

Abstract
BACKGROUND:
LMNA (lamin A/C)-related dilated cardiomyopathy is a rare genetic cause of heart failure. In a phase 2 trial and long-term extension, the selective p38α MAPK (mitogen-activated protein kinase) inhibitor, ARRY-371797 (PF-07265803), was associated with an improved 6-minute walk test at 12 weeks, which was preserved over 144 weeks.
METHODS:
REALM-DCM (NCT03439514) was a phase 3, randomized, double-blind, placebo-controlled trial in patients with symptomatic LMNA-related dilated cardiomyopathy. Patients with confirmed LMNA variants, New York Heart Association class II/III symptoms, left ventricular ejection fraction ≤50%, implanted cardioverter-defibrillator, and reduced 6-minute walk test distance were randomized to ARRY-371797 400 mg twice daily or placebo. The primary outcome was a change from baseline at week 24 in the 6-minute walk test distance using stratified Hodges-Lehmann estimation and the van Elteren test. Secondary outcomes using similar methodology included change from baseline at week 24 in the Kansas City Cardiomyopathy Questionnaire-physical limitation and total symptom scores, and NT-proBNP (N-terminal pro-B-type natriuretic peptide) concentration. Time to a composite outcome of worsening heart failure or all-cause mortality and overall survival were evaluated using Kaplan-Meier and Cox proportional hazards analyses.
RESULTS:
REALM-DCM was terminated after a planned interim analysis suggested futility. Between April 2018 and October 2022, 77 patients (aged 23–72 years) received ARRY-371797 (n=40) or placebo (n=37). No significant differences (P>0.05) between groups were observed in the change from baseline at week 24 for all outcomes: 6-minute walk test distance (median difference, 4.9 m [95% CI, −24.2 to 34.1]; P=0.82); Kansas City Cardiomyopathy Questionnaire-physical limitation score (2.4 [95% CI, −6.4 to 11.2]; P=0.54); Kansas City Cardiomyopathy Questionnaire-total symptom score (5.3 [95% CI, −4.3 to 14.9]; P=0.48); and NT-proBNP concentration (−339.4 pg/mL [95% CI, −1131.6 to 452.7]; P=0.17). The composite outcome of worsening heart failure or all-cause mortality (hazard ratio, 0.43 [95% CI, 0.11–1.74]; P=0.23) and overall survival (hazard ratio, 1.19 [95% CI, 0.23–6.02]; P=0.84) were similar between groups. No new safety findings were observed.
CONCLUSIONS:
Findings from REALM-DCM demonstrated futility without safety concerns. An unmet treatment need remains among patients with LMNA-related dilated cardiomyopathy.
REGISTRATION:
URL: https://classic.clinicaltrials.gov; Unique Identifiers: NCT03439514, NCT02057341, and NCT02351856.
Keywords: ARRY-371797; cardiomyopathy, dilated; heart failure; lamin type A; walk test
WHAT IS NEW?
REALM-DCM was the first phase 3, randomized, controlled, interventional trial of a disease-modifying therapy in patients with LMNA (lamin A/C)-related dilated cardiomyopathy.
The trial was terminated early after a planned interim analysis suggested futility.
WHAT ARE THE CLINICAL IMPLICATIONS?
An unmet treatment need remains among patients with LMNA-related dilated cardiomyopathy. Despite the early termination, the data gathered from REALM-DCM are valuable by forming the largest prospective data set to characterize LMNA-related dilated cardiomyopathy to date.
Findings from this study can potentially contribute to the development and design of future genetically informed clinical trials for LMNA-related cardiomyopathy and other rare cardiovascular diseases.
LMNA (lamin A/C)-related dilated cardiomyopathy (DCM) is a life-threatening condition arising from penetrant LMNA gene variants that are inherited in an autosomal dominant pattern.1 The estimated prevalence of DCM is 1:250 to 1:2500, with ≈35% of cases being familial.2,3 Among adult-onset DCM cases, LMNA variants have been identified in ≈0.5% to 5% of patients and are associated with a high risk of arrhythmias and progression to heart failure (HF).2,4,5 The typical onset of LMNA-related DCM is around 30 to 40 years, commonly presenting with atrioventricular conduction disease and atrial arrhythmias that later progress to DCM, ventricular arrhythmias that are poorly correlated with left ventricular ejection fraction (LVEF), and refractory HF. Sudden cardiac death may be the presenting symptom in some patients.6–8 One study showed that, by the age of 45 years, 69% of patients with symptomatic LMNA-related DCM had experienced a major cardiac event, received a heart transplant, or died.9
The exact pathophysiologic mechanisms of LMNA-related DCM are unclear, but cellular stress and hyperactivation of the p38 MAPK (mitogen-activated protein kinase) pathway have been observed in animal models and in biopsies from the hearts of patients with LMNA-related DCM.10,11 There are currently no disease-specific therapies for these patients.12 Current treatment recommendations rely on symptomatic management with guideline-directed medical therapy and disease-specific primary prevention of sudden cardiac death. However, in limited studies, the efficacy of even maximal guideline-directed medical therapy has been suboptimal in LMNA-related DCM, revealing an unmet clinical need.13,14
ARRY-371797 (also known as PF-07265803) is a potent and selective oral p38α MAPK inhibitor previously hypothesized as a potential treatment for LMNA-related DCM. In a completed 48-week, open-label, nonrandomized, phase 2 trial (NCT02057341) of 12 patients with New York Heart Association (NYHA) class II/III symptomatic LMNA-related DCM, treatment with ARRY-371797 (400 or 100 mg twice daily [BID]; pooled) was associated with significant improvement in functional capacity (assessed in the 6-minute walk test [6MWT]), reduction in NT-proBNP (N-terminal pro-B-type natriuretic peptide) concentration, and stable LVEF at 12 weeks.12,15 The improvements in 6MWT distance and reductions in NT-proBNP concentration were maintained in 8 patients who were treated continuously for up to 144 weeks in a long-term extension study (NCT02351856).16,17 Furthermore, no major side effects were associated with ARRY-371797 treatment.12,15–17
This article reports the findings of a subsequent multicenter, randomized, double-blind, placebo-controlled, phase 3 study (REALM-DCM [NCT03439514]) investigating the efficacy and safety of ARRY-371797 in patients with symptomatic LMNA-related DCM.
METHODS
The REALM-DCM study protocol was approved by the independent review board or ethics committee at each participating site and was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization–Good Clinical Practice guidelines. All patients provided written, informed consent. An independent end point adjudication committee, whose members were unaware of the trial group assignments, determined whether investigator-reported events met the definition of HF-related hospitalizations or urgent care visits.
Patients
Adult patients (≥18 years of age) were eligible to participate in REALM-DCM if they had symptomatic LMNA-related DCM with decreased LVEF ≤50% by echocardiography (adjudicated centrally, with or without LV dilatation); carried a confirmed LMNA variant categorized as pathogenic, likely pathogenic, or of uncertain significance but with a phenotype highly suggestive of LMNA-related DCM as assessed by the Investigator; NYHA functional class II/III HF symptoms that were stable for ≥3 months; reduced 6MWT distance (>100 to ≤450 m at screening and >100 to ≤485 m at day −1 and baseline); stable medical and device therapy consistent with regional guidelines18,19; and an implantable cardioverter-defibrillator implanted ≥4 weeks before initiation of study intervention. Upon protocol amendment, implantable cardioverter-defibrillator devices were required to have pacing capabilities, or a pacing-capable co-implant must have been present. The phenotypes of patients carrying LMNA variant of uncertain significance were assessed, and eligibility was confirmed by the investigators. Although patients with NYHA functional class IV HF symptoms were eligible to participate in this study, none were enrolled.
