

Abstract
Background:
The recently launched DNA methylation profiling platform, Illumina MethylationEPIC BeadChip Infinium microarray v2.0 (EPICv2), is highly correlated with measurements obtained from its predecessor MethylationEPIC BeadChip Infinium microarray v1.0 (EPICv1). However, the concordance between the two versions in the context of DNA methylation-based tools, including cell type deconvolution algorithms, epigenetic clocks, and inflammation and lifestyle biomarkers has not yet been investigated.
Findings:
We profiled DNA methylation on both EPIC versions using matched venous blood samples from individuals spanning early to late adulthood across three cohorts. On combining the DNA methylomes of the cohorts, we observed that samples primarily clustered by the EPIC version they were measured on. Within each cohort, when we calculated cell type proportions, epigenetic age acceleration (EAA), rate of aging estimates, and biomarker scores for the matched samples on each version, we noted significant differences between EPICv1 and EPICv2 in the majority of these estimates. These differences were not significant, however, when estimates were adjusted for EPIC version or when EAAs were calculated separately for each EPIC version.
Conclusions:
Our findings indicate that EPIC version differences predominantly explain DNA methylation variation and influence estimates of DNA methylation-based tools, and therefore we recommend caution when combining cohorts run on different versions. We demonstrate the importance of calculating DNA methylation-based estimates separately for each EPIC version or accounting for EPIC version either as a covariate in statistical models or by using version correction algorithms.
Keywords: Epigenetics, DNA methylation, Illumina EPIC array, epigenetic clocks, inflammation and lifestyle biomarkers, cell type deconvolution, replicate probes
Background
Infinium Methylation BeadChip microarrays have been widely used to cost-effectively measure the human DNA methylome in large scale and population-wide studies[1–3]. The recently developed Illumina MethylationEPIC BeadChip Infinium microarray v2.0 (900K, EPICv2) features a total of 936,866 probes, encompassing ~77% of the probes in the previous version, the MethylationEPIC BeadChip Infinium microarray v1.0 B5 (850K, EPICv1), and over 200,000 new probes designed for increased coverage of enhancers, open chromatin regions, and CTCF-binding domains[4]. EPICv2 also differs from its predecessor in the overall probe content and utility, with annotation to the most recent GRCh38/h38 human genome build, differences in probe design type and strand switches, incorporation of new “nv” probes targeting recurrent somatic cancer mutations, and 65 single nucleotide polymorphism (SNP) sites, compared to the 59 SNPs on EPICv1, for confirmation of sample identity. Unlike EPICv1 where each probe is unique, EPICv2 includes ~5100 probes that each have between 2–10 replicates, differentiated based on their probe names and sequences[5]. Approximately 143,000 poorly performing probes on the EPICv1 have been removed from the EPICv2, ~73% of which are likely to be influenced by underlying sequence polymorphisms[6,7]. Overall, these modifications in EPICv2 intend to provide wider coverage of the DNA methylome, with optimized performance across primary tissues and cancer cell lines, and extended reliability across diverse human populations[6,7].
Previous iterations of Illumina microarrays (27K, 450K and EPICv1) have been widely used to develop DNA methylation-based tools including cell type deconvolution algorithms[8–10], epigenetic clocks[11–18], interleukin 6 (IL-6) and C-reactive protein (CRP) inflammation markers[19,20], and lifestyle biomarker predictors such as smoking and alcohol use [21]. These tools have been trained on one or more of the previous generation of microarrays, and are based on the property that DNA methylation levels at specific cytosine-guanine dinucleotides (CpGs) highly correlate with cell types, chronological age, and biomarker measures, respectively[11–14,16,17], and a majority of predictive CpGs employed by these tools are retained on EPICv2[6]. It has been previously shown that estimates from some of these DNA methylation-based tools, while highly correlated, are significantly different between 450K and EPICv1[22–24]. With the launch of the recent version of the EPICv2 array, we thus sought to examine whether there are any such differences between EPICv1 and EPICv2, we systematically assessed the concordance of inferred estimates between the two EPIC versions. We also tested whether there were any differences in estimates between EPICv1 and EPICv2 when datasets profiled on the two versions are combined, given that this is an important consideration in meta-analyses which harness statistical power that comes from combining multiple cohorts, and longitudinal studies which include samples profiled on different arrays/versions.
To assess whether EPIC version differences have any implications on DNA methylation measures in the context of DNA methylation-based tools, we examined matched venous blood samples from individuals across three diverse populations spanning early to late adulthood, and profiled DNA methylome using the EPICv1 and EPICv2 arrays. To explore if the high agreement between EPICv1 and EPICv2 reported in cell lines is consistent in primary samples from human populations, we tested the concordance of the two EPIC versions at both array and probe levels and evaluated the potential contribution of EPIC version to overall DNA methylation variation. Next, to investigate whether there are differences in estimates between EPICv1 and EPICv2 from DNA methylation-based tools built on previous arrays/versions, including immune cell type deconvolution algorithms, epigenetic clocks, inflammation and lifestyle biomarkers, we compared their estimates between the two EPIC versions. We also examined whether EPIC version influenced these estimates by combining EPICv1 and EPICv2 samples, and demonstrated the significance of accounting for EPIC version in these DNA methylation-based estimates.
Results
Unsupervised clustering of samples was primarily determined by EPIC version
To compare the two most recent generations of Illumina MethylationEPIC BeadChip Infinium microarrays, we measured the DNA methylomes of a subset of venous whole blood samples on both EPICv1 and EPICv2 across three cohorts: (i) Vietnam Health and Aging Study (VHAS)[25], (ii) Cebu Longitudinal Health and Nutrition Survey (CLHNS)[26], and (iii) Comprehensive Assessment of Long-term Effects of Reducing Intake of Energy (CALERIE)[27] (Table 1, Figure 1). We first explored whether samples clustered depending on which EPIC version they were measured on or the cohort they belong to, given that the three cohorts were unique in their demographic and biological characteristics such as sex and age range. To do this, we carried out unsupervised hierarchical clustering on all samples from the three cohorts using the 721,378 probes shared between EPICv1 and EPICv2. We observed a clear demarcation of samples first by EPIC version despite cohort-specific characteristics (Figure 2A). When clustering was carried out on subsets of predictive CpGs employed by DNA methylation-based tools, we still noted some separation of samples by EPIC version, although not to the same extent as the clustering on shared probes (Supplementary Figure 1).
Table 1.
Demographics of the three investigated cohorts.
| VHAS | CLHNS | CALERIE | |
|---|---|---|---|
| Samples (n) | 48 (24 matched venous and capillary samples) | 16 | 24 |
| Country of origin | Vietnam | Philippines | United States of America |
| Biological sex (% Female) | 36.4% | 100% | 54.2% |
| Age in years (mean (SD, range)) | 74.6 (6.7, 66–86) | 48.4 (6.1, 40–63) | 36.7(8.1, 21.3–51) |
Figure 1.

Overview of study design and analyses.
Figure 2.

(A) Hierarchical clustering of 721,378 probes common between EPICv1 and EPICv2 of matched VHAS, CLHNS, and CALERIE samples; blue to red color range denotes Spearman correlation from 0.94–1.00. (B-D) Spearman correlation and root mean square error (RMSE) of 721,378 probes common between EPICv1 and EPICv2 investigated on (B) VHAS, (C) CLHNS, and (D) CALERIE. The X-axis represents the Spearman correlation and Y-axis represents the RMSE of probes common to both arrays. Dashed horizontal line indicates the RMSE threshold set at 0.05, and the vertical dashed line indicates the correlation threshold set at 0.70. The colors indicate the density of points, such that pink is low density and yellow is high density. Probes unique to either array are not shown.
