

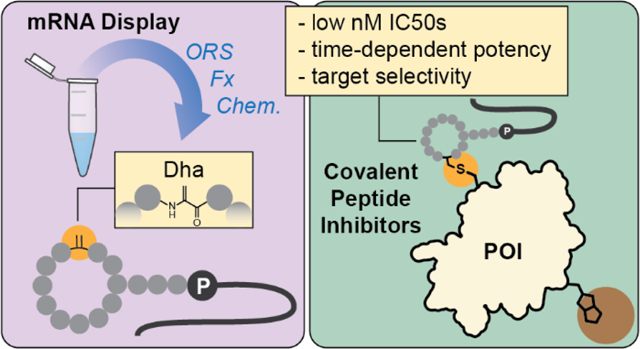
Abstract
Peptides have historically been underutilized for covalent inhibitor discovery despite their unique abilities to interact with protein surfaces and interfaces. This is in part due to a lack of methods for screening and identification of covalent peptide ligands. Here, we report a method to identify covalent cyclic peptide inhibitors in mRNA display. We combine co- and post-translational library diversification strategies to create cyclic libraries with reactive dehydroalanines (Dhas), which we employ in selections against two model targets. The most potent hits exhibit low nanomolar inhibitory activities and disrupt known protein-protein interactions of their selected targets. Overall, we establish Dhas as electrophiles for covalent inhibition and showcase how separate library diversification methods can work synergistically to dispose mRNA display to novel applications like covalent inhibitor discovery.
Graphical Abstract
Covalent inhibition has regained favor as a drug discovery strategy in recent years, as exemplified by a substantial increase in FDA approved covalent drugs in the last three decades.1,2 Many covalent inhibitors to date have relied on structural information to retrofit electrophiles onto reversible binders, optimize naturally occurring covalent ligands, or design covalent substrate analogs of a given protein target.2–5 However, when such information is not available, de novo covalent inhibitors must be discovered. In these endeavors, high throughput screening of electrophilic libraries can enable initial hit discovery. For instance, a small molecule acrylamide library screened against the G12C mutant of KRAS, which otherwise lacks distinct pockets for reversible ligand engagement, led to the discovery of a previously unknown binding site6—exploitation of which enabled the development of now FDA-approved Sotorasib.7,8 This success showcases the utility of electrophilic libraries for covalent drug development, and applications of this approach have continued to expand in recent years.9–14
Generation of electrophilic genetically encoded libraries (GELs), such as those in phage and mRNA display,15 could be significantly useful for covalent inhibitor discovery. GELs can be exceedingly large (>1013 molecules), are synthesized quickly and accurately by the ribosome, and are increasingly approaching the chemical space of natural products.16–18 Resulting peptide inhibitors have greater propensity to occupy shallower binding pockets,19,20 disrupt protein-protein interactions,21–23 and have high selectivity for their protein target;24–26 the last characteristic may be particularly desirable in covalent drug discovery, where non-specific reactivity is a unique concern. Despite these advantages, there are relatively few examples of electrophilic GELs, with a handful of reports in phage display,27–29 and only cofactor-targeted examples in mRNA display.30
Dehydroalanines (Dhas) are biologically occurring electrophiles31,32 that have previously been incorporated in mRNA display for peptide cyclization, side chain functionalization, or heterocycle formation.18,33,34 We anticipated that Dhas might also serve as competent cysteine electrophiles for covalent inhibitor discovery. Dhas can be accessed through protected precursors like phenylselenocysteine (PhSec), which can be revealed after peptide cyclization to allow compatibility with virtually all cysteine-based cyclization methods common to mRNA display. Thus, we envisioned a route to cyclic electrophilic mRNA display libraries involving 1) translation of PhSec with an orthogonal aminoacyl-tRNA synthetase (ORS),35 2) library cyclization through the robust Flexizyme-mediated thioether linkage,36,37 and 3) post-translational reveal of Dhas through oxidative elimination (Figure 1A; Figures S1-2). This elimination likely also results in oxidation of the macrocyclic linkage, however, herein we assume this static position has minimal impact on affinity and elect to synthesize hits as thioethers for simplicity. Importantly, this strategy allows positional variation and overrepresentation of the electrophile through strategic design of the mRNA library (Figure 1B), which contrasts prior phage display approaches where the chemistry restricts electrophile incorporation to a single position immediately flanked by cysteines. Lastly, we sought to denature the selection targets with guanidine after incubation with the electrophilic libraries to encourage recovery of covalent ligands, as previously done in phage display.28 Herein, we report the realization of this selection strategy against two model targets, calcium and integrin-binding protein 1 (CIB1) and melanoma-associated antigen 4 (MAGE-A4).
