

Abstract
Biomaterials are known to modulate immune cell functions, which subsequently determine the host inflammatory and immune responses. Poly(lactic-co-glycolic acid) or PLGA, a biodegradable and biocompatible biomaterial, induces a pro-inflammatory, mature phenotype in antigen presentation cells, namely dendritic cells (DCs) in vitro. In vivo, PLGA can boost the humoral immune response to a co-delivered model antigen, a phenomenon known as the PLGA-adjuvant effect. This study elucidates the link between PLGA’s effect on the DC phenotype in vitro and its adjuvant effect in vivo using the CD11c-DTR mouse model. These mice undergo conditional ablation of DCs upon treatment with diphtheria toxin (DT). To measure immune activation, the mice were first given OVA-reactive T cells from OT-II / OT-I mice. Later the same mice received subcutaneous OVA-loaded PLGA scaffold implants. In response to the scaffold implants, OVA-reactive OT-II CD4+ T cells showed decreased proliferation in the absence of CD11c+ DCs, indicating an attenuation of the PLGA-adjuvant effect. Furthermore, PLGA may also influence the antigen cross-presentation function of DCs, as evident with the lowered OVA-reactive OT-I CD8+ T-cell response. Understanding the immunomodulatory ability of biomaterials in the context of DCs will aid in designing improved DC-based immunotherapies against infectious diseases and cancer.
Keywords: Dendritic cells, biomaterials, PLGA scaffolds, CD11c-DTR, adjuvant effect
Introduction
Dendritic cells (DCs) are the most proficient antigen presenting cells (APCs) in the mammalian immune system. They process antigen in their microenvironment and present it on their surface for identification by naïve T cells. DCs express a wide array of pattern recognition receptors that can recognize various “antigen–motif” molecular patterns.1 As regulators of the immune system, DCs can either induce antigen-specific regulatory T cells or peripheral anergy of self-reactive T cells.2 On that account, the immune outcome can be tailored by presenting DCs with the appropriate biologics to induce either the immunostimulatory or immunosuppressive phenotype. Despite the recent surge of interest in the modulation of immune outcome, there is limited information about the influence biomaterial vehicles have on key fate-determining immune cells such as APCs.
A biodegradable copolymer of lactic acid (LA) and glycolic acid (GA) — poly(lactic-co-glycolic acid) or PLGA — has been extensively used in biomolecule delivery in therapeutic applications.3 As it is FDA approved for medical use,4,5 PLGA has been fast-tracked to several clinical trials. PLGA is not only easy to synthesize but also cost-efficient, making it favorable for commercial development. Depending on the application, the polymer composition can be modified to control the rate of in vivo degradation and can be formed into different geometries —films,6 scaffolds,7 matrices,8 microparticles,9 nanoparticles.10 Unlike commonly available synthetic polymers, PLGA exhibits a higher extent of biocompatibility and causes only minor toxicities since the degradation products are identical to body metabolites.4
Since PLGA vehicles are endocytosed by DCs, it is likely that the strong acidic environment inside phagosomes/endosomes increases its rate of degradation. This may result in altering the internal sensitivity of associated antigen peptides to the downstream cellular processing in the DC antigen-presentation machinery. Gaining a deeper insight into the host response that PLGA (with or without a biologic) may engender in vivo is extremely significant in improving our ability to design effective therapies that match a desired immune outcome.
Depending on the induced phenotype of the DCs, biomaterials can modify the overall host response by producing either an adjuvant/immunostimulatory effect or an immunosuppressive/tolerogenic effect.11,12 In vitro and in vivo immune response data for antigen-carrying PLGA suggests that the adjuvant effect induced by PLGA to co-delivered antigen in vivo is likely due to its effect on immune-fate determining DCs. Generally, PLGA alone triggers a non-specific immune/inflammatory response while the co-delivered biologics cause an antigen-specific adaptive immune response. PLGA can elicit an adjuvant effect in vivo by inducing the humoral immune Th2 response to a co-delivered model antigen.7 Studies have shown that PLGA in conjunction with model antigen ovalbumin (OVA) can induce maturation of human-derived or murine immune cells.13–15 Induced DCs shifted from the immature/untreated/quiescent phenotype to a more immunogenic phenotype. Although the level of DC maturation was less than that with toll-like receptor (TLR) agonists (e.g., lipopolysaccharide, LPS), PLGA could be used as an adjuvant to bolster the DC-induced immune response, such as in vaccine delivery. A change in DC phenotype is a crucial indicator of a potential change in DC function and the downstream adaptive response (stimulatory or regulatory).
PLGA can be modified in different ways to achieve immune-inert16 or even immunosuppressive17 behaviour in DCs. These functional differences can be imparted by varying the material characteristics of PLGA including molecular weight,16 lactide to glycolide ratios,15,18 chemical modifications with surfactants17 and differences in structural contact with DCs15,19 (nanoparticles, microparticles, films or scaffolds). While seemingly contradictory, these reports do not diminish the immunostimulatory or adjuvant effect of PLGA7,13,15,20–22 but, in fact, enhance the versatility of such a synthetic polymer system that can be functionally meaningful at opposing ends of the immunological spectrum.
This study assesses the correlation of the PLGA-adjuvant effect with the presence of CD11c+ DCs using 75:25 lactide:glycolide-based porous PLGA scaffolds of 8-mm diameter. CD11c+ is a type 1 transmembrane integrin, also known as integrin alpha ⍺x, that is highly expressed in DC cells;23 for this reason, it is often used as a marker for DC cells. The system used for the study was the CD11c-DTR transgenic mouse model, derived from C57BL/6 wild-type mice24. These mice express a simian DTR gene under the control of CD11c. As conventional DCs form the largest group of cells expressing CD11c in mice, conditional ablation of CD11c+ cells with DT also depletes the majority of DCs. The depletion of DCs is maintained only as long as there is circulating DT; DT has a relatively short half-life. This model allows DC depletion without lethality in mice for over a week. However, assessing the antibody response to OVA in the presence of PLGA requires several (up to 12) weeks of mouse survival. As this time frame was not possible with the CD11c-DTR mice, the effects of DC depletion in early T-cell responses, such as antigen-specific clonal expansion, were evaluated instead. It is important to mention that CD11c is also expressed in other leukocytes (monocytes, macrophage and neutrophils). CD11c is a part of Complement receptor 4 (CR4) that is involved in complement-mediated phagocytosis.25 It is possible that phagocytic capabilities of these leukocyte subtypes are diminished upon CD11c+ cell depletion. CD11c can also be expressed in unconventional B-cell26 and T-cell populations27,28 with specific functions such as immune memory, mucosal migration or regulatory function. However, DCs are the most potent antigen presenting cells that mediate adaptive immune priming against antigen.1 Hence, the depletion of CD11c+ DCs is expected to induce a stronger effect on antigen presentation than other types of CD11c+ cells.
