

Abstract
Chemical probes are invaluable tools for investigating essential biological processes. Understanding how small-molecule probes engage biomolecular conformations is critical to developing their functional selectivity. High-throughput solution X-ray scattering is well-positioned to profile target-ligand complexes during probe development, bringing conformational insight and selection to traditional ligand binding assays. Access to high-quality synchrotron SAXS datasets and high-throughput data analysis now allows routine academic users to incorporate conformational information into small-molecule development pipelines. Here we describe a general approach for benchmarking and preparing HT-SAXS chemical screens from small fragment libraries. Using the allosteric oxidoreductase Apoptosis-Inducing Factor (AIF) as an exemplary system, we illustrate how HT-SAXS efficiently identifies an allosteric candidate among hits of a microscale thermophoresis ligand screen. We discuss considerations for pursuing HT-SAXS chemical screening with other systems of interest and reflect on advances to extend screening throughput and sensitivity.
1. Introduction
As tools of research and the clinic, small-molecule compounds probe critical biological processes by modulating activities and architectures of their molecular targets (Arrowsmith et al., 2015). Notable examples from the field of DNA damage signaling and repair include Mre11 nuclease (Moiani et al., 2018; Shibata et al., 2014), DNA polymerase theta (Zhou et al., 2021), poly(ADP-ribose) glycohydrolase (PARG) (Houl et al., 2019), and O6-methylguanine DNA methyltransferase (MGMT) (Daniels et al., 2004). Understanding how these chemical probes impact biomolecular conformation, assembly formation, or allostery can lead to improved selectivity and specificity and advance new biology. High-throughput small-angle X-ray scattering (HT-SAXS) (Brosey & Tainer, 2019; Dyer et al., 2014; Hura et al., 2009; Putnam, Hammel, Hura, & Tainer, 2007; Rambo & Tainer, 2013a) is well positioned to report on global target-ligand conformations relevant to assembly and allostery and to complement information from other high-throughput binding and functional assays.
A key strength of SAXS is its translatability. Since SAXS is not limited by sample labeling or molecular weight requirements, optimized binding conditions and purified reagents can be applied to the solution format of SAXS assays. HT-SAXS sample preparation can also leverage established protocols to dispense and assemble target-ligand complexes in multi-well plates suitable for synchrotron data collection (Dyer et al., 2014). Ongoing development of HT-SAXS screening methods continues to expand sample throughput at synchrotron SAXS beamlines (Chen & Hennig, 2018; Chen, Masiewicz, Perez, & Hennig, 2020; Chen, Masiewicz, Rybin, Svergun, & Hennig, 2018; Dyer et al., 2014; Hura et al., 2013), allowing novice and experienced users to acquire high-quality target-ligand datasets. Importantly, SAXS enables access to conformational targets at the frontiers of drug discovery, including dynamically flexible systems (Rambo & Tainer, 2011), intrinsically disordered proteins (IDPs) (Joshi & Vendruscolo, 2015), nucleic acids (Warner, Hajdin, & Weeks, 2018), protein-protein interfaces (PPIs) (Scott, Bayly, Abell, & Skidmore, 2016) and allosteric assemblies (Brosey & Tainer, 2019).
Here, we present a methodological approach for screening target-ligand complexes with HT-SAXS. Our method is directed toward discovery and optimization of chemical probes but is relevant for investigating the conformational impact of any small-molecule panel. We demonstrate our approach with Apoptosis-Inducing Factor (AIF), a mitochondrial redox switch that allosterically dimerizes upon binding and reduction by NADH (Brosey et al., 2016; Sevrioukova, 2009). Dimeric AIF supports the mitochondrial disulfide import pathway and regulates biogenesis of respiratory complexes performing oxidative phosphorylation (OXPHOS) (Hangen et al., 2015), raising interest in its therapeutic potential for cancer (Ashton, McKenna, Kunz-Schughart, & Higgins, 2018; Lemberg, Gori, Tsukamoto, Rais, & Slusher, 2022). We describe parallel screening of AIF with microscale thermophoresis (MST) and HT-SAXS to identify chemical ligands and allosteric effectors from a specialized 96-compound library. Our HT-SAXS assay design leverages the accessible mail-in service offered by the SIBYLS SAXS (12.3.1) beamline at the Advanced Light Source (ALS) synchrotron (https://sibyls.als.lbl.gov/) (Classen et al., 2013) and utilizes standard analytic tools to assess and summarize target-ligand datasets. We also outline important considerations and concerns for applying and benchmarking HT-SAXS screening with other biomolecular systems and discuss priority areas for future development.
2. Considerations for SAXS target and library selection
An informative HT-SAXS assay relies on strategic selection of conformational targets and chemical libraries that are well suited for solution X-ray scattering (Fig. 1A). SAXS optimally detects global changes in biomolecular conformation; thus, monomer-dimer equilibria, protein assembly/disassembly, intra- or inter-domain rearrangements, and significant changes to molecular flexibility are well captured by X-ray scattering analysis (Brosey & Tainer, 2019; Nishimura et al., 2009; Rambo & Tainer, 2011). Improvements to the sensitivity and interpretation of high-resolution SAXS data hold the potential to monitor subtler changes in biomolecular architecture as the field continues to progress (Rambo & Tainer, 2013b).
Fig. 1.
