; 37.51; eBioscience) antibodies for 24 hours. Immediately after, cells were spin infected by centrifugation with Lib5 and Lib7 library supernatants supplemented with 1:200 LentiBoost (Sirion Biotech) and 55 μM beta-mercaptoethanol for 3h at 800G, followed by 4h incubation at 37°C 5% CO2. Immediately after, cells were washed and cultured with human IL-2 (20 IU/ml; Peprotech), mouse IL-7 (2.5 ng/ml; Peprotech) and IL-15 (25 ng/ml; Peprotech) for 4 days to expand and allow gene editing to occur. Next, cells were sorted based on the expression of Ametrine, and an aliquot of 5×106 transduced Cas9eGFP P14 cells was saved as “input” (around 500 × cell coverage per sgRNA). Transduced P14 cells (5×105) were then transferred IV to 7, and 9 naive C57BL/6 mice for Lib5 and Lib7, respectively, followed by LCMV infection (2×105 PFU) 1 h later. At day 13 and day 14 after LCMV infection for Lib 5 and Lib7, respectively, donor-derived TCM (CD62L+CD127+), TEM, and t-TEM from the spleen, TEM and TRM from the liver, and TRM from the SI were sorted into FBS and frozen at −80°C until genomic DNA extraction. Cell coverage per sgRNA per population and library were: Lib5: TEM = 110, TCM = 16, t-TEM = 66, Liver TEM = 28, Liver TRM = 8, SI TRM = 12, and Lib7: TEM = 141, TCM = 30, t-TEM = 77, Liver TEM = 50, Liver TRM = 17, Gut TRM = 31.
Targeted CRISPR Cas9 screen
sgRNAs targeting the 50 selected genes of the mevalonate and cholesterol synthesis pathway and adjacent enzymes were picked from either CRISPiCK or from the 3017 metabolic library and ordered as primers with BbsI overhangs from IDT as oPools Oligo Pools (IDT) and cloned into BbsI-linearized LsgA vector. Library preparation and equal sgRNA representation verification was done by subcloning into Stbl3 bacteria and sequencing of individual clones. Retroviral supernatant was titrated to achieve a targeted transduction efficiency of 30%, which is equivalent to an MOI of ~0.3. sgCd19 and sgFdft1 sgRNA were cloned into LMG retroviral plasmid. Double transduction of either LMG-sgCd19 or LMG-sgFdft1 with the targeted LsgA library was done via spinoculation of enriched and in vitro activated P14 Cas9eGFP T cells following by 24h expansion in hIl-2 (10 u/mL). LMG-derived eGFP expression is two orders of magnitude stronger than eGFP on P14 T cells, thus making the identification of LMG-transduced P14 Cas9eGFP cells possible by flow cytometry. After sorting double positive (GFP+Ame+) transduced P14 T cells, 5×105 cells from either sgCd19 and sgFdft1 groups were transferred into 6 male B6 mice each and infected with LCMV 1 hour later. Cell pellets from double-transduced sgCd19 and sgFdft1 were kept as input. TE and TMP populations were sorted based on CD127 and KLRG1 stain from blood pooled from 6 mice of each condition at day 7 after infection and cell pellets were kept at −80 C. Spleen TEM and liver TRM populations were sorted at day 14 after LCMV infection based on the expression of CD127+ and CD62L− stain (TEM), and IV− stain (TRM) of total P14 in the spleen, and liver, respectively, and pellets were stored at −80°C until processing. The SI TRM population included LP and IEL fractions and were similarly sorted at day 14 after LCMV infection by FACS. The dLN and TIL populations were sorted from single cell preparations from 5 pooled dLN and 5 pooled MC38-GP33–41 tumors 6 days after adoptive cell transfer. The cell yields and cell per sgRNA coverage for each sample were as follows: Input sgCd19, 1×105 total cells at 645 cells/sgRNA, Input sgFdft1, 1×105 total cells at 645 cells/sgRNA, sgCd19 TE, 2.39×105 total cells at 1542 cells/sgRNA, sgCd19 TMP, 1×104 total cells at 65 cells/sgRNA, sgCd19 Liver TRM, 1×104 total cells at 65 cells/sgRNA, sgCd19 Sp TEM, 1×104 total cells at 65 cells/sgRNA, sgFdft1 TE, 1.6×105 total cells at 1032 cells/sgRNA, sg Fdft1 TMP, 1×104 total cells at 65 cells/sgRNA, sgFdft1 Liver TRM, 6×103 total cells at 39 cells/sgRNA, and sgFdft1 Sp TEM: 2.5×103 at 16 cells/sgRNA. The cell yields and cell per sgRNA coverage of the SI TRM targeted screen were as follows: Input, 1×106 cells at 645 cells/sgRNA, SI TRM, 4.8×103 cells at 31 cells/sgRNA. The cell yields and cell per sgRNA coverage of the dLN and TIL screen in MC38-GP33–41 were as follows: Input, 5×104 total cells at 323 cells/sgRNA, dLN, 3×103 total cells at 19 cells/sgRNA. TIL fractions were collected in two subfractions: Tim3low, 4,425 cells at 29 cells/sgRNA, and Tim3high, 3,065 total cells at 20 cells/sgRNA. Frequency of Tim3high and Tim3low was ~50%. Fastq files from these two fractions were later combined to simplify analysis and increase coverage for TIL fraction. Genomic DNA was extracted according to the manufacturer’s instructions (DNeasy, Qiagen), Qiagen). The sgRNA-containing LsgA cassette was amplified with LV primers forward 5’ TTTCTTGGCTTTATATATCT 3’ and reverse 5’ GACTAGCCTTATTTTAACTT 3’. The PCR product was cleaned according to the manufacturer’s instructions (QIAquick PCR purification), and Nextera handles were added by PCR using Nextera Handle forward 5’- TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTTTCTTGGCTTTATATATCT 3’ and reverse 5’ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTAGCCTTATTTTAACTT 3’. After PCR product purification, PCR product was indexed with Illumina compatible indices. All PCR steps were done using proof-reading DNA polymerase Q5 (NEB). Library sizes were verified in the high sensitivity D1000 screen tape system (TapeStation, Agilent). Sequencing was performed in a Novaseq in a PE100 run format. sgRNA frequencies were obtained using Mageck58 count using the read 1 Fastq file and statistical testing against a background of six non-targeting sgRNAs using Mageck -test of each sample output to its corresponding input. Only the verified sgRNA for Srebf2 (Lib5_20788.3: ACTCCAGTGACAGTACACTG) was used for analysis.
Adoptive therapy tumor model
For adoptive therapy experiments, 5×105 B16-GP33–41 cells or MC38-GP33–41, which were verified Mycoplasma negative by PCR, and authenticated by in vitro killing assays, were transplanted subcutaneously into the right flank of wild-type B6 mice. After tumors became palpable, 7–8 days after transplantation, in vitro activated and expanded P14 CD8 T cells were transferred intravenously. For comparison of TIL accumulation in a mixed transfer setting, naive P14 CD8 T cells were activated, transduced and expanded with 50 U/mL of hIL-2 for 2–3 days; cells transduced with control constructs (Cd19 shRNAmir) or experimental constructs (Srebf2 shRNAmir) were mixed 1:1 and 1×106 P14 cells were transferred intravenously. For efficacy studies, transduced cells were expanded for 5–6 days; transduced cells were then sorted, and 1×106–2.5×106 cells were transferred intravenously into mice with established MC38-GP33–41 or B16-GP33–41 tumors. Tumors were monitored daily, and mice with ulcerated tumors or tumors exceeding 400 mm2 were euthanized, in accordance with UCSD IACUC.
Zaragozic Acid A (ZAA), anti-PD1 treatment, and CD8 depletion in mouse tumor models
At day 6 after tumor injection, the mice were randomized and treated every other day with ZAA (Santa Cruz Biotechnology) or vehicle control by alternating intravenous (200 μg/mouse) and intraperitoneal (100 μg/mouse) injections until day 16. For combination therapy with anti-PD1 antibody, ZAA was administered with the same schedule, and three consecutive treatments of anti-PD1 (InVivoMab anti-mouse PD-1 (CD279), BioXCell, BE0033–2) at 200 μg/mouse were delivered IP on days 9, 10, and 11 after tumor injection. Isotype control for Syrian IgG was used (InVivoMAb polyclonal Syrian hamster IgG, BioXCell). For CD8-depleting experiments in combination with ZAA, anti-CD8α Ab (2.43 BioXCell) was administered intraperitoneally at a dose of 200 μg/mouse starting 2 days prior to tumor implantation for three consecutive days (−2, −1, 0), followed by 2 additional doses of 100 μg/mouse on days 4 and 8, and ZAA treatment scheduled was started at day 4 after tumor implantation. Isotype control for Rat IgG2 was used (InVivoMAb rat IgG2a isotype control, anti-trinitrophenol, BioXCell).
Lovastatin treatment
2 days after LCMV Arm infection, mice were randomized into two groups and treated with either Vehicle (DMSO) or 10 mg/kg of Lovastatin (Calbiochem, 438185) in DMSO via IP injection daily for 5 consecutive days, followed by treatment once every three days after day 7 after LCMV infection until day 21 after LCMV infection.
Cloning of mouse Pdss2 CCDS in the pMIA vector
Total cDNA from liver homogenates of B6 WT mouse was used to PCR amplify Pdss2 CCDS by using the primers F (5’–3’): TCTGAGGGTTTGGAATCGCC, and R (5’–3’): CCGCCTCATTCTCTGGGTTT using a touch-down protocol starting at 62 with −0.5 C delta for 10 rounds, followed by 25 rounds of amplification at 58 C. PCR product was cleaned and reamplified with primers F (5’–3’): TAGCGGATCCCAATTGCTCGAGgccaccATGAGCCTCCGGCAGCTGCTGTTGCGCTTG and R (5’–3’): GGGCGGAATTGATCCCGCTCGAGTCAAGAAAATCTGGTCACAGCAAAC. This second set of primers adds complementary overhangs to the pMIA vector to allow cloning into Xho-I linearized pMIA plasmid using the NEBuilder HiFi DNA Assembly standard protocol (NEB), and add the Kozak sequence (gccacc) to promote gene expression. Cloned Pdss2 gene sequence was verified by full plasmid sequencing (Primordium).
