

Abstract
Postzygotic mutations (PZMs) begin to accrue in the human genome immediately after fertilization, but how and when PZMs affect development and lifetime health remains unclear. To study the origins and functional consequences of PZMs, we generated a multi-tissue atlas of PZMs spanning 54 tissue and cell types from 948 donors. Nearly half the variation in mutation burden among tissue samples can be explained by measured technical and biological effects, while 9% can be attributed to donor-specific effects. Through phylogenetic reconstruction of PZMs, we found that their type and predicted functional impact varies during prenatal development, across tissues and through the germ cell life cycle. Thus, methods for interpreting effects across the body and the lifespan are needed to fully understand the consequences of genetic variants.
One-Sentence Summary
The predicted burdens, functional effects and selection pressure of postzygotic mutations vary through the human life cycle.
The effects of age ravage all tissues of the body, but the pace and consequences of age-related decay vary among tissues and people. The accumulation of DNA damage is thought to be a primary agent of age-related disease (1), and surveys of postzygotic mutations (PZMs) in normal tissues (for example blood (2–4) brain (5), and skin (6, 7)), and across the body (8–10), have found PZMs to be pervasive across the genome and individuals. However, beyond cancer there are few conditions where PZMs are known to have a causal role. Due to the high cost and technological challenges of PZM studies, a general understanding of how and when mutation affects the function of specific cell and tissue types is essential for defining research priorities. One way to prioritize hypotheses about mutation and disease is to systematically characterize the consequences of PZMs on cellular fitness across a broad range of tissues. Surveys of normal tissues have found that PZMs appear to accrue neutrally (10, 11), but positive and negative selection do occur in specific genes and cellular contexts, suggesting PZMs affect cellular function.
Another fundamental question is how the timing of mutation modulates risk for diseases. As clearly demonstrated in oncology, it is possible to detect disease-causing PZMs and augment clinical care years before clinical disease is recognized (12, 13). If PZMs that confer risk for disease accrue across the lifespan, the PZM profile in a healthy individual could contain actionable prognostic information. While the relative contributions of prenatal and postnatal PZMs to disease risk are unclear, due to the massive cell proliferation during development, prenatal PZMs have the potential to affect many cells, and thus, play an important role in disease.
The vast majority of PZM research has been single-tissue studies largely focused on tissues that are easily accessible, such as blood, liver, skin and colon. An exciting next generation of PZM studies now examines PZMs across multiple tissues within an individual (8–10, 14). However, the relatively small numbers of individuals and tissue types used in such studies have limited the ability to ascribe sources of mutation variation among individuals or provide detailed descriptions of embryonic mutations that occur after the first few cell divisions. To expand our knowledge of PZMs in normal tissues, we developed a suite of methods called Lachesis to identify single-nucleotide PZMs from bulk RNA-seq data and predict when the mutations occurred during development and aging (Figs. S1 and S2). We ran the algorithm on the final major release of the Genotype Tissue Expression project (GTEx), a collection of RNA-seq data from 17,382 samples derived from 948 donors across 54 diverse tissues and cell types, to generate one of the most comprehensive databases of PZMs in normal tissues ((15), (16), Tables S1–S8). We used this atlas, and the rich metadata on GTEx donors, to characterize sources of variation in PZM burden among individuals and unveil the spatial, temporal, and functional variation of PZMs in normal development and aging.
Results
DNA PZMs are accurately detected in bulk tissue RNA-seq
We evaluated the accuracy of the algorithm using several in silico and experimental methods (Figs. S3, S4, Tables S3–S6). For experimental validation, we obtained four independent DNA- and RNA-based validation datasets generated from the same tissue samples as the primary data covering 296 unique genomic sites across 95 samples. The original PZM variant allele frequency (VAF) estimates from RNA-seq were well correlated with the VAFs from DNA-seq (Spearman’s ρ = 0.82, P-value = 2.3E-25) suggesting RNA-seq based VAFs are representative of true mutant cell frequencies. PZMs with VAFs as low as 0.16% and PZMs found in multiple tissues and multiple donors were validated. The average false discovery rate (FDR) across all validation datasets was 27% and was lower than with published methods for detecting PZMs from RNA-seq (34% - 82% (8, 9, 17)) (Fig. S1E, Table S3). Since mutations may fail to validate due to spatial variation in mosaicism, the FDRs may be overestimated. A small subset of samples (~5%) had an extraordinarily high number of detected PZMs; validation data from these samples produced an average FDR estimate of 98% (Table S3). We conclude that these outliers were likely technical artifacts, and not hypermutated tissues.
We used power simulations to estimate the algorithm’s sensitivity. As expected, simulated PZMs with larger VAFs and higher coverage had higher PZM detection power. At the middle quintile of coverage ([673, 1395) fold coverage), PZMs with VAFs as low as 0.66% could be detected in at least 90% of simulations, suggesting the method has reasonable sensitivity (Fig. S1F).
PZMs are pervasive and highly variable among donors and tissues
Following sample and PZM quality control, 56,585 PZMs were detected with variant allele frequencies (VAFs) as low as 0.04% and a median VAF of 0.5% (Table S7). These mutations are not a random sample of PZMs from the genome, but a critically important subset located in the “allowable transcriptome”: a filtered set of transcribed positions based on GENCODE 26 gene models ((16), Table S1). 100% of the donors and 77% of the tissue samples had detectable mosaicism (Table S2). We defined the mutation burden of a sample as the number of PZMs detected in a sample and the normalized mutation burden of a sample as the mutation burden normalized by the size of the sample’s transcriptome (the number of megabases with at least 20× total coverage). The median normalized mutation burden in a tissue ranged from 0.03 PZMs/expressed Mb in cerebellar hemisphere to 0.47 PZMs/expressed Mb in liver (Fig. 1A, Table S8). The observed normalized mutation burden was more variable within a tissue than between tissues (mean median absolute deviation (MAD) within a tissue = 0.07 PZMs/expressed Mb; MAD across tissues = 0.02 PZMs/expressed Mb). This observation suggests that processes generating detectable PZMs may be more variable across donors than across tissue types.
Fig. 1. PZM burden is correlated with biological and technical variables.