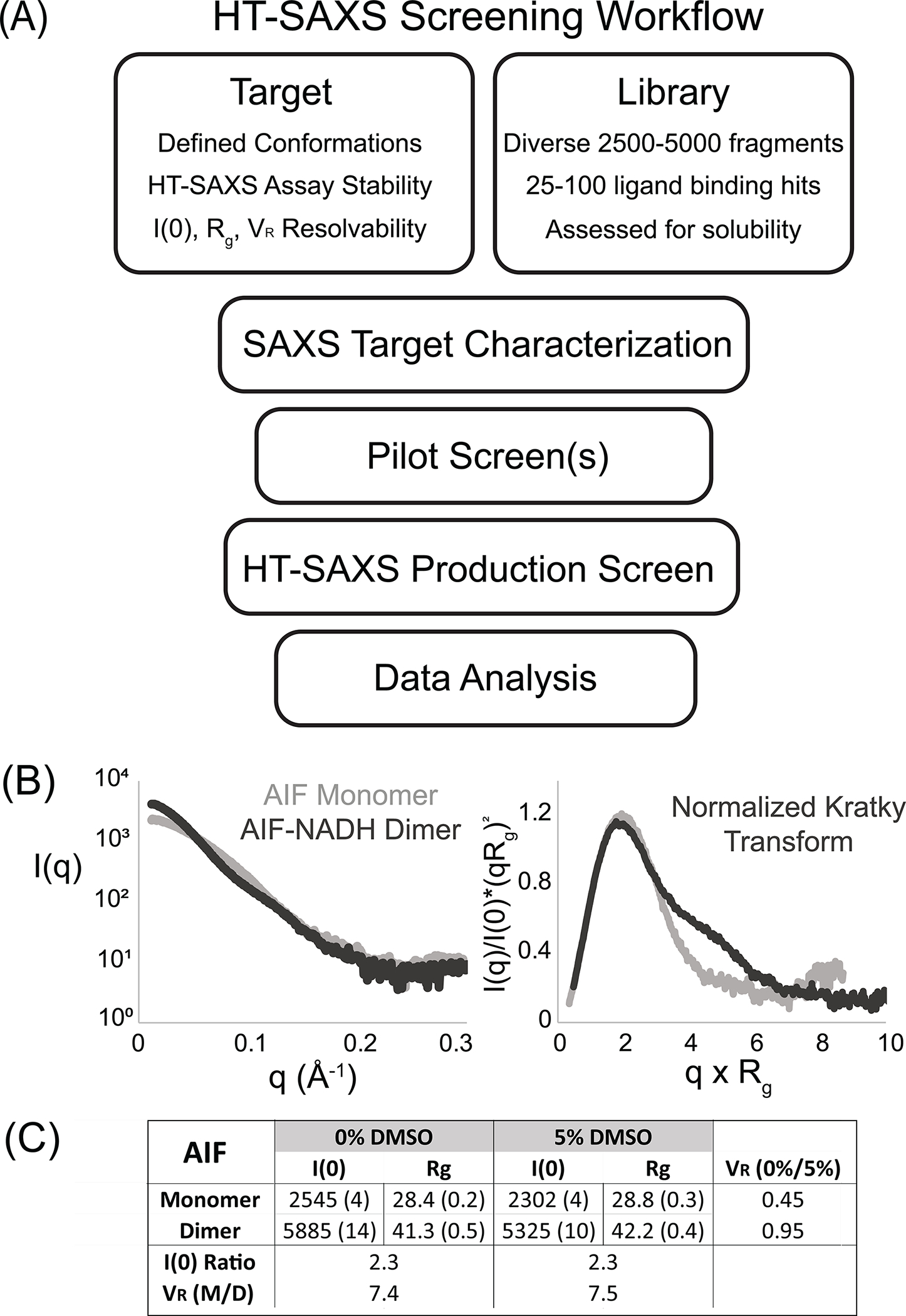
HT-SAXS Screening Work Flow. (A) Workflow of a prototypical HT-SAXS chemical screen with criteria for targets and libraries. (B) SAXS analysis of monomeric AIF and the NADH-stimulated dimer (AIF + 10-fold molar excess NADH) reveals well-resolved scattering curves, I(q). Kratky transformation of I(q) highlights distinct, folded monomer and dimer architectures. (C) Titration of 5% DMSO does not significantly alter AIF architecture or interfere with NADH-induced dimerization, as indicated by comparable I(0) and Rg values and VR < 1. The I(0) ratio and volatility-of-ratio (0.015–0.15 Å−1) between AIF monomer and dimer are unchanged by DMSO. Values in parentheses are errors of fitting.
To assess whether SAXS analysis can resolve known conformational endpoints of a potential target, scattering experiments on high-quality protein or nucleic acid reagents are necessary (Dyer et al., 2014; Graewert & Jeffries, 2017; Rambo, 2017). Such experiments will also establish whether a target is sufficiently stable during sample shipment and data acquisition to produce usable SAXS data. Key parameters to test at this stage are sample concentration (1–10 mg/mL), sample buffer (pH, salt, additives, see also https://bl1231.als.lbl.gov/saxs_protocols/saxs_sample_prep.php), the ability to match buffers containing native ligands, and shipping format (chilled or frozen). For AIF, HT-SAXS data were previously acquired on concentration series of the ligand-free monomeric (60 kDa) and NADH-bound dimeric (120 kDa) proteins (Brosey et al., 2016). These experiments established a target concentration (4 mg/mL), standard buffer (25 mM HEPES, pH 7.5, 150 mM NaCl), and shipping condition (dry ice) for SAXS analysis.
Visual comparison of AIF monomer and dimer X-ray scattering curves and their Kratky transforms highlights distinct scattering profiles for each allosteric state (Fig. 1B), while linear Guinier transforms support aggregation-free samples (data not shown). Our HT-SAXS analysis relies upon the extrapolated zero-angle scattering intensity, I(0), radius-of-gyration, Rg, and the volatility-of-ratio (VR) metric (Hura et al., 2013) (https://sibyls.als.lbl.gov/saxs-similarity/) to quantify and rank ligand-induced differences relative to a baseline state. VR captures differences between two SAXS curves by quantifying fluctuations in their normalized I(q) ratio or R(q). Perfect sample identity results in R(q) of 1 for the entire q-range and VR of 0; differences between samples cause R(q) deviations from unity and increase VR. To assess the resolving power of VR when applied to AIF’s allosteric states, one can calculate a Z-factor (Z) from replicate monomer and dimer VR values, referencing the AIF monomer as a baseline state.
| (1) |
where is the average VR of 2–3 replicate samples and is the associated standard deviation. We chose to focus VR calculation over the low- and mid-q region of the R(q) ratio (0.015–0.15 Å−1), the region of maximum monomer-dimer signal divergence and highest signal-to-noise within the I(q) curve. This yields AIF monomer and dimer VR values of 2.3 ± 0.2 and 8.0 ± 0.1, respectively. The resulting Z-factor is 0.83, where Z-factors within 0.5–1.0 are considered to provide good discrimination between baseline and maximum signal. Similar calculations for I(0) (22.6 ± 1.1Å vs 51.8 ± 1.7Å) and Rg (28.9 ± 0.9Å vs 43.2 ± 0.8 Å) yield Z-factors of 0.71 and 0.64, respectively.
For certain biomolecular systems, it may be desirable to estimate SAXS VR resolvability before investing in production of SAXS-quality protein or nucleic acid samples. If experimental structures of target conformational states are available, theoretical X-ray scattering curves can be calculated from three-dimensional coordinates using FoXS (Schneidman-Duhovny, Hammel, & Sali, 2010; Schneidman-Duhovny, Hammel, Tainer, & Sali, 2013, 2016) or CRYSOL (Franke et al., 2017; Svergun, Barberato, & Koch, 1995) webservers. These theoretical curves can be supplemented with a 1–5% Gaussian error to simulate replicate measurements, then used in Z-factor calculations to quantify I(0), Rg, and VR resolvability by SAXS. This approach, however, requires that known experimental structures accurately represent biomolecular conformations in solution.
Choice of chemical library also determines the effectiveness of HT-SAXS screening assays. Our screening efforts have relied on small, custom fragment libraries (Moiani et al., 2021) designed for chemical diversity, absence of problematic pan-assay interference (PAINS) (Baell & Holloway, 2010; Baell & Nissink, 2018), predicted favorable physicochemical properties, and tractability of chemical elaboration. These libraries are built from commercial fragments purchased from Life Chemicals and contain 2500–5000 fragments prepared as 10 mg/mL DMSO stocks (~20–70 mM). A ligand binding screen returning a 1–2% hit rate from this library yields 25–100 representative compounds. A single 96-well HT-SAXS screening plate can accommodate 28 compounds with controls (vide infra); thus, conformational characterization of projected library hits can be completed with 1–3 HT-SAXS plates, typically within 2 synchrotron SAXS shifts. Fragment pooling has the potential to increase throughput for other library formats while keeping HT-SAXS screening plates to a manageable number.
