

Abstract
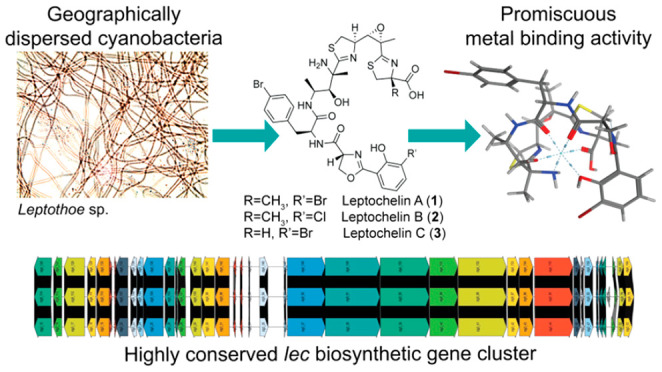
Metals are important cofactors in the metabolic processes of cyanobacteria, including photosynthesis, cellular respiration, DNA replication, and the biosynthesis of primary and secondary metabolites. In adaptation to the marine environment, cyanobacteria use metallophores to acquire trace metals when necessary as well as to reduce potential toxicity from excessive metal concentrations. Leptochelins A–C were identified as structurally novel metallophores from three geographically dispersed cyanobacteria of the genus Leptothoe. Determination of the complex structures of these metabolites presented numerous challenges, but they were ultimately solved using integrated data from NMR, mass spectrometry and deductions from the biosynthetic gene cluster. The leptochelins are comprised of halogenated linear NRPS-PKS hybrid products with multiple heterocycles that have potential for hexadentate and tetradentate coordination with metal ions. The genomes of the three leptochelin producers were sequenced, and retrobiosynthetic analysis revealed one candidate biosynthetic gene cluster (BGC) consistent with the structure of leptochelin. The putative BGC is highly homologous in all three Leptothoe strains, and all possess genetic signatures associated with metallophores. Postcolumn infusion of metals using an LC-MS metabolomics workflow performed with leptochelins A and B revealed promiscuous binding of iron, copper, cobalt, and zinc, with greatest preference for copper. Iron depletion and copper toxicity experiments support the hypothesis that leptochelin metallophores may play key ecological roles in iron acquisition and in copper detoxification. In addition, the leptochelins possess significant cytotoxicity against several cancer cell lines.
Introduction
Microorganisms have a remarkable ability to sense and adapt to their environment. As a result, they are ubiquitous and inhabit some of the most extreme habitats on Earth, from deep sea hydrothermal vents, desert crusts, high latitude ice floes, to acid lakes containing toxic levels of dissolved metals.1−4 This capacity for adaptation to diverse environmental conditions is also a characteristic of one of the most ancient groups of microorganisms, the Cyanobacteria. Arising some 2.5 billion years ago, these organisms have evolved specialized capacities to thrive in such diverse situations as sun-exposed tropical reef systems, concrete building walls, terrestrial hydrothermal vents, and oligotrophic ocean waters.5−7 Cyanobacteria have requirements for enzymatic metal cofactors that are in very limited supply in oligotrophic environments. To flourish in these habitats, they and other microorganisms produce a variety of small molecule natural products, known generally as metallophores, that assist in their acquisition of diverse metal ions required for enzymatic catalysis.8 Most metallophores exhibit tetradentate or hexadentate coordination when binding metal ions with high affinity. The best studied class of acquisition metallophores are those with a high-affinity for iron(III), known as siderophores.9 There are several classifications of siderophores based on the structural groups involved in metal ion coordination, including hydroxymates such as putrebactin and bisucabactin, catecholates such as bacillibactin, enterobactin, and vibriobactin, phenolates such as yersiniabactin and pyochelin, carboxylates such as staphyloferrins, and mixed type such as pyoverdine and aerobactin.10 Examples of previously reported acquisition metallophores from cyanobacteria include schizokinen and the synechobactins, both of which are hydroxamate metallophores, and anachelin, a mixed type metallophore.11−13
In environments where metal ion concentrations reach levels that would normally be toxic, microorganisms produce small molecules that are protective against this toxicity; examples are frankobactin, which was obtained from the actinobacterium Frankia sp. CH37, and a variety of metallothioneins isolated from diverse bacteria, including the cyanobacterium Synechococcus PCC 7942.14,15 The capacity of cyanobacteria to resist metal toxicity has been observed previously, especially for copper and iron.16−18
In the present work, we characterize the intriguingly complex structures of leptochelins A–C as novel metallophores produced by three strains of Leptothoe cyanobacteria (Figure 1). The leptochelins collectively possess remarkable selectivity for copper chelation and are proposed to allow Leptothoe species to persist in conditions of elevated copper ion concentrations. At the same time, promiscuous metal binding by leptochelins could enable Leptothoe cyanobacteria to thrive in conditions where essential metal cofactors are limiting. These unique leptochelin molecules are also potently toxic to mammalian cancer cells, and thus may have therapeutic potential.
Figure 1.

Structures of leptochelins A–C (1–3). Based on comparisons of 13C NMR chemical shifts (SI Appendix, Tables S5 and Figure S44), the absolute configurations for leptochelin B (2) and leptochelin C (3) are proposed to be the same as those of leptochelin A (1) at comparable centers. The configuration at C2 of compound 3, predicted to result from incorporation of l-cysteine, likely results in R configuration at this position.
Results
Geographically Dispersed Collections of Leptothoe
Living cultures of three strains of cyanobacteria of the genus Leptothoe (SI Appendix, Table S1) were collected over a 25-year period from geographically dispersed regions [Sulawesi, Indonesia (in 1994), El Aruk, Egypt (in 2007), and Baía das Gatas, Republic of Cape Verde (in 2018)], and were found to produce the same series of structurally unique natural products, named here as the leptochelins. The first two of these collections were subtidal and made using SCUBA whereas the latter was collected by hand from shallow tide pools. Despite the geographical dispersion and littoral versus benthic nature of the collection sites, the cyanobacterial cultures all produced the same or a very similar suite of leptochelin natural products, suggesting that their production is highly conserved, and the compounds possess important adaptive roles.
All three strains of Leptothoe examined in this study possessed thin filaments, were unbranched and nonheterocystous, and formed a pinkish-red to brownish-red biofilm in laboratory cultures, consistent with the description by Konstantinou et al. 2019.19 Phylogenetic analysis of the 16S rRNA gene (SI Appendix, Figure S1) revealed that these three cyanobacterial strains all belong to the recently described genus Leptothoe(19) and share 99.9%–100% sequence identity of this key phylogenetic gene (SI Appendix, Table S2). This genus comprises strains obtained from marine environments (supported by a bootstrap value of 100%) around the world. More distantly, the phylogenetic clade includes the genera Rhodoploca, Salileptolyngbya, Cymatolege, Halomicronema and Nodosilinea, which have less than 94% 16S rRNA sequence similarity to Leptothoe.
Isolation and Planar Structures of Leptochelins A–C (1–3)
Interestingly, laboratory cultures of all three Leptothoe strains produced leptochelins A–C (1–3). Although they were cultured in three different laboratories, the common general strategy for isolation of the leptochelins included extraction using CH2Cl2:MeOH, followed by an initial separation using normal phase chromatography on silica gel as the stationary phase, followed by repeated iterations of reversed phase High Performance Liquid Chromatography (HPLC) using C18 as the stationary phase. Zinc-bound leptochelins A–C were also isolated in parallel with leptochelins, especially when MS-grade water was not used in the HPLC purifications.