Spearman correlation between shared EPICv1 and EPICv2 probes was high at array level and low at the individual probe level
To compare the technical performance of EPICv1 and EPICv2, we first estimated Spearman correlation of probes between matched samples assessed on the two EPIC versions at the array level and at the individual probe level for each cohort independently. We identified overall high array level Spearman correlations between EPICv1 and EPICv2 in the three cohorts (VHAS: Spearman rho = 0.969–0.981, CLHNS: 0.973–0.979, and CALERIE: 0.968–0.981). As expected, technical replicate samples measured on the same EPIC version showed marginally higher array level Spearman correlation compared to those measured between versions (within EPICv1: 0.991, within EPICv2: 0.986, and between versions: 0.976). Similarly, samples from the same individual measured on EPICv1 and EPICv2 showed higher absolute β value differences for each probe when compared to differences calculated between technical replicates within each EPIC version. Specifically, we noted that ~75% of probes have absolute β value differences between EPIC versions higher than technical replicates within each EPIC version (VHAS: 74.258% (535,682 probes); CLHNS: 76.365% (550,880 probes); and CALERIE: 76.733% (553,821 probes)). On evaluating probe level Spearman correlations between the two versions, we identified that only a fourth of the investigated probes exhibited Spearman correlation greater than 0.70, specifically, VHAS: 29.220% (210,845 probes), CLHNS: 24.965% (180,093 probes), and CALERIE: 19.287% (139,134 probes)). The remaining ~75% of probes had a low median correlation between EPICv1 and EPICv2, specifically, VHAS: 0.390 (range: −0.648 to 0.991), CLHNS: 0.368 (range: −0.907 to 1.000), and CALERIE: 0.389 (range: −0.744–0.998). One cause for low probe level correlations may be low inter-sample variability[22]. To assess if observed low individual probe level correlations in the majority of investigated probes can indeed be attributed to low DNA methylation variability across samples, we used a RMSE threshold of < 5% to denote low variability (Figure 2B–D). We observed that of the probes with Spearman correlation < 0.70 between EPICv1 and EPICv2, more than 85% can be attributed to respective RMSE values estimated at less than 5% (VHAS: 89.752% (485,211 probes), CLHNS: 87.994% (476,299 probes), and CALERIE: 91.805% (534,530 probes)). In summary, ~96% of the shared probes between EPICv1 and EPICv2 had either modest-to-high correlation or low correlation explained by low DNA methylation variance, with 87.065% (628,069 probes) commonly identified across three cohorts. When we compared these ~96% probes to platform-bias free and high-confidence mapping probes (n = 542,491) recently identified in cell lines [28], we found that over 95% overlap across our investigated cohorts (VHAS: 96.642% (521,395 probes), CLHNS: 95.011% (512,598 probes), and CALERIE: 97.017% (523,418 probes)).
While the low correlation in the majority of shared EPICv1 and EPICv2 probes has been explained, ~4% probes still remain unexplained by DNA methylation variance (VHAS: 52,322 probes, CLHNS: 64,986 probes; and CALERIE: 47,640 probes; 25,058 common across all cohorts) (Supplementary Table S1). We noted that the vast majority of these probes which have low correlation and high RMSE had % absolute β value differences higher than technical noise measured within each EPIC version (VHAS: 70.217% (36,739 probes), CLHNS: 93.112% (60,510 probes), and CALERIE: 74.733% (35,658 probes)). On comparing these probes to our list of flagged probes with detection p-value > 0.01 or beadcount < 3 in greater than 1% of the samples, we identified an overlap of 4,977, 2,141, and 1,292 probes in the three cohorts, respectively. We next sought to determine whether design type switch or cross hybridization of probes to the genome can explain the observed low correlation and high RMSE. We noted that 14, 17, and 15 probes in VHAS, CLHNS, and CALERIE, respectively, overlapped with the 82 probes that have undergone design type switches in EPICv2[6], and a small fraction of these probes overlapped with previously identified as cross hybridizing to multiple genomic locations or mapped to genetic variant sites (742, 1132, and 633 probes in the Peters reference[5], 624, 980, and 537 probes in the Pidsley reference[29], 683, 1058, and 585 probes in the McCartney reference[30], and 1829, 2370, and 1659 probes in the Price reference[31], in the VHAS, CLHNS, and CALERIE cohorts, respectively).
High coverage of epigenetic clock, biomarker predictor, and cell type deconvolution CpGs was noted on EPICv2
To investigate the applicability of widely used DNA methylation-based tools, which were built on previous arrays/versions, on EPICv2, we noted the percentage of predictive CpGs which are absent in either of the EPIC versions and which are replicated on EPICv2. Across the seven epigenetic clocks investigated, we observed that ~77–96% predictive CpGs were retained on EPICv2, with a reintroduction of several predictive CpGs which were absent in EPICv1 on EPICv2 (Horvath pan-tissue: 14 CpGs; Hannum: 4 CpGs; and epigenetic timer of cancer (epiTOC): 25 CpGs). We noted that in all investigated clocks, except epiTOC, ~1–7% of the predictive clock CpGs have replicate probes on EPICv2. Similarly, across the four investigated biomarker predictors, we noted ~89–94% coverage of predictive CpGs on EPICv2, with 0.86–2.86% also including replicate probes. In the CRP predictor, we additionally observed that 5.84% predictive CpGs were absent in EPICv1, with 83 of these CpGs reintroduced in EPICv2. Next, on examining the 1200 pre-selected extended Identifying Optimal DNA methylation Libraries reference (IDOL) probes used in cell type deconvolution[10], we noted that 99.33% were represented on EPICv2, with 4% also having replicate probes on EPICv2 (Table 2). On estimating the correlation of β values of predictive CpGs employed by these DNA methylation-based tools between EPICv1 and EPICv2, weighted by corresponding coefficients, we identified high Spearman correlation ranging from 0.9615–0.9952 (Supplementary Figure 2). We also noted that average absolute differences in β values of these predictive CpGs between the EPIC versions range from 0.0157–0.0342, with the Horvath pan-tissue clock having the lowest average absolute difference (MAD) between the two versions (Supplementary Table S2–S3).
Table 2.
Summary of predictive CpGs of DNA methylation-based clocks, biomarker predictors, and cell type deconvolution in EPICv1 and EPICv2.
| Predictors | CpGs | Probes absent in EPICv1 (%) | Probes absent in EPICv2 (%) | Probes absent in EPICv1 and reintroduced in EPICv2 | EPICv2 replicate probes | Training arrays; training age range (year) | Training sample size | Probes absent in Infinium Methylation Screening Array (MSA) | Probes absent in 450K |
|---|---|---|---|---|---|---|---|---|---|
| Horvath pan-tissue* | 353 | 17 (4.82%) | 13 (3.68%) | 14 | 4(1.13%) | 27K and 450K Age: 0–100 | 7844 | 6(1.7%) | 0 |
| Hannum* | 71 | 6 (8.45%) | 7 (9.86%) | 4 | 5(7.04%) | 450K Age: 19–101 | 656 | 3(4.23%) | 0 |
| Horvath SkinBlood | 391 | 0 | 17 (4.35%) | 0 | 9(2.3%) | 450K and EPICv1 Age: 0–94 | 896 | 3(0.77%) | 0 |
| PhenoAge | 513 | 0 | 18 (3.51%) | 0 | 7(1.36%) | 27K, 450K, EPICv1 Age: 18–100 | 9926 | 3(0.58%) | 0 |
| GrimAge** | 1030 | na | na | 0 | na | 450K and EPICv1 Age: NA (mean 66) | 1731 | NA | na |
| PC Clocks*** | 78464 | 0 | 5801 (7.39%) | 0 | 650 (0.83%) | 450K and EPICv1 Age: 0–101 | na | 51108 (65.14%) | 0 |
| DunedinPACE | 173 | 0 | 29 (16.76%) | 0 | 2(1.16%) | EPICv1 Age 38 and 45 | 1037 | 2(1.16%) | 0 |
| DNAmTL | 140 | 0 | 31 (22.14%) | 0 | 3(2.14%) | 450K and EPICv1 Age: 22–93 | 2256 | 2(1.43%) | 0 |
| epiTOC* | 385 | 31 (8.05%) | 26 (6.75%) | 25 | 0 | 450K Age: 19–101 | 656 | 13(3.38%) | 0 |
| IL-6 score | 35 | 0 | 3 (8.57%) | 0 | 1(2.86%) | 450K and EPICv1 Age: 67–78 | 875 | 12(34.29%) | 0 |
| CRP score* | 1765 | 103 (5.84%) | 96 (5.44%) | 83 | 41 (2.32%) | 27K, 450K and EPICv1 Age: 16–75 | 22774 | 357 (20.23%) | 0 |
| Smoking score | 233 | 0 | 23 (9.87%) | 0 | 2(0.86%) | EPICv1 Age: 18–99 | 5087 | 27(11.59%) | 0 |
| Alcohol score | 450 | 0 | 49 (10.89%) | 0 | 6(1.33%) | EPICv1 Age: 18–99 | 5087 | 47(10.44%) | 0 |
| IDOL | 1200 | 0 | 8 (0.67%) | 0 | 48(4%) | EPICv1 Age: 19–58 | NA | 20(1.67%) | 741 (61.75%) |
CpGs commonly absent in both EPICv1 and EPICv2: Horvath pan-tissue; 3 CpGs; Hannum; 2 CpGs; EpiTOC; 6 CpGs; CRP; 20 CpGs.
Clock CpGs for GrimAge are not publicly available.
PC Clocks CpGs include the total CpGs required to calculate PC versions of Horvath pan-tissue, Hannum, Horvath SkinBlood, PhenoAge, and GrimAge.