shRNAmir sequences and sgRNA cloning in LsgA and LMG vectors
The following sequence was used for the shRNAmir targeting of Srebf2 with the pLMPd-Amt retroviral construct: 5’-TGCTGTTGACAGTGAGCGATGCTCTAGAGTATTTGAAATATAGTGAAGCCACAGATGTATATTTCAAATACTCTAGAGCACTGCCTACTGCCTCGGA-3’, and Cd19 (control): 5’- TGCTGTTGACAGTGAGCGAATGGATAAGTCTGACGACCTATAGTGAAGCCACAGATGTATAGGTCGTCAGACTTATCCATGTGCCTACTGCCTCGGA. Non-targeting (NT) shRNA (TRNSU6001), and shRNAmir targeting Fdft1 (ULTRA-3215720) were purchased from Transomics. The following primer pairs used for CRISPR/Cas9-mediated gene disruption were hybridized by heating at 95°C for 5 minutes followed by a ramp down of 1°C each 30s to 4°C in UltraPure Distilled Water (Invitrogen):
sgCd19 forward 5’- CACCGACCTTCACGTGCCTCTCGA
sgCd19 reverse 5’- AAACTCGAGAGGCACGTGAAGGTC-3’
sgSrebf2 (Lib5–20788.3) forward 5’-CACCGACTCCAGTGACAGTACACTG-3’
sgSrebf2 (Lib5–20788.3) reverse 5’-AAACCAGTGTACTGTCACTGGAGTC-3’
sgFdft1 (Lib7–14137.1) forward 5’-CACCCCATCCCACACCCCATCCGG-3’
sgFdft1 (Lib7–14137.1) reverse 5’-AAACCCGGATGGGGTGTGGGATGG-3’
sgHpd (Lib5–15445.3) forward 5’-CACCTGGAGATACCACACACACCC-3’
sgHpd (Lib5–15445.3) reverse 5’-AAACGGGTGTGTGTGGTATCTCCA-3’
sgPdss2 (Lib5–71365.1) forward 5’-AAACATGATATTGGAATCTCGACC-3’
sgPdss2 (Lib5–71365.1) reverse 5’-AAACGGTCGAGATTCCAATATCAT-3’
Hybridized primer pairs were cloned into BbsI-linearized LsgA or LMG constructs by overnight ligation (T4 DNA Ligase, NEB) at room temperature, following transformation into DH5α (ThermoFisher). In some instances, a “G” was added at the beginning of sgRNA sequences not naturally starting with a G to increase transcription efficiency.
qPCR and sanger sequencing validation of gene targeting
For validation of Srebf2, Fdft1, Pdss2 and Hpd gene targeting efficiencies with shRNAmir and sgRNA retroviral constructs, enriched CD8 T cells were activated, transduced, and expanded for 4–6 d in 25 mg/ml hIL-2 (Cetus). Ametrine+ cells (Srebf2 shRNAmir, Srebf2 sgRNA, Hpd sgRNA, Pdss2 sgRNA, Fdft1 sgRNA, Fdft1 shRNA, non-targeting shRNA, or control Cd19 shRNAmir and sgRNA) were sorted directly into TRIzol (Life Technologies) for RNA quantification of shRNAmir knockdown, or MACS buffer with 2% FBS for genomic DNA isolation. RNA, and DNA were extracted per manufacturer specifications (RNeasy, Qiagen for RNA, DNeasy, Qiagen for genomic DNA). cDNA was then synthesized using Superscript II (Life Technologies) and quantitative PCR was performed using the Stratagene Brilliant II Syber Green master mix (Agilent Technologies). Srebf2 and Hmgcr expression levels were normalized to the housekeeping genes Hprt1. The following primers were used for qPCR:
Srebf2 forward: 5′-GCGTTCTGGAGACCATGGA-3′
Srebf2 reverse 5′-ACAAAGTTGCTCTGAAAACAAATCA-3′
Hmgcr forward 5′-CTCGTGGAATGCCTTGTGATTG-3′
Hmgcr reverse 5′-AGCCGAAGCAGCACATGAT-3′
Hmgcs1 forward 5’-GCCGTGAACTGGGTCGAA-3’
Hmgcs1 reverse 5’-GCATATATAGCAATGTCTCCTGCAA-3’
Ldlr forward 5’-AGGCTGTGGGCTCCATAGG-3’
Ldlr reverse 5’-TGCGGTCCAGGGTCATCT-3’
Hprt1 forward 5′-TGAAGAGCTACTGTAATGATCAGTCAAC-3′
Hprt1 reverse: 5′-AGCAAGCTTGCAACCTTAACCA-3′
Pdss2 forward: 5’-CACCATGATATTGGAATCTCGACC-3’
Pdss2 reverse: 5’- AAACGGTCGAGATTCCAATATCAT-3’
Fdft1 forward: 5’-GGATGTGACCTCCAAACAGGAC-3’
Fdft1 reverse: 5’-CAGACCCATTGAGTTGGCACAC-3’
Targeting efficiency of sgSrebf2, sgFdft1, sgPdss2, and sgHpd constructs on genomic DNA was assessed by PCR amplification of 100 ng of genomic DNA using a high-fidelity polymerase (Q5 High Fidelity DNA polymerase 2x, New England Biolabs) using a touchdown protocol (63°C Tm −0.5 C/cycle for 10 cycles, followed by 25 cycles at 58 C). PCR products were purified according to the manufacturer’s instructions (QIAquick PCR purification, Qiagen). Genomic DNA was amplified with the following primers:
Srebf2 forward 5’-ATCATGGCTGTTCTGACTCGG-3’
Srebf2 reverse 5’-GAAGCCCATTTTTGCACAGGG-3’
Fdft1 forward 5’-CTTCATGGGTCCTTGGGGAAC-3’
Fdft1 reverse 5’-CCTGTGGGGCGTGTATTGTCA-3’
Hpd forward 5’-GCATCACAGTCTCCCATCCT-3’
Hpd reverse 5’-CTGTCTCACCTCCCGAAGTTT-3’
Pdss2 forward 5’-TTGTCCCCTGCTACCATGTTC-3’
Pdss2 reverse 5’-GAATGCCATGTCTTGGACCG-3’
Sanger sequencing of purified PCR products (Eton Biosciences) was done using the following primers:
Fdft1 5’-AACCATGAAGTGTGTCAT-3’
Srebf2 5’- AGCTGCTTTTCTGAAGGT
Hpd 5’-ACTACCGAGCAAAACGGCAG-3’
Pdss2 5’-GCCCTGTCTTCATGATGCCC-3’
Genome editing by CRISPR/Cas9 for each construct was calculated with TIDE59.
Curation of metabolic signatures
We constructed a list of minimally overlapping metabolic gene sets (Metab205) by aggregating custom gene lists, and all gene sets from KEGG, BIOCARTA, and REACTOME listed in the C2.CP.gmt file from MsigDb (Broad Institute), and iterating rounds of curation using the GSVA tool computeGeneSetsOverlap in R. To use Metab205 on mouse data sets, bulk microarray and RNAseq expression data sets were converted to human gene symbols using the GSEA Collapse Dataset tool with Mouse_Gene_Symbol_Remapping_Human_Orthologs_MSigDb.chip file with default options. For MAGECK analysis of pathway enrichments, and single cell RNAseq analysis, human Metab205 gene symbols were translated to mouse gene symbols using bioDBnet.
scRNAseq of P14 in the subcutaneous MC38-GP33–41 model
P14 from tumors and spleens of mice were profiled by scRNAseq in two independent repeats. For each repeat, 2×105 P14 cells coming from pooled spleens and MC38-GP33–41 tumors from three independent mice were sorted into T cell culture media. Samples were spun down at 500 rcf for 5 minutes and resuspended in PBS + 0.04% (w/v) bovine serum albumin. Samples were then loaded into Chromium Chip B (10X Genomics) and partitioned into Gel Bead In-Emulsions (GEMs) in a chromium controller (10X Genomics). Single cell RNA libraries were generated according to the Chromium Single Cell 3’ Reagent Kits v3 User Guide and sequenced on a HiSeq 4000. Reads were aligned to the mm10 genome using cellranger count and final data matrices for all samples were generated with cellranger aggregate. The resulting counts matrix was then processed using Seurat in RStudio and cells with mitochondrial read % greater than 7 were discarded. Data was normalized and scaled using NormalizeData and ScaleData followed by batch correction of biological replicates was corrected with Harmony60. The top 2000 most variable genes were calculated using FindVariableGenes and used then used in the PCA calculation using RunPCA. After adding Harmony embeddings, we applied dimensionality reduction by uMAP using harmony reduction, and 1:20 dimensions. Louvain clustering was performed with Seurat’s FindClusters based on the top 20 principal components with the resolution set to 0.5. Final gene expression values were normalized using sctransform in Seurat. Additionally, data imputation was performed using MAGIC61 with the log-normalized expression values and the default settings and the exact solver. Seurat’s AddModuleScore with default settings function was used to calculate scores for the mevalonate/cholesterol synthesis pathway gene list. Heatmap was generated by extracting average SCT gene expression values for each group, scaled gene expression values by row, and plotted with ComplexHeatmap62, installed from Bioconductor, with column clustering.
Bioinformatic analysis of publicly available single-cell RNA-seq datasets
Human single cell RNAseq of immune populations from PBMC, SI (Ileum) and colon of healthy patients:
Data were downloaded from GEO (GSE125527). Count matrices for each sample were acquired using cellranger “count” followed by cellranger aggregation to obtain a pooled data set. Filtered_feature_bc_matrix files were loaded into Seurat in RStudio to construct the Seurat object. Cells with nCount between 500 and 15000, and less than 25 % mitochondrial reads were retained. Gene expression was normalized with SCTransform, followed by standard Seurat clustering and dimensionality reduction steps with 40 dimensions and 0.8 resolution. CD8 T cells from healthy subjects were further subset using SingleR63. Imputation of gene expression was performed using MAGIC61. Core TRM scores17 were calculated with UCell64 in CD8 T cells from the spleen and the SI, and values were tested for statistical significance between statin and non-statin users for each tissue using the ggsignif package. The inclusion criteria for statin use included subjects who were actively taking statin medication at least two weeks before sampling.
Pan-cancer single-cell RNAseq atlas of tumor-infiltrating CD8 T cells39:
integrated SingleCellExperimnt .rds file (int.CD8.S35.sce.merged.rds) was retrieved from the Zenodo repository (10.5281/zenodo.5461803) and transformed to a Seurat Object in R. Mevalonate/cholesterol synthesis pathway scores were calculated using AddModuleScore. The heatmap was generated with ComplexHeatmap with Euclidean distances to cluster rows (cancer type) and columns (meta.cluster.coarse). DimPlot was used to plot umap.harmony embeddings in a 2D plot grouped by “meta.cluster.coarse” annotations. Data from the CellTypist resource was downloaded from (https://www.tissueimmunecellatlas.org/) as an h5ad file, converted to a Seurat object, and mevalonate/cholesterol signature scores were calculated with UCell. DotPlot using the original metadata were used to plot scores grouped by different categories. The human signature for the mevalonate cholesterol synthesis pathway used was: ACAT1, ACAT2, CYP51A1, EBP, FDFT1, FDPS, GGPS1, HMGCR, HMGCS1, HMGCS2, IDI1, LBR, LSS, MVD, MVK, PMVK, TM7SF2, DHCR7, DHCR24, NSDHL, HSD17B7, MSMO1, SC5DL, LOC651621, SQLE, LOC730412, IDI2. (Note; The SC5DL gene name was updated to SC5D on June 1, 2020.)