The OVA-reactive OT-II or OT-I model was used to investigate if conditional DC depletion in vivo influenced the proliferative response of adoptively transferred OVA-reactive T cells in mice that were pre-implanted with an OVA-loaded PLGA scaffold. As a read-out of the adaptive response to OVA, fluorescently labeled OVA-reactive CD8+ (from OT-I mice) or CD4+ (from OT-II mice) T cells were adoptively transferred into DC-depleted mice. Considering that antigen presentation by DCs initiates antigen-specific adaptive immune responses, such as a T-cell response and an antibody response,29 the expected outcome was a mitigated clonal expansion of OVA-reactive T cells in CD11c+ cell-depleted mice.
The study presented here furthers our understanding of PLGA’s immunostimulatory cellular influences on programmable DCs within the body and can help improve the design of biomaterial-based therapeutic vehicles for immune-related disorders
Materials and Methods
Animals:
Animal care and treatment were in compliance with the Institution Animal Care and Use Committee. Male C57BL/6 mice, CD11c-DTR [B6.FVB-Tg(Itgax-DTR/EGFP)57Lan/J], OT-I30 [C57BL/6-Tg(TcraTcrb)1100Mjb/J] and OT-II31 [B6.Cg-Tg(TcraTcrb)425Cbn/J] of 8–12 weeks were purchased from Jackson Labs, Bar Harbor, ME. Mice were housed at six or less per cage and allowed to acclimate for 1 week prior to receiving experimental treatments.
Characterization of CD11c+ Cell Depletion in CD11c-DTR Mice with Multiple Diphtheria Toxin (DT) Doses:
To determine the effect of repeated DT injections on CD11c-DTR, the time-points for injections and analysis were set up according to Figure 1. On day 0, mice either received adoptive transfer of OT-II T cells [2×107 cells/ 300 μL of sterile phosphate buffered saline (PBS)] via intravenous (I.V.) tail or penile vein injection. Control mice did not receive the adoptive transfer. Group 1 mice received 1 DT injection on day 1; Group 2 mice received 2 DT injections on days 1 and 3, and Group 3 mice received 3 DT injections on days 1, 3 and 5.
Figure 1. Timeline of repeated DT injections.

CD11c-DTR mice received adoptive transfer of OT-II T cells (2×107 cells/ 300 μL of sterile PBS) I.V. via tail vein or penile vein injection. Control mice did not receive any cells. Group 1 mice received 1x DT injection on day 1; group 2 mice received 2x DT injections on days 1 and 3, and Group 3 mice received 3x DT injections on days 1, 3 and 5. All DT injections were given I.P. at 4ng/g of body weight.
Each DT injection was given at 4 ng/g mouse body weight diluted in sterile PBS at a volume of 100 μL. Mice in Groups 1, 2 and 3 were sacrificed on day 2, 4 and 6, respectively. The body weights of the CD11c-DTR mice were measured before the first DT injection and 24 h after each DT injection to determine the effect of the three DT injections. Splenocytes were harvested and stained with fluorescently labeled antibodies against CD11c, CD3, CD8α, CD4, F4/80 or 33D1 (a DC marker) and analyzed by flow cytometry. Each group consisted of the following subgroups of mice — (i) C57BL/6 treated with DT (wild-type negative control), (ii) CD11c-DTR untreated by DT (transgenic negative control), (iii) CD11c-DTR treated with DT along with adoptively transferred OT-II T cells and (iv) CD11c-DTR treated with DT alone. Each subgroup consisted of 4 – 6 mice.
Preparation PLGA/OVA Scaffold:
All PLGA scaffolds were freshly prepared for each experimental procedure using the solvent casting, particulate leaching method previously published with minor modifications.19 Briefly, 75:25 PLGA (LACTEL Corporation) was dissolved in dichloromethane (DCM; Sigma-Aldrich) to obtain 83.3 mg/ml. For the OVA-loaded scaffold preparation, OVA was added to the PLGA-DCM solution at 166 mg/mL. The PLGA solutions were poured over sodium chloride (90–120 μm particle size; 0.3 g NaCl/mL PLGA solution). The DCM evaporated over time, and the salt was leached out into water over several days. The percent loading efficiency of OVA in the 8-mm diameter PLGA scaffolds was found to be 0.17 ± 0.06% with the total delivery polymer mass being 8.7 ± 1.3 mg. Dried PLGA scaffolds (8 mm in diameter) were cut and stored.
Adoptive Transfer of T cells:
T cells were obtained from splenocytes using a negative selection magnetic bead isolation kit (CD4+ or CD8+ or pan CD3+ T cells) (Miltenyi Biotec) according to manufacturer’s protocol with appropriate modifications. Briefly, harvested splenocytes were strained through 40 μm mesh and their red blood cells were lysed. The cells were treated with biotin-bound antibody cocktail, followed by anti-biotin magnetic beads and run through a magnetic LS column (Miltenyi); the effluent contained the unlabeled fraction of the respective enriched T cells. The cells were washed three times and prepared at 2×107/300 μL for adoptive transfer into recipient mice (C57BL/6 or CD11c-DTR) via either I.V. tail or penile vein injection.32,33 Adoptively transferred OT-I CD8+ T cells and pan CD3+ T cells were used in experiments described in Figure 6 and Figure S2, respectively. All other experiments used adoptively transferred OT-II CD4+ T cells (Figures 3, 4, 5).
Figure 6. Proliferation profile of OVA-reactive CD8+ T cells in the absence of CD11c+ cells.

Wild-type C57BL/6 or CD11c-DTR mice with adoptively transferred CFSE-labeled OT-I T cells, with or without 3 DT injections given 2 days apart, were implanted with OVA-loaded PLGA scaffolds as per the experimental design shown in Figure 2. OVA in CFA served as the positive control. Splenocytes obtained from mice on Day 6 were stained with antibodies against CD3, CD4 and CD8 and assessed by flow cytometry. (A) Gated region (representative of all groups) marked in red (top) was analyzed for CFSE fluorescence (bottom). The histograms are representative of 6 mice per group. The grey bar is representative of CFSE-labeled T cells prior to the adoptive transfer injection. While the blue bar represents the CFSE+ cells obtained from splenocytes post adoptive transfer. (B) Statistical comparison of DT treatments on OVA-reactive CD8+ T-cell proliferation using ANOVA followed by Holm-Sidak’s post-test. Error bars indicate standard error of mean of 6 mice per treatment or control group (* = p < 0.01). The inset image (right) shows a sample FI plot in which consecutive T-cell progeny is marked from P1-P6.
Figure 3. Effect of three DT injections on mouse body weight.

CD11c-DTR mice, with or without adoptively transferred (a.t.) OT-II T cells, were given either 1, 2 or 3 DT injections at 4ng/g body weight, spaced 2 days apart as per the timeline shown in Figure 1. Mice were weighed before the first DT injection and 24 h after each DT injection. Statistical analysis was done using ANOVA followed by Sidak’s post-test. No statistically significant differences in body were measured (p > 0.05). Error bars represent standard error of mean values of 4 – 6 mice per sub-group
Figure 4. Surface biomarker expression of CD11c-DTR murine splenocytes after repeated DT injections.