Patients were excluded if they met any of the following criteria: other forms of cardiomyopathy contributing to HF; clinically significant coronary artery disease; non-HF–related primary structural valvular disease; receiving intravenous inotrope therapy; initiation of cardiac resynchronization therapy in the prior 6 months before the initiation of study intervention; a history of heart transplantation or anticipated heart transplantation in the next 6 months; implantation of a ventricular assist device; myocardial infarction or cardiac surgical procedures in the 3 months before screening; anticipated to require renal replacement therapy within 6 months; or a life span–limiting (<1 year) noncardiac condition; an underlying condition that may impact the ability of the 6MWT to reflect changes in cardiovascular function (eg, orthopedic conditions that limit a patient’s walking abilities, significant musculoskeletal pathology, or any other condition that limits a patient’s performance on the 6MWT independently from their cardiomyopathy as assessed by the Investigator).
Treatment
Enrolled patients were randomized (1:1) to ARRY-371797 400-mg oral tablets BID or matching placebo, stratified by baseline 6MWT distance (average of day −1 and baseline: <320 or ≥320 m) and LMNA variant type (phenotype/likely pathogenic or, after protocol amendment, variant of uncertain significance). Throughout the study, dose reductions to 200 mg BID, or further to 100 mg BID, were allowed if the patient had safety or tolerability issues. The patient received all subsequent treatment at the reduced dose unless interruption or discontinuation of the study drug was warranted.
Outcomes and Statistical Analysis
Based on the findings from a planned interim futility analysis conducted on data from the first 68 randomized patients who completed week 24 or discontinued before week 24, the sponsor decided to terminate the study in August 2022 due to low conditional power to achieve the primary objective at the time of the final analysis.20 All ongoing patients discontinued treatment, attended a discontinuation visit, and underwent a subsequent safety follow-up visit 30 days later. The initial primary efficacy outcome was a change from baseline in 6MWT distance at week 12. The calculation of the initial sample size and expected effect size is described in the Supplemental Methods. Based on regulatory feedback, this was amended to week 24 in a blinded manner without alpha spending. The van Elteren test was used to assess the difference between ARRY-371797 and placebo groups stratified by baseline 6MWT quartiles. Patients who died before week 24 were given the lowest rank (from shortest to longest survival), followed by patients who discontinued from the study for any reason before week 24 (ordered based on their time to study discontinuation). For patients who remained in the trial at or beyond week 24 and had a missing assessment, missing values were imputed using the Markov chain Monte Carlo multiple imputation method. Treatment effect was estimated using the stratified Hodges-Lehmann method by baseline 6MWT quartiles based on patients who survived to week 24. For patients who discontinued before week 24 due to postrandomization events (eg, adverse events), missing values were imputed using copy-reference multiple imputation, assuming similarity between treatment groups.
Secondary efficacy outcomes included change from baseline at week 24 in the Kansas City Cardiomyopathy Questionnaire (KCCQ)-physical limitation (PL) and total symptom (TS) scores and NT-proBNP concentration, all analyzed with the same methodology as the primary outcome but with the van Elteren test stratified by baseline KCCQ-PL or TS scores or NT-proBNP concentration, respectively. Patient global impression of severity and change were summarized descriptively. Exploratory efficacy outcomes included the observed change from baseline in LVEF and right ventricular fractional area at week 24 assessed by echocardiography and read by a central laboratory.
Cardiovascular safety was assessed using a composite outcome of time-to-first event of adjudicated worsening heart failure (WHF), defined as HF-related hospitalization or urgent care visit, or all-cause mortality. Overall survival was also assessed. The composite outcome and overall survival were summarized using the Kaplan-Meier method. Hazard ratio (HR) was estimated using the Cox proportional hazards model stratified by baseline NYHA functional class (II or III) and 6MWT distance (<320 or ≥320 m) and included treatment arm as a covariate. Safety outcomes also included the incidence and severity of treatment-emergent adverse events (TEAEs).
Materials and Data Availability
Upon reasonable request and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions, and exceptions, Pfizer may also provide access to the related individual de-identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
RESULTS
Patients
In REALM-DCM, 485 patients suspected of having LMNA-related DCM were prospectively consented to and prescreened. The main reasons for prescreen and screen failures were the absence of an eligible LMNA variant (n=283, 58%) and asymptomatic status (defined as NYHA functional class I [n=125; 26%]). Between April 2018 and October 2022, 77 patients (ARRY-371797 [n=40], placebo [n=37]) aged 23 to 72 years with NYHA functional class II/III HF symptoms were enrolled at 31 sites in 6 countries (Figure 1; Table 1). Overall, baseline characteristics and demographics between the 2 treatment groups were broadly similar. Most of the enrolled patients were male (57%), White (96%), and carried a known phenotype or likely pathogenic LMNA variant (88%) or a variant of uncertain significance (12%). Of the 77 enrolled patients, 75 had LMNA variants with DNA sequence data determined by the central laboratory (ARRY-371797 [n=39], placebo [n=36]). In the ARRY-371797-treated group, 56% of patients (n=22/39) had a missense LMNA variant, and 44% (n=17/39) had a nonmissense variant. Respective proportions in the placebo group were 53% (n=19/36) and 47% (n=17/36; Table 2). Nonmissense variants included nonsense (ARRY-371797 [23%, n=9] versus placebo [17%, n=6]), frameshift (15% [n=6] versus 14% [n=5]), splice acceptor (3% [n=1] versus 14% [n=5]), and silent (3% [n=1] in each group) changes (Table 2). Baseline comorbidities included atrial fibrillation (ARRY-371797 58% [n=23] versus placebo 62% [n=23]), atrial flutter (10% [n=4] versus 8% [n=3]), and complete atrioventricular block (10% [n=4] versus 3% [n=1]).
Figure 1.

Study design and patient disposition. 6MWT indicates 6-minute walk test; AE, adverse event; BID, twice daily; DCM, dilated cardiomyopathy; KCCQ, Kansas City Cardiomyopathy Questionnaire; LMNA, lamin A/C; NT-proBNP, N-terminal pro-B-type natriuretic peptide; NYHA, New York Heart Association; PL, physical limitation; and TS, total symptom. *Prescreen failure included patients who signed the prescreening informed consent but did not sign the main study informed consent. †Screen failure included patients who signed the prescreening and main study informed consent but were not subsequently randomized. ‡Dose reduction for safety or tolerability were allowed throughout this study. The ARRY-371797 400 mg BID dose could be reduced to 200 mg BID and subsequently to 100 mg BID if necessary. §Patients who discontinued treatment can stay in the study for additional follow-up.
Table 1.
Baseline Demographics and Clinical Characteristics
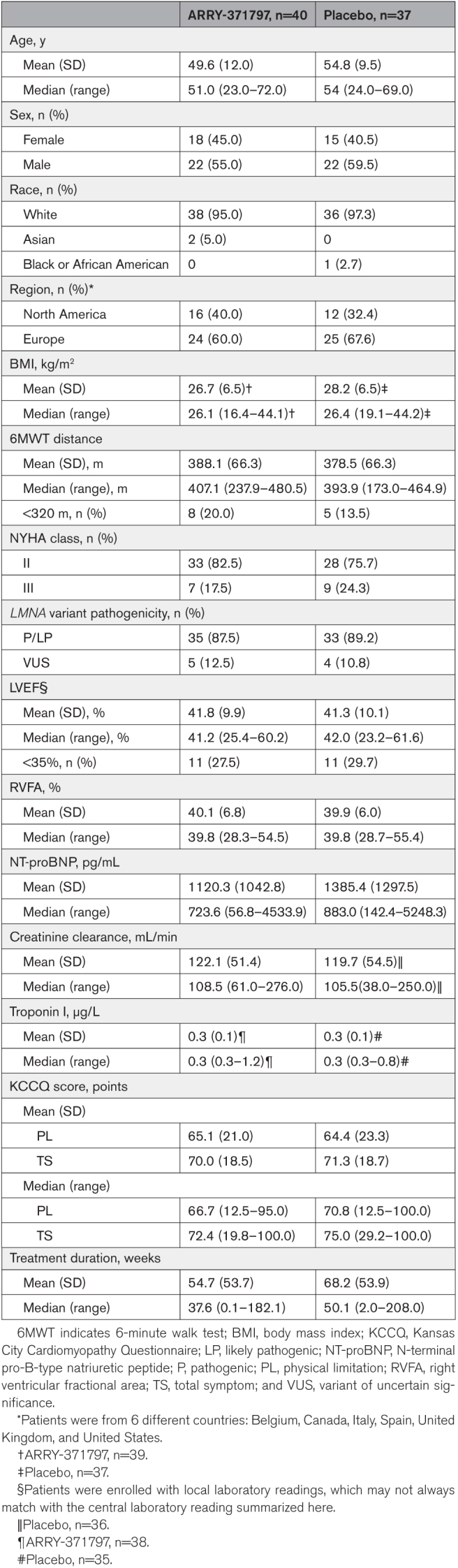
Table 2.
