

ABSTRACT
Intergenerational and transgenerational epigenetic effects resulting from conditions in previous generations can contribute to environmental adaptation as well as disease susceptibility. Previous studies in rodent and human models have shown that abnormal developmental exposure to thyroid hormone affects endocrine function and thyroid hormone sensitivity in later generations. Since the imprinted type 3 deiodinase gene (Dio3) regulates sensitivity to thyroid hormones, we hypothesize its epigenetic regulation is altered in descendants of thyroid hormone overexposed individuals. Using DIO3-deficient mice as a model of developmental thyrotoxicosis, we investigated Dio3 total and allelic expression and growth and endocrine phenotypes in descendants. We observed that male and female developmental overexposure to thyroid hormone altered total and allelic Dio3 expression in genetically intact descendants in a tissue-specific manner. This was associated with abnormal growth and neonatal levels of thyroid hormone and leptin. Descendant mice also exhibited molecular abnormalities in the Dlk1-Dio3 imprinted domain, including increased methylation in Meg3 and altered foetal brain expression of other genes of the Dlk1-Dio3 imprinted domain. These molecular abnormalities were also observed in the tissues and germ line of DIO3-deficient ancestors originally overexposed to thyroid hormone in utero. Our results provide a novel paradigm of epigenetic self-memory by which Dio3 gene dosage in a given individual, and its dependent developmental exposure to thyroid hormone, influences its own expression in future generations. This mechanism of epigenetic self-correction of Dio3 expression in each generation may be instrumental in descendants for their adaptive programming of developmental growth and adult endocrine function.
KEYWORDS: Thyroid hormone, Dlk1-Dio3 imprinted domain, Dio3, neonatal leptin, DNA methylation, transgenerational epigenetics, Meg3, developmental growth
Introduction
Investigations in recent years are increasingly showing that biological traits and pathologies can be inherited via epigenetic mechanisms resulting from the environmental circumstances to which progenitors or previous ancestors were exposed. Diet [1–4], exposure to chemicals, hormones and endocrine disruptors [5,6], stressful events [7–10], and social interactions [11–14] and drugs [15] are but a few of the environmental conditions that can affect phenotypes in descendants for one or more generations. Although the mechanisms underlying these intergenerational and transgenerational effects are not fully understood, they likely involve environmentally driven alterations in the epigenetic information carried by germ cells into the next generation [16–18]. It is then possible that intergenerational epigenetic effects induced by factors surrounding previous generations contribute to explain the ‘missing heritability’ in the aetiology of complex human disorders [19–22].
Interestingly, many of the models of intergenerational epigenetic effects described to date presumably involve signalling through members of the nuclear receptor superfamily [23]. Models of stress or glucocorticoid administration [24–28] or exposure to chemicals with oestrogenic or androgenic characteristics [29,30] may lead to altered signalling through the putative receptors of the corresponding hormones to directly or indirectly change the epigenetic information of the germ line.
Thyroid hormones also signal via members of the nuclear receptor superfamily. As others and we have shown, alterations in thyroid hormone levels result in abnormal biological traits in future generations [31–34]. Seminal work in rats made hypothyroid in adult life or overexposed to elevated thyroid hormone levels during neonatal development demonstrated that their offspring exhibits abnormalities in body weight endocrine tissue size and neuroendocrine physiology [34,35]. Humans overexposed to thyroid hormones in utero due to a genetic defect in the thyroid hormone receptor beta develop reduced pituitary sensitivity to thyroid hormone, a trait transmitted along the paternal lineage to genetically normal individuals at least for two generations [32]. In humans, the intergenerational effects of thyroid hormones could be of particular significance given the relatively high prevalence of thyroid disease [36] and the ubiquitous exposure to environmental chemicals that interfere with thyroid hormone physiology and action [37–39].
Transgenerational epigenetic effects on phenotypes have also been reported as a result of specific genotypes in previous generations [40]. We have also reported evidence of the intergenerational epigenetic effects of thyroid hormones using mice with an inactivation of Dio3 [33]. Dio3 is an imprinted gene in mice and humans [41–43] coding for the type 3 deiodinase (DIO3). This enzyme inactivates thyroid hormones [44] and has a critical modulatory role during development to prevent premature foetal exposure to adult hormone levels, ensuring appropriate timeliness in the tissue maturation effects of thyroid hormones [45]. Dio3-/- mice exhibit thyrotoxicosis during in utero and early neonatal life [46,47]. This insult causes alterations in the sperm methylome of Dio3-/- adult male mice [33]. These altered epigenetic signatures lead to pathology in genetically normal descendants, including abnormalities in gene expression profiles during brain development and adult behaviours concerning anxiety, locomotor activity [33] and sociability [48]. These observations support a role for ascendants’ thyroid hormone abnormalities in the non-genetic inheritance of susceptibility to neuroendocrine and neurological disease.
As with other models of intergenerational epigenetic effects, the underlying mechanisms remain poorly defined. The establishment of altered epigenetic information carried in germ cells is the likely first step, but how this information is interpreted during development in subsequent generations, and translated into particular adult traits and germ line epigenomes, is still largely unknown. Among the transgenerational epigenetic effects of thyroid hormone overexposure in humans [32] is decreased sensitivity to the more active thyroid hormone, T3. As Dio3 is responsible for regulating T3 sensitivity via inactivation of thyroid hormones, we hypothesize that alterations in Dio3 expression and its epigenetic regulation are part of the endocrine syndrome affecting descendants of mice overexposed to T3 during development.
Here we studied genetically intact F3 generation descendants of mice that experienced developmental thyrotoxicosis due to loss of DIO3 function. We show that transgenerational effects include changes in the total and allelic expression of Dio3, abnormalities in growth and neonatal levels of thyroid hormone and leptin, and changes in the methylation and gene expression in the Dlk1-Dio3 imprinted domain. These observations support a paradigm of intergenerational epigenetic self-memory of Dio3, which may partly mediate the inter- and trans-generational epigenetic effects of altered thyroid hormone states.
Materials and methods
Experimental animals
As a model of developmental overexposure to thyroid hormone (T3), we used mice genetically deficient in the type 3 deiodinase (DIO3). We have previously described that Dio3 -/- mice exhibit markedly elevated serum levels of T3 during foetal and early life [46]. All experimental mice were on an outbred CD-1 genetic background to overcome the severely impaired fertility of Dio3-/- mice on inbred genetic backgrounds [49]. The original mutant mouse strain was generated in a 129/SvJ genetic background [42,46] and has been backcrossed on a CD-1 background for more than 12 generations. Due to the genomic imprinting of the Dio3 gene [41,42,50], the colony has been maintained for more than 20 generations by crossing wild type males with heterozygous females, so that the heterozygous mice generated are phenotypically normal, as they carry the Dio3 mutation in the maternal allele, which is already largely suppressed due to genomic imprinting [41,42]. Approximately every six generations, the genetic background of the colony has been refreshed with a wild type CD-1 male purchased from Charles River. Dio3+/+ and Dio3-/- mice were littermates generated by crosses of heterozygous mice. For transgenerational experiments, and to avoid the influence of confounding factors and minimize variability, mothers of foetal and neonatal experimental animals were mated at two months of age and all experimental animals of neonatal age were born before the mother was five months old. For experimental foetuses, the morning a vaginal plug was noticed was considered embryonic day 0.5. Experimental mice represent only first litters from at least three different mothers per experimental group, and litter size was limited to 8–12 animals. For main F3-generation experimental groups, DIO3 enzymatic activity data represent nine to eleven different mothers form three different experiments, each experiment representing three or four different mothers per experimental group. Mothers of experimental mice of neonatal age were isolated before giving birth to raise the pups in the absence of the father and prevent a concurrent pregnancy when nursing the pups. All mice were maintained on a 12 h light/dark cycle and food and water were provided ad libitum. Tissues and blood samples were harvested between 2 and 5 h into the light cycle. Mice were euthanized by CO2 asphyxiation. All experiments were approved by the MaineHealth Institute for Research Institutional Animal Care and Use Committee (IACUC), under current protocol number 2112.
DNA methylation
DNA was isolated from tissues following standard methods of proteinase K digestion and isopropanol precipitation. Purified DNA was submitted to EpigenDX (Hopkinton, MA) for methylation quantification. This was achieved by bisulphite treatment of DNA to convert non-methylated cytosines, followed by PCR amplification of the small region of interest, and pyrosequencing [51], a sequencing method based on the release and quantification of pyrophosphate in each nucleotide incorporation during DNA synthesis. We utilized the company’s assays for the mouse IG-DMR (assay numbers ADS1452FS2 and ADS1452FS3) and the maternally expressed gene 3 (Meg3) promoter region (Assay number ADS1341FS3re and ADS1341FS3). These assays encompass the sequence and specific CpG methylation sites shown in Supporting Information (S1 and S2 Figs). For methylation of imprinted maternally expressed transcript (H19), used as a control, we used EpigenDX assays ADS438F1, ADS438FS4, ADS438FS2, ADS438FS3, ADS1702RS1, ADS1702RS2, ADS445FS, ADS447FS, ADS446FS1 and ADS446FS2, encompassing 40 individual CpG sites.
Real time quantitative PCR
Tissues were harvested and immediately frozen on dry ice and stored at −70 C. Total RNA was extracted using the RNeasy kit from Qiagen (Valencia, CA). Total RNA (1 µg) was reverse transcribed with M-MLV reverse transcriptase in the presence of random decamers (both from Thermo Fisher Scientific, Waltham, MA) at 65°C for 5 min, then 37°C for 50 min. The 20 μl reverse transcription reactions were diluted by adding 230 μl of DNase and RNase free water. An aliquot of each sample was mixed together for an internal standard and diluted fourfold. Real-time PCR reactions were set up in duplicate with gene-specific primers and SYBR Select Master Mix (Thermo Fisher Scientific, Waltham, MA) and run on the CFX Connect from Bio-Rad (Hercules, CA), where they underwent an initial 10 min denaturing step, followed by 36 cycles of a denaturing step (94°C for 30 s) and an annealing/extension step (60°C for 1 min). For each individual sample, expression was corrected by the expression of housekeeping gene Gapdh after establishing that there was not a statistically significant difference in its expression between experimental groups and that the means between experimental groups did not vary more than 10%. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) expression did not meet these standards in some tissues of Dio3-/- mice and 18S ribosomal RNA (Rn18s) was instead used. mRNA expression data are shown in arbitrary units and represented as fold-increase over the mean value in the control group. The sequences of the primers used for each gene are shown in Supporting Information (S1 Table). The specificity of all qPCR amplicons has been previously validated in our laboratory by sequencing and melting curve determinations.