In vivo Bodipy-Cholesterol, Bodipy-LDL uptake assays, and serum total cholesterol measurements
200 μl of a 10 mg/ml of Bodipy-Cholesterol in corn oil (Sigma) solution (2mg Bodipy-Cholesterol) was orally gavaged into C57B6 mice. After 16 minutes, mice were iv labeled with CD8α antibody, and 4 minutes later mice were taken down for immune cell profiling by flow cytometry. Bodipy-Cholesterol incorporation into spleen CD8 T cells was used as a negative control. One mouse per experiment was orally gavaged with 200 ul of corn oil to establish baseline signal. 200 μl of 1 mg/ml of Human Bodipy-LDL (Invitrogen) was delivered IV by RO injection. After 26 minutes, mice were iv labeled with 3 μg of CD8a antibody, and 4 minutes later mice were taken down for immune cell profiling by flow cytometry. One mouse per experiment was RO injected with PBS to establish baseline signal. Cholesterol measurement in mouse serum was done using the Amplex Red Cholesterol Assay (Thermo Fischer) according to the manufacturer’s instructions. Mouse serum was collected by submandibular bleeding followed by centrifugation at 7000 rcf for 15 minutes at 4C.
Intracellular Metabolite Isolation with Liquid chromatography-tandem mass spectrometry (LC-MS/MS) and analysis of metabolomics data.
To assess relative steady-state variation between metabolic intermediates in tissue resident and peripheral T cell populations, freshly sorted populations were washed twice with ice-cold phosphate-buffered saline, centrifuged at low speed to avoid lysis, and stored without buffer at −80°C prior to analysis. Intracellular metabolite isolates were prepared as previously described65. Briefly, pelleted cells were lysed in a cold solution of 4:1 methanol:water and rapidly cycled between −80°C for one minute and 37°C for one minute, three times. Lysates were centrifuged at 14,000 RPM for 10 minutes at 4°C to separate soluble metabolite extract from insoluble cell debris. Extracts were dried by vacuum concentrator and resuspended in a solution of cold 2:2:1 acetonitrile:methanol:water at 20,000 cell equivalents per microliter, and transferred to glass liquid chromatography vials for analysis. All solvents used were HPLC-grade (Fisher Scientific). Identification and quantification of extracted metabolites by LC-MS/MS analysis was described previously66. Sampling and liquid chromatography was performed on a Vanquish UHPLC system (Thermo Scientific) with Sequant ZIC-pHILIC polymeric column chromatography (100 mm × 2.1 mm, 5 μm) (EMD Millipore), maintained at 45°C. Cell extracts were separated by autosampler injection of 2 μL sample and eluting on a constant 0.4 mL/minute fluid flow rate. Elution followed a linear flow gradient of: (A) 20mM ammonium bicarbonate in water, pH 9.6, and (B) acetonitrile; 90% B for 0.25 minutes, a linear gradient to 55% B at 4 minutes, and maintained at 55% B until 6 minutes. Solid phase chromatography was then re-equilibrated for 2.5 minutes at 90% B between samples. Metabolite detection was performed via a coupled QExactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific). Samples were eluted in duplicate for acquisition in positive and negative ionization mode. Ionization was by a heated electrospray ionization (HESI) source: spray voltage of 3.5 kV; auxiliary gas, sheath gas, and sweep gas flow rates of 20, 40, and 2 AU, respectively. Capillary gas heater temperature was 275°C and auxiliary gas heater temperature was 350°C. MS1 profile spectra were acquired in a mass range of 67–1000 m/z, with resolution of 35,000, AGC volume of 1×106 maximum IT of 75 ms. Data-dependent tandem mass spectra were acquired to aid in metabolite identification, using a normalized collision energy of 25 eV, an isolation window of 1.5 m/z, resolution of 17,500, a loop count of 5, AGC volume of 1×105, and with maximum IT of 50 ms. Metabolite identifications were made against an in-house library of standards or against tandem spectral matching from publicly available databases housed in the GNPS Library (gnps.ucsd.edu) and meeting criteria for a match score of >0.70 and accurate mass window of 10 ppm. Tandem mass spectra matching against commercial standards of 4-hydroxybenzaldehyde (Millipore Sigma, 91554) and mevalonate (Millipore Sigma, 42147) were derived from the most abundant ion for each compound: m/z = 121.0295 for 4-hydroxybenzaldehyde [M-H], and m/z = 147.0663 for mevalonate [M-H]. Black spectra are derived from cell lysate, and red mirrored spectra from HPLC and GC-grade commercial standards. Intensity matrices for identified metabolites, or identified and annotated metabolites, were processed using Metaboanalyst 5.0 to perform clustered heatmaps and PCA.
Quantification of CoQ species in CD8 T cells
Enriched, in-vitro-activated CD8 T cells with CD3/CD28, as previously described, were expanded in 50 U/mL of human IL-2 for 24 and treated with ZAA at 1 μM for 48h. Similarly, in-vitro-activated CD8 T cells were transduced with LsgA-sgCd19, LsgA-sgFdft1, pMIA-Empty, or pMIA-Pdss2 constructs and expanded 48 h in 50 U/mL of human IL-2 before sorting 1–2×106 Ame+ cells per sample. 1–2×106 cells were pelleted and frozen at −80°C until processing. Lipids were extracted using a modified version of the Bligh-Dyer method. Briefly, frozen cell pellets were resuspended in 350 μL of milliQ water shaken in an eppendorf tube with 300 μL methanol and 1 mL chloroform containing internal standards (d7-Cholesterol and d6-CoQ10) for 30s. The resulting mixture was vortexed for 15s and centrifuged at 2600 × g for 8 minutes to induce phase separation. The organic (bottom) layer was retrieved using a Pasteur pipette, dried under a gentle stream of nitrogen, and reconstituted in 2:1 chloroform:methanol for LC/MS analysis. Lipidomic analysis was performed on a Vanquish HPLC online with a Q-Exactive quadrupole-orbitrap mass spectrometer equipped with an electrospray ion source (Thermo). Data was acquired in positive ionization mode. Solvent A consisted of 95:5 water:methanol, Solvent B was 70:25:5 isopropanol:methanol:water. Solvents A and B contained 5 mM ammonium formate with 0.1% formic acid. An XBridge (Waters) C8 column (5 μm, 4.6 mm × 50 mm) was used for separation. The gradient was held at 0% B between 0 and 5 min, raised to 20% B at 5.1 min, increased linearly from 20% to 100% B between 5.1 and 55 min, held at 100% B between 55 minutes and 63 min, returned to 0% B at 63.1 min, and held at 0% B until 70 min. Flow rate was 0.1 mL/minute from 0 to 5 min, 0.3 mL/minute between 5.1 minutes and 55 minutes, and 0.4 mL/minute between 55 minute and 70 min. Spray voltage was 3.5 kV; S-lens RF level is 65; Sheath, auxiliary, and sweep gases were 50, 10 and 1, respectively. Capillary temperature was 325°C and auxiliary gas heater temperature was 200°C. Data was collected in full MS/dd-MS2 (top 10). Full MS was acquired from 150–1500 m/z with resolution of 70,000, AGC target of 1×106 and a maximum injection time of 100 ms. MS2 was acquired with a resolution of 17,500, a fixed first mass of 50 m/z, AGC target of 1×105 and a maximum injection time of 200 ms. Stepped normalized collision energies were 20, 30 and 40%. Skyline67 was used to measure peak areas of a list of lipids of interest (CoQ9, CoQ10, DMQ9, DMQ10). Normalized peak areas were used in data reporting. Data were normalized using internal standards and total protein quantification.
Extended Data
Extended Data Figure 1. Functional genetics in vivo, metabolomics and transcriptional analysis of ex vivo populations of memory CD8 T cells identify a graded upregulation of a Srebp2-dependent metabolic programming across TRM.
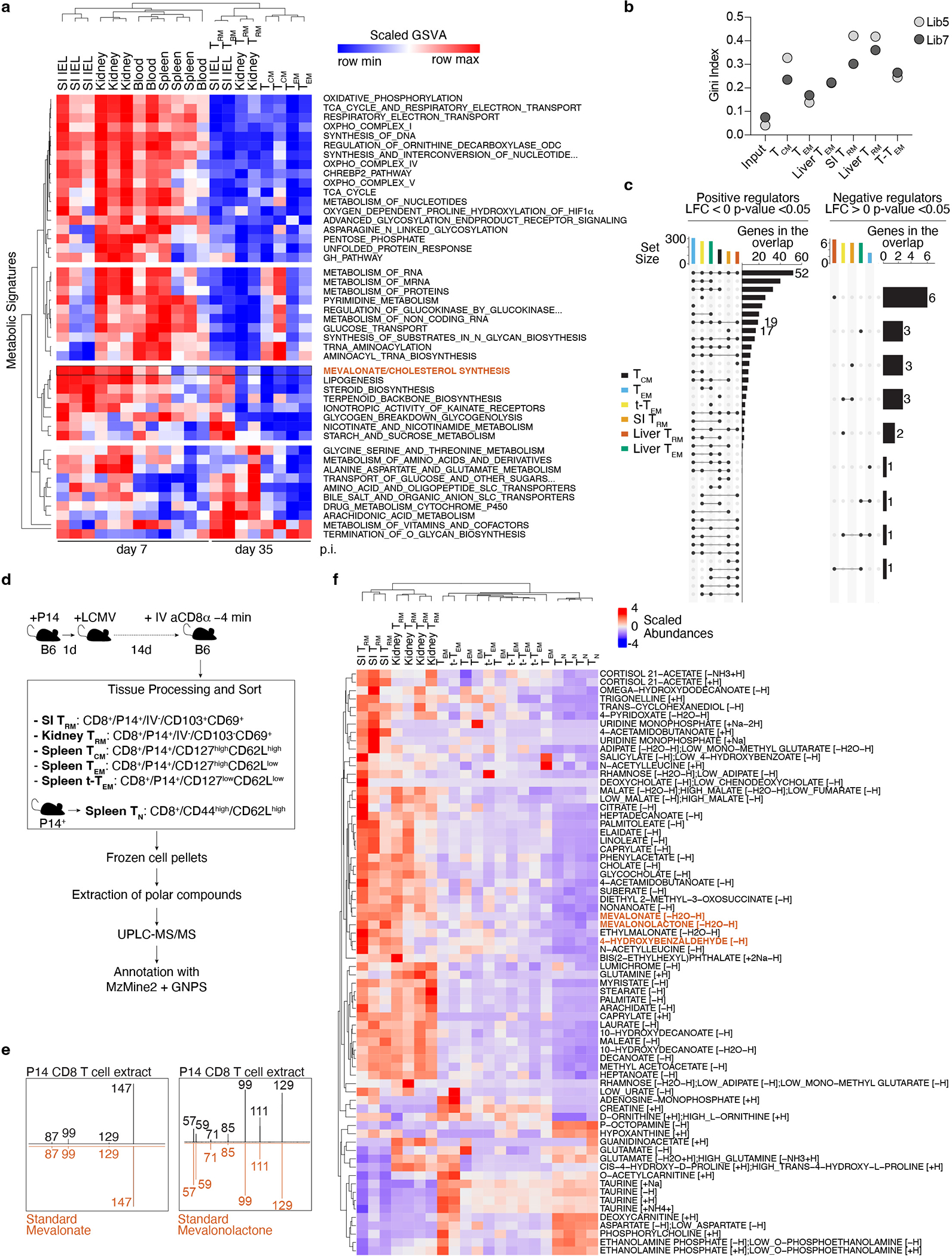
a, Unsupervised hierarchical clustering heatmap of scaled GSVA scores for metabolic signatures across samples, p.i., post-infection. GSE107278. b, sgRNA library heterogeneity reported as Gini Index values. c, Upset plot showing intersection sizes and set sizes for positive and negative regulators of each P14 CD8 T cell population in the in vivo CRISPR/Cas9-mediated loss-of-function screen. d, Sample acquisition and analysis workflow for LC-MS/MS-based untargeted metabolomics of ex vivo populations of CD8 T cells in the context of LCMV infection. Gating strategy included for each cell type analyzed. e, Tandem mass spectra matching against commercial standards of mevalonate and mevalonolactone. Tandem mass spectra derived from the most abundant ion for each compound. Black spectra are derived from cell lysate, red mirrored spectra from commercial standard. f, Unsupervised hierarchical clustering of scaled relative abundances of identified annotated metabolites of P14 cells profiled by untargeted metabolomics at day 13 post LCMV infection.