It is important to assess aqueous solubility of any candidate compounds before HT-SAXS screening, since highly insoluble material may clog SAXS sample loading and fluidics systems and/or produce unusable SAXS data. Complementary ligand binding assays should provide opportunity to assess and potentially optimize compound solubility (e.g., increasing DMSO). Preparing small test samples of compounds mixed with protein or nucleic acid at chosen SAXS concentrations can also flag potential problems with precipitation at room temperature and upon freeze-thaw treatment.
3. HT-SAXS sample preparation
There are many excellent resources describing preparation of high-quality protein reagents for X-ray scattering analysis (Dyer et al., 2014; Graewert & Jeffries, 2017; Jeffries et al., 2016; Rambo, 2017; Rambo & Tainer, 2010). Here, we focus on specific preparation of AIF samples with matched buffer for HT-SAXS using size-exclusion chromatography. Expression and purification of AIF are previously described (Brosey et al., 2016). Freshly purified or thawed aliquots of AIF protein are concentrated to ~300–700 μL at a final concentration of 30–40 mg/mL. Following a 5-min high-speed spin to pellet stray aggregation, the concentrate is loaded onto a Superdex 200 10/300 Increase column equilibrated in 25 mM HEPES, pH 7.5, 150 mM NaCl and eluted at 0.4 mL/min with 0.5 mL fractions. SEC fractions are scanned by UV–Vis spectroscopy to measure concentration and confirm the presence of AIF’s FAD cofactor (identified by A270/A450 ~ 7.5 and A260/A280 ~ 0.8). Fractions are also visualized by SDS-PAGE to confirm sample purity. Peak fractions free of contaminants are selected and pooled to 4.1 mg/mL (typically 1–2 mL) to provide the AIF protein stock to assemble the screen. Buffer fractions preceding the void volume (usually 2–4 mL into the elution) are also pooled for protein dilution and to prepare matched buffer samples. Using SEC-matched buffer for preparing AIF SAXS samples is crucial in order to ensure an accurate buffer subtraction between protein and buffer scattering curves. Prepped AIF protein and buffers can be kept on ice prior to assembling the plate or stored at 4 °C overnight if prepped the day before.
4. Benchmarking a pilot HT-SAXS screen
Performing at least two small trial screens is highly recommended prior to commencing a production HT-SAXS screen containing library compounds. A pilot screen serves several purposes. The first is to determine if titrated DMSO impacts target conformation or interferes with the binding of native ligands, providing a control for the library solvent. Our fragment libraries are usually screened with final concentrations of 3–10% DMSO. A second objective is testing the accuracy of buffer subtraction for a given sample assembly protocol. Because library compounds are often limited in material, the protocol reported here describes mixing DMSO controls and compounds into proteins and buffers manually. Since this may increase the margin of error for a poor background subtraction, it is critical to ensure the sample-buffer match is sufficient to produce usable data. Finally, pilot screens are useful for identifying and remedying any weaknesses in the logistics of preparing, shipping, and collecting SAXS screening plates at synchrotron beamlines.
A sample layout for a pilot SAXS screen in a 96-well PCR plate is presented in Fig. 2A, based upon SIBYLS recommendations for interleaving matched buffers with sample concentration series. DMSO is tested at 1–10% for three sample concentrations, where matched buffer wells contain identical DMSO concentrations. This layout can be repeated to include positive controls for known ligands or other conformational endpoints. All samples are prepared at 30 μL per well. Thus, a 10% DMSO AIF titration would involve combining 3 μL DMSO with 27 μL SEC-purified AIF (4.4 mg/mL stock) or 27 μL SEC-matched buffer. When manually adding titrant directly to samples, we recommend preparing the titrant concentration so that at least 1-μL volume is being added to the sample (i.e., adding 1 μL 30% DMSO to achieve 1% DMSO). If buffer subtraction becomes an issue, adjusting titrant concentration to allow larger, more accurate titrant volumes may help.
Fig. 2.
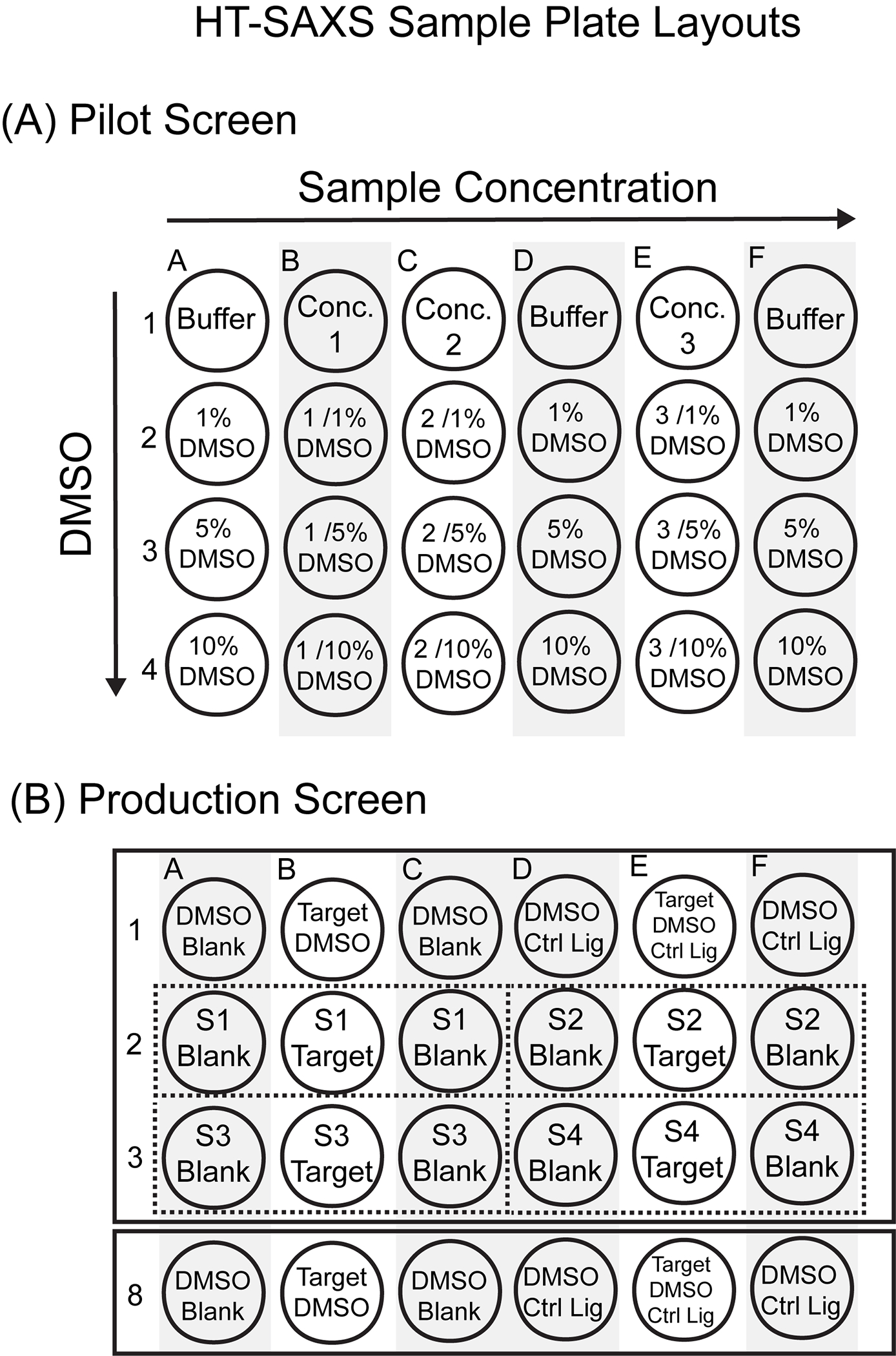
HT-SAXS Sample Plate Layouts. (A) Sample 96-well plate layout for a HT-SAXS pilot screen. (B) Sample 96-well plate layout for a HT-SAXS production screen with control samples (rows 1 and 8) and 4 screening compounds (S1-S4, rows 2 and 3).