DIO3 enzymatic activity
DIO3 enzymatic activities in tissues from experimental mice were determined as previously described [46]. In brief, tissues were homogenized in a 10 mM Tris-HCl, 0.25 M sucrose (pH 7.4) buffer. A suitable volume of tissue homogenate was used in the enzymatic reaction to ensure that total deiodination did not exceed 40% in the assay and was proportional to the protein content. Tissue homogenates were incubated at 37°C for an hour with 2 nM125I-labelled T3 (PerkinElmer) in the presence of 25 mM dithiothreitol. Deiodination was determined based on the percentage of125I-3,3-diiodothyronine produced. The latter was determined by measuring the amount of radioactivity associated with the reaction products after separation by paper chromatography as described [52].
Hormone determinations
Blood from P15 mice was collected from the inferior vena cava, while trunk blood was collected from P3 mice. Blood was allowed to clot at 4°C for at least four hours. After 10 minutes centrifugation at 3000 g, serum was taken from the supernatant and stored at −70°C until further used. Serum levels of T3 and T4 were measured in 10 and 1 μl of serum, respectively, as previously described [53–55] by highly sensitive specific radioimmunoassays using in-house generated antibodies. TSH determinations were performed in the laboratory of Samuel Refetoff at the University of Chicago, as previously described [56]. Serum determination of leptin was performed using commercially available ELISA kits according to the manufacturer’s instructions. Leptin kits were purchased from R&D Systems (Bio-Techne, Minneapolis, MN, USA).
Statistical analyses
Statistical analysis was performed using the statistical tools of GraphPad Prism 6 (GraphPad Software, Inc.). A Student’s t-test or a one-way ANOVA (ancestry as an independent variable) followed by Tukey’s post hoc test were used to determine statistical significance, which was defined as p < 0.05. Based on histograms of measured endpoints, we assumed normality and homocedasticity, and utilized the above tests in some experiments in which a low number of samples per experimental group were analysed. Unless otherwise stated, data are represented as the mean ± SEM.
Experimental design
We have recently shown that the developmental thyroid hormone excess (thyrotoxicosis) of Dio3-/- mice causes alterations in the sperm methylome, and this is associated in genetically intact descendants with changes in neonatal brain gene expression patterns and abnormal adult behaviour [33]. Using a similar experimental design (shown in Figure 1a), we studied F3-generation mice that were genetically intact (Dio3+/+), but had a Dio3-/- paternal grandfather (PGF mice) or paternal grandmother (PGM mice). Both experimental groups of mice referred to as PGF and PGM mice, in addition to be genetically intact (Dio3+/+), were born to genetically intact Dio3+/+ mothers (Figure 1a). Dio3+/+ mice with no Dio3-/- ancestry were used as the control group (Ctrl mice, Figure 1a). Note that the diagram in Figure 1 is designed to compare it to most prevalent transgenerational epigenetic inheritance models, in which pregnant mothers are exposed to an insult or chemical, and the foetus (F1 generation) and its germ line (F2 generation) are also exposed to the insult. In our model, because Dio3 is mostly expressed during development, Dio3-/- mice are only overexposed to thyroid hormone during development but in not adult life [57], so pregnancy in F1 generation females does not result in their foetuses being overexposed (Figure 1a).
Figure 1.
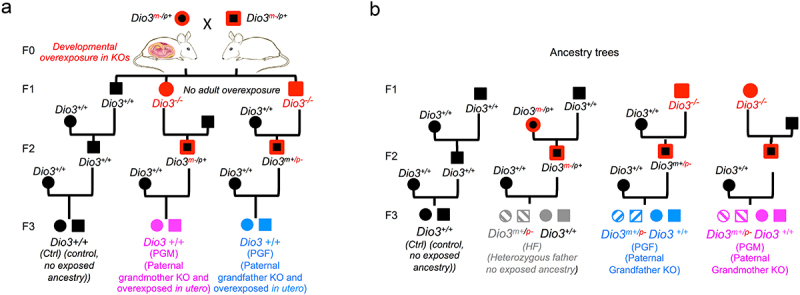
Ancestry trees of the F3 generation mice studied.
(a) Ancestry trees of mice studied in the experiments. (b) Ancestry trees of mice studied, detailing those heterozygous mice used to evaluate Dio3 allelic expression. Note that F1 generation Dio3-/- mice (males or females) are not overexposed to thyroid hormone in adult life. (Not all possible genotypes are shown).
To assess allelic Dio3 expression, we determined DIO3 enzymatic activity in the heterozygous littermates of PGF and PGM mice (Figure 1b). These heterozygous littermates (referred to as Het-PGF and Het-PGF) are subjected to the same transgenerational epigenetic effects as wild type PGF and PGM mice, but their paternally inherited allele is mutated, so only the maternal allele can produce a functional protein capable of enzymatic activity. As additional control groups for normal Dio3 allelic expression and for Het-PGF and Het-PGF mice, we used wild type and heterozygous mice littermates with no thyroid hormone overexposed ancestry. These were generated by crossing wild-type females and heterozygous fathers that were largely DIO3-sufficient because their mutated allele was maternally inherited [41,42,50]. These additional control groups (shown in grey colour in Figure 1b) are referred to as HF mice (genetically intact) and Het-HF mice (heterozygous littermates).
Results
Tissue-specific F3 generation changes in Dio3 expression and DNA methylation
We studied Dio3 expression in postnatal day 3 (P3) and P15 tissues of PGF and PGM mice. These two developmental stages were chosen because at P3 Dio3 expression is relatively high in most tissues examined, and P15 is a stage at which the thyroid endocrine axis matures and serum levels of thyroid hormones peak in the circulation. The tissues studied were selected to represent the range of previously observed variability in the percentage of Dio3 monoallelic expression [50]. We examined Dio3 expression at the level of both mRNA as well as protein by measuring DIO3 enzymatic activity. Compared to control mice, DIO3 activity in P3 PGF mice was significantly elevated in the hypothalamus and cerebellum, but not in the testis, liver, cerebral cortex or striatum (Figure 2a). At P15, DIO3 activity in PGF mice was increased compared to Ctrl mice in the hypothalamus, cerebral cortex and adrenal gland, reduced in the testis and ovary, and unchanged in the striatum (Figure 2a). At the mRNA level, Dio3 expression in PGF mice was higher than in Ctrl mice in P3 cerebellum and in P15 adrenal gland and hypothalamus (Figure 2b). Compared to Ctrl mice, Dio3 expression was also reduced in the ovary and testis of P15 PGF mice, while no changes were observed in the P3 liver and P15 striatum. These results at the mRNA level (Figure 2b) matched exactly those observed at the level of DIO3 enzymatic activity (Figure 2a).
Figure 2.
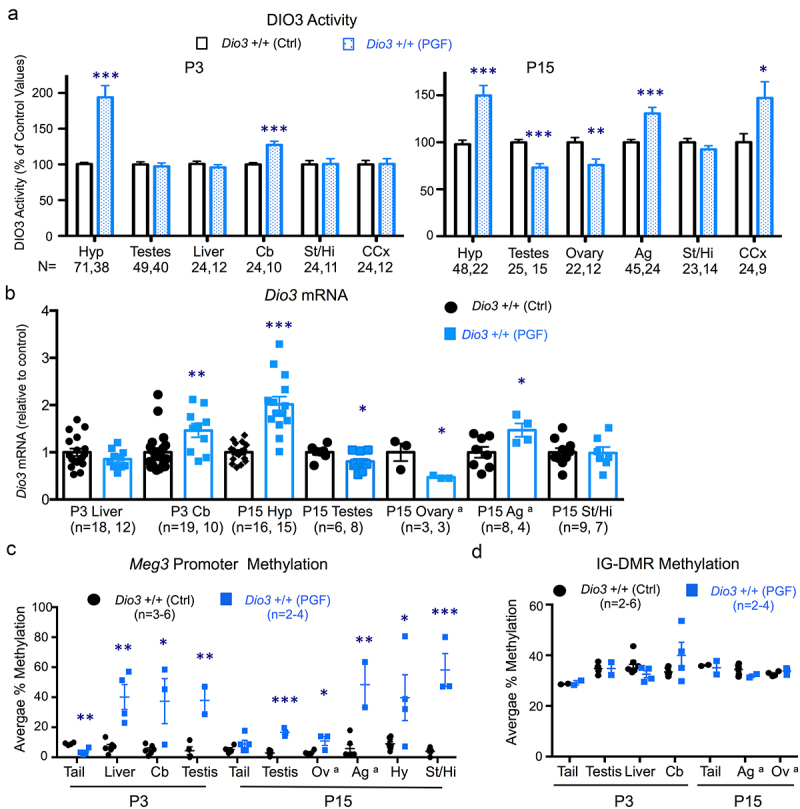
Dio3 expression and Meg3 and IG-DMR methylation in neonatal tissues of PGF mice.
(a) Enzymatic DIO3 activity in neonatal tissues. Data represent the mean ± SEM of the indicated number of mice per experimental group. These mice represent 9 to 11 different litters and three different experiments. (b), Dio3 mRNA expression in neonatal tissues. Data represent the mean ± SEM of the indicated number of mice per experimental group. (c), Meg3 promoter methylation in neonatal tissues. Data represent the mean ± SEM of 2 to 6 mice per experimental group. (d), IG-DMR promoter methylation in neonatal tissues. Data in (c and d) represent the mean ± SEM of the indicated number of mice per experimental group. Each data point for adrenal gland and ovary in (b, c and d) represent a pool of 2-3 mice. Data in (b, c and d) represent at least three different litters. *, ** and *** indicate p < 0.05, 0.01 and 0.001, respectively, as determined by the Student’s t test. Hy, hypothalamus; Cb, cerebellum, St/Hi, Striatum-hippocampus; Ag, adrenal gland; CCx, cerebral cortex; Ov, ovary. a, each of these samples represent a pool of two or three mice.