Immune cell type proportions inferred by IDOL and auto probe selection methods were significantly different between EPICv1 and EPICv2
Cellular composition is one of the key contributors to whole blood DNA methylation variation[32,33], and is often included as a covariate in statistical models to account for cell type heterogeneity. Besides that, cellular composition changes have been associated with phenotypes of interest and may in fact be integral in the understanding of underlying biological processes[34–36]. Most studies do not have actual cell counts and rely on predicted values from DNA methylation-based algorithms. Given that these algorithms use references profiled on previous arrays/versions, we tested whether there were differences in cell type proportion estimates obtained from the two EPIC versions using one of the most commonly used cell type deconvolution methods for whole blood[10]. We compared the proportions of twelve immune cell types, namely, basophils (Bas), naïve and memory B cells (Bnv, Bmem), naïve and memory CD4+ T cells (CD4nv, CD4mem), naïve and memory CD8+ T cells (CD8nv, CD8mem), eosinophils (Eos), monocytes (Mono), neutrophils (Neu), natural killer (NK), and T regulatory cells (Treg), inferred from DNA methylation measured on EPICv1 and EPICv2 using the estimatedCellCounts2 functions from FlowSorted.Blood.EPIC R package with two commonly used probe selection methods: IDOL and “auto”. The IDOL method includes 1200 pre-selected probes, eight of which are absent in EPICv2, and the “auto” method includes 1200 probes independently selected for each EPIC version as described in Methods. On comparing the probes selected for each EPIC version using the “auto” method, we identified a large overlap of 1090 probes (90.83%) between the two EPIC versions, with only 121 EPICv1 (10.983%) and 134 EPICv2 (11.167%) auto-selected probes overlapping with the 1200 pre-selected IDOL probes (Supplementary Figure 3).
Using the IDOL method, we found Pearson correlations between EPICv1 and EPICv2 to be greater than 0.80 for all predicted cell types, except for Tregs which showed slightly lower correlation (Pearson r = 0.628–0.715). The proportion of CD8nv was zero for all samples in VHAS and CLHNS, but averaged at 0.012 (EPICv1) and 0.010 (EPICv2) in CALERIE. On comparing inferred cell proportions between EPICv1 and EPICv2, we identified significantly higher estimated proportions of Bas, Mono, and NK cells, and lower proportions of CD8mem and Eos in EPICv2 compared to EPICv1, with Cohen’s d effect size ranging from small to large depending on the cell type. For all five cell types, MAD between EPIC versions was higher than technical replicate differences within EPIC versions. CD4nv and CD8nv proportions were not significantly different between EPIC versions in any of the investigated cohorts. As for the remaining five cell types, namely Bmem, Bnv, CD4mem, Neu and Tregs, we observed significant proportion differences between EPIC versions in at least one cohort, however, these findings were not consistent in all three cohorts (Figure 3, Supplementary Table S4).
Figure 3.

Differences in DNA methylation-based immune cell type proportions estimated using the IDOL reference on matched samples assessed on EPICv1 and EPICv2 in VHAS, CLHNS, and CALERIE. Statistical significance was defined as Bonferroni adjusted p-value <0.05. ** denotes Bonferroni p <0.05, *** denotes Bonferroni p <0.001, “ns” denotes “not significant”, and “d” denotes effect size measured using Cohen’s d. A positive Cohen’s d indicates higher estimates in EPICv2 compared to EPICv1.
Using the “auto” method, we found Pearson correlations between EPICv1 and EPICv2 to be greater than 0.80 for all predicted cell types, with the exception of CD4mem and CD4nv which showed only a modest Pearson r of 0.788 and 0.778 respectively in VHAS. For all EPICv1 samples, Treg proportions were estimated to be zero in all three cohorts, while CD8nv proportions were estimated to be zero for all samples in VHAS and CLHNS, and 87.5% in CALERIE. Paired t-tests showed significantly higher estimated proportions of Bas, Bmem, CD8mem, CD8nv, Mono, NK and Tregs, and lower proportions of CD4mem and CD4nv in EPICv2 compared to EPICv1. Meanwhile, we noted no significant differences in Eos between EPICv1 and EPICv2 across all three cohorts. For all cell types, the MAD between EPIC versions was higher than technical replicate differences within EPIC versions. Among the remaining cell types, we observed higher proportions higher proportions of Bnv (d = 0.13–0.22) and Neu (d = −0.06–0.08) in EPICv2 in CLHNS and CALERIE (Supplementary Figure 4, Supplementary Table S5).
Clock EAAs were significantly different between matched samples on EPICv1 and EPICv2 when samples were combined and EAAs were subsequently calculated
Epigenetic clocks are based on the property that DNA methylation levels at specific CpGs highly correlate with chronological age or age-related outcomes and are often estimated as a linear combination of weighted sum of the coefficients of predictive CpGs[11–14,16,17]. In order to evaluate the concordance between EPICv1 and EPICv2 in the context of DNA methylation-based tools, we compared estimates from widely used first- and second- generation epigenetic clocks, namely Horvath pan-tissue[11], Hannum[12], Horvath skin and blood clocks[13], PhenoAge[14], and GrimAge[15], and rate-based clocks, namely pace of aging calculated from the epigenome (DunedinPACE)[17], DNA methylation estimator of telomere length (DNAmTL)[16], and epiTOC[18] in the three cohorts VHAS, CLHNS, and CALERIE. We identified high Pearson correlations between chronological age and epigenetic age obtained from both EPICv1 (VHAS: Pearson r = 0.701–0.825, CLHNS: 0.819–0.912, and CALERIE: 0.853–0.951) and EPICv2 (VHAS: Pearson r = 0.583–0.818, CLHNS: 0.807–0.918, and CALERIE: 0.825–0.957) (Supplementary Table S6). Further, EPICv2 epigenetic ages for the first- and second-generation clocks were highly correlated with EPICv1 epigenetic ages (VHAS: Pearson r = 0.911–0.980, CLHNS: 0.879–0.990, and CALERIE: 0.915–0.996), with differences in epigenetic ages between technical replicate samples being 0.523–4.206 years and 0.546–2.994 years within EPICv1 and EPICv2 respectively in VHAS, 0.151–3.408 years within EPICv2 in CLHNS (no technical replicates within EPICv1 were available), and 0.13–2.25 years and 0.245–4.142 years within EPICv1 and EPICv2 respectively in CALERIE (Supplementary Table S2). In these three cohorts, we noted significant differences in epigenetic ages between EPICv2 and EPICv1 in all clocks, except for Horvath pan-tissue. We identified significantly lower estimated epigenetic ages for Hannum and Horvath skin and blood clocks and higher estimated epigenetic ages for PhenoAge in EPICv2 when compared to EPICv1, with mean absolute difference (MAD) greater than technical replicate differences. The Hannum clock showed the largest MAD and Cohen’s d effect size between EPICv1 and EPICv2 in all three cohorts (VHAS: MAD=21.87, d = 3.40, CLHNS: MAD = 19.81, d = 4.14, and CALERIE: MAD = 21.71, d = 2.86) (Supplementary Table S2, Supplementary Figure 5A).
We also estimated epigenetic age acceleration (EAA), a measure of rate of aging which has been associated with health outcomes, by considering samples profiled on each EPIC version separately, as well as by combining matched samples profiled on both versions. When EAA was calculated separately by versions (EPIC versions separate), we noted modest to high correlation of EAA (VHAS: Pearson r = 0.867–0.947, CLHNS: 0.577–0.976, and CLAERIE: 0.622–0.961) and no significant differences between EPICv1 and EPICv2 (Supplementary Table S7, Figure 4B, Supplementary Figure 6B, Supplementary Figure 7B). When clock estimates from the EPIC versions were combined prior to EAA calculation (EPIC versions combined), we observed similar modest to high correlation (VHAS: Pearson r = 0.862–0.947, CLHNS: 0.574–0.958, and CALERIE: 0.632–0.960), but with significant differences between EPICv1 and EPICv2 for all five clock EAAs, with the exception of Horvath pan-tissue (Supplementary Table S7, Figure 4C, Supplementary Figure 6C, Supplementary Figure 7C). Additionally, we identified significantly lower EAAs for Hannum and Horvath skin and blood, and higher EAAs for PhenoAge in EPICv2 when compared to EPICv1 (Supplementary Table S7). Next, we tested whether the different EPIC versions indeed contribute to observed EAA differences by first combining epigenetic ages in both EPIC versions and then calculating EAAs by including version as a covariate in the linear regression (EPIC versions combined and adjusted). After EPIC version adjustment, we noted modest to high correlation (VHAS: Pearson r = 0.862–0.947, CLHNS: 0.574–0.958, and CALERIE: 0.632–0.960) and no significant EAA differences between the two versions in the three cohorts (Supplementary Table S7, Figure 4D, Supplementary Figure 6D, Supplementary Figure 7D). We repeated the same analyses using epigenetic clocks estimated based on principal components (PC clocks) rather than individual predictive CpGs, as they have been shown to overcome unreliability in clock estimates due to technical noise[37]. We similarly noted that there were no significant EAA differences when EAA was calculated for each EPIC version separately. When EAAs were calculated on combined sets of matched samples, we noted significant differences in PCHorvath skin and blood and PCPhenoAge clocks in the three adult cohorts, and these differences were no longer significant with EPIC version adjustment (Supplementary Figures 8–10).
