

Abstract
The UL36 open reading frame (ORF) encodes the largest herpes simplex virus type 1 (HSV-1) protein, a 270-kDa polypeptide designated VP1/2, which is also a component of the virion tegument. A null mutation was generated in the UL36 gene to elucidate its role in the virus life cycle. Since the UL36 gene specifies an essential function, complementing cell lines transformed for sequences encoding the UL36 ORF were made. A mutant virus, designated KΔUL36, that encodes a null mutation in the UL36 gene was isolated and propagated in these cell lines. When noncomplementing cells infected with KΔUL36 were analyzed, both terminal genomic DNA fragments and DNA-containing capsids (C capsids) were detected; therefore, UL36 is not required for cleavage or packaging of DNA. Sedimentation analysis of lysates from mutant-infected cells revealed the presence of particles that have the physical characteristics of C capsids. In agreement with this, polypeptide profiles of the mutant particles revealed an absence of the major envelope and tegument components. Ultrastructural analysis revealed the presence of numerous unenveloped DNA containing capsids in the cytoplasm of KΔUL36-infected cells. The UL36 mutant particles were tagged with the VP26-green fluorescent protein marker, and their movement was monitored in living cells. In KΔUL36-infected cells, extensive particulate fluorescence corresponding to the capsid particles was observed throughout the cytosol. Accumulation of fluorescence at the plasma membrane which indicated maturation and egress of virions was observed in wild-type-infected cells but was absent in KΔUL36-infected cells. In the absence of UL36 function, DNA-filled capsids are produced; these capsids enter the cytosol after traversing the nuclear envelope and do not mature into enveloped virus. The maturation and egress of the UL36 mutant particles are abrogated, possibly due to a late function of this complex polypeptide, i.e., to target capsids to the correct maturation pathway.
The herpes simplex virus type 1 (HSV-1) virion is comprised of four structural elements: a DNA-containing core; an icosahedral capsid, which encloses the genome; a layer that immediately surrounds the capsid termed the tegument; and an outer membrane or envelope, which encloses the whole structure and in which are embedded the viral glycoproteins (39, 55; reviewed in references 40 and 47). The tegument represents the most diverse structural element of the virus particle in terms of both polypeptide composition and functions.
Virus-specified polypeptides that comprise the tegument structure include those that function to activate transcription, shut off host protein synthesis, and uncoat the virus genome, as well as others whose functions are not yet known (reviewed in references 40 and 47). The role of tegument is twofold. First, the tegument can be envisioned as a structure that delivers factors into the cytosol of the infected cell to facilitate the initiation of a successful infection. Components of the tegument that mediate this process include VP16, a potent viral transactivator of immediate-early genes (4, 8), and the virion host shutoff polypeptide (vhs), which is responsible for shutoff of host protein synthesis (28, 36). The second function of the tegument is structural. VP16 is also required for the structural integrity of the tegument; in its absence, enveloped particles are not formed (1, 52). Both VP22, a major tegument component, and vhs participate in direct physical interactions with VP16 (19, 43); therefore, VP16 may act as a nucleation factor for formation of the tegument, and incorporation of other proteins into the tegument layer may involve interaction with this multifunctional polypeptide (19, 43, 51). There are also a multitude of polypeptides that are minor components of the tegument. Their functions are varied, such as kinase activity (33), proteins that interact with ribosomes (41), proteins required for virus egress (2, 12), and others that are involved in DNA packaging (42). The function of these proteins may add yet greater complexity to the role of the tegument in the virus replication cycle.
The morphogenesis of the DNA-filled capsid into an enveloped virion is a complex and poorly understood process. Capsid assembly is a nuclear event resulting in the production of three types of capsids, A, B, and C (21). B capsids contain internal scaffold proteins 22a and 21, the viral protease VP24, and the capsid shell virion proteins VP5, VP19C, VP23, and VP26. For C capsids, genomic DNA replaces the scaffold proteins. A capsids are empty, i.e., devoid of any internal composition (reviewed in reference 47). Packaging of viral DNA into capsid shells is a complex process requiring the functions of several gene products, some of which remain capsid associated (reviewed in reference 24). Initial envelopment of the virion takes place at the inner nuclear membrane. The progression of this particle as it matures into an infectious virion is a contentious issue. Two pathways have been suggested for final maturation of the virus. In the first scenario, capsids are enveloped at the inner nuclear membrane and translocate through the periplasmic space to the endoplasmic reticulum and enter the cell secretory pathway (7, 13, 26). The other model, for which recent studies lends strong support, requires viruses to undergo initial envelopment at the inner nuclear membrane but then fuse with the outer membrane to release naked capsids into the cytosol. These capsids are transported to the Golgi compartment or other cytoplasmic organelles, where they are enveloped (5, 11, 20, 22, 34, 46, 49, 53, 54). These two opposing ideas raise the question of where tegument proteins accumulate prior to their incorporation into the maturing virus and the viral factors that traffic particles to the maturation compartment.
UL36 encodes the largest HSV-1 polypeptide, the tegument protein VP1/2 (23, 25, 29, 30, 31, 39, 45). The UL36 gene product is a 273-kDa polypeptide that is expressed as a true-late gene (30). There are approximately 100 to 150 copies of this protein per virion (23); however, in terms of protein mass it is a significant component of the tegument, as much as 50% of the VP16 mass. It has been implicated in uncoating of the viral genome based on the characterization of a temperature-sensitive mutant in this gene, tsB7 (3, 4, 27). The phenotype of this virus at the restrictive temperature is the accumulation of DNA-filled capsids at the nuclear membrane. The DNA is released into the nucleus only after a shift-down to the permissive temperature (3). Studies have also shown that it binds the packaging sequence of the HSV-1 genome and therefore may play some role in DNA packaging/transport (10). An intimate association of VP1/2 with the capsid is suggested by reports in the literature that demonstrate interaction of VP1/2 with VP5 (30) and the tight association of this polypeptide with the capsid structure (21). The human cytomegalovirus homologue of UL36, HMWP, has been shown to interact with the counterpart of the HSV-1 UL37 gene (M. E. Harmon and W. G. Gibson, unpublished data). In addition, cryoelectron microscopy of virions has recently identified tegument-capsid interactions (9, 48, 59).
The aim of this study was to isolate a null mutant in the UL36 gene in order to characterize its function in the virus replication cycle. Since UL36 specifies an essential function, transformed cell lines that expressed the gene in trans were derived. These cell lines permitted the isolation of a null mutant in the UL36 gene. The phenotype of this mutant demonstrates the complexity of tegument protein functions. The absence of the UL36-encoded polypeptide results in the accumulation of numerous cytosolic DNA-containing capsids that do not mature into enveloped virus.
MATERIALS AND METHODS
Cells and viruses.
Human embryonic lung (HEL) cells, Vero cells, and transformed Vero cell lines were grown in minimum essential medium (alpha medium supplemented with 10% fetal calf serum [Life Technologies]) and passaged as described by Desai et al. (16) Virus stocks of the KOS strain of HSV-1 and the mutant viruses were prepared as previously described (16). The HS30 cell line was used to propagate KΔUL36 virus, and typical yields were approximately 600 PFU/cell during a single cycle of growth. David Knipe (Harvard Medical School, Boston, Mass.) kindly provided the temperature-sensitive mutant tsB7.
Plasmids.