Extended Data Figure 2. Upregulation of the mevalonate/cholesterol synthesis pathway in mouse and human TRM.
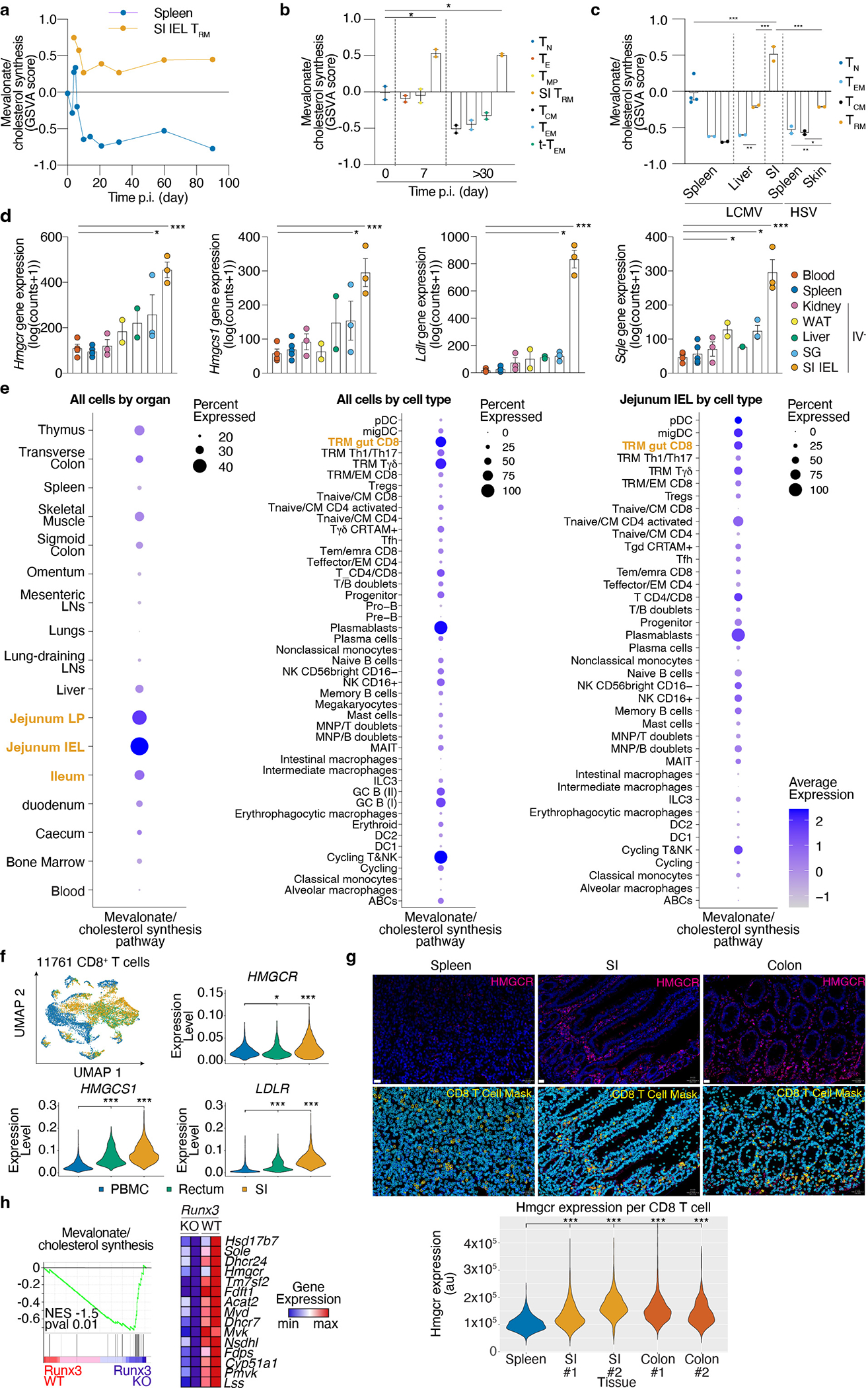
a, Mevalonate/cholesterol synthesis GSVA scores from averaged single cell expression of spleen and SI P14 at indicated time points after LCMV infection profiled by scRNASeq, GSE131847. b, Mevalonate/cholesterol synthesis GSVA scores for indicated subsets of CD8 T cells in the spleen or SI in the context of LCMV infection, GSE157072. c, Mevalonate/cholesterol synthesis GSVA scores for indicated subsets of CD8 T cells at the indicated tissues in the context of LCMV and Herpes Simplex Virus (HSV), GSE70813. d, Gene expression of indicated genes in P14 CD8 T cell populations from indicated tissues >30 days after LCMV infection, GSE182276 e, Mevalonate/cholesterol synthesis pathway scores for all human immune cells grouped by indicated tissues (left), cell types (middle), or cell types of the Jejunum SI (right), from the tissue immune cell scRNAseq atlas. f, Expression of selected genes on human CD8 T cells from PBMC, rectum, and SI samples, pooled from 13 healthy donors, GSE125527. g, HMGCR protein expression in CD8 T cells from human spleen, SI, and colon by immunofluorescence staining. Quantification of total HMGCR by CD8 T cell area. Donor ID (#). Scale bar, 20 μm. h, GSEA of the mevalonate/cholesterol synthesis in Runx3 KO or control CD8 T cells GSE106107. Data are pseudo-bulk averaged values (a), mean +/− s.e.m. (b, c, and d), or geometric distribution (f and g), with a total of n=2 (b), n=2–4 (c), n=2–5 (d) mice, and n=1–2 (g) samples. Two-sided unpaired t-Test (b, c, d, f, and g), and Gene Set Enrichment Analysis (h). *P<0.05, **P<0.01, ***P< 0.005.
Extended Data Figure 3. A Srebp2-dependent metabolic program controls SI TRM formation.
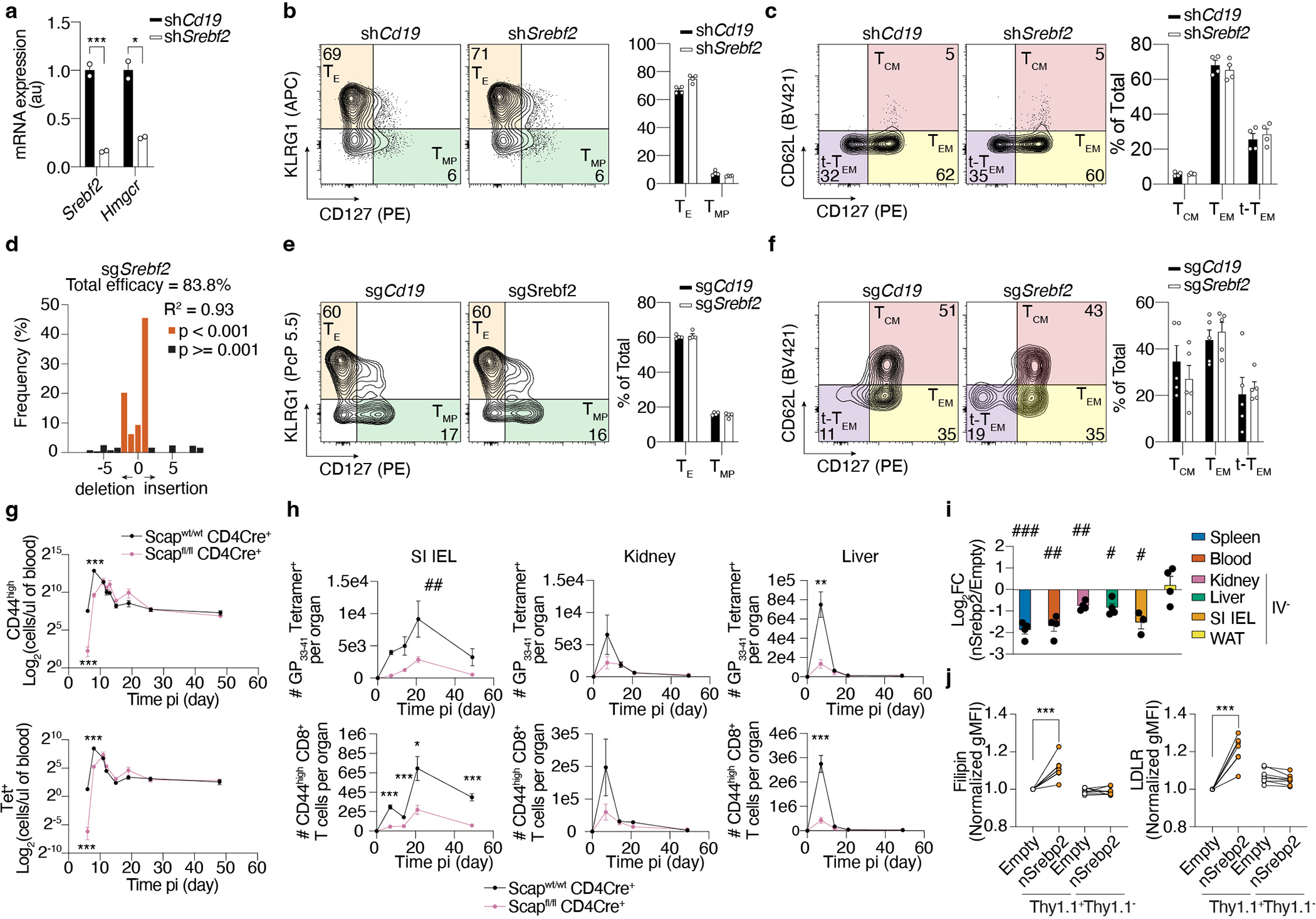
a, Quantification of gene expression by qPCR of shRNAmir control (shCd19) or shSrebf2 shRNAmir P14 CD8 T cells before adoptive transfer. b, Frequency of effector shRNAmir control (shCd19) or shSrebf2 shRNAmir P14 CD8 T cell populations in the spleen 7 days after LCMV infection. c, Frequency of memory shRNAmir control (shCd19) or shSrebf2 shRNAmir P14 CD8 T cell populations in the spleen 21 days after LCMV infection. d, CRISPR/Cas9-mediated indel efficiency of the Srebf2 sgRNA construct on sorted transduced P14 cells before adoptive transfer. e, Frequency of effector sgCd19 or sgSrebf2 P14 cell populations in the spleen 7 days after LCMV infection. f, Frequency of memory sgCd19 or sgSrebf2 P14 cell populations in the spleen 21 days after LCMV infection. g, Total cell quantification of CD44high CD8 T cells (left) and Tet+ CD8 T cells (right) in the blood of Scap WT or Scap KO at different time points after LCMV infection. h, Total cell quantification of Tet+ CD8 T cells (upper) and CD44high CD8 T cells (lower) in SI, kidney, and liver of Scap WT or Scap KO at different time points after LCMV infection. i, Ratio of congenically distinct P14 CD8 T cells transduced with a constitutively active nuclear form of human Srebp268 (nSrebp2) or an empty vector (Empty) from indicated tissues 7 days after LCMV infection. j, Flow cytometry analysis of cholesterol content by Filipin stain (left), and LDLR expression in P14 CD8 T cells transduced with a bicistronic construct encoding Thy1.1 alone or Thy1.1 and nSrebp2 (right). P14 CD8 T cells from the kidney, liver, WAT, and SI were gated on the IV− population (h and i). Data are mean +/− s.e.m. and representative of at least two independent experiments, with a total of n=2 (a), n=4 (b-e), n=5 (f), n=6 (WT) and n=7 (Scap KO) (g and h), n=4 (i and j) mice and n=2 (d) cell replicates. Two-sided unpaired (a, d, g, and h) and paired (j) t-Test. *P<0.05, **P<0.01, ***P< 0.005. Two-sided one-sample t-Test (i) #P<0.05, ##P<0.01, ###P< 0.005. Two-way ANOVA (h) ##P<0.01.