Pure leptochelin A (1), isolated as an optically active off-white amorphous solid [α]24D −89.1 (c 0.1, CH2Cl2), possessed a monoisotopic mass of m/z 895.0790 [M + H]+ by high resolution ESIMS. While the isotope pattern suggested two halogen atoms (either 2 Br, or 1 Br and 1 Cl), the mass accuracy of the Thermo QExactive (∼1 ppm) did not allow for unambiguous calculation of the molecular formula. However, a 21 T Fourier transform ion cyclotron resonance (21 T FT-ICR) mass spectrometer permitted resolution of the isotopic fine structure and resultant high-confidence elemental composition for 1 as C35H41Br2N6O8S2+, matching the experimentally observed mass with 0.1 ppm mass accuracy. This molecular formula indicated that leptochelin A (1) possessed 18 degrees of unsaturation, and by NMR analysis these were present as two substituted phenyl rings and six ester/amide/carboxyl-type carbons with chemical shifts between δC 165–187, leaving the presence of four additional rings to be determined. HMBC correlations established one of the rings as a trisubstituted epoxide (δC 61.5 qC and 64.4 CH). A combination of NMR chemical shift and 2D NMR data were subsequently used to construct 6 partial structures, A–F (Figure 2), which accounted for all atoms in the molecular formula as well as the three remaining rings.
Figure 2.
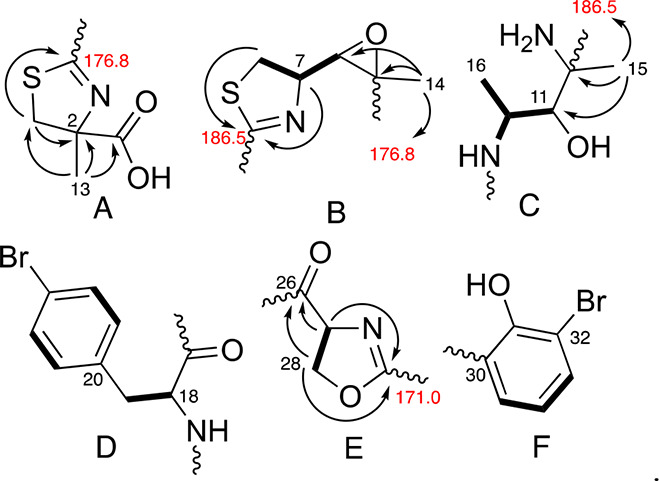
Partial structures A–F determined by 1H, 13C and 2D NMR experiments for leptochelin A (1). Bolded bonds show connectivity deduced by 1H–1H COSY and arrows depict selected 1H–13C HMBC correlations.
Partial structure A (Figure 2) possessed a distinctive singlet methyl group with a shift of δH 1.46 that exhibited 1H–13C HMBC correlations to a nonprotonated carbon signal at δC 84.4, an isolated methylene at δC 41.8 and a deshielded carboxylic acid carbon signal at δC 178.9. The terminal nature of this carboxyl group was demonstrated by treatment of compound 1 with diazomethane (CH2N2), resulting in the observation of a new dibromo isotopic cluster [M]+/[M + 2]+/[M + 4]+ at m/z 909.19/911.17/913.08 displaying a series of fragments diagnostic for methylation at the carboxyl terminus of compound 1 to yield compound 4 (SI Appendix, Table S8 and Figure S5). The methylene chemical shift (δC 41.8) was consistent with its attachment to a sulfur atom. This methylene also showed HMBC correlations to the nonprotonated carbon signal δC 85.4 and a deshielded carbon signal (δC 176.8) that was assigned as a nonprotonated carbon attached by a single bond to sulfur, a double bond to nitrogen, and a final single bond to a distal carbon atom. As a result, this constellation of atoms was formulated as a thiazoline ring with α-methyl and α-carboxylic acid functionalities (Figure 2A, partial structure A).
In partial structure B (Figure 2B), a −CH2–CH–CH– motif was defined by 1H–1H COSY correlations. The methylene group possessed 1H and 13C NMR shifts consistent with an attached sulfur atom while the central methine had shifts indicative of an attached nitrogen atom. HMBC correlations from H-7 and H-8b to a downfield carbonyl-like shifted resonance (C-9) identified this as a disubstituted thiazoline ring. The terminal methine of this motif was the protonated component of the trisubstituted epoxide described above. 1H–13C HMBC correlations from H3-14 to C-5 and C-6 indicated that the nonprotonated carbon of the epoxide bore a methyl group (δH 1.57).
The delineation of partial structure C (Figure 2C) commenced with a methyl doublet resonance coupled to a midfield methine multiplet at δH 4.51 that was HSQC-correlated with a 13C resonance at δC 50.2. These midfield chemical shifts implied a N-bound methine that was confirmed by observing homonuclear coupling between the methine multiplet and an NH doublet at δH 10.08 (3JH,H = 7.7 Hz). The δH 4.51 multiplet was also coupled to an oxymethine 1H doublet (δH 3.83, δC 77.9). A methyl singlet at δH 1.53 displayed HMBC correlations to the oxymethine 13C resonance, as well as to two carbon atoms without attached hydrogen atoms, one resonating at δC 186.5 (C-9, part of the thiazoline ring system in partial structure B discussed above) and a second midfield 13C signal at δC 62.7, consistent with its attachment to another nitrogen atom.
One of the two phenyl rings was 1,4-disubstituted as determined by a pair of 2H doublets at δH 7.22 (H-21/25) and δH 7.46 (H-22/24). A benzylic CH2 moiety was indicated by reciprocal HMBC correlations between the diastereotopic H2-19 signals and C-21/25 and H-21/25 and C-19. The para substituent was assigned as Br by virtue of a characteristically shielded 13C chemical shift of δC 121.0. Homonuclear coupling was also observed between H2-19 and the adjacent H-18 methine (δH 4.40, δC 57.8), for which the midfield 13C NMR shift, and an HMBC correlation from H-18 to an amide type carbonyl at δC 175.2 (C-17), indicated an alpha amino acid motif. Taken together, these moieties constituted a para-bromo-phenylalanine residue (Figure 2D, partial structure D).
Partial structure E (Figure 2) possessed a motif of −O–CH2–CH–N–, in which the oxymethylene was supported by a notably deshielded methylene 13C NMR shift (CH2-28, δC 69.4, δH 4.33 and 4.45), while the comparable methine chemical shifts of CH-27 were consistent with an attached nitrogen atom (δC 67.4, δH 4.57). HMBC correlations from both H-27 and H2-28 led to assignment of an amide-type carbonyl C-26 (δC 173.0) as a substituent on C-27. The H-27 and H-28 protons also showed HMBC correlations to a second deshielded signal at δC 171.0, which allowed formulation of this oxazoline partial structure by comparison with literature values.12,20
For the second phenyl ring (Figure 2F, partial structure F), a 1,2,3-trisubstitution pattern was interpreted from the mutually coupled 1H NMR spin system with relatively large ortho-couplings between protons (7.9 Hz). Based on 13C NMR chemical shifts, hydroxy (δC 165.0), bromo (δC 111.2) and carbon (δC 118.0) substituents were inferred. HMBC correlations from H-34 to C-30/C-32 and H-33 to C-31/C-35 placed the oxygenated carbon between the brominated and carbon-substituted positions (Table 1).
Table 1. NMR Data for Leptochelin A (1) (CDCl3, 400 MHz)a.