LMNA Variants as Determined by the Central Laboratory

The medications relevant to the management of LMNA-related DCM taken by patients at any time alongside study treatment (concomitant) are presented in Table S1. Most notably, nearly all patients were concomitantly receiving a beta-blocker (ARRY-371797 [95%] versus placebo [100%]), a renin-angiotensin system-acting agent (90% versus 92%), or an antithrombotic agent (either an antiplatelet agent or anticoagulant: 83% versus 92%). During the study, 40% of patients in the ARRY-371797 group and 46% in the placebo group were reported to have received a concomitant procedure (Table S2), including cardioversion (ARRY-371797 [15%] versus placebo [24%]), implantable defibrillator replacement (13% versus 5%), or cardiac ablation (3% versus 8%).
Primary Efficacy Outcome: Change From Baseline in 6MWT Distance at Week 24
Five participants (ARRY-371797 [n=3], placebo [n=2]) discontinued the study before week 24 due to the sponsor’s decision to terminate the study for futility and were excluded from the primary analysis. Of the 72 patients included in the primary analysis, 4 died (2 in each treatment group). Four discontinued from the study (ARRY-371797 [n=3], placebo [n=1]) for reasons other than death before week 24, and the values for these patients were imputed.
The median observed 6MWT distance at baseline and week 24 is plotted in Figure S1. The median change from baseline in 6MWT distance at week 24 was 21 m (95% CI, −22.8 to 51.5) in the ARRY-371797 group and 3 m (95% CI, −11.5 to 33.7) in the placebo group (Figure 2A). No significant difference between groups was found in the van Elteren rank-sum test (2-sided P=0.82). The treatment difference, as calculated using the Hodges-Lehmann estimation, was 4.9 m (95% CI, −24.2 to 34.1).
Figure 2.

Primary and secondary efficacy outcomes. Change from baseline in median 6MWT distance (A), KCCQ-PL score (B), KCCQ-TS score (C), and NT-proBNP concentration (D) at week 24. Vertical bars indicate the IQR (25th and 75th percentile). 6MWT indicates 6-minute walk test; HL, Hodges-Lehmann; IQR, interquartile range; KCCQ, Kansas City Cardiomyopathy Questionnaire; NT-proBNP, N-terminal pro-B-type natriuretic peptide; PL, physical limitation; and TS, total symptom.
Secondary Efficacy Outcomes
The median observed KCCQ-PL score, KCCQ-TS score, and NT-proBNP concentration at baseline and week 24 are plotted in Figure S1. The median change from baseline at week 24 in the ARRY-371797 and placebo groups is plotted in Figure 2B through 2D. For all 3 outcomes, no significant differences between treatment groups were found in the van Elteren rank-sum test (2-sided P values: KCCQ-PL 0.54; KCCQ-TS 0.48; NT-proBNP 0.17). Treatment differences, as calculated using the Hodges-Lehmann estimation of treatment effect, were 2.4 points (95% CI, −6.4 to 11.2), 5.3 points (95% CI, −4.3 to 14.9), and −339.4 pg/mL (95% CI, −1131.6 to 452.7), respectively (Figure 2B through 2D).
In the patient global impression survey at week 24, 75% of patients in the ARRY-371797 group and 71% in the placebo group reported mild to moderate HF symptom severity (Table S3), whereas 11% and 10%, respectively, reported severe symptoms. When considering change since the start of the study, 39% of ARRY-371797-treated and 31% of placebo-treated patients reported overall improvement in their HF symptoms (“a little better” to “very much better”), whereas 4% versus 7% reported worsening (“moderately” to “very much” worse) at week 24.
Exploratory Efficacy Outcomes: Change From Baseline in LVEF and Right Ventricular Fractional Area at Week 24
The observed median changes from baseline (range) in LVEF at week 24 were 1.9% (−11.2% to 9.6%) in the ARRY-371797 group and −0.8% (−13.3% to 6.1%) in the placebo group. The respective observed median (range) changes from baseline in the right ventricular fractional area were −2.6% (−8.7% to 19.1%) and −1.5% (−12.2% to 10.3%; Figure S2).
Safety Outcomes
Patients were treated with ARRY-371797 for a median (range) of 37.6 (0.1–182.1) weeks or placebo for 50.1 (2.0–208.0) weeks (Table 1). Kaplan-Meier and Cox proportional hazard analyses showed no significant difference in the composite outcome of first WHF or all-cause mortality in the ARRY-371797 (no WHF, 3 deaths) and placebo (6 WHF, 1 death) groups. The composite HR (ARRY-371797 versus placebo) was 0.43 ([95% CI, 0.11–1.74]; P=0.23; Figure 3A). Throughout the study, 3 deaths were reported in each group, with an HR for all-cause mortality of 1.19 ([95% CI, 0.23–6.02]; P=0.84; Figure 3B). No deaths were considered by the investigator to be related to the study drug. The reported causes of death in the ARRY-371797 group were pancreatic neoplasia, ventricular tachycardia, and anoxic brain injury from sequelae of primary graft dysfunction after heart transplantation due to refractory ventricular arrythmias, and in the placebo group, bacterial pneumonia, respiratory failure, and disease progression.
Figure 3.

Composite outcome and survival analyses. Kaplan-Meier plots of the composite outcome of all-cause mortality or WHF (A) and overall survival (B). Cox proportional hazard analysis was stratified by New York Heart Association functional class (II vs III) and baseline 6-minute walk test (6MWT) distance (<320 m vs ≥320 m). WHF indicates worsening heart failure.