These data suggested altered epigenetic inheritance affecting Dio3. We then determined the methylation status of two differentially methylated regions with well-established roles in the regulation of allelic expression in the domain, Meg3 and the intergenic differentially-methylated region (IG-DMR) [58,59]. The specific sequence and detailed methylation data are shown in Supplementary Figure 1. Meg3 methylation was approximately 10% in all tissues examined from control mice, but was markedly elevated (between 20–60% methylation) in PGF tissues, including P3 cerebellum, liver and testis, and P15 testis, ovary, adrenal gland, hypothalamus and striatum (Figure 2c). Interestingly, Meg3 promoter methylation was lower in tail snips DNA of P3 PGF mice than in Ctrl mice (Figure 2c). In contrast, no significant methylation changes were observed in the IG-DMR (specific sequence analysed and detailed methylation data is provided in Supplementary Figure 2). IG-DMR methylation was in the 30–40% range and we observed no significant differences between PGF and control mice in any of the tissues examined, including P3 tail, testis, liver and cerebellum and P15 tail, adrenal gland and ovary (Figure 2d).
With limited differences, the analysis of Dio3 expression and imprinted domain methylation showed comparable results in tissues of PGM and PGF mice. Compared to Ctrl mice, P3 DIO3 enzymatic activity was elevated in P3 hypotalamus and cerebellum, and reduced in P3 testis of PGM mice, while unchanged in the striatum and liver (Figure 3a). At P15, DIO3 activity was elevated in the PGM adrenal gland, but reduced in the hypothalamus, striatum, testis and ovary of PGM mice (Figure 3a). These abnormal enzymatic activities were consistent with expression alterations at the mRNA level. Thus, in PGM mice, Dio3 expression was elevated compared to controls in the P3 cerebellum and P15 adrenal gland, reduced in P15 hypothalamus, striatum, ovary and testis and unchanged in P3 liver (Figure 3b). Similar to PGF tissues, we observed markedly higher Meg3 promoter methylation in most PGM tissues compared to Ctrl tissues, with approximately 10% methylation levels in controls and 20–50% methylation in PGM tissues including P3 cerebellum, liver and testis, and P15 testis, ovary, adrenal gland, hypothalamus and striatum (Figure 3c and Supplementary Figure 3). Methylation of the IG-DMR was not determined in PGM tissues after results from a few samples suggested no methylation changes, as observed in PGF mice.
Figure 3.
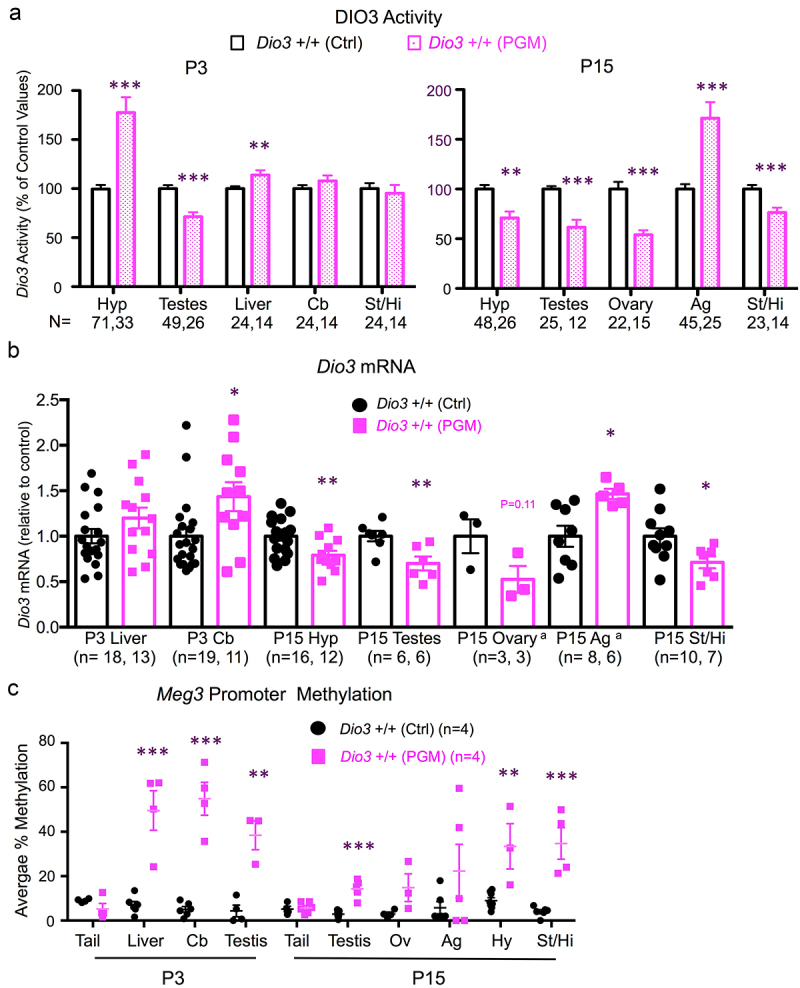
Dio3 expression and Meg3 and IG-DMR methylation in neonatal tissues of PGM mice.
(a) Enzymatic DIO3 activity in neonatal tissues. Data represent the mean ± SEM of the indicated number of mice per experimental group. These mice represent 9 to 11 different litters and three different experiments. (b), Dio3 mRNA expression in neonatal tissues. Data represent the mean ± SEM of the indicated number of animals per experimental group from three different litters. (c), Meg3 promoter methylation in neonatal tissues. Data represent the mean ± SEM of the indicated number of mice per experimental group. Each data point for adrenal gland and ovary in (b and c) represent a pool of 2-3 mice. *, ** and *** indicate p < 0.05, 0.01 and 0.001, respectively, as determined by the Student’s t test. Hy, hypothalamus; Cb, cerebellum, St/Hi, Striatum-hippocampus; Ag, adrenal gland; Ov, ovary.a, each of these samples represent a pool of two or three mice.
F3 generation fetal brain gene expression and methylation at the Dlk1-Dio3 domain
Considering the behavioural abnormalities in PGF mice we have previously reported [33], we also examined the foetal brain at embryonic day 13.5 (E13.5) for gene expression and methylation at the Dlk1-Dio3 imprinted domain, a simplified diagram of which is shown in Figure 4a. This embryonic stage was selected as an early milestone in brain development and corticogenesis, and we have recently demonstrated that at this stage the brain is already sensitive to thyroid hormones and DIO3 deficiency [60]. We observed that genes that are expressed from the maternal allele, Meg3, Rian (RNA imprinted and accumulated in nucleus) and Mirg (miRNA containing gene), showed significantly increased expression in the E13.5 brain of PGF foetuses, but not in the brain of PGM foetuses (Figure 4b). This pattern of expression was different for genes that are expressed from the paternal allele: Dlk1 (delta like non-canonical Notch ligand 1), Rtl1/Rtl1as (retrotransposon Gaglike 1) and Dio3. Compared to Ctrl mice, these genes showed increased expression in the brain of both PGF and PGM foetuses (Figure 4c). These alterations in gene expression were associated with increases in DNA methylation. Methylation of the IG-DMR was slightly increased (but not significantly so) and more variable in the brain of PGF and PGM foetuses (Figure 4d). Concerning Meg3, we observed that methylation substantially increased from ~ 10% in control foetuses to ~ 70% and 55%, respectively, in the brain of PGF and PGM foetuses (Figure 4e). In contrast, as a control, methylation of H19, a differentially methylated and imprinted gene, showed the same tight methylation levels in control and PGF and PGM mice (Figure 4e).
Figure 4.
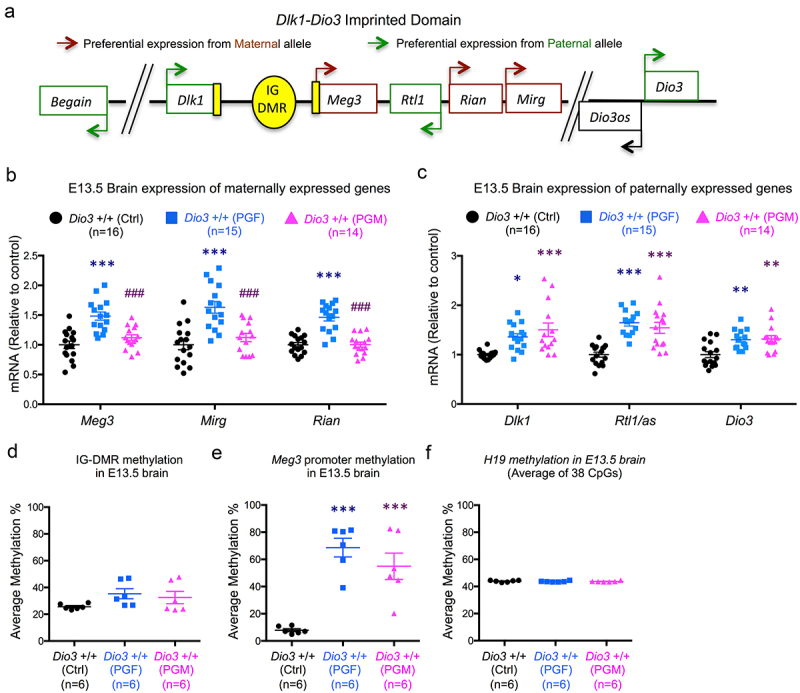
Gene expression and DNA methylation in F3 generation foetal brain.