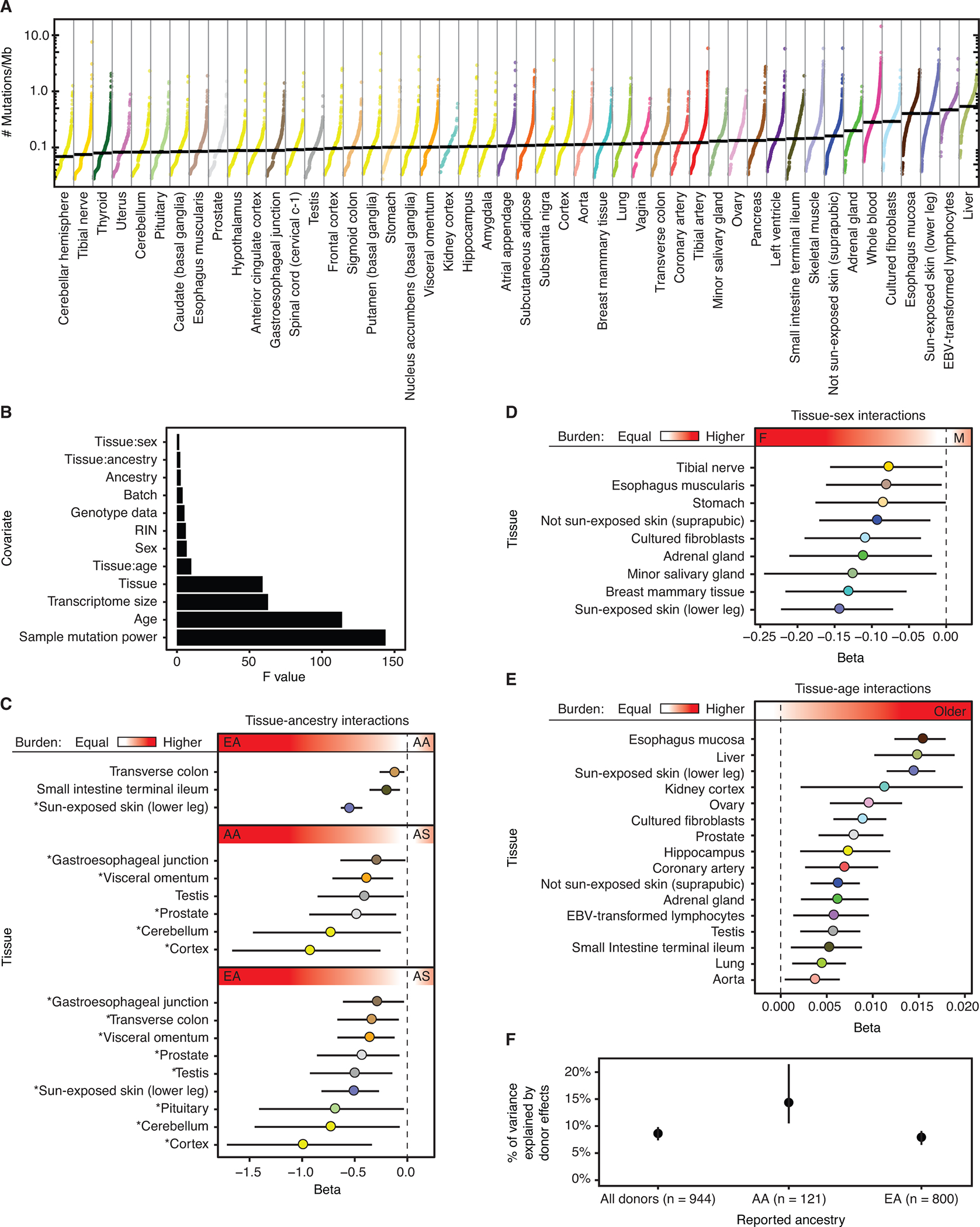
Each datapoint represents a single tissue sample and is colored by tissue. Median normalized PZM burden in a tissue denoted by horizontal black line. Tissues are sorted by increasing median normalized PZM burden. A pseudocount of 1 mutation was added to each sample before normalization and log transformation for visualization. (B) We fit a regression model for single-tissue PZM burden using 12 covariates and 48 tissues. Shown here are the Type II ANOVA F statistics for each covariate in the model. Larger F statistics correspond to greater explanatory power of the covariate. (C) Regression coefficients of tissue-ancestry interactions and (D) tissue-sex interactions indicate strong effects of ancestry and sex on PZM burden. AA = African American. AS = Asian American. EA = European American. * in C denote differences in mutation burden among ancestry groups that are consistent with cancer incidence trends (18) (E) Significant positive tissue-age interaction effects were detected for 16/48 (33%) tissues. In C-E, the red gradient and text labels within indicate the meaning of the regression coefficients’ sign and magnitude. (F) Variance component estimates of donor-specific random effects on PZM burden indicate that 8%−15% of variation among tissues can be ascribed to donor effects, which could be genetic and environmental. Dashed vertical lines at beta = 0 in interaction plots denote no association between mutation burden and interaction. C,D,E,F: Error bars represent 95% CIs. A,C,D,E: Tissues are colored using the GTEx coloring convention (see Table S8 for a complete legend).
To build further support for the validity of our per-tissue estimates of mutation burden, we compared our data to a recent multi-tissue survey of PZMs based on DNA sequencing of three donors (10, 14). Encouragingly, when comparing 12 tissues assessed by both studies, we found reasonably high correlation in estimated PZM burden ( (16), Fig. S5). The Pearson correlation for the average burden was 0.8 (P-value = 0.0018, Pearson’s correlation test).
PZM burden is correlated with biological and technical variables
To partition and quantify potential sources of single-tissue PZM burden, we fit linear models relating technical and biological metadata to single-tissue PZM burdens and selected the best fitting model identified from detailed model comparisons (16). The final model contained twelve covariates and explained 48% of the variation in mutation burden. All covariates yielded F-test P-values < 0.05 in a Type II Analysis of Variance and included both biological (age, tissue, and interactions of tissue with age, sex, and self-reported ancestry) and technical (for example, mutation detection power and RNA extraction batch) sources of variation (Fig. 1B, Table S9). 20.8% (10/48) of tissues showed significant (Wald-test q-value < 0.05, ) associations with self-reported ancestry, including, as expected, a much lower burden of mutation in sun-exposed skin in African Americans and Asian Americans compared to European Americans (8). The incidence rates of cancer types affecting these tissues have ancestry associations that are consistent with (in the same direction as) the mutation burden associations in 83% (15/18) of comparisons (18), suggesting that variation in PZM burden in normal tissues may contribute to differences in cancer risk among ancestries (Fig. 1C, Table S10). Unexpectedly, males had lower burden in all three skin-related sample types compared to females (Fig. 1D). This result was essentially unchanged when removing genes inferred to have sex-biased expression (Fig. S6). Age was positively associated with 33% (16/48) of tissues and was the strongest for esophagus mucosa, liver, and sun-exposed skin (Fig. 1E). We note that power may have been too low to detect some associations; for example, there were few young GTEx brain donors.
Extending this model to include a random donor effect, we estimated that 8.8% of variation in PZM burden can be attributed to systematic properties of donors that extend across some or all tissues of a donor, even after controlling for metadata such as age and sex. This donor variance component estimate was larger in African Americans (14.1%; 95% confidence interval (CI): 10.5–21.5%) than in European Americans (8%; 95% CI: 6.5–9.1%) (Fig. 1F). These unexplained donor-specific effects could have both genetic and environmental bases. Notably, a recent study estimated that 5.2% of variance in germline mutation rate could be attributed to family-specific effects (19). In total, our results indicate that variation in PZM rate among individuals is less constrained than variation in germline mutation rate and that there is considerable scope for heritable variation in observable PZM burden. The inability of the models to explain all variation imply there are additional factors associated with detectable mutation burden and/or stochasticity plays a major role in mosaicism (20, 21). A reanalysis of the data that incorporates information on the apparent clonality of mutations produced models with similar biological conclusions and less explanatory power ((16), Fig. S7, Table S11).