The 25.9-kb BglII D fragment of KOS (Fig. 1) which encodes UL31 to UL38 (29) was cloned into a modified pUC19 plasmid containing a BglII restriction site. This plasmid (pKBGD) was used to obtain a 13.4-kb XbaI-to-SnaB1 fragment (Fig. 1), which was cloned into the XbaI site and a filled-in EcoRI site of pUC19 to give pKXSB. A 3.6-kb deletion in the UL36 gene was generated by digestion of pKXSB with KpnI and EcoRV; the KpnI site was blunt ended and ligated with the EcoRV end to generate pKΔUL36. To generate a plasmid expressing just UL36, pKXSB was digested with XbaI, the overhang was filled in with Klenow enzyme, and the fragment was digested with BstBI to generate the pKXSB:XB vector. A plasmid specifying a deletion in the UL35 gene, pKΔ26 (16), was digested with HpaI and BstB1; the 400-bp fragment was isolated and cloned into the pKXSB:XB vector. This plasmid was designated pKUL36.
FIG. 1.

Schematic representation of the BglII D region of the KOS genome. The BglII D (25.9-kb) fragment encodes UL31 to UL38 (29). The direction of the UL36 transcript is indicated below the ORF. The UL36 ORF starts at genome nucleotide 80543 and stops at 71051. Three cell lines that express UL36 were made: B80, which was transformed with the BglII D fragment (pKBGD); XS13, transformed with the XbaI-to-SnaB1 fragment (pKXSB); and HS30, transformed with the HpaI-to-SnaB1 fragment containing a deletion in the UL35 gene (pKUL36). The deletion in pKΔUL36 spans from the KpnI to EcoRV sites in UL36. Relevant restriction enzyme sites and genome nucleotide numbers (29) in parentheses are indicated at the top.
Construction of transformed Vero cell lines.
The procedure of DeLuca et al. (14), also described by Desai et al. (15), was used for transformation of Vero cells. Subconfluent monolayers of Vero cells (106 cells in 100-mm-diameter-dishes) were cotransfected with pSV2-neo (1.0 μg) (44) and a molar three- or fivefold excess of the plasmid carrying the UL36 gene. For propagation of the UL36 mutant, Vero cells were transformed with the BglII D fragment of HSV-1 (pKBGD). This fragment encodes UL31 to UL38 (Fig. 1) (29). G418-resistant cell lines were selected for UL36 expression by the ability to plaque tsB7 (27) at the nonpermissive temperature. One cell line, designated B80, gave the highest complementing activity and was used for subsequent experiments. Cells were also transformed with pKXSB, which carries genes UL34 to UL36 (Fig. 1). Out of 57 G418-resistant transformants obtained, 5 were able to plaque tsB7 at the nonpermissive temperature. Cell line XS13 was chosen for further characterization. Finally, cells were also transformed with pKUL36 (Fig. 1), which carries only UL36. Cells were again selected for the ability to support the replication of tsB7 at 39.5°C; out of 65 cell lines derived, 12 transformants exhibited this property. Cell line HS30 was used for all subsequent experiments.
Radiolabelling of infected cells.
Confluent monolayers of Vero cells in two 100-mm-diameter petri dishes (approximately 107 cells) were infected at a multiplicity of infection (MOI) of 10 PFU/cell. After adsorption, 5 ml of labeling medium was added to each culture. Labeling medium for [35S]methionine consisted of 70% Dulbecco's modified Eagle medium without methionine, 25% F12 (contains 5 μg of methionine/ml), and 5% fetal bovine serum (Life Technologies). Labeling medium for [3H]thymidine consisted of F12 medium supplemented with 5% fetal bovine serum. Eight hours after infection, 150 μCi of [35S]methionine or 150 μCi of [3H]thymidine (DuPont-NEN) was added to each culture, and incubation continued until 24 h postinfection. Cells were scraped into phosphate-buffered saline (PBS); cultures from duplicate plates were combined and pelleted at 3,500 × g for 5 min at 4°C.
Sedimentation analysis of capsids.
Sedimentation analysis of capsids from radiolabeled extracts derived from infected Vero cells was performed as described by Desai et al. (15) and Person and Desai (35).
Virus purification.
Intracellular viruses were purified by rate velocity sedimentation in sucrose gradients. Radiolabeled infected cells were subjected to three freeze-thaw cycles and sonicated. Virus was pelleted in a Beckman SW41 rotor at 24,000 rpm for 45 min. Virus was resuspended in a small volume of growth medium and layered onto 20 to 50% (wt/vol in PBS) sucrose gradients. Centrifugation was performed in the SW41 rotor for 60 min at 34,000 rpm. Radioactivity present in the fractions collected was determined by liquid scintillation counting. In some cases the sedimented particles were visualized as light-scattering bands. All gradients were made using a BioComp Gradient Mate (BioComp).
TEM.
Vero cells (5 × 105) in 35-mm-diameter dishes were infected at an MOI of 10 PFU/cell. Infected cells were fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer containing 250 mM CaCl2. The samples were then postfixed in 1% osmium tetroxide (reduced with 1% KFeCN) and stained with 2% uranyl acetate, dehydrated through a graded series of ethanol, and embedded in Epon resin. Thin sections (70 to 90 nm) were cut, mounted on carbon-coated grids, stained with lead citrate (0.3%) and uranyl acetate (2%), and examined at 60 kV in a Philips transmission electron microscope (TEM).
Confocal microscopy.
Confluent monolayers of cells in eight-well LabTek chamber slides (2.5 × 105 cells per tray) were infected at an MOI of 10 PFU/cell. At various times postinfection, the cells were rinsed twice in PBS and overlaid with PBS for microscopy. Confocal analysis was carried with a Noran Oz confocal microscope, which consists of an Olympus inverted fluorescence microscope using a Kr-Ar ion laser. Cells were viewed with 60× and 100× (oil) objectives and the fluorescein isothiocyanate-narrow channel. The full area was scanned in slow mode, with medium resolution and a sampling time of 400 ns (2 s/image). The confocal slit was set at 10 μm in most cases. Images of the cells were collected in several focal planes, usually through the middle of the cells and in planes above and below the cell. The thickness of the optical section was 0.52 μm.
Data preparation.
Autoradiographs were scanned on a Umax Powerlook II scanner. The images were scanned at 300 dots/in. into Adobe Photoshop 3.0 and were transported as PICT files into Microsoft Powerpoint for presentation and printing. The confocal images presented in Fig. 8 were saved as TIFF files (24 bit) transferred into Adobe Photoshop 5.0 for final presentation. The electron microscope (EM) negatives for Fig. 7A to C were also scanned into Adobe Photoshop 5.0 for final presentation.
FIG. 8.

Analysis in living cells of the replication of GFP-tagged KΔUL36. Cells infected with K26GFP (A, C, and E) and KΔUL36-GFP (B, D, and F) at an MOI of 10 PFU/cell were visualized live in a confocal microscope at 8 (A and B), 12 (C and D), and 18 (E and F) h after infection. Magnification was ×60 (A, B, C, E, and F) or ×100 (D).
FIG. 7.
