

Abstract
The histone H2A variant H2A.W occupies transposons and thus prevents access to them in Arabidopsis thaliana. H2A.W is deposited by the chromatin remodeler DDM1, which also promotes the accessibility of chromatin writers to heterochromatin by an unknown mechanism. To shed light on this question, we solve the cryo-EM structures of nucleosomes containing H2A and H2A.W, and the DDM1-H2A.W nucleosome complex. These structures show that the DNA end flexibility of the H2A nucleosome is higher than that of the H2A.W nucleosome. In the DDM1-H2A.W nucleosome complex, DDM1 binds to the N-terminal tail of H4 and the nucleosomal DNA and increases the DNA end flexibility of H2A.W nucleosomes. Based on these biochemical and structural results, we propose that DDM1 counters the low accessibility caused by nucleosomes containing H2A.W to enable the maintenance of repressive epigenetic marks on transposons and prevent their activity.
Subject terms: Cryoelectron microscopy, DNA methylation, Chromatin remodelling, Histone variants
Osakabe and colleagues report the cryo-EM structures of Arabidopsis nucleosomes and how they complex with the chromatin remodeler DDM1, providing insights into how DDM1 promotes the access of chromatin writers to heterochromatin.
Introduction
Transposons are mobile DNA elements that contribute to genetic evolution in animals and plants1–11. The expression of transposons that results in their mobilization (referred to as transposition) can potentially disrupt gene function and threaten the integrity of the host genome. Therefore, transposons are generally silenced by heterochromatin formation with the contributions of repressive epigenetic modifications, such as cytosine methylation within DNA and histone H3K9 and H3K27 methylations12–14. In plants, several DNA methyltransferases methylate DNA in CG and non-CG contexts, and DNA methylation provides a feedback loop to recruit H3K9 methyltransferases15–23, which participate in transposon silencing.
In eukaryotes, paralogs of each core histone H2A, H2B, and H3 have been identified as histone variants24,25, with characteristic genome distributions, production patterns, and functions26–33. In Arabidopsis, four classes of H2A variants have been identified, and they occupy particular functional chromatin domains34. Among them, H2A.W evolved in land plants and is specifically localized in pericentromeric heterochromatin34,35. H2A.W is distinguished from other H2A variants by its unique C-terminal tail containing the KSPKK motif, which stabilizes heterochromatin via interactions with linker DNA34,36–38, and it cooperates with H3 lysine 9 dimethylation (H3K9me2) to silence transposons39.
Chromatin remodeling factors are proteins harboring an ATPase domain, and promote the translocation of nucleosomal DNA by distorting histone-DNA interactions. Consequently, DNA sliding and histone removal/exchange/replacement are facilitated within the nucleosome40,41. In Arabidopsis, 41 proteins belong to the family of sucrose non-fermenting 2 (Snf2) chromatin remodeling factors42. Their functions are associated with various physiological pathways, including the control of flowering, flower development, and resistance to pathogens43–52. An Snf2-type chromatin remodeling factor named DECREASE IN DNA METHYLATION 1 (DDM1) reportedly functions in DNA methylation maintenance53–56 and transposon silencing57–62 in the Arabidopsis genome. DDM1 specifically binds H2A.W and mediates its deposition over transposons for silencing35,63. However, the synergistic action of H2A.W and H3K9me2 accounts for the silencing of less than half of the transposons that are silenced by DDM139, suggesting additional modes of action for DDM1, such as allowing DNA methyltransferases to access heterochromatin64,65. Nucleosomal DNA is highly protected against DNA methylation66,67, and thus the chromatin remodeling activity by DDM1 would be required for DNA methylation in the context of heterochromatin. Other than the deposition of H2A.W, in the absence of further biochemical and structural studies, the mechanisms by which DDM1 functions in transposon silencing have remained unclear.
We now report the cryo-electron microscopy (cryo-EM) structures of nucleosomes containing H2A and H2A.W, and the DDM1-H2A.W nucleosome complex. Our cryo-EM structures revealed the flexible entry/exit DNA regions in the H2A nucleosome, possibly caused by its specific amino acid substitutions in the C-terminal docking domain. By contrast, the entry/exit regions of nucleosomal DNA were tightly associated with histone in the H2A.W nucleosome, suggesting the low accessibility to DNA. The DDM1-H2A.W nucleosome structure revealed that DDM1 contacts the N-terminal tail of H4 and the DNA within the H2A.W nucleosome. Crosslinking mass spectrometry and biochemical analyses suggested that DDM1 contacts the H2A.W-specific C-terminal tail and increases the flexibility of the entry/exit DNA regions in the H2A.W nucleosome, leading it to resemble the features of the H2A nucleosome. Based on these results, we propose that DDM1 counteracts the DNA end stability of the H2A.W nucleosome and enables chromatin modifiers to access the H2A.W nucleosome on transposons for repressive mark deposition, promoting their silencing in heterochromatin.
Results
Cryo-EM structures of nucleosomes containing H2A and H2A.W
In wild type plants, H2A.W is enriched in the pericentromeric heterochromatin34,68. Previous genomic analyses have shown that a moderate decrease, but not complete loss, of non-CG methylation was observed in h2a.w mutant plants68. The h2a.w mutant plants also exhibited ectopic localization of canonical H2A (hereafter referred to as H2A) in pericentromeric heterochromatin. These findings suggested that ectopically incorporated H2A in heterochromatin complements the function of H2A.W for the maintenance of DNA methylation, and prompted us to compare the structures of nucleosomes containing these H2A variants.
We reconstituted nucleosomes composed of a 169 base-pair DNA fragment with the Widom 601 sequence69 and H2A or H2A.W, together with Arabidopsis histones H2B, H3.1, and H4 (Supplementary Fig. 1). We used H3.1 for the nucleosome reconstitution because it is relatively enriched in heterochromatin compared to H3.335,70–72. We determined the cryo-EM structures of nucleosomes containing H2A and H2A.W (Supplementary Table 1 and Fig. 1). A single-particle cryo-EM workflow was performed on the H2A- and H2A.W-nucleosomes, and both structures of nucleosomes containing H2A and H2A.W were determined at 2.9 Å resolution (Supplementary Figs. 2–4). Interestingly, in the H2A nucleosome, the entry/exit regions of nucleosomal DNA are disordered and only 113 base pairs (bps) of DNA are bound to the histone octamer (Fig. 1a). By contrast, in the H2A.W nucleosome, the entry/exit regions are fully wrapped around the histone octamer, and 145 bps of DNA are visualized (Fig. 1a, b). These results suggest that the entry/exit DNA ends of the H2A nucleosome are flexible, as compared to those of the H2A.W nucleosome.
Fig. 1. Cryo-EM structures of nucleosomes containing H2A and H2A.W.

Cryo-EM structures of nucleosome containing AtH2A (a) and AtH2A.W (b). Arrowheads indicate the terminal DNA detected by cryo-EM density maps. Structures of AtH2A (c) and AtH2A.W (d) complexed with H3. Red and yellow dashed lines indicate the disordered regions in AtH2A nucleosomes (c). The two contact sites discussed in panel e are enclosed in dashed boxes (i) and (ii). e Close-up views of regions (i) and (ii) from panel d, where AtH2A.W and H3 are colored green and yellow, respectively. Map-to-density figures of region (i) in the nucleosomes containing AtH2A and AtH2A.W are shown in Supplementary Fig. 4. f Graphical summary for the flexibility of the entry/exit nucleosomal DNA ends in the nucleosomes containing H2A.W (upper) and H2A (lower).
In the H2A nucleosome, we also found ambiguous cryo-EM density maps of the H2A C-terminal docking domain and the H3 αN helix (Fig. 1c and Supplementary Fig. 4). The histone-DNA contacts at the entry/exit regions of the nucleosomal DNA by the H3 αN helix are known to be supported by interactions with the H2A C-terminal docking domain73–76 (Fig. 1c, d and Supplementary Fig. 4). The H2A Met109, His113, and Leu115 residues are substituted by Leu117, Asn121, and Val123 in H2A.W, respectively (Supplementary Fig. 5a). Notably, we observed that the H2A.W Asn121 and Val123 residues contact the H3 Ile112 residue (Fig. 1e). In addition, the H2A.W Leu117 residue, which is not conserved in H2A, contacts the Asn98 residue of the same H2A.W, and may stabilize the docking domain of H2A.W (Fig. 1e and Supplementary Figs. 4 and 5). These residues correspond to Leu108, Gln112, and Val114 in human H2A, and disordered regions of the Arabidopsis H2A in the nucleosome are clearly visualized in the human H2A nucleosome (Supplementary Figs. 4 and 5). We thus conclude that Arabidopsis H2A might weaken the intra- and inter-histone interactions in the nucleosome, resulting in destabilized interactions between the H3 αN region and the entry/exit nucleosomal DNA in the H2A nucleosome (Fig. 1f). By contrast, H2A.W interacts with the H3 αN and α2 regions, resulting in the tight wrapping of the entry/exit DNA regions around histone octamer in the H2A.W nucleosome.