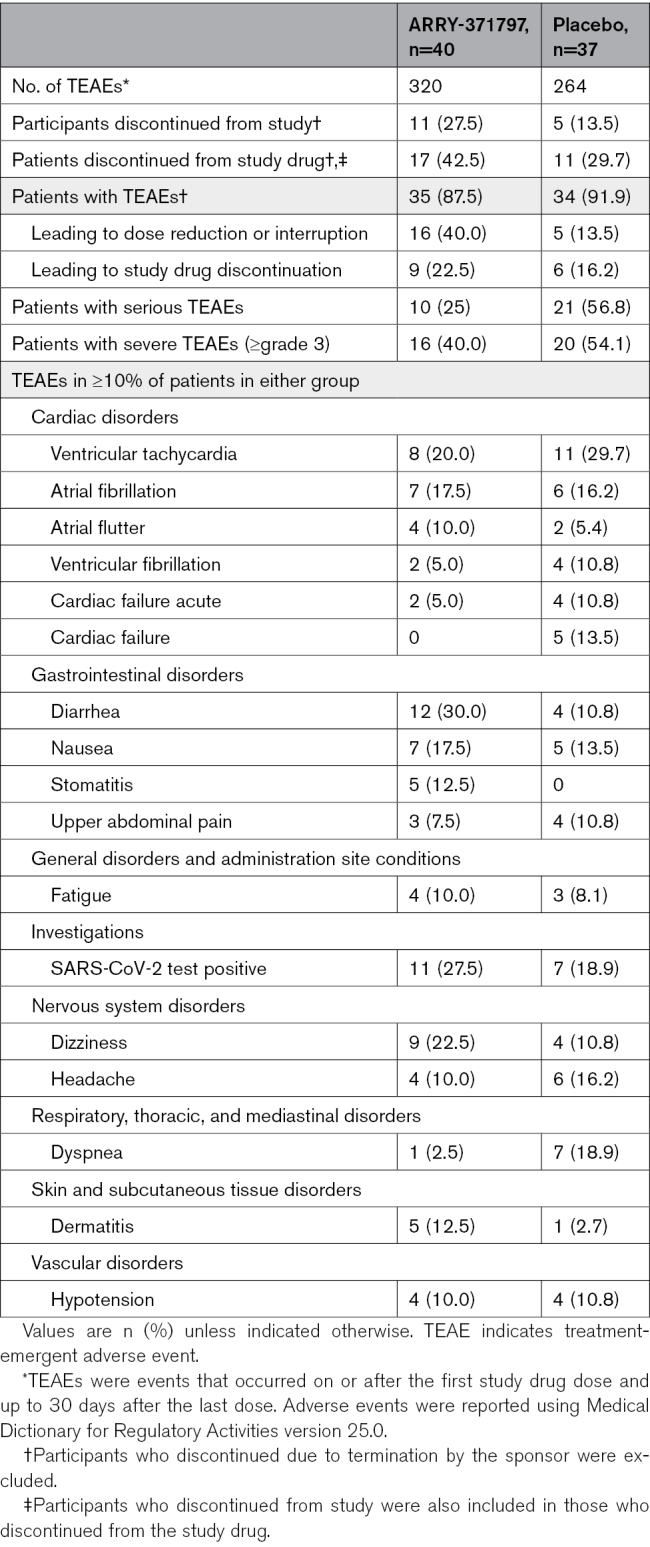
The overall safety profile of ARRY-371797 was similar to that observed in previous studies of the drug.12,16 Dose reductions for any reason (28% versus 8%) and due to TEAEs (13% versus 3%) were more common in patients treated with ARRY-371797 versus placebo. Excluding patients who discontinued due to termination of the study by the sponsor, a higher proportion of patients treated with ARRY-371797 versus placebo discontinued from the study (28% versus 14%) and from the study drug (43% versus 30%; Table 3). Sixty-nine patients reported TEAEs, including 88% (35/40) in the ARRY-371797 group and 92% (34/37) in the placebo group (Table 3). The most commonly reported TEAEs in the placebo versus ARRY-371797 group were ventricular tachycardia (30% versus 20%), SARS-CoV-2 test positive (19% versus 28%), and dyspnea (19% versus 3%). The most commonly reported TEAEs in the ARRY-371797 versus placebo group were diarrhea (30% versus 11%), SARS-CoV-2 test positive, and dizziness (23% versus 11%). Patients treated with ARRY-371797 reported fewer serious (25% versus 57%) or severe (≥grade 3: 40% versus 54%) TEAEs compared with those treated with placebo (Table 3; Table S4).
Table 3.
Study Discontinuations and TEAEs, Including All Causalities, by System Organ Class and Preferred Term
DISCUSSION
The phase 3 REALM-DCM study evaluated the efficacy and safety of ARRY-371797 in patients with symptomatic LMNA-related DCM carrying LMNA variants that are either phenotype/likely pathogenic (88%, n=68) or of uncertain significance (12%, n=9). Based on the findings of a planned interim analysis, this study was terminated early due to futility.
REALM-DCM enrolled 77 patients aged 23 to 72 years from Europe (64%) or North America (36%) with NYHA functional class II (79%) or III (21%) HF symptoms. Of the 75 patients with a central laboratory-determined LMNA variant, 55% had a missense change and 45% a nonmissense change.
There was no significant difference between the ARRY-371797 and placebo groups in the primary efficacy outcome of median change from baseline in 6MWT distance after 24 weeks of treatment (2-sided P=0.82; Hodges-Lehmann median difference: 4.9 m). Similarly, there were no significant differences between treatment groups in the median change from baseline at week 24 in KCCQ-PL score, KCCQ-TS score, NT-proBNP concentration, LVEF, and right ventricular fractional area, or between groups in the patient global impression survey at week 24. The composite end point of first WHF or all-cause mortality had a nonsignificant HR of 0.43 ([95% CI, 0.11–1.74]; P=0.23), and overall survival was similar between groups (HR, 1.19 [95% CI, 0.23–6.02]; P=0.84). Overall findings from the REALM-DCM trial demonstrated similar efficacy with ARRY-371797 and placebo. The safety findings for ARRY-371797 were consistent with previous studies.12,16
The limited data on LMNA genotype-phenotype associations and the natural history of LMNA-related DCM impede the diagnosis of patients and limit their effective treatment.21–24 Some previous studies have suggested that nonmissense LMNA variants may be associated with a worse prognosis than missense variants, but such studies are readily confounded in rare familial conditions.23,25–27 A mechanistic understanding of LMNA-related DCM will be necessary to understand any variant-specific effects on the natural history and prognosis of the disease.
REALM-DCM was the first phase 3, randomized, controlled interventional trial of a disease-modifying treatment in patients with LMNA-related DCM, and despite the early termination, the data gathered are valuable in the characterization of this disease and, pivotally, will provide benefit to study patients and their families.
For studies on rare diseases such as LMNA-related DCM, identifying eligible participants and determining clinically relevant outcome measures are challenging.28 It is inevitable that sample sizes will be relatively modest and potentially constrain adequate statistical powering, particularly in patient populations with variable phenotypes and no quantitative mechanistic end points. Genetic evaluation of patients with nonischemic DCM is recommended for cascade screening but is not widely used outside of tertiary medical centers. There are many pathological LMNA variants, and an understanding of detailed genotype-phenotype associations is still emerging, which contributes to the heterogeneity of patient study populations. In this study, the observed median change from baseline in 6MWT distance, KCCQ-PL and -TS scores, and NT-proBNP concentration were highly variable, possibly due to this heterogeneity and the small sample size. Moreover, the most appropriate primary efficacy outcome measures for this population have not been established. Although this trial was focused on outcomes commonly used in trials of patients with HF, patients with LMNA-related DCM are also at high risk of clinically significant arrythmias, and arrythmia burden may be a key efficacy outcome for consideration in future trials.5,8,29 In addition, extracardiac manifestations of LMNA-related disease may be useful adjunct end points in future studies of LMNA-related DCM.
Limitations
The limitations of this study, as detailed above, include the early termination, modest enrollment, and the heterogeneous nature of the patient population, which further constrained the ability to detect treatment-related changes. The COVID-19 pandemic posed additional challenges to patient enrollment and follow-up. REALM-DCM enrolled a subset of patients with symptomatic, stable LMNA-related DCM who were receiving standard-of-care therapy. As cardiac impairment in enrolled patients was determined using LVEF ≤50% (with or without LV dilatation), some patients might have hypokinetic nondilated cardiomyopathy (HNDCM), defined as LV or biventricular global systolic dysfunction without dilatation (LVEF <45%), that is not explained by abnormal loading conditions or coronary artery disease.30 However, HNDCM was not a widely used term during study inception.31 Also, the study cohort included patients with LMNA variants of uncertain significance and excluded patients with LMNA-related muscular dystrophy that limited their ability to perform the 6MWT. Overlap of the LMNA cardiac phenotype with coexisting limb-girdle muscular dystrophy type 1B symptoms may still have confounded exercise capacity and stamina. For these reasons, the study cohort may not be reflective of the broader patient population with LMNA-related DCM. Furthermore, the measures or biomarkers used in this study to determine efficacy have not been validated in patients with LMNA-related DCM and may be suboptimal.32–35 In some studies of disease-modifying therapies in patients with HF with reduced ejection fraction, no significant improvements in 6MWT distance or KCCQ scores were observed, despite reductions in morbidity or mortality.36,37 Although no significant differences between the ARRY-371797 and placebo groups were observed in all outcomes in this study, future trials may further investigate the effects of p38α MAPK inhibition in patients with LMNA-related DCM.