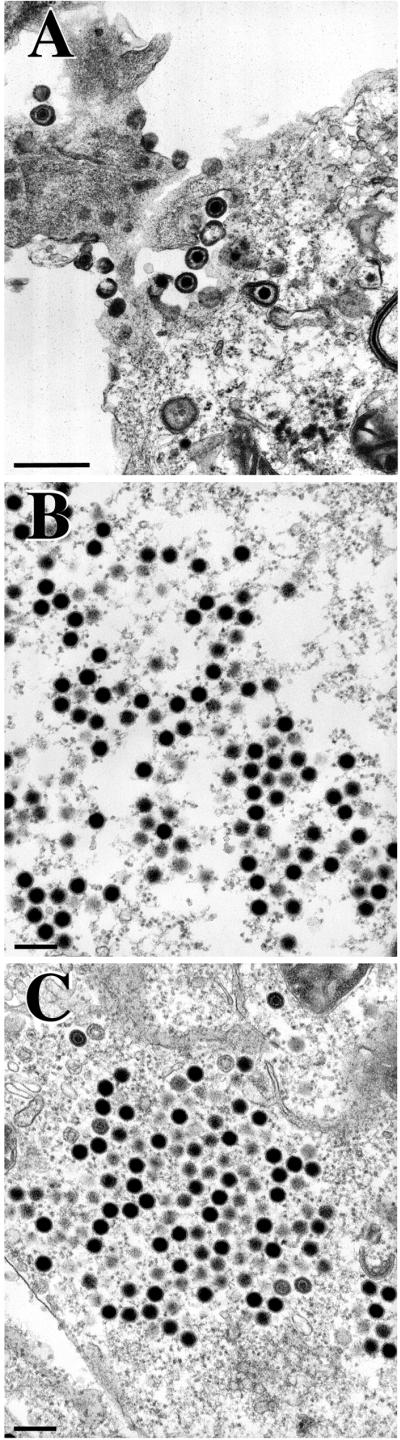

TEM analysis of KΔUL36-infected cells. Vero cells infected with KOS (A) and KΔUL36 (B to E) at an MOI of 10 PFU/cell were fixed at 16 h postinfection, thin sectioned, and processed for TEM. Magnifications were ×27,500 (A), ×37,000 (B and C), ×13,600 (D), and ×20,500 (E). Bars = 500 nm (A and E), 200 nm (B and C), and 2,000 nm (D).
RESULTS
Isolation of transformed cell lines that express the UL36 ORF.
Since UL36 specifies an essential function (3, 27), isolation of a null mutant in this gene requires a transformed cell line that expresses this protein in trans. Several cell lines have been derived that were cotransformed with the neomycin gene and sequences encoding the UL36 gene. These cell lines were selected for the ability to complement the growth of tsB7 (27), which encodes a temperature-sensitive lesion in the UL36 gene, at the nonpermissive temperature. Cell line B80 was transformed with the BglII D fragment (26 kb) of HSV-1 strain KOS, which carries genes UL31 to UL38 (Fig. 1) (29). In addition to supporting the growth of a virus specifying a lesion in UL36, this cell line also supported the growth of mutants in UL37 and UL38. The expression in trans of the other genes present in this sequence was not tested for. This cell line was used for initial marker transfer experiments but was not useful for isolation of the UL36 null mutant because the large size of the transfected sequence resulted in a high yield of background wild-type virus. Therefore, cell lines transformed with smaller DNA fragments were isolated. Cell line XS13 was transformed with sequences spanning from the XbaI site to the SnaB1 site in UL37 (13.4 kb [Fig. 1]). Since both B80 and XS13 cell lines expressed additional gene products, a cell line that expressed just the UL36 gene was derived. Manipulation of the existing plasmids was carried out to generate the sequences encoding the UL36 open reading frame (ORF) and the 5′ and 3′ flanking sequences required for its expression. Since the 3′ flanking sequences includes the UL35 ORF, a deletion that abrogated UL35 expression (16) was transferred into a plasmid that spans from the HpaI site to the SnaB1 site (Fig. 1). This plasmid encodes just UL36 (12 kb) and was used to transform Vero cells. Out of 65 neomycin-resistant clones isolated, 12 complemented the growth of tsB7 at the nonpermissive temperature. One cell line, designated HS30, was used to isolate and propagate a UL36 null mutant virus.
Construction and isolation of a null mutation in the UL36 gene.
A null mutation in UL36 was generated by digestion of pKXSB with KpnI (79456) at the 5′ end of the UL36 ORF and EcoRV (75877) (Fig. 1). This resulted in a deletion of 3,600 nucleotides and of amino acid residues 362 to 1555 (total UL36 residues = 3,164 [29]). The deletion also created a frameshift and premature translation termination 42 amino acid residues beyond the junction of the deletion. This plasmid, designated pKΔUL36, was used in marker transfer experiments (35) using KOS genomic DNA and the HS30 cell line. Single plaques derived from the transfection progeny were tested for the ability to replicate on HS30 cells but not on Vero cells. A number of isolates that exhibited this phenotype were identified, and initial Southern blot analysis of their genomes indicated correct transfer of the UL36-specified deletion into the virus genome. One isolate was further purified, and Southern blot analysis of the virus genome was carried out to confirm the introduction of the deletion (data not shown). All studies presented below were carried out with this virus, designated KΔUL36. This virus usually gave a burst size of 600 PFU/cell when grown in HS30 cells (KOS gave a burst size of approximately 1,000 PFU/cell). Virus stocks of KΔUL36 grown in HS30 cells contained approximately 0.003% recombinant wild-type virus. Since it was possible to isolate and propagate the UL36 null mutant on a cell line that expresses only UL36, the resulting phenotype of this virus must therefore be due to the mutation in UL36 alone. In addition, a marker-rescued virus was made by cotransfecting HS30 cells with KΔUL36 genomic DNA and pKUL36. When the cotransfection progeny were plated on Vero cells, several plaques formed on these monolayers. One such plaque was further purified and designated KΔUL36R. This marker-rescued virus had growth properties similar to those of wild-type virus. The burst sizes of wild-type virus and KΔUL36R, determined in Vero cells following a single 24-h cycle of growth, were 1,435 and 1,400 PFU/cell (averages of four separate infections), respectively.
The UL36 polypeptide was not detected in Vero cells infected with KΔUL36.
To confirm the absence of the UL36 polypeptide in Vero cells infected with KΔUL36, [35S]methionine-labeled infected cell polypeptides were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The result (Fig. 2) shows that the UL36 null mutant (lane 3) synthesizes the whole spectrum of infected cell polypeptides during the course of the replication cycle, similar to that observed in wild-type-infected cells (lane 2). The only detectable difference was the absence of a polypeptide which has a mobility greater than that of the 220-kDa protein standard and which presumably corresponds to UL36. This polypeptide was readily visible in the KOS-infected cell lysates (lane 2). There were additional, less abundant proteins that migrated in the gel with mobilities greater than that of the full-length UL36 protein. These were UL36 specified because they were absent in the null mutant (lane 3); some probably correspond to the smaller form of UL36 that migrates with a mobility of 260 kDa and is designated ICP3 (23, 25, 45).
FIG. 2.