Mutation spectra is variable across tissues and reflect known biological processes
Diverse processes mutate the human genome with characteristic mutational signatures (22). Thus, the observed mutation spectra can provide insight on the types and relative activities of the unobserved mutation processes that occurred. We estimated the contribution of canonical mutation signatures for each tissue. Due to the relatively low number of detected mutations, mutation spectra were reliably deconstructed for only four tissues/cell types ((16), Fig. S8). Consistent with expectations and previous studies (3, 6, 7), the mutations were resolved into mutational signatures associated with age in all tissues and ultraviolet light exposure in skin-related tissues (Fig. S9).
For a higher powered, but coarser-grained analysis of mutation spectra, we assessed the frequency of the six base substitutions across all tissues (Fig. S8). Mutation spectra were highly variable across tissues suggesting that mutational mechanisms and their relative activity may vary across the human body. C>T was the most common mutation type across tissues whereas C>G and T>A were the least common. Hierarchical clustering of the mutation types revealed two significant large clusters (P-value < 1E-3, bootstrap resampling). We denoted these cluster A (marked by depleted T>G) and cluster B (marked by elevated T>G). Cluster membership was associated with mutation burden suggesting the underlying mutation mechanisms may be coupled to the frequency of mutagenic events (P-value = 3.8E-2, Mann-Whitney U test). Additionally, Cluster B was enriched with neural ectoderm tissues compared to cluster A (P-value = 7.7E-6, Fisher’s exact test). These clusters could not be attributed to differences in sample processing ((16), Fig. S10). We speculated that these clusters may reflect differences in the relative contributions of mutations acquired during prenatal development and mutations that accrue during age-related tissue renewal. To further study the properties of prenatal and postnatal PZMs, we developed methods to define the developmental origin of each PZM.
The developmental origins of prenatal PZMs
Multi-tissue PZMs exhibit prenatal properties
We defined a multi-tissue PZM as a PZM that was detected in at least two tissues from the same donor. Since the PZM burden was relatively low across tissues (Fig. 1) and PZMs are predominantly under neutral selection (11), we hypothesized that a multi-tissue PZM was the result of a single PZM that occurred in a common ancestor of the mutated tissues. Since the common ancestors of any set of GTEx tissues (excluding cell lines) occurred before the end of organogenesis, multi-tissue PZMs may have occurred prenatally. Consistent with this hypothesis, we found several lines of evidence suggesting the multi-tissue PZMs occurred prenatally ((16), Figs. S11, S12). We found a significant positive correlation between VAF and the fraction of the donor’s tissues that had the multi-tissue mutation detected (Spearman’s ρ = 0.34, P-value = 9.7E-56, Spearman’s rank correlation test, Fig. S11A). Controlling for technical and biological confounders, age was not significantly associated with multi-tissue mutation burden for the majority of tissues but was significantly associated with single tissue mutation burden for a large number of tissues (Figs. S11B–11C). Additionally, the multi-tissue age regression coefficients were significantly smaller than the single tissue age regression coefficients (P-value = 0.016, Wilcoxon signed-rank test) (Fig. S11D). We denoted these multi-tissue mutations as prenatal PZMs, while all other mutations were called postnatal PZMs. We note that there may be an error rate associated with this classification as some mutations labeled as postnatal may have been prenatal mutations lost in some tissues or were undetected in some donors due to limited samples.
PZM burden and spectra vary throughout prenatal development with most mutations occurring during early embryogenesis
To determine when and where PZMs occur in prenatal development, we developed a method called LachesisMap to map the origin of 1,864 prenatal mutation events (Fig. 2A, Figs. S13–S16, Table S12, (16)). Briefly, the method takes as input a directed rooted tree representing the developmental relationships among the tissues and a list of multi-tissue PZMs and maps the PZMs to the tree while accounting for differential mutation detection power across the genome, human body, and developmental tree. The algorithm outputs a list of edge weights that represent the estimated fraction of PZMs that occurred in that spatiotemporal window of development.
Fig. 2. Mutation burden and spectra of prenatal PZMs across time and space.
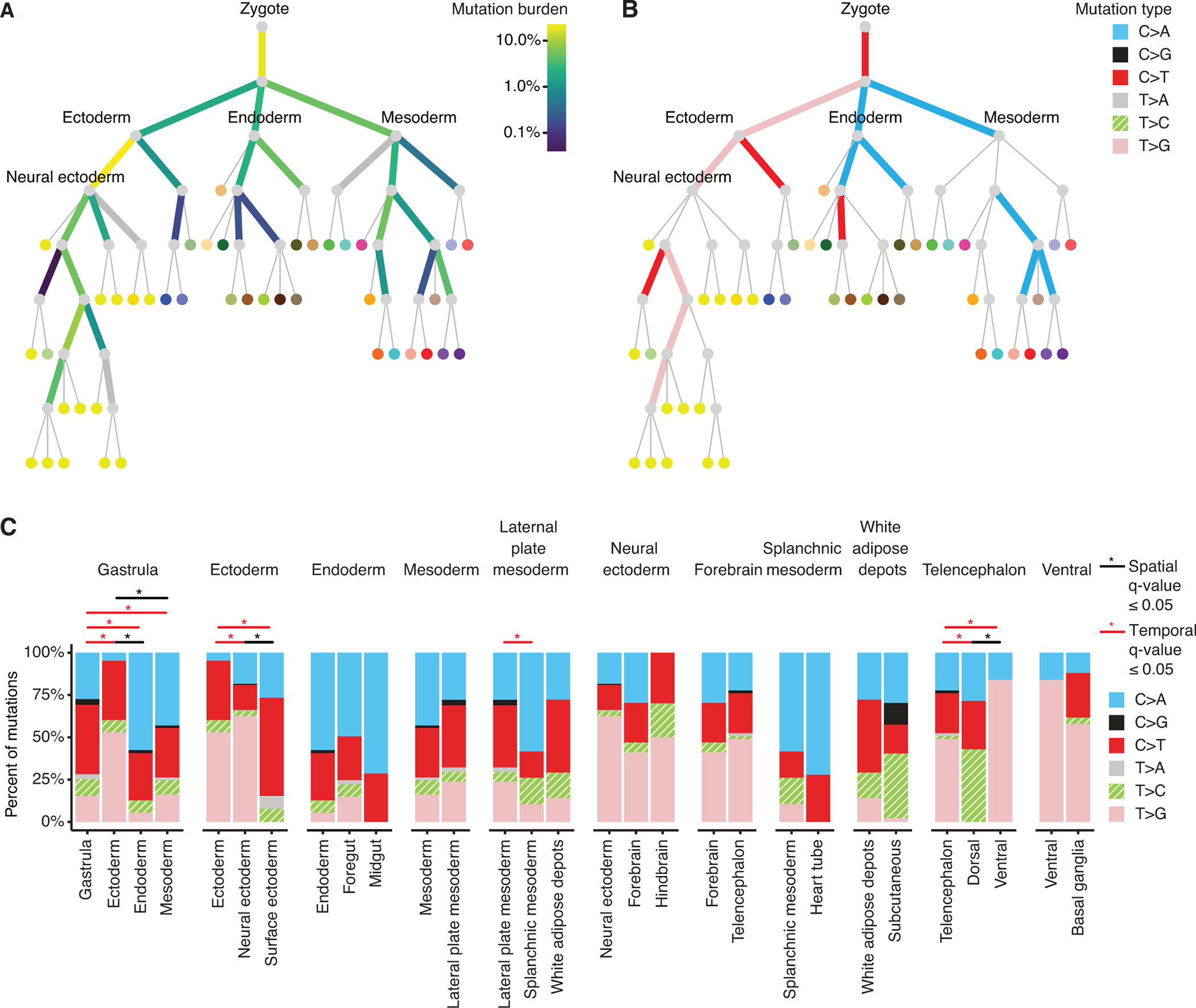
(A) Prenatal PZM mutation burden. Edge color represents the percent of prenatal PZMs mapped to that period in development. Thick gray edges are edges with limited mutation detection power. (B) Edge color represents the predominant mutation type of mutations mapped to that edge, as established by binomial testing. Thin gray edges are edges with no predominant mutation type. See Fig. S13A for the full set of vertex labels. Adult tissues (leaves of tree) are colored using the GTEx coloring convention (see Table S8 for a complete legend). (C) Local variation in mutation spectra across developmental space and time. Each facet represents the mutation spectra observed in a parent edge (leftmost barplot) and its children’s edges. Statistically significant differences in mutation spectra are annotated with “*”.