Figure 1.

A) Strategy for Dha incorporation in mRNA display. B) Adjustments to standard selection process to facilitate identification of covalent binders.
We first confirmed the compatibility of 5 M guanidinium with cDNA amplification and the biotin-streptavidin interaction, then showed that guanidine could ablate the recovery of a previously selected non-covalent CIB1 inhibitor in mRNA display (Figure S3).38 CIB1 is a well-behaved protein with two surface-exposed cysteines,39 making it a desirable first target for covalent inhibitor discovery. We designed a 9-mer mRNA library with a cysteine-deficient NWW randomization to prevent the potential consumption of Dhas in additional macrocycles. Notably, this randomization also removes several amino acid residues and the TAG codon, resulting in a reduced theoretical library diversity of ~2E8 molecules. The TAG codon was scanned across all positions of the randomized library (Figure 1B), resulting in library members each with one Dha.
We conducted selections with denaturation against CIB1 and analyzed results by next generation sequencing (NGS). Significant convergence appeared in round four, with one sequence comprising >20% of NGS reads (Figures 2A-B). We synthesized this peptide as CCP1 (Figure 2C) and tested its ability to covalently modify CIB1 via MALDI-MS. Pleasingly, we observed a dose-dependent shift in CIB1 mass that roughly corresponds to CCP1 addition (Figure 2D). We did not observe CIB1 modification with a non-specific electrophile, iodoacetamide (Figure S4), or when the Dha of CCP1 was mutated to non-reactive L- or D-Ala. These results suggest specificity toward CIB1 and implicate the Dha in covalent modification. Trypsin-MALDI-MS of the CIB1-CCP1 complex suggested that CCP1 modifies a protein fragment containing Cys134 (Figure S4), which was confirmed when CCP1 did not modify a CIB1 C134A mutant (Figure 2D).
Figure 2.

A) Top families from round 4 of Dha selections with guanidine denaturation against CIB1, annotated by similar (blue) and significant (red) mutations from top sequence in each family. B) NGS % composition of CCP1 tracked over selection rounds. C) Structure of CCP1 with Dha highlighted. D) MALDI-MS of CIB1 when incubated with CCP1, its L/D-Ala mutants, or CCP1 + CIB1 C134A mutant. E) Time-dependent TR-FRET between CIB1 and AF647-phage peptide in the presence of CCP1, its L/D-Ala mutants, and non-covalent CIB1 inhibitors UNC10245131 and UNC10245231. F) Calculated IC50s from TR-FRET in part E. G) Western blot of biotin-CCP1 titration in MDA-MB-453 cell lysates spiked with CIB1 (6 μM). Detection enabled by neutravidin-HRP.
We next sought to assess whether CCP1 can inhibit interactions in the C-terminal binding pocket of CIB1.40 To test this activity, we used a previously developed TR-FRET assay between CIB1 and a known phage display peptide.41 In this assay, CCP1 shows modest inhibitory activity, with an IC50 of ~0.5 μM after 1 hour incubation (Figure 2E-F; Figure S5). In contrast, non-covalent cyclic peptide inhibitors UNC10245131 and UNC1024523138 display low nanomolar IC50s at this time point. The potency of CCP1 increases significantly over time—a hallmark of covalent inhibitors—with IC50 improving almost ten-fold after 24 hours. In contrast, the non-covalent inhibitors largely sustain potency on longer incubation. L- and D-Ala mutants of CCP1 do not display any inhibition in this assay, and CCP1 is inactive against the CIB1 C134A mutant (Figure S5). Notably, C134 is located ~7Å from the surface of the phage peptide binding pocket (Figure S6).41 Thus, lack of inhibition against C134A suggests that CIB1 undergoes a conformational change upon reaction with CCP1 that prevents access to the phage peptide binding site, or that binding is lost in absence of the cysteine target of CCP1. We also evaluated the ability of CCP1 to label CIB1 when spiked into cellular lysates (MDA-MB-453); CCP1 only labels CIB1 with no additional off-target modifications, demonstrating its selectivity toward CIB1 in a complex environment (Figure 2G; Figure S7).