| Position | δC, mult | δH, mult | HMBC | ROESY |
|---|---|---|---|---|
| 1 | 178.9, C | |||
| 2 | 85.4, C | |||
| 3a | 41.8, CH2 | 3.43, d (11.7 Hz) | 2, 4, 13 | 3b, 13 |
| 3b | 3.58, d (11.7 Hz) | 1, 2, 13 | 3a | |
| 4 | 176.8, C | |||
| 5 | 61.5, C | |||
| 6 | 64.4, CH | 3.68, d (7.8 Hz) | 7, 8 | 14 |
| 7 | 75.4, CH | 3.93, ovlp | 6, 9 | 8a, 8b, 16 |
| 8a | 37.8, CH2 | 3.72, ovlp | 6, 7 | 7 |
| 8b | 3.88, ovlp | 6, 7, 9 | 7 | |
| 9 | 186.5, C | |||
| 10 | 62.7, C | |||
| NH2-10 | not observed | |||
| 11 | 77.9, CH | 3.83, bs | 12, 15, 16 | |
| 12 | 50.2, CH | 4.51, ovlp | 11, 16, NH-12 | |
| NH-12 | 10.08, d (7.7 Hz) | 12, 16, 19a, 18 wk | ||
| 13 | 22.9, CH3 | 1.46, s | 1, 2, 3 | 3a, 27 |
| 14 | 21.1, CH3 | 1.57, s | 4, 5, 6 | ovlp |
| 15 | 31.1, CH3 | 1.53, s | 9, 10, 11 | ovlp |
| 16 | 15.4, CH3 | 1.14, d (7.4 Hz) | 11, 12 | 7, 11, 12, NH-12 |
| 17 | 175.2, C | |||
| 18 | 57.8, CH | 4.40, ovlp | 19a, 19b, 21/25 | |
| NH-18 | 8.06, d (7.0 Hz) | 18, 19a, 21/25, 27, NH-18, 28b wk, 19b wk | ||
| 19a | 37.1, CH2 | 2.91, dd (14.3, 11.5 Hz) | 17, 18, 20, 21/25 | 21/25, NH-18 |
| 19b | 3.24, dd (14.3, 4.1 Hz) | 18, 20, 21/25 | 18, 21/25 | |
| 20 | 135.7, C | |||
| 21/25 | 130.9, CH | 7.22, d (8.3 Hz) | 19, 22/24, 23 | 18, 19a, 19b, 22/24, |
| 22/24 | 131.9, CH | 7.46, d (8.3 Hz) | 20, 21/25,23 | 21/25 |
| 23 | 121.0, C | |||
| 26 | 173.0, C | |||
| 27 | 67.4, CH | 4.57, ovlp | 13, 28a, 28b, NH-18 | |
| 28a | 69.4, CH2 | 4.33, t (7.8 Hz) | 26, 27, 29 | 27 |
| 28b | 4.45, ovlp | 26, 27, 29 | 13 wk | |
| 29 | 171.0, C | |||
| 30 | 118.0, C | |||
| 31 | 165.0, C | |||
| 32 | 111.2, C | |||
| 33 | 137.5, CH | 7.58, dd (7.9, 1.8 Hz) | 30, 31, 35 | 34 |
| 34 | 112.8, CH | 6.29, t (7.9 Hz) | 30, 32 | 33, 35 |
| 35 | 130.2, CH | 7.61, dd (7.9, 1.8 Hz) | 29, 33 | 34 |
ROESY spectrum was obtained in CDCl3 on a 500 MHz instrument. Note: “ovlp” denotes signal overlap and “wk” indicates a weak but notable correlation. 15N NMR shifts from a 1H–15N gHMBC experiment (500 MHz, CDCl3) (SI Appendix, Figure S13): δN 285.8 (N-2), 122.5 (NH-18), 119.5 (NH-12), 37.9 (NH2-10). N-27 not observed.
Partial structures A–F (Figure 2) were assembled into an overall linear structure using a combination of NMR (Table 1) and MS2 data (Figure 3). Partial structures A and B were connected by observation of HMBC correlations between H3-14 and the deshielded imino carbon C-4. In turn, H3-15 in partial structure C was correlated to the downfield thiazoline carbon of partial structure B, connecting these two through a C-9/C-10 bond. ROESY correlations between highly deshielded NH-17 to several resonances (H-18, H-19a, H-19b, H-21/25) associated with the bromophenylalanine residue of partial structure D connected these moieties. Partial structures D and E could be connected by HMBC correlations between α-proton H-18 and carbonyl C-26, as well as ROESY correlations between H-27 and NH-26. Finally, connection of remaining partial structure F was enabled by an HMBC correlation observed between H-35 and C-29.
Figure 3.

Exact mass, isotope pattern and MS2 fragmentation patterns for leptochelins A (1) and B (2). (A, B) 21 T FT-ICR spectra showing the isotopic fine structure for the monoisotopic molecular ions of leptochelin A (1) and leptochelin B (2) compared with the calculated values. (C) MS2 fragments deriving from fragment mass m/z 851 that results from neutral loss of CO2 from leptochelin A (1), with fragments from HCD in black and CID in red. Roman numerals refer to fragmentation tree depicted in panel E. (D) MS2 fragments deriving from fragment mass m/z 807 that results from neutral loss of CO2 from leptochelin B (2), with fragments from HCD in black and CID in red. Roman numerals refer to fragmentation tree depicted in panel E. (E) Depiction of proposed fragmentations for leptochelins A (1) and B (2). Proposed mechanisms for fragmentation of leptochelin A (1) and leptochelin B (2) are indicated by the blue arrows within the gray box in panel E.
A range of MS2 and MS3 experiments were obtained from higher-energy C-trap dissociation (HCD) using a QExactive orbitrap MS; collision-induced dissociation (CID) with a 21 T FT-ICR; and CID and HCD with an Orbitrap Elite MS (Figure 3D). With the precursor molecular formulas in hand, the mass differences in the MS2 spectra were calculated and MS3 fragmentation series were developed (SI Appendix, Figure S14) with calculated product ion molecular formulas. Using SIRIUS and MSFinder, we constructed fragmentation trees for the different activation methods and derivatives (SI Appendix, Figure S3 and Figure S4). Characteristic b-ion fragments were observed for the brominated salicylate unit. An internal C5H10NO+ fragment corresponded to neutral losses between major b- and y-ion fragments, indicating that this residue was located between the bromophenylalanine and thiazole moieties. With most ion molecular formulas identified, a fragmentation series of b- and y-ions was proposed (Figure 3E). Notably, a diaminopropionyl moiety in the fragmentation series was incongruent with the NMR derived structure. Guided by the NMR data, the diaminopropionyl moiety is proposed to derive from the oxazoline residue and the C-10 amino group. We hypothesize that a gas-phase transamination rearrangement between the C-10 amino group and the oxazoline occurs prior to MS2 and MS3 fragmentation (Figure 3E, products 3–8), which corresponds to the downstream b- and y-ion fragmentation series. Overall, this analysis provides additional support for the identity and sequence of residues that comprise leptochelin A (1).
Leptochelin B (2) analyzed for m/z 851.1294 for the [M + H]+, and by 21 T FT-ICR MS analysis, the nature of the halogen atoms was unambiguously resolved (Figure 3B), yielding a molecular formula of C35H41BrClN6O8S2. NMR analysis (SI Appendix, Tables S3–S6) of 2 yielded nearly identical spectra to 1, with small differences observed for the halogenated salicylate group (e.g., H-33 to H-35). From these data along with the comparable molecular formula to leptochelin A, we reasoned that the bromosalicylate group in 1 was replaced by a chlorosalicylate residue in 2. Again, using a range of MS2 and MS3 experiments as described above (Figure 3C), a fragmentation pathway highly comparable to that for 1 was observed for 2 (SI Appendix, Figure S41 and Figure S42). Notably, the sequence of fragments for 2 differed from the sequence for 1 by −44.451 amu, the mass difference between 35Cl and 79Br. Thus, leptochelin B (2) was assigned as the chlorosalicylate equivalent of leptochelin A (1), and by its co-occurrence, we propose it to be of the same absolute configuration, as described below.
Leptochelin C (3) displayed a prominent [M + H]+ ion peak by (+)-HRMS at m/z 881.0643, indicative of a molecular formula of C34H39Br2N6O8S2. Since the NMR data for compound 3 was very similar to those of 1 and 2, an analogous elucidation strategy was used to assign its structure (SI Appendix, Tables S3–S6). The mass difference of 14.01 Da between compounds 1 and 3 suggested the absence of a methyl group in leptochelin C (3). This variation was confirmed in the terminal thiazoline of 3, for which the α-carbon C-2 (δC 78.4), was protonated (δH 5.16, dd, J = 11.7, 9.3 Hz) instead of being fully substituted, and resonating at a more shielded chemical shift than C-2 in 1 (ΔδC = 7.0 ppm). The 1H–1H COSY correlations between H-2 and the diastereotopic protons H-3a/H-3b (δH 3.67; 3.83), together with HMBC correlations to the C-1 carboxylic acid carbon (δC 176.7) and the methylene carbon C-3 (δC 35.0) confirmed the proposed planar structure of leptochelin C (3).