Extended Figure 4. Limited effect of Srebp2 on transcriptional programs of CD8 T cell differentiation.
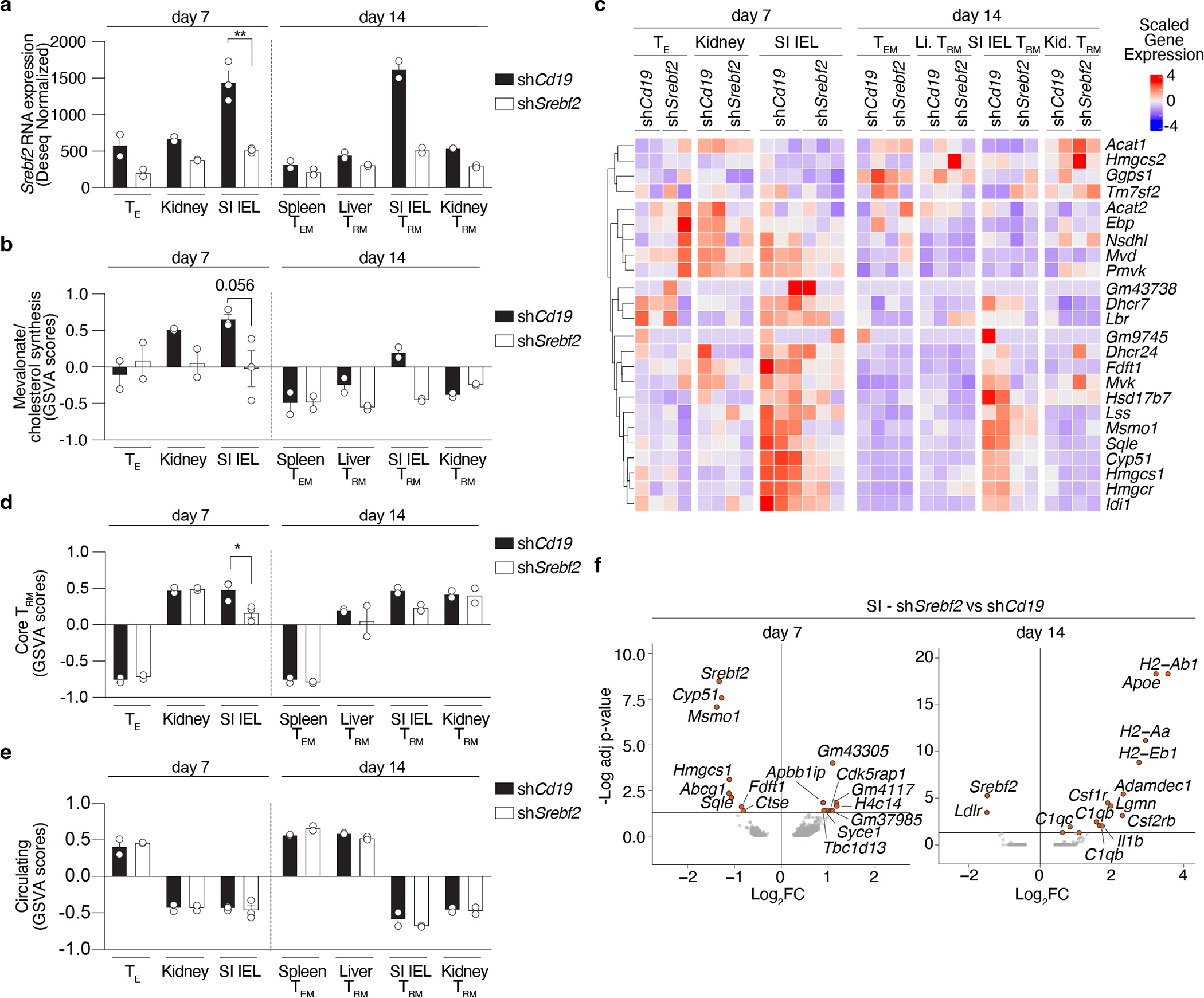
a, Srebf2 expression values from the bulk ULI-RNAseq analysis of shRNAmir control (shCd19) and Srebf2 shRNAmir P14 CD8 T cells from the spleen, kidney, liver, and SI from day 7 and 14 after LCMV infection. b, Mevalonate/cholesterol synthesis pathway GSVA scores from the bulk ULI-RNAseq analysis of shRNAmir control (shCd19) and Srebf2 shRNAmir P14 CD8 T cells from the spleen, kidney, liver, and SI from day 7 and 14 after LCMV infection. c, Unsupervised hierarchically clustered heatmap of mevalonate/cholesterol synthesis pathway gene expression values from the bulk ULI-RNAseq analysis of shRNAmir control (shCd19) and Srebf2 shRNAmir P14 CD8 T cells from the spleen, kidney, liver, and SI from day 7 and 14 after LCMV infection. d, Core TRM GSVA scores from the bulk ULI-RNAseq analysis of shRNAmir control (shCd19) and Srebf2 shRNAmir P14 CD8 T cells from the spleen, kidney, liver, and SI from day 7 and 14 after LCMV infection. e, Circulating GSVA scores from the bulk ULI-RNAseq analysis of shRNAmir control (shCd19) and Srebf2 shRNAmir P14 CD8 T cells from the spleen, kidney, liver, and SI from day 7 and 14 after LCMV infection. f, Volcano plots of differentially expressed genes between shCd19 and shSrebf2 SI P14 CD8 T cells at days 7 and 14 after LCMV infection, adjusted (adj) p-value. P14 CD8 T cells from the kidney, liver and SI were gated on the IV− population (a-f). TE were defined as KLRG1highCD127low P14 CD8 T cells. Data are mean +/− s.e.m.. (a, b, d, and e) from one experiment with n=2–3 (a-f), where every sample is 1×103 cells pooled from 2~3 mice. Two-sided unpaired t-Test (a, b, d, and e), and Deseq2 DEG testing with Benjamini-Hochberg multiple test correction. *P<0.05, **P<0.01, ***P< 0.005.
Extended Data Figure 5. SI TRM uptake and sense dietary cholesterol.