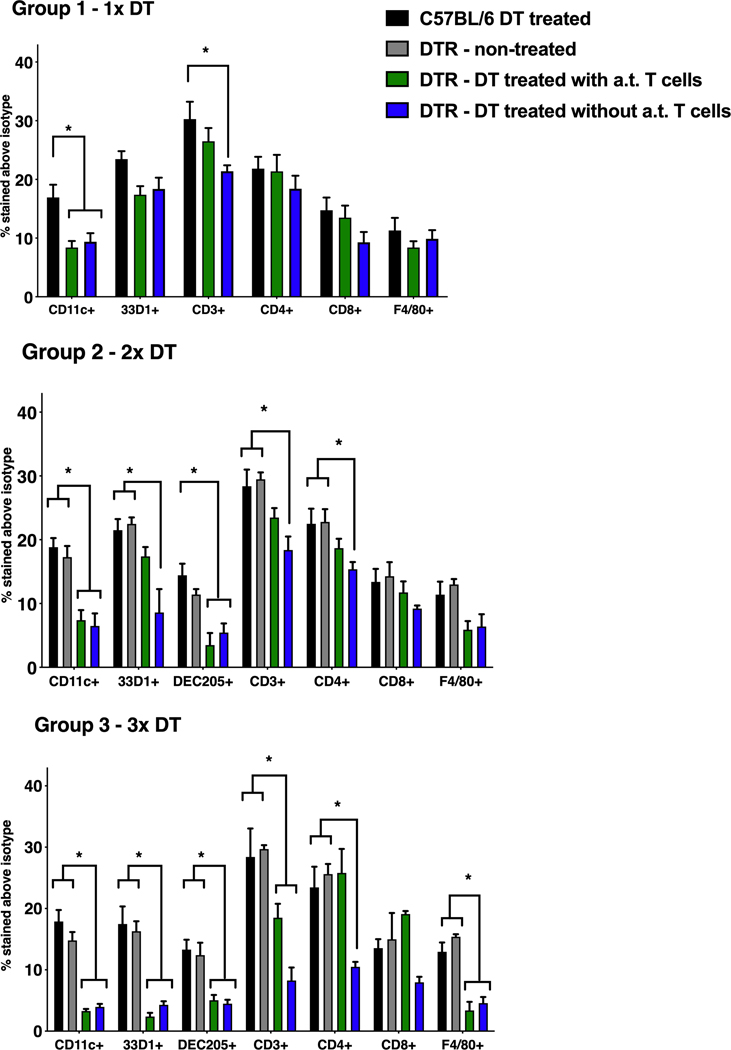
CD11c-DTR mice, with or without adoptively transferred (a.t.) OT-II T cells, were given either 1, 2 or 3 DT injections at 4ng/g body weight, spaced 2 days apart as per the timeline shown in Figure 1. (A) Group 1 received 1x DT injection, (B) group 2 – 2x DT injections and (C) group 3 – 3x DT injections. Statistical analysis was done using ANOVA followed by Sidak’s post-test and differences within 95% confidence intervals were accepted as significant (* = p < 0.05). Error bars represent standard error of mean values of 4 – 6 mice per group.
Figure 5. Proliferation profile of OVA-reactive CD4+ T cells in the absence of CD11c+ cells.

Wild-type C57BL/6 or CD11c-DTR mice with adoptively transferred CFSE-labeled OT-II T cells, with or without 3 DT injections given 2 days apart, were implanted with OVA-loaded PLGA scaffolds as per the experimental design shown in Figure 2. OVA in CFA served as the positive control. Splenocytes obtained from mice of Day 6 were stained with antibodies against CD3, CD4 and CD8 and assessed by flow cytometry. (A) Gated region (representative of all groups) marked in red (top) was analyzed for CFSE fluorescence (bottom). The histograms are representative of 6 mice per group. The grey bar is representative of CFSE-labeled T cells prior to the adoptive transfer injection. While the blue bar represents the CFSE+ cells obtained from splenocytes post adoptive transfer. (B) Statistical comparison of DT treatments on OVA-reactive CD4+ T-cell proliferation using ANOVA followed by Holm-Sidak’s post-test (* = p < 0.001). Error bars indicate standard error of mean of n=6 mice per treatment or control group. The inset image (right) shows a sample FI plot in which consecutive T-cell progeny is marked from P1-P6.
In vivo effect of PLGA with OVA on Antigen-Specific Clonal Expansion of T Cells in the Absence of Dendritic Cells in CD11c-DTR mice:
To determine the outcome of DT-mediated DC depletion in CD11c-DTR mice, the time-points for injections, implantation and analyses were setup according to Figure 2. On day 0, mice either received adoptive transfer of carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled OT-II or OT-I T cells [2×107 cells/ 300 μL of sterile phosphate buffered saline (PBS)] via I.V. tail or penile vein injection. On days 1, 3 and 5, a DT injection, diluted in sterile PBS at a volume of 100 μL, was given intraperitoneally (I.P.) at 4ng/g body weight. On day 2, an 8-mm diameter scaffold of 16 mg PLGA containing 12.8 mg OVA was implanted dorsally, subcutaneously. The control groups received an equivalent quantity of OVA in PBS emulsified in a 1:1 dilution with Complete Freund’s Adjuvant (CFA, positive control) via a subcutaneous injection as previously described7. On day 6, the mice were sacrificed; the T cells harvested from their spleens were subsequently analyzed by flow cytometry.
Figure 2. Schematic of DT injections and OVA-loaded PLGA scaffolds implantation.

Wild-type C57BL/6 or CD11c-DTR mice received adoptive transfer of CFSE-labeled OT-II T cells (2×107 cells/ 300 μL of sterile PBS) I.V. via tail vein or penile vein injection. Mice were then treated with 3 DT injections given I.P. (4 ng/g of body weight) on Days 1, 3 and 5, or left untreated. On Day 2, mice were subcutaneously given an OVA-loaded PLGA scaffold implant or OVA in CFA (positive control). On Day 6, mice were sacrificed and their splenocytes were assessed using flow cytometry.
CFSE Labeling and In Vivo Tracking of T-Cell Proliferation:
The method was adapted from a previously published protocol.32,33 Briefly, splenic T cells were harvested on day 6 (Figure 2) and labeled with antibodies against T-cell antigen CD3, CD4 and/or CD8. The cells were analyzed using flow cytometry. A single-cell suspension of 5×106 cells/ml CFSE-labeled T cells was prepared (un-proliferated control). ‘%Divided cells’ defined as the percentage of cells from the original sample and the fluorescence intensity (FI) histograms were gated into 6 FI-distinct populations that were compared across treatment groups by statistical analysis of 6 mice per group.
Flow Cytometry
Splenocytes obtained from mice were stained with antibodies against CD11c, 33D1, DEC205, CD3, CD4, CD8, F4/80. Dying cells were excluded from analysis using a propidium iodide stain. Cells with or without fluorescent labels were prepared in a single-cell suspension and analyzed with a flow cytometer (BD LSR II) and FlowJo software (TreeStar LLC). The CFSE fluorescence profiles of T cells, exposed to OVA either in the presence of PLGA or CFA as adjuvant, were allotted to 6 consecutive gates (P1 to P6). Proliferation profiles were obtained by observing the decreasing FIs, and these were compared across all treatment and control groups.