Figure 4.

Epigenetic ages on matched samples assessed on EPICv1 and EPICv2 in VHAS. (A) Scatter plot of Horvath pan-tissue, Hannum, Horvath skin and blood, PhenoAge, and GrimAge clock ages (Y axis) and chronological age (X axis) with dotted line indicating x=y, coloured by EPIC version. (B-D) Boxplots comparing EPICv1 and EPICv2 EAAs calculated by considering (B) EPIC versions separately, (C) combined, and (D) combined and EPIC version adjusted. Statistical significance was defined as Bonferroni adjusted p-value <0.05. ** denotes Bonferroni p <0.05, *** denotes Bonferroni p <0.001, “ns” denotes “not significant”, and “d” denotes effect size measured using Cohen’s d. A positive Cohen’s d indicates higher estimates in EPICv2 compared to EPICv1.
Given the discrepancies known to exist in epigenetic clock estimations using different normalization methods[23,38,39], we sought to examine whether our results, observed in data normalized by functional normalization, remained consistent irrespective of the choice of normalization method. To test this, we combined the common probes on the two EPIC versions and applied Beta-MIxture Quantile (BMIQ) normalization, and subsequently calculated epigenetic ages and EAAs. We still noted significant differences in epigenetic age and EAA between EPICv1 and EPICv2 in Horvath pan-tissue, Horvath skin and blood, PhenoAge, and GrimAge clocks in at least two out of the three cohorts when the two EPIC versions were combined and analyzed (Supplementary Figure 5B and 11C, 12C, and 13C). No significant differences in EAA between the versions were observed when EAAs were calculated separately for EPICv1 and EPICv2 or when the versions were combined and EAAs were adjusted for EPIC version (Supplementary Figure 11–13). Given the contribution of EPIC version in explaining DNA methylation variation, we also examined if applying version correction using batch correction algorithms can circumvent version-specific effects and eliminate the observed differences in DNA methylation-based estimates between EPICv1 and EPICv2. We tested this by combining common probes on EPICv1 and EPICv2 after functional normalization, and sequentially correcting for EPIC version, chip and sample position on chip (row) effects as applicable using the ComBat function implemented in the sva R package[40]. As expected, when epigenetic ages were calculated using version-corrected input β values, there were no significant differences between the EPIC versions (Supplementary Figure 5C).
On evaluating the DunedinPACE, DNAmTL, and epiTOC rate-based clocks, we observed a high correlation between EPICv1 and EPICv2 when the two were considered separately, as well as when they were combined and adjusted for EPIC versions (VHAS: Pearson r = 0.936–0.983, CLHNS: 0.838–0.967, and CALERIE: 0.808–0.941) (Supplementary Table S8). When we examined the EPIC versions separately, we noted that DNAmTL estimates were significantly higher in EPICv2 when compared to EPICv1 in all three cohorts and the differences were higher than technical replicate differences within EPIC versions (VHAS: d = 0.96, CLHNS: d = 1.50, and CALERIE: d = 0.65). DunedinPACE estimates were significantly higher in EPICv2 in VHAS (d = 0.47) and CALERIE (d = 0.65), and epiTOC estimates were significantly lower in EPICv2 in CLHNS (d = 0.43) but higher in CALERIE (d = 0.60) (Supplementary Table S8, Figure 5A, Supplementary Figure 6E, and Supplementary Figure 7E). As expected, we observed no significant differences between EPICv1 and EPICv2 when the rate-based clock estimates were adjusted for EPIC version (Supplementary Table S8, Figure 5B, Supplementary Figure 6F, and Supplementary Figure 7F). We observed similar differences when BMIQ normalized data was used as input for rate-based clocks. When EPICv1 and EPICv2 were considered separately, there were significant differences in DunedinPACE and DNAmTL estimates between versions in all three cohorts, and significant differences in epiTOC estimates in CLHNS and CALERIE, and these differences were no longer significant when EPIC version was accounted for as a covariate (Supplementary Figure 14) or when input β values were adjusted using version correction algorithms upstream (Supplementary Figure 15A). We also repeated the same analyses using matched capillary blood samples available in the VHAS cohort, as both venous and capillary blood sampling are common in epigenetic research[41]. Consistent with our findings in venous blood samples, we observed significant differences between EPIC versions in epigenetic ages as well as EAAs when EPIC versions were combined and EAAs were calculated in four of the investigated first- and second-generation clocks, two PC clocks, and all three rate-based clocks. When EAAs were calculated separately for each version and when EAAs and rate-based clock estimates were adjusted for EPIC versions, there were no significant differences between the EPIC versions (Supplementary Figures 16–19).
Figure 5.
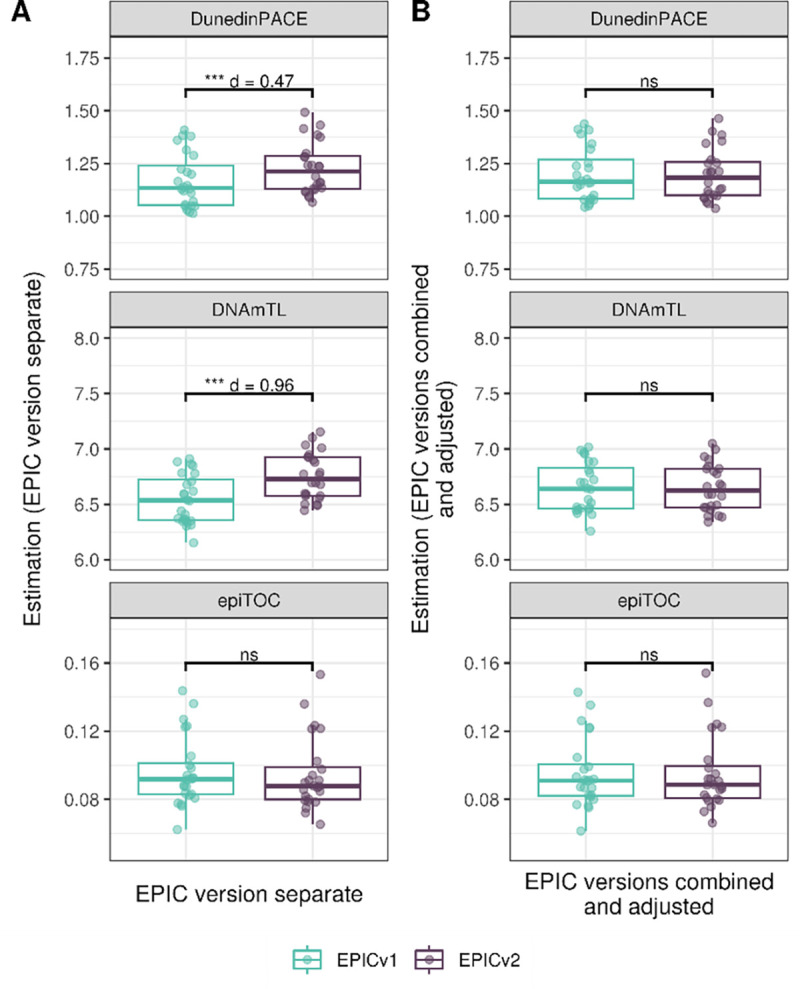
Rate-based clock estimates on matched samples assessed on EPICv1 and EPICv2 in VHAS. Boxplots comparing DunedinPACE, DNAmTL and epiTOC estimates calculated by considering EPIC versions separately (A) and combined and EPIC version adjusted (B), between EPICv1 and EPICv2. Statistical significance was defined as Bonferroni adjusted p-value <0.05. ** denotes Bonferroni p <0.05, *** denotes Bonferroni p <0.001, “ns” denotes “not significant”, and “d” denotes effect size measured using Cohen’s d. A positive Cohen’s d indicates estimates in EPICv2 compared to EPICv1.
