

Abstract
Equine rhinitis A virus (ERAV) has recently been classified as an aphthovirus, a genus otherwise comprised of the different serotypes of Foot-and-mouth disease virus (FMDV). FMDV initiates translation via a type II internal ribosomal entry site (IRES) and utilizes two in-frame AUG codons to produce the leader proteinases Lab and Lb. Here we show that the ERAV 5′ nontranslated region also possesses the core structures of a type II IRES. The functional activity of this region was characterized by transfection of bicistronic plasmids into BHK-21 cells. In this system the core type II structures, stem-loops D to L, in addition to a stem-loop (termed M) downstream of the first putative initiation codon, are required for translation of the second reporter gene. In FMDV, translation of Lb is more efficient than that of Lab despite the downstream location of the Lb AUG codon. The ERAV genome also has putative initiation sites in positions similar to those utilized in FMDV, except that in ERAV these are present as two AUG pairs (AUGAUG). Using the bicistronic expression system, we detected initiation from both AUG pairs, although in contrast to FMDV, the first site is strongly favored over the second. Mutational analysis of the AUG codons indicated that AUG2 is the major initiation site, although AUG1 can be accessed, albeit inefficiently, in the absence of AUG2. Further mutational analysis indicated that codons downstream of AUG2 appear to be accessed by a mechanism other than leaky scanning. Furthermore, we present preliminary evidence that it is possible for ribosomes to access downstream of the two AUG pairs. This study reveals important differences in IRES function between aphthoviruses.
Equine rhinitis A virus (ERAV), formerly known as Equine rhinovirus 1, is a member of the Picornaviridae family and has been recently reclassified as the only non-foot-and-mouth disease virus (non-FMDV) member of the genus Aphthovirus (26). This reclassification was based largely on nucleotide sequence determination of the ERAV genome (14, 32), although it is also consistent with many of the known physicochemical and biological properties of the virus (20). ERAV infection of horses results in an acute febrile respiratory disease that is accompanied by viremia and persistent virus shedding in the urine and feces (see reference 30 for a review). It is also pathogenic for a broad range of other animal species, including humans (24, 25). The recent finding that strains of ERAV which are noncytopathic in in vitro-cultured cells are responsible for outbreaks of febrile respiratory disease in horses suggests that the virus has been underdiagnosed and that its relative significance as an equine pathogen may have been underestimated (15). Further investigation into the epidemiology and pathogenesis of this virus is clearly required. In this paper we characterize the role of the ERAV 5′ nontranslated region (5′-NTR) in translation initiation, an important pathogenic determinant in picornaviruses.
Picornaviruses initiate translation in a cap-independent manner and require an internal ribosome entry site (IRES) for this process. These IRESs form stable secondary structures, but the nature of the predicted RNA fold and of the start codon usage differs between genera. At present, the IRESs of different picornaviruses conform to one of three models. A type I IRES is found in enterovirus and rhinovirus genomes and is characterized by translation initiation of the polyprotein at an AUG codon located a considerable distance downstream of the distinct RNA structure that forms the IRES. Type II IRESs are found in cardioviruses and aphthoviruses, and these have a very different predicted secondary structure that is characterized by the presence of core stem-loops D to L (21, 23, 29). In this model, translation is initiated 12 to 15 nucleotides (nt), downstream of a polypyrimidine tract in a location that is immediately 3′ to the core structural elements that define the IRES (2, 3, 23). A type III IRES is found in hepatoviruses, and although this model is much less studied, it appears to share features of both types I and II (4).
A conserved feature among the FMDV serotypes is that translation of the polyprotein is initiated at two different AUG codons, one at the 3′ end of the IRES and another located 84 nt downstream (1, 5). This results in the production of two forms of the leader (L) proteinase, Lab and Lb, in infected cells (28). The smaller FMDV Lb species is consistently synthesized in excess of Lab despite the downstream location of the Lb initiation codon. The ERAV genome also possesses putative start sites in positions similar to those utilized in FMDV, except that in ERAV these are present as two AUG pairs that are separated by 57 nt (14, 32). Translation initiation from these sites would result in the synthesis of L proteinases of similar sizes to the FMDV Lab and Lb species. In FMDV it is not known why two forms of L proteinase are synthesized, as the two species possess similar proteolytic activities (18). It has been determined, however, that functional FMDV L proteinase is not essential for replication, as deletion of L sequence downstream of the Lb AUG in an infectious clone resulted in the production of viable virus, albeit with a small-plaque phenotype (22). Some uncertainty remains as to the mechanism(s) by which the different AUG codons are accessed in FMDV. Current evidence suggests direct entry of the ribosome at or immediately upstream of the Lab AUG into what is described as the “starting window.” It was originally proposed that the Lb AUG is accessed by leaky scanning, and this is consistent with the poor Kozak sequence context surrounding the Lab AUG (2). However, more recent studies are not totally consistent with this mechanism and suggest that the Lb AUG may also be accessed by direct ribosomal entry (8, 22).
In this report, we describe secondary structure modelling and functional characterization of the ERAV IRES using a bicistronic system. This analysis predicts that ERAV possesses a type II IRES and reveals that full translation activity requires nt 245 to 961 downstream of the poly(C) tract. This region includes the core type II stem-loops as well as additional sequence downstream of these structures. The sequence between the AUG codon pairs, which is predicted to form a stable hairpin, is clearly crucial to IRES activity in this system. Using mutational analysis, we show that the second AUG of the first pair, AUG2, appears to be the dominant start site, although translation also initiates from the second AUG pair. This implies that ERAV-infected cells may also produce two forms of the L proteinase, but, in contrast to FMDV, the ERAV Lab species is likely to be produced in vast excess of Lb.
MATERIALS AND METHODS
Secondary-structure determination.
An RNA secondary structure of the 5′-NTR of ERAV strain 393/76 downstream of the poly(C) tract was predicted through the use of the mFOLD program (17, 33). The minimum free-energy structures were determined for the first 961 nt. Although structures that were either at or very close to the minimum free-energy values were always used, distal loops of known function (7, 11) that have been found to be conserved throughout the viruses were utilized to discern between different thermodynamically stable structures. For the identification of these conserved sequences, the corresponding nucleotide sequences of encephalomyocarditis virus (EMCV) strain B, EMCV strain R, Theiler's murine encephalomyelitis virus (TMEV) strain DA, FMDV strain A12, ERAV 393/76, and equine rhinitis B virus (ERBV) (32) were obtained from GenBank and aligned by use of Eclustalw software (31) (data not shown).