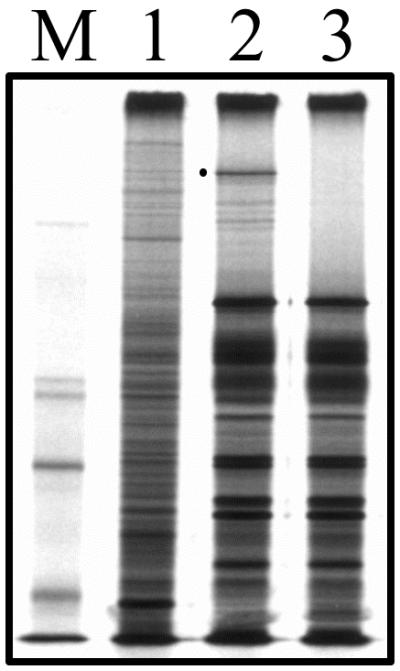
Infected cell polypeptide synthesis in KΔUL36-infected cells. Vero cells (106 in 35-mm-diameter dishes) were infected with KOS (lane 2) and KΔUL36 (lane 3) at an MOI of 10 PFU/cell or mock infected (lane 1). The infected cells were metabolically labeled with [35S]methionine from 9 to 24 h postinfection. Cells were solubilized in Laemmli sample buffer, and the proteins were analyzed by SDS-PAGE (9% acrylamide). Protein standards (lane M) correspond to 220, 97.4, 66, and 46 kDa (the 97.4-kDa marker migrates as a doublet in our gels). The closed circle indicates the position of the UL36 protein in KOS lysates.
Cleavage of viral DNA in KΔUL36-infected cells.
Southern blot analysis was carried out to determine the processing of replicated DNA into unit-length molecules in the absence of the UL36 gene product. Cells were infected with KOS and KΔUL36, and infected cell DNA was extracted 24 h after infection (18). The DNA was analyzed by Southern blot hybridization using the BamHI K junction fragment as a probe (Fig. 3). The probe hybridized to the K junction fragment and the two end (Q and S) fragments of KOS DNA extracted from Vero infected cells (lane 1). This result was consistent with the presence of linear unit-length genomes. This same pattern of hybridization was observed in DNA extracted from Vero cells infected with KΔUL36 (lane 2). Therefore, cleavage of viral DNA, and presumably DNA packaging into capsids, occurs in KΔUL36-infected cells. The presence of multiple bands of the S fragment is due to the heterogeneity of a sequences at the left end of the genome.
FIG. 3.

Cleavage of viral genomic DNA in KΔUL36-infected cells. Vero (lanes 1 and 2) and HS30 (lanes 3 and 4) cells (106 in 35-mm-diameter dishes) were infected with KOS (lanes 1 and 3) and KΔUL36 (lanes 2 and 4) at an MOI of 10 PFU/cell. Infected cell DNA was prepared 24 h postinfection, and 2 μg was digested with BamHI. The restriction fragments were analyzed by Southern blot hybridization using a 32P-labeled DNA fragment corresponding to the BamHI K junction fragment. The BamHI K junction fragment and terminal Q and S fragments are indicated at the right; shown below the blot is a schematic representation of the HSV-1 genome and locations of the BamHI junction and terminal fragments.
Capsid formation in KΔUL36-infected cells.
The state of viral DNA, examined as described above, suggested that the DNA would be packaged into capsids (15). To determine whether this was the case and to study the assembly process, capsid formation was studied by sedimentation of radiolabeled nuclear extracts through sucrose gradients. Cell monolayers were infected with KOS or the mutant virus and metabolically labeled with [35S]methionine from 8 to 24 h postinfection. Nuclear lysates were prepared and layered onto sucrose gradients. After sedimentation, fractions were collected and analyzed by SDS-PAGE (Fig. 4). The three peaks of radioactivity detected for KOS-infected cell extracts (Fig. 4A) corresponded to the faster-sedimenting C capsids (fraction 4 to 5) and the scaffold-filled B (fraction 8) and empty A (fraction 10) capsids. A, B, and C capsids contain VP5, VP19C, VP23, VP24, and VP26. Protein 22a was detected in B capsids, as expected. Some 22a was observed in the fraction corresponding to A capsids due to contamination with B capsids. Capsids detected in KΔUL36 extracts (Fig. 4B) were similar in composition and sedimentation profile to wild-type capsids. Therefore, the absence of the UL36-specified function does not abrogate capsid assembly.
FIG. 4.

Capsid formation in KΔUL36-infected cells. Vero cell monolayers (107 cells) were infected with KOS (A) and KΔUL36 (B) at an MOI of 10 PFU/cell and labeled with [35S]methionine from 8 to 24 h postinfection. Nuclear extracts were prepared and layered onto 20 to 50% sucrose gradients. Fractions collected after sedimentation were TCA precipitated, and the proteins in the fractions were resolved by SDS-PAGE (17% acrylamide). Direction of sedimentation was from right to left. The positions of capsid proteins are indicated at the left for KOS; the positions at which A, B, and C capsids sediment are indicated at the bottom.
Virion morphogenesis in KΔUL36-infected cells.
Since DNA-filled capsids were present in noncomplementing cells infected with the UL36 null mutant, the next step was to determine whether they mature into virions in these infected cells. In initial studies using sedimentation assays, HEL cell monolayers were infected with KOS and KΔUL36, and then intracellular virus was harvested and purified by rate velocity sedimentation through 20 to 50% sucrose gradients. Nuclear lysates prepared from KOS-infected cells were similarly sedimented. Virus and capsids were visualized as light-scattering bands in the gradients. This analysis revealed the presence of a thick light-scattering band for the mutant that had sedimentation properties similar to those of C capsids observed in nuclear lysates. A broad light-scattering band corresponding to enveloped virions was detected in gradients of KOS lysates but not in gradients from mutant lysates. A light-scattering band which sedimented at the position of C capsids was also observed in gradients of KOS-infected cell lysates, but at much lower levels. Light-scattering bands corresponding to A and B capsids were also visible in both KOS and KΔUL36 gradients. The sedimentation properties of these mutant particles were investigated using [3H] thymidine-labeled extracts, which were subjected to similar sedimentation conditions in sucrose gradients (Fig. 5A). Maximal incorporation of radioactivity into particles was detected in fractions 3 and 4 for KΔUL36. This coincides with the position at which C capsids sediment. For KOS, the radioactivity corresponding to labeled virions peaked in fraction 10 of the gradient and for C capsids in fractions 3 and 4. The amounts of the UL36 particles as judged by both incorporation of radioactivity and the intensity of the light-scattering band was always much greater than for wild-type particles. Therefore, no enveloped virions were detected in KΔUL36-infected cells. Sedimentation experiments were also performed for the rescued virus, KΔUL36R, using [3H]thymidine-labeled extracts prepared in Vero cells. In this experiment, radioactivity corresponding to C capsids peaked in fraction 3, and that for virions peaked in fraction 5 (Fig. 5B). This was due to differences in the time the particles were sedimented and fractionation of the gradient. The light-scattering band corresponding to enveloped virus was much broader in these extracts, and the levels of unenveloped C capsids detected in KOS extracts were consistently lower in Vero cells than in HEL cells. This appeared to be a cell type phenotype. The data presented in Fig. 5B show that the sedimentation profile of KΔUL36R DNA-containing particles was similar to that of wild-type virus.
FIG. 5.

Particle formation in KΔUL36-infected cells. HEL (2 × 107) (A) and Vero (107) (B) cells infected with KOS (A and B), KΔUL36 (A and B), and KΔUL36R (B) at an MOI of 10 PFU/cell were metabolically labeled with [3H]thymidine from 8 to 24 h postinfection. Intracellular virus was pelleted, loaded onto 20 to 50% sucrose gradients, and subjected to rate velocity sedimentation. The radioactivity present in the fractions collected was determined by liquid scintillation. Fraction 1 corresponds to the bottom of the tube.