To demonstrate the robustness of this covalent selection strategy, we performed a second selection against another protein target, MAGE-A4. In light of the CIB1 selection, we made adjustments to the selection protocol: 1) we extended incubation times of the first two selection rounds because these conditions appear to improve reaction between CCP1 and CIB1 (Figure S8); 2) we employed a more comprehensive NNK library randomization because guanidine denaturation appears sufficient to prevent enrichment of non-covalent binders. We also hypothesized that the lower NWW library diversity might be partly responsible for the moderate activity of CCP1. With these changes, parallel selections with (+G) or without (-G) guanidine denaturation were performed against MAGE-A4. After four rounds, NGS data revealed convergence upon several families (Figure 3A). The top families in each selection differed significantly, and we did not observe enrichment of any multi-Cys families in the +G NGS data. The most enriched -G family, which appears similar to a previous non-covalent selection against MAGE-A4,24 was absent from the top 200 sequences of the +G selections, demonstrating the ability of guanidine to hinder the emergence of non-covalent ligands by >100-fold.
Figure 3.

A) Top families from +G (round 4) or -G (round 3) Dha selections, annotated by similar (blue) and significant (red) mutations from top sequence in each family. B) Structures of the selected peptides from MAGE-A4 +G selections with Dhas highlighted and accompanying MALDI-MS spectra of MAGE-A4 when incubated with each peptide, its L/D-Ala mutants, or when peptides are incubated with MAGE-A4 C80A. C) Time-dependent TR-FRET between AF647-MTP-1 and MAGE-A4 or MAGE-A4 C80A in the presence of covalent peptides. D) Calculated IC50s from TR-FRET curves in part C. E) Western blots of MAGE-A4 co-immunoprecipitation with RAD18 at 10 μM peptides, including an MTP-1 Y5A negative control.
We synthesized three representative peptides, MCP1, MCP2, and MCP3 (Figure 3B), from the +G selections, all of which are distinct from previously selected MAGE-A4 inhibitors. All peptides modify MAGE-A4 by MALDI-MS, with MCP1 and MCP2 performing more efficiently than MCP3 (Figure 3B). MCP3 may modify a second site over an extended time, though we note the emergent peak is several hundred Daltons heavier than MCP3. To crudely gauge peptide selectivity, we tested if any of the MCPs modify CIB1 and only observed any significant labeling with MCP3 (Figure S9). The more selective and complete labeling efficiencies of MCP1 and MCP2 correlate well with their increased enrichment over MCP3 in the NGS data. L- and D-Ala mutants of MCP1 and MCP2 did not modify MAGE-A4 by MALDI-MS, confirming their modifications as Dha-dependent.
MAGE-A4 has four cysteines: two within the MAGE homology domain (MHD) and two in the structurally undefined N-terminal region.42 We initially tested if any of the covalent peptides modify the MHD alone, but did not observe substantial modification by MALDI-MS (Figure S9). GluC-MALDI-MS digests of MAGE-A4 adducts with MCP1 or MCP2 suggested that the peptides modify a fragment containing Cys80 (Figure S10), which was confirmed when MCP1 and MCP2 did not label a MAGE-A4 C80A mutant (Figure 3B). Some modification of C80A by MCP3 was still observed, further suggesting that MCP3 may be capable of modifying two sites.