Plasmids.
All plasmids were constructed using standard cloning methods (27). The parental plasmid pT7CG was derived from the pTM-1 vector kindly provided by B. Moss (19). The bicistronic cassette contains the chloramphenicol acetyltransferase (CAT) and green fluorescent protein (GFP) reporter genes under the control of the T7 promoter. The cassette was constructed by first inserting an NheI/XbaI fragment containing the GFP gene from the pEGFP-C2 vector (Clontech, Palo Alto, Calif.) into the XbaI site of the pCAT3 control vector (Promega, Madison, Wis. to produce pSV40CG. This ligation placed a unique AgeI restriction site between the CAT and GFP genes. The CAT-GFP cassette was removed from this vector using BamHI and partial NcoI digestion and inserted into the equivalent restriction sites in the pTM-1 multicloning site to produce pT7CG. pT7GFP was formed by insertion of the GFP-containing NcoI/BamHI digestion fragment from pSV40CG into the same sites of pTM-1.
The 5′-NTR regions of ERAV 393/76 were subjected to reverse transcription followed by PCR amplification with specific oligonucleotides (shown in Table 1) that each contained an AgeI restriction site. Briefly, purified ERAV RNA was copied into cDNA using Superscript II reverse transcriptase according to the manufacturer's instructions (Gibco-BRL, Gaithersburg, Md.). The cDNA was subjected to PCR amplification with relevant oligonucleotides using 35 cycles at 94°C for 45, 58°C for 45, and 72°C for 45 with Platinum Taq Hi-Fi polymerase according to instructions (Gibco-BRL). PCR products were digested with AgeI before ligation into pT7CG. Plasmids containing AUG mutations were constructed by site-directed mutagenesis of pE1(245–961) through overlap extension PCR. Oligonucleotides used and mutations constructed are shown in Table 2. These PCR products were digested with AgeI and ligated into pT7CG. To produce plasmids with the GFP gene fused to the different AUG codons, the pE1(245–961) vector was mutagenized by overlap extension PCR using the oligonucleotides shown in Table 2. These PCR products were digested with AgeI and BsrGI, a site located within the GFP coding sequence, before ligation into the same sites of pT7CG. All inserts were completely sequenced by primer extension and BigDye chain termination chemistry (Applied Biosystems, Foster City, Calif.). Each ERAV insert had an identical insert in the overlapping regions except where shown.
TABLE 1.
Oligonucleotides used to amplify regions of the ERAV 5′-NTR
| Oligonucleotide | Location on genomea | Sequence (5′→3′)b |
|---|---|---|
| TH1-F1 | Sense/1 | TAC CGG TCT TAA GCT TGT ACC TGT AGC |
| TH1-F245 | Sense/245 | CTG TCA CCG GTG AGA GGA GCC CG |
| TH1-F338 | Sense/338 | ATT GAC CGG TCG AAG CGT TGC C |
| TH1-F434 | Sense/434 | CCC GAC CGG TGT ATA GCG CCA GG |
| TH1-F508 | Sense/508 | TGT CAC CGG TCT GAC AAA TGC GGA G |
| TH1-R961 | Antisense/961 | TTT GAC CGG TTG GGG GAC AAA AAC AGT TCT C |
| TH1-R1299 | Antisense/1299 | GAA TAC TGC ATG TAC CGG TTT TGC |
Direction of oligonucleotide with respect to the genome/nucleotide number downstream of the poly(C) tract.
Nucleotides in boldface indicate an AgeI restriction site.
TABLE 2.
Oligonucleotides used to modify or delete regions of the ERAV 5′-NTR
| Oligonucleotide | Mutation | Sequence (5′→3′)c |
|---|---|---|
| TH-GFPR | 3′ end of GFP | TTG AGC TCG AGA TCA GAG TCC |
| TH1-omA1Fa | Alter ATG1 to ATA | TAC TGA CAT AAT GGC GGC G |
| TH1-omA1Rb | Alter ATG1 to ATA | GAC GCC GCC ATT ATG TCA GTA |
| TH1-omA2Fa | Alter ATG2 to ATA | GTA CTG ACA TGA TAG CGG CGT CTA AG |
| TH1-omA2Rb | Alter ATG2 to ATA | GAC GCC GCT ATC ATG TCA GTA |
| TH1-omA1/2Fa | Alter ATG1 and ATG2 to ATA | GTA CTG ACA TAA TAG CGG CGT C |
| TH1-omA1/2Rb | Alter ATG1 and ATG2 to ATA | GAC GCC GCT ATT ATG TCA GTA |
| TH1-omA4Fa | Alter ATG4 to ATA | GGT GCC GTT CGC ATG ATA GAC AAA TTC TTG |
| TH1-omA4Rb | Alter ATG4 to ATA | TCT ATC ATG CGA ACG GCA CCT GC |
| TH1-omA3/4Fa | Alter ATG3 and ATG4 to ATA | GGT GCC GTT CGC CAT AAT AGA CAA ATT CTT G |
| TH1-omA3/4Rb | Alter ATG3 and ATG4 to ATA | TCT ATT ATG CGA ACG GCA CCT GC |
| TH1-omTAFa | ATG2 Kozak modification | CTG ACT TGA TGA CGG CGT CTA AGG |
| TH1-omTARb | ATG2 Kozak modification | CGC CGT CAT CAA GTC AGT ACA AAA G |
| TH1-GFPMFa | 5′ end of GFP | ATG GTG AGC AAG GGC GAG GAG CTG TTC |
| TH1-odA1Rb | Delete sequence in pE1(245–961) between ATG1 and GFP ATG | CTC GCC CTT GCT CAC CAT GTC AGT ACA AAA GGA AAA GAA ACG C |
| TH1-odA2Rb | Delete sequence in pE1(245–961) between ATG2 and GFP ATG | CGC CCT TGC TCA CCA TCA TGT CAG TAC AAA AGG AAA AGA AAC GC |
| TH1-odA3Rb | Delete sequence in pE1(245–961) between ATG2 and GFP ATG | CGC CCT TGC TCA CCA TGC GAA CGG CAC CTG CCA GAG TCT GCT C |
| TH1-odA4Rb | Delete sequence in pE1(245–961) between ATG4 and GFP ATG | CGC CCT TGC TCA CAT CAT GCG AAC GGC ACC TGC CAG AGT CTG CTC |
Oligonucleotides used with TH-GFPR for first-round PCR.