The polypeptide composition of the UL36 particles was determined by similar sedimentation experiments using virus labeled with [35S]methionine. Infected cell lysates were subjected to rate velocity sedimentation through 20 to 50% sucrose gradients; the light-scattering bands visualized corresponded to all three capsid types for KOS and KΔUL36 gradients. A light-scattering band corresponding to enveloped virions was evident only for the KOS infections. The most abundant particles evident in the gradients of KOS were enveloped viruses, and those of the UL36 mutant were C capsid particles. Thus, these two particles were harvested by side puncture and resedimented through 20 to 50% sucrose gradients. This second sedimentation was necessary to obtain relatively pure preparations of these particles. The gradient was fractionated, and the fractions were analyzed by SDS-PAGE. The results shown in Fig. 6B demonstrate that the UL36 mutant particles (fraction 5) obtained from infected cell lysates have a composition similar to that of C capsids detected in nuclear lysates (Fig. 4B, fractions 4 and 5). The shell proteins VP5, VP19C, VP23, and VP26 are evident, as is the protease (VP24). These proteins are also present in the KOS (Fig. 6A) peak fractions (7 to 9). However, major envelope proteins, such as glycoproteins B and C (gB and gC) or the major tegument component such as VP16 which are readily observed in KOS fractions (Fig. 6A) are absent in the UL36 mutant particles (Fig. 6B). Therefore, the UL36 particles are similar in polypeptide composition to C capsids; that is, they lack both the tegument and envelope proteins.
FIG. 6.

SDS-PAGE analysis of KΔUL36 particles. HEL cells (2 × 107) infected with KOS and KΔUL36 at an MOI of 10 PFU/cell were labeled with [35S]methionine from 8 to 24 h after infection. Cell lysates were sedimented through 20 to 50% sucrose gradients, the light-scattering bands were harvested by side puncture, and the particles were pelleted and again sedimented through 20 to 50% sucrose gradients. Fractions collected after sedimentation were TCA precipitated, and the proteins in the fractions were resolved by SDS-PAGE (17% acrylamide). Fraction 1 corresponds to the bottom of the tube. Relevant virion proteins are indicated at the right; protein standards (lane M) correspond to 220, 97.4, 66, 46, 30, and 14.3 kDa (indicated by the closed circles).
The UL36 null mutant accumulates unenveloped capsids in the cytoplasm of infected cells.
Ultrastructural analysis of cells infected with the UL36 null mutant was carried out to obtain a graphic picture of the mutant particles detected by sedimentation analysis. Vero cell monolayers were infected with KOS or KΔUL36, and the cells were fixed at 16 h postinfection; thin sections were prepared, stained, and examined by TEM (Fig. 7). In KOS-infected cells, enveloped (Fig. 7A) and in some instances unenveloped (data not shown) capsids were observed in the cytoplasm. The latter result is consistent with reports of the presence of cytoplasmic unenveloped particles (11). In cells infected with KΔUL36, numerous particles that appear to be DNA-filled capsids were observed in the cytoplasm (Fig. 7B and C). These particles lack both the tegument layer and an envelope structure. The DNA present in these capsids binds to the heavy metal stains used, resulting in the greater electron density observed in these micrographs. Some capsids do not contain DNA and appear to contain the scaffold within the core. The reason why these were present in the cytoplasm was not clear; however, the majority of the particles detected contain DNA. In a cell infected with KΔUL36, numerous particles between the inner and outer nuclear membranes were evident (Fig. 7D). These particles were enveloped or in the process of envelopment. In the higher magnification shown in Fig. 7E are three particles in different cellular locations: an intranuclear capsid, a capsid that is enveloped, and an unenveloped capsid in the cytoplasm. This rare image of a cell displaying viruses traversing the nuclear envelope demonstrates that it is possible for the UL36 mutant capsids to traverse the nuclear envelope by envelopment at the inner nuclear membrane followed by deenvelopment at the outer nuclear membrane, resulting in the release of naked capsids into the cytosol. Thus, the absence of UL36 function results in the accumulation of unenveloped capsids in the cytoplasm. In addition, the mutation in UL36 appears not to hinder capsids from enveloping at the inner nuclear membrane, as judged by this ultrastructural assay.
Analysis in living cells of the replication of KΔUL36 tagged with VP26-GFP.
Due to the static nature of the EM analysis and the fixation process used, an assay was required to visualize the fate of the UL36 mutant particles in living cells. Recently a virus, K26GFP, that contained the green fluorescent protein (GFP) fused to VP26 was constructed (17). VP26 is the smallest capsid protein; a component of the shell, it decorates the outer surface of the capsid structure by virtue of its interaction with the major capsid protein, VP5 (38, 56, 58). When GFP was fused to the N terminus of VP26, the fusion protein retained the ability to bind to the capsid shell. The virus capsid, and consequently the mature virion, is fluorescently tagged and can be visualized in living cells, allowing the progression of virions during the maturation process to be followed. The VP26-GFP marker was crossed into the genome of the UL36 mutant. This was achieved by coinfecting HS30 cells with K26GFP and KΔUL36 viruses. K26GFP contains the VP26-GFP fusion construct in the KOS background. Individual plaques were scored in the fluorescence microscope and for a host range phenotype. Viruses that replicated on the complementing cell line but not Vero cells and that were fluorescent were isolated. The UL36 null mutant virus containing the VP26-GFP marker was designated KΔUL36-GFP. Capsids isolated from this virus contained the VP26-GFP protein (data not shown); therefore, the absence of UL36 does not alter the ability of the GFP fusion to bind to capsids. The VP26-GFP marker was also crossed into the gB null mutant virus (K082). K082 undergoes a complete cycle of maturation in Vero cells and produces enveloped particles that lack infectivity (6). Cells were infected at high MOI with the GFP-tagged viruses, and confocal analysis on living cells was performed (Fig. 8). At 8 h following infection (Fig. 8A and B), the fluorescence observed was predominantly nuclear for both viruses, consistent with the assembly of capsids in the nucleus. However, at late times (12 and 18 h postinfection) in wild-type-infected cells (Fig. 8C and E), fluorescence began to appear and accumulate at the plasma membrane, indicative of maturing viruses that have translocated to the cell surface. In the UL36 null mutant infected cells (Fig. 8D and F), fluorescence did not accumulate at the plasma membrane even at late times in infection. Therefore, virus egress to the cell surface was disrupted. What was observed was intense particulate fluorescence throughout the cytoplasm. This fluorescence corresponds to the cytoplasmic capsids observed by EM experiments. This observation in living cells gave a more accurate picture of the numbers and cellular location of UL36 mutant capsids. It was also interesting that a number of the nuclei in cells infected with KAUL36 (Fig. 8D and F) exhibited less fluorescence than wild-type-infected cells; the reason for this is not known. In addition, some wild-type-infected cells usually displayed an accumulation of fluorescence around the nucleus (Fig. 8E). This presumably corresponds to particles in the process of envelopment at the nuclear membrane. This pattern of fluorescence was never seen in KΔUL36-infected cells. Yet as seen by EM (Fig. 7D and E), the UL36 mutant particles traversed the nuclear envelope and were deposited in the cytoplasm. It is possible that this event occurred rapidly in KΔUL36-infected cells and hence there was generally less nuclear fluorescence. In HS30 cells infected with KΔUL36 or Vero cells infected with K082-GFP, the pattern of fluorescence observed was similar to that for wild-type-infected cells (data not shown). Therefore, in the absence of the UL36-encoded function, virus maturation and egress were arrested; consequently unenveloped capsids accumulated in infected cells and were present throughout the cytoplasm.