For AIF, manual titration of DMSO into ligand-free monomer or NADH-reduced dimer does not cause major disruptions in the buffer subtraction. Importantly, DMSO neither alters protein conformation nor interferes with NADH-induced dimerization (Fig. 1C). This is initially assessed by overlay and visual inspection of scattering curves, then quantitatively established by comparing extrapolated zero-angle scattering intensity I(0), radius-of-gyration (Rg), and VR with reference to DMSO-free controls.
5. Ligand screen design and assembly
Once pilot HT-SAXS screens have identified reliable DMSO concentrations and benchmarked the sample shipping/data collection pipeline, preparing the production HT-SAXS screen should be a streamlined process. Before setting up a production screen, we recommend reaching out to your local SAXS beamline scientist to confirm shipment plans and to ensure available shift times will cover the planned data collection.
An exemplary layout of an HT-SAXS production screen is shown in Fig. 2B. Internal controls (DMSO, native ligand with DMSO) are placed at the beginning and end of each plate. Target-ligand samples are prepared with flanking ligand-matched buffers collected before and after the target-ligand complex. If a plate contains 4 control sections (12 wells), the remaining wells can accommodate 28 target-ligand complexes with matched-buffers. Overall, this plate design will require 3–9 μL compound for each target-ligand complex (one target, two matched buffers). If library materials are not limited, this design can be expanded to include duplicate target-ligand wells (two targets flanked by two matched buffers) to provide redundancy should bubbles or poor sample transfer result in unusable scattering profiles.
In this example, we screened AIF with a focused 96-fragment library, created by similarity search of high-ranking scaffolds from a previous discovery screen (Brosey et al., 2020). Ligand binding was initially assessed by microscale thermophoresis (MST) with Atto488-labeled AIF (Fig. 3A). Briefly, 100nM Atto488-AIF was incubated with 0.3 mg/mL compound or DMSO in MST buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 0.01% Tween-20) for 15 min at room temperature. Thermophoresis curves were measured in triplicate at 25°C using a Monolith NT.115 system (NanoTemper) with 30% LED power and 40% infrared excitation for 20 s (3-s equilibration and 1-s recovery periods). Time-averaged amplitudes were calculated over a 1-s window using NanoTemper’s MO.Affnity Analysis software and exported to Excel for analysis. Screening hits were defined as fragments increasing Atto488-AIF MST amplitude three standard deviations above averaged DMSO controls (Fig. 3A). Twenty-five fragments qualified for this cutoff and were verified by MST titration experiments, which report micromolar affinity among the top candidates (Fig. 3B). Inspection of these top fragments reveals an enrichment in benzothiazolamine scaffolds, lending confidence to the MST results (Fig. 3C).
Fig. 3.
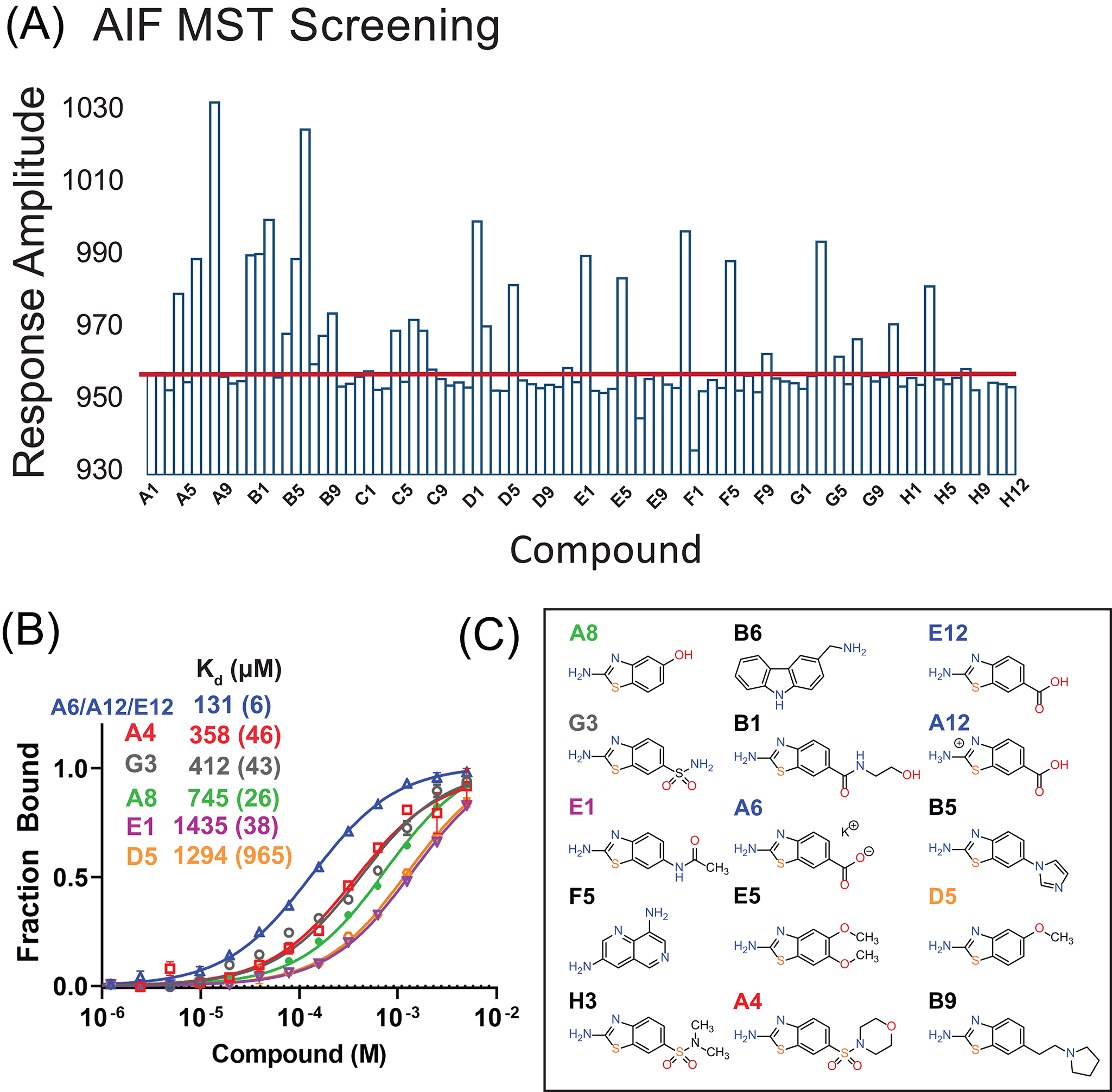
AIF MST Screen. (A) AIF MST screening results from the focused 96-fragment library. Red line indicates three standard deviations from the average DMSO response amplitude. (B) MST titration curves and affinities for the top fragment ligands. Response amplitudes were exported and fit for one-site binding in GraphPad Prism 9. Values in parentheses are fitting errors. (C) The top MST candidates reveal an enrichment of benzothiazolamine scaffolds.