Oligonucleotides used with TH1-F245 for first-round PCR. Overlapping first-round PCR products were combined and subjected to second-round PCR with TH1-F245 (Table1) and TH-GFPR.
Underlined nucleotides, mutated bases; boldfaced nucleotides, overlaps with TH1-GFPMF.
Transient expression assays.
For transcription by T7 polymerase, plasmid constructs were transfected into BHK-21 cells that had been infected with recombinant vaccinia virus vTF7-3 (10) 1 h previously. Transfections were performed using Lipofectamine Plus reagent essentially as described by the manufacturer (Gibco-BRL); 0.5 μg of the appropriate plasmid DNA was transfected into semiconfluent monolayers of approximately 2.5 × 105 cells. Each experiment was repeated at least twice. At 20 h posttransfection, cells were released with trypsin and resuspended in phosphate-buffered saline (PBS) for determination of CAT activity and GFP fluorescence. Cell extracts were prepared for CAT assay by lysis in CAT reporter lysis buffer (Promega), and CAT activity was determined by measuring [14C]chloramphenicol (Amersham Pharmacia, Little Chalfont, Buckinghamshire, United Kingdom) acetylation according to the manufacturer's instructions (Promega). Acetylated and nonacetylated forms of [14C]chloramphenicol were separated by thin-layer chromatography, and spots were quantitated by phosphorimager analysis (Molecular Dynamics). Samples were serially diluted so that CAT activity was always measured in the linear range. Fluorescence produced by GFP was determined by fluorescence-activated cell sorter (FACS) analysis. For this, cells were centrifuged at 1,500 × g for 2 min, resuspended in FACS fix buffer (PBS containing 1% paraformaldehyde, 1% fetal calf serum [FCS], and 0.1% sodium azide), and injected into a Becton Dickinson FACSort machine set for detection of fluorescein isothiocyanate (FITC). For each sample, the GFP/CAT ratio was determined as a ratio of total GFP fluorescence (geometric mean) to CAT activity from an equivalent number of cells.
RIP assays.
Cell extracts were prepared from transfected cells as described above with some alterations. At 12 h posttransfection, the medium was replaced with methionine-free medium for 1 h, after which 30 μCi of [35S]methionine (Amersham Pharmacia) was added. Three hours later, the cells were washed twice with PBS and harvested in 500 μl of radioimmunoprecipitation (RIP) lysis buffer (0.05 M Tris-HCl, 1% Triton X, 0.6 M KCl). The lysate was incubated on ice for 30 min, vortexed for 30 s, and then clarified by centrifugation (∼16,000 × g) for 5 min. Cell lysates were precleared for 1 h at 4°C with 20 μl of a 50% suspension of protein A-Sepharose beads (Amersham Pharmacia). Rabbit anti-GFP (kindly provided by P. Silver) and rabbit anti-CAT immunoglobulins (5 Prime-3 Prime, Boulder, Colo.) were added separately to a 50% suspension of Sepharose A beads in PBS at 0.1 or 0.5 μg, respectively, per 20-μl bead suspension. These were incubated at 4°C with continuous mixing. Antibody-coated beads were washed once with PBS and reconstituted to 50% suspensions in PBS. CAT- and GFP-coated bead suspensions were combined, and 40 μl was added to each 500 μl of cell lysate. These were incubated for 2 h at 4°C with continuous mixing. Beads were washed four times with RIP wash buffer (0.05 M Tris-HCl, 1 mM EDTA, 0.15 M NaCl, 0.25% bovine serum albumin [BSA], 1% Triton X) and then twice with PBS before the addition of 30 μl of 5 × reducing sodium dodecyl sulfate (SDS) sample buffer to the bead pellet. Samples were run on SDS-polyacrylamide gel electrophoresis (PAGE) gels (12% polyacrylamide). Gels were fixed for 30 min at room temperature in 7% acetic acid–10% methanol and scintillated for 30 min at room temperature in 1 M sodium salicylate–50% methanol. Gels were dried and subjected to phosphorimager analysis for isotope quantitation or exposed to X-ray film for preparation of figures.
RESULTS
The ERAV IRES folds into a type II secondary structure.
We present an RNA secondary structure prediction for the ERAV 5′-NTR downstream of the poly(C) tract (Fig. 1). The model incorporates 18 stem-loop structures upstream of the polypyrimidine tract and shows features characteristic of a type II IRES. Structures that resemble the D, E, F, G, H, I, J, K, and L stem-loops found in other type II IRESs were identified. In FMDV and the cardioviruses, stem-loop I is proposed to be a single large stem-loop; however, such a structure was not thermodynamically stable by our analysis. Instead, this region consistently resolved into three smaller stem-loops, Ia, Ib, and Ic. A similar structural prediction has recently been described for another type II IRES (21) (Fig. 1). Multiple sequence alignments of the D-through-L stem-loop regions of other type II IRESs reveal that the unpaired regions are generally more conserved than stem structures (data not shown). The most conserved loops are highlighted in Fig. 1. The sequence further upstream appears less conserved both in sequence and in folding pattern than that found in FMDV. It should be noted that the ERAV 5′-NTR between the poly(C) and polypyrimidine tracts is ∼150 nt longer than the corresponding region of FMDV.
FIG. 1.

Proposed secondary structure of ERAV RNA downstream from the poly(C) tract. Major structural domains are labeled according to the work of Palmenberg and Sgro (21). The four potential initiation codons are boxed, and the polypyrimidine tract is indicated by the dashed line. The positions of oligonucleotides used in several experiments described below are indicated (arrows). Shaded boxes highlight predicted unpaired sequences that are conserved in the cardiovirus and aphthovirus genomes. The position of the functionally relevant GNRA loop is indicated by the asterisk. Numbering begins at the first base after the poly(C) tract.
Identification of the ERAV IRES and of 5′ cis-acting elements.