Statistical Analysis
Statistical analysis was done using ANOVA followed by Sidak’s or Holm-Sidak’s post-test. Differences within 95% confidence intervals were accepted as significant. The p-values of each experiment are denoted in the respective figure descriptions. Error bars represent standard error of mean values of 4 – 6 mice per group.
Results
Effect of Repeated DT Treatments on CD11c-DTR
Figure 1 describes the timeline for testing the effects of repeated DT injections in CD11c-DTR mice with or without adoptive transfer of T cells. Figure 2 shows the experimental set up for testing the effects of DC depletion on the antigen-specific T-cell proliferative response in the presence of the PLGA scaffold. The magnitude of the T-cell response in vivo depends on the duration of antigen availability and the sufficiency of well-equipped antigen presenting cells (e.g., levels of MHC or co-stimulatory molecule expression).34 Since a minimum of 24 – 48 h is necessary for T cells to undergo detectable levels of proliferation,35 cells were harvested ~96 h after antigen exposure to give sufficient time for antigen and T cell contact (Figure 2). A single DT injection depletes CD11c+ cells for up to 48 h after which the amount of circulating CD11c+ cells gradually increase.24 It was necessary to maintain DC depletion throughout the period of T-cell antigen contact; therefore, three consecutive injections of DT were administered spaced two days apart from each other.
Figure 3 shows that the mouse body weights 24 h after being treated with one, two or three DT injections did not undergo significant changes compared to the untreated control group. Also, the introduction of adoptively transferred OT-II T cells did not influence any changes in weight either. Body weight is a reliable indicator of immediate health status in animals.36 As the administration of three DT injections did not adversely affect CD11c-DTR health, it was deemed safe for use in subsequent experiments.
Along with assessing body weights, the effect of these repeated DT injections on the immune system of CD11c-DTR mice (with or without the adoptively transferred T cells) was determined by analyzing cell surface biomarkers on harvested splenocytes. Figure 4 shows that the total CD11c content was significantly lowered upon one or more DT doses and the magnitude of CD11c loss increased with each additional dose. Murine lymphoid-circulating conventional DCs are primarily composed of CD11c+ CD8+ and CD11c+ CD8– cells; the CD8+ subset is largely associated with the expression of DEC205 (CD205) while the CD8– subset expresses 33D1 (DCIR2).37–39 The decrease in CD11c was coupled with a significant loss of 33D1 (a DC marker) as well as DEC205, suggesting that different DC subsets were depleted in response to the DT injections. Importantly, the magnitude of the decrease in expression of these biomarkers in Group 3 (3x DT) indicated that the mice were, in part, losing immunocompetency with each repeated dose of DT. Contrary to previously published work40,41, a relative change in CD3, CD4 and F4/80 expression profiles was also observed in addition to the typical set of DC-associated biomarkers. The presence of adoptively transferred T cells prior to DT administration showed a noticeable effect on the surface biomarker profile especially during the early stages of DC depletion, i.e., for Groups 1 and 2. These results indicate that the three DT injections significantly lowered the number of CD11c+ cells, predominantly DCs, in CD11c-DTR mice during the 6-day period. Moreover, the presence of adoptively transferred T cells minimally aided in the general immunocompetency of the animals.
CD4+ T-Cell Response to Antigen-Loaded PLGA in the Absence of DCs
PLGA’s influence on the antigen-specific T-cell proliferation was assessed in the depleted DC environment. DT-treated CD11c-DTR mice, with adoptively transferred CFSE-labeled OVA-reactive OT-II cells, were given a dorsal subcutaneous implant of a PLGA scaffold co-delivering the model antigen OVA (Figure 2). The proliferation profiles of the T cells assessed on day 6 as an early surrogate read-out of the adjuvant effect of PLGA, depended on the presence of CD11c+ DCs. In a DC-intact environment with an implanted OVA-loaded PLGA scaffold, a dilution of CFSE fluorescence signal was observed with each subsequent cell division. Up to 6 consecutive FI peaks would be visible until the fluorescence of CFSE lowered to background levels (Figure 5).32,33 Whereas in DT-treated CD11c-DTR mice, the CFSE FI of CD3+ CD4+ CD8– splenocytes at the end of day 6 was nearly as high as the originally injected CFSE-labeled T cells for both OVA/PLGA and OVA/CFA administered mice (diminished proliferation was detected in either case). A high FI was also observed at early division (P1 and P2) for these cells while all other groups showed negligible FI. The DC-intact mice showed higher FI than DC-depleted mice in P4 (mid-stage), suggesting that DC-depleted mice underwent minimal or no mid-stage cell division or rather their cell division was still in the early phases. Despite allowing 4 days of antigen contact (from day 2 to day 6), there were only 1 or 2 visible peaks of FI from these DC-depleted mice. The adoptively transferred T cells likely underwent 1–2 rounds of cell division, indicating that the T-cell response was greatly diminished in the absence of sufficient DCs to effectively present the antigen to T cells for the effective proliferation. Collectively these results indicate that administration of DT to CD11c-DTR mice receiving OVA with PLGA or CFA led to an altered proliferation profile of OVA-reactive OT-II T cells. Since OT-II T cells are primarily CD4+, the significant loss of DCs in vivo likely diminished the otherwise strong antigen-specific MHC-II – CD4+ T cell antigen recognition involved in such models.
CD8+ T-Cell Response to Antigen-Loaded PLGA in the Absence of DCs
To assess the effect of PLGA on the cross-presentation of antigen to CD8+ T cells in a DC-depleted environment, a similar setup as before was used replacing OT-II T cells with OT-I T cells (Figure 2). On day 6, CD3+ CD8+ CD4– splenocytes from DT-treated CD11c-DTR mice showed an altered T-cell proliferation profile compared to control groups. The multiple short peaks shown for these mice in Figure 6 suggest that diminished of OT-I T-cell division occurred in response to OVA in PLGA or CFA. The peaks were visibly higher than that of their OT-II counterparts from Figure 5 (compare P1 and P2). In P3 (mid-stage), the CFSE intensity from DC-depleted mice was higher than in the controls, which is probably due to associated early-stage cell divisions. As before, control groups with intact DCs showed consecutive dilution of CFSE peaks suggesting normal proliferation. Conversely in P5 (late-stage), the CFSE signal for DT-treated CD11c-DTR mouse with OVA/CFA was significantly lower than that of the DC-intact controls. Together, these results indicate that the mechanism of cross presentation of OVA to OT-I CD8+ T cells in the presence of co-delivered PLGA was decreased when the DC numbers in vivo were greatly diminished.