Conclusions
The phase 3 REALM-DCM study of ARRY-371797, a p38α MAPK inhibitor, was terminated early due to futility for the primary efficacy outcome. An unmet need for genetically informed treatment persists for patients with LMNA-related DCM. Despite the lack of treatment differences identified, findings from REALM-DCM represent the largest and most robust prospectively gathered clinical data set for genotype-phenotype correlations and natural history evaluations of LMNA-related DCM captured to date. These will contribute toward a better disease understanding and optimized design for future trials.
ARTICLE INFORMATION
REALM-DCM Investigators
Tomas V. Ripoll Vera, Hospital Universitario Son Llatzer, Palma de Mallorca, Spain; Thomas V. McDonald, University of South Florida, Tampa, FL; Juan Carlos Castillo Dominguez, C.H. Regional Reina Sofia, Córdoba, Spain; Stuart D. Katz and Stephen Pan, NYU Langone Health, New York, NY; Juan R. Gimeno Blanes, Hospital Universitario Virgen de la Arrixaca, Murcia, Spain; Dinesh K. Gupta, Tennessee Center for Clinical Trials, Tullahoma, TN; Matthew R.G. Taylor, University of Colorado, Aurora, CO; Cinzia Forleo, Azienda Ospedaliero Universitaria Consorziale Policlinico di Bari, Bari, Italy; Kia Afshar, Intermountain Medical Center, Murray, UT; Matthew T. Wheeler, Stanford University School of Medicine, Stanford, CA; Silvia G. Priori, IRCCS Istituti Clinici Scientifici Maugeri Spa Società Benefit, Pavia, Italy; Marc Vanderheyden, Onze-Lieve-Vrouwziekenhuis, Aalst, Belgium; Iacopo Olivotto, Azienda Ospedaliera Universitaria Careggi, Florence, Italy; Jose A. Tallaj, The Kirklin Clinic of University of Alabama at Birmingham, Birmingham, AL; Johan Van Cleemput, UZ Leuven, Leuven, Belgium; Ana Garcia Alvarez, Hospital Clinic de Barcelona, Barcelona, Spain; April C. Stempien-Otero, University of Washington Medical Center, Seattle, WA; Victor Alfonso Jimenez Diaz, Complejo Hospitalario Universitario de Vigo - H. Alvaro Cunqueiro, Vigo, Spain; Maria Beatrice Musumeci, Azienda Ospedaliera Sant’Andrea, Rome, Italy; James Ware, Royal Brompton Hospital, London, United Kingdom; Anjali T. Owens, University of Pennsylvania Heart and Vascular Center, Philadelphia, PA; Julia Cadrin-Tourigny, Clinical Laboratories of Montreal Heart Institute, Montreal, Canada; W. H. Wilson Tang, The Cleveland Clinic Foundation, Cleveland, OH; Gregory A. Ewald, Washington University Center for Advanced Medicine, Saint Louis, MO; Vito Maurizio Parato, Presidio Ospedaliero Madonna del Soccorso, San Benedetto del Tronto, Italy; Alonzo E. Jones, Columbus Regional Research Institute, Columbus, GA; Giuseppe Ambrosio, Ospedale Santa Maria Della Misericordia, Perugia, Italy; Cristian E. Botta, Hospital Provincial Dr. Jose Maria Cullen, Santa Fe, Argentina; Kristina H. Haugaa, Oslo University Hospital, Oslo, Norway; Kjell Andersen, Sykehuset Innlandet HF Hamar, Hamar, Norway; Alejandro R. Hershson, Fundacion Favaloro para la Docencia e Investigacion Medica, Buenos Aires, Argentina; Nabil M. Dib, Dignity Health Medical Group Cardiology Mercy Gilbert, Gilbert, AZ; Horacio A. Avaca, Hospital Británico de Buenos Aires, Buenos Aires, Argentina; Serge Lepage, Centre Intégré Universitaire de santé et de services sociaux de l’Estrie Centre hospitalier universitaire de Sherbrooke, Sherbrooke, Quebec, Canada; Jeffrey Martindale, MyMichigan Medical Center Midland, Midland, MI; Miguel E. Trevino, Innovative Research of West Florida, Clearwater, FL; David A. Hinchman, Saint Luke’s Idaho Cardiology Associates, Boise, ID; Juan Loureyro, Instituto CAICI, Santa Fe, Argentina; Frank A. McGrew, Stern Cardiovascular Foundation Inc, Germantown, TN; Saurabh Kapoor, Newark Beth Israel Medical Center, Newark, NJ; Michael A. Burke, Emory University Hospital, Atlanta, GA; Joshua Williams and Armando Garcia Castillo, Cardiolink Clin Trials S.C., Monterrey, Nuevo León, Mexico; Farhana Latif, Columbia University Irving Medical Center, New York, NY; John LeDoux, CB Flock Research Corporation, Mobile, AL; Yigal Pinto, Amsterdam UMC, Amsterdam, The Netherlands; Eloisa Arbustini, Fondazione IRCCS Policlinico San Matteo di Pavia, Pavia, Italy; Caroline Coats and Pierpaolo Pellicori, Queen Elizabeth University Hospital, Glasgow, United Kingdom; Ray Hershberger, Ohio State University Wexner Medical Center, Columbus, OH; John Moses, Meriter Hospital, Madison, WI; Martin Gardner, NSHA QEII Health Sciences Halifax Infirmary, Halifax, Nova Scotia, Canada; Michael C. Hartleib, Kawartha Cardiology Clinical Trials, Peterborough, Ontario, Canada; Mustafa Toma, St. Paul’s Hospital, Vancouver, British Columbia, Canada; Darryl Davis, University of Ottawa Heart Institute, Ottawa, Ontario, Canada; Mark Hofmeyer and Farooq Sheikh, Medstar Washington Hospital Center, Washington, DC; Marc Klapholz, Rutgers New Jersey Medical School, Newark, NJ; Marco Metra, Azienda Ospedaliera Spedali Civili di Brescia, Brescia, Italy; Eliud Samuel Montes Cruz, Fundación de Atención e Investigación Médica Lindavista S.C., Ciudad de México, Mexico; J R. Pineda and Nancy Sweitzer, University of Arizona Sarver Heart Center, Tucson, AZ.
Acknowledgments
The authors thank everyone who has contributed to the REALM-DCM study, including the study participants and clinical investigators. This study was sponsored by Pfizer. Medical writing support was provided by April Cabang and Jennifer Bodkin of Engage Scientific Solutions and was funded by Pfizer. Dr Angeli and H. Li take responsibility for all aspects of this study including study design, data acquisition, integrity, analysis, and interpretation, manuscript preparation, and revision. Drs Garcia-Pavia, Palomares, Sinagra, Barriales-Villa, Lakdawala, Gottlieb, Goldberg, Elliott, Lee, Judge, and MacRae contributed to data acquisition, interpretation, manuscript preparation, and revision. All authors reviewed the manuscript and approved the final version.
Sources of Funding
This study was sponsored by Pfizer.