One objective of this study was to define the boundaries of the ERAV IRES. To perform this analysis a plasmid (pT7CG) that comprised the CAT and GFP reporter genes under the control of the T7 promoter was constructed (Fig. 2). Initially the entire 5′-NTR sequence downstream of the poly(C) tract was inserted as an intergenic spacer between the two reporter genes. The 3′ boundary of this insert was placed at 104 nt downstream of the first putative initiation codon, as this region included other potential initiation codons, as well as two further predicted stem-loops, here named M and N (Fig. 1). The GFP gene was in frame with the putative initiation codons. Inserts containing progressive 5′ deletions of the ERAV 5′-NTR were also inserted into the pT7CG plasmid between the two reporter genes (Fig. 3A). The nucleotide sequence of each insert was determined in order to confirm that the overlapping ERAV sequence was identical in each plasmid. When transfected into BHK-21 cells infected with a recombinant vaccinia virus that expresses T7 polymerase (vTF7-3), the full-length construct pE1(1–961) produced high levels of GFP compared to the parental plasmid pT7CG, indicating the presence of an active IRES in this sequence (Fig. 3B). Deletion of the first 245 nt to produce pE1(245–961) had no apparent effect on GFP translation, indicating that this sequence is not required for internal initiation. Truncation of the 5′-NTR to position 338, pE1(338–961), resulted in a fourfold decrease in GFP expression (Fig. 3B), demonstrating that nt 245 to 338 contain an element(s), probably a full stem-loop D, important for full IRES function in the bicistronic system. Plasmids containing inserts further truncated at the 5′ end, pE1(434–961) and pE1(508–961), failed to produce detectable GFP expression when transfected into vTF7-3-infected BHK-21 cells. Therefore, nt 338 to 434, which contain partial D, E, F, and G stem-loops, appear to contain a separate element(s) important to IRES function. This experiment was repeated on numerous occasions, and the results shown in Fig. 3B are entirely representative of these analyses.
FIG. 2.

Diagram of the parental bicistronic plasmid pT7CG. The vector is a derivative of pTM1 (19) and contains two reporter genes, CAT and GFP, that are separated by a short stretch of sequence (top) containing a unique AgeI site (underlined) used to insert ERAV sequences in frame with the GFP gene. The locations of the T7 promoter (T7) and terminator (T7 term) are indicated. Note that the CAT gene is also translated by an IRES (EMCV IRES).
FIG. 3.

Functional analysis of the ERAV IRES using a bicistronic plasmid. (A) Diagram showing the ERAV AgeI fragments ligated into plasmid pT7CG between the CAT and GFP reporter genes. The names of the plasmids are indicated to the right, and the numbering begins from the poly(C) tract. (B) GFP-to-CAT ratios in VTF7-3-infected BHK-21 cells (expressing T7 RNA polymerase) following transfection with the plasmids shown at the left. GFP fluorescence was measured by FACS analysis of whole cells, while CAT enzyme activity was determined from cell extracts as detailed in Materials and Methods. Values are represented as a percentages of the  ratio determined for pE1(1–961). The dashed vertical line indicates the value obtained for the GFP negative control plasmid pT7CG.
ratio determined for pE1(1–961). The dashed vertical line indicates the value obtained for the GFP negative control plasmid pT7CG.
Identification of ERAV initiation codons.
Extracts of pE1(245–961)-transfected cells metabolically labeled with [35S]methionine were immune precipitated with GFP-specific antibodies to identify the GFP polypeptides produced by the ERAV IRES (Fig. 4). Three GFP-related bands were observed, a major species at 33 kDa (termed Lab-GFP [see below]), a more rapidly migrating but relatively minor species at 31 kDa (termed Lb-GFP), and a 30-kDa species that comigrated with the authentic GFP protein precipitated from cells transfected with the GFP control plasmid pT7GFP. Although the Lb-GFP band was weak, it was observed in each experiment performed and was sometimes more obvious (see below). This presence of the GFP-comigrating species suggested that in addition to the ERAV codons present upstream, translation initiates at the GFP AUG in this system. This possibility is explored further below. The bands were quantitated by phosphorimager analysis and corrected for methionine content and for levels of CAT expression. This analysis revealed that in cells transfected with the parental pE1(245–961) plasmid, Lab-GFP is consistently found in an 8- to 12-fold excess over Lb-GFP.
FIG. 4.

Identification of the GFP fusion proteins synthesised by the ERAV IRES. Plasmid pE1(245–961) and a GFP-expressing plasmid, pT7GFP, constructed by deleting the CAT gene in pT7CG, were transfected into vTF7-3-infected BHK-21 cells. Following metabolic labeling with [35S]methionine, cell extracts were immune precipitated with polyclonal rabbit anti-GFP antibodies and analyzed by SDS-PAGE and autoradiography. Molecular size standards (in kilodaltons) are shown on the right.
A site-directed mutational analysis was performed to determine which of the four AUG codons at the 3′ end of the IRES are utilized to initiate translation of the ERAV polyprotein. The complete nucleotide sequence of each ERAV insert was determined following mutagenesis to confirm that the sequence possessed only the desired change. In order to identify which AUG pair gave rise to the GFP-related species observed in Fig. 4, each AUG pair in pE1(245–961) was mutated separately to an AUA pair to give rise to the plasmids pE1(245–961) ΔA1/2 and pE1(245–961)ΔA3/4 (Fig. 5A). Immune precipitation of extracts from cells transfected with pE1 (245– 961)ΔA1/2 showed that the Lab-GFP species clearly initiates at AUG1 and AUG2, as loss of these AUG codons caused the disappearance of this species (Fig. 5B). These mutations did not result in a noticeable loss of IRES activity but instead resulted in a shift in translation initiation to the more rapidly migrating Lb-GFP species (Fig. 5B). To confirm that this effect was due solely to the absence of AUG codons required to produce Lab, the second AUA in pE1(245–961)ΔA1/2 was mutated back to AUG. The resulting revertant, pE1(245–961)rΔA1, restored the wild-type expression levels of the Lab- and Lb-GFP species (data not shown). Cells transfected with pE1(245–961)ΔA3/4 did not express Lb-GFP, confirming that this species is initiated at the second AUG pair (Fig. 5B). In these cells, no effect on Lab-GFP expression was observed, although the level of the GFP-comigrating species was reduced.