Estimated CRP and smoking scores were significantly different between EPICv1 and EPICv2
DNA methylation-based estimates of IL-6, CRP, smoking, and alcohol use are widely used as reliable proxies of actual measurements in epigenetic studies[39], and we tested if there were any significant differences in these estimates between the two EPIC versions. We observed high correlation of IL-6 (VHAS: Pearson r = 0.902, CLHNS: 0.912, and CALERIE: 0.85), CRP (VHAS: Pearson r = 0.993, CLHNS: 0.992, and CALERIE: 0.944), and smoking scores (VHAS: Pearson r = 0.981, CLHNS: 0.99, and CALERIE: 0.72), and modest correlation of alcohol scores (VHAS: Pearson r = 0.789, CLHNS: 0.76, and CALERIE: 0.75) between EPICv1 and EPICv2. When estimates were calculated separately by versions (EPIC versions separate), we identified significantly higher CRP scores in EPICv2 compared to EPICv1 in VHAS (d = 0.26) and CLHNS (d = 0.27), with differences higher than technical replicate differences, while there were no significant differences in IL-6 and alcohol scores in either cohort. We noted significantly higher IL-6 score and alcohol score in EPICv2 compared to EPICv1 in CALERIE (d = 0.49 and 1.28, respectively), and significantly lower smoking scores in CLHNS (d = 0.30). When estimates from the EPIC versions were first combined and then adjusted for version, as expected, we noted no significant differences between EPICv1 and EPICv2 in any of the DNA methylation-based predictors tested in the three cohorts (Supplementary Table S3, Supplementary Figure 20). Our observation of significant EPIC version differences in estimates remained consistent when BMIQ normalized β was used as input (Supplementary Figure 21), and these differences were no longer significant when EPIC version was adjusted for as a covariate during estimate calculation or when version-correction was applied upstream (Supplementary Figure 15B).
Discussion
The newly developed Illumina EPICv2 microarray provides wider coverage of the DNA methylome compared to its predecessor EPICv1, with some reported differences in overall probe content. While good correlations between the two EPIC versions have been previously reported [5–7], a recent study demonstrated that differing probe content between versions influences epigenetic age estimates, although this was not evaluated in matched samples profiled on EPICv1 and EPICv2[42]. To that end, we measured DNA methylation on EPICv1 and EPICv2 in matched venous blood samples from adults across three geographically diverse populations, and comprehensively examined the concordance between versions in a wide range of DNA methylation-based tools.
We performed unsupervised hierarchical clustering on all samples across the three cohorts using the shared probes between EPICv1 and EPICv2 and observed that samples clustered based on the EPIC version they were measured on, despite cohort-specific characteristics. While examining subsets of probes employed by the different DNA methylation-based tools, we noted a similar clustering pattern indicating EPIC version as the predominant driver of DNA methylation variation, followed by biological characteristics such as sex and age, which are known to influence DNA methylation. Further, when we combined matched EPICv1 and EPICv2 samples in each cohort and calculated cell type proportions, epigenetic ages, and biomarker scores, we observed significant systematic differences in the majority of these estimated measures between the EPIC versions, although some of these differences and their corresponding effect sizes were cohort-specific. These differences were no longer significant when EPIC version was adjusted for during the calculation of estimates, and in the case of epigenetic clocks specifically, when EAAs were calculated in an EPIC version-specific manner.
On comparing the two EPIC versions, we observed overall high correlations at the array level and low correlation at the individual probe level, in agreement with previous findings[5–7]. Only ~25% of the shared probes between EPICv1 and EPICv2 showed modest-to-high probe level correlation, with an additional ~67% showing low correlation explained by low DNA methylation variance. The majority of these probes have also been reported previously as platform-bias free and high-confidence mapping in cell lines[28], indicating their reliability between the two EPIC versions and potentially across different sample types. In the remaining ~4% of probes, the vast majority had notable differences in β values between EPICv2 and EPICv1, suggesting that these observed differences may stem from EPIC version beyond technical noise, consistent with previous findings comparing 450K and EPICv1[22,43,44]. While non-specific probe hybridization to the genome, underlying genetic variation, or technical differences such as altered EPICv2 probe design may explain the low concordance between the two EPIC versions[5,6,29–31], such probes accounted for only a small proportion. We therefore recommend cautious interpretation of DNA methylation readouts from these probes owing to their differences between the versions, specifically when integrating EPICv1 and EPICv2 data for analyses.
We observed generally high correlation of cell type proportion estimates between EPICv1 and EPICv2, however, significant proportion differences in specific immune cell types were observed. Using pre-selected IDOL and auto methods of probe selection, we found that the proportions of five to nine cell types were significantly different between EPICv1 and EPICv2, with monocytes and NK cells consistently identified in both methods. Within each probe selection method, our observations were generally consistent across all three investigated cohorts indicating systematic differences between the two EPIC versions that are not driven by cohort-specific characteristics. Given that EPICv2 cell type proportions were predicted using the reference library created by profiling purified immune cell types from peripheral blood on EPICv1[10], and similar differences in predicted cell type proportions have been observed previously between 450K and EPICv1[22], the observed differences are likely due to version incompatibility between the reference library used and input samples for EPICv2. These effects may be compounded by the proportional nature of cell type estimates, implying that any changes in the proportion of even one cell type will influence the proportions of other cell types. Together, our findings indicate inherent EPIC version differences influencing cell type proportion estimations, and the need to investigate whether EPIC version-specific cell type reference libraries would enhance the accuracy of cell type deconvolution in EPICv2, and whether the detected EPIC version differences in cell type proportions would affect downstream analyses such as epigenome wide association studies.
Our epigenetic clock analyses showed significant differences in estimates of epigenetic age between EPICv1 and EPICv2 in five investigated clocks despite the high correlation of shared clock CpGs, suggesting systematic differences in epigenetic ages depending on which EPIC version the samples were profiled on. Given that there are varying clock CpGs absent on either EPIC version, it is possible that the subsequent imputation performed by the clock algorithms could influence obtained epigenetic ages and explain some of the observed variation. While using common probes between the two versions as input for clock calculation, however, the variation in epigenetic age estimates was still observed, suggesting that the proportion of missing clock CpGs does not account for the EPIC version differences. This is in line with a previous study that missing values do not compromise clock estimates in first-generation clocks[23]. Although in our analyses the differences in Horvath pan-tissue epigenetic ages between EPICv1 and EPICv2 did not reach statistical significance with functional normalization, these differences were significant when we applied BMIQ normalization, and in both normalization methods, epigenetic ages were higher in EPICv2 compared to EPICv1. Our Horvath pan-tissue clock findings therefore affirm previous reports that choice of normalization method influences clock estimates[23,38,39]. Nevertheless, we still observe significant epigenetic age differences in all other investigated first- and second-generation clocks in both functional and BMIQ normalization, highlighting the contribution of systematic EPIC version differences to epigenetic ages irrespective of the normalization method used. It has been noted that the stochastic changes to single-CpG DNA methylation level as a function of aging explain a non-negligible amount of variation in established epigenetic clocks[45]. However, the individual-level stochastic variation in epigenetic ages is not the focus of our analyses and cannot be measured using the methods employed in this study, and is likely to be minimal relative to cohort and other batch effects.
We also tested whether there were EPIC version differences in EAAs, which are more robust to data preprocessing methods compared to epigenetic ages[23,38,39]. We noted no EAA differences in the investigated first- and second-generation clocks when EAAs were calculated separately for the two EPIC versions, regardless of observed systematic shifts in epigenetic ages, lending additional support to the robustness of EAA as an epigenetic age measure when calculating in a version-specific manner. On the other hand, with EAA calculation being relative to input samples and observed epigenetic age differences between EPICv1 and EPICv2, we expectedly observed EAA differences when samples from both EPIC versions were combined and then EAA was calculated. These differences were no longer significant when we calculated EAA by regressing epigenetic age on chronological age and additionally adjusting for EPIC version. In contrast, calculating epigenetic age differences, by subtracting chronological age from epigenetic age, would yield significant differences between EPICv1 and EPICv2 due to the observed systematic EPIC version differences in epigenetic ages, once again highlighting the appropriateness of EAA in epigenetic age analyses. Our findings of significant differences between EPIC versions hold true in rate-based clock estimates and biomarker scores as well, and these differences were no longer significant with EPIC version adjustment, thus emphasizing correcting for EPIC version in these tools as an effective strategy to obtain comparable estimates.
Overall, our results indicate that epigenetic age differences can be ascribed to EPIC version, which will influence interpretation of findings if not accounted for. While adding EPIC version as a covariate in statistical models may be an option to handle version-specific discrepancies, this approach is not as straightforward in cases when EPIC version is confounded with the variable of interest. Alternatively, as noted in our investigations, version correction by ComBat may also circumvent observed version differences. However, employing such tools when study designs are unbalanced can introduce false biological signals, and is not appropriate when biological variables are confounded with EPIC versions[46–49]. Although the impact of applying batch correction on DNA methylation-based estimates is not well studied, it is known that data normalization methods can introduce variation in epigenetic clock estimates[23,39]. Therefore, it is reasonable to speculate that implementing additional data preprocessing steps such as version correction will result in biased estimates, specifically when biological variables are not evenly distributed across the two versions, highlighting an area that warrants further investigation.