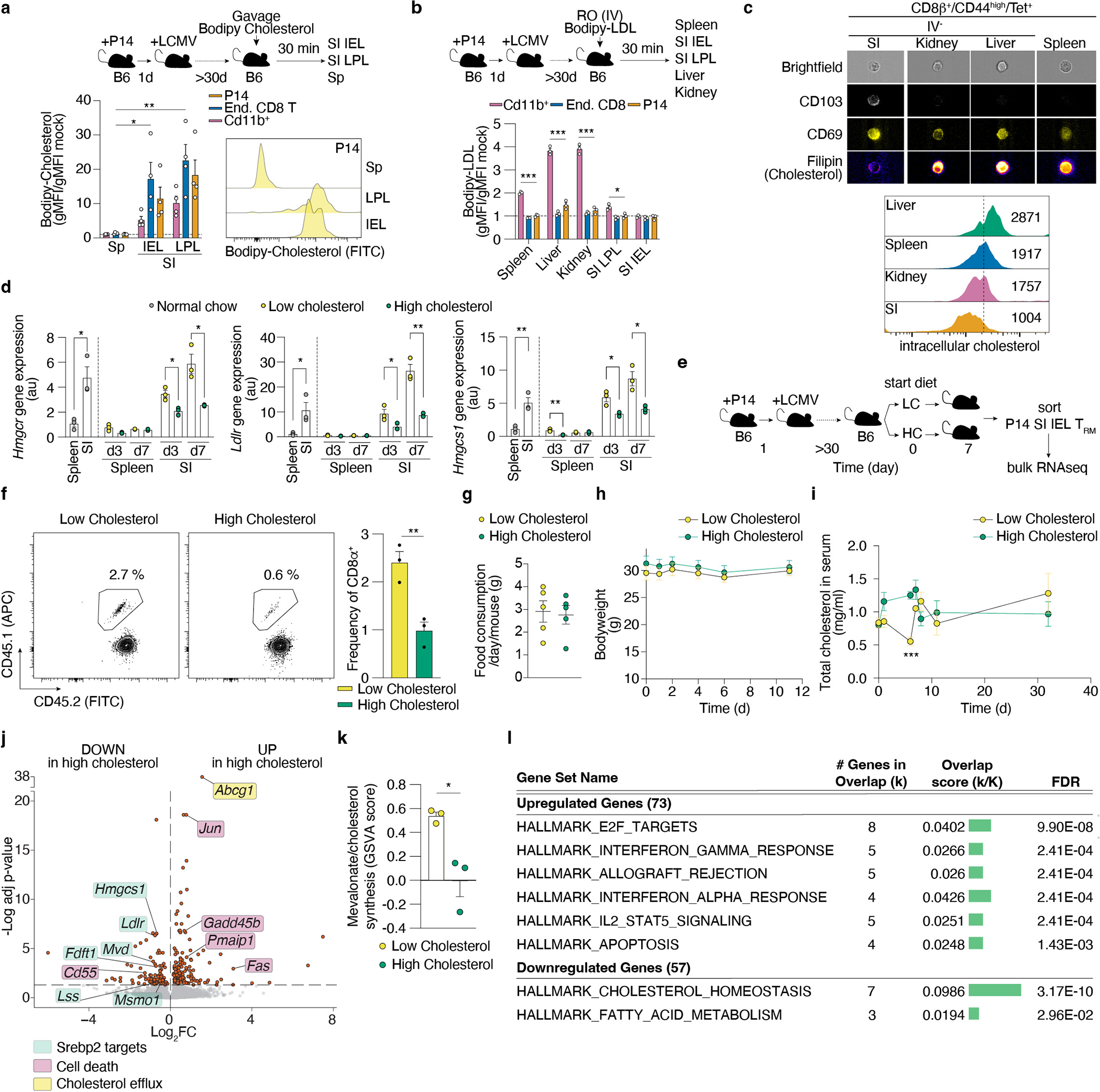
a, Quantification by flow cytometry of in vivo incorporation of Bodipy-cholesterol on indicated cell populations after 10 minutes after oral administration of Bodipy-cholesterol to memory P14 mice. b, Quantification by flow cytometry of incorporation of Bodipy-LDL on indicated cell types of the spleen 20 minutes (min) after IV delivery to memory P14 mice. c, Quantification of intracellular cholesterol by filipin staining in Tet+ populations of CD8 T cells for various tissues >30 days post LCMV infection measured by imaging flow cytometry. d, Gene expression analysis by qPCR of selected Srebp2 targets on CD8 T cells from the spleen and SI of mice on regular chow or subjected to low and high cholesterol-containing diets. e, Experimental design to measure transcriptional changes induced by dietary cholesterol on established SI TRM by ULI-RNAseq, f, SI TRM frequencies from total CD8 T cells after 7 days of dietary intervention. g, Food consumption per day per mouse subjected to a low or high-cholesterol diet. h, Bodyweight of mice subjected to low or high cholesterol-containing diets. i, Total cholesterol in serum in mice subjected to low or high cholesterol-containing diets. j, Volcano plot of differentially expressed genes in SI TRM from mice fed a high vs a low cholesterol-containing diet. k, Mevalonate/cholesterol synthesis GSVA scores in SI TRM from mice fed a high versus low cholesterol-containing diet. l, Gene pathway analysis of top upregulated and top downregulated genes between SI TRM from mice fed a high versus low cholesterol-containing diet (MsigDb, Hallmark gene sets). P14 CD8 T cells and CD8β+/CD44high/Tet+ from the kidney, liver and SI were gated on the IV− population (a, b, c, e, f, j, k, and l). Data are mean +/− s.e.m. and representative of at least two independent experiments, with a total of n=4 (a), n=3 (b, d), n=5 (c), n=3 (f) mice, or pooled from at least two independent experiments with a total of n=5–10 (g-i) mice, or performed once (j-l) with a total of n=3 samples each sample is 1×103 SI TRM pooled from 2~3 mice (j, k). Two-sided unpaired t-Test (a, b, d, f, i, and k), and Deseq2 DEG testing with Benjamini-Hochberg multiple test correction (j). *P<0.05, **P<0.01, ***P< 0.005.
Extended Data Figure 6. Mevalonate/cholesterol synthesis pathway intermediates sustain the production of non-steroidal metabolites.
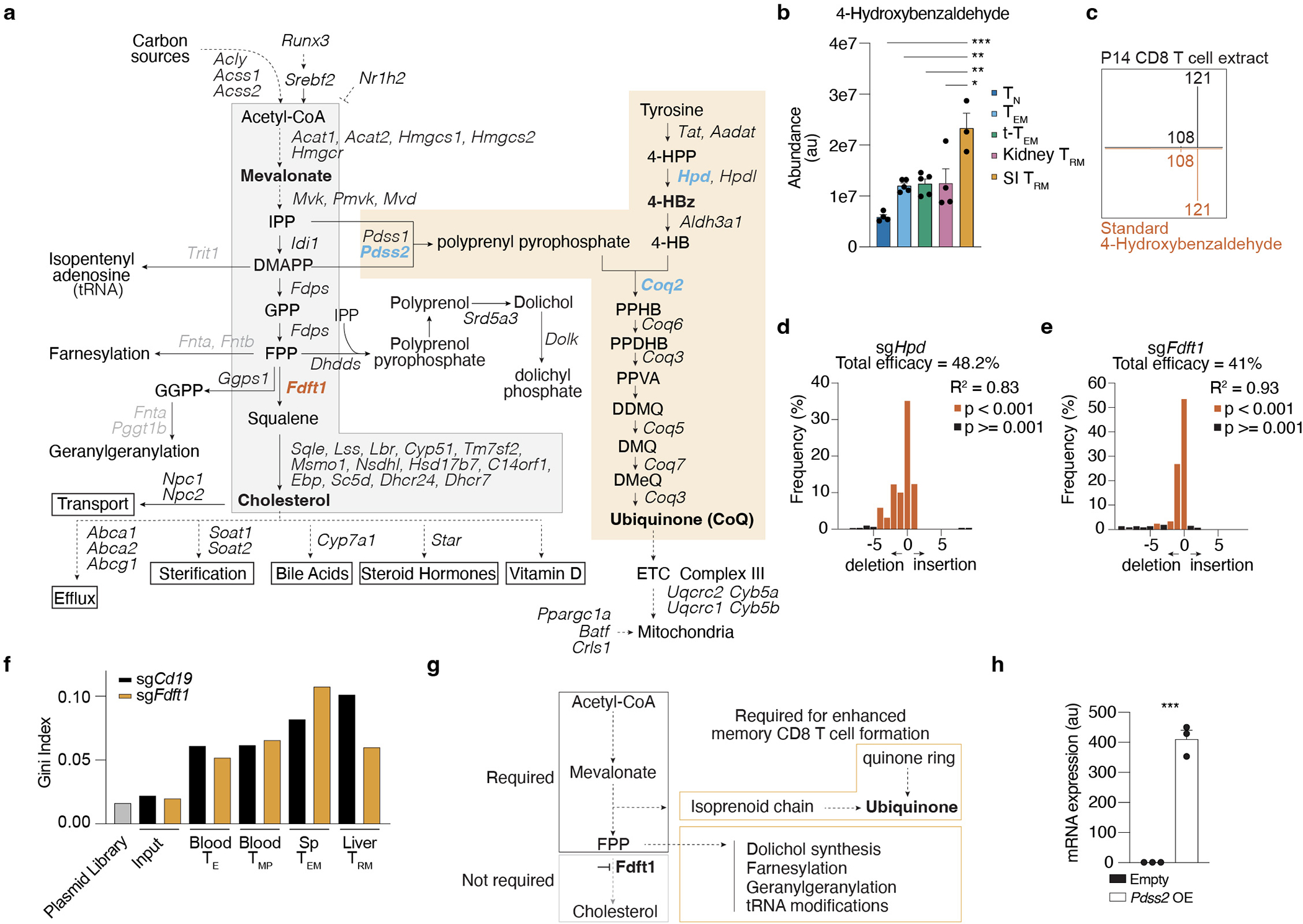
a, The mevalonate/cholesterol synthesis pathway with adjacent and downstream metabolic routes. Enzymes not included in the first CRISPR/Cas9-mediated loss-of-function screen in grey, downstream products and/or fates of cholesterol are boxed, key measured metabolites in bold, non-steroidal metabolites in blue, key enzymes are bold and colored by in vivo effect of deletion in P14 CD8 T cells, gain-of-function in red, loss-of-function in blue, dashed lines indicate indirect or unknown mode of regulation. b, Abundance of 4-hydroxybenzaldehyde profiled by LC-MS/MS of ex vivo populations of P14 cells 13 days after LCMV infection. c, Tandem mass spectra matching against commercial standards of 4-hydroxybenzaldehyde. Tandem mass spectra derived from the most abundant ion for each compound: m/z = 121.0295 for 4-hydroxybenzaldehyde [M-H], black spectra are derived from cell lysate, red mirrored spectra from commercial standard. d, CRISPR/Cas9-mediated indel efficiency of the Hpd sgRNA construct on sorted transduced P14 cells before adoptive transfer. e, CRISPR/Cas9-mediated indel efficiency of the Fdft1 sgRNA construct on sorted transduced P14 CD8 T cells before adoptive transfer. f, Gini index scores of sgRNA representation in each library. g, Schematic illustration of the contribution of mevalonate/cholesterol pathway intermediates to non-steroidal products. h, Quantification of Pdss2 mRNA by qPCR in transduced Empty and Pdss2 OE P14 CD8 T cells. Data are mean +/− s.e.m. (b and h), and representative of at least two independent experiments (d, e, and h), with a total of n=2 (d, e), and n = 3 (h) cell replicates, or one experiment (b), with n=3 (SI), n=4 (TN), and n=5 (rest) (b) samples, where each sample contains cells pooled from 2 to 5 mice. Two-sided unpaired t-Test (b, d, e and h). *P<0.05, **P<0.01, ***P< 0.005.
Extended Data Figure 7. Increased production of non-steroidal products of the mevalonate/cholesterol synthesis pathway is a common requirement between SI TRM and TIL.