The Leptochelin (lec) Biosynthetic Gene Cluster, Biosynthesis, and Genetic Insights on Configuration
To further confirm the structure of leptochelin A and explore its biosynthetic assembly process, we set out to identify the candidate genes responsible for its production. The biosynthetic gene clusters (BGCs) responsible for siderophore and other metallophore production often arise from nonribosomal peptide synthetases (NRPSs) and NRPS-independent siderophore synthetases (NISs), and these are frequently associated with distinct transcriptional and other regulatory genes, such as TonB transporters and AraC transcriptional regulators.10,21−23 The isolated DNA from Leptothoe sp. ISB3NOV94-8A was sequenced using Nanopore and Illumina technologies, and a hybrid assembly resulted in eight contigs with a total length of 8.51 Mbp; the longest contig was 7,062,414 bp and the GC content was 47.4%. The isolated DNA from Leptothoe sp. EHU-05/26/07-4 was sequenced using Nanopore technology, and the resulting 8.85 Mbp assembly was comprised of 11 contigs and the GC content was 47.4%. The final genome, Leptothoe sp. LEGE181152, also had a GC content of 47.4%, and was sequenced using Illumina technology, resulting in an 8.36 Mbp assembly comprised of 68 contigs (SI Appendix, Table S7). Genetic signatures of siderophore production as well as a retrobiosynthetic analysis were used to screen the three cyanobacterial genomes for a candidate leptochelin BGC (lec BGC). Of the 18 BGCs identified in the genome assembly for Leptothoe sp. ISB3NOV94-8A, three were NRPS-PKS hybrid systems and only one contained the requisite isochorismate synthase needed for biosynthesis of the salicylate group found in the leptochelins. This hybrid NRPS-PKS pathway (Figure 4A) had excellent congruence with the retrobiosynthetic scheme for leptochelin A (SI Appendix, Figure S45) as well as regulation and transport genetic signatures for siderophore production. A nearly identical hybrid PKS-NRPS BGC was present in the genomes of both Leptothoe sp. EHU-05/26/07–4 and Leptothoe sp. LEGE181152, and all three showed remarkable synteny and a high degree of nucleotide identity for each gene (98–100%). This is notable as the three collections were obtained from geographically dispersed regions (Celebes Sea, South Pacific Ocean; Red Sea, Indian Ocean; Republic of Cabo Verde, Eastern Mid-Atlantic Ocean) over a 30-year period (Figure 4B, SI Appendix, Table S9). Such a profound preservation of the BGC and secondary metabolite production despite the divergent temporal and geographic distribution of the three collections indicates that the leptochelins must confer a strong adaptive advantage. Additionally, modules 5 and 7 within the cluster appear to be the result of a gene duplication event with neofunctionalization (amino acid percent identity of 48% and similarity of 63%). Also present in the lec BGC are two genes for AraC family transcriptional regulators and a TonB-dependent receptor transporter domain (PF07715.17), which provide insight into how the lec BGC is transcriptionally regulated as well as how the expressed molecule is transported through the cell membrane.21−23
Figure 4.

Putative lec BGC for the biosynthesis of the leptochelins. A. The 71.6 kbp gene cluster (54 kbp of which represents core biosynthetic genes) is an NRPS-PKS hybrid system that begins with a cassette containing an isochorismate synthase followed by a series of NRPS and PKS modules to form the elongated chain. A proposed trans-acting halogenase is likely responsible for at least one of the halogenations seen on the aromatic rings. Additionally, a proposed trans-acting cytochrome P450 is likely responsible for one or two epoxidations (C-5–C-6 and possibly C-10–C-11). A TonB-dependent transporter associated with metallophore-specific transport and several regulatory proteins are encoded. Also encoded are RTX calcium-binding nonapeptide repeats (PF00353.21) (ctg4_128), a sulfite exporter with homology to TauE and SafE (PF01925.21) (ctg4_129), an oxoacyl-ACP synthase III (ctg4_130), chlorophyllase (ctg4_133), along with several hypothetical proteins which fall within the designated gene neighborhood. In the schematic above, dark blue arrows represent core biosynthetic genes. Green arrows represent genes with ancillary functions (e.g., transcriptional regulation). White arrows represent genes that encode hypothetical proteins. The light blue arrow represents a gene associated with transport, in this case the TonB-dependent transporter. B. Clinker analysis of the putative lec BGC from the assembled genomes of the three leptochelin producers showing high synteny and near 100% identity between the BGCs. Similarities between genes are indicated by the shaded links between the genes, where the gradient ranges from 0% identity (white) to 100% identity (black). The figure shows only links for genes with >50% identity, which in the case of the three putative leptochelin BGCs is at least 94% for all of the core biosynthetic and tailoring genes (SI Appendix, Table S9). Intergenic regions are shown by lines without arrows and without links.
The first complete biosynthetic module of the lec BGC (Figure 4A, module 1) encodes a cassette consistent with the formation of salicylic acid, and includes an isochorismate synthase, chorismate binding enzyme, and salicylate synthase, along with a benzoate-CoA ligase that is proposed to tether the salicylate to a carrier protein via a phosphopantetheinyl arm. The second AraC family transcriptional regulator is then encoded before a series of modules responsible for production of the remaining sections of leptochelin A (1). In module 2, an adenylation domain specific for serine (cysteine) is followed by a C-methyl transferase (cMT) that likely serves as a methyltransferase-like epimerase (MTe) (see below).24,25 This is followed by a heterocyclic condensation domain that forms the oxazoline ring from the tethered serine and salicylate residues. A hydrophobic amino acid, phenylalanine, is subsequently incorporated into module 3. It is uncertain if this is incorporated as bromophenylalanine, or if the bromination occurs later in the assembly process. Next, an alanine subunit is incorporated followed by elongation with acetate from an PKS module. This latter module contains a C-methyltransferase domain, placing a methyl group at the α-position between the two transient carbonyl functionalities. The β-carbonyl is next reduced to a hydroxy group by a ketoreductase, which may be transformed to an intermediate 10,11-epoxy species that is subsequently opened by an ammonolysis reaction.26 In module 6, an adenylation domain with specificity for cysteine (SI Appendix, Table S10) is paired with a heterocyclic condensation domain, producing a thiazoline ring. This is followed by another PKS elongation in module 7 that also adds a methyl group to the α-position from a C-methyl transferase. This module again contains a ketoreductase that results in a C-6 hydroxy group, and then this is subsequently transformed into a 5,6-epoxide, possibly by action of the adjacent CyP450 gene. This CyP450 may be responsible for both the enzymatic epoxidation proposed between C-10 and C-11 and the observed epoxidation between C-5 and C-6. A second and possibly trans acting tryptophan halogenase is encoded adjacent to the CyP450. This may be responsible for both aromatic ring halogenations27,28 (the salicylic acid monomer and the benzyl ring of phenylalanine) resulting in dibromination for leptochelins A (1) and C (3), and in monobromination and monochlorination for leptochelin B (2). Alternatively, a second currently unrecognized trans acting halogenase may be present in the genome. The final elongation step is encoded by NRPS module 8 in which cysteine is incorporated followed by α-methylation from SAM and heterocyclization to form a methyl- and carboxyl-substituted thiazoline ring in leptochelins A (1) and B (2). These core biosynthetic genes terminate with a thioesterase (TE) domain that is proposed to hydrolytically liberate the completed leptochelin molecule. A phylogenetic analysis of hydrolyzing versus cyclizing TEs from cyanobacteria indicates that the leptochelin TE catalyzes hydrolysis, consistent with the results of CH2N2 treatment of leptochelin A (1) to produce methyl leptochelin A (4), and providing another level of confirmation that the leptochelins have an overall linear architecture (SI Appendix, Figure S46).