(a), Simplified diagram of the genes analysed within the Dlk1-Dio3 imprinted domain indicating preferential maternal (brown arrows) or paternal (green arrows) expression. Shown genomic spacers represent approximately 320 and 650 kB, respectively. Shaded in yellow are regions differentially methylated between alleles, including the regulatory regions in the Meg 3 promoter and the intergenic differentially methylated region (IG-DMR). (b and c), Gene expression of maternal (b) and paternally expressed (c) genes in E13.5 brains of PGF and PGM mice. (d, e and f), Methylation status of 29 CpGs in the IG-DMR (d), 7 CpGs of the Meg3 promoter (e) and 40 CpGs in the H19 gene (f) in PGF and PGM E13.5 brains. Data represent the mean ± SEM of the indicated number of animals from at least three different litters. *, ** and *** indicate p < 0.05, 0.01 and 0.001, respectively, when compared to controls; and ### indicate p < 0.001 when compared to PGF, as determined by one-way ANOVA and Tukey’s post hoc test.
Tissue-specific transgenerational effects on Dio3 allelic expression in the F3 generation
Our data (Figures 2, 3, and 4) indicated changes in Dio3 expression across tissues of PGF and PGM mice. To assess if one particular allele was responsible for these changes, we determined DIO3 enzymatic activity also in the heterozygous littermates of PGF and PGM mice (Figure 1b). In the P3 hypothalamus, DIO3 activity in wild type HF mice was the same as that in wild-type controls (Figure 5a, left), while that in wild-type PGF and PGF mice was significantly higher, as also shown in Figures 2 and 3. However, DIO3 activity in heterozygous mice (i.e., maternal allele contribution) showed similar levels in Het-HF and Het-PGM heterozygous, while it was markedly increased in Het-PGF heterozygous (Figure 5a, centre). Making a composite of the maternal allele contribution to total DIO3 activity, we observed that in HF mice (mice with a heterozygous father but no T3-overexposure in past generations), as previously described [50], there is predominant expression from the paternal allele (Figure 5a, right). However, in PGF mice, there is a bi-allelic contribution to DIO3 activity, and the increase in total DIO3 activity can be accounted by a de-repression of the maternal allele (Figure 5a, right). In contrast, the increase in total DIO3 activity in PGM mice compared to Ctrl mice is the result of increased contribution from the paternal allele, causing a more pronounced pattern of preferential expression from the paternal allele (Figure 5a, right). As a negative control DIO3 enzymatic activities in the same tissues are undetectable or lower than 3% of control values (not shown).
Figure 5.
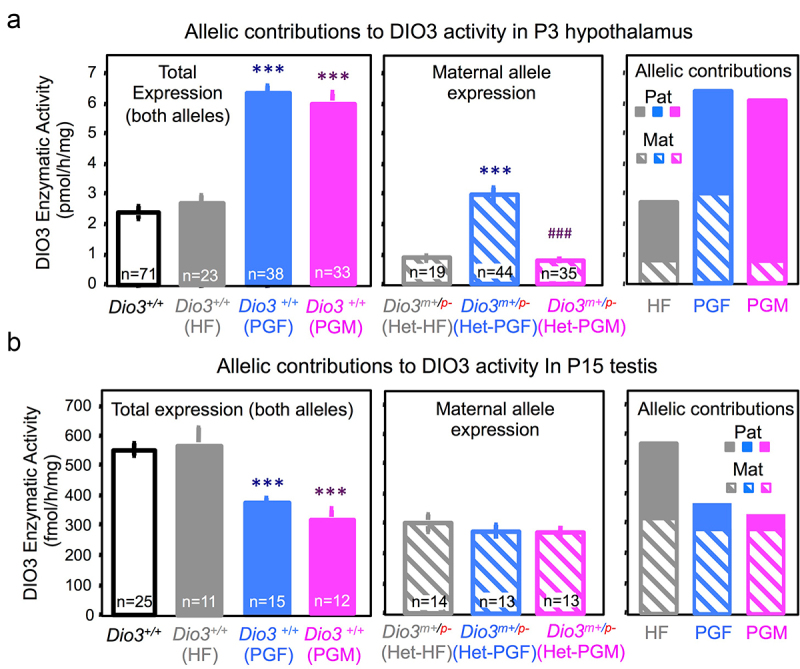
Allele-specific contributions to Dio3 expression changes in PGF and PGM mice.
(a) Total (both alleles) and maternal allele contributions, and composite of allelic contribution to DIO3 activity in the PGF and PGM P3 hypothalamus. (b) Total (both alleles) and maternal allele contributions, and composite of allelic contribution to DIO3 activity in the PGF and PGM P15 testis. Data represent the mean ± SEM of the number of animals indicated in the bars. Number of experimental mice are indicated in the bars and represent 5-11 litters. *** and ### indicate p < 0.001 when compared, respectively, with control and PGF mice, as determined by one-way ANOVA and Tukey’s post hoc test.
These allelic expression effects appear tissue-specific, as similar analyses in P15 testis yielded different effects. In this tissue, wild-type HF mice showed similar DIO3 activity as wild-type controls, but both wild-type PGF and wild-type PGM mice showed significantly lower total DIO3 expression than Ctrl mice (Figure 5b, left). We observed no differences between heterozygous HF, PGF and PGM mice in the contributions from the maternal allele (Figure 5b, centre). In this case, the composite of contributions to DIO3 activity showed that while in the testis of HF mice, as previously described [50], the expression is bi-allelic, in both PGF and PGM testis the reduction in DIO3 expression is largely due to repression of the paternal allele, with total expression showing now a pattern of preferential expression from the maternal allele (Figure 5b, right).
Neonatal tissue expression of other genes in the Dlk1-Dio3 domain
Despite the very distinctive patterns of altered expression in paternally- and maternally expressed genes that PGF and PGM mice exhibited in the foetal brain, we observed no such patterns – just a few expression changes- in neonatal tissues. In the P15 hypothalamus, Meg3 expression was elevated in PGF and PGM mice compared to Ctrl mice, but no changes were observed in the other maternally expressed genes, Mirg and Rian (Figure 6a). Paternally expressed genes Dlk1 and Rtl1/Rtl1as showed decreased expression in the P15 hypothalamus of PGF and PGM mice, although this was not statistically significant in all cases (Figure 6b).
Figure 6.
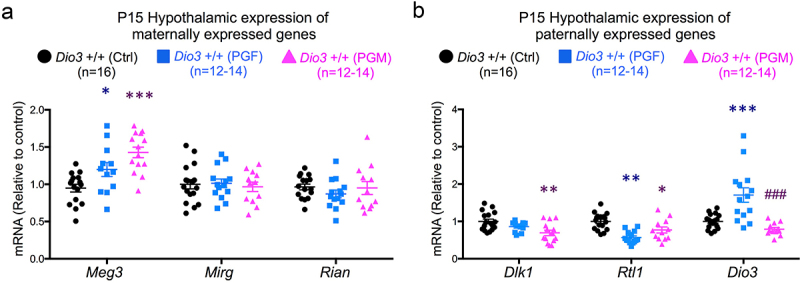
Expression of Dlk1-Dio3 domain genes in PGF and PGM tissues.
(a) Expression of maternally expressed genes in the P15 hypothalamus. (b) Expression of paternally expressed genes in the P15 hypothalamus. Data represent the mean ± SEM of the indicated number of animals representing at least three different litters. *, ** and *** indicate p < 0.05, p < 0.01 and p < 0.001, respectively, when compared to controls; and ### indicate p < 0.001 when compared to PGF, as determined by one-way ANOVA and Tukey’s post hoc test.
In P15 testis (Supplementary Figure 4A), Mirg expression was repressed in PGM mice, and Rian expression was elevated in P15 adrenal glands of PGF mice (supplementary Figure 4C) but we observed no additional alterations other than those described above for Dio3 (Supplementary Figure 4B and 4D). We noted no other abnormalities in the expression of genes from the Dlk1-Dio3 imprinted domain (other than Dio3) in the P3 liver and cerebellum of PGF and PGM mice (Supplementary Figure 5).
Alterations in developmental growth and neonatal hormones
The epigenetic abnormalities and altered allelic expression of Dio3 in PGF and PGM mice were associated with abnormal developmental phenotypes. At E13.5, the weight of PGF and PGM foetuses was significantly reduced compared to controls (Figure 7a). This growth retardation in both experimental groups was still observed on P3 (Figure 7b). However, we observed a neonatal catch-up in growth, as both PGF and PGM mice showed increased weight at P15 compared to control mice (Figure 7c).
Figure 7.
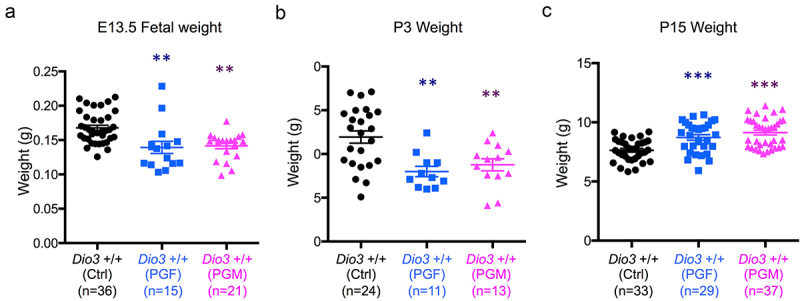
Fetal and neonatal growth of PGF and PGM mice.
(a, b and c), Body weight of PGF and PGM mice at E13.5 foetal age (A), P3 (B) and P15 (C). Each point represents one mice. Data represent the mean ± SEM of the indicated number of animals representing at 3 to 6 different litters. ** and *** indicate p < 0.01 and p < 0.001, respectively, when compared to controls.