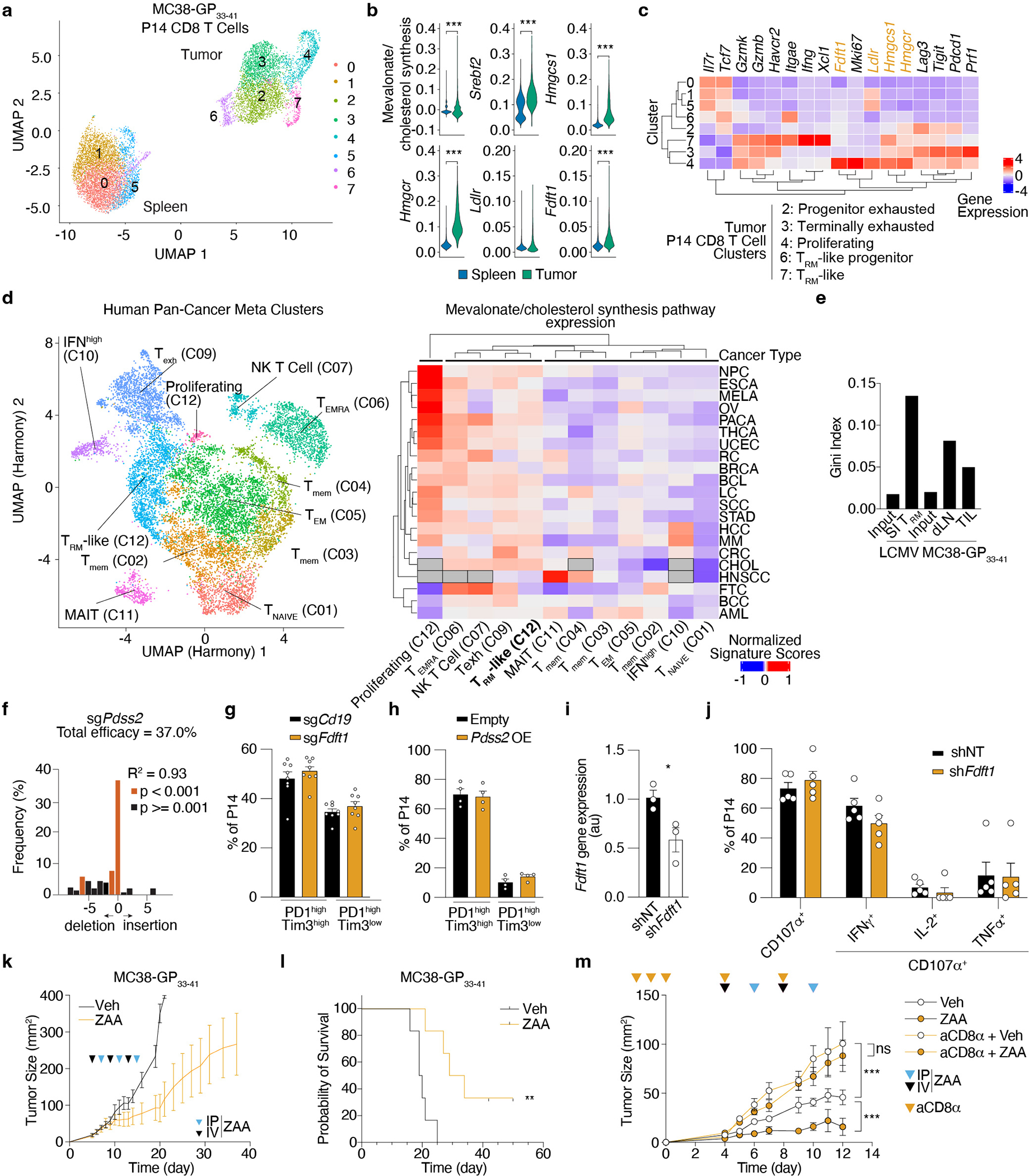
a, Dimensionality reduction plot by UMAP of P14 CD8 T cells profiled from spleens and subcutaneously implanted MC38-GP33–41 tumors by scRNAseq 7 days after ACT. b, Mevalonate/cholesterol synthesis signature values and expression of indicated genes in P14 CD8 T cells from the spleen and MC38-GP33–41 tumors profiled by scRNAseq. MAGIC-imputed gene values are shown. c, Hierarchically clustered heatmap of scaled averaged gene expression values by cluster of P14 cells profiled from spleens and MC38-GP33–41 tumors by scRNAseq 7 days after adoptive transfer. d, Dimensionality reduction plot by UMAP of CD8 T cell metaclusters (left) with “meta.cluster.coarse” labels and hierarchically clustered heatmap of mevalonate/cholesterol synthesis pathway scores grouped by “meta.cluster.coarse” labels and cell type. NPC, nasopharingeal carcinoma, ESCA, esophageal cancer, LC, lung cancer, HCC, hepatocellular carcinoma, CHOL, cholangiocarcinoma, RC, renal carcinoma, CRC, colorectal cancer, AML, acute myeloid leukemia, BCL, B-cell lymphoma, MM, multiple myeloma, HNSCC, head and neck squamous cell carcinoma, THCA, thyroid carcinoma, MELA, melanoma, BCC, basal cell carcinoma, SCC, squamous cell carcinoma, BRCA, breast cancer, STAD, stomach adenocarcinoma, PACA, pancreatic cancer, OV, ovarian cancer, FTC, fallopian tube carcinoma, UCEC, uterine corpus endometrial carcinoma. EMRA, effector memory CD45RA+, mem, memory. e, sgRNA library heterogeneity reported as Gini Index values. f, CRISPR/Cas9-mediated indel efficiency of the Pdss2 sgRNA construct on sorted transduced P14 CD8 T cells. g, Frequency of PD1highTim3high and PD1highTim3low subpopulations for sgCd19 and sgFdft1 Cas9 P14 CD8 T cells in MC38-GP33–41 tumors at day 5–7 after adoptive transfer. h, Frequency of PD1highTim3high and PD1highTim3low subpopulations for Empty and Pdss2 OE P14 CD8 T cells in MC38-GP33–41 tumors at day 5–7 after adoptive transfer. i, Quantification of gene expression by qPCR of control (shNT) and Fdft1 deficient (shFdft1) P14 CD8 T cells before adoptive transfer. j, Restimulation capacity of control (shNT) and Fdft1 deficient (shFdft1) P14 TIL 4 days after in vivo transfer treated with GP33–41 peptide ex vivo. k, MC38-GP33–41 tumor growth curves of mice receiving ZAA or vehicle. l, Survival of mice implanted with MC38-GP33–41 tumors receiving ZAA or vehicle. m, MC38-GP33–41 tumor growth curves of mice receiving ZAA in combination with CD8 T cell depletion. Data are mean +/− s.e.m.. (g, h, i, j, k, and m), raw values (d), geometrical distributions of single-cell gene expression values (b) or averaged gene expression (c and e) and pooled or representative of at least two independent experiments, with a total of n=2 (f) cell replicates, and n=8 (g), n=9 (h), n=3 (i), n=10 (j), n=12/group (k, l), n=5/group (m) mice. Two-sided unpaired t-Test (b, f and i). Log-rank test (l). Two-way ANOVA (m). *P<0.05, **P<0.01, ***P< 0.005.
Extended Data Figure 8. A requirement for the mevalonate/cholesterol synthesis enzymes for TIL persistence and heightened antitumor activity in a melanoma model.
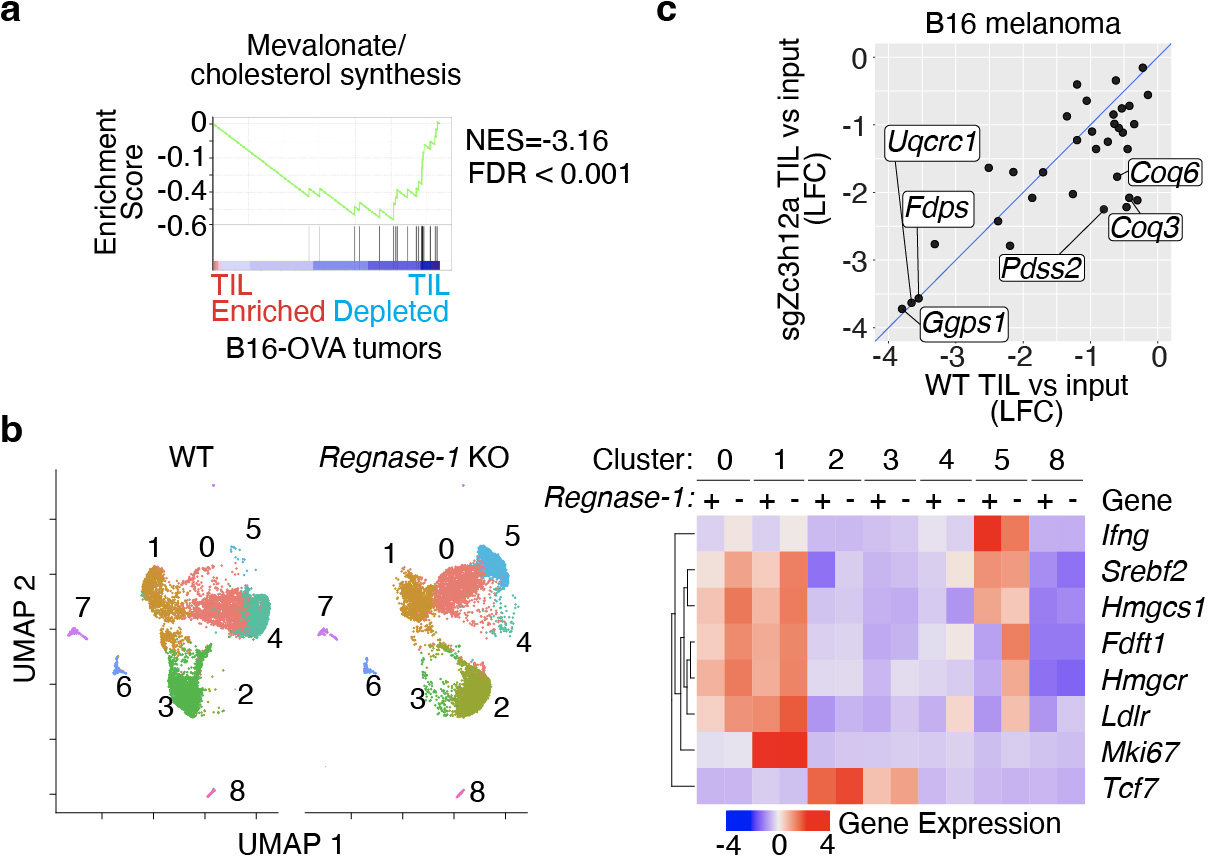
a, GSEA of the mevalonate/cholesterol synthesis pathway in genes pre-ranked by their impact on TIL accumulation in B16-OVA tumors from a published CRISPR/Cas9-mediated loss-of-function screen. b, Dimensionality reduction plot by UMAP of WT and Regnase-1 KO OT-I cells profiled from MC38-GP33–41 tumors by scRNAseq 7 days after adoptive transfer (left) and unsupervised hierarchical clustering heatmap of selected genes related to CD8 T cell cytotoxicity (Ifng) stemness (Tcf7), proliferation (Mki67), and the mevalonate/cholesterol synthesis pathway (Srebf2, Hmgcs1, Fdft1, Hmgcr, and Ldlr) grouped by cell type and cluster. c, Comparison of the effect of targeting the 50 genes of the mevalonate/cholesterol pathway included in our targeted screen in WT TIL or Regnase-1 KO TIL (sgZc3h12a) from a published CRISPR/Cas9-mediated loss-of-function screen in B16 melanoma tumors32. Relevant enzymes are labeled.