The mutation burdens across developmental time and space were highly variable, with edge weights ranging from 0.04% to 23%, and appeared compatible with an exponential distribution (P-value = 0.56, Kruskal-Wallis test). The ensemble of observed edge weights was significantly different from random (P-value = 2.2E-308, multinomial goodness-of-fit test) and the majority of individual edge weights (56%, 14/25) were significantly different from random after Benjamini-Hochberg correction (permutation tests) (Fig. S17). The top two edge weights, representing 41% of prenatal mutation events, were the zygote to gastrula transition and the ectoderm to neural ectoderm transition, suggesting that most detectable prenatal mutations occur during early embryogenesis (14, 23). Of critical note, the edge mutation burdens were not explained by differential edge mapping power across the developmental tissue tree ((16), Fig. S17). It is also important to note that these are estimates for mutations that are detectable in adulthood — the data does not allow for extrapolating to all developmentally acquired mutations since some fraction is likely lost through cell death, revertant mosaicism, etc.
We next asked if the mutational processes, as proxied by their mutation spectra, varied over development, using binomial tests to establish the “predominant” mutation type on each edge. There was a strong dichotomy between ectoderm lineages, which tended to have T>G mutations, and endoderm and mesoderm lineages which tended to have C>A mutations (Fig. 2B). These observations could not be attributed to differences in sample processing ((16), Fig. S10).
In addition to global changes in mutation across the tree, we also examined local changes by comparing mutation spectra between sibling edges (local spatial differences) and parent-child edges (local temporal differences) (Fig. 2C). Significant spatial and temporal variation was detected during gastrulation and in ectodermal lineages (q-value < 0.05, Multinomial goodness-of-fit test). Differences in mutation spectra across developmental space (n = 4/8 (50%) sibling edge comparisons) occurred at similar rates as differences along developmental time (n = 8/18 (44%) parent-child comparisons) (P-value = 1.00, Fisher’s exact test).
Together, these results suggest that the mutational mechanisms that operate during development may vary across space and time. Although published data are limited, others have also detected variations in mutation spectra in fetal stem cells in humans (24) and during early embryogenesis and gametogenesis in mice (25).
We repeated these analyses using a simplified germ layer tree and observed similar results as the full developmental tissue tree (Fig. S18), suggesting that the development tree definition does not substantially affect the results.
The functional consequences of PZMs across the human lifespan
The GTEx PZM atlas provides a great opportunity to compare the quality and fitness consequences of mutations that arise at different stages of the human life cycle. First, we annotated the PZM atlas with Combined Annotation Dependent Depletion (CADD), a widely used machine learning classifier of genetic variation (26). The CADD score of a genetic variant is a quantitative prediction of deleteriousness, measured on an evolutionary timescale. Here, a mutation was defined as deleterious if the PHRED-scaled CADD score ≥ 20. We performed a series of systematic comparisons of PZM CADD scores to identify differences across mutation VAF, developmental time, developmental location, and tissue type.
When comparing prenatal and postnatal PZMs, we found a major effect of VAF on the distribution of CADD scores (Fig. 3A and Fig. S19). For prenatal PZMs, low VAF PZMs were much more deleterious than high VAF PZMs (odds ratio = 1.9, P-value = 2.6E-7, Fisher’s exact test), while no such difference was observed for postnatal PZMs (P-value = 0.24, Wald Test). Furthermore, we found that for low VAF PZMs, deleteriousness decreased over time (odds ratio = 0.58, P-value = 1.4E-9) but remained constant for high VAF PZMs (P-value = 0.15). These results suggests that mutations that appear deleterious on an evolutionary timescale may be benign or even beneficial to a growing fetus so long as the mutation remains in a small fraction of cells.
Fig. 3.