We have previously shown that DIO3 deficiency blunts neonatal leptin and exerts intergenerational effects on the offspring neonatal levels of both leptin and thyroid hormones [61]. Thus we measured levels of thyroid hormones and leptin in PGF and PGM mice during neonatal life. At P3, due to the limited volume of serum samples, we only measured thyroxine (T4) and thyroid stimulating hormone (TSH). T4 was significantly reduced in PGF at P3, but not in PGM mice (Figure 8a), while TSH levels were not different between groups (Figure 8b). Compared to Ctrl mice, serum T3 (3,5,3’-triiodothyronine) was significantly reduced in PGM mice, but in not in PGF mice (Figure 8c). At this age, serum T4 in both PGF and PGM mice was significantly lower than in Ctrl mice (Figure 8d). These thyroid hormone values indicated an increase serum T3/T4 ratio in PGF mice, but this ratio was normal in PGM mice (Figure 8e). The abnormal levels of thyroid hormones at P15 were not accompanied by any significant changes in serum TSH (Figure 8f). Interestingly, P15 serum levels of leptin were markedly higher in PGF and PGM mice than in Ctrl mice, with this effect being significantly more pronounced in PGF mice than in PGM mice (Figure 8g).
Figure 8.
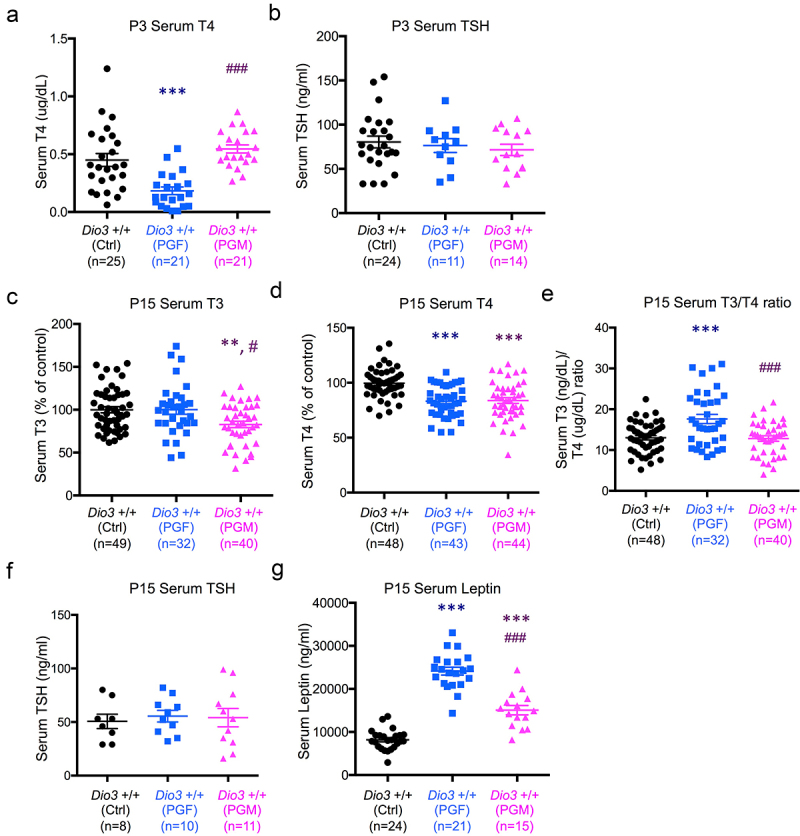
Neonatal levels of thyroid hormones and leptin in PGF and PGM mice.
Serum T4 at P3. (b), Serum TSH at P3. (c), Serum T3 at P15. (d), Serum T4 at P15. (e), serum T3/T4 ratio at P15. (f), Serum TSH at P15. (g), Serum leptin at P15. Each point represents one mice. Data represent the mean ± SEM of the indicated mouse samples representing at 3 to 9 different litters. T3 and T4 data at P15 represents three different experiments and the data is expressed as a percentage of control values to avoid inter-assay variations. ** and *** indicate p < 0.01 and p < 0.001, respectively, when compared to control group; # and ### indicate p < 0.05 and p < 0.001 when compared to PGF group, as determined by one-way ANOVA and Tukey’s post hoc test.
Dlk1-Dio3 imprinted domain gene expression in Dio3-/- mice
To determine if some of the molecular abnormalities in the Dlk1-Dio3 domain were present in the original exposed ascendants, we analysed the expression of genes located within the Dlk1-Dio3 imprinted domain (Figure 9a) in tissues of Dio3-/- mice and wild type littermates. We used mice at P4, as it is during the early neonatal period that thyrotoxicosis is most severe [46]. This day also represents a day older that the mice studied above for transgenerational effects on Dio3 expression, as T3 excess due to loss of DIO3 activity will need about 24 h to exert its full effect on gene expression programs. We also measured Dio3 mRNA expression, as Dio3-/- mice carry a triple point mutation that renders the enzyme inactive [46] but leaves the expression of the mutated mRNA intact. Enhanced thyroid hormone action in each tissue examined was demonstrated by elevated expression of a well-established marker of T3 action. We used Hairless (Hr) in hypothalamus, cerebral cortex and testes (Figure 9a-c) and Dio1 (type 1 deiodinase) in liver (Figure 9d), as hepatic expression of Hr is very low. DIO3 inactivation lead to markedly elevated expression of Dio3 and Dio3os in neonatal hypothalamus, cerebral cortex and testes, but decreased or unchanged in the liver (Figure 9b-e). Compared to wild type mice, Rian expression was significantly reduced in all Dio3-/- tissues, while the expression of Mirg trended lower but was only significantly decreased in the cerebral cortex (Figure 9b-e). The expression of Rtl1/Rtl1as and Begain (brain-enriched guanylate kinase-associated) was not altered in any tissue examined, while Dlk1 expression was significantly reduced in the testes. Finally, Meg3 expression was significantly elevated in hypothalamus and cerebral cortex (Figure 9b,c), but was unchanged in testes and liver (Figure 9d,e). These results indicate that the thyrotoxicosis caused by DIO3 deficiency alters the developmental gene expression in the Dlk1-Dio3 imprinted domain in a gene- and tissue-specific manner.
Figure 9.
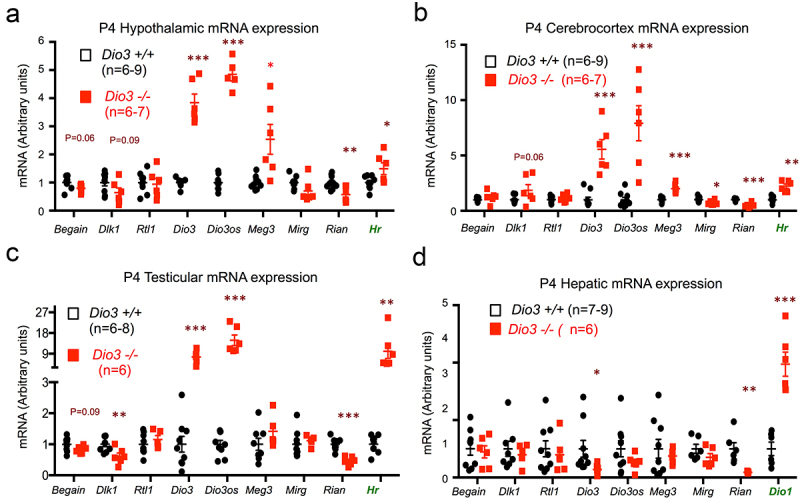
Dio3 deficiency influences gene expression in the Dlk1-Dio3 imprinted domain.
(a-d) Postnatal day 4 (P4) gene expression in hypothalamus (a), cerebrocortex (b), testis (c) and Liver (d). (Note that Dio3 -/- mice carry a mutation that completely inactivates the DIO3 protein, but they still express a full Dio3 mRNA). Data represent the mean ± SEM of mouse samples representing at least three different litters. ***, ** and * indicate p < 0.001, p < 0.01 and p < 0.05, respectively, Dio3 +/+ vs. Dio3 -/-, as determined by the Student’s t-test.
Methylation status in Dio3-/- P4 tissues and adult sperm of ascendant Dio3-/- mice
The observed gene expression changes in the Dlk1-Dio3 imprinted of Dio3-/- ascendants suggested the possibility of aberrant methylation Meg3 as observed in descendants. In Dio3+/+ P4 tissues, the average methylation of the Meg3 CpGs analyzes varied between 3 and 20% across testis, heart, liver, hypothalamus and cerebral cortex. However, a substantial increase in methylation was observed in tissues of Dio3-/- mice, with methylation levels ranging between 40 and 45% in the same tissues (Figure 10a). In the sperm of adult Dio3-/- males, we observed similar methylation changes in Meg3, which was 45% methylated in Dio3-/- sperm compared to 5% in Dio3+/+ sperm (Figure 10b). In contrast, we observed a moderate but significant methylation reduction at the Dio3 promoter in Dio3-/- sperm. The average methylation (n = 4) of 72 CpG sites in this region was approximately 5% in Dio3-/- sperm vs 10% in Dio3+/+ sperm (Figure 10c).
Figure 10.
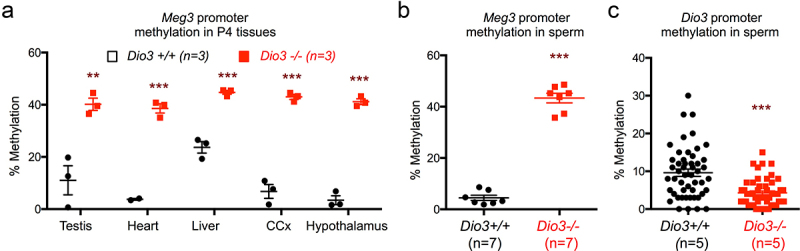
Neonatal tissue and adult sperm methylation in Dio3-/- mice.
(a and b), Meg3 promoter methylation in P4 tissues (a) and adult sperm (b). Each point represents the average methylation of 7 CpG sites in a given mouse. (c) Dio3 promoter methylation in adult sperm. Each point represents the average methylation of a specific CpG in 5 different animals. Data represent the mean ± SEM and *** and ** indicate p < 0.001, p < 0.01, respectively, Dio3 +/+ vs. Dio3 -/-, as determined by the Student’s t-test.