Cryo-EM structure of the DDM1-H2A.W nucleosome complex
Previous reports have shown that DDM1 changes the DNA register in nucleosomes and functions to maintain DNA methylation in pericentromeric regions65. To investigate the mechanism of DDM1 binding to the nucleosome, we determined the cryo-EM structure of the H2A.W nucleosome complexed with full-length DDM1 (Supplementary Table 1, and Fig. 2a, b). We performed a single-particle cryo-EM workflow on the DDM1-nucleosome complex. Three-dimensional (3D) classifications identified the class of DDM1 bound to the H2A.W nucleosome, and the DDM1-H2A.W nucleosome structure was determined at 4.7 Å resolution, in which the local resolution ranges of DDM1 and nucleosome in the complex are approximately 5.2-6.2 Å and 4.2-6.2 Å, respectively (Supplementary Fig. 6). The central nucleosomal DNA region is located on the dyad axis of the nucleosome, and termed superhelical location (SHL0). The nucleosomal DNA locations are named every 10 base pairs from SHL0, as SHL±1, SHL±2, SHL±3, SHL±4, SHL±5, SHL±6, and SHL±7. In the DDM1-H2A.W nucleosome structure, DDM1 binds the nucleosomal DNA at SHL-2 and SHL+6 by crossing the superhelical DNA gyres (Fig. 2b–d). The nucleosomal DNA at SHL-2 is substantially distorted by DDM1 binding (Fig. 2d and Supplementary Fig. 7), in a similar manner to the nucleosomal DNA distortion induced by the yeast Snf2 nucleosome remodeler41,77–79 (Fig. 2d and Supplementary Fig. 8). Hence this result suggests that DDM1 interacts with the nucleosome through a widely conserved mechanism for nucleosome remodeling.
Fig. 2. Cryo-EM structure of the AtDDM1-nucleosome complex.

a Schematic representation of the full-length AtDDM1 (upper) and the AtDDM1 fragment observed by cryo-EM (lower). The ATPase core domains 1 and 2 are colored cyan and magenta, respectively. The amino acid sequence alignment between AtDDM1 and Saccharomyces cerevisiae Snf2 is presented in Supplementary Fig. 8. b Cryo-EM structure of the AtDDM1-nucleosome complex. The atomic structure model of the AtDDM1-nucleosome complex is fitted to the transparent cryo-EM density map. The ATPase core domains 1 and 2 of AtDDM1 are colored cyan and magenta, respectively. c Structure of nucleosomal DNA bound by AtDDM1. The dashed box corresponds to the nucleosomal DNA around SHL-2, where DNA distortion was observed. d Structural comparison of nucleosomal DNAs bound by AtDDM1 (light orange and gray), ScSnf2 in the absence of ADP (yellow and red, PDB ID: 5X0Y77 (Snf2-nucleosome complex)), or H2A.W nucleosome (white and green). Map-to-density figures of the nucleosomal DNA around SHL-2 of the H2A.W nucleosome and the DDM1-H2A.W nucleosome complex are shown in Supplementary Fig. 7.
DDM1 increases the flexibility of nucleosomal entry/exit DNA regions
In the DDM1-H2A.W nucleosome complex, only 111 base pairs of DNA are bound to the histone octamer, and the entry/exit regions from SHL-5 to SHL-7 and from SHL+6 to SHL+7 are disordered (Figs. 2 and 3). These results suggest that DDM1 binding to the H2A.W nucleosome causes the nucleosomal DNA ends to become more flexible, as in the Arabidopsis H2A nucleosome (Fig. 1a).
Fig. 3. Structural comparison of AtDDM1-bound and AtDDM1-free nucleosomes.

Cryo-EM structures of the AtDDM1-AtH2A.W nucleosome complex (a) and the AtH2A.W nucleosome (b). Structures of nucleosomal DNA are compared at SHL −7 to 0 (middle) and SHL 0 to +7 (right). Blue arrowheads indicate the dyad axis. Black arrowheads indicate the terminal DNA detected by cryo-EM density maps. Dashed red circles indicate the AtH2A.W docking domain and the H3 αN helix disordered in the AtDDM1-AtH2A.W nucleosome complex.
To test if DDM1 increases the flexibility of nucleosomal entry/exit DNA regions in solution, we conducted a restriction enzyme susceptibility assay. We used the MspI and RsaI restriction enzymes, which recognize the nucleosomal DNA regions 60–64 and 5–8 bases away from the dyad axis of the nucleosome, respectively (Fig. 4a and Supplementary Fig. 1a). MspI cleaves its target site if the DNA ends become accessible in the nucleosome; however, RsaI cleavage efficiency may be unchanged (Fig. 4a). In the absence of DDM1, both RsaI and MspI poorly digested the nucleosomal DNA, reflecting the tight wrapping of the entry/exit DNA region around histone octamer containing H2A.W (Figs. 1b and 4b–g). By contrast, in the presence of DDM1, the susceptibility of nucleosomal DNA to MspI was enhanced, but not to RsaI (Fig. 4b–g). These results suggest that DDM1 changes the flexibility of the entry/exit regions of nucleosomal DNA without nucleosome disassembly.
Fig. 4. Analyses of the nucleosomal DNA end flexibility by the restriction enzyme susceptibility assay.

a Graphical presentation of the relevant restriction enzyme recognition sites within the DNA fragment used in this experiment. The sequence of the DNA fragment is shown in Supplementary Fig. 1a. Cryo-EM structures of the AtH2A.W nucleosome with the locations of the MspI (b) and RsaI (e) sites. Native-PAGE analyses of DNA fragments after the restriction enzyme susceptibility assays with MspI (c) and RsaI (f). Graphical presentations of the restriction enzyme susceptibility assay results with MspI (d) and RsaI (g). Means and error bars represent SD from five independent experiments. The statistical significance (P) was determined using a t-test. NS not significant. Source data are provided as a Source Data file.
We further tested the flexibility of the nucleosomal entry/exit DNA regions by fluorescence resonance energy transfer (FRET), using fluorescein and BHQ-1 as its quencher (Fig. 5 and Supplementary Fig. 9). The fluorescein-labeled base and BHQ-1 quencher-labeled base were introduced 57 bases from the 5’ end of the forward strand and 16 bases from the 5’ end of the complementary strand in 145 base pair DNA fragments (Fig. 5a and Supplementary Fig. 9a). The fluorescein signal was detected with the naked DNA, but was substantially suppressed in the nucleosome because of the proximity of BHQ-1 (Supplementary Fig. 9b). The fluorescence signal becomes enhanced when the DNA ends are flexibly disordered, because the BHQ-1 quencher located near a nucleosomal DNA end is farther from the fluorescein in the nucleosome.
Fig. 5. Analyses of nucleosomal DNA end flexibility by the FRET assay.

a Graphical summary of the FRET assay in this study. The emission of fluorescein is inhibited by nucleosome formation because the neighboring BHQ-1 quenches fluorophores, resulting in low fluorescence signals. When the entry/exit nucleosomal DNA ends are unwrapped, fluorescence signals can be detected. b Graphical presentation of the FRET assay results with DDM1. Means and error bars represent SD from four independent experiments. Source data are provided as a Source Data file.
We reconstituted the nucleosome containing H2A.W and performed the FRET assay. As expected, the fluorescence signals of the H2A.W nucleosome were drastically enhanced when the nucleosome was disrupted by increasing the NaCl concentration (Supplementary Fig. 9c, d). We found that DDM1 enhanced the fluorescence signal of the H2A.W nucleosome in a concentration-dependent manner (Fig. 5b). This result further supports that DDM1 increases the flexibility of nucleosomal entry/exit DNA regions of H2A.W nucleosome such that it resembles the Arabidopsis H2A nucleosome (Fig. 1a).
Crosslinking mass spectrometric analyses of DDM1-nucleosome complexes
Our biochemical and structural analyses demonstrated that DDM1 binding increased the flexibility of the entry/exit nucleosomal DNA regions of the H2A.W nucleosome. These results suggest that DDM1 binding changes the structure of the H2A.W nucleosome, although we did not detect a direct interaction between H2A.W and DDM1 in our cryo-EM structure of the DDM1-H2A.W nucleosome complex (Figs. 2 and 3). Therefore, we speculated that some disordered regions that were not detected by cryo-EM contribute to the flexible entry/exit DNA ends of the DDM1-bound H2A.W nucleosome.
To identify the specific interactions between DDM1 and H2A.W, we next performed a crosslinking mass spectrometric analysis of the DDM1-nucleosome complex (Fig. 6a and Supplementary Fig. 10). We employed disuccinimidyl suberate (DSS-H12/D12), which crosslinks inter- and intra-molecular lysine residues, as the crosslinker. In our crosslinking mass spectrometric analyses, crosslinked peptides corresponding to the flexible tails of histone proteins and DDM1 were detected (Supplementary Fig. 10). Intriguingly, the DDM1 Lys208 and Lys342 residues were crosslinked with the H2A.W Lys147 and Lys140 residues, which are both located in the flexible C-terminal tail (Fig. 6a). To determine whether the DDM1 Lys208 and Lys342 residues directly interact with the disordered H2A.W Lys147 and Lys140 residues, we present the possible crosslinking areas with colored circles centered on the Cα atom of His113 of H2A.W, with a 115.35 Å radius corresponding to the possible length of the H2A.W 113–140 peptide (Fig. 6b). As shown in Fig. 6b, the DDM1 Lys208 and Lys342 residues are located within this possible crosslinking area, and therefore could directly interact with the H2A.W Lys147 and Lys140 residues in the DDM1-H2A.W nucleosome complex.
Fig. 6. C-terminal tail of AtH2A.W binds DDM1.

a Schematic representation (left) and sequence information (right) for the results obtained by crosslinking mass spectrometry of the DDM1-bound nucleosomes containing AtH2A.W. Two contacts between the H2A.W C-terminal tail and DDM1 are shown. Other crosslinks of the DDM1-bound nucleosomes containing AtH2A.W in the top 25% of ld-scores are shown in Supplementary Fig. 10. b Graphical summary of the contacts between the H2A.W C-terminal tail and DDM1, identified by crosslinking mass spectrometry. The yellow circle represents the 115.35 Å radius, corresponding to residues 113–140 of H2A.W (the central point is the Cα atom of His113 of H2A.W), which indicates the possible crosslinking area of H2A.W Lys140 by DSS-H12/D12. The residues of DDM1 contacting the H2A.W C-terminal tail are shown in red with orange circles.