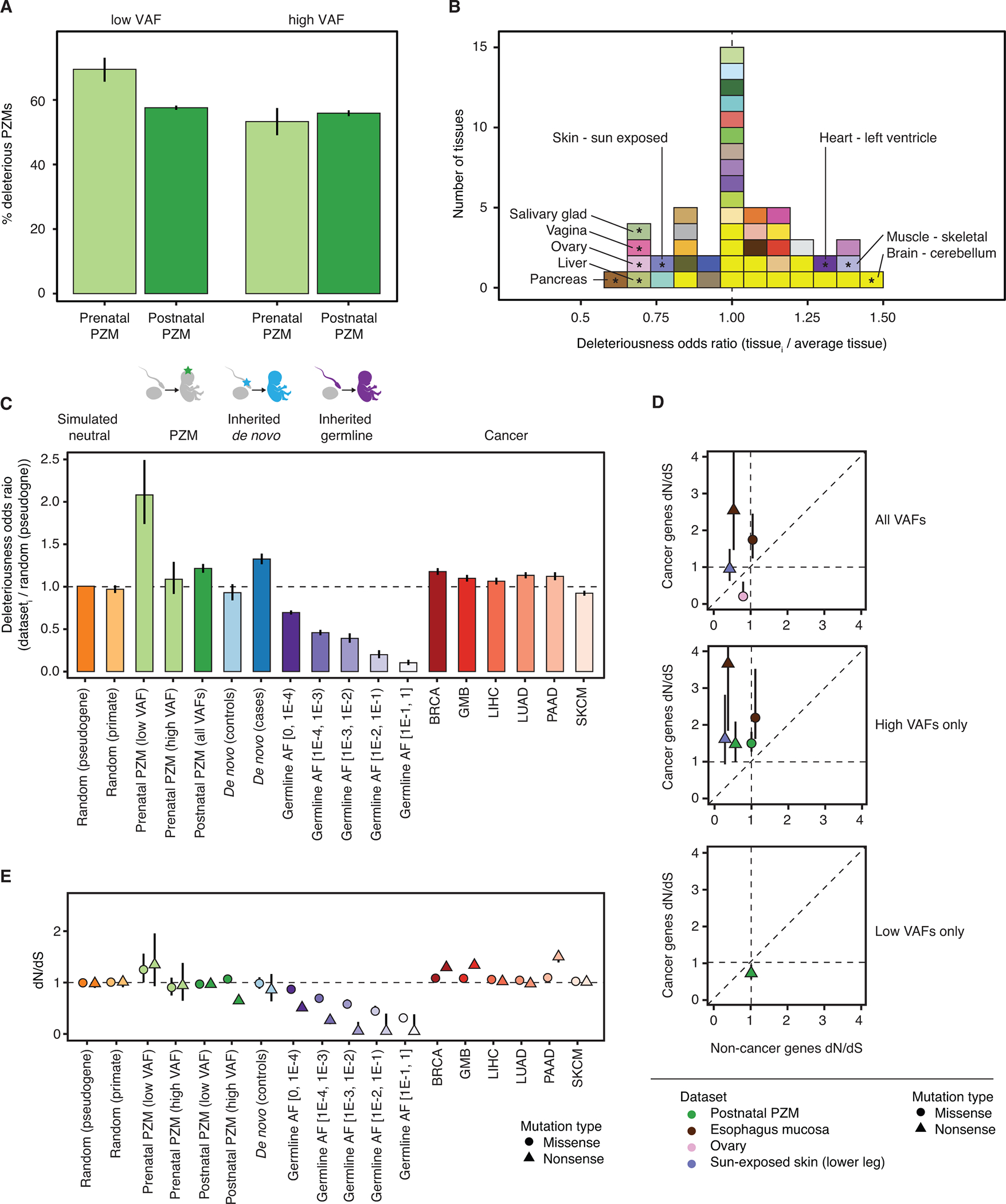
Deleteriousness and selective pressure changes as a function of VAF, space, time, and classes of genetic variation. (A) Relative odds of detecting deleterious mutations across developmental time (gray bars) and VAF bins (green bars). (B) Histogram of the odds of detecting deleterious postnatal PZMs in each tissue compared to the average tissue. Tissues are colored using the GTEx coloring convention (see Table S8 for a complete legend). Tissues with significant odds ratios (at q-value ≤ 0.05) are marked with “*” and labeled with their names. Vertical dashed line at odds ratio = 1 indicates no difference in odds. (C) Relative odds of detecting deleterious PZM mutations compared to different classes of genetic variation. Dashed line at odds ratio = 1 indicates no difference in odds of detecting deleterious mutations compared to reference group. Error bars represent 95% CIs. (D) Comparison of postnatal PZM selection pressure in cancer and non-cancer genes. For clarity, only PZM datasets that had different selection pressure between cancer and non-cancer genes are shown. Top: PZM datasets that had variable selection when using all mutations; middle: high VAF mutations; bottom: low VAF mutations. Error bars represent 95% CIs. Some CIs are smaller than the datapoint so are not directly visible. (E) dN/dS values for classes of genetic variation, as in (C). CIs are plotted behind each datapoint and are sometimes smaller than the datapoint size. dN/dS = 1 indicates neutral expectation. AF = allele frequency. BRCA = breast invasive carcinoma. GBM = glioblastoma multiforme. LIHC = liver hepatocellular carcinoma. PAAD = pancreatic adenocarcinoma. SKCM = skin cutaneous melanoma.
Next, we asked if deleteriousness varied across the adult human body by comparing postnatal PZMs in each adult tissue. PZM deleteriousness was similar across tissues; however, there were a few exceptions (Fig. 3B). PZMs in 6/48 (13%) tissues were significantly less deleterious than the average tissue and 3/48 (6%) tissues were more deleterious (q-value <0.05, Wald test). When analyzed together, the PZMs from all brain regions were also more deleterious than average (P-value = 0.02, Fisher’s exact test).
Finally, to provide context for our results, we compared the deleteriousness of GTEx PZMs to other classes of single-nucleotide genetic variation: 1) random mutations (simulated from two different models of neutral evolution), 2) standing germline variation (from gnomAD, a comprehensive database of germline genetic variation (27)), 3) inherited de novo mutations from cases of disease and controls (from denovo-db, a curated database of de novo mutations (28)) and 4) somatic mutations observed in cancer (from TCGA, a comprehensive database of cancer somatic mutations) (29).
The low VAF prenatal PZMs were the most deleterious class of genetic variation investigated (Fig. 3C, Fig. S19). Using the simulated random mutations as a reference, we found that postnatal PZMs, de novo mutations in cases, and somatic cancer mutations to be significantly enriched for deleterious mutations (q-value < 0.05, Fisher’s exact test). De novo mutations in controls and high VAF prenatal PZMs were not statistically different from simulated random mutations. Inherited germline variants were depleted of deleterious mutations, with the extent of depletion increasing with population frequency. These observations were recapitulated in 3 validation datasets that used a variety of nucleic acid sources and variant calling methods ((16), Fig. S20–S23, Table S13).
The selective constraint on the transcribed exome varies throughout the human lifespan
The deleteriousness results suggest that selection pressure may be different across classes of genetic variation. We investigated this hypothesis by estimating the selection pressure on PZMs and other classes of genetic variation using dN/dS, a normalized rate of nonsynonymous to synonymous mutations (30). dN/dS values greater than one were interpreted as evidence for positive selection, while negative selection can lead to dN/dS values less than one. Using dNdScv, a method for the study of somatic evolution (11), we assessed dN/dS across VAF, developmental time, developmental location, and tissue type, and contextualized the results by comparing selection pressures on PZMs to other classes of genetic variation as before (Fig. 3D–E, (16), Figs. S24–S26).
For most tissues of the body, single-tissue dN/dS was not significantly different from 1, consistent with previous work (11). However, for postnatal missense mutations, dN/dS was higher for high VAF PZMs compared to low VAF PZMs for all tissues en masse and for three tissues/cell types individually (whole blood, EBV-transformed lymphocytes, and adrenal gland) (Fig. S24). Additionally, dN/dS estimates for high VAF postnatal PZMs were higher in cancer driver genes than non-cancer driver genes for all tissues en masse, sun-exposed skin and esophagus mucosa, tissues where the action of adaptive evolution has already been documented (7, 31) (Fig. 3D). These observations are consistent with the expectation that positive selection on a mutation may result in clonal growth, and indeed, we detected mutations associated with clonal hematopoiesis of indeterminant potential in the blood of individuals without apparent hematological malignancies ((16), Fig. S27). Six unique CHIP mutations were detected in 7 samples (Table S14). Two of the mutations (IDH2 R140Q and MYD88 L273P) are in the 99.99th percentile of recurrent mutations in hematopoietic and lymphoid cancers and have been shown to have gain-of-function properties (32, 33). 0.1% (1/746) of whole blood donors and 3.5% (6/174) of EBV-transformed lymphocyte donors had a CHIP mutation. Of note, none of the CHIP-positive donors had a history of cancer. The observed CHIP prevalence in GTEx is similar to what we would expect given the age demographics of the cohort and published prevalence rates (2).
dN/dS for the low-VAF prenatal PZM class was nominally greater than 1 (missense dN/dS = 1.25, P-value = 0.047) (Fig. 3E). The high VAF postnatal nonsense mutations showed dN/dS much less than 1, which can be attributed to sampling bias against transcripts carrying premature stop codons, due to nonsense-mediated decay (34). Altogether, the deleteriousness and selection results suggest a dichotomy between growth within an individual versus growth within a population: mutations that are selected for within parts of an individual may be detrimental when considered across the entire lifespan.