Discussion
Inter- and transgenerational epigenetic effects elicited by environmental conditions in previous generations are increasingly appreciated as important factors contributing to environmental adaptation [62] and the non-genetic inheritance of disease susceptibility [63–65]. Here we present evidence for the transgenerational epigenetic self-memory of the mouse Dio3, a highly conserved gene in mammals that is subject to genomic imprinting and locates within the Dlk1-Dio3 imprinted domain on mouse chromosome 12 and syntenic region on human chromosome 14 [66]. Given the critical role of Dio3 in modulating the transition of developing mammalian tissues from a low to a high, adult-like thyroid hormone signalling environment [45], and the broad consequences of DIO3 deficiency for adult neurological and endocrine pathophysiology [47,49,67], the intergenerational memory of Dio3 regulation may be a critical component of environmental adaptation and evolution, as well as an important contributor to the non-genetic heritable aetiology of disease susceptibility.
We find that the loss of DIO3 function influences the Dlk1-Dio3 domain. The expression of most genes in the domain is altered in several Dio3-/- neonatal tissues studied. Furthermore, this is accompanied by marked increases in methylation at the Meg3 promoter, a region that, together with the IG-DMR [68], regulates allelic expression of genes in the domain [59], including that of Dio3 [69,70]. These observations suggest that a developmental excess of thyroid hormones alters gene expression at the Dlk1-Dio3 locus partly by regulating the methylation of the Meg3-DMR, which is critical for gene expression regulation in the Dlk1-Dio3 imprinted domain [58].
Importantly, the developmental excess of T3 caused by loss of DIO3 function also leads to specific DNA methylation differences in the adult sperm. The robust increase in Meg3 methylation in sperm DNA, as well as the significantly lower methylation in the Dio3 promoter, a genomic region has been shown to have low methylation levels in mouse foetuses [59], are both consistent with the altered methylome we have recently reported in these mice [33], and point to a specific epigenetic effect on the Dlk1-Dio3 locus. As methylation levels of the Meg3 promoter region, and not Meg3 expression itself, regulate Dio3 allelic expression [58], our observations support the hypothesis that these epigenetic marks are maintained in the tissues of descendants, influencing the developmental expression of Dio3, and impacting DIO3-associated pathophysiological endpoints.
Our results in tissues from F3 generation mice indicate that the increases in Meg3 methylation observed in the tissues and sperm of ancestors are largely maintained in descendants, with most foetal and neonatal tissues studied exhibiting comparable hypermethylation of Meg3. Meg3 hypermethylation occurs without concomitant methylation changes in the other main regulatory region, the IG-DMR [59]. Furthermore, Meg3 hypermethylation in the tissues of PGF and PGM descendants was associated with gene expression changes in Dio3 and other genes in this imprinted locus. Dio3 mRNA expression in neonatal tissues of PGF and PGM mice was altered in a manner that depended on the tissue, the neonatal age, and the sex of the ancestor exposed to thyroid hormone excess. The changes in tissue Dio3 mRNA expression in descendants were accurately reproduced by changes in DIO3 enzymatic activity, indicating a tight correlation between altered gene expression and biological function.
The variability of Dio3 expression (up, down or no change) across the tissues of descendants is not particularly surprising considering the broad tissue variability in the relative allelic expression of Dio3 we previously reported [50]. While most imprinted genes in the Dlk1-Dio3 locus exhibit strict monoallelic expression [68], the preferential expression of Dio3 from the paternal allele in the mouse foetus (~80% from the paternal allele) is not strict [42], with some tissues like testis [50], placenta [68,70] and regions of the central nervous system, especially in later life [50], showing relaxed imprinting patterns of allelic expression or biallelic expression of Dio3. Although the determinants of the tissue-specific Dio3 allele expression ratios have not been identified, this tissue variability is consistent with the Dio3 expression abnormalities observed in PGF and PGM mice, and suggest a remarkable plasticity in Dio3 gene dosage across tissues and generations.
In addition to their tissue specificity, our data also demonstrate that the intergenerational epigenetic effects observed on Dio3 expression are largely the result of allele-specific effects. The marked and comparable increases in Dio3 expression in the P3 hypothalamus of PGF and PGM animals originate from very different effects on Dio3 alleles. The increase in PGF mice is due to a de-repression of the maternal Dio3 allele, making Dio3 expression bi-allelic, while the increase in PGM mice is due to additional Dio3 expression from the paternal allele, making Dio3 more strictly monoallelic. Interestingly, in the P15 testis, where Dio3 expression is normally biallelic, the decreased expression observed in PGF and PGM mice is the result in both cases of repression of the Dio3 paternal allele, making testicular Dio3 preferentially expressed from the maternal allele.
In both PGF and PGM mice the epigenetic alterations should be last transmitted by the paternal allele. However, our results on Meg3 methylation and allelic Dio3 expression indicate that the maternal allele is affected. This suggests that epigenetic abnormalities in the paternal allele, which may also occur in other parts of the genome, exert a regulatory function in the maternal Dio3 allele, a mechanism that has been described in other imprinted domains and may be mediated by non-coding RNAs [71,72].
Although the similarities in some of the results from PGF and PGM mice suggest that the male and female germ lines share common response mechanisms to thyroid hormone excess [33] that trigger specific germ line epigenetic signatures (like Meg3 methylation), our findings also reveal differential Dio3 expression outcomes depending on the sex of the exposed ancestor. This suggests that other unidentified altered epigenetic signatures specific to the sex of the originally exposed individual also play a role in the implementation of a sex-specific agenda for the epigenetic transmission of particular traits across generations.
It is important to note that the hypothalamus and gonads of the mice studied consistently show abnormalities in Dio3 expression. Given the roles of these tissues, respectively, in regulating important endocrine systems, energy balance, and in the establishment of the epigenetic information of the germ line, our observations suggest an important role for Dio3 dosage in the intergenerational adaptation to thyroid hormone states in previous generations. This idea, postulated for genomic imprinting [73,74], is not surprising. Many imprinted genes have important roles in the regulation of developmental growth, endocrine functions and behaviour [75–77], and the pathophysiological importance of Dio3, as evidenced in Dio3 null mice, is tightly aligned with those roles [46,47,49,78–80].
Although our study focused on Dio3, we also observed abnormalities in the total expression of other genes in the Dlk1-Dio3 imprinted domain. Most remarkable in this regard is the expression pattern in the foetal brain, in which the expression of paternally expressed genes, including Dio3, follows abnormal patterns that are distinct from those of maternally expressed genes. This further reinforces the notion that the trans-generational effects of thyroid hormone excess are allele-specific and partly depend on the sex of the individual exposed. The distinct and well-defined expression pattern observed at this developmental stage also suggests that the transgenerational effects on the domain are already established – and more tightly defined- at early embryonic stages, partly fading later in development, as we only observed minor and limited changes in the expression of other genes in the domain at late neonatal stages.
Of note, the developmental gene expression abnormalities in the Dlk1-Dio3 domain in PGF and PGM mice are of significant consequence for developmental growth and endocrine phenotypes. PGF and PGM mice exhibit a distinctive profile of growth retardation in utero and shortly after birth, but a growth spurt later in neonatal life that surpasses that of littermate controls. This altered growth profile may have later life implications for body composition and susceptibility to metabolic disease and obesity. PGF and PGM mice also show different degrees of reduction in their levels of thyroid hormones as well as different abnormal increases of neonatal leptin. Given the critical importance of developmental levels of leptin [81,82] and thyroid hormones in the programming of multiple neuroendocrine systems, energy balance and brain functions [83–85], our results suggest that the transgenerational effects elicited by thyroid hormone excess in ascendants broadly affects the development of multiple physiological systems, with consequences for adult susceptibility to endocrine and neurological disorders, as we have specifically described for the neuroendocrine traits in the F2 generation [61], and behavioural alterations of F3 generation PGF mice [33,48].
Overall, our results show that thyroid hormone status, which is controlled during development by DIO3 activity, influences Meg3 methylation. This epigenetic signature is maintained in descendants, in which Dio3 allelic expression and dosage are affected in a tissue-specific manner that also depends on the sex of the exposed ancestor, impacting developmental growth and endocrine outcomes. Our work describes a rather unique example of an imprinted gene with variable allelic expression ratios across tissues, for which allelic expression and dosage may be continuously adapting according to perceived developmental Dio3 dosages in previous generations. We are not aware of a similar paradigm described in the literature for another gene. The transgenerational epigenetic self-memory of Dio3 opens novel prespectives into the role of genomic imprinting in environmental adaptation and evolution. Our findings may also have important clinical implications, as changes in thyroid status due to highly prevalent thyroid conditions may modify the epigenetic information of the germ line and determine physiological programming and disease susceptibility in future generations.
Supplementary Material
Acknowledgments
We are grateful to Xiao-Hui Liao and Samuel Refetoff for TSH measurements, and to Jean Jamo for thoughtfulness and dedication to animal care. This work utilized the MaineHealth Institute for Research Molecular Phenotyping Core and Physiology Core, which are supported by grants P30GM106391, U54GM115516, P20GM121301 from the National Institute of General Medical Sciences.
Funding Statement
This work was supported by grants DK095908 from the National Institute of Diabetes, Digestive and Kidney Diseases and MH096050 from the National Institute of Mental Health.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Ethics approval and consent to participate
All experiments were approved by the MaineHealth Institute for Research Institutional Animal Care and Use Committee (IACUC), under current protocol number 2112.
Availability of data and materials
All data generated are included in the article and supplementary materials.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2024.2376948.