FIG. 5.

Identification of functional ERAV AUG codons by mutational analysis. (A) Representation of ERAV sequence surrounding the ATG codons (boxed) downstream of the polypyrimidine tract (indicated to the left of the sequence) in a wild-type ERAV vector, pE1(245–961), and in derivatives of this vector. These derivatives contain point mutations at the positions shown. The minimum free energy M and N stem-loops predicted in each mutant are shown on the right. Plasmid names are given at the extreme left. (B) RIP of vTF7-3-infected BHK-21 cell extracts following transfection with ERAV mutant plasmids and metabolic labeling with [35S]methionine. Extracts were immune precipitated with a mixture of GFP and CAT antibody-coated protein A beads. The lanes are labeled with shortened plasmid names. The WT lane represents the parental pE1(245–961) plasmid containing the wild-type IRES, while the Δ2/ΔGNRA lane represents pE1(245–961)Δ2/ΔGNRA, a plasmid that contains a PCR-induced mutation in the GNRA tetraloop (converting it to GNRG) in addition to the AUG2 mutation. The intensities of the bands were determined by phosphorimager analysis, and the  ratio was determined for each GFP band following correction for methionine content (bottom). (C) Results of an experiment similar to that in panel B, but using the pE1(245–961)Δ2 mutant.
ratio was determined for each GFP band following correction for methionine content (bottom). (C) Results of an experiment similar to that in panel B, but using the pE1(245–961)Δ2 mutant.
To further examine initiation codon usage, mutagenesis of individual AUG codons was performed on the parental pE1(245–961) plasmid. Mutation of AUG1 to AUA resulted in the plasmid pE1(245–961)Δ1. This mutation had no significant effect on initiation codon usage or translation efficiency, indicating that AUG2 can be efficiently utilized (Fig. 5B). An initial attempt to mutate AUG2 to AUA resulted in a plasmid with a single PCR-induced mutation in the IRES that converted the GNRA loop found within stem-loop Ib to GNRG. Transfection with this plasmid, pE1(245–961)Δ2/ΔGNRA, showed no IRES activity (Fig. 5B). This finding is consistent with the known importance of the GNRA tetraloop to internal ribosomal entry (7) and provided some functional validation of the predicted secondary-structure model shown in Fig. 1. Transfection of an authentic AUG2-mutagenized plasmid, pE1(245–961)Δ2 (Fig. 5A and C), resulted in substantially reduced expression of Lab but had little effect on expression levels of the Lb and GFP species. Also, the Lab-GFP species observed here consistently migrated more slowly than the Lab-GFP species expressed from the wild-type IRES consistent with a shift from initiation from AUG2 to AUG1 in this mutant. This finding indicates that although AUG1 can be utilized, albeit inefficiently, in the absence of AUG2, AUG2 appears to be the major initiation codon utilized in the synthesis of the Lab species.
To address which of AUG3 or AUG4 is utilized in the synthesis of the minor Lb species, AUG4 was mutated to AUA in pE1(245–961)Δ1/2 to give rise to pE1(245–961)Δ1/2/4 (Fig. 5A). When assayed, as expected, there was no detectable Lab species; however, translation of the Lb species was markedly reduced compared to the levels of this protein in pE1(245–961)Δ1/2-transfected cells. This suggests that AUG4 may be favored over AUG3, consistent with the better Kozak consensus surrounding AUG4. Interestingly, the level of the GFP-comigrating species was increased in cells transfected with pE1(245–961)Δ1/2/4.
The major FMDV initiation codon is the second of two utilized AUG codons, which, unlike AUG1, is in an optimal Kozak context. We constructed a plasmid, pE1(245–961)A2con, that mimics the FMDV AUG1 context. Here the Kozak consensus sequence surrounding AUG2 was weakened, which resulted in the mutation of AUG1 to TUG (Fig. 5A). Transfection with pE1(245–961)A2con resulted in a marked reduction in translation of Lab-GFP; however, this was not accompanied by a concomitant increase in translation of Lb-AUG, as would be expected if AUG3 and/or AUG4 are accessed by a leaky scanning mechanism (Fig. 5B). Translation of the GFP species in these cells was similar to that observed in cells transfected with parental pE1(245–961).
We also addressed whether extending the ERAV sequence at the 3′ end had an effect on codon usage. A plasmid, pE1(245–1299), was constructed by insertion of an AgeI fragment comprising nt 245 to 1299 into the pT7CG plasmid. The 3′ end of this fragment extended to a position downstream of the next in-frame AUG codon, AUG5, in the ERAV genome (Fig. 6A). Immune precipitation with an anti-GFP antibody revealed that the dominant GFP-related protein produced is the most slowly migrating species (46 kDa), consistent with initiation from AUG2 (Fig. 6B). Several faster-migrating GFP fusion proteins were also detected. The molecular weight of the smallest of these species (Fig. 6B) may represent initiation from AUG5. Such a result is consistent with the observation that translation initiates from the nonviral GFP codon when this AUG is positioned downstream of the two AUG pairs [see results for pE1(245– 961) and derivative plasmids]. Together, these data suggest that it is possible for ribosomes to initiate downstream of the first four AUGs. Several other minor bands were observed in cells transfected with pE1(245–1299), one of which presumably represents the Lb-GFP species, while the others may represent degradation products.
FIG. 6.

Extension of the ERAV IRES at the 3′ end does not dramatically effect codon usage. (A) Diagram of AgeI insert in plasmid pE1(245–1299), highlighting the positions of the AUG codons. The initiation codons in frame with the GFP gene are shown (arrows). (B) Immune precipitation with GFP antibody of extracts of vTF7-3-infected BHK-21 cell extracts following transfection with plasmid pE1(245–1299) (1299) (WT), or pE1(245–961)Δ2 (Δ2). Labels to the left refer to the identities of bands in the WT and Δ2 lanes only. The small arrows indicate the locations of the slowest- and fastest-migrating species in lane 1299. Molecular size standards in kilodaltons are shown on the right.
Sequence downstream of the major initiation site is required for IRES activity.