Altogether, our cryo-EM structure and crosslinking mass spectrometric analyses suggest that DDM1 opens the entry/exit nucleosomal DNA regions of the H2A.W nucleosome through the interaction with the flexible tails of H2A.W, and this activity possibly promotes the accessibility to DNA in constitutive heterochromatin.
DDM1 slides nucleosomes regardless of H2A variants
Previous biochemical studies revealed that DDM1 has nucleosome sliding activity72,80, which is required for the maintenance of DNA methylation in heterochromatin65. However, it remained unclear if DDM1 shows a preference for specific histone variants. To clarify this, we performed the nucleosome sliding assay with nucleosomes containing H2A and H2A.W (Fig. 7 and Supplementary Fig. 11). Nucleosome sliding activity can be monitored by migration distance in native-PAGE gels, where faster and slower migrations correspond to the end and middle positions of nucleosomes, respectively (Supplementary Fig. 11b). Under these experimental conditions, the band shift was detected by the addition of DDM1, suggesting that DDM1 has nucleosome sliding activity as previously reported72,80. Notably, the sliding activity was detected in both H2A and H2A.W nucleosomes (Fig. 7a, b). Consistent with the nucleosome sliding activity, DDM1 did not show specific ATPase activity for histone variants, but displayed higher ATPase activity with nucleosomes rather than free DNA (Supplementary Fig. 12).
Fig. 7. Nucleosome sliding assay.

a Native-PAGE analyses of the nucleosomes containing AtH2A and AtH2A.W after the sliding assay. b, e Graphical presentation of the nucleosome sliding assay results. Means and error bars represent SD from three independent experiments. The efficiency of the remodeled nucleosome was calculated by the ratio of the intensity of each band and normalized to the ratio obtained at 0 min. Source data are provided as a Source Data file. c Overall structures of the AtDDM1-AtH2A.W nucleosome (middle) and the AtH2A.W nucleosome (right). The calculated electrostatic potential of the atomic surfaces of AtDDM1 and the H4 N-terminal tail (residues 19–24) molecules are presented (left). The dashed red circle indicates the disordered regions of the H4 N-terminal tail (residues 19–24) in the AtH2A.W nucleosome. Map to density figures of the H4 N-terminal tail of the AtDDM1-AtH2A.W nucleosome and the AtH2A.W nucleosome are shown in Supplementary Fig. 13. d Native-PAGE analyses of the nucleosomes after the sliding assay with nucleosomes containing AtH2A.W, with or without the N-terminal tail of AtH4.
We further investigated the mechanism by which DDM1 targets and slides nucleosomes. The density of the H4 N-terminal tail was not detected in our cryo-EM structure of the H2A.W nucleosome (Fig. 7c). By contrast, our cryo-EM structure of the DDM1-H2A.W nucleosome complex revealed that the N-terminal tail of H4 is located near the ATPase core domain of DDM1, and is captured by its acidic pocket (Fig. 7c and Supplementary Fig. 13). The binding of the H4 N-terminal tail within the acidic pocket was also reported for the yeast Snf2 protein41,77–79. To test the biological significance of the H4 N-terminal tail binding to DDM1, we performed the nucleosome sliding assay with nucleosomes lacking this tail (residues 1–24) (Fig. 7d, e and Supplementary Fig. 14). Consistent with the conserved role of the H4 N-terminal tail in the Snf2-mediated nucleosome remodeling activity77, the deletion of the H4 N-terminal tail drastically reduced the DDM1-mediated nucleosome sliding (Fig. 7d, e). We thus conclude that DDM1 binds and remodels nucleosomes by a mechanism similar to that described for other chromatin remodelers of the Snf2 family.
Discussion
In the present study, the cryo-EM structure of the DDM1-H2A.W nucleosome complex revealed that DDM1 binds the nucleosomal DNA at the SHL-2 and SHL+6 positions (Fig. 2). This structure is similar to those reported recently by other groups72,81. In addition, a structural comparison between the DDM1-bound H2A.W nucleosome and the DDM1-free H2A.W nucleosome suggested that DDM1 binding caused the H2A.W nucleosome to adopt DNA end flexibility, like the H2A nucleosome (Figs. 2–5). The structural comparison of the H2A and H2A.W nucleosomes suggested that the unique residues of Arabidopsis H2A might contribute to the unstable interaction between the docking domain and the H3 αN and α2 helices, resulting in the flexible nucleosomal DNA entry/exit regions in the H2A nucleosome compared to the H2A.W nucleosomes (Fig. 1). Therefore, the DDM1 binding could perturb the histone-DNA contacts around the entry/exit regions of the H2A.W nucleosome. This correlates with the increased accessibility of DNA methyltransferases to DNA and unchanged DNA methylation levels in h2aw mutant plants, in which H2A.W is replaced by H2A68. The flexibility of the entry/exit nucleosomal DNA does not affect the ATPase activity of DDM1, resulting in the equally efficient sliding of both H2A and H2A.W nucleosomes. These results suggest that DDM1 still functions with the H2A nucleosome for the maintenance of DNA methylation in h2aw mutant plants. These findings explain the mechanism by which DDM1 counteracts the low DNA accessibility of H2A.W nucleosomes and enables chromatin modifiers to access heterochromatin (modeled in Fig. 8).
Fig. 8. Model of DDM1 activity for the maintenance of repressive marks.
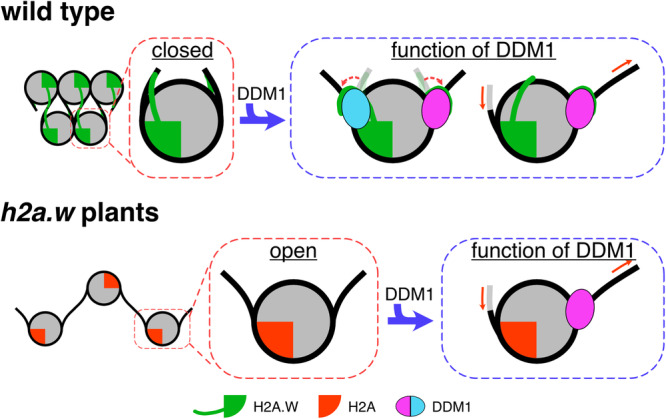
The pericentromeric heterochromatin is occupied with H2A.W, which forms a condensed structure caused by the extended C-terminal tail of H2A.W interacting with the linker DNA, and the H3 αN and α2 helices. In the absence of H2A.W, the pericentromeric heterochromatin is occupied with H2A. The nucleosome containing H2A forms an open structure, caused by the loss of the interactions with the H3 αN and α2 helices. In the presence of DDM1, the C-terminal tail of H2A.W might dissociate from the linker DNA and H3 by interacting with DDM1. In addition, DDM1 slides nucleosomes containing H2A.W and H2A with identical efficiency. These activities might promote the increased accessibility of the heterochromatin to DNA methyltransferases.
Previous studies have shown that the ATPase activity of the chromatin remodeler ISWI is inhibited by its internal AutoN domain, which is enriched with basic residues and thus resembles the N-terminal tail of H4. This autoinhibition is released via competitive binding with the N-terminal tail of H482–85. Our cryo-EM structures and biochemical analyses demonstrated that the interaction between the N-terminal tail of H4 and DDM1 plays important roles in the DDM1-mediated nucleosome sliding (Fig. 7). We noticed the sequence similarities of conserved regions in angiosperm DDM1 with the AutoN domain of ISWI and the N-terminal tail of H4 (Supplementary Fig. 15a, b). Accordingly, like the ISWI AutoN domain, we propose that the conserved DDM1 region also serves as a negative regulator and function via competitive binding with the H4 N-terminal tail. Note that we did not detect an interaction between the N-terminal tail of H4 and DDM1 in our crosslinking mass spectrometric analyses of the DDM1-nucleosome complex (Fig. 6 and Supplementary Fig. 10). This result might be due to the enrichment of acidic residues and the lack of lysine residues within the DDM1 acidic pocket, resulting in the interruption of crosslinking by steric hindrance (Fig. 7c).
Previous genomic analyses have shown the synergistic action of H2A.W and H3K9me2 for transposon silencing39. In this study, our crosslinking mass spectrometric analyses identified an interaction between the C-terminal tail of H2A.W and the N-terminal tail of H3 (Supplementary Fig. 10). These regions are located close to the preferred linker DNA for DNA methylation86, suggesting that the interaction between H2A.W and H3 might contribute to the establishment or maintenance of the repressive marks for transposon silencing. Further structural and biochemical analyses will be required to clarify this issue.
In addition to our determination of the DDM1 binding mechanism to the H2A.W nucleosome, we also found that DDM1 increases the flexibility of the entry/exit nucleosomal DNAs (Figs. 2–5). This may enhance the accessibility of DNA-binding proteins, including DNA methyltransferases. The binding of the pioneer transcription factor SOX11 to nucleosomal DNA at SHL-2 reportedly leads to a clash with the secondary DNA gyre around SHL+6, inducing the detachment of the DNAs around the entry/exit regions of the nucleosome87. In contrast to SOX11, DDM1 bound to SHL-2 interacts with the nucleosomal DNA around SHL+6 without steric clashes (Supplementary Fig. 16). DDM1 may directly interact with the H2A.W C-terminal tails including the docking domains, which are located near the DNA entry/exit regions, and the N-terminal region of H3. These interactions would contribute to the flexible entry/exit nucleosomal DNA ends in the DDM1-H2A.W nucleosome complex.