Discussion
The PLGA-adjuvant effect was previously identified in mice in the context of co-delivered model antigen OVA, wherein the humoral OVA-specific immune response was enhanced in the presence of PLGA compared to adjuvant-free controls.7 This enhanced immune response was also observed when human or murine primary culture DCs underwent in vitro treatment with PLGA films. The resulting mature DCs had a stronger stimulus on antigen-specific T-cell responses.14 To confirm that these observations were a result of the biomaterial, it was required to examine PLGA’s effect on co-delivered antigen-specific T-cell proliferation in a mouse model with reliably depleted DCs. Since the antigen-specific T-cell proliferation depends on the presence of CD11c+ DC, by depleting these cells the extent of PLGA’s influence on an antigen induced immune response can be evaluated.
The ability of PLGA to enhance antigen-specific T-cell proliferation against the co-delivered model antigen OVA concomitant with DC depletion was assessed. The OVA-reactive T-cell proliferation model was used as an early time point read-out and is known to be necessary for subsequent humoral immune responses.
Antigen presentation by DCs is a crucial first step in the OVA-reactive immune response and is dependent on the state of DC phenotype.2 Antigen (OVA) processed and presented by DCs stimulate naïve or OVA-reactive T cells to clonally expand. Clonal expansion is another important step required to initiate strong T-cell responses. OVA-reactive T cells are expected to proliferate excessively in the presence of DCs and OVA-loaded PLGA scaffolds, the latter serving as a maturation stimulus. In contrast, T-cell proliferation is expected to diminish in the absence of DCs. Therefore, the extent of proliferation of OVA-reactive T cells in the context of PLGA was utilized as a measure consistent with its adjuvant effect.
The DC-depleted in vivo environment, used for the duration of the study, was obtained using the CD11c-DTR model. As a single DT injection depletes CD11c+ cells for up to 48 h,24 three DT injections were administered every other day to maintain a DC-depleted environment for 6 days. By pre-delivering CFSE-labeled OVA-reactive T cells into these mice, any severe immunodeficiencies caused by repetitive DT doses were circumvented. An OVA-loaded PLGA scaffold was implanted 24 h after the administration of the first DT dose since CD11c+ cell depletion is known to be maximal around this time-point.40 Analysis of the surface biomarkers and CFSE fluorescence of harvested splenocytes from DT-treated CD11c-DTR mice (i.e., DC-depleted environment), showed a significantly lowered antigen-specific CD4+ T-cell proliferative response and a slightly less affected CD8+ T-cell proliferative response. Classic presentation of exogenous antigen through MHC-II molecules as well as cross-presentation of antigen through MHC-I mechanisms were likely diminished due to the DC-deficient environment. All things considered, DCs are the main in vivo mediators of PLGA’s enhancing effect on the antigen-specific proliferative T-cell response to the co-delivered model antigen OVA. Although the scheme of T-cell response was significantly decreased in this study, some proliferation of OVA-reactive T cells is observed indicating that complete elimination of in vivo antigen presentation with CD11c had not taken place.
A small percentage of CD11c+ cells remained even after three consecutive injections — suggesting that some conventional DCs were still in circulation (Figure 4) and were likely unaffected by DT treatments. There are documented inherent limitations of the CD11c-DTR mouse model that restrict its use beyond a short testing period (1 week)24. Repeated injections of DT over several weeks are known to cause lethality of the CD11c-DTR mice and require the use of bone marrow chimeras to reverse DT-induced immunodeficiency. An alternative model known as CD11c-DOG may be used. In this model the simian DTR of CD11c-DTR model is replaced with a human DTR. This change eliminates lethality of the repeated DT delivery in study mice even when administered repeatedly for over two weeks. However, both models are capable of achieving only incomplete DC depletion and improved models of DC depletion are yet to be developed. To truly assess the dependence of PLGA on DCs in the antibody response, a model that allows for DC depletion over several weeks would be required. For this study, the DC depletion was only required for 6 days. Therefore, the commercially available CD11c-DTR mouse model was chosen due to its known tolerance to repeated DT treatments.
Another possible reason for the incomplete elimination of antigen presentation (i.e., low levels of T-cell proliferation in DT-treated CD11c-DTR mice) is the presence of macrophages and antigen-presenting B-cells. Although previously unreported in literature,24,40,41 our results showed a decrease in the expression of surface CD3+, CD4+ and F4/80 cells in some treatment groups. It is not entirely clear why DT treatments would induce a loss in the CD3+ cell population. There is some recent evidence that CD11c and CD3 are co-expressed on T cells in mouse spleens.42 Given the potent adjuvant capabilities of CFA or PLGA, it is expected this loss only minimally impacted the proliferations of adoptively transferred OT-II/OT-I cell proliferation.
A decrease in CD4+ cells was seen in mice that received ≥ 2 DT injections without adoptively transferred OT-II CD4+ T cells (Figure 4). This may have been caused by a depletion in the CD11c+ CD4+ DCs, that are known to exist in germinal centers such as the spleen43. On the other hand, mice that received the adoptively transferred OT-II T cells after ≥2 DT injections, did not show a significant decrease in the CD4+ cells. This was likely because the OT-II T cells increased the total number of available splenic CD4+ cells, such that CD11c+ CD4+ cells only formed a small percentage of them. While a decrease in the proliferative behaviour of CD4+ T cells is expected after repeated DT injections (shown in Figure 5), it is unlikely that this was because of a depletion of CD4+ T cells in the spleen. Hence, this response would not have affected the outcomes described herein.
F4/80 loss in cell surface biomarkers is associated with the loss of red pulp splenic macrophages.44 Red pulp macrophages, primarily CD11c+ F4/80hi, are known to maintain blood homeostasis by actively phagocytosing erythrocytes and blood-borne particulates.45 While they do not seem to play a role in CD4+ or CD8+ T-cell proliferation, recent evidence indicates that they may induce differentiation of CD4+ T cells to regulatory T cells as part of their homeostasis maintenance.46 Relatedly, histological analysis on the spleens of DT-treated CD11c-DTR mice in another study indicated a loss of marginal zone macrophages and metallophillic macrophages that are predominantly F4/80–.41 The loss of F4/80+ cells in our results seems to be associated with an overall loss of splenic macrophages. Although DT treatments depleted cells other than CD11c+ cells, for the specific goal of identifying the effect of PLGA on the adaptive immune response in the absence of antigen presentation, the CD11c-DTR model was still suitable for use.
To further abate the antigen presentation system, CD11c-DTR-based DC depletion can potentially be combined with in vivo ablation of macrophages using clodronate liposome microspheres.47 It may be valuable to understand the effect of PLGA on the host immune response in the absence of antigen presentation by DCs and macrophages (or macrophages alone for comparison).