Our study improves the current understanding of the applicability of DNA methylation-based tools for EPICv1 and EPICv2, however, there are several limitations. While our analyses may be limited by the modest sample sizes, our study includes matched samples collected from adults with wide age range and across diverse geographical regions. We also recognize that the CLHNS cohort contains female only, however, the consistency of our findings across these demographically heterogeneous cohorts provides support that the identified differences were not driven by any cohort-specific characteristics. We also acknowledge that the time between EPICv1 and EPICv2 measurement varied by approximately one month for VHAS and two years in CLHNS. While the time between array quantification was not consistent, it offers a more realistic representation of research settings where samples are collected and quantified across multiple batches on different EPIC versions. Next, given that the training datasets of the investigated epigenetic clocks were predominantly composed of European populations, the suitability of investigated clocks may not be well established in our investigated Asian and African cohorts[50–52]. However, the differences in epigenetic clock estimates we observed in the VHAS and CLHNS cohorts was similar to our observations in the CALERIE cohort comprising individuals of European and African descent, indicating that our findings were not entirely driven by population-specific effects. Additionally, any population-specific effects which may influence obtained estimates are likely to influence both EPIC versions in a comparable manner. In our analyses, we observed high correlations between clock estimates and chronological age and performed all comparisons between EPICv1 and EPICv2 in a cohort-specific manner, thereby circumventing any potential population-specific effects on calculated clock estimates. Further, while we compared EPICv2 cell type proportions and biomarker scores to EPICv1, we recognize that actual cell counts and biomarker measurements would be more appropriate as the ground truth and would be useful in validating our DNA methylation-based estimates. However, the primary aim of the current work is to investigate the concordance in estimates obtained from EPICv2 and EPICv1. Furthermore, high concordance between actual cell counts or biomarker scores and bioinformatically predicted estimates for EPICv1 has been previously reported[10,19–21], therefore it may be reasonable to rely on these DNA methylation-based estimates. We are also cognizant of the influence of preprocessing methods including normalization and batch correction options on DNA methylation-based estimates[23,38,39]. We have addressed this to a certain degree by employing two different normalization methods, namely functional normalization and BMIQ, as input in our analyses. Our findings in the majority of the tested epigenetic clocks and biomarker predictors remained significant with BMIQ normalization, implying that EPIC version effects still persisted across the two investigated normalization methods. While applying version correction resolved some of these EPIC version differences, it is plausible that currently available batch correction algorithms can result in biased estimates as previously mentioned. Future work to investigate or develop other preprocessing methods could be helpful in harmonizing EPICv1 and EPCIv2 and potentially resolving observed EPIC version differences.
Conclusions
With the rapid generation of DNA methylation data profiled on the newer iterations of the Illumina microarrays, such as EPICv2 and Infinium Methylation Screening Array (MSA), integrating samples across these platforms poses a challenge, owing to discrepancies in probe content among arrays/versions. Our findings highlight significant differences in the new EPICv2 compared to EPICv1, demonstrate the influence of EPIC version on DNA methylation-based tools, and provide recommendations to minimize technical variation which may arise from inconsistencies between versions. We therefore encourage caution when harmonizing and interpreting DNA methylation data when combining multiple arrays/versions to ensure reliability and reproducibility in epigenetics analyses.
Methods
Description of cohorts
To compare the performance of the two most recent generations of MethylationEPIC BeadChip Infinium microarrays in the context of DNA methylation-based clocks, biomarkers and cell type proportion estimates, we measured the DNA methylomes of a subset of venous whole blood samples on both EPICv1 and EPICv2 selected from three cohorts: (i) Vietnam Health and Aging Study (VHAS)[25], (ii) Cebu Longitudinal Health and Nutrition Survey (CLHNS)[26], and (iii) Comprehensive Assessment of Long-term Effects of Reducing Intake of Energy (CALERIE)[27]. The VHAS cohort additionally includes matched capillary blood samples, randomized using the same array design on both EPIC versions, such that the concordance between EPICv1 and EPICv2 can be assessed in capillary blood as well (n = 24 × 2 blood collection methods × 2 versions). The time between EPICv1 and EPICv2 array quantification was approximately one month and two years for VHAS and CLHNS respectively. EPICv1 and EPICv2 array quantification was carried out at the same time for CALERIE. Given that the demographic characteristics across the cohorts are different (Table 1), we performed all analyses independently on the cohorts and reported the findings in a cohort-specific manner, excluding the unsupervised hierarchical clustering analyses as described below.
DNA methylation profiling, sample and probe quality control
Using similar protocols for all three cohorts, DNA was extracted from samples, bisulfite converted using EZ-96 DNA Methylation kits (Zymo Research, Irvine, CA), hybridized to the EPICv1 and EPICv2 arrays, and scanned with the Illumina iScan 2000 to obtain IDAT files that capture raw DNA methylation intensities. IDATs were read using minfi R package to obtain β values that represent DNA methylation intensities for each CpG site ranging from 0 (fully unmethylated) to 1 (fully methylated). Technical replicates derived from the same sample were quantified to monitor technical variation within each EPIC version, independently for each cohort. Sample quality control checks were performed as described in previous publications[34,53]. Blood samples collected in the three cohorts passed all 17 Illumina quality control metrics in the ewastools R package[54,55], and detection p-value, beadcount, average methylated and unmethylated intensity metrics in the minfi R package (Supplementary Table S9). We also performed sample identity checks using unsupervised hierarchical clustering analysis on the 57 SNP probes that are common to both EPIC versions, and noted that matched samples from the same individual assessed on EPICv1 and EPICv2 grouped together.
In order to identify EPICv1 and EPICv2 probes of poor quality, we performed quality control checks using the detectionP and beadcount functions in the minfi and wateRmelon R packages, respectively. Probes with detection p-value > 0.01 or beadcount < 3 in greater than 1% of the samples were flagged (VHAS- EPICv1: 59,233, EPICv2: 46,735, common to both versions: 4,826; CLHNS-EPICv1: 23,793, EPICv2: 17,587, common to both versions: 991; CALERIE-EPICv1: 29,608, EPICv2: 24,872, common to both versions: 1,292), but all probes in EPICv1 and EPICv2 were retained for subsequent analyses.
DNA methylation data preprocessing and replicate probe analyses
To account for color and probe-type bias, we performed functional normalization (funnorm) with background correction and dye-bias normalization (noob) in the minfi R package[56,57] independently on EPICv1 and EPICv2 samples for each cohort. Cohort-specific technical replicate sample correlations were used to monitor preprocessing. Between technical replicate samples on the same EPIC version, improved Spearman correlations of whole array β values and reduced root mean square error (RMSE) were observed as processing progressed from raw (VHAS: Spearman rho = 0.9832, RMSE = 0.0306; CLHNS: Spearman rho = 0.9890, RMSE = 0.0235, CALERIE: Spearman rho = 0.9859, RMSE = 0.0349) to funnorm normalized data (VHAS: Spearman rho = 0.9851, RMSE=0.0288; CLHNS: Spearman rho = 0.9917, RMSE = 0.0231, CALERIE: Spearman rho = 0.9887, RMSE = 0.0223).
To calculate estimates of DNA methylation-based tools, noob corrected data was used as input for cell type deconvolution, and funnorm normalized data was used as input for epigenetic clocks and biomarkers. In addition, to test whether normalization methods influence DNA methylation-based tools, we compared estimates calculated by combining the common probes on EPICv1 and EPICv2 after noob correction in a cohort-wise manner for VHAS, CLHNS, and CALERIE, and subsequently applying Beta-MIxture Quantile (BMIQ) normalization implemented in the wateRmelon R package[58]. Due to their type I and type II design switch between EPICv1 and EPICv2[6], 82 probes were removed prior to BMIQ normalization. To account for any systematic bias in DNA methylation measurements[40] and subsequently test whether there are differences in DNA methylation-based estimates between EPIC versions, we applied batch correction for EPIC version, chip and row on funnorm normalized data using the ComBat function implemented in the sva R package[40]. We applied Spearman correlation to examine concordance of probes between the two EPIC versions, while in all other analyses we applied Pearson correlation to evaluate linear relationships between variables.
Given that there are certain probes on EPICv2 having two or more replicates (replicate probes), we characterized their distribution across the genome, and compared three strategies to collapse them into a single β value (based on detection p-value, mean and median). Our analyses identified that collapsed β values of EPICv2 replicate probes obtained using all three methods were highly correlated to corresponding EPICv1 probes, therefore EPICv2 replicate probes with lowest detection p-value were chosen as the representative probe based on previous recommendation[6] (Supplementary Material), and this approach was used for all the reported subsequent analyses.