Stereochemical Insights from the Putative Leptochelin BGC and Marfey’s Analysis
Informatic analyses of the biosynthetic gene cluster provided stereochemical insights at 7 of the 9 asymmetric centers in leptochelin A (1). In module 2, the first amino acid monomer, l-serine, is selected and undergoes heterocyclization and epimerization from a MTe domain that would result in R-configuration at C-27.24,25 This is consistent with the Marfey’s analysis that established the presence of d-serine in the hydrolysis product of 1 (SI Appendix, Figure S47). The condensation domains in modules 3 and 4 specify for the incorporation of l-phenylalanine and l-alanine, corresponding with S configurations at C-12 and C-18. Marfey’s analysis of hydrolyzed 1, in comparison with authentic standards, confirmed the S configuration of the bromophenylalanine residue (SI Appendix, Figure S48). The ketoreductases in modules 5 and 7 both have active site predictions for l-hydroxy products at C-11 and C-6, and transiently, an l-methyl at C-10 with the KR in module 5 identified as a C2-Type.29 The result is an S configuration at C-11, and predicting that there is no alteration of the C-6 configuration upon conversion to a 5,6-epoxide by a trans-acting CypP450 enzyme, an S-configuration at C-6. The relative configuration of C-6 and the fully substituted C-5 center was demonstrated by ROESY analysis to be cis, and thus the C-5 center is also predicted to be of S configuration. There are no epimerases present in the sequences for modules 6 or 8, and the adenylation domain specificities have consensus predictions for l-cysteine in both cases, resulting in an initial assignment of R configurations for C-7 and C-2; however, subsequent methylation at C-2 by a cMT make its absolute configuration uncertain from this biosynthetic analysis. Marfey’s analysis after sequential ozonolysis (SI Appendix, Figure S49) and hydrolysis of 1, revealed the presence of 2-Me-l-cysteic acid in the reaction product, supporting an R configuration for C-2, and implying that the C-methylation event occurs with retention of configuration. The S configuration at C-10 was inferred from observation of NOE correlations between H3-15/H-11, H3-16/H-8, H3-16/H-11, H-12/H-11, combined with an intense HMBC correlation from H-12 to C-10 (SI Appendix, Figure S50). These various chemical, spectroscopic and bioinformatic analyses support a 2R,5S,6S,7R,10S,11S,12S,18S,27R configuration for leptochelin A (1).
Discovery of Chelating Properties of Leptochelin A (1) and Subsequent Metal-Based Culture Studies to Increase Leptochelin Production and Measure Strain Resilience
The leptochelins were isolated over a span of 25 years from three geographically distinct regions. While leptochelin A (1) was first isolated and identified around 2001 from the Indonesian strain (Leptothoe sp. ISB3NOV94-8A), it initially appeared absent from the Red Sea strain (Leptothoe sp. EHU-05/26/07-4). Instead, the Red Sea strain contained only the brominated macrolide, phormidolide,30−32 which was also isolated from Leptothoe sp. ISB3NOV94-8A. However, a careful inspection of the (+)-HRESIMS isotope pattern for a minor metabolite (m/z 956.9924) from the Red Sea isolate led to the discovery of a zinc complexed form of leptochelin A (SI Appendix, Figure S43). A notable difference between the culture media for these two samples was the inclusion of an uncharacterized soil extract that was used in the Red Sea strain culture medium to enrich and potentially induce the production of secondary metabolites.33 Based on these observations, in addition to the presence of iron, copper and cobalt-complexed leptochelins in subsequent LCMS analyses, several metal-based culture experiments were designed to explore the role of these metabolites as metallophores.
In an iron-depleted medium, the Leptothoe sp. ISB3NOV94–8A strain exhibited a substantial enhancement in production of leptochelin A as detected by LCMS analysis, indicating its potential to function as a siderophore. Interestingly, in media possessing a range of copper concentrations (0 to 1,000 ppb) compared with standard SWBG11 medium (20 ppb)34 or native coastal seawater (2 ppb and as high as 25 ppb for anthropogenically affected sites), no enhancement in leptochelin production was noted. Alternatively, as leptochelin has bromine incorporated into its structure, we also evaluated the effect of bromide concentration in the growth medium on leptochelin production. When the normal SWBG11 medium with ∼5.4 ppm of bromide (with normal seawater concentration ∼65 ppm) was enriched to 0.5 g/L (500 ppm) and 1.0 g/L (1,000 ppm), this also led to a roughly 4-fold enhancement in the production of leptochelin A from 4 mg/L to 15 mg/L and 16 mg/L, respectively.
A native metabolomics analysis35,36 and post-LC infusion of metals was used to explore the binding of metals to leptochelin A (Figure 5A) as well as leptochelin B and a mixture of leptochelins A and B (SI Appendix, Figures S54 and S55). In one experimental arrangement, native metabolomics were performed that included postcolumn pH adjustment and dilution of acetonitrile content, followed by metal salt infusion, a process which has been shown to improve the conservation of weak ionophore-metal complexes (SI Appendix, Figure S57).35 Iron, copper, cobalt, and zinc were separately infused as well as an equimolar mixture of metal salts. When each metal was infused separately, the corresponding metal adduct along with the protonated adduct were observed for both leptochelins A and B by LC-MS2. In the mixed metal experiment, the copper-bound species clearly predominated, indicating a strong preference for binding of Cu by leptochelins A and B. However, zinc and cobalt adduct peaks were also observed in this mixed metal infusion experiment, indicating that the leptochelins are promiscuous in their metal binding ability. This effect was least prominent with iron; an iron adduct was observed most clearly when it was the only metal infused.
Figure 5.

Metal binding properties of leptochelin A (1). A. Extracted ion chromatograms from metal-binding studies using pure leptochelin A (1). Note that the cobalt injection shows small peaks for copper and zinc adducts. These latter peaks may result from in-instrument contamination. Molecular modeling in MOE (Amber10:EHT) as seen in B - E indicates that the carbonyl groups, amine group, phenolic oxygen, and nitrogen atoms of the thiazoline and oxazoline rings participate in the hexadentate and tetradentate coordination of metals. B. Modeled pose of iron(III) complexed with leptochelin A (1); coordinating residues: C-7 N, C-10 N, C-17 O, and C-26 O. C. Modeled pose of cobalt(II) complexed with leptochelin A (1); coordinating residues: C-7 N, C-10 N, C-17 O, C-26 O, C-27 N, and C-31 O. D. Modeled pose of copper(II) complexed with leptochelin A (1); coordinating residues: C-1 O, C-7 N, C-10 N, and C-17 O. E. Modeled pose of zinc(II) complexed with leptochelin A (1); coordinating residues: C-7 N, C-10 N, C-17 O, and C-26 O. Additionally, when coordinated with metals, the leptochelins produce a pseudocyclic conformation, an observation that is consistent with through-space correlations observed by ROESY NMR with zinc-bound leptochelin A (1) (e.g., ROE from H3-13 to H-27, Table 1, SI Appendix, Figure S51).
The observed selective copper binding but lack of enhanced leptochelin production when copper concentrations were reduced led us to consider other potential roles for the leptochelins. Cyanobacteria are quite sensitive to copper-induced toxicity,16,18,37 and several commercial “algicides” that target “blue green algae” (= cyanobacteria) contain various formulations of copper. Therefore, we evaluated the ability of the Indonesian leptochelin-producing strain of Leptothoe sp. for its ability to tolerate elevated levels of copper, present as copper(II) sulfate. The visually apparent health of cultures of Leptothoe in culture media containing elevated levels of copper was charted (SI Appendix, Table S12 and Figures S58–S66). Additionally, the relative concentrations of intracellular and extracellular leptochelin production with and without trace metal complexation were measured by LC-MS analysis. A range of copper concentrations were evaluated in SWBG11 media, with native SWBG11 media forming the control condition. These studies were performed with Leptothoe sp. ISB3NOV94-8A compared to other taxonomically similar strains for which we possessed genome sequence information. A bioinformatic analysis of these other strains indicated that the strain demonstrating susceptibility to elevations in copper levels lacked genes for siderophore production (Leptolyngbya sp. PAP09SEP10-2A), while the other three organisms with siderophore-related genes (e.g., TonB-dependent receptor genes) demonstrated resistance to elevated copper levels. Remarkably, Leptothoe sp. ISB3NOV94-8A has the ability to withstand elevated copper concentrations that are 125–250 times the average coastal seawater concentrations of 2 μg/L38,39 (SI Appendix, Figures S58–S66), while the strain without siderophore-like biosynthetic genes was negatively impacted in its growth and visible health at elevations of only 1.5–2 times the average coastal seawater concentration. Elevation of the copper concentration in the culture medium did not result in an observable change in leptochelin levels, based on LC-MS analysis of the media and the biomass of the resultant cultures (see SI Appendix, Figure S67).