Disclosures
Dr Garcia-Pavia reports speaking fees from Alnylam Pharmaceuticals, AstraZeneca, Bristol Myers Squibb (BMS), Bridgebio, Ionis Pharmaceuticals, NovoNordisk, and Pfizer; consulting fees from Alexion, Alnylam Pharmaceuticals, AstraZeneca, ATTRalus, BMS, Bridgebio, Cytokinetics, General Electric, Intellia, Lexeo Therapeutics, Neurimmune, NovoNordisk, Pfizer, and Rocket Pharmaceuticals; reports research/educational support to his institution from Alnylam Pharmaceuticals, AstraZeneca, Bridgebio, Intellia, NovoNordisk, and Pfizer. Dr Barriales-Villa received consultancy fees from Alnylam, Amicus, BMS, Chiesi, Cytokinetics, Pfizer, and Sanofi. Dr Lakdawala has received consulting fees from Array BioPharma, BMS, MyoKardia, Pfizer, and Tenaya Therapeutics. Dr Gottlieb consulted for Alnylam and Gilead Sciences; participated in scientific advisory board meetings for AbbVie, Alnylam, AstraZeneca, Eli Lilly, Gilead Sciences, GlaxoSmithKline, and Roche; received research support from CareDx; has been a member of a speaker’s bureau for Alnylam and Pfizer; and has been a National Principal Investigator (unrelated field) for Johnson & Johnson. Dr Elliott has received consultancy fees from Alnylam and Pfizer and educational grants from Pfizer. Dr Lee, H. Li, and Dr Angeli are full-time employees of Pfizer and hold stock and/or stock options. Dr Judge received consultancy fees from Alexion, Alleviant Medical, Cytokinetics, Novo Nordisk, Pfizer, Renovacor, and Tenaya Therapeutics. Dr MacRae consulted for Adrestia, Affinia, Array BioPharma, AstraZeneca, Bayer, BMS, Design Therapeutics, Dewpoint Therapeutics, DINAQOR, Merck, MyoKardia, Novartis, Novo Nordisk, Nuevocor, and Pfizer; and received grant support from Apple, AstraZeneca, Bayer, Janssen, Merck, Microsoft, Novartis, Quest Diagnostics, Sanofi, and Verily. The other authors report no conflicts.
Supplemental Material
Supplemental Methods
Tables S1–S4
Figures S1 and S2
Supplementary Material
Nonstandard Abbreviations and Acronyms
- 6MWT
- 6-minute walk test
- AE
- adverse event
- BID
- twice daily
- DCM
- dilated cardiomyopathy
- HF
- heart failure
- HR
- hazard ratio
- IQR
- interquartile range
- KCCQ
- Kansas City Cardiomyopathy Questionnaire
- LMNA-DCM
- lamin A/C-related dilated cardiomyopathy
- LVEF
- left ventricular ejection fraction
- MAPK
- mitogen-activated protein kinase
- NT-proBNP
- N-terminal pro-B-type natriuretic peptide
- NYHA
- New York Heart Association
- PL
- physical limitation
- TEAE
- treatment-emergent adverse event
- TS
- total symptom
For Sources of Funding and Disclosures, see page 654.
Full list of the investigators are given in Article Information section (under REALM-DCM Investigators).
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCHEARTFAILURE.123.011548.
Contributor Information
Pablo Garcia-Pavia, Email: pablogpavia@yahoo.es.
Gianfranco Sinagra, Email: Gianfranco.sinagra@asugi.sanita.fvg.it.
Roberto Barriales-Villa, Email: rbarrialesv@gmail.com.
Neal K. Lakdawala, Email: nlakdawala@bwh.harvard.edu.
Robert L. Gottlieb, Email: robert.gottlieb@bswhealth.org.
Perry Elliott, Email: perry.elliott@ucl.ac.uk.
Patrice Lee, Email: paleedddc@gmail.com.
Huihua Li, Email: Huihua.Li@pfizer.com.
Franca S. Angeli, Email: franca.stedileangeli@pfizer.com.
Tomas V. Ripoll Vera, Hospital Universitario Son Llatzer, Palma de Mallorca, Spain
Thomas V. McDonald, University of South Florida, Tampa, FL
Juan Carlos Castillo Dominguez, C.H. Regional Reina Sofia, Córdoba Spain.
Stuart D. Katz, NYU Langone Health, New York, NY
Stephen Pan, NYU Langone Health, New York, NY.
Juan R. Gimeno Blanes, Hospital Universitario Virgen de la Arrixaca, Murcia, Spain
Dinesh K. Gupta, Tennessee Center for Clinical Trials, Tullahoma, TN
Matthew R.G. Taylor, University of Colorado, Aurora, CO
Cinzia Forleo, Azienda Ospedaliero Universitaria Consorziale Policlinico di Bari, Bari, Italy.
Kia Afshar, Intermountain Medical Center, Murray, UT.
Matthew T. Wheeler, Stanford University School of Medicine, Stanford, CA
Silvia G. Priori, IRCCS Istituti Clinici Scientifici Maugeri Spa Società Benefit, Pavia, Italy
Marc Vanderheyden, Onze-Lieve-Vrouwziekenhuis, Aalst, Belgium.
Iacopo Olivotto, Azienda Ospedaliera Universitaria Careggi, Florence, Italy.
Jose A. Tallaj, The Kirklin Clinic of University of Alabama at Birmingham, Birmingham, AL
Johan Van Cleemput, UZ Leuven, Leuven, Belgium.
Ana Garcia Alvarez, Hospital Clinic de Barcelona, Barcelona, Spain.
April C. Stempien-Otero, University of Washington Medical Center, Seattle, WA
Victor Alfonso Jimenez Diaz, Complejo Hospitalario Universitario de Vigo - H. Alvaro Cunqueiro, Vigo, Spain.
Maria Beatrice Musumeci, Azienda Ospedaliera Sant’Andrea, Rome, Italy.
James Ware, Royal Brompton Hospital, London, United Kingdom.
Anjali T. Owens, University of Pennsylvania Heart and Vascular Center, Philadelphia, PA
Julia Cadrin-Tourigny, Clinical Laboratories of Montreal Heart Institute, Montreal, Canada.
W. H. Wilson Tang, The Cleveland Clinic Foundation, Cleveland, OH.
Gregory A. Ewald, Washington University Center for Advanced Medicine, Saint Louis, MO
Vito Maurizio Parato, Presidio Ospedaliero Madonna del Soccorso, San Benedetto del Tronto, Italy.
Alonzo E. Jones, Columbus Regional Research Institute, Columbus, GA
Giuseppe Ambrosio, Ospedale Santa Maria Della Misericordia, Perugia, Italy.
Cristian E. Botta, Hospital Provincial Dr. Jose Maria Cullen, Santa Fe, Argentina
Kristina H. Haugaa, Oslo University Hospital, Oslo, Norway
Kjell Andersen, Sykehuset Innlandet HF Hamar, Hamar, Norway.
Alejandro R. Hershson, Fundacion Favaloro para la Docencia e Investigacion Medica, Buenos Aires, Argentina
Nabil M. Dib, Dignity Health Medical Group Cardiology Mercy Gilbert, Gilbert, AZ
Horacio A. Avaca, Hospital Británico de Buenos Aires, Buenos Aires, Argentina
Serge Lepage, Centre Intégré Universitaire de santé et de services sociaux de l’Estrie Centre hospitalier universitaire de Sherbrooke, Sherbrooke, Quebec, Canada.
Jeffrey Martindale, MyMichigan Medical Center Midland, Midland, MI.
Miguel E. Trevino, Innovative Research of West Florida, Clearwater, FL
David A. Hinchman, Saint Luke’s Idaho Cardiology Associates, Boise, ID
Juan Loureyro, Instituto CAICI, Santa Fe, Argentina.
Frank A. McGrew, Stern Cardiovascular Foundation Inc, Germantown, TN
Saurabh Kapoor, Newark Beth Israel Medical Center, Newark, NJ.
Michael A. Burke, Emory University Hospital, Atlanta, GA
Joshua Williams, Cardiolink Clin Trials S.C., Monterrey, Nuevo León, Mexico.