Supplementary Material
Acknowledgments
We thank Dr. Matthew Pipkin for generously sharing the shSrebf2 plasmid, Dr. Andrea Reboldi for generously sharing the nSrebp2 OE plasmid, Dr. Shane Crotty and Dr. Katia Faliti for generously sharing the LsgA vector backbone, and Guillermina Garcia and Monica Sevilla for technical histological assistance. We also thank the Center for Advanced Technology at UCSF for sequencing support, Dr. Lara Labarta-Bajo and the Goldrath laboratory members for technical advice, helpful discussion and critical reading of the manuscript, and the Immunological Genome Project (ImmGen) for reagents and sample/data processing. We thank Dr. Antonio M. Pinto and the Mass Spectrometry Core of the Salk Institute with funding from NIH-NCI CCSG: P30 014195 and the Helmsley Center for Genomic Medicine. The MS data described here were gathered on a ThermoFisher Q Exactive Hybrid Quadrupole Orbitrap mass spectrometer funded by NIH grant (1S10OD021815–01).
Funding
National Institutes of Health grant R01 AI067545 (AG)
National Institutes of Health grant P01 AI132122 (AG)
National Institutes of Health grant R01 AI150282 (AG)
National Institutes of Health grant R01 AI072117 (AG)
National Institutes of Health grant R01 CA197363 (MK)
National Institutes of Health grant P01 HL146358 (SJB)
National Institutes of Health grant R01 HL157710 (SJB)
Cancer Research Institute Postdoctoral Fellowship CRI2943 (MR-C)
Cancer Research Institute Postdoctoral Fellowship CRI4145 (GG)
HHMI fellowship GT14887 (KTK)
NCI grant P30 CA030199 (SBP Flow Cytometry Core)
NIH grant P30 DK063720 (UCSF Parnassus Flow CoLab)
National Institutes of Health SIG grant S10 OD026929 (UCSD IGM Core)
Footnotes
Competing interests
A.W.G. serves on the scientific advisory boards of ArsenalBio and Foundery Innovations. H.C. is a consultant for Kumquat Biosciences. M.F.K. is a founder and shareholder of PIONYR Immunotherapeutic and Foundery Innovations. A provisional patent application has been filed by M.R-C., G.G, and A.G. covering the methods of use of FDFT1 and PDSS2 manipulation for “ENHANCING CAR-T CELL PERFORMANCE FOR CANCER IMMUNOTHERAPY” (U.S. Provisional Application Serial No. 63/449,701). The rest of the authors declare that they have no competing interests.
Data and materials availability
Bulk RNA-seq, and single-cell RNA-seq have been deposited to GEO under the reference series GSE207044. In addition, the following published datasets were used: GSE10727817, GSE10610717, GSE18227624, GSE7081347, GSE15707248, GSE12552749, GSE13184748, GSE13701528, GSE7081347, and GSE15707250.
References
- 1.Masopust D & Soerens AG Tissue-Resident T Cells and Other Resident Leukocytes. Annu Rev Immunol (2019) doi: 10.1146/annurev-immunol-042617-053214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park SL, Gebhardt T & Mackay LK Tissue-Resident Memory T Cells in Cancer Immunosurveillance. Trends Immunol 40, 735–747 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Byrne A et al. Tissue-resident memory T cells in breast cancer control and immunotherapy responses. Nature Reviews Clinical Oncology Preprint at 10.1038/s41571-020-0333-y (2020). [DOI] [PubMed] [Google Scholar]
- 4.Konjar Š et al. Mitochondria maintain controlled activation state of epithelial-resident T lymphocytes. Sci Immunol 3, 2543 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinert EM et al. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 161, 737–749 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wijeyesinghe S et al. Expansible residence decentralizes immune homeostasis. Nature 592, 457–462 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soerens AG et al. Functional T cells are capable of supernumerary cell division and longevity. | Nature | 614, (2023). [DOI] [PubMed] [Google Scholar]
- 8.Gebhardt T et al. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nature Immunology 2009 10:5 10, 524–530 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Iijima N & Iwasaki A A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science (1979) 346, 93–98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang X et al. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature 2012 483:7388 483, 227–231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teijaro JR et al. Cutting Edge: Tissue-Retentive Lung Memory CD4 T Cells Mediate Optimal Protection to Respiratory Virus Infection. The Journal of Immunology 187, 5510–5514 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ariotti S et al. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science (1979) 346, 101–105 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Schenkel JM et al. Resident memory CD8 t cells trigger protective innate and adaptive immune responses. Science (1979) 346, 98–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mackay LK et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A 109, 7037–7042 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shin H & Iwasaki A A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 2012 491:7424 491, 463–467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu T et al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol 95, 215–224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milner JJ et al. Runx3 programs CD8 + T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nizard M et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nature Communications 2017 8:1 8, 1–11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malik BT et al. Resident memory T cells in the skin mediate durable immunity to melanoma. Sci Immunol 2, 6346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ganesan AP et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nature Immunology 2017 18:8 18, 940–950 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Djenidi F et al. CD8 + CD103 + Tumor–Infiltrating Lymphocytes Are Tumor-Specific Tissue-Resident Memory T Cells and a Prognostic Factor for Survival in Lung Cancer Patients. The Journal of Immunology 194, 3475–3486 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Reina-Campos M, Scharping NE & Goldrath AW CD8+ T cell metabolism in infection and cancer. Nat Rev Immunol 21, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang H et al. In vivo CRISPR screening reveals nutrient signaling processes underpinning CD8 + T cell fate decisions. Cell 184, 1245–1261.e21 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crowl J et al. Tissue-resident memory CD8+ T cells possess unique transcriptional, epigenetic and functional adaptations to different tissue environments. Nat Immunol (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan Y et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252–256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milner JJ & Goldrath AW Transcriptional programming of tissue-resident memory CD8+ T cells. Curr Opin Immunol 51, 162–169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frizzell H et al. Organ-specific isoform selection of fatty acid-binding proteins in tissue-resident lymphocytes. Sci Immunol 5, 9283 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Wei J et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 576, 471–476 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Locke FL, Zha Y, Zheng Y, Driessens G & Gajewski TF Conditional deletion of PTEN in peripheral T cells augments TCR-mediated activation but does not abrogate CD28 dependency or prevent anergy induction. J Immunol 191, 1677 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pauls SD & Marshall AJ Regulation of immune cell signaling by SHIP1: A phosphatase, scaffold protein, and potential therapeutic target. Eur J Immunol 47, 932–945 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Kidani Y et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol 14, 489–499 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu B et al. Epigenetic landscapes reveal transcription factors that regulate CD8 + T cell differentiation. Nat Immunol 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fernandez-Ruiz D et al. Liver-Resident Memory CD8+ T Cells Form a Front-Line Defense against Malaria Liver-Stage Infection. Immunity 45, 889–902 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Shimano H & Sato R SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol 13, 710–730 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Zeiser R Immune modulatory effects of statins. Immunology 154, 69–75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sullivan ZA et al. γδ T cells regulate the intestinal response to nutrient sensing. Science (1979) 371, (2021). [DOI] [PubMed] [Google Scholar]
- 37.Menk AV et al. 4–1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. Journal of Experimental Medicine 215, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scharping NE et al. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity (2016) doi: 10.1016/j.immuni.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng L et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science (1979) 374, (2021). [DOI] [PubMed] [Google Scholar]
- 40.Lanterna C et al. The administration of drugs inhibiting cholesterol/oxysterol synthesis is safe and increases the efficacy of immunotherapeutic regimens in tumor-bearing mice. Cancer Immunol Immunother 65, 1303–1315 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shires J, Theodoridis E & Hayday AC Biological Insights into TCRγδ+ and TCRαβ+ Intraepithelial Lymphocytes Provided by Serial Analysis of Gene Expression (SAGE). Immunity 15, 419–434 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Fahrer AM et al. Attributes of γδ intraepithelial lymphocytes as suggested by their transcriptional profile. Proc Natl Acad Sci U S A 98, 10261–10266 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoytema van Konijnenburg DP et al. Intestinal Epithelial and Intraepithelial T Cell Crosstalk Mediates a Dynamic Response to Infection. Cell 171, 783–794.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Passi S, Stancato A, Aleo E, Dmitrieva A & Littarru GP Statins lower plasma and lymphocyte ubiquinol/ubiquinone without affecting other antioxidants and PUFA. BioFactors 18, 113–124 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Xu S et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8 + T cells in tumors. Immunity 54, 1561–1577.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma X et al. Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab 30, 143–156.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mackay LK et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Kurd NS et al. Early precursors and molecular determinants of tissue-resident memory CD8 + T lymphocytes revealed by single-cell RNA sequencing. Sci Immunol 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boland BS et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci Immunol 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Milner JJ et al. Delineation of a molecularly distinct terminally differentiated memory CD8 T cell population. Proc Natl Acad Sci U S A 117, 25667–25678 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 51.O’Connor A, Quizon PM, Albright JE, Lin FT & Bennett BJ Responsiveness of cardiometabolic-related microbiota to diet is influenced by host genetics. Mamm Genome 25, 583–599 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anderson KG et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nature Protocols 2013 9:1 9, 209–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCausland MM & Crotty S Quantitative PCR technique for detecting lymphocytic choriomeningitis virus in vivo. J Virol Methods 147, 167–176 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bankhead P et al. QuPath: Open source software for digital pathology image analysis. Scientific Reports 2017 7:1 7, 1–7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 1–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hänzelmann S, Castelo R & Guinney J GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14, 1–15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li W et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brinkman EK, Chen T, Amendola M & Van Steensel B Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168–e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nature Methods 2019 16:12 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Dijk D et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 174, 716–729.e27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gu Z, Eils R & Schlesner M Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Aran D et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nature Immunology 2019 20:2 20, 163–172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Andreatta M & Carmona SJ UCell: Robust and scalable single-cell gene signature scoring. Comput Struct Biotechnol J 19, 3796–3798 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kushnareva Y et al. Functional Analysis of Immune Signature Genes in Th1* Memory Cells Links ISOC1 and Pyrimidine Metabolism to IFN-γ and IL-17 Production. J Immunol 206, 1181–1193 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ye Q et al. HORMA Domain Proteins and a Trip13-like ATPase Regulate Bacterial cGAS-like Enzymes to Mediate Bacteriophage Immunity. Mol Cell 77, 709–722.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.MacLean B et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trindade BC et al. The cholesterol metabolite 25-hydroxycholesterol restrains the transcriptional regulator SREBP2 and limits intestinal IgA plasma cell differentiation. Immunity 54, 2273–2287.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNA-seq, and single-cell RNA-seq have been deposited to GEO under the reference series GSE207044. In addition, the following published datasets were used: GSE10727817, GSE10610717, GSE18227624, GSE7081347, GSE15707248, GSE12552749, GSE13184748, GSE13701528, GSE7081347, and GSE15707250.