Using an adoptive transfer of CFSE-labeled OT-II or OT-I T cells allowed us to determine the antigen-specific immune response within a short time frame (1 week). Although other studies using this type of analysis48 have shown successive T-cell divisions on FI plots with distinct consecutive peaks, we observed a collected pooling of FI in our DC-intact control groups (Figures 5, 6). The lack of distinct peaks may have been caused by excessive T-cell division in response to OVA delivered with potent adjuvant effects of CFA or PLGA. Another cause for the lack of distinct peaks is that CFSE and CFDA-SE dyes are vulnerable to photobleaching49 that may have occurred during adoptive transfer or sample processing. To ensure that the issue was not due to OVA insensitivity, we tested the proliferation of OT-II T cells in vitro in the presence of the OVA peptide (323–339) in a light-protected environment which showed successive distinct FI peaks (Figure S1). No photobleaching issues were seen in the in vitro model further suggesting that photobleaching may have occurred during adoptive transfer in the in vivo model. Additionally, to confirm that the adoptive transfer procedure itself was being done correctly, naïve T cells (pan CD3+ T cells from C57BL/6) were labeled with CFSE and injected I.V. into wild-type mice. A portion of the splenocytes harvested after 24 h showed distinct CFSE fluorescence corresponding to the injected cells (Figure S2, blue peak to the right). The remaining splenocytes were unlabeled and did not exhibit any fluorescence (blue peak to the left.) Both these supporting experiments confirmed that the in vivo proliferation tracking of CFSE-labeled OVA-reactive T cells was functional.
The adjuvant effect of PLGA has also been examined in tissue engineering.7,13,50 Delivery of PLGA microparticles (MPs) or scaffolds by injection or implantation, respectively, with a model shed antigen has resulted in increased production of antigen-specific antibody.20 This increased humoral response appeared to be dependent on the form of PLGA.19 Furthermore, the humoral response was unique for the material in use. PLGA and not agarose induced an increased response to the model shed antigen7, as predicted based on PLGA’s in vitro effects on DC maturation.14
PLGA’s interaction with the immune system has also been characterized along with other materials such as agarose, alginate, chitosan, etc. PLGA-induced DC maturation is similar to that of standard TLR agonist (like LPS), resulting in a phenotype associated with promoting antigen-specific adaptive immune response. In contrast, agarose maintains DCs in an immature phenotype,14,51 which are capable of priming co-cultured autologous T cells into regulatory T cells.52 Furthermore, the possibility that activation is due to a non-specific antigen depot effect is unlikely for two reasons. First, PLGA but not agarose scaffold of similar mass, dimensions and antigen load, resulted in high levels of antibody production against co-delivered OVA.7 Second, as shown here, the OVA-specific T-cell proliferation was greatly diminished in CD11c+ cells despite the presence of PLGA.
PLGA’s adjuvant effect is being studied for both vaccine delivery and tissue engineering applications in preclinical and clinical studies. In one application an enhanced immune response is desired; however, in the other an enhanced immune response is less desirable. Another application is in mimicking clinically relevant autoimmune diseases. For example, the model antigen can be replaced with a Multiple Sclerosis-relevant antigen, Myelin oligodendrocyte glycoprotein (MOG) peptide and its cognate 2D2 T cells for tracking induced extent of proliferation. Verification of the observed differential adjuvant effect (mirroring the in vitro effects on DC maturation) for antigen delivered with agarose scaffolds11 would also be of interest.
The evidence presented in this study suggests a complex interaction between biomaterials and immune cells. For future development of enhanced immunotherapies, a deeper understanding of biomaterials’ immunomodulatory ability and their effects on DCs is needed and important.
Conclusion
PLGA facilitates DCs to induce a proliferative antigen-specific T-cell response to co-delivered antigen in vivo. This is an early precursor to the previously seen PLGA adjuvant effect. This effect is, at least in part, due to the influence of PLGA on DC phenotype and the subsequent antigen presentation ability. In the absence of CD11c+ DCs, PLGA failed to induce a strong antigen-specific CD4+ T-cell response. Further, it is shown that PLGA may play a role in enhancing the antigen cross presentation in DCs. In conclusion, biomaterials such as PLGA, possess immunomodulatory capabilities in vivo due to their interaction with DCs and can be used for therapeutic immune outcomes requiring activated DC phenotypes.
Supplementary Material
Acknowledgements
This research was funded by the Georgia Tech/Emory Center (GTEC) for Engineering of Living Tissues: Individual Investigator-Initiated Seed Grant in Regenerative Medicine and the National Institutes of Health (NIBIB/NIAID) 1R21EB019166–01A1. The authors would also like to acknowledge undergraduate researchers Gopi Patel and Gargi Mukherjee for their technical assistance during this work.
References
- (1).Banchereau J; Briere F; Caux C; Davoust J; Lebecque S; Liu YJ; Pulendran B; Palucka K. Immunobiology of Dendritic Cells. Annual Review of Immunology. Annu Rev Immunol 2000, pp 767–811. 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- (2).Banchereau J; Steinman RM Dendritic Cells and the Control of Immunity. Nature. Nature March 19, 1998, pp 245–252. 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- (3).Kapoor DN; Bhatia A; Kaur R; Sharma R; Kaur G; Dhawan S. PLGA: A Unique Polymer for Drug Delivery. Therapeutic Delivery. Future Science Ltd; January 1, 2015, pp 41–58. 10.4155/tde.14.91. [DOI] [PubMed] [Google Scholar]
- (4).Blasi P. Poly(Lactic Acid)/Poly(Lactic-Co-Glycolic Acid)-Based Microparticles: An Overview. Journal of Pharmaceutical Investigation. Springer; Netherlands: July 1, 2019, pp 337–346. 10.1007/s40005-019-00453-z. [DOI] [Google Scholar]
- (5).Search of: PLGA | Recruiting Studies - List Results -ClinicalTrials.gov https://clinicaltrials.gov/ct2/results?term=PLGA&Search=Apply&recrs=a&age_v=&gndr=&type=&rslt= (accessed Nov 1, 2020). [Google Scholar]