Two HT-SAXS screens were prepared in parallel to the MST experiments. The first focused upon assessing AIF conformational impact among the highest affinity fragment binders (14 samples). The second, broader screen examined the entire 96-fragment library in order to probe for cryptic ligands undetectable by MST. SAXS samples were prepared by combining library fragments or DMSO with SEC-purified AIF stock or SEC-matched buffer. Final concentrations were 4 mg/mL AIF, 1–5 mM library fragments, and 3–6.7% DMSO. For the broad 96-fragment screen, AIF-ligand samples were prepared in duplicate with flanking ligand-matched buffers. In order to conserve material and extend screening boundaries, the 96-fragment screen was prepared with a 15-μL sample volume; however, the prevalence of interfering bubbles during data collection supports the recommended 30-μL sample volume used in the focused 14-fragment screen. With DMSO controls, the focused and broad HT-SAXS screens required 1 and 4.5 96-well sample plates, respectively. Once assembled, SAXS plates were sealed, flash frozen by setting the plate in a shallow layer of liquid nitrogen and shipped on dry ice to the ALS SIBYLS beamline, where they were stored at −80 °C prior to data collection. For the user-provided data collection instructions (https://bl1231.als.lbl.gov/htsaxs/instructions/htsaxs), we specified a needle wash after collection of the target-ligand complex and also after the final ligand-matched buffer for each compound.
Ideally, problematic, precipitating compounds would be flagged and mitigated prior to preparing the production screen. Should ligand-induced precipitation occur once the plate is at the beamline, a high-speed centrifuge spin can pellet insoluble material and allow loading and data collection of the soluble remainder. This also is a point to discuss with a local beamline contact when planning the screening shipment.
6. Analysis of HT-SAXS screening datasets
The SIBYLS mail-in HT-SAXS program provides a package of processed, buffer-subtracted I(q) scattering curves with annotations on aggregated samples or problematic background subtractions. Data are collected as 200–300-millisecond frames over a 10-s exposure; thus each sample contains 30–50 time-resolved I(q) curves. We recommend an initial visual inspection of the I(q) data to sort out any samples with unusable curves—those with poor buffer subtraction or missing data (Fig. 4A). This can be achieved efficiently for the 25–100 compound screens described here using the drag-and-drop feature of ScÅtter (https://bl1231.als.lbl.gov/scatter/) to load and graph each sample time series.
Fig. 4.
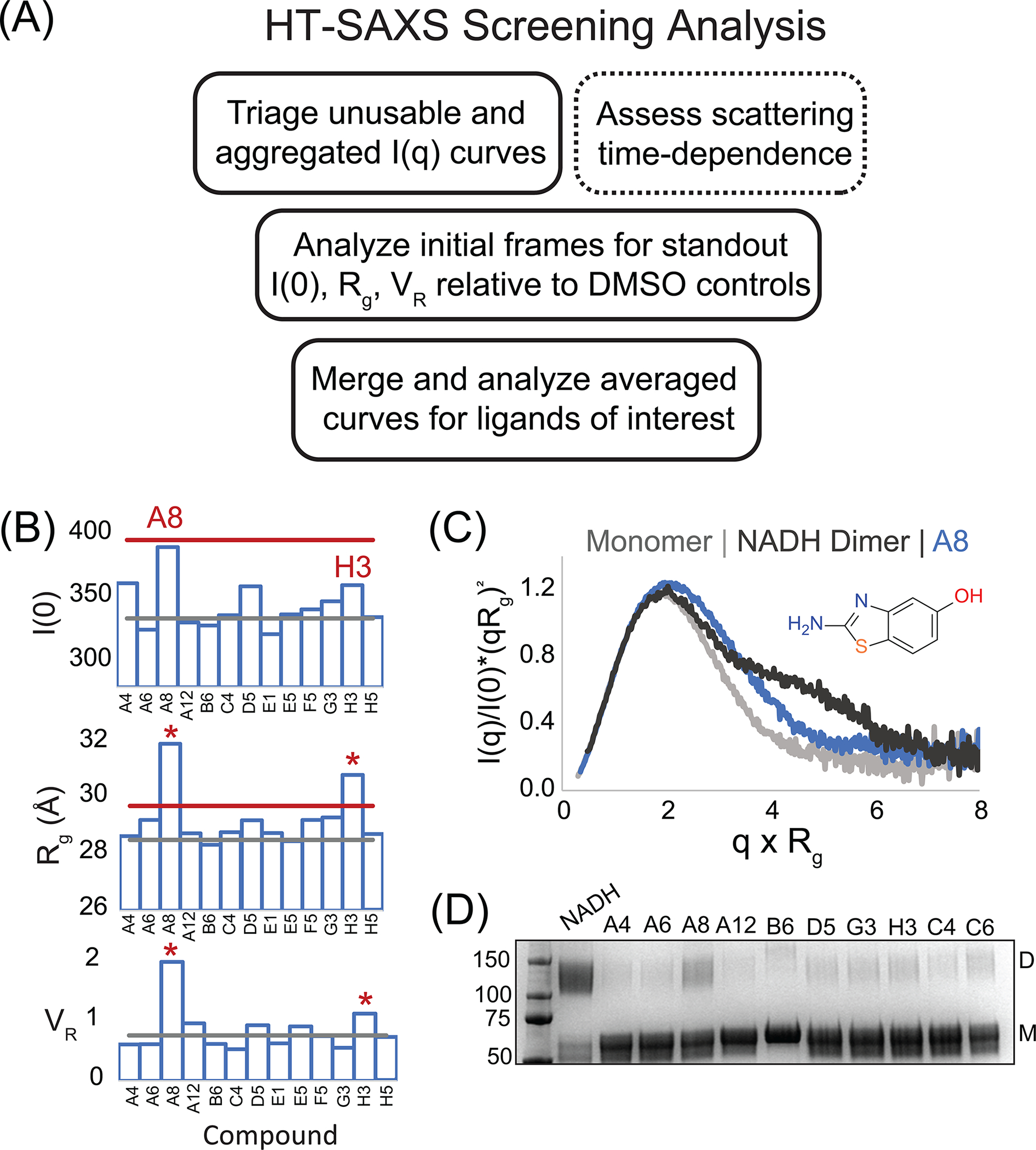
AIF HT-SAXS Screening Analysis of 14 Focused Hits. (A) Workflow for triaging and analyzing large HT-SAXS screening datasets. (B) I(0), Rg, and VR results from the focused 14-fragment AIF SAXS screen. Gray and red lines indicate average and three-standard deviations from AIF-DMSO controls. (C) Overlay of normalized Kratky transforms from A8 (blue) and AIF monomer (gray) and dimer (black) benchmarks. (D) SDS-PAGE of BS3 amine-crosslinking reactions that capture AIF dimers in the presence of NADH among top candidates from MST screening. D, dimer; M, monomer.