We investigated the role of sequence between the two pairs of initiation codons in determining IRES activity. Deletions at the 3′ end of the IRES element in pE1(245–961) were carried out to produce plasmids pE1(245–855), pE1(245–858), pE1(245–918), and pE1(245–921), in which the GFP gene was placed adjacent to AUG codons 1, 2, 3, and 4, respectively (Fig. 7A). When assayed in the transient expression system, the constructs with deletions up to AUG3 and AUG4, pE1(245–918) and pE1(245–921), respectively, demonstrated approximately 60% of full IRES activity (Fig. 7B). This indicated that sequence downstream of AUG4, which comprises the N stem-loop, is required for full activity in this system. Constructs lacking nucleotides between the AUG pairs pE1(245–855) and pE1(245–858) had no detectable GFP expression by FACS analysis (Fig. 7B). This indicated that some sequence between the AUG pairs is crucial for IRES activity and for initiation from upstream codons, suggesting a role for stem-loop M in this process.
FIG. 7.

Sequence between the two pairs of AUG codons is important for ERAV IRES activity. (A) Diagram of plasmids containing 3′ truncations of the ERAV IRES, highlighting the positions of the AUG codons (arrows). Note that in these plasmids there is no additional sequence between the GFP AUG and the ERAV sequence. (B) GFP-to-CAT ratios in VTF7-3 infected BHK-21 cells (T7 expressing) following transfection with the plasmids named on the left. GFP was measured by FACS analysis of whole cells, while CAT enzyme activity was determined from cell extracts as detailed in Materials and Methods. Values are expressed as percentages of the  ratio determined for pE1(245–961). The vertical line indicates the value obtained for the GFP-negative control plasmid pT7CG. (C) RIP analysis of vTF7-3-infected BHK-21 cell extracts following transfection with the plasmids diagramed in panel A and metabolic labeling with [35S] methionine. Extracts were immune precipitated with GFP antibody-coated protein A beads. Labeling for the pE1(245–961) lane is on the left.
ratio determined for pE1(245–961). The vertical line indicates the value obtained for the GFP-negative control plasmid pT7CG. (C) RIP analysis of vTF7-3-infected BHK-21 cell extracts following transfection with the plasmids diagramed in panel A and metabolic labeling with [35S] methionine. Extracts were immune precipitated with GFP antibody-coated protein A beads. Labeling for the pE1(245–961) lane is on the left.
RIP analysis of these samples revealed an identical pattern of expression (Fig. 7C), although a faint band corresponding to GFP was detected in pE1(245–855)- and pE1(245–858)-transfected cells. Importantly, in cells transfected with pE1(245–918) or pE1(245–921), only one species was detected, and this corresponded in size to the Lab polypeptide initiated from AUG2. The absence of a band comigrating with GFP suggests that elements downstream of AUG3 and AUG4 are required for initiation from these codons and perhaps also from an additional downstream codon(s).
In summary, nt 245 to 961 are required for full ERAV IRES activity in the bicistronic system, nt 338 to 918 appear to comprise a minimal IRES element. AUG2 at position 858 appears to be the major start site. A downstream AUG codon, probably that at position 921 (AUG4), is able to initiate translation, but only to about 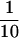 the level of AUG2. This suggests that, in contrast to FMDV, ERAV-infected cells are likely to contain the Lab proteinase in vast excess of the Lb proteinase, although this remains to be shown directly.
the level of AUG2. This suggests that, in contrast to FMDV, ERAV-infected cells are likely to contain the Lab proteinase in vast excess of the Lb proteinase, although this remains to be shown directly.
DISCUSSION
In this study we show that ERAV nt 245 to 961 downstream of the poly (C) tract are sufficient to confer efficient internal initiation of protein synthesis within a bicistronic construct. All the elements of a picornavirus type II IRES are found within this region, including the predicted presence of characteristic stem-loop structures (23, 29), the location of a potential start codon close to the polypyrimidine tract, and the presence of motifs known to be crucial to the function of type II IRES's such as the GNRA loop (7). The requirement of ERAV stem loops D through L for maximal IRES activity is consistent with results obtained with other type II IRES elements (3, 9, 12). In ERAV a minimal region of 570 nt (positions 338 to 918) does appear to contain elements sufficient for partial IRES activity. This region does not possess a full D stem-loop at the 5′ end or the N stem-loop at the 3′ end. It has been reported for EMCV that the H stem-loop at the 5′ boundary of the IRES is sufficient for activity in a bicistronic plasmid (12) but that additional upstream sequence is required for IRES activity in its native context (9, 13). In the bicistronic system used here, however, the presence of the ERAV H stem-loop at the 5′ end was not sufficient for activity [see pE1(434–961)], implying a role for the small hairpin structures E, F, and G in IRES activity. Importantly, the 3′ boundary of the ERAV IRES includes sequence downstream of the major start codon. This region is predicted to fold into a stable hairpin structure, stem-loop M. One study of FMDV (22) highlighted a role for sequence downstream of the first functional initiation codon in IRES activity. These authors showed that deletion of L sequence downstream of the Lab AUG in an infectious clone dramatically reduced the translation efficiency of the polyprotein. The requirement of sequence between the two functional AUG codons may be an important determinant of IRES activity in the aphthoviruses.
Our results indicate that translation initiation in the bicistronic system predominantly occurs from the first pair of AUG codons and that AUG2 appears to be the major initiator. We do show, however, that AUG1 can be accessed in the absence of AUG2 but that initiation in this instance is less efficient. Why then is AUG2 utilized in preference to AUG1? The most likely reason is that the sequence context surrounding AUG2 is more favorable for the direct entry and positioning of the small ribosomal subunit. That is, ribosomes do not enter upstream and scan down to AUG2; rather, they most likely enter directly and initiate at AUG2, bypassing AUG1. With respect to this, AUG2 is present in a more favorable Kozak consensus than AUG1. Also, in contrast to AUG1, AUG2 is appropriately positioned 12 nt downstream of the polypyrimidine tract. Both of these features are characteristic of functional AUG codons in other type II IRESs (6, 16). It appears that in the absence of AUG2, the small ribosomal subunit is repositioned upstream to the less-favorable AUG codon.