References
- [1].Soubry A. Epigenetic inheritance and evolution: a paternal perspective on dietary influences. Prog Biophys Mol Biol. 2015;118(1–2):79–20. doi: 10.1016/j.pbiomolbio.2015.02.008 [DOI] [PubMed] [Google Scholar]
- [2].Dunn GA, Bale TL. Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology. 2011;152(6):2228–2236. doi: 10.1210/en.2010-1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dunn GA, Bale TL. Maternal high-fat diet promotes body length increases and insulin insensitivity in second-generation mice. Endocrinology. 2009;150(11):4999–5009. doi: 10.1210/en.2009-0500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zambrano E, Martinez-Samayoa PM, Bautista CJ, et al. Sex differences in transgenerational alterations of growth and metabolism in progeny (F2) of female offspring (F1) of rats fed a low protein diet during pregnancy and lactation. J Physiol. 2005;566(1):225–236. doi: 10.1113/jphysiol.2005.086462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Walker DM, Gore AC. Transgenerational neuroendocrine disruption of reproduction. Nat Rev Endocrinol. 2011;7(4):197–207. doi: 10.1038/nrendo.2010.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Skinner MK. Endocrine disruptors in 2015: epigenetic transgenerational inheritance. Nat Rev Endocrinol. 2016;12(2):68–70. doi: 10.1038/nrendo.2015.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rodgers AB, Morgan CP, Bronson SL, et al. Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neurosci. 2013;33(21):9003–9012. doi: 10.1523/JNEUROSCI.0914-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rodgers AB, Morgan CP, Leu NA, et al. Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proc Natl Acad Sci USA. 2015;112(44):13699–13704. doi: 10.1073/pnas.1508347112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Morgan CP, Bale TL. Early prenatal stress epigenetically programs dysmasculinization in second-generation offspring via the paternal lineage. J Neurosci. 2011;31(33):11748–11755. doi: 10.1523/JNEUROSCI.1887-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jawaid A, Roszkowski M, Mansuy IM. Transgenerational epigenetics of traumatic stress. Prog Mol Biol Transl Sci. 2018;158:273–298. [DOI] [PubMed] [Google Scholar]
- [11].Weaver ICG, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2006;7(8):847–854. doi: 10.1038/nn1276 [DOI] [PubMed] [Google Scholar]
- [12].Krishnan K, Rahman S, Hasbum A, et al. Maternal care modulates transgenerational effects of endocrine-disrupting chemicals on offspring pup vocalizations and adult behaviors. Horm Behav. 2019;107:96–109. doi: 10.1016/j.yhbeh.2018.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dickson DA, Stohn P, Saavedra Rodriguez L, et al. Involvement of early embryonic miR-409-3p in the establishment of anxiety levels in female mice. Dev Neurobiol. 2020;80(5–6):160–167. doi: 10.1002/dneu.22756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Saavedra-Rodríguez L, Feig LA. Chronic social instability induces anxiety and defective social interactions across generations. Biol Psychiatry. 2013;73(1):44–53. doi: 10.1016/j.biopsych.2012.06.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Escher J, Ford D, LaSalle J. General anesthesia, germ cells and the missing heritability of autism: an urgent need for research. Environ Epigenet. 2020;6(1):dvaa007. doi: 10.1093/eep/dvaa007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chang HS, Anway MD, Rekow SS, et al. Transgenerational epigenetic imprinting of the male germline by endocrine disruptor exposure during gonadal sex determination.[see comment]. Endocrinology. 2006;147(12):5524–5541. doi: 10.1210/en.2006-0987 [DOI] [PubMed] [Google Scholar]
- [17].Soubry A, Jirtle RL, Murphy SK. A paternal environmental legacy: evidence for epigenetic inheritance through the male germ line. BioEssays: news and reviews in molecular, cellular and developmental biology. Bioessays. 2014;36(4):359–371. doi: 10.1002/bies.201300113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Skinner C, Haque M, Nilsson E, et al. Environmentally induced transgenerational epigenetic reprogramming of primordial germ cells and the subsequent germ line. PLOS ONE [Electron Resource]. 2013;8(7):e66318. doi: 10.1371/journal.pone.0066318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Girirajan S. Missing heritability and where to find it. Genome Biol. 2017;18(1):89. doi: 10.1186/s13059-017-1227-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Trerotola M, Relli V, Simeone P, et al. Epigenetic inheritance and the missing heritability. Hum Genomics. 2015;9(1):17. doi: 10.1186/s40246-015-0041-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Koch L. Disease genetics: insights into missing heritability. Nat Rev Genet. 2014;15(4):218. doi: 10.1038/nrg3713 [DOI] [PubMed] [Google Scholar]
- [22].Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ozgyin L, Erdos E, Bojcsuk D, et al. Nuclear receptors in transgenerational epigenetic inheritance. Prog Biophys Mol Biol. 2015;118(1–2):34–43. doi: 10.1016/j.pbiomolbio.2015.02.012 [DOI] [PubMed] [Google Scholar]
- [24].Weaver IC, Szyf M, Meaney MJ. From maternal care to gene expression: DNA methylation and the maternal programming of stress responses. Endocr Res. 2002;28(4):699. doi: 10.1081/ERC-120016989 [DOI] [PubMed] [Google Scholar]
- [25].Ruf M, Gunter HM, Schauer K, et al. Transgenerational impact of intimate partner violence on methylation in the promoter of the glucocorticoid receptor. Transl Psychiatr Psychiatr. 2011;1(7):e21. doi: 10.1038/tp.2011.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yeshurun S, Rogers J, Short AK, et al. Elevated paternal glucocorticoid exposure modifies memory retention in female offspring. Psychoneuroendocrinology. 2017;83:9–18. doi: 10.1016/j.psyneuen.2017.05.014 [DOI] [PubMed] [Google Scholar]
- [27].Moisiadis VG, Constantinof A, Kostaki A, et al. Prenatal glucocorticoid exposure modifies endocrine function and behaviour for 3 generations following maternal and paternal transmission. Sci Rep. 2017;7(1):11814. doi: 10.1038/s41598-017-11635-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Short AK, Fennell KA, Perreau VM, et al. Elevated paternal glucocorticoid exposure alters the small noncoding RNA profile in sperm and modifies anxiety and depressive phenotypes in the offspring. Transl Psychiatry. 2016;6(6):e837. doi: 10.1038/tp.2016.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Crews D, Gore AC, Hsu TS, et al. Transgenerational epigenetic imprints on mate preference. Proc Natl Acad Sci USA. 2007;104(14):5942–5946. doi: 10.1073/pnas.0610410104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Anway MD, Cupp AS, Uzumcu M, et al. Epigenetic transgenerational actions of endocrine disruptors and male fertility.[see comment]. Science (New York, NY). 2006;308(5727):1466–1469. doi: 10.1126/science.1108190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hernandez A, Martinez ME, Chaves C, et al. Epigenetic developmental programming and intergenerational effects of thyroid hormones. Vitam Horm. 2023;122:23–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Anselmo J, Scherberg NH, Dumitrescu AM, et al. Reduced sensitivity to thyroid hormone as a transgenerational epigenetic Marker transmitted along the human male line. Thyroid. 2019;29(6):778–782. doi: 10.1089/thy.2019.0080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Martinez ME, Duarte CW, Stohn JP, et al. Thyroid hormone influences brain gene expression programs and behaviors in later generations by altering germ line epigenetic information. Mol Psychiatry. 2020;25(5):939–950. doi: 10.1038/s41380-018-0281-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bakke JL, Lawrence NL, Robinson S, et al. Endocrine studies in the untreated F1 and F2 progeny of rats treated neonatally with thyroxine. Biol Neonate. 1977;31(1–2):71–83. doi: 10.1159/000240946 [DOI] [PubMed] [Google Scholar]
- [35].Bakke JL, Lawrence NL, Robinson S, et al. Observations on the untreated progeny of hypothyroid male rats. Metab Clin & Exp. 1976;25(4):437–444. doi: 10.1016/0026-0495(76)90076-7 [DOI] [PubMed] [Google Scholar]
- [36].McLachlan SM, Rapoport B. Breaking tolerance to thyroid antigens: changing concepts in thyroid autoimmunity. Endocr Rev. 2014;35(1):59–105. doi: 10.1210/er.2013-1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Brent GA, Braverman LE, Zoeller RT. Thyroid health and the environment. Thyroid. 2007;17:807–809. [DOI] [PubMed] [Google Scholar]
- [38].Wadzinski TL, Geromini K, McKinley Brewer J, et al. Endocrine disruption in human placenta: expression of the dioxin-inducible enzyme, CYP1A1, is correlated with that of thyroid hormone-regulated genes. J Clin Endocrinol Metab. 2014;99(12):E2735–43. doi: 10.1210/jc.2014-2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vuong AM, Webster GM, Romano ME, et al. Maternal polybrominated diphenyl ether (PBDE) exposure and thyroid hormones in maternal and cord sera: the HOME study, Cincinnati, USA. Environ Health Perspect. 2015;123(10):1079–1085. doi: 10.1289/ehp.1408996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cullen SM, Hassan N, Smith-Raska M. Effects of noninherited ancestral genotypes on offspring phenotypes†. Biol Reprod. 2021;105(3):747–760. doi: 10.1093/biolre/ioab120 [DOI] [PubMed] [Google Scholar]
- [41].Tsai CE, Lin SP, Ito M, et al. Genomic imprinting contributes to thyroid hormone metabolism in the mouse embryo. Curr Biol. 2002;12(14):1221–1226. doi: 10.1016/S0960-9822(02)00951-X [DOI] [PubMed] [Google Scholar]
- [42].Hernandez A, Fiering S, Martinez E, et al. The gene locus encoding iodothyronine deiodinase type 3 (Dio3) is imprinted in the fetus and expresses antisense transcripts. Endocrinology. 2002;143(11):4483–4486. doi: 10.1210/en.2002-220800 [DOI] [PubMed] [Google Scholar]