Once unusable I(q) curves have been excluded from the screening dataset, a second triage step is applied to remove aggregated samples exhibiting dramatic upward deflection in the low-angle I(q) signal or non-linear Guinier regions. Here, initial I(q) curves (t = 0.2 or 0.3 s) from each sample are copied and organized into a directory, then uploaded into ScÅtter for visualization and comparison to DMSO or other controls. ScÅtter automatically calculates I(0) and Rg values, allowing for rapid quantitative comparison to a DMSO baseline. Guinier plots with residuals are also automatically calculated and can be assessed for linearity.
Once problematic and aggregated datasets have been excluded, one must decide whether (1) to continue analysis with all initial frames (higher throughput), (2) to average all or part of each time-series to produce a single I(q) curve (higher signal-to-noise), or (3) to analyze the SAXS data as time-resolved I(q) series (time-dependent or kinetic effects). Because AIF is a redox-regulated flavoprotein, X-ray-induced reduction unlocks its response to certain classes of allosteric fragment effectors (Brosey et al., 2020). Thus, some ligand impacts on AIF conformation are most efficiently revealed by time-resolved HT-SAXS analysis. The expectation is that X-ray-induced redox effects will primarily be relevant for redox-sensitive protein targets, e.g., those containing metal centers, functional disulfides, or redox-related cofactors such as FAD or FMN.
If a target system has a likelihood of containing time-dependent information, one can quantitatively assess this by comparing time-evolved I(0) and Rg values relative to time-invariant controls using ScÅtter. Calculations of VR relative to the initial frame or a control series may also be useful for detecting time-evolution within low- or mid-q intensities (0.015–0.15 Å). The SIBYLS SAXS similarity web application offers a drag-and-drop interface for rapidly calculating and ranking VR values in structural similarity maps (Hura et al., 2013) (https://sibyls.als.lbl.gov/saxs-similarity/). As a caveat, radiation damage can also cause time-dependent aggregation and should be ruled out before kinetic analysis. This can usually be diagnosed by appearance of upward deflection in low-angle I(q) intensities, non-linear Guinier plots, and a continuous dose-dependent increase in I(0) and Rg values in later I(q) frames.
Time-resolved assessment of the AIF HT-SAXS datasets reveals radiation-induced aggregation in several samples. Thus, we chose to focus upon high-throughput analyses of the initial, aggregation-free I(q) snapshots. I(0) and Rg values were calculated with ScÅtter, then exported to Excel and GraphPad Prism for visualization and statistical analysis. VR values were calculated using the SIBYLS SAXS similarity web application over q-ranges matched to exposure time (0.02–0.15 Å−1 for 300-ms frames, 14-compound screen; 0.02–0.13 Å−1 for 200-ms frames, 96-compound screen) to minimize high-q noise. VR values were copied into Excel using the “Metric Matrix” option under the Operations menu.
Initial analysis focused upon assessing the consistency of replicate DMSO controls from each screen. For the focused 14-compound screen, the coefficient-of-variation among replicate DMSO samples was 6% and 1% for I(0) and Rg, respectively, while VR was 0.76. For the broader 96-compound HT-SAXS screen, plotting AIF-DMSO I(0) and Rg indicates plate-to-plate consistency with the exception of I(0) outliers in Plates 2 and 3 (Fig. 5A). These screening plates required a longer collection period; we surmise that the outlier samples, which were located at the end of each plate, experienced evaporative concentration and corresponding elevation of concentration-dependent I(0). Coefficients-of-variation for I(0) and Rg excluding I(0) outliers are 1–5%, reflecting robust internal consistency of the screening assay. VR, as referenced to the first AIF-DMSO sample of Plate 1, aligns with the pilot DMSO measurements (VR 1.9 (0.6)).
Fig. 5.
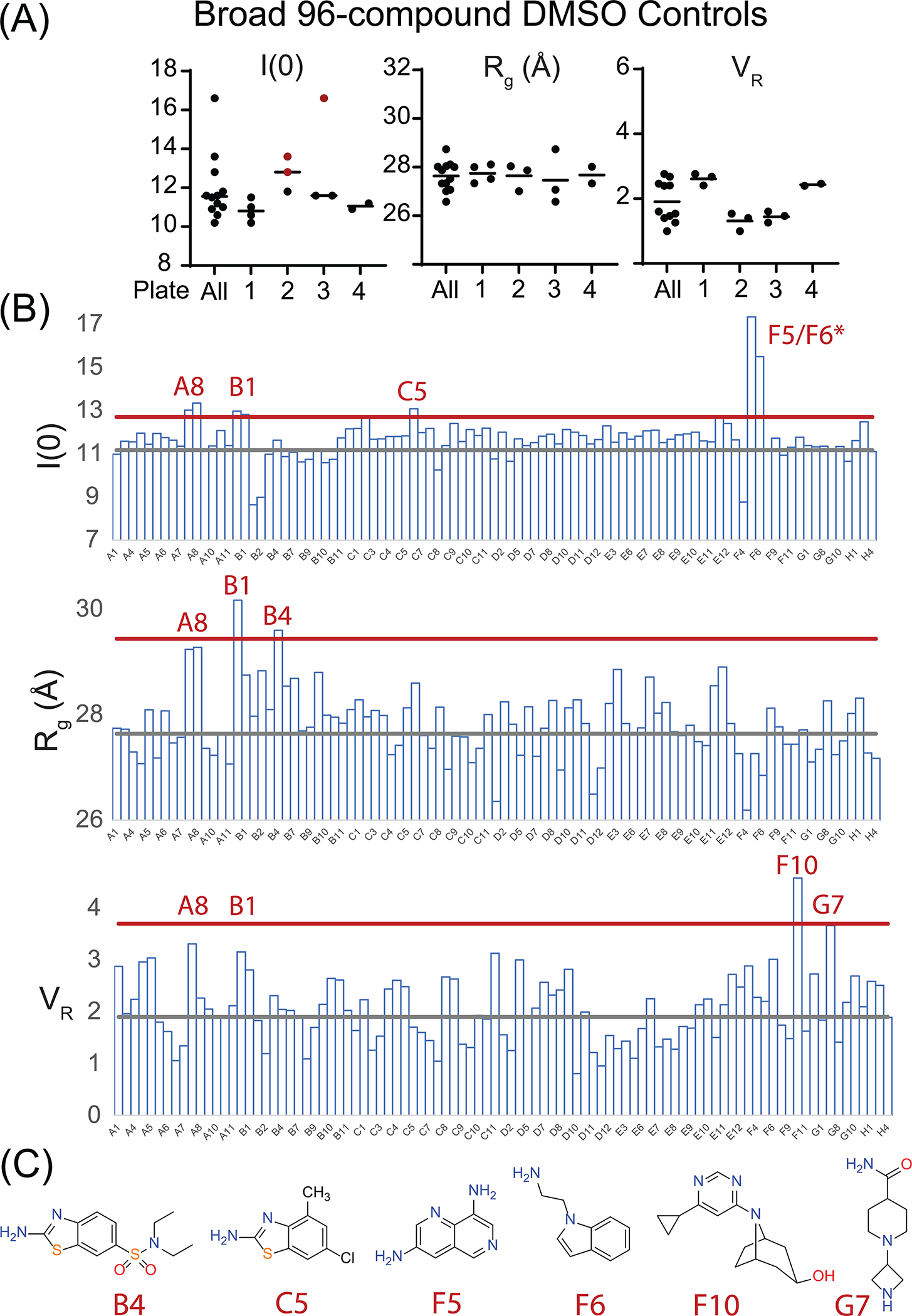
AIF HT-SAXS Screening Analysis of 96-Fragment Library. (A) I(0), Rg, and VR values from AIF-DMSO controls demonstrating consistency across screening plates. (B) I(0), Rg, and VR analysis from curated 64-compound dataset. Gray and red lines indicate average and three-standard deviations from AIF-DMSO controls. (C) Novel library fragments exhibiting significant change in at least one SAXS metric.