Additional Biological Properties of Leptochelins A–C
The cytotoxicity of metal-free leptochelin A (1) was evaluated against a panel of human cancer cell lines, including NCI-H460 lung carcinoma, HeLa cervical carcinoma, SF188 glioblastoma, D283-med medulloblastoma, and HCT116 colon carcinoma (SI Appendix, Table S13 and Figures S69–S72). Free leptochelin A (1) was cytotoxic to all the cell lines tested with dose-dependent cytotoxicity. D283-med medulloblastoma cells (IC50 = 390 ± 20 nM) and monolayers of HCT116 colon carcinoma cells (IC50 = 400 ± 69 nM) were the most sensitive to leptochelin A. The cytotoxic effects of free and zinc-bound forms of leptochelins A–C (1–3) were compared in HCT 116 cells; the zinc-bound form of each was significantly less cytotoxic (SI Appendix, Table 3). In addition to evaluation of the leptochelins in 2D monolayer cultures, we also evaluated their activity in three-dimensional (3D) multicellular tumor spheroids (SI Appendix, Table S13 and Figure S73). Metal free leptochelins caused a decrease in proliferating and quiescent cells and an increase in the necrotic core in three-dimensional multicellular tumor spheroids of HCT 116 cells (see SI Appendix, Section 2).
Discussion
Despite investigations of the unique natural products of marine cyanobacteria for the past 50 years, they continue to be a rich source of structurally novel and biologically active metabolites.40−42 In the last few decades, this has been assisted and enriched by biosynthetic investigations, and most recently, the genetic basis for their production has been pursued.25 Indeed, genome sequencing projects have revealed some strains of marine cyanobacteria to use over 20% of their relatively large genomes (7–10 MB) to potentially encode for over 40 different natural products.43 They are especially rich in hybrid NRPS-PKS pathways which display a strong collinearity between gene order and the assembly process, allowing for their relatively accurate deciphering into predicted chemical structures. Additionally, it is quite frequent that unique biochemical transformations are observed in these otherwise interpretable pathways, and these distinctive enzymatic reactions are enriching the tool kits for synthetic chemical biology.44−46
In the current work, we discovered a series of structurally novel natural products named leptochelins A–C (1–3) from three geographically dispersed cyanobacterial collections of the genus Leptothoe. Complete structure elucidation of these complex molecules was challenging, and required multiple orthogonal strategies including NMR, mass spectrometry, bioinformatic deduced stereochemical insights, Marfey’s analysis, and chemical reactivities, in concert with molecular modeling. Moreover, genome sequencing of all three of these collections resulted in identification of the putative biosynthetic gene cluster that encodes production of the leptochelins. Interestingly, this hybrid NRPS-PKS biosynthetic gene cluster is extremely well conserved across these geographically disparate Leptothoe collections, signifying the importance of the encoded metabolites to the ecology and physiology of the producing strains. The genome sequence data also revealed signatures for metal-dependent regulation in close proximity to the BGC and suggested that the leptochelins could have metallophore properties.
This recognition of the potential metallophore properties of the leptochelins inspired a deeper investigation of their capacity to bind metals. This property was initially recognized by a careful inspection of LC-MS traces of the leptochelins, and recognition that iron, cobalt and zinc adducts were present. Subsequently, culture experiments were conducted with reduced available iron, leading to an increase in production of leptochelin A. This capacity to bind metal ions was probed in a native metabolomics investigation with introduction of individual metal species as well as mixed metal formulations into the flow system following HPLC separation. This was highly insightful as it revealed a strong preference of leptochelin A (1) for the binding of copper. However, a culture experiment with media deficient in copper failed to induce upregulation of leptochelin A production, suggesting that leptochelin A might be protective of copper toxicity by sequestration and detoxification of this metal species, thus serving multiple roles in the regulation of local metal ion concentrations. A range of copper concentrations were formulated in liquid SWBG11 media and cultures were grown and evaluated for growth. This revealed that Leptothoe possesses a remarkable tolerance to elevated copper concentrations up to 125–250-fold over those of normal coastal seawater, and that the leptochelins can therefore potentially serve diverse roles such as helping to acquire trace metals needed for growth, such as iron, and protecting against the toxic effects of elevated copper levels. Thus, the leptochelins join a growing number of metallophores that possess a variety of roles in microbial physiology and ecology.47,48 As anthropomorphic impacts increase in coastal waters, metallophore production may be a critical mechanism for microbial resilience against elevated toxic metal concentrations, such as seen here with these three cyanobacterial strains of Leptothoe.
Distinctive features of the structure and biosynthesis of leptochelin A include the two aromatic positions of halogenation, the alpha-methylation of the terminal cysteine-derived thiazoline ring, and two six-carbon moieties that appear to result from a gene duplication of an NRPS-PKS bimodular duet and subsequent neofunctionalization to create distinct yet recognizably similar fragments (C4–8 + 14; C9–12 + 15,16). Also present in the leptochelins is a salicylate-oxazoline fragment, along with two additional thiazoline motifs, that represent signature moieties found in several other metallophores such as amychelin and yersiniabactin.49,50 In addition to their metal binding properties, the leptochelins have potential pharmaceutical value in that they show relatively potent cancer cell toxicity to multiple cancer cell lines. The potential utility of the leptochelins as cancer chemotherapeutics is under continuing investigation.
Acknowledgments
We thank N. Arakawa (UCSD – ECAL) for assistance with MS3 experiments;T. Leão (UCSD) for assistance with the genome sequencing of Leptothoe sp. ISB3NOV94-8; A.B. Duggan, X. Huang, and A. Mrse for NMR experiment assistance (UCSD); and the University of California Davis Genomics Core for the sequencing and assembly of Leptothoe sp. EHU-05/26/07-4. We also appreciate the contributions for biological activity testing at OSU (T. Okino, D. Goeger, A.M. Hau, J.D. Serrill, J.M. Malmo, J. Sikorska, and J. Ishmael) and at the University of Szeged (N. Szemerédi and G. Tóth). We thank J. Diaz for use of the SpectraMax microplate reader for cytotoxicity assays. We thank T. Hall and T. Berger for laboratory assistance. We thank B.J. Philmus for advice on Marfey’s analysis and M.K. Gilson for advice on molecular modeling. We thank Raquel Silva for assistance with DNA extraction and amplification at CIIMAR.
Data Availability Statement
Raw and processed LC-MS2 and MS3 data are available through the MassIVE repository (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) under the following identifier: MSV000084765. The genome assemblies have been submitted to NCBI with accession numbers SAMN38524433 for Leptothoe sp. ISB3NOV94-8A, SAMN38764028 for Leptothoe sp. EHU-05/26/07-4 and SAMN34340027 for Leptothoe sp. LEGE 181152. Additionally, the putative BGCs from each genome assembly have been submitted to the MIBiG repository (https://mibig.secondarymetabolites.org/) under accession numbers BGC0002819, BGC0002820 and BGC0002821. The NMR data for the following compounds has been deposited in the Natural Products Magnetic Resonance Database (NP-MRD; www.np-mrd.org) and can be found at NP0332615 (Leptochelin A), NP0332616 (Leptochelin B), NP0332617 (Leptochelin C), NP0332618 (Leptochelin A - Zn Bound), NP0332619 (Leptochelin B - Zn Bound) and NP0332620 (Leptochelin C - Zn Bound).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c05399.
Detailed description of materials and methods, genomics and bioinformatics, chemical data, mass spectrometry, NMR spectra, cytotoxicity assays, and biochemical and metal binding assays (PDF)
Author Contributions
+ N.E.A., M.A.R., and C.C.T. contributed equally to the project.