Despite covalently modifying the N-terminal region, MCPs may still be able to block access to the MHD. Thus, we tested if the MCPs could inhibit association of a previously selected non-covalent MHD binder, MTP-1, in TR-FRET.24 Indeed, MCP1 and MCP2 exhibit single digit nanomolar IC50s of 9.0 and 6.4 nM, respectively, after 1 hour (Figure 3C-D). As expected, MCP3 exhibits much weaker inhibition. The L- and D- Ala mutants of MCP1 and MCP2 are much less potent than their Dha-containing counterparts. However, the D-Ala mutants of both peptides are more potent than L-Ala, suggesting D-Ala stereochemistry may be adopted upon nucleophilic attack by C80. We observed small yet significant increases in potency over time only for MCP1 and MCP2, which might be further differentiated in an assay with sub-nanomolar sensitivity. In the presence of MAGE-A4 C80A, IC50s of MCP1 and MCP2 fell in between those of their L- and D-Ala mutants, while MCP3 activity was ablated (Figure 3C-D). We also determined kinetic parameters for MCP1 through a mass dilution TR-FRET assay (Figure S11). MCP1 exhibits a kinact of 3.4 ± 0.3 min−1 and KI of 7.3 ± 1.1 nM, for an inactivation efficiency (kinact/KI) of 4.9 × 108 ± 5.2 × 107 M−1 min-1. These values align well with the observed IC50s for MCP1 and its L- and D-Ala mutants, further supportting that MCP1 inhibition is reliant on covalent modification. Lastly, we tested if the MCPs could disrupt MAGE-A4 engagement with one of its E3 ligase binding partners, RAD18, in cell lysates. In a co-immunoprecipitation of the RAD18-MAGE-A4 complex, all MCPs were able to disrupt MAGE-A4 pulldown, with MCP3 being less effective than MCP1 and MCP2 (Figure 3E). In contrast, none of the peptides disrupted the interaction between E2 ligase RAD6 and RAD18, showcasing selectivity for MAGE-A4.
In summary, we have combined multiple library diversification techniques—ORSs, post-translational chemistry, and Flexizymes—to incorporate electrophiles into macrocyclic mRNA display libraries for selection of covalent binders. This strategy is theoretically compatible with other cysteine-based cyclizations, including chemical cyclizations such as dibromoxylene. We establish Dhas as electrophiles for covalent inhibitor discovery and demonstrate the ability of guanidine to bias mRNA display selections toward covalent ligands. Additionally, we show this method can identify covalent inhibitors that exhibit selectivity in complex environments and disrupt known protein-protein interactions. Although the current method incurs potential background oxidation and requires targets with surface exposed cysteines, future optimization of electrophile identity and selection conditions (e.g. time, temperature) may fine-tune the resultant covalent binders for affinity, expedient reactivity, and target class.
Supplementary Material
ACKNOWLEDGMENT
The authors would like to thank Victoria Haberman for providing UNC10245131, UNC10245231, and fluorescently labeled CIB1 phage peptide, Matthew Fleming for providing MTP-1, MTP-1 Y5A, and fluorescently labeled MTP-1, and Steven Fleming for recombinant PhSec-RS expression. This work was supported in large part by a grant from the National Institutes of Health, NIH R35GM125005 (to A.A.B). Additional support was provided by grants from the National Cancer Institute (NCI, R01CA229530) and the National Institute of Environmental Health Sciences (NIEHS, R01ES029079) both to C.V. and K.H.P. L.F.C. was supported in part by NIGMS training grant T32GM135128.
Footnotes
ASSOCIATED CONTENT
Experimental details, lists of gene fragments and primers, supplementary figures, and peptide characterizations (PDF)
The authors declare no competing financial interests.