- (6).Pang J; Luan Y; Li F; Cai X; Du J; Li Z. Ibuprofen-Loaded Poly(Lactic-Co-Glycolic Acid) Films for Controlled Drug Release. International journal of nanomedicine 2011, 6, 659–665. 10.2147/ijn.s17011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Norton LW; Park J; Babensee JE Biomaterial Adjuvant Effect Is Attenuated by Anti-Inflammatory Drug Delivery or Material Selection. Journal of Controlled Release 2010, 146 (3), 341–348. 10.1016/j.jconrel.2010.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ali OA; Huebsch N; Cao L; Dranoff G; Mooney DJ Infection-Mimicking Materials to Program Dendritic Cells in Situ. Nature Materials 2009, 8 (2), 151–158. 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gross BP; Wongrakpanich A; Francis MB; Salem AK; Norian LA A Therapeutic Microparticle-Based Tumor Lysate Vaccine Reduces Spontaneous Metastases in Murine Breast Cancer. AAPS Journal 2014, 16 (6), 1194–1203. 10.1208/s12248-014-9662-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Householder KT; Diperna DM; Chung EP; Wohlleb GM; Dhruv HD; Berens ME; Sirianni RW Intravenous Delivery of Camptothecin-Loaded PLGA Nanoparticles for the Treatment of Intracranial Glioma. International Journal of Pharmaceutics 2015, 479 (2), 374–380. 10.1016/j.ijpharm.2015.01.002. [DOI] [PubMed] [Google Scholar]
- (11).Srinivasan S; Babensee JE Controlled Delivery of Immunomodulators from a Biomaterial Scaffold Niche to Induce a Tolerogenic Phenotype in Human Dendritic Cells. ACS Biomaterials Science and Engineering 2020, 6 (7), 4062–4076. 10.1021/acsbiomaterials.0c00439. [DOI] [PubMed] [Google Scholar]
- (12).Srinivasan S; Babensee J. Host Response to Biomaterials. In Scaffolds for Tissue Engineering; Pan Stanford Publishing, 2014; pp 115–171. 10.1201/b15649-6. [DOI] [Google Scholar]
- (13).Yoshida M; Babensee JE Poly(Lactic-Co-Glycolic Acid) Enhances Maturation of Human Monocyte-Derived Dendritic Cells. Journal of Biomedical Materials Research - Part A 2004, 71 (1), 45–54. 10.1002/jbm.a.30131. [DOI] [PubMed] [Google Scholar]
- (14).Yoshida M; Babensee JE Differential Effects of Agarose and Poly(Lactic-Co-Glycolic Acid) on Dendritic Cell Maturation. Journal of Biomedical Materials Research - Part A 2006, 79 (2), 393–408. 10.1002/jbm.a.30798. [DOI] [PubMed] [Google Scholar]
- (15).Yoshida M; Mata J; Babensee JE Effect of Poly(Lactic-Co-Glycolic Acid) Contact on Maturation of Murine Bone Marrow-Derived Dendritic Cells. Journal of Biomedical Materials Research Part A 2007, 80A (1), 7–12. 10.1002/jbm.a.30832. [DOI] [PubMed] [Google Scholar]
- (16).Allen RP; Bolandparvaz A; Ma JA; Manickam VA; Lewis JS Latent, Immunosuppressive Nature of Poly(Lactic- Co -Glycolic Acid) Microparticles. ACS Biomaterials Science and Engineering 2018, 4 (3), 900–918. 10.1021/acsbiomaterials.7b00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hunter Z; McCarthy DP; Yap WT; Harp CT; Getts DR; Shea LD; Miller SD A Biodegradable Nanoparticle Platform for the Induction of Antigen-Specific Immune Tolerance for Treatment of Autoimmune Disease. ACS Nano 2014, 8 (3), 2148–2160. 10.1021/nn405033r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Fischer S; Uetz-von Allmen E; Waeckerle-Men Y; Groettrup M; Merkle HP; Gander B. The Preservation of Phenotype and Functionality of Dendritic Cells upon Phagocytosis of Polyelectrolyte-Coated PLGA Microparticles. Biomaterials 2007, 28 (6), 994–1004. 10.1016/j.biomaterials.2006.10.034. [DOI] [PubMed] [Google Scholar]
- (19).Bennewitz NL; Babensee JE The Effect of the Physical Form of Poly(Lactic-Co-Glycolic Acid) Carriers on the Humoral Immune Response to Co-Delivered Antigen. Biomaterials 2005, 26 (16), 2991–2999. 10.1016/j.biomaterials.2004.08.023. [DOI] [PubMed] [Google Scholar]
- (20).Matzelle MM; Babensee JE Humoral Immune Responses to Model Antigen Co-Delivered with Biomaterials Used in Tissue Engineering. Biomaterials 2004, 25 (2), 295–304. 10.1016/S0142-9612(03)00531-3. [DOI] [PubMed] [Google Scholar]
- (21).Keselowsky BG; Lewis JS Dendritic Cells in the Host Response to Implanted Materials. Seminars in Immunology. Academic Press; February 1, 2017, pp 33–40. 10.1016/j.smim.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Koerner J; Horvath D; Groettrup M. Harnessing Dendritic Cells for Poly (D,L-Lactide-Co-Glycolide) Microspheres (PLGA MS)-Mediated Anti-Tumor Therapy. Frontiers in Immunology. Frontiers Media S.A. 2019, p 707. 10.3389/fimmu.2019.00707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wu J; Wu H; An J; Ballantyne CM; Cyster JG Critical Role of Integrin CD11c in Splenic Dendritic Cell Capture of Missing-Self CD47 Cells to Induce Adaptive Immunity. Proceedings of the National Academy of Sciences of the United States of America 2018, 115 (26), 6786–6791. 10.1073/pnas.1805542115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Jung S; Unutmaz D; Wong P; Sano GI; de Los Santos K; Sparwasser T; Wu S; Vuthoori S; Ko K; Zavala F; Pamer EG; Littman DR; Lang RA In Vivo Depletion of CD11c+ Dendritic Cells Abrogates Priming of CD8+ T Cells by Exogenous Cell-Associated Antigens. Immunity 2002, 17 (2), 211–220. 10.1016/S1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lukácsi S; Nagy-Baló Z; Erdei A; Sándor N; Bajtay Z. The Role of CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in Complement-Mediated Phagocytosis and Podosome Formation by Human Phagocytes. Immunology Letters 2017, 189, 64–72. 10.1016/j.imlet.2017.05.014. [DOI] [PubMed] [Google Scholar]
- (26).Du SW; Arkatkar T; al Qureshah F; Jacobs HM; Thouvenel CD; Chiang K; Largent AD; Li Q-Z; Hou B; Rawlings DJ; Jackson SW Functional Characterization of CD11c + Age-Associated B Cells as Memory B Cells. The Journal of Immunology 2019, 203 (11), 2817–2826. 10.4049/jimmunol.1900404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Qualai J; Li L-X; Cantero J; Tarrats A; Fernández MA; Sumoy L; Rodolosse A; McSorley SJ; Genescà M. Expression of CD11c Is Associated with Unconventional Activated T Cell Subsets with High Migratory Potential. PLOS ONE 2016, 11 (4), e0154253. 10.1371/journal.pone.0154253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Vinay DS; Kim CH; Choi BK; Kwon BS Origins and Functional Basis of Regulatory CD11c+CD8+ T Cells. European Journal of Immunology 2009, 39 (6), 1552–1563. 10.1002/eji.200839057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ueno H; Schmitt N; Palucka AK; Banchereau J. Dendritic Cells and Humoral Immunity in Humans. Immunology and Cell Biology. NIH Public; Access May 2010, pp 376–380. 10.1038/icb.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Clarke S. Rm K; Barnden M; Kurts C; Carbone FR; Miller JF; Heath WR Characterization of the Ovalbumin-Specific TCR Transgenic Line OT-I: MHC Elements for Positive and Negative Selection. Immunology and Cell Biology 2000, 78 (2), 110–117. 10.1046/j.1440-1711.2000.00889.x. [DOI] [PubMed] [Google Scholar]