DISCUSSION
The UL36 gene product is the largest structural component of the virion particle; specifically, it is localized in the tegument layer of the virion (23, 25, 29, 30, 31, 39, 45). The functional role of this gene product in the virus replication cycle was determined by the isolation of a null mutant in the gene. Complementing cell lines were derived that express the UL36 gene product in trans, as judged by their ability to support the growth of tsB7 (27) at the nonpermissive temperature. The null mutation constructed in plasmid DNA was then transferred into the virus genome by homologous recombination using these complementing cell lines. A virus designated KΔUL36 was isolated, purified, and found to be unable to replicate on noncomplementing Vero cells. The genotype of the null mutant was confirmed by Southern blot analysis, which showed that the virus contained the null mutation.
The full spectrum of infected cell polypeptides are synthesized in KΔUL36-infected cells, as judged by SDS-PAGE analysis of radiolabeled protein lysates, the only exception being the UL36 polypeptide. In addition, viral DNA was cleaved and packaged into capsid structures, since both terminal end fragments of the virus genome were detected and C capsids were observed in nuclear extracts. However, these capsids do not mature into enveloped viruses. This was demonstrated by sedimentation analysis of infected cell lysates, which showed that the mutant virus particles have sedimentation properties similar to those of C capsids. In addition, SDS-PAGE analysis of the polypeptide composition of these particles showed that they are devoid of the major envelope and tegument proteins. This was confirmed by ultrastructural analysis of infected cells, which revealed the presence of numerous unenveloped DNA-filled capsids in the cytoplasm. Progression of the UL36 mutant particles in living cells was followed by using the VP26-GFP tag. At early times after infection, fluorescence corresponding to intranuclear particles was observed. As the infection progressed, fluorescence corresponding to wild-type viruses was observed to accumulate at the plasma membrane, indicative of virus maturation and egress to the cell surface. In contrast, intense particulate fluorescence corresponding to the UL36 mutant particles was distributed throughout the cytoplasm. Plasma membrane (cell surface) fluorescence indicative of mature viruses was never detected. Therefore, the UL36 gene specifies a function required for the maturation of capsids into enveloped virus and consequently their egress to the cell surface, possibly by actively translocating particles to a cytoplasmic maturation compartment.
A number of constituents of the tegument, including vhs (36, 37), VP11/12, VP13/14 (57), and others (50), can be lost without significant effect on the virus replicative cycle in cell culture. The role of the major tegument protein VP16 in the structural integrity of the tegument and the mature virion is clearly critical (1, 52). Other tegument proteins that specify maturation functions include the products of the UL11 (2), UL14 (12), and UL37 (P. Desai, unpublished data) ORFs. The function encoded by the UL36 gene is also essential for virus replication. Its role in the virus life cycle is complex: it acts at the onset of the life cycle to uncoat the virus genome (3, 27); late in infection it may be required to target capsids to a cytoplasmic maturation compartment for final envelopment. This dual functionality both early and late in infection is typical of many tegument proteins.
The most dramatic phenotype of the UL36 mutation is the inability of DNA-filled capsids to acquire a tegument/envelope structure and consequently abrogation of infectious virus production. The numerous particles present in the cytoplasm are not membrane bound, since they are easily isolated in large quantities from infected cell lysates in the absence of detergent. Since the particles are present in the cytoplasm, the capsids must have traversed the nuclear membrane, presumably by budding into the inner nuclear membrane followed by fusion of these particles with the outer nuclear membrane to release naked capsids into the cytosol. This was observed visually; hence, UL36 may not be required for initial envelopment at the nucleus. The mutant particles progress beyond the nucleus but are randomly distributed throughout the cytoplasm. Therefore, a second late function of UL36 may be to direct these capsids to a cytoplasmic maturation compartment for final envelopment. The cytoplasmic site would serve to add cytosolic tegument proteins to the maturing capsid and envelop this structure. This cytoplasmic compartment would be a critical maturation point for the capsids as they traverse to the cell surface. In the absence of UL36, the numerous C capsids produced are not transported to this cytoplasmic site; they do not become enveloped and are therefore never detected at the cell surface. Immunofluorescence studies using antibodies to UL36 have shown that the protein is concentrated at the nuclear periphery late in infection (32). In a normal infection, UL36 maybe added to capsids as they exit the nucleus, and they would then translocate to a cytoplasmic compartment for final envelopment. How UL36 directs this translocation is unclear at present, but it may do so by interacting with the host cell cytoskeleton and the secretory pathway. This speculation is of course relevant only if HSV uses a two-stage method of envelopment.
How the herpes simplex virion acquires its envelope is a controversial issue. The two models proposed to explain how a virus becomes enveloped, single envelopment versus the two-stage envelopment process, each have supporters and evidence in the literature. An elegant discussion by Whitely et al. (54) of the evidence in favor of the two-stage mode of virus maturation was published recently. Their studies involved an analysis of the envelope composition of virions following restriction of the essential glycoproteins gH (5) and gD (54) to different cellular organelles. Their results showed that endoplasmic reticulum-restricted glycoproteins are not incorporated into the mature virion (5, 54), whereas Golgi-localized glycoproteins are part of the mature envelope (54). These data strongly suggest that HSV-1 uses membrane exchange during the maturation process. The Golgi compartment is a crucial organelle for the biogenesis of the infectious virion (20, 26, 49, 53, 54). This compartment or a post-Golgi organelle may act as the site where final maturation of the HSV-1 virus takes place. The phenotype of the UL36 mutant does not definitively support this model of maturation, since it has been argued that cytoplasmic unenveloped capsids observed for another mutant virus represent dead ends (7). It is possible that the location of the UL36 mutant capsids in the cytoplasm is due to aberrant fusion events that occur at the nuclear membrane. Nevertheless, it is compelling to argue that the function of the UL36 gene product may be to actively direct capsids to a maturation compartment. In the absence of UL36, capsids acquire their initial envelope from the inner nuclear membrane and then by fusing with the outer membrane are released into the cytosol. However, the mutant particles are not transported to the correct compartment for maturation, envelopment, and egress to the plasma membrane.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant AI33077 from the National Institutes of Health.
I acknowledge the generous support, both scientifically and personally, of Stan Person over the years. He has taught me a lot during our time together, even when I thought my learning days were over. As a mentor and friend he has done a great job. In addition, I acknowledge scientific discussions with and support from Wade Gibson and members of his laboratory. I acknowledge Neal A. DeLuca for discussions of the data. The virus tsB7 was generously provided by David Knipe. Finally, I thank members of the Johns Hopkins School of Medicine microscope facility: Mike Delannoy for help with the confocal and EM studies and Carol Cooke and Brad Harris for help with the EM experiments.