Armando Garcia Castillo, Cardiolink Clin Trials S.C., Monterrey, Nuevo León, Mexico.
Farhana Latif, Columbia University Irving Medical Center, New York, NY.
John LeDoux, CB Flock Research Corporation, Mobile, AL.
Yigal Pinto, Amsterdam UMC, Amsterdam, The Netherlands.
Eloisa Arbustini, Fondazione IRCCS Policlinico San Matteo di Pavia, Pavia, Italy.
Caroline Coats, Queen Elizabeth University Hospital, Glasgow, United Kingdom.
Pierpaolo Pellicori, Queen Elizabeth University Hospital, Glasgow, United Kingdom.
Ray Hershberger, Ohio State University Wexner Medical Center, Columbus, OH.
John Moses, Meriter Hospital, Madison, WI.
Martin Gardner, NSHA QEII Health Sciences Halifax Infirmary, Halifax, Nova Scotia, Canada.
Michael C. Hartleib, Kawartha Cardiology Clinical Trials, Peterborough, Ontario, Canada
Mustafa Toma, St. Paul’s Hospital, Vancouver, British Columbia, Canada.
Darryl Davis, University of Ottawa Heart Institute, Ottawa, Ontario, Canada.
Mark Hofmeyer, Medstar Washington Hospital Center, Washington, DC.
Farooq Sheikh, Medstar Washington Hospital Center, Washington, DC.
Marc Klapholz, Rutgers New Jersey Medical School, Newark, NJ.
Marco Metra, Azienda Ospedaliera Spedali Civili di Brescia, Brescia, Italy.
Eliud Samuel Montes Cruz, Fundación de Atención e Investigación Médica Lindavista S.C., Ciudad de México, Mexico.
J R. Pineda, University of Arizona Sarver Heart Center, Tucson, AZ
Collaborators: Tomas V. Ripoll Vera, Thomas V. McDonald, Juan Carlos Castillo Dominguez, Stuart D. Katz, Stephen Pan, Juan R. Gimeno Blanes, Dinesh K. Gupta, Matthew R.G. Taylor, Cinzia Forleo, Kia Afshar, Matthew T. Wheeler, Silvia G. Priori, Marc Vanderheyden, Iacopo Olivotto, Jose A. Tallaj, Johan Van Cleemput, Ana Garcia Alvarez, April C. Stempien-Otero, Victor Alfonso Jimenez Diaz, Maria Beatrice Musumeci, James Ware, Anjali T. Owens, Julia Cadrin-Tourigny, W. H. Wilson Tang, Gregory A. Ewald, Vito Maurizio Parato, Alonzo E. Jones, Giuseppe Ambrosio, Cristian E. Botta, Kristina H. Haugaa, Kjell Andersen, Alejandro R. Hershson, Nabil M. Dib, Horacio A. Avaca, Serge Lepage, Jeffrey Martindale, Miguel E. Trevino, David A. Hinchman, Juan Loureyro, Frank A. McGrew, Saurabh Kapoor, Michael A. Burke, Joshua Williams, Armando Garcia Castillo, Farhana Latif, John LeDoux, Yigal Pinto, Eloisa Arbustini, Caroline Coats, Pierpaolo Pellicori, Ray Hershberger, John Moses, Martin Gardner, Michael C. Hartleib, Mustafa Toma, Darryl Davis, Mark Hofmeyer, Farooq Sheikh, Marc Klapholz, Marco Metra, Eliud Samuel Montes Cruz, and J R. Pineda
REFERENCES
- 1.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Jr, Spudich S, De Girolami U, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302 [DOI] [PubMed] [Google Scholar]
- 2.Reichart D, Magnussen C, Zeller T, Blankenberg S. Dilated cardiomyopathy: from epidemiologic to genetic phenotypes: a translational review of current literature. J Intern Med. 2019;286:362–372. doi: 10.1111/joim.12944 [DOI] [PubMed] [Google Scholar]
- 3.Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet. 2017;390:400–414. doi: 10.1016/S0140-6736(16)31713-5 [DOI] [PubMed] [Google Scholar]
- 4.Escobar-Lopez L, Ochoa JP, Mirelis JG, Espinosa MA, Navarro M, Gallego-Delgado M, Barriales-Villa R, Robles-Mezcua A, Basurte-Elorz MT, Gutiérrez García-Moreno L, et al. Association of genetic variants with outcomes in patients with nonischemic dilated cardiomyopathy. J Am Coll Cardiol. 2021;78:1682–1699. doi: 10.1016/j.jacc.2021.08.039 [DOI] [PubMed] [Google Scholar]
- 5.Ferradini V, Cosma J, Romeo F, De Masi C, Murdocca M, Spitalieri P, Mannucci S, Parlapiano G, Di Lorenzo F, Martino A, et al. Clinical features of LMNA-related cardiomyopathy in 18 patients and characterization of two novel variants. J Clin Med. 2021;10:5075. doi: 10.3390/jcm10215075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Captur G, Arbustini E, Bonne G, Syrris P, Mills K, Wahbi K, Mohiddin SA, McKenna WJ, Pettit S, Ho CY, et al. Lamin and the heart. Heart. 2018;104:468–479. doi: 10.1136/heartjnl-2017-312338 [DOI] [PubMed] [Google Scholar]
- 7.Hasselberg NE, Haland TF, Saberniak J, Brekke PH, Berge KE, Leren TP, Edvardsen T, Haugaa KH. Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur Heart J. 2018;39:853–860. doi: 10.1093/eurheartj/ehx596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar S, Baldinger SH, Gandjbakhch E, Maury P, Sellal JM, Androulakis AF, Waintraub X, Charron P, Rollin A, Richard P, et al. Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. J Am Coll Cardiol. 2016;68:2299–2307. doi: 10.1016/j.jacc.2016.08.058 [DOI] [PubMed] [Google Scholar]
- 9.Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, Di Lenarda A, Bohlmeyer TJ, Ferguson DA, Brodsky GL, et al. ; Familial Dilated Cardiomyopathy Registry Research Group. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol. 2003;41:771–780. doi: 10.1016/s0735-1097(02)02954-6 [DOI] [PubMed] [Google Scholar]
- 10.Muchir A, Wu W, Choi JC, Iwata S, Morrow J, Homma S, Worman HJ. Abnormal p38alpha mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum Mol Genet. 2012;21:4325–4333. doi: 10.1093/hmg/dds265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerbino A, Procino G, Svelto M, Carmosino M. Role of lamin A/C gene mutations in the signaling defects leading to cardiomyopathies. Front Physiol. 2018;9:1356. doi: 10.3389/fphys.2018.01356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacRae CA, Taylor MRG, Mestroni L, Moses J, Ashley EA, Wheeler MT, Lakdawala NK, Hershberger RE, Sandor V, Saunders ME, et al. Efficacy and safety of ARRY-371797 in LMNA-related dilated cardiomyopathy: a phase 2 study. Circ Genom Precis Med. 2023;16:e003730. doi: 10.1161/CIRCGEN.122.003730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jansweijer JA, Nieuwhof K, Russo F, Hoorntje ET, Jongbloed JD, Lekanne Deprez RH, Postma AV, Bronk M, van Rijsingen IA, de Haij S, et al. Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. Eur J Heart Fail. 2017;19:512–521. doi: 10.1002/ejhf.673 [DOI] [PubMed] [Google Scholar]
- 14.Ferreira A, Ferreira V, Antunes MM, Lousinha A, Pereira-da-Silva T, Antunes D, Cunha PS, Oliveira M, Ferreira RC, Rosa SA. Dilated cardiomyopathy: a comprehensive approach to diagnosis and risk stratification. Biomedicines. 2023;11:834. doi: 10.3390/biomedicines11030834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacRae CA, Taylor MR, Mestroni L, Moses J, Ashley EA, Wheeler MT, Lakdawala NK, Hershberger RE, Sandor V, Saunders ME, et al. Plain language summary of publication of the safety and efficacy of ARRY-371797 in people with dilated cardiomyopathy and a faulty LMNA gene. Future Cardiol. 2023;19:55–63. doi: 10.2217/fca-2022-0099 [DOI] [PubMed] [Google Scholar]