Estimation of immune cell type proportions using DNA methylation-based cell type deconvolution
Cellular composition in heterogeneous tissue such as whole blood is one of the key contributors to the variation in DNA methylation profiles of bulk tissue[32,33]. In the absence of complete cell count data for the study samples, we estimated proportions of twelve immune cell types, basophils (Bas), naïve and memory B cells (Bnv, Bmem), naïve and memory CD4+ T cells (CD4nv, CD4mem), naïve and memory CD8+ T cells (CD8nv, CD8mem), eosinophils (Eos), monocytes (Mono), neutrophils (Neu), natural killer (NK), and T regulatory cells (Treg) from matched venous blood samples measured on the two EPIC versions. We used two methods of probe selection to estimate these cell type proportions: (i) the extended Identifying Optimal DNA methylation Libraries reference (IDOL), with probes not represented on EPICv2 removed from the reference before cell type proportions estimation in EPICv2 samples, and (ii) the “auto” method which selects the top 100 probes with F-stat p-value < 1E-8 for each cell type with the greatest magnitude of methylation difference, both implemented in the FlowSorted.Blood.EPIC R package with noob corrected values as recommended[10].
Estimation of epigenetic age and epigenetic age acceleration
We compared the performance of eight commonly used epigenetic clocks between EPICv1 and EPICv2 in VHAS, CLHNS, and CALERIE. Epigenetic age of first generation clocks including Horvath pan-tissue[11], Hannum[12], and Horvath skin and blood clocks[13], and second generation clocks including PhenoAge[14], and GrimAge[15] were obtained from the online DNA Methylation Age Calculator (https://dnamage.genetics.ucla.edu/new). For these clocks, we calculated epigenetic age acceleration (EAA), a measure of rate of aging commonly used in epigenetic clock investigations, by employing three approaches independently for each cohort.
EPIC version separate: we separated epigenetic ages of EPICv1 and EPICv2 samples and then calculated EAA independently on EPICv1 and EPICv2 samples by extracting residuals from the linear regression model: Epigenetic age ~ chronological age.
EPIC versions combined: we first combined epigenetic ages of EPICv1 and EPICv2 samples and then calculated EAA by extracting residuals from the linear regression model: Epigenetic age ~ chronological age.
EPIC versions combined and version adjusted: we first combined epigenetic ages of EPICv1 and EPICv2 samples and calculated EAA by extracting residuals from the linear regression model: Epigenetic age ~ chronological age + EPIC version.
To evaluate if there are significant differences in principal components (PC) clock[37] estimates, which are more robust to technical noise, by EPIC version, we first estimated epigenetic ages by the R script provided (https://github.com/MorganLevineLab/PC-Clocks) and then calculated EAAs using the approaches mentioned above.
As for the rate-based clocks, DunedinPACE was calculated using the DunedinPACE R package[17], epiTOC was obtained using the getEpiTOC function in the cgageR R package[18], and DNA methylation-based estimator of telomere length (DNAmTL)[16] were obtained from the online DNA Methylation Age Calculator (https://dnamage.genetics.ucla.edu/new). Unlike EAA calculation, there were no secondary measures calculated from these rate-based estimates in our analyses, estimates remained the same when calculated by the EPIC versions separate or combined approach, therefore, we employed only two approaches for each cohort:
EPIC version separate: rate-based clock estimates were calculated for EPICv1 and EPICv2 samples without EPIC version adjustment.
EPIC versions combined and version adjusted: rate-based clock estimates were first calculated for EPICv1 and EPICv2 samples, and were subsequently adjusted for EPIC versions by regressing out EPIC versions using linear regression model: Rate estimate ~ EPIC version.
Using paired t-tests and applying a Bonferroni multiple test correction, we evaluated differences in epigenetic clock estimations and EAAs (as well as biomarker predictor scores and cell type proportions described below) between matched samples assessed on EPICv1 and EPICv2 in a cohort-specific manner. Statistical significance was defined as Bonferroni adjusted p-value < 0.05. Effect sizes were measured by Cohen’s d, and classified as “small” (d = 0.2–0.49), “medium” (d = 0.5–0.79), and “large” (d ≥ 0.8) based on recommended benchmarks [59].
Estimation of DNA methylation-based inflammation, smoking, and alcohol scores
Among other DNA methylation-based tools are inflammation, smoking and alcohol score predictors, which provide biomarker measures that correlate with levels of inflammatory markers (IL-6, CRP), smoking and alcohol use, respectively. DNA methylation-derived scores of IL-6, CRP, smoking and alcohol use were calculated as a weighted sum of coefficients derived from published lists of predictive CpGs[19–21]. Being that these biomarkers were trained on previous Illumina arrays, we sought out to determine the correlation of derived DNA methylation-based scores between EPICv1 and EPICv2, and compare these scores without version adjustment (EPIC version separate) and with version adjustment (EPIC versions combined and adjusted) using approaches similar to the rate-based epigenetic clocks.
Supplementary Material
Acknowledgements
We thank Dr. Sarah Merrill, Maggie Fu, and Dr. Kimberly Schmidt for their helpful feedback during manuscript preparation. We would also like to thank all cohort participants for their involvement and providing biological samples for research. This research utilized the FlowSorted.BloodExtended.EPIC software packages developed at Dartmouth College, which are governed by the licensing terms provided by Dartmouth Technology Transfer. D.W.B. and M.S.K. are fellows of the CIFAR CBD Network.
Funding
This work received supported from the Canadian Institutes of Health Research (CIHR) / Project Grant (Sponsor Reference Number: PJT-175309), National Institute on Aging of the National Institutes of Health under Award Number R01AG052537, National Institutes of Health / Research Project Grant (R01) (Sponsor Reference Number: 5R01AG061006–05), and US National Institute on Aging Grants R01AG061378.
Footnotes
Competing interests
D.W.B. is listed as an inventor of the Duke University and University of Otago invention DunedinPACE, which is licensed to TruDiagnostic. The other authors declare that they have no competing interests.
Ethics approval and consent to participate
This study was approved by the University of British Columbia Research Ethics Boards (H18–03136) and the University of Utah Institutional Review Board (IRB 00098861).