REFERENCES
- (1).Sutanto F; Konstantinidou M; Dömling A. Covalent Inhibitors: A Rational Approach to Drug Discovery. RSC Med. Chem 2020, 11 (8), 876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Boike L; Henning NJ; Nomura DK Advances in Covalent Drug Discovery. Nat. Rev. Drug Discov. 2022, 21 (12), 881–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Anand K; Anand K; Ziebuhr J; Wadhwani P. Coronavirus Main Proteinase (3CLpro) Structure : Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1768. [DOI] [PubMed] [Google Scholar]
- (4).Saito Y; Kawashima S. The Neurite-Initiating Effect of a Tripeptide Aldehyde Protease Inhibitor on PC12h Cells. J. Biochem 1989, 106 (6), 1035–1040. [DOI] [PubMed] [Google Scholar]
- (5).Yoo DY; Hauser AD; Joy ST; Bar-sagi D; Arora PS Covalent Targeting of Ras G12C by Rationally Designed Peptidomimetics. ACS Chem. Biol 2020, 15, 1604–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Shin Y; Jeong JW; Wurz RP; Achanta P; Arvedson T; Bartberger MD; Campuzano IDG; Fucini R; Hansen SK; Ingersoll J; Iwig JS; Lipford JR; Ma V; Kopecky DJ; McCarter J; San Miguel T; Mohr C; Sabet S; Saiki AY; Sawayama A; Sethofer S; Tegley CM; Volak LP; Yang K; Lanman BA; Erlanson DA; Cee VJ Discovery of N-(1-Acryloylazetidin-3-Yl)-2-(1 H-Indol-1-Yl)Acetamides as Covalent Inhibitors of KRASG12C. ACS Med. Chem. Lett 2019, 10 (9), 1302–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Skoulidis F; Li BT; Dy GK; Price TJ; Falchook GS; Wolf J; Italiano A; Schuler M; Borghaei H; Barlesi F; Kato T; Curioni-Fontecedro A; Sacher A; Spira A; Ramalingam SS; Takahashi T; Besse B; Anderson A; Ang A; Tran Q; Mather O; Henary H; Ngarmchamnanrith G; Friberg G; Velcheti V; Govindan R. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med 2021, 384 (25), 2371–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lanman BA; Allen JR; Allen JG; Amegadzie AK; Ashton KS; Booker SK; Chen JJ; Chen N; Frohn MJ; Goodman G; Kopecky DJ; Liu L; Lopez P; Low JD; Ma V; Minatti AE; Nguyen TT; Nishimura N; Pickrell AJ; Reed AB; Shin Y; Siegmund AC; Tamayo NA; Tegley CM; Walton MC; Wang HL; Wurz RP; Xue M; Yang KC; Achanta P; Bartberger MD; Canon J; Hollis LS; McCarter JD; Mohr C; Rex K; Saiki AY; San Miguel T; Volak LP; Wang KH; Whittington DA; Zech SG; Lipford JR; Cee VJ Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem 2020, 63 (1), 52–65. [DOI] [PubMed] [Google Scholar]
- (9).Kathman SG; Xu Z; Statsyuk AV A Fragment-Based Method to Discover Irreversible Covalent Inhibitors of Cysteine Proteases. J. Med. Chem 2014, 57, 4969–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Resnick E; Bradley A; Gan J; Douangamath A; Krojer T; Sethi R; Geurink PP; Aimon A; Amitai G; Bellini D; Bennett J; Fairhead M; Fedorov O; Gabizon R; Gan J; Guo J; Plotnikov A; Straub VM; Szommer T; Velupillai S; Zaidman D; Zhang Y; Coker AR; Brennan PE; Ovaa H; Delft F. Von; London N. Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc 2019, 141, 8951–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zambaldo C; Daguer JP; Saarbach J; Barluenga S; Winssinger N. Screening for Covalent Inhibitors Using DNA-Display of Small Molecule Libraries Functionalized with Cysteine Reactive Moieties. Medchemcomm 2016, 7 (7), 1340–1351. [Google Scholar]
- (12).Daguer J; Zambaldo C; Abegg D; Barluenga S; Tallant C; Müller S; Adibekian A; Winssinger N. Identification of Covalent Bromodomain Binders through DNA Display of Small Molecules**. Angew. Chemie - Int. Ed 2015, 54, 6057–6061. [DOI] [PubMed] [Google Scholar]