Inspection of the 14-compound focused screening results highlights AIF-A8 as consistently increased across all three SAXS metrics (Fig. 4B). AIF-H3 exhibits an elevated Rg but does not differ significantly among the other SAXS metrics. In contrast, fragments with higher micromolar affinities for AIF (A6/A12/E12, A4, G3) do not cause significant deviation from AIF-DMSO controls. Normalized Kratky plots, which detect AIF dimerization as a shoulder of the primary maximum, suggest that A8 is stimulating weak dimerization (Fig. 4C). This is confirmed by amine-crosslinking, which captures a minor dimeric species when AIF is saturated with A8 (Fig. 4D). Fragments with highest affinities do not induce detectable dimerization. Thus, the SAXS counter screen usefully selects for fragments with allosteric properties among AIF ligands identified by MST.
After elimination of unusable and aggregated curves, the curated HT-SAXS dataset from the broad 96-compound screen resulted in 64 unique AIF-ligand scattering curves (Fig. 5B). Sample bubbles and AIF aggregation accounted for the majority of eliminated curves. Among I(0), Rg, and VR values, fragments A8 and B1 are consistently elevated. Additional fragments are highlighted among individual metrics (B4, C5, D5/D6, F10, G7) and include additional benzothiazolamine scaffolds (Fig. 5C); however, these do not appear as consistently as the MST-identified fragments. B1 was originally excluded from focused screening, due to signs of aggregation in the original MST data. The aggregation-free SAXS data suggests that this candidate is well behaved and may provide a second allosteric lead for optimization with A8.
7. Summary and future perspectives
Screening approaches using HT-SAXS continue to advance and promise to provide unique, complementary insights to traditional ligand binding assays. In this application, we have paired HT-SAXS chemical screening with MST to identify potential allosteric effectors among ligands of Apoptosis-Inducing Factor. This protocol and data collection strategy are readily accessible to SAXS academic users. Importantly, our approach can be customized toward any biological system experiencing well-resolved conformational changes. Efforts continue to reduce sample volume and concentration through optimization of HT-SAXS sample loading systems and hold the potential to reduce sample consumption by half. Future improvements in detector hardware and analysis algorithms are expected to extend the throughput, sensitivity, and resolution of HT-SAXS chemical screening as a robust and practical approach.
Key Benchmarks for SAXS Chemical Screens
Target
-
Are structural states of the target resolvable with SAXS parameters—VR, Rg, I(0)? This can be assessed with...
Simulated SAXS data from available atomic structures
Experimental SAXS data
Can the target be produced at sufficient quantities and purity for SAXS experiments?
Is the target resilient during shipment to SAXS synchrotron beamlines?
Does the target produce high-quality, aggregation-free HT-SAXS data?
What are optimal SAXS sample concentrations and buffer conditions for screening?
Library
Is the library soluble in aqueous screening buffer (as assessed by complementary ligand binding experiments)?
Does the target remain soluble when mixed with candidate library hits at SAXS concentrations?
Do target-ligand samples remain soluble after a freeze-thaw cycle (if required for shipment)?
Pilot Screen
Does DMSO affect sample conformation or stability?
Is DMSO buffer matching optimal?
Are there weaknesses in the pipeline of screen preparation, shipping, and collection?
Production Screen
Are shipment plans and shift scheduling confirmed with SAXS beamline staff?
Are sufficient shifts available to collect the production screen?
Simple Scattering deposition
Curated, aggregation-free I(q) scattering curves of AIF-ligand complexes from the focused screen of Fig. 4 are deposited under XSLGMJSW in the Simple Scattering database (https://simplescattering.com/).
Acknowledgments
This work was supported by the National Institutes of Health (NIH) (P01 CA92584, R35 CA220430), the Cancer Prevention and Research Institute of Texas (RP180813), and a Robert A. Welch Chemistry Chair. SAXS experiments were conducted at the SIBYLS beamline (12.3.1) at the Advanced Light Source (ALS), a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the Department of Energy, Office of Basic Energy Sciences, through the Integrated Diffraction Analysis Technologies (IDAT) program, supported by DOE Office of Biological and Environmental Research. Additional support comes from the National Institute of Health project ALS-ENABLE (P30 GM124169) and a High-End Instrumentation Grant S10OD018483.