Translation in ERAV also appears to initiate from the second pair of AUG codons, probably AUG4, but this occurs at only about 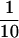 of the level of initiation from AUG2. Although FMDV also initiates translation from two different sites, in this case both codons are well used, and it is the second, located 84 nt downstream from the first, that predominates. We attempted to address why there was such a marked difference in translation initiation between these aphthoviruses and also to shed light on the mechanisms by which the downstream codons are utilized. To this end, we generated a mutant [pE1(245–961)A2con] whereby the Kozak context surrounding AUG2 was weakened to resemble that of FMDV AUG1. This mutation, which was not predicted to impact on the thermodynamic stability of stem-loop M, dramatically reduced translation initiation from AUG2 but had no effect on initiation from downstream AUG codons. A very similar result was observed with the AUG2 mutant [pE1(245–961)Δ2]. Therefore, downstream AUG codons do not appear to be accessed by leaky scanning. This is consistent with the findings of recent work with FMDV, where it has been shown that the mechanism by which the downstream AUG is accessed appears to be independent of the functionality of the first AUG (8). It is possible that in both FMDV and ERAV the downstream AUG may be accessed by direct entry, and that normal scanning may not follow internal ribosomal entry in these viruses. The presence of a second polypyrimidine tract immediately upstream of the FMDV Lb codon may at least in part explain why it is more efficiently used than its ERAV counterpart.
of the level of initiation from AUG2. Although FMDV also initiates translation from two different sites, in this case both codons are well used, and it is the second, located 84 nt downstream from the first, that predominates. We attempted to address why there was such a marked difference in translation initiation between these aphthoviruses and also to shed light on the mechanisms by which the downstream codons are utilized. To this end, we generated a mutant [pE1(245–961)A2con] whereby the Kozak context surrounding AUG2 was weakened to resemble that of FMDV AUG1. This mutation, which was not predicted to impact on the thermodynamic stability of stem-loop M, dramatically reduced translation initiation from AUG2 but had no effect on initiation from downstream AUG codons. A very similar result was observed with the AUG2 mutant [pE1(245–961)Δ2]. Therefore, downstream AUG codons do not appear to be accessed by leaky scanning. This is consistent with the findings of recent work with FMDV, where it has been shown that the mechanism by which the downstream AUG is accessed appears to be independent of the functionality of the first AUG (8). It is possible that in both FMDV and ERAV the downstream AUG may be accessed by direct entry, and that normal scanning may not follow internal ribosomal entry in these viruses. The presence of a second polypyrimidine tract immediately upstream of the FMDV Lb codon may at least in part explain why it is more efficiently used than its ERAV counterpart.
In the light of these results, it was somewhat surprising that mutation of both AUG1 and AUG2 [pE1(245–961)Δ1/2] did allow efficient initiation from the second AUG pair. In this instance, as expected, no initiation was evident from AUG1 or AUG2, in contrast to the lower level of initiation from these positions observed with pE1(245–961)A2con and pE1(245–961)Δ2. It is possible that entry of ribosomes onto the favored “upstream” position of viral RNA, even if this results in inefficient initiation, may alter RNA structure and/or the binding of associated molecules such that ribosomal entry at downstream codons is restricted. We speculate that in the absence of upstream ribosomal entry and initiation, such as may be the case with pE1(245–961)Δ1/2, a structural context is maintained in the RNA that allows ribosomes to efficiently access downstream codons.
Why have two AUG pairs if only some of these codons are utilized? It is noteworthy that the nucleotides of AUG1 and AUG3, which are not predicted to be involved in initiation, are involved in base pairing at the base of stem-loop M (see Fig. 1). The potential importance of this stem-loop to translation initiation in ERAV suggests that a purpose of these AUGs may be to maintain the structure of this stem-loop. It would be of interest to test the effect on translation initiation of mutating these codons to other codons that are predicted to either maintain or disrupt stem-loop M.
It remains to be determined if the codon usage data obtained here using the bicistronic plasmid concur with the different forms of the leader protein produced in ERAV-infected cells. Such a correlation is evident in FMDV (8). Our results predict that ERAV produces homologues of the FMDV Lab and Lb proteinases but that, unlike FMDV, the Lab species is probably produced in vast excess of the Lb species. The reason for the presence of the different forms of L proteinase and how, if at all, different ratios of Lab to Lb impact on aphthoviral pathogenesis require investigation.
ACKNOWLEDGMENTS
We thank Bernard Moss for the provision of the recombinant vaccinia virus vTF7-3 and the pTM-1 plasmid and Pamela Silver for the GFP antibody. We thank Carol Hartley, Nino Ficorilli, and Michael Studdert for the provision of ERAV 393/76 stocks and cell lines and for helpful advice throughout the course of this study.
T.M.H. is the recipient of an Australian Postdoctoral Research Award and receives scholarship support from the CRC for Vaccine Technology. F.L. was the recipient of a scholarship from the Australian International Developmental Assistance Bureau (AIDAB) during his involvement in this study.