The cryo-EM structures of the DDM1-nucleosome (H3.3/H2A.W72 or H3.1/H2A81) complex have been reported. The structure of DDM1 in this study is similar to the published structures except for the loop close to H3, which was disordered in our cryo-EM structure (Supplementary Fig. 17). Intriguingly, we found that the H2A.W docking domain and entry/exit regions of nucleosomal DNA regions were disordered, in contrast to a previous cryo-EM structure (Supplementary Fig. 17). This difference might arise from the use of Xenopus H4 in the previous structural analysis. Especially, SWI/SNF-independent (SIN) mutations present in H4 reportedly alter the nucleosome structure88,89. Importantly, two amino acid residues, Ile60 and Arg77, are not conserved in Xenopus H4. The Arabidopsis H4 Ile60 residue exists in the central helix, which could be perturbed in the SIN mutants, and the Arabidopsis H4 Arg77 residue is in the loop region located near the nucleosomal DNA backbone. Therefore, it is possible that the structural differences of the nucleosomal DNA ends between the present and previous structures may be due to the use of H4 from different species.
Our structural and biochemical data revealed the mechanism by which the chromatin remodeling factor DDM1 promotes accessibility in constitutive heterochromatin, which is likely crucial for the maintenance of DNA methylation. These results provide important information for future studies of the mammalian DDM1 homolog HELLS/LSH, which mediates the deposition of a histone variant, macroH2A90,91. MacroH2A is major component of heterochromatin in mammals92, and shares sequence similarity and functions with plant H2A.W38. HELLS/LSH also controls DNA methylation deposition and transposon silencing93–95. HELLS/LSH possesses the basic patch and shares sequence similarities with possible regulatory regions in DDM1 and the AutoN domain of ISWI (Supplementary Fig. 15c). Therefore, it is possible that HELLS/LSH has similar activity to DDM1, thus providing clues about the mechanism of HELLS/LSH and its role in epigenetic human diseases96.
Methods
Preparation and purification of the nucleosome containing Arabidopsis histones
The DNA fragments encoding Arabidopsis thaliana AtH2A.13, AtH2A.W.6, AtH3.1, and AtH4 were inserted into the pET-15b vector (Novagen). The DNA fragment encoding AtH2B.9 was inserted into the pET-15b vector, in which the sequence of the thrombin recognition site (Leu-Val-Pro-Arg-Gly-Ser) was substituted with that of the TEV protease recognition site (Glu-Asn-Leu-Tyr-Phe-Gln-Gly-Ser). The expression and purification of the recombinant Arabidopsis thaliana histones were performed by following the methods37,63,97–99. Briefly, histones except for H4 were expressed in the BL21(DE3) containing minor tRNA expression vector (Codon (+) RIL) and H4 was expressed in the JM109(DE3) containing minor tRNA expression vector (Codon (+) RIL) as N-terminally hexa-histidine (His6)-tagged proteins. After purification of all histones using nickel-nitrilotriacetic acid agarose (Ni-NTA) resin (QIAGEN) under denaturing conditions, the His6-tag portion was removed by thrombin protease for AtH2A.13, AtH2A.W.6, AtH3.1, and AtH4, and TEV protease for AtH2B.9, respectively. All histones were then purified on a HiPrep SP HP 16/10 cation exchange column (Cytiva) under denaturing conditions with a linear gradient of 200–800 mM NaCl. The DNA fragment encoding truncated AtH4 (aa 25–102) was inserted into pET-15b. Methods for the expression and purification of the truncated AtH4 protein were the same as AtH4. After purification using a cation exchange column, histone proteins were dialyzed against water and freeze-dried. The histone octamers were reconstituted and purified by Superdex 200 gel filtration column (Cytiva) as described37,98,99. Briefly, lyophilized histones were mixed at an equal molar ratio and dissolved under denaturing conditions. Then histone octamers were dialyzed against refolding buffer (10 mM Tris-HCl (pH 7.5), 2 M NaCl, 1 mM EDTA, and 5 mM 2-mercaptoethanol). After the dialysis, refolded histone octamers were then purified on a Superdex 200 gel filtration column (Cytiva) under the refolding buffer. The purified histone octamers were frozen in liquid nitrogen and stored at −80 ˚C until the reconstitution of nucleosomes.
The sequences of the DNA fragments used for nucleosome reconstitution are shown in Supplementary Figs. 1a, 9a, and 11a. The 169 base-pair (24N0) and 193 base-pair (24N24) DNA fragments containing the Widom 601 sequence were purified by polyacrylamide gel electrophoresis (PAGE), using a Prep Cell apparatus as described100. Briefly, the DNA fragments were obtained by the excision from the plasmid DNA using EcoRV restriction enzyme and then purified by polyethylene glycol precipitation. The DNA fragments were then further purified by PAGE using a Prep Cell apparatus. The 145 base-pair DNA fragment containing fluorescein and its quencher (BHQ-1) and the 193 base-pair (48N0) DNA fragment containing the Widom 601 sequence were amplified by PCR and purified using a Prep Cell apparatus. The amplification of 145 base-pair DNA fragment containing fluorescein and its quencher (BHQ-1) was performed using primers containing fluorescein or BHQ-1 (FASMAC) at the position described in Supplementary Fig. 9a. The nucleosomes composed of DNA and histone octamers were reconstituted by the salt dialysis method, and then purified by PAGE using a Prep Cell apparatus, as described37,98,99. Briefly, the DNA fragments and histone octamers were mixed, and the nucleosomes were reconstituted by the salt dialysis method. The resulting nucleosomes were then purified using a Prep Cell apparatus. The buffer of purified nucleosomes was then exchanged with the storage buffer (20 mM Tris-HCl (pH 7.5), 5% glycerol, and 1 mM DTT) for storage at −80 ˚C.
Purification of recombinant Arabidopsis DDM1
The DNA fragment encoding AtDDM1 was inserted into the pET-15b vector (Novagen), in which the sequence of the thrombin recognition site (Leu-Val-Pro-Arg-Gly-Ser-His) was substituted with that of the human rhinovirus (HRV) 3C protease (Leu-Glu-Val-Leu-Phe-Gln-Gly-Pro), and a SUMO tag was integrated between the His6-tag and the HRV 3C protease recognition site. Expression and purification of AtDDM1 were performed by the method described previously63. Briefly, AtDDM1 protein was expressed in the BL21(DE3) containing minor tRNA expression vector (Codon (+) RIL) as an N-terminally His6-tagged protein. After purification using Ni-NTA resin, GST-tagged HRV 3C protease (0.1 mg/mg of His6-SUMO-AtDDM1) was added to remove His6-tagged SUMO from the DDM1 portion. AtDDM1 was subjected to a RESOURCE Q anion exchange column (Cytiva) and collected from the flow-through fractions, and then purified on a RESOURCE S cation exchange column (Cytiva). AtDDM1 protein was further purified on a Superdex 200 gel filtration column using the DDM1 storage buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10% glycerol, and 2 mM 2-mercaptoethanol).
Preparation of nucleosomes and DDM1-nucleosome complexes for cryo-EM
The nucleosomes containing AtH2A or AtH2A.W (150 µg) were crosslinked and purified by the GraFix method101, using a gradient prepared with buffer A (10 mM HEPES-NaOH (pH 7.5), 1 mM DTT, and 5% (w/v) sucrose) and buffer B (10 mM HEPES-NaOH (pH 7.5), 1 mM DTT, 20% (w/v) sucrose, and 0.1% glutaraldehyde). The nucleosome containing AtH2A.W (2.39 µM) was mixed with the full-length AtDDM1 (14.34 µM) in a total volume of 300 µl reaction buffer (20 mM HEPES-NaOH (pH 7.5), 14.7 mM Tris-HCl (pH 7.5), 150 mM NaCl, 3 mM MgCl2, 1.2 mM DTT, 1 mM ADP, 6.2% glycerol, and 1 mM 2-mercaptoethanol), and incubated at 30 ˚C for 30 min. Afterwards, the sample was crosslinked and fractionated by the GraFix method, using a gradient prepared with buffer A containing 150 mM NaCl and buffer B containing 150 mM NaCl. The samples were applied to the top of the gradient solution and then centrifuged at 4 ˚C for 16 h at 125,000 × g, using an SW 41 Ti rotor (Beckman Coulter). After centrifugation, aliquots were collected from the top of the solution and analyzed by 6% (nucleosomes) or 4% (AtDDM1-nucleosome complex) non-denaturing PAGE in 0.5 × TBE (44.5 mM Tris-Borate (pH 8.3) and 1 mM EDTA), followed by ethidium bromide staining. The fractions containing the nucleosomes or AtDDM1-nucleosome complex were collected and then desalted on a PD-10 column (Cytiva) by elution with elution buffer (10 mM HEPES-NaOH (pH 7.5) and 2 mM TCEP). The eluted samples were concentrated using an Amicon Ultra-2 centrifugal filter unit (Merck) and stored on ice.
Preparation of grids for cryo-EM
For the AtDDM1-nucleosome complex (0.25 mg/ml), the nucleosome containing AtH2A (2.0 mg/ml), and the nucleosome containing AtH2A.W (2.0 mg/ml), 2.5 μl portions of samples were applied onto freshly glow-discharged Quantifoil R1.2/1.3, Cu, 200-mesh grids. The grids were blotted for 8 sec at 4 ˚C in 100% humidity, and then plunge-frozen in liquid ethane by using a Vitrobot Mark IV (Thermo Fisher Scientific).
Cryo-EM data collection
The AtDDM1-nucleosome complex, the nucleosome containing AtH2A, and the nucleosome containing AtH2A.W were imaged on a Krios G4 microscope (Thermo Fisher Scientific), operated at 300 kV and equipped with a BioQuantum energy filter and a K3 direct electron detector (Gatan) with a slit width of 20 eV, operated in the counting mode at a calibrated pixel size of 1.06 Å. Images of the AtDDM1-nucleosome complex, the nucleosome containing AtH2A, and the nucleosome containing AtH2A.W were recorded at a frame rate of 150 ms for 4.5 s. A nominal defocus range of −1 to −2.5 μm was employed, and the movies were automatically acquired using the EPU software (Thermo Fisher Scientific).