Characterization of germ cell PZMs
Construction of a catalog of germ cell PZMs throughout the germ cell life cycle
While a great deal is known about germline variation (27) and de novo mutations (25, 35–39), much less is known about the PZMs that seed these forms of inherited genetic variation. To better understand PZMs in germ cells, we characterized and contrasted the mutation burden, spectra, and deleteriousness of germ cell PZMs across the germ cell life cycle.
Due to cell composition differences between male and female gonads, PZMs in testes samples could be confidently mapped to germ cells but PZMs in ovary samples could not ((16), Fig. S28, and Table S15). Therefore, only testicular germ cell PZMs were analyzed further. Germ cell PZMs were classified into “gonosomal” (present in somatic and germ cells) and “germ cell-specific”. 571 germ cell PZMs were identified in bulk testis from 281 testis donors of which 12% were putative gonosomal PZMs and the remaining 88% were putative germ cell-specific PZMs. As expected, germ cell-specific PZM burden was positively associated with donor age (P-value = 0.03) but gonosomal mutation burden was not (P-value = 0.28). Additionally, as expected, germ cell-specific PZMs had lower VAFs than gonosomal PZMs (P-value = 1.3E-14, Mann-Whitney U test, Fig. S28E).
Testicular germ cell PZMs represent the full reservoir of mutations that can be passed to progeny. We hypothesized that the selection pressures on spermatogenesis, fertilization, and prenatal development may alter the types of mutations that pass through each of these bottlenecks of life. To examine germ cell PZMs that passed the spermatogenesis bottleneck, we generated whole exome sequencing data on small 200-cell pools of ejaculated sperm and identified and validated 83 PZMs in the same genomic regions that we assessed in the GTEx RNA-seq samples (defined as the “allowable transcriptome”) (Table S1, Table S16, Fig. S29 (16)). To examine germ cell PZMs that completed prenatal development, we used ~17,000 de novo mutations in the allowable transcriptome from denovo-db (28).
The mutation spectra for each germ cell mutation dataset were statistically different from the others (Fig. 4A and Table S17, Chi-square test). While C>T was the most common mutation type in all datasets, C>A was the most variable. Hierarchical clustering of the spectra nested the classes in developmental order, indicating that the mutation spectra shift during development (Fig. 4A inset). Given the complex ascertainment of these diverse mutation callsets, we cannot exclude the possibility that some of the apparent structure is attributable to differences in mutation detection among sources, either due to bioinformatic or experimental effects.
Fig. 4. Germ cell PZM characteristics.
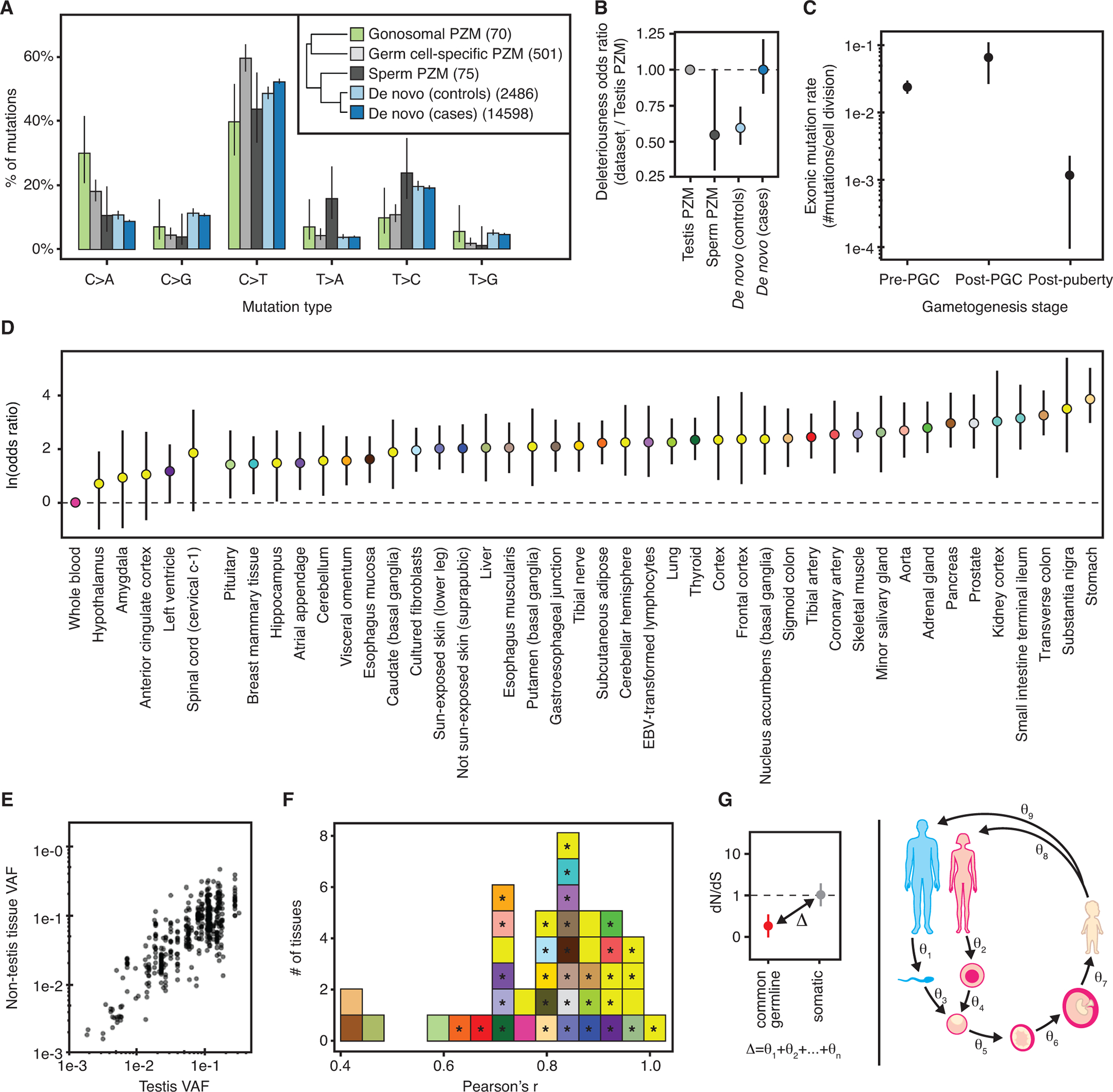
(A) Mutation spectra of different germ cell mutation classes. Number of mutations used in each dataset is listed in the inset. Inset: Hierarchical clustering of germ cell mutation spectra. (B) Relative odds of detecting deleterious mutations across germ cell datasets compared to testis PZMs. Bars colored by dataset. Horizontal black line at odds ratio = 1 denotes no difference in odds. (C) Germ cell mutation rate varies during gametogenesis in males. (D) Majority of somatic tissues have a higher odds of detecting a gonosomal PZM than blood. Natural log odds ratio for detecting a gonosomal PZM in each somatic tissue compared to blood. Dashed line at Y = 0 denotes no difference in odds. (E) Comparison of gonosomal PZM VAF in non-testis tissues versus testis tissue. (F) Distribution of tissue-specific Pearson correlations of log10-transformed gonosomal PZM VAFs in each somatic tissue and testis. Significant correlations at q-value ≤ 0.05 marked with “*”. (G) Schematic of the difference in selective constraint between germline and somatic genetic variation partitioned into discrete stages of the life cycle. A,B,C,D: Error bars denote 95% CIs.