REFERENCES
- 1.Ace C I, Dalrymple M A, Ramsay F H, Preston V G, Preston C M. Mutational analysis of the herpes simplex virus type 1 trans-inducing factor Vmw65. J Gen Virol. 1988;69:2595–2605. doi: 10.1099/0022-1317-69-10-2595. [DOI] [PubMed] [Google Scholar]
- 2.Baines J D, Roizman B. The UL11 gene of herpes simplex virus type 1 encodes a function that facilitates nucleocapsid envelopment and egress from cells. J Virol. 1992;66:5168–5174. doi: 10.1128/jvi.66.8.5168-5174.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batterson W, Furlong D, Roizman B. Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J Virol. 1983;45:397–407. doi: 10.1128/jvi.45.1.397-407.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batterson W, Roizman B. Characterization of the herpes simplex virion-associated factor responsible for the induction of α genes. J Virol. 1983;46:371–377. doi: 10.1128/jvi.46.2.371-377.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Browne H B, Bell S, Minson T, Wilson D W. An endoplasmic reticulum-retained herpes simplex virus glycoportein H is absent from secreted virions: evidence for reenvelopment during egress. J Virol. 1996;70:4311–4316. doi: 10.1128/jvi.70.7.4311-4316.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai W, Person S, Warner S C, Zhou J, DeLuca N A. Linker-insertion nonsense and restriction-site deletion mutations of the gB glycoprotein gene of herpes simplex virus type 1. J Virol. 1987;61:714–721. doi: 10.1128/jvi.61.3.714-721.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campadelli-Fiume G, Farabegoli F, Di Gaeta S, Roizman B. Origin of unenveloped capsids in the cytoplasm of cells infected with herpes simplex virus 1. J Virol. 1991;65:1589–1595. doi: 10.1128/jvi.65.3.1589-1595.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell M E M, Palfreyman J W, Preston C M. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for the stimulation of immediate early transcription. J Mol Biol. 1984;180:1–19. doi: 10.1016/0022-2836(84)90427-3. [DOI] [PubMed] [Google Scholar]
- 9.Chen D H, Jiang H, Lee M, Liu F, Zhou Z H. Three-dimensional visualization of tegument/capsid interactions in the intact human cytomegalovirus. Virology. 1999;260:10–16. doi: 10.1006/viro.1999.9791. [DOI] [PubMed] [Google Scholar]
- 10.Chou J, Roizman B. Characterization of DNA sequence-common and sequence-specific proteins binding to cis-acting sites for cleavage of the terminal a sequence of the herpes simplex virus 1 genome. J Virol. 1993;63:1059–1068. doi: 10.1128/jvi.63.3.1059-1068.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Church G A, Wilson D W. Study of herpes simplex virus maturation during a synchronous wave of assembly. J Virol. 1997;71:3603–3612. doi: 10.1128/jvi.71.5.3603-3612.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunningham C, Davison A J, MacLean A R, Taus N S, Baines J D. Herpes simplex virus type 1 gene UL14: phenotype of a null mutant and identification of the encoded protein. J Virol. 2000;74:33–41. doi: 10.1128/jvi.74.1.33-41.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darlington R W, Moss L H., III Herpesvirus envelopement. J Virol. 1968;2:48–55. doi: 10.1128/jvi.2.1.48-55.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeLuca N A, McCarthy A M, Schaffer P A. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J Virol. 1985;56:558–570. doi: 10.1128/jvi.56.2.558-570.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desai P, DeLuca N A, Glorioso J C, Person S. Mutations in herpes simplex virus type 1 genes encoding VP5 and VP23 abrogate capsid formation and cleavage of replicated DNA. J Virol. 1993;67:1357–1364. doi: 10.1128/jvi.67.3.1357-1364.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desai P, DeLuca N A, Person S. Herpes simplex virus type 1 VP26 is not essential for replication in cell culture but influences production of infectious virus in the nervous system of infected mice. Virology. 1998;247:115–124. doi: 10.1006/viro.1998.9230. [DOI] [PubMed] [Google Scholar]
- 17.Desai P, Person S. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J Virol. 1998;72:7563–7568. doi: 10.1128/jvi.72.9.7563-7568.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desai P, Person S. Second site mutations in the N-terminus of the major capsid protein (VP5) overcome a block at the maturation cleavage site of the capsid scaffold proteins of herpes simplex virus type 1. Virology. 1999;261:357–366. doi: 10.1006/viro.1999.9877. [DOI] [PubMed] [Google Scholar]
- 19.Elliot G, Mouzakitis G, O'Hare P. VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J Virol. 1995;69:7932–7941. doi: 10.1128/jvi.69.12.7932-7941.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gershon A A, Sherman D L, Zhu Z, Gabel C A, Ambron R T, Gershon M D. Intracellular transport of newly synthesized varicella-zoster virus: final envelopment in the trans-Golgi network. J Virol. 1994;68:6372–6390. doi: 10.1128/jvi.68.10.6372-6390.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibson W, Roizman B. Proteins specified by herpes simplex virus. VIII. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J Virol. 1972;10:1044–1052. doi: 10.1128/jvi.10.5.1044-1052.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Granzow H, Weiland F, Jons A, Klupp B G, Karger A, Mettenleiter T C. Ultrastructural analysis of the replication cycle of pseudorabies virus in cell culture: a reassessment. J Virol. 1997;71:2072–2082. doi: 10.1128/jvi.71.3.2072-2082.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heine J W, Honess R W, Cassai E, Roizman B. Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J Virol. 1974;14:640–651. doi: 10.1128/jvi.14.3.640-651.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Homa F L, Brown J C. Capsid assembly and DNA packaging in herpes simplex virus. Med Virol. 1997;7:107–122. doi: 10.1002/(sici)1099-1654(199707)7:2<107::aid-rmv191>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 25.Honess R W, Roizman B. Proteins specified by herpes simplex virus. XI. Identification and relative molar rates of synthesis of structural and nonstructural herpes virus polypeptides in the infected cell. J Virol. 1973;12:1347–1365. doi: 10.1128/jvi.12.6.1347-1365.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson D C, Spear P G. Monensin inhibits the processing of herpes simplex virus glycoproteins, their transport to the cell surface, and the egress of virions from infected cells. J Virol. 1982;43:1102–1112. doi: 10.1128/jvi.43.3.1102-1112.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knipe D M, Ruyechan W T, Roizman B. Molecular genetics of herpes simplex virus. VI. Characterization of a temperature-sensitive mutant defective in the expression of all early viral gene products. J Virol. 1979;38:539–547. doi: 10.1128/jvi.38.2.539-547.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwong A D, Kruper J A, Frenkel N. Herpes simplex virus virion host shutoff function. J Virol. 1988;62:912–921. doi: 10.1128/jvi.62.3.912-921.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McGeoch D J, Dalrymple M A, Davison A J, Dolan A, Frame M C, McNab D, Perry L J, Scott J E, Taylor P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol. 1988;69:1531–1574. doi: 10.1099/0022-1317-69-7-1531. [DOI] [PubMed] [Google Scholar]
- 30.McNabb D S, Courtney R J. Characterization of the large tegument protein (ICP1/2) of herpes simplex virus type 1. Virology. 1992;190:221–232. doi: 10.1016/0042-6822(92)91208-c. [DOI] [PubMed] [Google Scholar]