Image processing
The frames of the movies for the AtDDM1-nucleosome complex, the nucleosome containing AtH2A, and the nucleosome containing AtH2A.W were subjected to motion correction using MOTIONCOR2, with dose weighting102. The contrast transfer function (CTF) was estimated using CTFFIND4103, and RELION4104 was used for the following image processing. For the AtDDM1-nucleosome complex, a total of 7,004,935 particles from dataset1 and 2,125,419 particles from dataset2 were picked by template-based auto-picking, using the 2D class averages of auto-picked particles based on a Laplacian-of-Gaussian filter as templates, followed by a few rounds of 2D classification to remove junk particles, resulting in the selection of 895,881 and 898,969 particles, respectively. The two datasets were combined, and a de novo initial model generated by Relion4 was low-pass filtered to 60 Å and used as the initial model for the 3D classification. The 3D class with the density map of AtDDM1 containing 128,471 particles was selected, and subjected to focused 3D classification without alignment using the AtDDM1 mask. Subsequently, 34,559 particles selected from the best classes were subjected to Bayesian polishing and CTF refinement. The final postprocessing yielded a cryo-EM map of the AtDDM1-nucleosome complex with a global resolution of 4.71 Å, with the gold standard Fourier Shell Correlation (FSC = 0.143) criteria105. The cryo-EM map of the AtDDM1-nucleosome complex was post-processed with the DeepEMhancer software106.
For the AtH2A and AtH2A.W nucleosomes, 3,651,924 particles and 4,254,793 particles were picked by template-based auto-picking, respectively, using the 2D class averages of auto-picked particles based on a Laplacian-of-Gaussian filter as templates. After 2D classification to remove junk particles, 2,141,126 and 2,579,646 particles were selected for the nucleosomes containing AtH2A and AtH2A.W, respectively. The crystal structure of the nucleosome107 (PDB ID: 3LZ0 (Nucleosome core particle composed of the Widom 601 DNA sequence)) was low-pass filtered to 60 Å and used as the initial model for the 3D classification. The selected particles were subjected to 3D classification. Subsequently, the best classes from the 3D classifications of the AtH2A and AtH2A.W nucleosomes, containing 411,432 and 196,430 particles, respectively, were subjected to Bayesian polishing and CTF refinement. The final postprocessing yielded cryo-EM maps of the AtH2A and AtH2A.W nucleosomes with global resolutions of 2.94 Å and 2.94 Å, respectively, with the gold standard Fourier Shell Correlation (FSC = 0.143) criteria105. The cryo-EM map of the nucleosome containing AtH2A.W was post-processed with the DeepEMhancer software106.
The local resolutions of the AtDDM1-nucleosome complex, the nucleosome containing AtH2A, and the nucleosome containing AtH2A.W were calculated by RELION-4. Visualization and rendering of all cryo-EM maps were performed with UCSF ChimeraX108.
Model building and refinement
Model building was performed with COOT109, using the crystal structure of the nucleosome107 (PDB ID: 3LZ0 (Nucleosome core particle composed of the Widom 601 DNA sequence)) and the AtDDM1 structure generated by AlphaFold2110. The nucleosomal DNA was automatically fitted into the vacant volume with ISOLDE111. The structural models of the AtDDM1-nucleosome complex and the nucleosome containing AtH2A.W were refined by real-space refinement in Phenix112,113, and validation was performed with MolProbity114. The data collection and statistics for the 3D reconstruction and model refinement are shown in Supplementary Table 1.
Restriction enzyme susceptibility assay
The nucleosomes (0.2 µM) were mixed with DDM1 (1.6 µM) in a total volume of 10 µl reaction solution, containing 20 mM HEPES-NaOH (pH 7.5), 12 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5% glycerol, 5 mM MgCl2, 1.2 mM DTT, 0.8 mM 2-mercaptoethanol, 0.1 mg/ml BSA, and 0 or 1 mM ADP. The restriction enzyme MspI (10 units) or RsaI (10 units) was added to the mixtures and incubated at 30 ˚C for 60 min. After restriction enzyme digestion, the reaction was terminated by the addition of 5 µl deproteinization solution (20 mM Tris-HCl (pH 8.0), 20 mM EDTA, 0.1% SDS, and 0.5 mg/ml Proteinase K). The resulting DNA was extracted with phenol-chloroform and then analyzed by 10% non-denaturing PAGE in 0.5× TBE. The gel was stained with SYBR Green I solution and DNA was visualized by iBright Imaging Systems.
FRET assay
The nucleosome (0.05 µM), composed of histones and the DNA fragment containing fluorescein and BHQ-1, was incubated with different concentrations of DDM1 (0, 0.05, 0.1, 0.2, 0.4, and 0.8 µM). The reaction was performed in a 20 µL total volume, containing 23 mM Tris-HCl (pH 7.5), 75 mM NaCl, 1.2 mM DTT, 1 mM 2-mercaptoethanol, 6% glycerol, 0.03% NP-40, and 3 mM MgCl2, and was incubated at 30 ˚C for 30 min within a 384-well microplate (Greiner Bio-One). After the incubation, the fluorescence intensity of each sample was measured using a Synergy H1M2F (BioTek), with the excitation and emission wavelengths set to 467 nm and 528 nm, respectively.
Crosslinking mass spectrometry
Nucleosomes (0.2 µM) were mixed with AtDDM1 (0.4 µM) in reaction buffer (20 mM HEPES-NaOH (pH 7.5), 60 mM NaCl, 1 mM MgCl2, 1.1 mM DTT, 1 mM ADP, 4.5% glycerol, 0.03% NP-40, and 0.8 mM 2-mercaptoethanol), and incubated at 25 ˚C for 30 min. The samples were then crosslinked with 1.6 mM DSS-H12/D12 (Creative Molecules) at 25 ˚C for 30 min, and the reaction was quenched by the addition of 50 mM Tris-HCl (pH 7.5) followed by an incubation at 25 ˚C for 15 min. Crosslinking mass spectrometry was performed as described previously115,116. Briefly, the samples were dried and dissolved in an 8 M urea solution to a final protein concentration of 1.0 mg/ml. Re-dissolved samples were reduced by 2.5 mM TCEP, followed by alkylation with 5 mM iodoacetamide. For tryptic digestion, the samples were diluted with a 50 mM ammonium bicarbonate solution to a final concentration of 1 M urea, and then sequencing-grade endopeptidase Trypsin/Lys-C Mix (Promega) was added at an enzyme–substrate ratio of 1:50 wt/wt. The digested samples were applied to a Superdex 30 Increase 3.2/300 (GE Healthcare) column, using buffer containing 25% acetonitrile and 0.1% TFA. The eluted fractions (100 μl) were collected and dried completely. The residues were dissolved in 0.1% TFA and analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS), using an Orbitrap Fusion mass spectrometer equipped with an Ultimate3000 nano-HPLC system (Thermo Fisher Scientific). The crosslinked peptides were identified using the xQuest/xProphet software (version 2.1.5)115, and the following criteria were applied to the xProphet results filter: maximum border of MS1 tolerance = 7 ppm, minimum border of MS1 tolerance = −4 ppm, false discovery rate (FDR) <0.05, minimum d-score = 0.95. The crosslinks listed in the top 25% of ld-scores were visualized using the webserver xVis117. The LC-MS/MS was performed in 2 technical replicates.
Nucleosome sliding assay
The nucleosomes (0.22 µM) were mixed with DDM1 (1.77 µM), in a reaction solution containing 13.3 mM Tris-HCl (pH 7.5), 67 mM NaCl, 0.2 mM DTT, 0.9 mM 2-mercaptoethanol, and 5.6% glycerol, and incubated at 30 ˚C for 15 min. The reaction was then initiated by the addition of ATP, in a reaction solution containing 20 mM HEPES-NaOH (pH 7.5), 12 mM Tris-HCl (pH 7.5), 75 mM NaCl, 0.1 mg/ml BSA, 2.5 mM MgCl2, 1.4 mM DTT, 0.8 mM 2-mercaptoethanol, 5% glycerol, and 1 mM ATP, and incubated at 30 ˚C for 5, 10, 30, and 60 min. The reaction was stopped by the addition of pUC19 plasmid DNA (0.042 µM) in the presence of 15 mM EDTA. The samples were analyzed by 6% non-denaturing PAGE in 0.5× TBE. The gel was stained with SYBR Green I solution, and the DNA was visualized by an iBright Imaging System (Thermo Fisher Scientific). The quantification was performed with the iBright Analysis Software (Thermo Fisher Scientific). The efficiency of the remodeled nucleosome was calculated by the ratio of the intensity of each band and normalized to the ratio obtained at 0 min.
DDM1-nucleosome binding assay
The 48N0 nucleosomes (0.2 µM) were mixed with AtDDM1 (0, 0.1, 0.2, 0.4, 0.8, and 1.6 µM) in a total volume of 10 µl reaction buffer, containing 20 mM HEPES-NaOH (pH 7.5), 12 mM Tris-HCl (pH 7.5), 60 mM NaCl, 0.5% glycerol, 1 mM MgCl2, 0.03% NP-40, 1.2 mM DTT, 0.8 mM 2-mercaptoethanol, and 1 mM ATP. The samples were incubated at 25 ˚C for 30 min, and then analyzed by 4% non-denaturing PAGE in 0.5× TBE. The gel was stained with SYBR Green I solution, and the DNA was visualized by an iBright Imaging System (Thermo Fisher Scientific).