- (13).Guilinger JP; Archna A; Augustin M; Bergmann A; Centrella PA; Clark MA; Cuozzo JW; Däther M; Guié MA; Habeshian S; Kiefersauer R; Krapp S; Lammens A; Lercher L; Liu J; Liu Y; Maskos K; Mrosek M; Pflügler K; Siegert M; Thomson HA; Tian X; Zhang Y; Konz Makino DL; Keefe AD Novel Irreversible Covalent BTK Inhibitors Discovered Using DNA-Encoded Chemistry. Bioorganic Med. Chem 2021, 42. [DOI] [PubMed] [Google Scholar]
- (14).Li L; Su M; Lu W; Song H; Liu J; Wen X; Suo Y; Qi J; Luo X; Zhou Y; Liao X; Li J; Lu X. Triazine-Based Covalent DNA-Encoded Libraries for Discovery of Covalent Inhibitors of Target Proteins. ACS Med. Chem. Lett 2022, 13, 1574–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wilson DS; Keefe AD; Szostak JW The Use of mRNA Display to Select High-Affinity Protein-Binding Peptides. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (7), 3750–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Iskandar SE; Haberman VA; Bowers AA Expanding the Chemical Diversity of Genetically Encoded Libraries. ACS Comb. Sci 2020, 22 (12), 712–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Iskandar SE; Pelton JM; Wick ET; Bolhuis DL; Baldwin AS; Emanuele MJ; Brown NG; Bowers AA Enabling Genetic Code Expansion and Peptide Macrocyclization in mRNA Display via a Promiscuous Orthogonal Aminoacyl-tRNA Synthetase. J. Am. Chem. Soc 2023, 145 (3), 1512–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Vinogradov AA; Zhang Y; Hamada K; Chang JS; Okada C; Nishimura H; Terasaka N; Goto Y; Sengoku T; Ogata K; Onaka H; Suga H. De Novo Discovery of Thiopeptide Pseudo-Natural Products Acting as Potent and Selective TNIK Kinase Inhibitors. J. Am. Chem. Soc 2022, 144, 20332–20341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tucker TJ; Embrey MW; Alleyne C; Amin RP; Bass A; Bhatt B; Bianchi E; Branca D; Bueters T; Buist N; Ha SN; Hafey M; He H; Higgins J; Johns DG; Kerekes AD; Koeplinger KA; Kuethe JT; Li N; Murphy B; Orth P; Salowe S; Shahripour A; Tracy R; Wang W; Wu C; Xiong Y; Zokian HJ; Wood HB; Walji A. A Series of Novel, Highly Potent, and Orally Bioavailable Next-Generation Tricyclic Peptide PCSK9 Inhibitors. J. Med. Chem 2021, 64 (22), 16770–16800. [DOI] [PubMed] [Google Scholar]
- (20).Iskandar SE; Bowers AA MRNA Display Reaches for the Clinic with New PCSK9 Inhibitor. ACS Med. Chem. Lett 2022, 13 (9), 1379–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yang X; Lennard KR; He C; Walker MC; Ball AT; Doigneaux C; Tavassoli A; van der Donk WA A Lanthipeptide Library Used to Identify a Protein–Protein Interaction Inhibitor. Nat. Chem. Biol 2018, 14 (4), 375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Male AL; Forafonov F; Cuda F; Zhang G; Zheng S; Oyston PCF; Chen PR; Williamson ED; Tavassoli A. Targeting Bacillus Anthracis Toxicity with a Genetically Selected Inhibitor of the PA/CMG2 Protein-Protein Interaction. Sci. Rep 2017, 7 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Song X; Lu LY; Passioura T; Suga H. Macrocyclic Peptide Inhibitors for the Protein-Protein Interaction of Zaire Ebola Virus Protein 24 and Karyopherin Alpha 5. Org. Biomol. Chem 2017, 15 (24), 5155–5160. [DOI] [PubMed] [Google Scholar]
- (24).Fleming MC; Chiou LF; Tumbale PP; Droby GN; Lim J; Norris-Drouin JL; Williams JG; Pearce KH; Williams RS; Vaziri C; Bowers AA Discovery and Structural Basis of the Selectivity of Potent Cyclic Peptide Inhibitors of MAGE-A4. J. Med. Chem 2022, 65 (10), 7231–7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhang Z; Gao R; Hu Q; Peacock H; Peacock DM; Dai S; Shokat KM; Suga H. GTP-State-Selective Cyclic Peptide Ligands of K-Ras(G12D) Block Its Interaction with Raf. ACS Cent. Sci 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kawamura A; Münzel M; Kojima T; Yapp C; Bhushan B; Goto Y; Tumber A; Katoh T; King ONF; Passioura T; Walport LJ; Hatch SB; Madden S; Müller S; Brennan PE; Chowdhury R; Hopkinson RJ; Suga H; Schofield CJ Highly