REFERENCES
- 1.Beck E, Fross S, Strebel K, Cattaneo R, Feil G. Structure of the FMDV translation initiation site and of the structural proteins. Nucleic Acids Res. 1983;11:7873–7885. doi: 10.1093/nar/11.22.7873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belsham G J. Dual initiation sites of protein synthesis on foot-and-mouth disease virus RNA are selected following internal ribosome entry and scanning of ribosomes in vivo. EMBO J. 1992;11:1105–1110. doi: 10.1002/j.1460-2075.1992.tb05150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belsham GJ, Brangwyn J K. A region of the 5′ noncoding region of foot-and-mouth disease virus RNA directs efficient internal initiation of protein synthesis within cells: involvement with the role of L protease in translational control. J Virol. 1990;64:5389–5395. doi: 10.1128/jvi.64.11.5389-5395.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown E A, Day S P, Jansen R W, Lemon S M. The 5′ nontranslated region of hepatitis A virus RNA: secondary structure and elements required for translation in vitro. J Virol. 1991;65:5828–5838. doi: 10.1128/jvi.65.11.5828-5838.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarke B E, Sangar D V, Burroughs J N, Newton S E, Carroll A R, Rowlands D J. Two initiation sites for foot-and-mouth disease virus polyprotein in vivo. J Gen Virol. 1985;66:2615–2626. doi: 10.1099/0022-1317-66-12-2615. [DOI] [PubMed] [Google Scholar]
- 6.Davies M V, Kaufman R J. The sequence context of the initiation codon in the encephalomyocarditis virus leader modulates efficiency of internal translation initiation. J Virol. 1992;66:1924–1932. doi: 10.1128/jvi.66.4.1924-1932.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Quinto S L, Martinez-Salas E. Conserved structural motifs located in distal loops of aphthovirus internal ribosome entry site domain 3 are required for internal initiation of translation. J Virol. 1997;71:4171–4175. doi: 10.1128/jvi.71.5.4171-4175.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Quinto S L, Martinez-Salas E. Involvement of the aphthovirus RNA region located between the two functional AUGs in start codon selection. Virology. 1999;255:324–336. doi: 10.1006/viro.1999.9598. [DOI] [PubMed] [Google Scholar]
- 9.Duke G M, Hoffman M A, Palmenberg A C. Sequence and structural elements that contribute to efficient encephalomyocarditis virus RNA translation. J Virol. 1992;66:1602–1609. doi: 10.1128/jvi.66.3.1602-1609.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuerst T R, Niles E G, Studier W, Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffman M A, Palmenberg A C. Mutational analysis of the J-K stem-loop region of the encephalomyocarditis virus IRES. J Virol. 1995;69:4399–4406. doi: 10.1128/jvi.69.7.4399-4406.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jang S K, Monique V D, Kaufman R J, Wimmer E. Initiation of protein synthesis by internal entry of ribosomes into the 5′ nontranslated region of encephalomyocarditis virus RNA in vivo. J Virol. 1989;63:1651–1660. doi: 10.1128/jvi.63.4.1651-1660.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jang S K, Wimmer E. Cap-independent translation of encephalomyocarditis virus RNA: structural elements of the internal ribosomal entry site and involvement of a cellular 57-kD RNA-binding protein. Genes Dev. 1990;4:1560–1572. doi: 10.1101/gad.4.9.1560. [DOI] [PubMed] [Google Scholar]
- 14.Li F, Browning G F, Studdert M J, Crabb B S. Equine rhinovirus 1 is more closely related to foot-and-mouth disease virus than to other picornaviruses. Proc Natl Acad Sci USA. 1996;93:990–995. doi: 10.1073/pnas.93.3.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li F, Drummer H E, Ficorilli N, Studdert M J, Crabb B S. Identification of noncytopathic equine rhinovirus 1 as a cause of acute febrile respiratory disease in horses. J Clin Microbiol. 1997;35:937–943. doi: 10.1128/jcm.35.4.937-943.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez S E, Saiz J C, Davila M, Belsham G J, Domingo E. A single nucleotide substitution in the internal ribosome entry site of foot-and-mouth disease virus leads to enhanced cap-independent translation in vivo. J Virol. 1993;67:3748–3755. doi: 10.1128/jvi.67.7.3748-3755.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathews D H, Sabina J, Zuker M, Turner D H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 18.Medina M, Domingo E, Brangwyn J K, Belsham G J. The two species of the foot-and-mouth disease virus leader protein, expressed individually, exhibit the same activities. Virology. 1993;194:355–359. doi: 10.1006/viro.1993.1267. [DOI] [PubMed] [Google Scholar]
- 19.Moss B, Elroy-Stein O, Mizukami T, Alexander W A, Fuerst T R. New mammalian expression vectors. Nature. 1990;348:91–92. doi: 10.1038/348091a0. [DOI] [PubMed] [Google Scholar]
- 20.Newman J, Rowlands D J, Brown F. A physico-chemical sub-grouping of the mammalian picornaviruses. J Gen Virol. 1973;18:171–180. doi: 10.1099/0022-1317-18-2-171. [DOI] [PubMed] [Google Scholar]
- 21.Palmenberg A C, Sgro J-Y. Topological organization of picornaviral genomes: statistical prediction of RNA structural signals. Semin Virol. 1997;8:231–241. [Google Scholar]
- 22.Piccone M E, Rieder E, Mason P W, Grubman M J. The foot-and-mouth disease virus leader proteinase gene is not required for viral replication. J Virol. 1995;69:5376–5382. doi: 10.1128/jvi.69.9.5376-5382.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pilipenko E V, Blinov V M, Dmitrieva T M, Agol V I. Conservation of the secondary structure elements of the 5′-untranslated region of cardio- and aphthovirus RNAs. Nucleic Acids Res. 1989;17:5701–5711. doi: 10.1093/nar/17.14.5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plummer G. An equine respiratory enterovirus: some biological and physical properties. Arch Gesamte Virusforsch. 1963;12:694–700. doi: 10.1007/BF01246390. [DOI] [PubMed] [Google Scholar]
- 25.Plummer G. An equine respiratory virus with enterovirus properties. Nature. 1962;195:519–520. doi: 10.1038/195519a0. [DOI] [PubMed] [Google Scholar]
- 26.Pringle C R. Virus taxonomy—1997. Proceedings of the 26th Meeting of the Executive Committee of the ICTV, Strasbourg, France, May 10–11, 1997. Arch Virol. 1997;142:1727–1733. [Google Scholar]
- 27.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 28.Sangar D V, Newton S E, Rowlands D J, Clarke B E. All foot-and-mouth disease virus serotypes initiate protein synthesis at two separate AUGs. Nucleic Acids Res. 1987;15:3305–3315. doi: 10.1093/nar/15.8.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stewart S R, Semler B L. RNA determinants of Picornavirus cap-independent translation initiation. Semin Virol. 1997;8:242–255. [Google Scholar]
- 30.Studdert M J. Equine rhinovirus infections. In: Studdert M J, editor. Virus infections of equines. 1st ed. Vol. 6. Amsterdam, The Netherlands: Elsevier Science B. V.; 1996. pp. 213–217. [Google Scholar]
- 31.Thompson J D, Higgins D G, Gibson T J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wutz G, Auer H, Nowotny N, Grosse B, Skern T, Kuechler E. Equine rhinovirus serotypes 1 and 2: relationship to each other and to aphthoviruses and cardioviruses. J Gen Virol. 1996;77:1719–1730. doi: 10.1099/0022-1317-77-8-1719. [DOI] [PubMed] [Google Scholar]
- 33.Zuker M, Mathews D H, Turner D H. Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide. In: Barciszewski J, Clark B F C, editors. RNA biochemistry and biotechnology. Vol. 70. Dordrecht, The Netherlands: Kluwer Academic Publishers; 1998. pp. 11–43. [Google Scholar]