ATPase assay
The 169 base-pair DNA fragments or 24N0 nucleosomes (0.06 µM) were mixed with DDM1 (0.48 µM) in a 50 µL total reaction volume, containing 54 mM Tris-HCl (pH 7.5), 15 mM NaCl, 0.1 mM DTT, 0.2 mM 2-mercaptoethanol, 1.5% glycerol, 0.5 mM ATP, and 2.5 mM MgCl2, and incubated at 30 ˚C for 5, 15, 30, and 60 min. ATPase assays were performed using a colorimetric kit (abcam) and ATPase activity was calculated from optical density values at 600 nm using a Synergy H1M2F.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Source data
Acknowledgements
We thank all members of the Berger, Kakutani, and Kurumizaka laboratories, and especially Mitsuo Ogasawara (Univ. Tokyo) for technical assistance with cryo-EM data collection, and Y. Iikura, M. Dacher, and Y. Takeda (Univ. Tokyo) for their assistance. This work was supported in part by JSPS KAKENHI Grant Numbers JP21K20628, JP22H05172, and JP22H05178 [to A.O.], JP23H05475 [to H.K.], JP22K06098 [to Y.T.], and 21H04977 and 23H00365 [to T.K.], Research Support Project for Life Science and Drug Discovery (BINDS) from AMED under Grant Number JP23ama121009 [to H.K.], HFSP Grant Number RGP0025/2021 [to T.K.], JST ERATO Grant Number JPMJER1901 [to H.K.], and JST PRESTO Grant Number JPMJPR20K3 [to A.O.]. This work was also supported by the Austrian Science Fund (FWF): P32054 and P33380 [to F.B.].
Author contributions
A.O., F.B., T.K., and H.K. conceived, designed, and supervised all the work. A.O. performed all the biochemical analyses. A.O., Y.T., and N.H. prepared the DDM1-nucleosome complex for cryo-EM. A.O. and Y.T. performed cryo-EM analyses. A.O., S.H., and L.N. performed crosslinking mass spectrometry. A.O. and S.S. performed the FRET assay. A.O., Y.T., F.B., T.K., and H.K. prepared all figures and wrote the manuscript. All of the authors discussed the results and commented on the manuscript.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
The cryo-EM maps and atomic models in this study have been deposited in the Electron Microscopy Data Bank and the Protein Data Bank, under the accession codes EMD- 36083 (DDM1-nucleosome complex) and PDB ID 8J90 (DDM1-nucleosome complex) for the AtDDM1-nucleosome complex, EMD-36084 (H2A nucleosome) and PDB ID 8J91 (H2A nucleosome) for the nucleosome containing AtH2A, and EMD-36085 (H2A.W nucleosome) and PDB ID 8J92 (H2A.W nucleosome) for the nucleosome containing AtH2A.W, respectively. The raw mass spectrometry data used in this study have been deposited to the proteomeXchange Consortium under accession code PXD043417 (Crosslinking mass spectrometry of DDM1 complexed with the nucleosome) via the Japan ProteOme STandard (JPOST) repository (JPST002218 (Crosslinking mass spectrometry of DDM1 complexed with the nucleosome))118. The structures of the nucleosome composed of the Widom 601 DNA sequence, human nucleosome core particle, Snf2-nucleosome complex, and DDM1-nucleosome complex used in this study can be found in the Protein Data Bank under the accession codes 3LZ0 (Nucleosome core particle composed of the Widom 601 DNA sequence), 7VZ4 (human nucleosome core particle), 5X0Y (Snf2-nucleosome complex), and 7UX9 (DDM1-nucleosome complex), respectively. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Akihisa Osakabe, Yoshimasa Takizawa
Contributor Information
Akihisa Osakabe, Email: akihisa-osakabe@g.ecc.u-tokyo.ac.jp.
Tetsuji Kakutani, Email: tkak@bs.s.u-tokyo.ac.jp.
Hitoshi Kurumizaka, Email: kurumizaka@iqb.u-tokyo.ac.jp.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-49465-w.
References
- 1.Cosby RL, Chang N-C, Feschotte C. Host-transposon interactions: conflict, cooperation, and cooption. Genes Dev. 2019;33:1098–1116. doi: 10.1101/gad.327312.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bourque G, et al. Ten things you should know about transposable elements. Genome Biol. 2018;19:199. doi: 10.1186/s13059-018-1577-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wicker T, et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 4.Gagnier L, Belancio VP, Mager DL. Mouse germ line mutations due to retrotransposon insertions. Mob. DNA. 2019;10:15. doi: 10.1186/s13100-019-0157-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kidwell MG, Lisch D. Transposable elements as sources of variation in animals and plants. Proc. Natl Acad. Sci. USA. 1997;94:7704–7711. doi: 10.1073/pnas.94.15.7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chuong EB, Elde NC, Feschotte C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science. 2016;351:1083–1087. doi: 10.1126/science.aad5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jangam D, Feschotte C, Betrán E. Transposable Element Domestication As an Adaptation to Evolutionary Conflicts. Trends Genet. 2017;33:817–831. doi: 10.1016/j.tig.2017.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Velanis CN, et al. The domesticated transposase ALP2 mediates formation of a novel Polycomb protein complex by direct interaction with MSI1, a core subunit of Polycomb Repressive Complex 2 (PRC2) PLoS Genet. 2020;16:e1008681. doi: 10.1371/journal.pgen.1008681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rishishwar L, et al. Evidence for positive selection on recent human transposable element insertions. Gene. 2018;675:69–79. doi: 10.1016/j.gene.2018.06.077. [DOI] [PubMed] [Google Scholar]
- 10.Maksakova IA, et al. Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLoS Genet. 2006;2:e2. doi: 10.1371/journal.pgen.0020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quadrana L, et al. The Arabidopsis thaliana mobilome and its impact at the species level. Elife. 2016;5:e15716. doi: 10.7554/eLife.15716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boissinot S, Davis J, Entezam A, Petrov D, Furano AV. Fitness cost of LINE-1 (L1) activity in humans. Proc. Natl Acad. Sci. USA. 2006;103:9590–9594. doi: 10.1073/pnas.0603334103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Payer LM, Burns KH. Transposable elements in human genetic disease. Nat. Rev. Genet. 2019;20:760–772. doi: 10.1038/s41576-019-0165-8. [DOI] [PubMed] [Google Scholar]
- 14.Déléris A, Berger F, Duharcourt S. Role of Polycomb in the control of transposable elements. Trends Genet. 2021;37:882–889. doi: 10.1016/j.tig.2021.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010;11:204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henderson IR, Jacobsen SE. Epigenetic inheritance in plants. Nature. 2007;447:418–424. doi: 10.1038/nature05917. [DOI] [PubMed] [Google Scholar]
- 17.Chan SW-L, Henderson IR, Jacobsen SE. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005;6:351–360. doi: 10.1038/nrg1601. [DOI] [PubMed] [Google Scholar]
- 18.Finnegan EJ, Dennis ES. Isolation and identification by sequence homology of a putative cytosine methyltransferase from Arabidopsis thaliana. Nucleic Acids Res. 1993;21:2383–2388. doi: 10.1093/nar/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao X, Jacobsen SE. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 2002;12:1138–1144. doi: 10.1016/S0960-9822(02)00925-9. [DOI] [PubMed] [Google Scholar]
- 20.Stroud H, et al. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014;21:64–72. doi: 10.1038/nsmb.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackson JP, Lindroth AM, Cao X, Jacobsen SE. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature. 2002;416:556–560. doi: 10.1038/nature731. [DOI] [PubMed] [Google Scholar]
- 22.Woo HR, Dittmer TA, Richards EJ. Three SRA-domain methylcytosine-binding proteins cooperate to maintain global CpG methylation and epigenetic silencing in Arabidopsis. PLoS Genet. 2008;4:e1000156. doi: 10.1371/journal.pgen.1000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawashima T, Berger F. Epigenetic reprogramming in plant sexual reproduction. Nat. Rev. Genet. 2014;15:613–624. doi: 10.1038/nrg3685. [DOI] [PubMed] [Google Scholar]
- 24.Loppin B, Berger F. Histone Variants: The Nexus of Developmental Decisions and Epigenetic Memory. Annu Rev. Genet. 2020;54:121–149. doi: 10.1146/annurev-genet-022620-100039. [DOI] [PubMed] [Google Scholar]
- 25.Talbert PB, Henikoff S. Histone variants at a glance. J. Cell Sci. 2021;134:jcs244749. doi: 10.1242/jcs.244749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Talbert PB, et al. A unified phylogeny-based nomenclature for histone variants. Epigenetics Chromatin. 2012;5:7. doi: 10.1186/1756-8935-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Talbert PB, Henikoff S. Histone variants on the move: substrates for chromatin dynamics. Nat. Rev. Mol. Cell Biol. 2017;18:115–126. doi: 10.1038/nrm.2016.148. [DOI] [PubMed] [Google Scholar]
- 28.Bonisch C, et al. Histone H2A variants in nucleosomes and chromatin: more or less stable? Nucleic Acids Res. 2012;40:10719–10741. doi: 10.1093/nar/gks865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hake SB, Allis CD. Histone H3 variants and their potential role in indexing mammalian genomes: the ‘H3 barcode hypothesis’. Proc. Natl Acad. Sci. USA. 2006;103:6428–6435. doi: 10.1073/pnas.0600803103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ingouff M, Berger FF. Histone3 variants in plants. Chromosoma. 2010;119:27–33. doi: 10.1007/s00412-009-0237-1. [DOI] [PubMed] [Google Scholar]
- 31.Kurumizaka H, Horikoshi N, Tachiwana H, Kagawa W. Current progress on structural studies of nucleosomes containing histone H3 variants. Curr. Opin. Struct. Biol. 2013;23:109–115. doi: 10.1016/j.sbi.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 32.Loyola A, Almouzni G. Marking histone H3 variants: how, when and why? Trends Biochem Sci. 2007;32:425–433. doi: 10.1016/j.tibs.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 33.Borg M, Jiang D, Berger F. Histone variants take center stage in shaping the epigenome. Curr. Opin. Plant Biol. 2021;61:101991. doi: 10.1016/j.pbi.2020.101991. [DOI] [PubMed] [Google Scholar]
- 34.Yelagandula R, et al. The histone variant H2A.W defines heterochromatin and promotes chromatin condensation in. Arabidopsis. Cell. 2014;158:98–109. doi: 10.1016/j.cell.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jamge B, et al. Histone variants shape chromatin states in Arabidopsis. Elife. 2023;12:RP87714. doi: 10.7554/eLife.87714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawashima T, et al. Diversification of histone H2A variants during plant evolution. Trends Plant Sci. 2015;20:419–425. doi: 10.1016/j.tplants.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Osakabe A, et al. Histone H2A variants confer specific properties to nucleosomes and impact on chromatin accessibility. Nucleic Acids Res. 2018;46:7675–7685. doi: 10.1093/nar/gky540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osakabe A, Molaro A. Histone renegades: Unusual H2A histone variants in plants and animals. Semin Cell Dev. Biol. 2023;135:35–42. doi: 10.1016/j.semcdb.2022.05.001. [DOI] [PubMed] [Google Scholar]
- 39.Bourguet, P. et al. The histone variant H2A.W cooperates with chromatin modifications and linker histone H1 to maintain transcriptional silencing of transposons in Arabidopsis. bioRxiv, 10.1101/2022.05.31.493688 (2022).