Selective Inhibition of Histone Demethylases by de Novo Macrocyclic Peptides. Nat. Commun 2017, 8 (1), 14773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).McCarthy KA; Kelly MA; Li K; Cambray S; Hosseini AS; Van Opijnen T; Gao J. Phage Display of Dynamic Covalent Binding Motifs Enables Facile Development of Targeted Antibiotics. J. Am. Chem. Soc 2018, 140 (19), 6137–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chen S; Lovell S; Lee S; Fellner M; Mace PD; Bogyo M. Identification of Highly Selective Covalent Inhibitors by Phage Display. Nat. Biotechnol 2020, 39 (490–498). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zheng M; Chen FJ; Li K; Reja RM; Haeffner F; Gao J. Lysine-Targeted Reversible Covalent Ligand Discovery for Proteins via Phage Display. J. Am. Chem. Soc 2022, 144 (34), 15885–15893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Morimoto J; Hayashi Y; Suga H. Discovery of Macrocyclic Peptides Armed with a Mechanism-Based Warhead: Isoform-Selective Inhibition of Human Deacetylase SIRT2. Angew. Chemie - Int. Ed 2012, 51 (14), 3423–3427. [DOI] [PubMed] [Google Scholar]
- (31).Chatterjee C; Paul M; Xie L; Donk W. A. van der. Biosynthesis and Mode of Action of Lantibiotics. Chem. Rev 2005, 105, 633–683. [DOI] [PubMed] [Google Scholar]
- (32).Cao L; Do T; James Link A. Mechanisms of Action of Ribosomally Synthesized and Posttranslationally Modified Peptides (RiPPs). J. Ind. Microbiol. Biotechnol 2021, 48 (3–4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hofmann FT; Szostak JW; Seebeck FP In Vitro Selection of Functional Lantipeptides. J. Am. Chem. Soc 2012, 134 (19), 8038–8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Jongkees SAK; Umemoto S; Suga H. Linker-Free Incorporation of Carbohydrates into in Vitro Displayed Macrocyclic Peptides. Chem. Sci 2017, 8 (2), 1474–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wang J; Schiller SM; Schultz PG A Biosynthetic Route to Dehydroalanine-Containing Proteins. Angew. Chemie - Int. Ed 2007, 119, 6973–6975. [DOI] [PubMed] [Google Scholar]
- (36).Ohuchi M; Murakami H; Suga H. The Flexizyme System: A Highly Flexible TRNA Aminoacylation Tool for the Translation Apparatus. Curr. Opin. Chem. Biol 2007, 11 (5), 537–542. [DOI] [PubMed] [Google Scholar]
- (37).Goto Y; Katoh T; Suga H. Flexizymes for Genetic Code Reprogramming. Nat. Protoc 2011, 6 (6), 779–790. [DOI] [PubMed] [Google Scholar]
- (38).Haberman VA; Fleming SR; Leisner TM; Puhl AC; Feng E; Xie L; Chen X; Goto Y; Suga H; Parise LV; Kireev D; Pearce KH; Bowers AA Discovery and Development of Cyclic Peptide Inhibitors of CIB1. ACS Med. Chem. Lett 2021, 12 (11), 1832–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Blamey CJ; Ceccarelli C; Naik UP; Bahnson BJ The Crystal Structure of Calcium- and Integrin-Binding Protein 1: Insights into Redox Regulated Functions. Protein Sci. 2005, 14 (5), 1214–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Leisner TM; Freeman TC; Black JL; Parise LV CIB1 : A Small Protein with Big Ambitions. FASEB J. 2016, 30 (8), 2640–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Puhl AC; Bogart JW; Haberman VA; Larson JE; Godoy AS; Norris-Drouin JL; Cholensky SH; Leisner TM; Frye SV; Frye SV; Parise LV; Parise LV; Bowers AA; Bowers AA; Bowers AA; Pearce KH; Pearce KH Discovery and Characterization of Peptide Inhibitors for Calcium and Integrin Binding Protein 1. ACS Chem. Biol 2020, 15 (6), 1505–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Newman JA; Cooper CDO; Roos AK; Aitkenhead H; Oppermann UCT; Cho HJ; Osman R; Gileadi O. Structures of Two Melanoma-Associated Antigens Suggest Allosteric Regulation of Effector Binding. PLoS One 2016, 11 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.