Deleterious mutations are likely purged during the germ cell life cycle.
Consistent with the action of purifying selection on male germ cells, we found that mutation deleteriousness decreased over the germ cell life cycle when comparing testicular germ cell PZMs and de novo mutations in controls (Fig. 4B). In contrast, de novo mutations from cases of disease were just as likely to be deleterious as testis PZMs. To replicate these observations, we performed a similar analysis using only DNA-based measurements from published datasets ((16), Fig. S30) (10, 40). Both the fraction of coding mutations and the odds of detecting a deleterious mutation decreased over the germ cell life cycle in the independent datasets (Fig. S30). Donor age was not associated with PZM deleteriousness in each dataset.
The mutation rate during male gametogenesis is dynamic
We estimated the mutation rate (the number of mutations in the transcriptome per cell division) for each of three major stages during male gametogenesis (16).Consistent with previous work (37), the observed mutation rate was higher in prenatal timepoints than the postnatal timepoint (Fig. 4C). The observed lower mutation rate during adulthood may be a strategy to limit the number of deleterious mutations that are passed to the next generation. Unlike (37) and other studies that use transmitted de novo mutations to measure mutation rates (35, 36, 38, 39), these estimates reflect mutation rates in germ cells in the testis and thus offer insight on germ cell mutagenesis.
Blood is a poor surrogate for measuring mosaicism of gonosomal PZMs
Motivated by the fact that only a small subset of tissue types is easily and ethically accessible in antemortem human subjects research, we hypothesized that more accessible tissues may be useful surrogates for examining prenatal PZMs in less accessible tissues. The results of such analyses may shed light on the cellular dynamics of human development and implications for preconception genetic counseling and de novo mutation discovery.
We fit a mixed-effects model to predict whether a gonosomal PZM was detected in a somatic tissue while controlling for technical effects (16). Surprisingly, 88% (38/43) of tissues had significantly higher odds of detecting gonosomal PZMs than in blood (Fig. 4D), suggesting that blood is a poor surrogate for detecting gonosomal PZMs. Additionally, 76% (32/42) of somatic tissues had a significant linear correlation between the somatic VAF and the germ cell VAF (Fig. 4E and F; q-value <0.05, Pearson’s correlation test), suggesting that somatic tissues may offer a faithful representation of gonosomal PZMs in germ cells. These observations were not an artificial result of germline variant filtering or our cross-sample mutation calling strategy ((16), Fig. S31). While 82% of GTEx donors were genotyped using blood, for 6% of GTEx donors, a non-blood tissue was used for genotyping, and for 12% of donors, no genotyping data were available. There was no detectable difference among these three groups in the probability of detecting a prenatal PZM in blood, while controlling for other confounders; this suggests that poor detection of gonosomal PZMs in blood is not simply the result of aggressive germline filtering using genotype calls from blood-derived DNA (16).
Discussion
Here, we present one of the most comprehensive and diverse surveys of PZM variation in normal individuals, which should prove a valuable resource for understanding the causes and consequences of PZMs across the body. By linking these mutation calls to the vast data and tissue resources of the GTEx project, there are a number of analyses that could be attempted. First, if there is a heritable component to PZM burden, variants modulating this burden may be detectable using GWAS (41, 42). Second, the impact of PZMs on gene expression traits, both in cis and trans, can be directly assessed (9, 43). Third, the spatial and cell-type distribution of the mutations reported here could be mapped in banked tissue samples from the GTEx donors (7, 44), and the mutation type and burden of each sample associated with histology images collected by the GTEx project. We performed extensive validation of our PZM callset, and these validation data will be helpful in training algorithms for PZM detection.
We observed a number of striking features regarding the developmental origins of mutations that deserve follow-up. Most intriguing is a class of low VAF prenatal mutations that appear to have the highest fraction of deleterious mutation across the human lifespan, even considering disease states. This observation, based on a definition of deleteriousness on an evolutionary timescale, suggests that the functional consequences of mutation can have opposite fitness effects at different stages of the life cycle of genomes and in different cellular contexts. One well established example of dramatic differences in fitness effects between somatic and germline cells is the RAS-MAPK pathway, in which gain-of-function mutations provide a transmission advantage to male germ cells, but are often reproductively lethal for the resulting conceptus (45, 46). While some parallels have been noted between molecular mechanisms of carcinogenesis and normal embryogenesis (47, 48), there are essentially no data on the potential adaptive effects of PZMs on embryonic or fetal development in healthy individuals.
We advise caution in the interpretation of the dN/dS values for multi-tissue PZMs. Although we have evaluated obvious sources of technical error, such as the multi-tissue ascertainment (Fig. S25) and small sample size (Fig. S26), there may be other complexities influencing this rather general statistic, including recurrent mutation, and changes in mutation processes throughout development. Clearly it will be important to continue research into appropriate statistical methods for assessing fitness consequences of PZMs from multi-tissue datasets.
We found that blood-derived RNA appeared to be a poor proxy for detection of gonosomal mutations. Based on these results, for trio studies, we recommend sperm (a direct readout of germ cells) should be profiled in males, and skin (which is predicted to be over 5× more likely than blood to contain a gonosomal PZM) should be profiled in females. It should be noted that these findings on gonosomal PZMs were based on analysis of data exclusively from male tissues. We are optimistic that this conclusion will hold for female gonosomal mutations. In humans, male and female germ cells are both formed from a common progenitor cell type: primordial germ cells (PGCs). Early embryonic development, up to and including the formation of PGCs, is the time frame in which gonosomal mutations occur, and is thought to occur identically in males and females (49). The developmental phylogeny that relates PGCs and the three germ layers is unclear, and the patterns of gonosomal mutations observed across human tissues may yield important insight into the matter. Some studies indicate that PGCs may be most related to mesoderm: incipient mesoderm or mesendoderm cells can be induced to form PGC-like cells in vitro (50, 51) and PGCs may share expression markers with mesoderm/primitive streak (52). However, loss of BLIMP1, a key driver of germline identity, from germline competent cells leads to activation of a default neuronal differentiation program (50). When mapping gonosomal mutations frequencies across the body, we found that brain tissues were most similar to testis (Fig. 4F). This might be an indication that PGCs and ectoderm share a closer developmental origin.