- (31).Barnden MJ; Allison J; Heath WR; Carbone FR Defective TCR Expression in Transgenic Mice Constructed Using CDNA- Based α- and β-Chain Genes under the Control of Heterologous Regulatory Elements. Immunology and Cell Biology 1998, 76 (1), 34–40. 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- (32).Parish CR; Glidden MH; Quah BJC; Warren HS Use of the Intracellular Fluorescent Dye CFSE to Monitor Lymphocyte Migration and Proliferation. Current Protocols in Immunology. Curr Protoc Immunol 2009. 10.1002/0471142735.im0409s84. [DOI] [PubMed] [Google Scholar]
- (33).Quah BJC; Warren HS; Parish CR Monitoring Lymphocyte Proliferation in Vitro and in Vivo with the Intracellular Fluorescent Dye Carboxyfluorescein Diacetate Succinimidyl Ester. Nature Protocols 2007, 2 (9), 2049–2056. 10.1038/nprot.2007.296. [DOI] [PubMed] [Google Scholar]
- (34).Blair DA; Turner DL; Bose TO; Pham Q-M; Bouchard KR; Williams KJ; McAleer JP; Cauley LS; Vella AT; Lefrançois L. Duration of Antigen Availability Influences the Expansion and Memory Differentiation of T Cells. The Journal of Immunology 2011, 187 (5), 2310–2321. 10.4049/jimmunol.1100363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Serre K; Mohr E; Gaspal F; Lane PJL; Bird R; Cunningham AF; MacLennan ICM IL-4 Directs Both CD4 and CD8 T Cells to Produce Th2 Cytokines in Vitro, but Only CD4 T Cells Produce These Cytokines in Response to Alum-Precipitated Protein in Vivo. Molecular Immunology 2010, 47 (10), 1914–1922. 10.1016/j.molimm.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ullman-Culleré MH; Foltz CJ Body Condition Scoring: A Rapid and Accurate Method for Assessing Health Status in Mice. Laboratory Animal Science 1999, 49 (3), 319–323. [PubMed] [Google Scholar]
- (37).Crowley M; Inaba K; Witmer-Pack M; Steinman RM The Cell Surface of Mouse Dendritic Cells: FACS Analyses of Dendritic Cells from Different Tissues Including Thymus. Cellular Immunology 1989, 118 (1), 108–125. 10.1016/0008-8749(89)90361-4. [DOI] [PubMed] [Google Scholar]
- (38).Dudziak D; Kamphorst AO; Heidkamp GF; Buchholz VR; Trumpfheller C; Yamazaki S; Cheong C; Liu K; Lee HW; Chae GP; Steinman RM; Nussenzweig MC Differential Antigen Processing by Dendritic Cell Subsets in Vivo. Science 2007, 315 (5808), 107–111. 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- (39).Jiang W; Swiggard WJ; Heufler C; Peng M; Mirza A; Steinman RM; Nussenzweig MC The Receptor DEC-205 Expressed by Dendritic Cells and Thymic Epithelial Cells Is Involved in Antigen Processing. Nature. Nature May 11, 1995, pp 151–155. 10.1038/375151a0. [DOI] [PubMed] [Google Scholar]
- (40).Bar-On L; Jung S. Defining in Vivo Dendritic Cell Functions Using CD11c-DTR Transgenic Mice. Methods in molecular biology (Clifton, N.J.) 2010, 595, 429–442. 10.1007/978-1-60761-421-0_28. [DOI] [PubMed] [Google Scholar]
- (41).Probst HC; Tschannen K; Odermatt B; Schwendener R; Zinkernagel RM; van den Broek M. Histological Analysis of CD11c-DTR/GFP Mice after in Vivo Depletion of Dendritic Cells. Clinical and Experimental Immunology 2005, 141 (3), 398–404. 10.1111/j.1365-2249.2005.02868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Rostamzadeh D; Haghshenas MR; Daryanoosh F; Samadi M; Hosseini A; Ghaderi A; Mojtahedi Z; Babaloo Z. Altered Frequency of CD8+ CD11c+ T Cells and Expression of Immunosuppressive Molecules in Lymphoid Organs of Mouse Model of Colorectal Cancer. Journal of Cellular Physiology 2019, 234 (7), 11986–11998. 10.1002/jcp.27856. [DOI] [PubMed] [Google Scholar]
- (43).Grouard G; Durand I; Fligueira L; Banchereau J; Liu YJ Dendritic Cells Capable of Stimulating T Cells in Germinal Centres. Nature 1996, 384 (6607), 364–367. 10.1038/384364a0. [DOI] [PubMed] [Google Scholar]
- (44).Hey YY; O’Neill HC Murine Spleen Contains a Diversity of Myeloid and Dendritic Cells Distinct in Antigen Presenting Function. Journal of Cellular and Molecular Medicine 2012, 16 (11), 2611–2619. 10.1111/j.1582-4934.2012.01608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kurotaki D; Uede T; Tamura T. Functions and Development of Red Pulp Macrophages. Microbiology and Immunology 2015, 59 (2), 55–62. 10.1111/1348-0421.12228. [DOI] [PubMed] [Google Scholar]
- (46).Kurotaki D; Kon S; Bae K; Ito K; Matsui Y; Nakayama Y; Kanayama M; Kimura C; Narita Y; Nishimura T; Iwabuchi K; Mack M; van Rooijen N; Sakaguchi S; Uede T; Morimoto J. CSF-1–Dependent Red Pulp Macrophages Regulate CD4 T Cell Responses. The Journal of Immunology 2011, 186 (4), 2229–2237. 10.4049/jimmunol.1001345. [DOI] [PubMed] [Google Scholar]
- (47).Zeisberger SM; Odermatt B; Marty C; Zehnder-Fjällman AHM; Ballmer-Hofer K; Schwendener RA Clodronate-Liposome-Mediated Depletion of Tumour-Associated Macrophages: A New and Highly Effective Antiangiogenic Therapy Approach. British Journal of Cancer 2006, 95 (3), 272–281. 10.1038/sj.bjc.6603240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Bertholet S; Debrabant A; Afrin F; Caler E; Mendez S; Tabbara KS; Belkaid Y; Sacks DL Antigen Requirements for Efficient Priming of CD8+ T Cells by Leishmania Major-Infected Dendritic Cells. Infection and Immunity 2005, 73 (10), 6620–6628. 10.1128/IAI.73.10.6620-6628.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Progatzky F; Dallman MJ; Celso C. lo. From Seeing to Believing: Labelling Strategies for in Vivo Cell-Tracking Experiments. Interface Focus. Royal Society June 6, 2013. 10.1098/rsfs.2013.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Babensee JE; Paranjpe A. Differential Levels of Dendritic Cell Maturation on Different Biomaterials Used in Combination Products. In Journal of Biomedical Materials Research - Part A; 2005; Vol. 74, pp 503–510. 10.1002/jbm.a.30429. [DOI] [PubMed] [Google Scholar]
- (51).Park J; Babensee JE Differential Functional Effects of Biomaterials on Dendritic Cell Maturation. Acta Biomaterialia 2012, 8 (10), 3606–3617. 10.1016/j.actbio.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Park J; Gerber MH; Babensee JE Phenotype and Polarization of Autologous T Cells by Biomaterial-Treated Dendritic Cells. Journal of Biomedical Materials Research - Part A 2015, 103 (1), 170–184. 10.1002/jbm.a.35150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.