- 40.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi W, Kurumizaka H. Structural transition of the nucleosome during chromatin remodeling and transcription. Curr. Opin. Struct. Biol. 2019;59:107–114. doi: 10.1016/j.sbi.2019.07.011. [DOI] [PubMed] [Google Scholar]
- 42.Knizewski L, Ginalski K, Jerzmanowski A. Snf2 proteins in plants: gene silencing and beyond. Trends Plant Sci. 2008;13:557–565. doi: 10.1016/j.tplants.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 43.Noh Y-S, Amasino RM. PIE1, an ISWI family gene, is required for FLC activation and floral repression in Arabidopsis. Plant Cell. 2003;15:1671–1682. doi: 10.1105/tpc.012161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farrona S, Hurtado L, Bowman JL, Reyes JC. The Arabidopsis thaliana SNF2 homolog AtBRM controls shoot development and flowering. Development. 2004;131:4965–4975. doi: 10.1242/dev.01363. [DOI] [PubMed] [Google Scholar]
- 45.Hurtado L, Farrona S, Reyes JC. The putative SWI/SNF complex subunit BRAHMA activates flower homeotic genes in Arabidopsis thaliana. Plant Mol. Biol. 2006;62:291–304. doi: 10.1007/s11103-006-9021-2. [DOI] [PubMed] [Google Scholar]
- 46.Walley JW, et al. The chromatin remodeler SPLAYED regulates specific stress signaling pathways. PLoS Pathog. 2008;4:e1000237. doi: 10.1371/journal.ppat.1000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aichinger E, Villar CBR, Di Mambro R, Sabatini S, Köhler C. The CHD3 chromatin remodeler PICKLE and polycomb group proteins antagonistically regulate meristem activity in the Arabidopsis root. Plant Cell. 2011;23:1047–1060. doi: 10.1105/tpc.111.083352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song Z-T, Liu J-X, Han J-J. Chromatin remodeling factors regulate environmental stress responses in plants. J. Integr. Plant Biol. 2021;63:438–450. doi: 10.1111/jipb.13064. [DOI] [PubMed] [Google Scholar]
- 49.Zou B, et al. The Arabidopsis Chromatin-Remodeling Factor CHR5 Regulates Plant Immune Responses and Nucleosome Occupancy. Plant Cell Physiol. 2017;58:2202–2216. doi: 10.1093/pcp/pcx155. [DOI] [PubMed] [Google Scholar]
- 50.Huanca-Mamani W, Garcia-Aguilar M, León-Martínez G, Grossniklaus U, Vielle-Calzada J-P. CHR11, a chromatin-remodeling factor essential for nuclear proliferation during female gametogenesis in Arabidopsis thaliana. Proc. Natl Acad. Sci. USA. 2005;102:17231–17236. doi: 10.1073/pnas.0508186102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luo Y-X, et al. A plant-specific SWR1 chromatin-remodeling complex couples histone H2A.Z deposition with nucleosome sliding. EMBO J. 2020;39:e102008. doi: 10.15252/embj.2019102008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li D, et al. The ISWI remodeler in plants: protein complexes, biochemical functions, and developmental roles. Chromosoma. 2017;126:365–373. doi: 10.1007/s00412-017-0626-9. [DOI] [PubMed] [Google Scholar]
- 53.Mittelsten Scheid O, Afsar K, Paszkowski J. Release of epigenetic gene silencing by trans-acting mutations in Arabidopsis. Proc. Natl Acad. Sci. USA. 1998;95:632–637. doi: 10.1073/pnas.95.2.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kakutani T, Jeddeloh JA, Richards EJ. Characterization of an Arabidopsis thaliana DNA hypomethylation mutant. Nucleic Acids Res. 1995;23:130–137. doi: 10.1093/nar/23.1.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jeddeloh JA, Stokes TL, Richards EJ. Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat. Genet. 1999;22:94–97. doi: 10.1038/8803. [DOI] [PubMed] [Google Scholar]
- 56.Vongs A, Kakutani T, Martienssen RA, Richards EJ. Arabidopsis thaliana DNA methylation mutants. Science. 1993;260:1926–1928. doi: 10.1126/science.8316832. [DOI] [PubMed] [Google Scholar]
- 57.Kato M, Miura A, Bender J, Jacobsen SE, Kakutani T. Role of CG and non-CG methylation in immobilization of transposons in Arabidopsis. Curr. Biol. 2003;13:421–426. doi: 10.1016/S0960-9822(03)00106-4. [DOI] [PubMed] [Google Scholar]
- 58.Miura A, et al. Mobilization of transposons by a mutation abolishing full DNA methylation in. Arabidopsis. Nat. 2001;411:212–214. doi: 10.1038/35075612. [DOI] [PubMed] [Google Scholar]
- 59.Tsukahara S, et al. Bursts of retrotransposition reproduced in. Arabidopsis. Nat. 2009;461:423–426. doi: 10.1038/nature08351. [DOI] [PubMed] [Google Scholar]
- 60.Tan F, et al. DDM1 Represses Noncoding RNA Expression and RNA-Directed DNA Methylation in Heterochromatin. Plant Physiol. 2018;177:1187–1197. doi: 10.1104/pp.18.00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Singer T, Yordan C, Martienssen RA. Robertson’s Mutator transposons in A. thaliana are regulated by the chromatin-remodeling gene Decrease in DNA Methylation (DDM1) Genes Dev. 2001;15:591–602. doi: 10.1101/gad.193701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hirochika H, Okamoto H, Kakutani T. Silencing of retrotransposons in Arabidopsis and reactivation by the ddm1 mutation. Plant Cell. 2000;12:357–369. doi: 10.1105/tpc.12.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Osakabe A, et al. The chromatin remodeler DDM1 prevents transposon mobility through deposition of histone variant H2A.W. Nat. Cell Biol. 2021;23:391–400. doi: 10.1038/s41556-021-00658-1. [DOI] [PubMed] [Google Scholar]
- 64.Zemach A, et al. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell. 2013;153:193–205. doi: 10.1016/j.cell.2013.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lyons DB, Zilberman D. DDM1 and Lsh remodelers allow methylation of DNA wrapped in nucleosomes. Elife. 2017;6:e30674. doi: 10.7554/eLife.30674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bröhm A, et al. Methylation of recombinant mononucleosomes by DNMT3A demonstrates efficient linker DNA methylation and a role of H3K36me3. Commun. Biol. 2022;5:192. doi: 10.1038/s42003-022-03119-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schrader A, Gross T, Thalhammer V, Längst G. Characterization of Dnmt1 Binding and DNA Methylation on Nucleosomes and Nucleosomal Arrays. PLoS One. 2015;10:e0140076. doi: 10.1371/journal.pone.0140076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bourguet P, et al. The histone variant H2A.W and linker histone H1 co-regulate heterochromatin accessibility and DNA methylation. Nat. Commun. 2021;12:2683. doi: 10.1038/s41467-021-22993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 70.Johnson L, et al. Mass spectrometry analysis of Arabidopsis histone H3 reveals distinct combinations of post-translational modifications. Nucleic Acids Res. 2004;32:6511–6518. doi: 10.1093/nar/gkh992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang K, Sridhar VV, Zhu J, Kapoor A, Zhu J-K. Distinctive core histone post-translational modification patterns in Arabidopsis thaliana. PLoS One. 2007;2:e1210. doi: 10.1371/journal.pone.0001210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee SC, et al. Chromatin remodeling of histone H3 variants by DDM1 underlies epigenetic inheritance of DNA methylation. Cell. 2023;186:4100–4116.e15. doi: 10.1016/j.cell.2023.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kono H, Shirayama K, Arimura Y, Tachiwana H, Kurumizaka H. Two arginine residues suppress the flexibility of nucleosomal DNA in the canonical nucleosome core. PLoS One. 2015;10:e0120635. doi: 10.1371/journal.pone.0120635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mariño-Ramírez L, Kann MG, Shoemaker BA, Landsman D. Histone structure and nucleosome stability. Expert Rev. Proteom. 2005;2:719–729. doi: 10.1586/14789450.2.5.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Armeev GA, Kniazeva AS, Komarova GA, Kirpichnikov MP, Shaytan AK. Histone dynamics mediate DNA unwrapping and sliding in nucleosomes. Nat. Commun. 2021;12:2387. doi: 10.1038/s41467-021-22636-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hirai S, et al. Unusual nucleosome formation and transcriptome influence by the histone H3mm18 variant. Nucleic Acids Res. 2022;50:72–91. doi: 10.1093/nar/gkab1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu X, Li M, Xia X, Li X, Chen Z. Mechanism of chromatin remodelling revealed by the Snf2-nucleosome structure. Nature. 2017;544:440–445. doi: 10.1038/nature22036. [DOI] [PubMed] [Google Scholar]
- 78.Li M, et al. Mechanism of DNA translocation underlying chromatin remodelling by Snf2. Nature. 2019;567:409–413. doi: 10.1038/s41586-019-1029-2. [DOI] [PubMed] [Google Scholar]
- 79.Yan L, Wang L, Tian Y, Xia X, Chen Z. Structure and regulation of the chromatin remodeller ISWI. Nature. 2016;540:466–469. doi: 10.1038/nature20590. [DOI] [PubMed] [Google Scholar]