- [43].Martinez ME, Cox DF, Youth BP, et al. Genomic imprinting of DIO3, a candidate gene for the syndrome associated with human uniparental disomy of chromosome 14. Eur J Hum Genet. 2016;24(11):1617–1621. doi: 10.1038/ejhg.2016.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Charalambous M, Hernandez A. Genomic imprinting of the type 3 thyroid hormone deiodinase gene: regulation and developmental implications. Biochim et Biophys Acta (BBA) - Gener Subj. 2013;1830(7):3946–3955. doi: 10.1016/j.bbagen.2012.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hernandez A, Martinez ME, Ng L, et al. Thyroid hormone deiodinases: dynamic switches in developmental transitions. Endocrinology. 2021;162(8):162. doi: 10.1210/endocr/bqab091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hernandez A, Martinez ME, Fiering S, et al. Type 3 deiodinase is critical for the maturation and function of the thyroid axis. J Clin Investig. 2006;116(2):476–484. doi: 10.1172/JCI26240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Martinez ME, Pinz I, Preda M, et al. DIO3 protects against thyrotoxicosis-derived cranio-encephalic and cardiac congenital abnormalities. JCI Insight. 2022;7(21). doi: 10.1172/jci.insight.161214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Martinez ME, Stohn JP, Mutina EM, et al. Thyroid hormone elicits intergenerational epigenetic effects on adult social behavior and fetal brain expression of autism susceptibility genes. Front Neurosci. 2022;16:1055116. doi: 10.3389/fnins.2022.1055116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Martinez ME, Karaczyn A, Stohn JP, et al. The type 3 deiodinase is a critical determinant of appropriate thyroid hormone action in the developing testis. Endocrinology. 2016;157(3):1276–1288. doi: 10.1210/en.2015-1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Martinez ME, Charalambous M, Saferali A, et al. Genomic imprinting variations in the mouse type 3 deiodinase gene between tissues and brain regions. Mol Endocrinol (Baltim, Md). 2014;28(11):1875–1886. doi: 10.1210/me.2014-1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhu Y, Mordaunt CE, Yasui DH, et al. Placental DNA methylation levels at CYP2E1 and IRS2 are associated with child outcome in a prospective autism study. Hum Mol Genet. 2019;28(16):2659–2674. doi: 10.1093/hmg/ddz084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Galton VA, Hiebert A. Hepatic iodothyronine 5-deiodinase activity in Rana catesbeiana tadpoles at different stages of the life cycle. Endocrinology. 1987;121(1):42–47. doi: 10.1210/endo-121-1-42 [DOI] [PubMed] [Google Scholar]
- [53].Gil-Ibanez P, Belinchon MM, Morte B, et al. Is the intrinsic genomic activity of thyroxine relevant in vivo? Effects on gene expression in primary cerebrocortical and neuroblastoma cells. Thyroid. 2017;27(8):1092–1098. doi: 10.1089/thy.2017.0024 [DOI] [PubMed] [Google Scholar]
- [54].Bárez-López S, Obregon MJ, Martínez-de-Mena R, et al. Effect of triiodothyroacetic acid treatment in Mct8 deficiency: a word of caution. Thyroid. 2016;26(5):618–626. doi: 10.1089/thy.2015.0388 [DOI] [PubMed] [Google Scholar]
- [55].Escobar-Morreale HF, Obregon MJ, Hernandez A, et al. Regulation of iodothyronine deiodinase activity as studied in thyroidectomized rats infused with thyroxine or triiodothyronine 1. Endocrinology. 1997;138(6):2559–2568. doi: 10.1210/endo.138.6.5212 [DOI] [PubMed] [Google Scholar]
- [56].Pohlenz J, Maqueem A, Cua K, et al. Improved radioimmunoassay for measurement of mouse thyrotropin in serum: strain differences in thyrotropin concentration and thyrotroph sensitivity to thyroid hormone. Thyroid. 1999;9(12):1265–1271. doi: 10.1089/thy.1999.9.1265 [DOI] [PubMed] [Google Scholar]
- [57].Hernandez A, Quignodon L, Martinez ME, et al. Type 3 deiodinase deficiency causes spatial and temporal alterations in brain T3 signaling that are dissociated from serum thyroid hormone levels. Endocrinology. 2010;151(11):5550–5558. doi: 10.1210/en.2010-0450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhu W, Botticelli EM, Kery RE, et al. Meg3-DMR, not the Meg3 gene, regulates imprinting of the Dlk1-Dio3 locus. Dev Biol. 2019;455(1):10–18. doi: 10.1016/j.ydbio.2019.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].da Rocha St, Edwards CA, Ito M, et al. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008;24(6):306–316. doi: 10.1016/j.tig.2008.03.011 [DOI] [PubMed] [Google Scholar]
- [60].Martinez ME, Hernandez A. The type 3 deiodinase is acritical modulator of thyroid hormone sensitivity in the fetal brain. Front Neurosci. 2021;15:703–730. doi: 10.3389/fnins.2021.703730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Martinez ME, Wu Z, Hernandez A. Paternal developmental thyrotoxicosis disrupts neonatal leptin leading to increased adiposity and altered physiology of the melanocortin system. Front Endocrinol (Lausanne). 2023;14:1210414. doi: 10.3389/fendo.2023.1210414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Prokopuk L, Western PS, Stringer JM. Transgenerational epigenetic inheritance: adaptation through the germline epigenome? Epigenomics. 2015;7(5):829–846. doi: 10.2217/epi.15.36 [DOI] [PubMed] [Google Scholar]
- [63].Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8(4):253–262. doi: 10.1038/nrg2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Jirtle RL, Sander M, Barrett JC. Genomic imprinting and environmental disease susceptibility. Environ Health Perspect. 2000;108(3):271–278. doi: 10.1289/ehp.00108271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Feinberg AP. Mendel stayed home. Trends in Genetics. 1999;15(2):46. doi: 10.1016/SO168-9525(98)01663-1 [DOI] [PubMed] [Google Scholar]
- [66].Hernandez A, Park JP, Lyon GJ, et al. Localization of the type 3 iodothyronine deiodinase (DIO3) gene to human chromosome 14q32 and mouse chromosome 12F1. Genomics. 1998;53(1):119–121. doi: 10.1006/geno.1998.5505 [DOI] [PubMed] [Google Scholar]
- [67].Hernandez A, Stohn JP. The type 3 deiodinase: epigenetic control of brain thyroid hormone action and neurological function. Int J Mol Sci. 2018;19(6):1804. doi: 10.3390/ijms19061804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lin SP, Youngson N, Takada S, et al. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nat Genet. 2003;35(1):97–102. doi: 10.1038/ng1233 [DOI] [PubMed] [Google Scholar]
- [69].Charalambous M, da Rocha St, Hernandez A, et al. Perturbations to the IGF1 growth pathway and adult energy homeostasis following disruption of mouse chromosome 12 imprinting. Acta Physiol (Oxf). 2014;210(1):174–187. doi: 10.1111/apha.12160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Charalambous M, Ferron SR, da Rocha St, et al. Imprinted gene dosage is critical for the transition to independent life. Cell metabolism 2012; 15(2):209–221. doi: 10.1016/j.cmet.2012.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Cavaillé J. Box C/D small nucleolar RNA genes and the Prader-Willi syndrome: a complex interplay. WIREs RNA. 2017;8(4). doi: 10.1002/wrna.1417 [DOI] [PubMed] [Google Scholar]
- [72].Stelzer Y, Sagi I, Yanuka O, et al. The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome. Nat Genet. 2014;46(6):551–557. doi: 10.1038/ng.2968 [DOI] [PubMed] [Google Scholar]
- [73].Wilkins JF. Genomic imprinting of Grb10: coadaptation or conflict? PLOS Biol. 2014;12(2):e1001800. doi: 10.1371/journal.pbio.1001800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Haig D. Coadaptation and conflict, misconception and muddle, in the evolution of genomic imprinting. Heredity (Edinb). 2014;113(2):96–103. doi: 10.1038/hdy.2013.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Weinstein LS, Yu S, Warner DR, et al. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. [Review] [416 refs]. Endocr Rev. 2001. Oct;22(5):675–705. doi: 10.1210/edrv.22.5.0439 [DOI] [PubMed] [Google Scholar]
- [76].Preece MA, Moore GE. Genomic imprinting, uniparental disomy and foetal growth. Trends Endocrinol & Metab. 2000;11(7):270–275. doi: 10.1016/S1043-2760(00)00277-0 [DOI] [PubMed] [Google Scholar]
- [77].Nicholls RD. The impact of genomic imprinting for neurobehavioral and developmental disorders. J Clin Invest. 2000;105(4):413–418. doi: 10.1172/JCI9460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wu Z, Martinez ME, St Germain DL, et al. Type 3 deiodinase role on central thyroid hormone action affects the leptin-melanocortin system and circadian activity. Endocrinology. 2017;158(2):419–430. doi: 10.1210/en.2016-1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Stohn JP, Martinez ME, Hernandez A. Decreased anxiety- and depression-like behaviors and hyperactivity in a type 3 deiodinase- deficient mouse showing brain thyrotoxicosis and peripheral hypothyroidism. Psychoneuroendocrinology. 2016;74:46–56. doi: 10.1016/j.psyneuen.2016.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Stohn JP, Martinez ME, Zafer M, et al. Increased aggression and lack of maternal behavior in Dio3-deficient mice are associated with abnormalities in oxytocin and vasopressin systems. Genes Brain Behav. 2018;17(1):23–35. doi: 10.1111/gbb.12400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Vickers MH, Gluckman PD, Coveny AH, et al. Neonatal leptin treatment reverses developmental programming. Endocrinology. 2005;146(10):4211–4216. doi: 10.1210/en.2005-0581 [DOI] [PubMed] [Google Scholar]
- [82].Childs GV, Odle AK, MacNicol MC, et al. The importance of leptin to reproduction. Endocrinology. 2021;162(2):162. doi: 10.1210/endocr/bqaa204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Fonseca TL, Garcia T, Fernandes GW, et al. Neonatal thyroxine activation modifies epigenetic programming of the liver. Nat Commun. 2021;12(1):4446. doi: 10.1038/s41467-021-24748-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Hernandez A, Martinez ME, Liao XH, et al. Type 3 deiodinase deficiency results in functional abnormalities at multiple levels of the thyroid axis. Endocrinology. 2007;148(12):5680–5687. doi: 10.1210/en.2007-0652 [DOI] [PubMed] [Google Scholar]
- [85].Stohn JP, Martinez ME, St Germain DL, et al. Adult onset of type 3 deiodinase deficiency in mice alters brain gene expression and increases locomotor activity. Psychoneuroendocrinology. 2019;110:104439. doi: 10.1016/j.psyneuen.2019.104439 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated are included in the article and supplementary materials.