Abbreviations
- AIF
Apoptosis-Inducing Factor
- ALS
Advanced Light Source
- FAD
flavin adenine dinucleotide
- FMN
flavin adenine mononucleotide
- HT
high-throughput
- IDP
intrinsically disordered protein
- MST
microscale thermophoresis
- NADH
nicotinamide adenine dinucleotide
- PPI
protein-protein interface
- Rg
radius-of-gyration
- RNA
ribonucleic acid
- SAXS
small-angle X-ray scattering
- SEC
size-exclusion chromatography
- SSM
structural similarity map
- VR
volatility-of-ratio
References
- Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, et al. (2015). The promise and peril of chemical probes. Nature Chemical Biology, 11(8), 536–541. 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton TM, McKenna WG, Kunz-Schughart LA, & Higgins GS (2018). Oxidative phosphorylation as an emerging target in cancer therapy. Clinical Cancer Research, 24(11), 2482–2490. 10.1158/1078-0432.CCR-17-3070. [DOI] [PubMed] [Google Scholar]
- Baell JB, & Holloway GA (2010). New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. Journal of Medicinal Chemistry, 53(7), 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Baell JB, & Nissink JWM (2018). Seven year itch: Pan-assay interference compounds (PAINS) in 2017-utility and limitations. ACS Chemical Biology, 13(1), 36–44. 10.1021/acschembio.7b00903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosey CA, Ho C, Long WZ, Singh S, Burnett K, Hura GL, et al. (2016). Defining NADH-driven allostery regulating apoptosis-inducing factor. Structure, 24(12), 2067–2079. 10.1016/j.str.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosey CA, Shen R, Burnett K, Hura G, Moiani D, Jones D, et al. (2020). Incorporating HT-SAXS into drug-discovery pipelines. Acta Crystallogr. Sect. A, 76(a1), a139. 10.1107/S010876732009861X. [DOI] [Google Scholar]
- Brosey CA, & Tainer JA (2019). Evolving SAXS versatility: Solution X-ray scattering for macromolecular architecture, functional landscapes, and integrative structural biology. Current Opinion in Structural Biology, 58, 197–213. 10.1016/j.sbi.2019.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PC, & Hennig J (2018). The role of small-angle scattering in structure-based screening applications. Biophysical Reviews, 10, 1295–1310. 10.1007/s12551-018-0464-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PC, Masiewicz P, Perez K, & Hennig J (2020). Structure-based screening of binding affinities via small-angle X-ray scattering. IUCrJ, 7(Pt. 4), 644–655. 10.1107/S2052252520004169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PC, Masiewicz P, Rybin V, Svergun D, & Hennig J (2018). A general small-angle X-ray scattering-based screening protocol validated for protein-RNA interactions. ACS Combinatorial Science, 20(4), 197–202. 10.1021/acscombsci.8b00007. [DOI] [PubMed] [Google Scholar]
- Classen S, Hura GL, Holton JM, Rambo RP, Rodic I, McGuire PJ, et al. (2013). Implementation and performance of SIBYLS: A dual endstation small-angle X-ray scattering and macromolecular crystallography beamline at the advanced light source. Journal of Applied Crystallography, 46, 1–13. 10.1107/S0021889812048698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels DS, Woo TT, Luu KX, Noll DM, Clarke ND, Pegg AE, et al. (2004). DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nature Structural & Molecular Biology, 11(8), 714–720. 10.1038/nsmb791. [DOI] [PubMed] [Google Scholar]
- Dyer KN, Hammel M, Rambo RP, Tsutakawa SE, Rodic I, Classen S, et al. (2014). High-throughput SAXS for the characterization of biomolecules in solution: A practical approach. Methods in Molecular Biology, 1091, 245–258. 10.1007/978-1-62703-691-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Petoukhov MV, Konarev PV, Panjkovich A, Tuukkanen A, Mertens HDT, et al. (2017). ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. Journal of Applied Crystallography, 50(Pt 4), 1212–1225. 10.1107/S1600576717007786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graewert MA, & Jeffries CM (2017). Sample and buffer preparation for SAXS. Advances in Experimental Medicine and Biology, 1009, 11–30. 10.1007/978-981-10-6038-0_2. [DOI] [PubMed] [Google Scholar]
- Hangen E, Feraud O, Lachkar S, Mou H, Doti N, Fimia GM, et al. (2015). Interaction between AIF and CHCHD4 regulates respiratory chain biogenesis. Molecular Cell, 58(6), 1001–1014. 10.1016/j.molcel.2015.04.020. [DOI] [PubMed] [Google Scholar]
- Houl JH, Ye Z, Brosey CA, Balapiti-Modarage LPF, Namjoshi S, Bacolla A, et al. (2019). Selective small molecule PARG inhibitor causes replication fork stalling and cancer cell death. Nature Communications, 10(1), 5654. 10.1038/s41467-019-13508-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hura GL, Budworth H, Dyer KN, Rambo RP, Hammel M, McMurray CT, et al. (2013). Comprehensive macromolecular conformations mapped by quantitative SAXS analyses. Nature Methods, 10(6), 453–454. 10.1038/nmeth.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL 2nd, Tsutakawa SE., et al. (2009). Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nature Methods, 6(8), 606–612. 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffries CM, Graewert MA, Blanchet CE, Langley DB, Whitten AE, & Svergun DI (2016). Preparing monodisperse macromolecular samples for successful biological small-angle X-ray and neutron-scattering experiments. Nature Protocols, 11(11), 2122–2153. 10.1038/nprot.2016.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P, & Vendruscolo M (2015). Druggability of intrinsically disordered proteins. Advances in Experimental Medicine and Biology, 870, 383–400. 10.1007/978-3-319-20164-1_13. [DOI] [PubMed] [Google Scholar]
- Lemberg KM, Gori SS, Tsukamoto T, Rais R, & Slusher BS (2022). Clinical development of metabolic inhibitors for oncology. The Journal of Clinical Investigation, 132(1), e148550. 10.1172/JCI148550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiani D, Link TM, Brosey CA, Katsonis P, Lichtarge O, Kim Y, et al. (2021). An efficient chemical screening method for structure-based inhibitors to nucleic acid enzymes targeting the DNA repair-replication interface and SARS CoV-2. Methods in Enzymology, 661, 407–431. 10.1016/bs.mie.2021.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiani D, Ronato DA, Brosey CA, Arvai AS, Syed A, Masson JY, et al. (2018). Targeting allostery with avatars to design inhibitors assessed by cell activity: Dissecting MRE11 endo- and exonuclease activities. Methods in Enzymology, 601, 205–241. 10.1016/bs.mie.2017.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura N, Hitomi K, Arvai AS, Rambo RP, Hitomi C, Cutler SR, et al. (2009). Structural mechanism of abscisic acid binding and signaling by dimeric PYR1. Science, 326(5958), 1373–1379. 10.1126/science.1181829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Hammel M, Hura GL, & Tainer JA (2007). X-ray solution scattering (SAXS) combined with crystallography and computation: Defining accurate macromolecular structures, conformations and assemblies in solution. Quarterly Reviews of Biophysics, 40(3), 191–285. 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- Rambo RP (2017). Considerations for sample preparation using size-exclusion chromatography for home and synchrotron sources. Advances in Experimental Medicine and Biology, 1009, 31–45. 10.1007/978-981-10-6038-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, & Tainer JA (2010). Improving small-angle X-ray scattering data for structural analyses of the RNA world. RNA, 16(3), 638–646. 10.1261/rna.1946310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, & Tainer JA (2011). Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers, 95(8), 559–571. 10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, & Tainer JA (2013a). Accurate assessment of mass, models and resolution by small-angle scattering. Nature, 496(7446), 477–481. 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, & Tainer JA (2013b). Super-resolution in solution X-ray scattering and its applications to structural systems biology. Annual Review of Biophysics, 42, 415–441. 10.1146/annurev-biophys-083012-130301. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, & Sali A (2010). FoXS: A web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Research, 38(Web Server issue), W540–W544. 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Tainer JA, & Sali A (2013). Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophysical Journal, 105(4), 962–974. 10.1016/j.bpj.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Tainer JA, & Sali A (2016). FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Research, 44(W1), W424–W429. 10.1093/nar/gkw389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DE, Bayly AR, Abell C, & Skidmore J (2016). Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nature Reviews. Drug Discovery, 15(8), 533–550. 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- Sevrioukova IF (2009). Redox-linked conformational dynamics in apoptosis-inducing factor. Journal of Molecular Biology, 390(5), 924–938. 10.1016/j.jmb.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, et al. (2014). DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Molecular Cell, 53(1), 7–18. 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D, Barberato C, & Koch MHJ (1995). CRYSOL—A program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. Journal of Applied Crystallography, 28(6), 768–773. 10.1107/S0021889895007047. [DOI] [Google Scholar]
- Warner KD, Hajdin CE, & Weeks KM (2018). Principles for targeting RNA with drug-like small molecules. Nature Reviews. Drug Discovery, 17(8), 547–558. 10.1038/nrd.2018.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Gelot C, Pantelidou C, Li A, Yücel H, Davis RE, et al. (2021). A first-in-class polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. Nature Cancer, 2(6), 598–610. 10.1038/s43018-021-00203-x. [DOI] [PMC free article] [PubMed] [Google Scholar]