- 80.Brzeski J, Jerzmanowski A. Deficient in DNA methylation 1 (DDM1) defines a novel family of chromatin-remodeling factors. J. Biol. Chem. 2003;278:823–828. doi: 10.1074/jbc.M209260200. [DOI] [PubMed] [Google Scholar]
- 81.Liu Y, et al. Molecular basis of chromatin remodelling by DDM1 involved in plant DNA methylation. Nat. Plants. 2024 doi: 10.1038/s41477-024-01640-z. [DOI] [PubMed] [Google Scholar]
- 82.Clapier CR, Cairns BR. Regulation of ISWI involves inhibitory modules antagonized by nucleosomal epitopes. Nature. 2012;492:280–284. doi: 10.1038/nature11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mueller-Planitz F, Klinker H, Ludwigsen J, Becker PB. The ATPase domain of ISWI is an autonomous nucleosome remodeling machine. Nat. Struct. Mol. Biol. 2013;20:82–89. doi: 10.1038/nsmb.2457. [DOI] [PubMed] [Google Scholar]
- 84.Hwang WL, Deindl S, Harada BT, Zhuang X. Histone H4 tail mediates allosteric regulation of nucleosome remodelling by linker DNA. Nature. 2014;512:213–217. doi: 10.1038/nature13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ludwigsen J, et al. Concerted regulation of ISWI by an autoinhibitory domain and the H4 N-terminal tail. Elife. 2017;6:e21477. doi: 10.7554/eLife.21477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Felle M, et al. Nucleosomes protect DNA from DNA methylation in vivo and in vitro. Nucleic Acids Res. 2011;39:6956–6969. doi: 10.1093/nar/gkr263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dodonova SO, Zhu F, Dienemann C, Taipale J. & Cramer, P. Nucleosome-bound SOX2 and SOX11 structures elucidate pioneer factor function. Nature. 2020;580:669–672. doi: 10.1038/s41586-020-2195-y. [DOI] [PubMed] [Google Scholar]
- 88.Santisteban MS, Arents G, Moudrianakis EN, Smith MM. Histone octamer function in vivo: mutations in the dimer-tetramer interfaces disrupt both gene activation and repression. EMBO J. 1997;16:2493–2506. doi: 10.1093/emboj/16.9.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kruger W, et al. Amino acid substitutions in the structured domains of histones H3 and H4 partially relieve the requirement of the yeast SWI/SNF complex for transcription. Genes Dev. 1995;9:2770–2779. doi: 10.1101/gad.9.22.2770. [DOI] [PubMed] [Google Scholar]
- 90.Ni K, et al. LSH mediates gene repression through macroH2A deposition. Nat. Commun. 2020;11:5647. doi: 10.1038/s41467-020-19159-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ni K, Muegge K. LSH catalyzes ATP-driven exchange of histone variants macroH2A1 and macroH2A2. Nucleic Acids Res. 2021;49:8024–8036. doi: 10.1093/nar/gkab588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Douet J, et al. MacroH2A histone variants maintain nuclear organization and heterochromatin architecture. J. Cell Sci. 2017;130:1570–1582. doi: 10.1242/jcs.199216. [DOI] [PubMed] [Google Scholar]
- 93.Han M, et al. A role for LSH in facilitating DNA methylation by DNMT1 through enhancing UHRF1 chromatin association. Nucleic Acids Res. 2021;48:12116–12134. doi: 10.1093/nar/gkaa1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dunican DS, et al. Lsh regulates LTR retrotransposon repression independently of Dnmt3b function. Genome Biol. 2013;14:R146. doi: 10.1186/gb-2013-14-12-r146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Berger F, Muegge K, Richards EJ. Seminars in cell and development biology on histone variants remodelers of H2A variants associated with heterochromatin. Semin Cell Dev. Biol. 2023;135:93–101. doi: 10.1016/j.semcdb.2022.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Thijssen PE, et al. Mutations in CDCA7 and HELLS cause immunodeficiency-centromeric instability-facial anomalies syndrome. Nat. Commun. 2015;6:7870. doi: 10.1038/ncomms8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tanaka Y, et al. Expression and purification of recombinant human histones. Methods. 2004;33:3–11. doi: 10.1016/j.ymeth.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 98.Kujirai T, et al. Methods for Preparing Nucleosomes Containing Histone Variants. Methods Mol. Biol. 2018;1832:3–20. doi: 10.1007/978-1-4939-8663-7_1. [DOI] [PubMed] [Google Scholar]
- 99.Tachiwana H, et al. Structural basis of instability of the nucleosome containing a testis-specific histone variant, human H3T. Proc. Natl Acad. Sci. USA. 2010;107:10454–10459. doi: 10.1073/pnas.1003064107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Arimura Y, Tachiwana H, Oda T, Sato M, Kurumizaka H. Structural Analysis of the Hexasome, Lacking One Histone H2A/H2B Dimer from the Conventional Nucleosome. Biochemistry. 2012;51:3302–3309. doi: 10.1021/bi300129b. [DOI] [PubMed] [Google Scholar]
- 101.Kastner B, et al. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat. Methods. 2008;5:53–55. doi: 10.1038/nmeth1139. [DOI] [PubMed] [Google Scholar]
- 102.Zheng SQ, et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kimanius D, Dong L, Sharov G, Nakane T, Scheres SHW. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem J. 2021;478:4169–4185. doi: 10.1042/BCJ20210708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Scheres SHW. Processing of Structurally Heterogeneous Cryo-EM Data in RELION. Methods Enzymol. 2016;579:125–157. doi: 10.1016/bs.mie.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 106.Sanchez-Garcia R, et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 2021;4:874. doi: 10.1038/s42003-021-02399-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vasudevan D, Chua EYD, Davey CA. Crystal structures of nucleosome core particles containing the ‘601’ strong positioning sequence. J. Mol. Biol. 2010;403:1–10. doi: 10.1016/j.jmb.2010.08.039. [DOI] [PubMed] [Google Scholar]
- 108.Goddard TD, et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. doi: 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 110.Jumper J, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Croll TI. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D. Struct. Biol. 2018;74:519–530. doi: 10.1107/S2059798318002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Afonine PV, et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D. Struct. Biol. 2018;74:531–544. doi: 10.1107/S2059798318006551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liebschner D, et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D. Struct. Biol. 2019;75:861–877. doi: 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Leitner A, Walzthoeni T, Aebersold R. Lysine-specific chemical cross-linking of protein complexes and identification of cross-linking sites using LC-MS/MS and the xQuest/xProphet software pipeline. Nat. Protoc. 2014;9:120–137. doi: 10.1038/nprot.2013.168. [DOI] [PubMed] [Google Scholar]
- 116.Hatazawa S, et al. Structural basis for binding diversity of acetyltransferase p300 to the nucleosome. iScience. 2022;25:104563. doi: 10.1016/j.isci.2022.104563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Grimm M, Zimniak T, Kahraman A, Herzog F. xVis: a web server for the schematic visualization and interpretation of crosslink-derived spatial restraints. Nucleic Acids Res. 2015;43:W362–W369. doi: 10.1093/nar/gkv463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Okuda S, et al. jPOSTrepo: an international standard data repository for proteomes. Nucleic Acids Res. 2017;45:D1107–D1111. doi: 10.1093/nar/gkw1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM maps and atomic models in this study have been deposited in the Electron Microscopy Data Bank and the Protein Data Bank, under the accession codes EMD- 36083 (DDM1-nucleosome complex) and PDB ID 8J90 (DDM1-nucleosome complex) for the AtDDM1-nucleosome complex, EMD-36084 (H2A nucleosome) and PDB ID 8J91 (H2A nucleosome) for the nucleosome containing AtH2A, and EMD-36085 (H2A.W nucleosome) and PDB ID 8J92 (H2A.W nucleosome) for the nucleosome containing AtH2A.W, respectively. The raw mass spectrometry data used in this study have been deposited to the proteomeXchange Consortium under accession code PXD043417 (Crosslinking mass spectrometry of DDM1 complexed with the nucleosome) via the Japan ProteOme STandard (JPOST) repository (JPST002218 (Crosslinking mass spectrometry of DDM1 complexed with the nucleosome))118. The structures of the nucleosome composed of the Widom 601 DNA sequence, human nucleosome core particle, Snf2-nucleosome complex, and DDM1-nucleosome complex used in this study can be found in the Protein Data Bank under the accession codes 3LZ0 (Nucleosome core particle composed of the Widom 601 DNA sequence), 7VZ4 (human nucleosome core particle), 5X0Y (Snf2-nucleosome complex), and 7UX9 (DDM1-nucleosome complex), respectively. Source data are provided with this paper.