Research reported in this publication was supported in part by the National Center for Complementary and Integrative Health of the NIH under award number F32AT011475 to N.E.A., and NIH 5R01GM107550-10 to L.G., W.H.G., and P.D. D.P. was supported by the Deutsche Forschungsgemeinschaft through the CMFI Cluster of Excellence (EXC 2124) and the Collaborative Research Center CellMap (TRR 261). M.A.R., L.F., J. Morais, and V.V. were supported by FCT (Fundação para a Ciência e a Tecnologia) through CIIMAR research unit strategic fund support (UIDB/04423/2020 and UIDP/04423/2020) and also by the Innovation Pact, Project No. C644915664–00000026 (WP9- Portuguese Blue Biobank under the Blue Economy Pact), known as the “Blue Bioeconomy Pact”, resulting from the submission of the application to Notice No. 02/C05-i01/2022, within the scope of the Recovery and Resilience Plan (PRR) cofunded by the Portuguese Republic and the European Union. L.F. is also supported by the Fundação para a Ciência e Tecnologia grant 2022.11979.BD. G.S. was supported by the János Bolyai Research Scholarship (BO/00158/22/5) of the Hungarian Academy of Sciences. K.L.A. was funded by the NIH training grants T32 CA009523 and T32 GM067550. Y.E.A. was funded by NIH 1R03OD034493–01. A portion of this research was performed at the Environmental Molecular Sciences Laboratory, a DOE Office of Science User Facility sponsored by the BER program under Contract No. DE-AC05–76RL01830. A portion of this research was performed under the U.S.-Egypt Science and Technology joint fund number BIO6–002–010 to D.T.A.Y. and the Institutional Fund Projects number IFPIP-601–166–1443 to A.M.A. We acknowledge NSF grant 2116395 for the NMR spectrometer utilized in this work.
The authors declare the following competing financial interest(s): P. Dorrestein is an advisor and holds equity in Sirenas and Cybele, consulted for MSD animal health in 2023, and he is a Co-founder, scientific advisor and holds equity in Ometa Labs, Arome, and Enveda with prior approval by UC San Diego. W. Gerwick is an advisor and holds equity in Sirenas.
Supplementary Material
References
- Jannasch H. W.; Mottl M. J. Geomicrobiology of Deep-Sea Hydrothermal Vents. Science 1985, 229, 717–725. 10.1126/science.229.4715.717. [DOI] [PubMed] [Google Scholar]
- Perera I.; Subashchandrabose S. R.; Venkateswarlu K.; Naidu R.; Megharaj M. Consortia of cyanobacteria/microalgae and bacteria in desert soils: an underexplored microbiota. Appl. Microbiol. Biotechnol. 2018, 102, 7351–7363. 10.1007/s00253-018-9192-1. [DOI] [PubMed] [Google Scholar]
- Hauptmann A. L.; et al. Bacterial diversity in snow on North Pole ice floes. Extremophiles 2014, 18, 945–951. 10.1007/s00792-014-0660-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghori N.-U.-H.; Wise M. J.; Whiteley A. S. Temporal Microbial Community Dynamics Within a Unique Acid Saline Lake. Front. Microbiol. 2021, 12, 649594. 10.3389/fmicb.2021.649594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopf J. W. Disparate rates, differing fates: tempo and mode of evolution changed from the Precambrian to the Phanerozoic. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 6735–6742. 10.1073/pnas.91.15.6735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank C. E. Evolutionary timing of the origins of mesophilic sulphate reduction and oxygenic photosynthesis: a phylogenomic dating approach. Geobiology 2004, 2, 1–20. 10.1111/j.1472-4677.2004.00020.x. [DOI] [Google Scholar]
- Tice M. M.; Lowe D. R. Photosynthetic microbial mats in the 3,416-Myr-old ocean. Nature 2004, 431, 549–552. 10.1038/nature02888. [DOI] [PubMed] [Google Scholar]
- Hider R. C.; Kong X. Chemistry and biology of siderophores. Nat. Prod. Rep. 2010, 27, 637. 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- Raymond K. N.; Müller G.; Matzanke B. F.. Complexation of iron by siderophores a review of their solution and structural chemistry and biological function. Structural Chemistry, Topics in Current Chemistry; Springer: Berlin Heidelberg, 1984; pp 49–102. [Google Scholar]
- Miethke M.; Marahiel M. A. Siderophore-Based Iron Acquisition and Pathogen Control. Microbiol Mol. Biol. Rev. 2007, 71, 413–451. 10.1128/MMBR.00012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y.; Butler A. Structure of synechobactins, new siderophores of the marine cyanobacterium Synechococcus sp. PCC 7002. Limnol. Oceanogr. 2005, 50, 1918–1923. 10.4319/lo.2005.50.6.1918. [DOI] [Google Scholar]
- Beiderbeck H.; Taraz K.; Budzikiewicz H.; Walsby A. E. Anachelin, the Siderophore of the Cyanobacterium Anabaena cylindrica CCAP 1403/2A. Zeitschrift für Naturforschung C 2000, 55, 681–687. 10.1515/znc-2000-9-1002. [DOI] [PubMed] [Google Scholar]
- Mullis K. B.; Pollack J. R.; Neilands J. B. Structure of schizokinen, An iron-transport compound from Bacillus megaterium. Biochemistry 1971, 10, 4894–4898. 10.1021/bi00802a010. [DOI] [PubMed] [Google Scholar]
- Mohr J. F.; et al. Frankobactin Metallophores Produced by Nitrogen-Fixing Frankia Actinobacteria Function in Toxic Metal Sequestration. J. Nat. Prod. 2021, 84, 1216–1225. 10.1021/acs.jnatprod.0c01291. [DOI] [PubMed] [Google Scholar]
- Blindauer C. A. Bacterial metallothioneins: past, present, and questions for the future. J. Biol. Inorg. Chem. 2011, 16, 1011–1024. 10.1007/s00775-011-0790-y. [DOI] [PubMed] [Google Scholar]
- McKnight D. M.; Morel F. M. M. Copper complexation by siderophores from filamentous blue-green algae1: Copper complexation. Limnol. Oceanogr. 1980, 25, 62–71. 10.4319/lo.1980.25.1.0062. [DOI] [Google Scholar]
- Wilhelm S. W.; Trick C. G. Iron-limited growth of cyanobacteria: Multiple siderophore production is a common response. Limnol. Oceanogr. 1994, 39, 1979–1984. 10.4319/lo.1994.39.8.1979. [DOI] [Google Scholar]
- Huertas M.; López-Maury L.; Giner-Lamia J.; Sánchez-Riego A.; Florencio F. Metals in Cyanobacteria: Analysis of the Copper, Nickel, Cobalt and Arsenic Homeostasis Mechanisms. Life 2014, 4, 865–886. 10.3390/life4040865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinou D.; et al. Leptothoe, a new genus of marine cyanobacteria (Synechococcales) and three new species associated with sponges from the Aegean Sea. J. Phycol. 2019, 55, 882–897. 10.1111/jpy.12866. [DOI] [PubMed] [Google Scholar]
- Okujo N.; et al. Structure of vulnibactin, a new polyamine-containing siderophore from Vibrio vulnificus. Biometals 1994, 7, 109–116. 10.1007/BF00140480. [DOI] [PubMed] [Google Scholar]
- Mirus O.; Strauss S.; Nicolaisen K.; Von Haeseler A.; Schleiff E. TonB-dependent transporters and their occurrence in cyanobacteria. BMC Biol. 2009, 7, 68. 10.1186/1741-7007-7-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont F. C.; Kang H. Y.; Brickman T. J.; Armstrong S. K. Identification and Characterization of alcR, a Gene Encoding an AraC-Like Regulator of Alcaligin Siderophore Biosynthesis and Transport in Bordetella pertussis and Bordetella bronchiseptica. J. Bacteriol. 1998, 180, 862–870. 10.1128/JB.180.4.862-870.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel L.; González N.; Jagdeep S.; Nguyen-Ngoc T.; Reimmann C. PchR-box recognition by the AraC-type regulator PchR of Pseudomonas aeruginosa requires the siderophore pyochelin as an effector: Pyochelin-dependent PchR-box recognition. Mol. Microbiol. 2005, 58, 495–509. 10.1111/j.1365-2958.2005.04837.x. [DOI] [PubMed] [Google Scholar]