References
- 1.Battram T, Yousefi P, Crawford G, Prince C, Sheikhali Babaei M, Sharp G, et al. The EWAS Catalog: a database of epigenome-wide association studies. Wellcome Open Res. 2022;7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuzawa CW, Ryan CP, Adair LS, Lee NR, Carba DB, MacIsaac JL, et al. Birth weight and maternal energy status during pregnancy as predictors of epigenetic age acceleration in young adults from metropolitan Cebu, Philippines. Epigenetics. 2022;17:1535–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li M, Zou D, Li Z, Gao R, Sang J, Zhang Y, et al. EWAS Atlas: a curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res. 2019;47:D983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Infinium MethylationEPIC v2.0 BeadChip. Infinium MethylationEPIC v2.0 BeadChip. 2023; [Google Scholar]
- 5.Peters TJ, Meyer B, Ryan L, Achinger-Kawecka J, Song J, Campbell EM, et al. Characterisation and reproducibility of the HumanMethylationEPIC v2.0 BeadChip for DNA methylation profiling. BMC Genomics. 2024;25:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaur D, Lee SM, Goldberg D, Spix NJ, Hinoue T, Li H-T, et al. Comprehensive evaluation of the Infinium human MethylationEPIC v2 BeadChip. Epigenetics Commun. 2023;3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noguera-Castells A, García-Prieto CA, Álvarez-Errico D, Esteller M. Validation of the new EPIC DNA methylation microarray (900K EPIC v2) for high-throughput profiling of the human DNA methylome. Epigenetics. 2023;18:2185742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinforma Oxf Engl. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salas LA, Koestler DC, Butler RA, Hansen HM, Wiencke JK, Kelsey KT, et al. An optimized library for reference-based deconvolution of whole-blood biospecimens assayed using the Illumina HumanMethylationEPIC BeadArray. Genome Biol. 2018;19:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salas LA, Zhang Z, Koestler DC, Butler RA, Hansen HM, Molinaro AM, et al. Enhanced cell deconvolution of peripheral blood using DNA methylation for high-resolution immune profiling. Nat Commun. 2022;13:761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol Cell. 2013;49:359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horvath S, Oshima J, Martin GM, Lu AT, Quach A, Cohen H, et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging. 2018;10:1758–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging. 2018;10:573–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu AT, Quach A, Wilson JG, Reiner AP, Aviv A, Raj K, et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging. 2019;11:303–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu AT, Seeboth A, Tsai P-C, Sun D, Quach A, Reiner AP, et al. DNA methylation-based estimator of telomere length. Aging. 2019;11:5895–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belsky DW, Caspi A, Corcoran DL, Sugden K, Poulton R, Arseneault L, et al. DunedinPACE, a DNA methylation biomarker of the pace of aging. Deelen J, Tyler JK, Suderman M, Deelen J, editors. eLife. 2022;11:e73420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Z, Wong A, Kuh D, Paul DS, Rakyan VK, Leslie RD, et al. Correlation of an epigenetic mitotic clock with cancer risk. Genome Biol. 2016;17:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stevenson AJ, Gadd DA, Hillary RF, McCartney DL, Campbell A, Walker RM, et al. Creating and Validating a DNA Methylation-Based Proxy for Interleukin-6. J Gerontol A Biol Sci Med Sci. 2021;76:2284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wielscher M, Mandaviya PR, Kuehnel B, Joehanes R, Mustafa R, Robinson O, et al. DNA methylation signature of chronic low-grade inflammation and its role in cardio-respiratory diseases. Nat Commun. 2022;13:2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCartney DL, Hillary RF, Stevenson AJ, Ritchie SJ, Walker RM, Zhang Q, et al. Epigenetic prediction of complex traits and death. Genome Biol. 2018;19:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solomon O, MacIsaac J, Quach H, Tindula G, Kobor MS, Huen K, et al. Comparison of DNA methylation measured by Illumina 450K and EPIC BeadChips in blood of newborns and 14-year-old children. Epigenetics. 2018;13:655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McEwen LM, Jones MJ, Lin DTS, Edgar RD, Husquin LT, MacIsaac JL, et al. Systematic evaluation of DNA methylation age estimation with common preprocessing methods and the Infinium MethylationEPIC BeadChip array. Clin Epigenetics. 2018;10:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dhingra R, Kwee LC, Diaz-Sanchez D, Devlin RB, Cascio W, Hauser ER, et al. Evaluating DNA methylation age on the Illumina MethylationEPIC Bead Chip. PLOS ONE. 2019;14:e0207834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korinek K, Teerawichitchainan B, Zimmer Z, Brindle E, Nguyen TKC, Nguyen HM, et al. Design and measurement in a study of war exposure, health, and aging: protocol for the Vietnam health and aging study. BMC Public Health. 2019;19:1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adair LS, Popkin BM, Akin JS, Guilkey DK, Gultiano S, Borja J, et al. Cohort Profile: The Cebu Longitudinal Health and Nutrition Survey. Int J Epidemiol. 2011;40:619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rochon J, Bales CW, Ravussin E, Redman LM, Holloszy JO, Racette SB, et al. Design and conduct of the CALERIE study: comprehensive assessment of the long-term effects of reducing intake of energy. J Gerontol A Biol Sci Med Sci. 2011;66:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen BH, Zhou W. mLiftOver: Harmonizing Data Across Infinium DNA Methylation Platforms [Internet]. bioRxiv; 2024. [cited 2024 Apr 8]. p. 2024.03.18.585415. Available from: 10.1101/2024.03.18.585415v1 [DOI] [PMC free article] [PubMed]
- 29.Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCartney DL, Walker RM, Morris SW, McIntosh AM, Porteous DJ, Evans KL. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genomics Data. 2016;9:22–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price EM, Cotton AM, Lam LL, Farré P, Emberly E, Brown CJ, et al. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013;6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones MJ, Moore SR, Kobor MS. Principles and Challenges of Applying Epigenetic Epidemiology to Psychology. 2017;30. [DOI] [PubMed] [Google Scholar]
- 33.Zheng SC, Beck S, Jaffe AE, Koestler DC, Hansen KD, Houseman AE, et al. Correcting for cell-type heterogeneity in epigenome-wide association studies: revisiting previous analyses. Nat Methods. 2017;14:216–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merrill SM, Gladish N, Fu MP, Moore SR, Konwar C, Giesbrecht GF, et al. Associations of peripheral blood DNA methylation and estimated monocyte proportion differences during infancy with toddler attachment style. Attach Hum Dev. 2023;25:132–61. [DOI] [PubMed] [Google Scholar]
- 35.Ryan CP, Jones MJ, Edgar RD, Lee NR, Kobor MS, McDade TW, et al. Immune cell type and DNA methylation vary with reproductive status in women: possible pathways for costs of reproduction. Evol Med Public Health. 2022;10:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kong Y, Rastogi D, Seoighe C, Greally JM, Suzuki M. Insights from deconvolution of cell subtype proportions enhance the interpretation of functional genomic data. PLOS ONE. 2019;14:e0215987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Higgins-Chen AT, Thrush KL, Wang Y, Minteer CJ, Kuo P-L, Wang M, et al. A computational solution for bolstering reliability of epigenetic clocks: Implications for clinical trials and longitudinal tracking. Nat Aging. 2022;2:644–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ori APS, Lu AT, Horvath S, Ophoff RA. Significant variation in the performance of DNA methylation predictors across data preprocessing and normalization strategies. Genome Biol. 2022;23:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engelbrecht H-R, Merrill SM, Gladish N, MacIsaac JL, Lin DTS, Ecker S, et al. Sex differences in epigenetic age in Mediterranean high longevity regions. Front Aging. 2022;3:1007098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinforma Oxf Engl. 2012;28:882–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hubens WHG, Maié T, Schnitker M, Bocova L, Puri D, Wessiepe M, et al. Targeted DNA Methylation Analysis Facilitates Leukocyte Counts in Dried Blood Samples. Clin Chem. 2023;69:1283–94. [DOI] [PubMed] [Google Scholar]
- 42.Garma LD, Quintela-Fandino M. Epigenetic age prediction drifts resulting from next-generation methylation arrays [Internet]. 2024. [cited 2024 Jul 1]. Available from: 10.1101/2024.06.07.597709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sugden K, Hannon EJ, Arseneault L, Belsky DW, Corcoran DL, Fisher HL, et al. Patterns of Reliability: Assessing the Reproducibility and Integrity of DNA Methylation Measurement. Patterns. 2020;1:100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olstad EW, Nordeng HME, Sandve GK, Lyle R, Gervin K. Low reliability of DNA methylation across Illumina Infinium platforms in cord blood: implications for replication studies and meta-analyses of prenatal exposures. Clin Epigenetics. 2022;14:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyer DH, Schumacher B. Aging clocks based on accumulating stochastic variation. Nat Aging. 2024;1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Price EM, Robinson WP. Adjusting for Batch Effects in DNA Methylation Microarray Data, a Lesson Learned. Front Genet [Internet]. 2018. [cited 2021 Jun 22];9. Available from: 10.3389/fgene.2018.00083/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buhule OD, Minster RL, Hawley NL, Medvedovic M, Sun G, Viali S, et al. Stratified randomization controls better for batch effects in 450K methylation analysis: a cautionary tale. Front Genet. 2014;5:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nygaard V, Rødland EA, Hovig E. Methods that remove batch effects while retaining group differences may lead to exaggerated confidence in downstream analyses. Biostat Oxf Engl. 2016;17:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zindler T, Frieling H, Neyazi A, Bleich S, Friedel E. Simulating ComBat: how batch correction can lead to the systematic introduction of false positive results in DNA methylation microarray studies. BMC Bioinformatics. 2020;21:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin W-Y. Epigenetic clocks derived from western samples differentially reflect Taiwanese health outcomes. Front Genet. 2023;14:1089819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Horvath S, Gurven M, Levine ME, Trumble BC, Kaplan H, Allayee H, et al. An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biol. 2016;17:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cronjé HT, Nienaber-Rousseau C, Min JL, Green FR, Elliott HR, Pieters M. Comparison of DNA methylation clocks in Black South African men. Epigenomics. 2021;13:437–49. [DOI] [PubMed] [Google Scholar]
- 53.Konwar C, Asiimwe R, Inkster AM, Merrill SM, Negri GL, Aristizabal MJ, et al. Risk-focused differences in molecular processes implicated in SARS-CoV-2 infection: corollaries in DNA methylation and gene expression. Epigenetics Chromatin. 2021;14:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murat K, Grüning B, Poterlowicz PW, Westgate G, Tobin DJ, Poterlowicz K. Ewastools: Infinium Human Methylation BeadChip pipeline for population epigenetics integrated into Galaxy. GigaScience. 2020;9:giaa049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Illumina Inc. BeadArray Controls Reporter Software Guide. 2015; [Google Scholar]
- 56.Fortin J-P, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Triche TJ, Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013;41:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29:189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cohen J. Statistical Power Analysis for the Behavioral Sciences. 2nd ed. New York: Routledge; 1988. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.