- 31.McNabb D S, Courtney R J. Analysis of the UL36 open reading frame encoding the large tegument protein (ICP1/2) of herpes simplex virus type 1. J Virol. 1992;66:7581–7584. doi: 10.1128/jvi.66.12.7581-7584.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morrison E E, Stevenson A J, Wang Y-F, Meredith D M. Differences in the intracellular localization and fate of herpes simplex virus tegument proteins early in the infection of Vero cells. J Gen Virol. 1998;79:2517–2528. doi: 10.1099/0022-1317-79-10-2517. [DOI] [PubMed] [Google Scholar]
- 33.Overton H A, McMillan D J, Klavinskis L S, Hope L, Ritchie A J, Wong-kai-in P. Herpes simplex virus type 1 gene UL13 encodes a phosphoprotein that is a component of the virion. Virology. 1992;190:184–192. doi: 10.1016/0042-6822(92)91204-8. [DOI] [PubMed] [Google Scholar]
- 34.Penfold M E T, Armati P, Cunningham A L. Axonal transport of herpes simplex virions to epidermal cells: evidence for a specialized mode of virus transport and assembly. Proc Natl Acad Sci USA. 1994;91:6529–6533. doi: 10.1073/pnas.91.14.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Person S, Desai P. Capsids are formed in a mutant virus blocked at the maturation site of the UL26 and UL26.5 open reading frame of herpes simplex virus type 1 but are not formed in a null mutant of UL38 (VP19C) Virology. 1998;242:193–203. doi: 10.1006/viro.1997.9005. [DOI] [PubMed] [Google Scholar]
- 36.Read G S, Frenkel N. Herpes simplex virus mutants defective in the virion shut-off of host polypeptide synthesis and exhibiting abnormal synthesis of α (immediate-early) viral polypeptides. J Virol. 1983;46:498–512. doi: 10.1128/jvi.46.2.498-512.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Read G S, Karr B M, Knight K. Isolation of a herpes simplex virus type 1 mutant with a deletion in the virion host shutoff gene and identification of multiple forms of the vhs (UL41) polypeptide. J Virol. 1993;67:7149–7160. doi: 10.1128/jvi.67.12.7149-7160.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rixon F J, Addison C, McGregor A, McNab S J, Nicholson P, Preston V G, Tatman J D. Multiple interactions control the intracellular localization of the herpes simplex virus type 1 capsid proteins. J Gen Virol. 1996;77:2251–2260. doi: 10.1099/0022-1317-77-9-2251. [DOI] [PubMed] [Google Scholar]
- 39.Roizman B, Furlong D. The replication of herpesviruses. In: Fraenkel-Conrat H, Wagner R R, editors. Comprehensive virology. New York, N.Y: Plenum Press; 1974. pp. 11–68. [Google Scholar]
- 40.Roizman B, Sears A. Herpes simplex viruses and their replication. In: Fields B N, Knipe D M, Howley P M, et al., editors. Fields virology. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 2231–2295. [Google Scholar]
- 41.Roller R J, Roizman B. The herpes simplex virus 1 RNA binding protein UL11 is a virion component and associates with ribosomal 60S subunits. J Virol. 1992;66:3624–3632. doi: 10.1128/jvi.66.6.3624-3632.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salmon B, Cunningham C, Davison A J, Harris W J, Baines J D. The herpes simplex virus type 1 UL17 gene encodes virion tegument proteins that are required for cleavage and packaging of viral DNA. J Virol. 1998;72:3779–3788. doi: 10.1128/jvi.72.5.3779-3788.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smibert C A, Popova B, Xiao P, Capone J P, Smiley J R. Herpes simplex virus VP16 forms a complex with the virion host shutoff protein Vhs. J Virol. 1994;68:2339–2346. doi: 10.1128/jvi.68.4.2339-2346.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Southern P J, Berg P. Transformation of mammalian cells to antibiotic resistance with a bacterial gene under the control of the SV40 early region promoter. J Mol Appl Genet. 1982;1:327–341. [PubMed] [Google Scholar]
- 45.Spear P G, Roizman B. Proteins specified by herpes simplex virus. V. Purification and structural proteins of the herpes virion. J Virol. 1972;9:143–159. doi: 10.1128/jvi.9.1.143-159.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stackpole C W. Herpes-type virus of the frog renal adenocarcinoma. I. Virus development in tumor transplants maintained at low temperature. J Virol. 1969;4:75–93. doi: 10.1128/jvi.4.1.75-93.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steven A C, Spear P G. Herpesvirus capsid assembly and envelopment. In: Burnett R, Chiu W, Garcea R, editors. Structural biology of viruses. New York, N.Y: Oxford University Press; 1996. pp. 312–351. [Google Scholar]
- 48.Trus B L, Gibson W, Cheng N, Steven A C. Capsid structure of simian cytomegalovirus from cryoelectron microscopy: evidence for tegument attachment sites. J Virol. 1999;73:2181–2192. doi: 10.1128/jvi.73.3.2181-2192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Genderen I L, Brandimarti R, Torrisi M R, Campadelli-Fiume G, van Meer G. The phospholipid concentration of extracellular herpes simplex virions differs from that of the host cell nuclei. Virology. 1994;200:831–836. doi: 10.1006/viro.1994.1252. [DOI] [PubMed] [Google Scholar]
- 50.Ward P L, Roizman B. Herpes simplex genes: the blueprint of a successful human pathogen. Trends Genet. 1994;10:267–275. doi: 10.1016/0168-9525(90)90009-u. [DOI] [PubMed] [Google Scholar]
- 51.Ward P L, Ogle W O, Roizman B. Assemblons: nuclear structures defined by aggregation of immature capsids and some tegument proteins of herpes simplex virus type 1. J Virol. 1996;70:4623–4631. doi: 10.1128/jvi.70.7.4623-4631.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weinheimer S P, Boyd B A, Durham S K, Resnick J L, O'Boyle D R. Deletion of the VP16 open reading frame of herpes simplex virus type 1. J Virol. 1992;66:258–269. doi: 10.1128/jvi.66.1.258-269.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Whealy M E, Card J P, Meade R P, Robbins A K, Enquist L W. Effect of brefeldin A on alphaherpesvirus membrane protein glycosylation and virus egress. J Virol. 1991;65:1006–1081. doi: 10.1128/jvi.65.3.1066-1081.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whitely A, Bruun B, Minson T, Browne H. Effects of targeting herpes simplex virus type 1 gD to the endoplasmic reticulum and trans-Golgi network. J Virol. 1999;73:9515–9520. doi: 10.1128/jvi.73.11.9515-9520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wildy P, Watson D H. Electron microscopic studies on the architecture of animal viruses. Cold Spring Harbor Symp Quant Biol. 1963;27:25–47. doi: 10.1101/sqb.1962.027.001.006. [DOI] [PubMed] [Google Scholar]
- 56.Wingfield P T, Stahl S J, Thomsen D R, Homa F L, Booy F P, Trus B L, Steven A C. Hexon-only binding of VP26 reflects differences between the hexon and penton conformations of VP5, the major capsid protein of herpes simplex virus. J Virol. 1997;71:8955–8961. doi: 10.1128/jvi.71.12.8955-8961.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y, Sirko D A, McKnight J L C. Role of the herpes simplex virus type 1 UL46 and UL47 in αTIF-mediated transcriptional induction: characterization of three viral deletion mutants. J Virol. 1991;65:829–841. doi: 10.1128/jvi.65.2.829-841.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou Z H, He J, Jakana J, Tatman J, Rixon F J, Chiu W. Assembly of VP26 in herpes simplex virus-1 inferred from structures of wild-type and recombinant capsids. Nat Struct Biol. 1995;2:1026–1030. doi: 10.1038/nsb1195-1026. [DOI] [PubMed] [Google Scholar]
- 59.Zhou Z H, Chen D H, Jakana J, Rixon F J, Chiu W. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J Virol. 1999;73:3210–3218. doi: 10.1128/jvi.73.4.3210-3218.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]