- Patel H. M.; Tao J.; Walsh C. T. Epimerization of an l -Cysteinyl to a d -Cysteinyl Residue during Thiazoline Ring Formation in Siderophore Chain Elongation by Pyochelin Synthetase from Pseudomonas aeruginosa. Biochemistry 2003, 42, 10514–10527. 10.1021/bi034840c. [DOI] [PubMed] [Google Scholar]
- Wu Q.; et al. Metabolomics and Genomics Enable the Discovery of a New Class of Nonribosomal Peptidic Metallophores from a Marine Micromonospora. J. Am. Chem. Soc. 2023, 145, 58–69. 10.1021/jacs.2c06410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries E. J.; Janssen D. B. Biocatalytic conversion of epoxides. Curr. Opin. Biotechnol. 2003, 14, 414–420. 10.1016/S0958-1669(03)00102-2. [DOI] [PubMed] [Google Scholar]
- Mori S.; Pang A. H.; Thamban Chandrika N.; Garneau-Tsodikova S.; Tsodikov O. V. Unusual substrate and halide versatility of phenolic halogenase PltM. Nat. Commun. 2019, 10, 1255. 10.1038/s41467-019-09215-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gäfe S.; Niemann H. H. Structural basis of regioselective tryptophan dibromination by the single-component flavin-dependent halogenase AetF. Acta Crystallogr. D Struct Biol. 2023, 79, 596–609. 10.1107/S2059798323004254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J.; Keatinge-Clay A. T. Structural and Functional Analysis of C2-Type Ketoreductases from Modular Polyketide Synthases. J. Mol. Biol. 2011, 410, 105–117. 10.1016/j.jmb.2011.04.065. [DOI] [PubMed] [Google Scholar]
- Williamson R. T.; Boulanger A.; Vulpanovici A.; Roberts M. A.; Gerwick W. H. Structure and Absolute Stereochemistry of Phormidolide, a New Toxic Metabolite from the Marine Cyanobacterium Phormidium sp. J. Org. Chem. 2002, 67, 7927–7936. 10.1021/jo020240s. [DOI] [PubMed] [Google Scholar]
- Ndukwe I. E.; et al. Synergism of anisotropic and computational NMR methods reveals the likely configuration of phormidolide A. Chem. Commun. 2020, 56, 7565–7568. 10.1039/D0CC03055D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin M. J.; et al. The Phormidolide Biosynthetic Gene Cluster: A trans -AT PKS Pathway Encoding a Toxic Macrocyclic Polyketide. ChemBioChem. 2016, 17, 164–173. 10.1002/cbic.201500467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg C. C.; et al. Cyclic Depsipeptides, Grassypeptolides D and E and Ibu-epidemethoxylyngbyastatin 3, from a Red Sea Leptolyngbya Cyanobacterium. J. Nat. Prod. 2011, 74, 1677–1685. 10.1021/np200270d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss N. A.; Leao T.; Glukhov E.; Gerwick L.; Gerwick W. H.. Collection, Culturing, and Genome Analyses of Tropical Marine Filamentous Benthic Cyanobacteria. Methods in Enzymology; Elsevier, 2018; pp 3–43. [DOI] [PubMed] [Google Scholar]
- Aron A. T.; et al. Native mass spectrometry-based metabolomics identifies metal-binding compounds. Nat. Chem. 2022, 14, 100–109. 10.1038/s41557-021-00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reher R.; et al. Native metabolomics identifies the rivulariapeptolide family of protease inhibitors. Nat. Commun. 2022, 13, 4619. 10.1038/s41467-022-32016-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima S.; et al. Extracellular vesicles as an alternative copper-secretion mechanism in bacteria. Journal of Hazardous Materials 2022, 431, 128594. 10.1016/j.jhazmat.2022.128594. [DOI] [PubMed] [Google Scholar]
- Lewis A. G.; Whitfield P. H.. The biological importance of copper in the sea. A literature review, No. 223; International Copper Research Association Inc.: New York, 1974.
- McNeely R. N.; Neimanis V. P.; Dwyer L.. Water Quality Sourcebook: A Guide to Water Quality Parameters; Inland Waters Directorate, Water Quality Branch, 1979. [Google Scholar]
- Jones M. R.; et al. CyanoMetDB, a comprehensive public database of secondary metabolites from cyanobacteria. Water Res. 2021, 196, 117017. 10.1016/j.watres.2021.117017. [DOI] [PubMed] [Google Scholar]
- Nandagopal P.; et al. Bioactive Metabolites Produced by Cyanobacteria for Growth Adaptation and Their Pharmacological Properties. Biology 2021, 10, 1061. 10.3390/biology10101061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpine R.; Sieber S. Antibacterial and antiviral metabolites from cyanobacteria: Their application and their impact on human health. Current Research in Biotechnology 2021, 3, 65–81. 10.1016/j.crbiot.2021.03.001. [DOI] [Google Scholar]
- Leão T.; et al. A Multi-Omics Characterization of the Natural Product Potential of Tropical Filamentous Marine Cyanobacteria. Marine Drugs 2021, 19, 20. 10.3390/md19010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda K.; Kubota N.; Hamada H.; Kaji M.; Hirata T. Asymmetric reduction of enones with Synechococcus sp. PCC 7942. Tetrahedron: Asymmetry 2004, 15, 1677–1679. 10.1016/j.tetasy.2004.04.024. [DOI] [Google Scholar]
- Górak M.; Żymańczyk-Duda E. Application of cyanobacteria for chiral phosphonate synthesis. Green Chem. 2015, 17, 4570–4578. 10.1039/C5GC01195G. [DOI] [Google Scholar]
- Lukowski A. L.; et al. Enzymatic Halogenation of Terminal Alkynes. J. Am. Chem. Soc. 2023, 145, 18716. 10.1021/jacs.3c05750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone T. C.; Nolan E. M. Beyond iron: non-classical biological functions of bacterial siderophores. Dalton Trans. 2015, 44, 6320–6339. 10.1039/C4DT03559C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRose D. L.; Seyedsayamdost M. R.; Morel F. M. M. Multiple siderophores: bug or feature?. J. Biol. Inorg. Chem. 2018, 23, 983–993. 10.1007/s00775-018-1617-x. [DOI] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Traxler M. F.; Zheng S.-L.; Kolter R.; Clardy J. Structure and Biosynthesis of Amychelin, an Unusual Mixed-Ligand Siderophore from Amycolatopsis sp. AA4. J. Am. Chem. Soc. 2011, 133, 11434–11437. 10.1021/ja203577e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer B. A.; Wang C. C. C.; Walsh C. T.; Khosla C. Biosynthesis of Yersiniabactin, a Complex Polyketide-Nonribosomal Peptide, Using Escherichia coli as a Heterologous Host. Appl. Environ. Microbiol. 2003, 69, 6698–6702. 10.1128/AEM.69.11.6698-6702.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed LC-MS2 and MS3 data are available through the MassIVE repository (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) under the following identifier: MSV000084765. The genome assemblies have been submitted to NCBI with accession numbers SAMN38524433 for Leptothoe sp. ISB3NOV94-8A, SAMN38764028 for Leptothoe sp. EHU-05/26/07-4 and SAMN34340027 for Leptothoe sp. LEGE 181152. Additionally, the putative BGCs from each genome assembly have been submitted to the MIBiG repository (https://mibig.secondarymetabolites.org/) under accession numbers BGC0002819, BGC0002820 and BGC0002821. The NMR data for the following compounds has been deposited in the Natural Products Magnetic Resonance Database (NP-MRD; www.np-mrd.org) and can be found at NP0332615 (Leptochelin A), NP0332616 (Leptochelin B), NP0332617 (Leptochelin C), NP0332618 (Leptochelin A - Zn Bound), NP0332619 (Leptochelin B - Zn Bound) and NP0332620 (Leptochelin C - Zn Bound).