- 16.Judge DP, Lakdawala NK, Taylor MRG, Mestroni L, Li H, Oliver C, Angeli FS, Lee PA, MacRae CA. Long-term efficacy and safety of ARRY-371797 (PF-07265803) in patients with lamin A/C-related dilated cardiomyopathy. Am J Cardiol. 2022;183:93–98. doi: 10.1016/j.amjcard.2022.08.001 [DOI] [PubMed] [Google Scholar]
- 17.Judge DP, Taylor MR, Li H, Oliver C, Angeli FS, Lee PA, MacRae CA. Long-term effectiveness of ARRY-371797 in people with dilated cardiomyopathy and a faulty LMNA gene: a plain language summary. Future Cardiol. 2023;19:117–126. doi: 10.2217/fca-2022-0125 [DOI] [PubMed] [Google Scholar]
- 18.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Jr, Colvin MM, Drazner MH, Filippatos GS, Fonarow GC, Givertz MM, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2017;136:e137–e161. doi: 10.1161/CIR.0000000000000509 [DOI] [PubMed] [Google Scholar]
- 19.Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, Gonzalez-Juanatey JR, Harjola VP, Jankowska EA, et al. ; ESC Scientific Document Group. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37:2129–2200. doi: 10.1093/eurheartj/ehw128 [DOI] [PubMed] [Google Scholar]
- 20.Pfizer Inc. Pfizer to discontinue development program for PF-07265803 for LMNA-related dilated cardiomyopathy. 2022. Accessed April 1, 2023. https://www.pfizer.com/news/announcements/pfizer-discontinue-development-program-pf-07265803-lmna-related-dilated. [Google Scholar]
- 21.Crasto S, My I, Di Pasquale E. The broad spectrum of LMNA cardiac diseases: from molecular mechanisms to clinical phenotype. Front Physiol. 2020;11:761. doi: 10.3389/fphys.2020.00761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ollila LH, Nikus K, Parikka H, Weckstrom S, Tiina H. Timing of pacemaker and ICD implantation in LMNA mutation carriers. Open Heart. 2021;8:e001622. doi: 10.1136/openhrt-2021-001622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Rijsingen IA, Nannenberg EA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, van Tintelen JP, van den Berg MP, Grasso M, et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur J Heart Fail. 2013;15:376–384. doi: 10.1093/eurjhf/hfs191 [DOI] [PubMed] [Google Scholar]
- 24.Wahbi K, Ben Yaou R, Gandjbakhch E, Anselme F, Gossios T, Lakdawala NK, Stalens C, Sacher F, Babuty D, Trochu JN, et al. Development and validation of a new risk prediction score for life-threatening ventricular tachyarrhythmias in laminopathies. Circulation. 2019;140:293–302. doi: 10.1161/CIRCULATIONAHA.118.039410 [DOI] [PubMed] [Google Scholar]
- 25.Hoorntje ET, Bollen IA, Barge-Schaapveld DQ, van Tienen FH, Te Meerman GJ, Jansweijer JA, van Essen AJ, Volders PG, Constantinescu AA, van den Akker PC, et al. Lamin A/C-related cardiac disease: late onset with a variable and mild phenotype in a large cohort of patients with the lamin A/C p.(Arg331Gln) founder mutation. Circ Cardiovasc Genet. 2017;10:e001631. doi: 10.1161/CIRCGENETICS.116.001631 [DOI] [PubMed] [Google Scholar]
- 26.Barriales-Villa R, Ochoa JP, Larranaga-Moreira JM, Salazar-Mendiguchia J, Diez-Lopez C, Restrepo-Cordoba MA, Alvarez-Rubio J, Robles-Mezcua A, Olmo-Conesa MC, Nicolas-Rocamora E, et al. Risk predictors in a Spanish cohort with cardiac laminopathies. The REDLAMINA registry. Rev Esp Cardiol (Engl Ed). 2021;74:216–224. doi: 10.1016/j.rec.2020.03.026 [DOI] [PubMed] [Google Scholar]
- 27.Captur G, Arbustini E, Syrris P, Radenkovic D, O’Brien B, McKenna WJ, Moon JC. Lamin mutation location predicts cardiac phenotype severity: combined analysis of the published literature. Open Heart. 2018;5:e000915. doi: 10.1136/openhrt-2018-000915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mellerio JE. The challenges of clinical trials in rare diseases. Br J Dermatol. 2022;187:453–454. doi: 10.1111/bjd.21686 [DOI] [PubMed] [Google Scholar]
- 29.van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T, Heidbüchel H, de Visser M, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med (Berl). 2005;83:79–83. doi: 10.1007/s00109-004-0589-1 [DOI] [PubMed] [Google Scholar]
- 30.Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Bohm M, Duboc D, Gimeno J, de Groote P, Imazio M, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016;37:1850–1858. doi: 10.1093/eurheartj/ehv727 [DOI] [PubMed] [Google Scholar]
- 31.Arbelo E, Protonotarios A, Gimeno JR, Arbustini E, Barriales-Villa R, Basso C, Bezzina CR, Biagini E, Blom NA, de Boer RA, et al. ; ESC Scientific Document Group. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023;44:3503–3626. doi: 10.1093/eurheartj/ehad194 [DOI] [PubMed] [Google Scholar]
- 32.Brunner-La Rocca HP, Sanders-van Wijk S. Natriuretic peptides in chronic heart failure. Card Fail Rev. 2019;5:44–49. doi: 10.15420/cfr.2018.26.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uszko-Lencer N, Mesquita R, Janssen E, Werter C, Brunner-La Rocca HP, Pitta F, Wouters EFM, Spruit MA. Reliability, construct validity and determinants of 6-minute walk test performance in patients with chronic heart failure. Int J Cardiol. 2017;240:285–290. doi: 10.1016/j.ijcard.2017.02.109 [DOI] [PubMed] [Google Scholar]
- 34.Green CP, Porter CB, Bresnahan DR, Spertus JA. Development and evaluation of the Kansas City Cardiomyopathy Questionnaire: a new health status measure for heart failure. J Am Coll Cardiol. 2000;35:1245–1255. doi: 10.1016/s0735-1097(00)00531-3 [DOI] [PubMed] [Google Scholar]
- 35.Nassif M, Fine JT, Dolan C, Reaney M, Addepalli P, Allen VD, Sehnert AJ, Gosch K, Spertus JA. Validation of the Kansas City Cardiomyopathy Questionnaire in symptomatic obstructive hypertrophic cardiomyopathy. JACC Heart Fail. 2022;10:531–539. doi: 10.1016/j.jchf.2022.03.002 [DOI] [PubMed] [Google Scholar]
- 36.Abraham WT, Lindenfeld J, Ponikowski P, Agostoni P, Butler J, Desai AS, Filippatos G, Gniot J, Fu M, Gullestad L, et al. Effect of empagliflozin on exercise ability and symptoms in heart failure patients with reduced and preserved ejection fraction, with and without type 2 diabetes. Eur Heart J. 2021;42:700–710. doi: 10.1093/eurheartj/ehaa943 [DOI] [PubMed] [Google Scholar]
- 37.Lee MMY, Brooksbank KJM, Wetherall K, Mangion K, Roditi G, Campbell RT, Berry C, Chong V, Coyle L, Docherty KF, et al. Effect of empagliflozin on left ventricular volumes in patients with type 2 diabetes, or prediabetes, and heart failure with reduced ejection fraction (SUGAR-DM-HF). Circulation. 2021;143:516–525. doi: 10.1161/CIRCULATIONAHA.120.052186 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.