We reported a large difference in deleteriousness and dN/dS inferred from PZMs and inherited germline variants, consistent with strong purifying selection reducing the transmission of deleterious mutation across generations. An important future direction is to dissect and quantify the physiological basis of this purifying selection (Fig. 4G). With careful thought and experimental design, it should be possible to model the steps of the human life cycle where purifying selection can occur, estimate the strength of selection at each step, and translate these data into life stage-specific measures of selective constraint for each gene in the genome. This would be of great benefit to human geneticists, who rely heavily on selective-constraint measures aggregated across the life cycle (such as CADD) for interpretation of genetic variants in the context of disease (53, 54). Stage-specific constraint metrics could augment current methods for variant interpretation to be more relevant to the tissue and developmental time affected by a disease.
Methods summary
GTEx Data
We detected PZMs in the GTEx v8 dataset. To achieve a high-quality dataset, we removed RNA-seq samples that had RNA integrity number (RIN) < 6, were derived from tissues with overall poor quality (8) or had an extremely high PZM mutation burden (Table S2). We also confirmed that none of the analyzed samples were from transplanted tissue. After our quality control, there were 14,672 samples from 944 donors from 48 diverse tissue and cell types. Library preparation, sequencing, alignment, and GTEx quality control are described in detail in (15).
Algorithms for detecting PZMs
LachesisDetect contains four basic steps. First, alignment files are filtered for extremely high-quality alignments. Next, the algorithm leverages cohort-wide information by simultaneously analyzing all samples to estimate position-specific error models for over 115 Mb of the transcriptome. LachesisDetect uses these models to detect putative postzygotic mutations (PZMs) with single-sample calling. Third, the method removes sources of false positive PZMs such as RNA editing and allele-specific expression of germline variants using > 15 filters based on theoretical and experimental validation metrics. In the last step, the method leverages donor information by jointly analyzing all samples in a donor to detect mutations with low power and estimate empirical false positive rates (Fig. S1).
PZM Validation
We performed several orthogonal validation experiments to quantify the FDR of the mutation calling algorithm. These efforts included both in silico and experimental approaches and involved analysis of both DNA and RNA from the tissues used for mutation detection. A summary of the validation results is in Table S3. We analyzed independent genomics datasets generated by the ENCODE project on four GTEx donors, encompassing 245 DNA assays and 67 RNA assays generated from 4 GTEx donors. Finally, we generated our own validation data by performing targeted DNA sequencing of over 1,650 putative PZMs using DNA from GTEx donors.
Mutation burden modeling
In order to evaluate biological and technical sources of variation in mutation burden, we used linear mixed effect models. We explored a large variety of model choices to arrive at our final modeling framework, comparing modeling choices using deviance, stability of model fitting, and other diagnostics. We used type II ANOVA to summarize the relative contributions of covariates.
Algorithm for mapping PZMs to a developmental tree
We manually derived two developmental tissue trees that represent the phylogenetic relationships among GTEx tissues during human development, using information from the literature: the full tree, and the simplified germ layer tree. We then developed an algorithm, LachesisMap, to reconstruct the phylogenetic history of multi-tissue PZMs. The algorithm jointly analyzes all multi-tissue PZMs and accounts for missing data as well as differential PZM detection power due to differences in VAF, expression level, and tissue profiling in the dataset.
Sperm Sequencing Experiments
Ejaculated sperm and venous blood were collected from a European American. Sperm samples had normal sperm density, sperm motility and morphology. Fresh ejaculates were stained using the LIVE/DEAD Sperm Viability Kit (Invitrogen) and propidium iodide (PI). Sperm samples were then selectively sorted via fluorescence-activated cell sorting (FACS) into 96 well plates (~200 sperm cells per well) and 5 ml Falcon tubes based on their staining. We used MALBAC amplification (61) to prepare up to 1.5 μg of DNA from each pool of sperm using a kit, and 6 pools were selected for sequencing. Exome library preparation was performed according to the manufacturer’s protocol using 50 ng of pre-amplified MALBAC reactions or DNA extracted from blood.
Supplementary Material
Acknowledgements
We would like to thank the GTEx donors and families for their generous and selfless donation of tissues and organs for the advancement of science. We thank T. Lappalainen (New York Genome Center & KTH Royal Institute of Technology), H. Leitch (London Institute of Medical Sciences), G. Coop (UC Davis), I. Martincorena (Sanger Institute), the M. Griffiths lab (Washington University), S. Montgomery lab (Stanford University), the Conrad lab and the Cohen lab (Washington University) for data sharing and helpful discussions. Sequencing for the validation experiments was performed by the Genome Technology Access Center (GTAC) in the Department of Genetics at Washington University School of Medicine. We appreciate obtaining access to de novo mutations on SFARI base (https://base.sfari.org).
We would like to thank the following funders for their support of the TwinsUK resource: Wellcome Trust, Medical Research Council, European Union, Chronic Disease Research Foundation (CDRF), the National Institute for Health Research (NIHR)-funded BioResource Clinical Research Facility and Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London.
We would also like to acknowledge that this manuscript includes several analyses that focus on GTEx donors with European and African American ancestry and less so on other ancestries in GTEx. This decision was made so that we’d have higher statistical power to detect potentially rare genetic signals that may vary across populations. We hope that in future studies, larger numbers of Black, Indigenous, and People of Color are included so that we can learn about all populations in an equitable manner.
Funding
National Institutes of Health grant R01MH101810 (to DFC), R01HG007178 (to DFC), and R01HD078641 (to DFC)
National Human Genome Research Institute grant T32HG000045 (to BAC)
Medical Research Council MR/M004422/1(to KSS) and MR/R023131/1 (to KSS)
Footnotes
Competing interests
D.F.C. is an advisor to Paterna Biosciences. All other authors declare that they have no competing interests.
Data and materials availability
All GTEx protected data are available through the database of Genotypes and Phenotypes (dbGaP) (accession no. phs000424.v8). Access to the raw sequence data is provided through the AnVIL platform (https://gtexportal.org/home/protectedDataAccess).
The cancer results are based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. de novo mutations were obtained from denovo-db (https://denovo-db.gs.washington.edu/denovo-db/).
Data generated on GTEx tissues by the ENCODE project are available from http://www.encodeproject.org
Source code used in this study is available from https://github.com/conradlab/RockweilerEtAl.Archived source code at time of publication: doi:10.5281/zenodo.7378922
REFERENCES AND NOTES
- 1.Melzer D, Pilling LC, Ferrucci L, The genetics of human ageing. Nat. Rev. Genet. 21, 88–101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, Purcell SM, Svantesson O, Landén M, Höglund M, Lehmann S, Gabriel SB, Moran JL, Lander ES, Sullivan PF, Sklar P, Grönberg H, Hultman CM, McCarroll SA, Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, Ozenberger BA, Welch JS, Link DC, Walter MJ, Mardis ER, Dipersio JF, Chen F, Wilson RK, Ley TJ, Ding L, Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 20, 1472–1478 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Young AL, Challen GA, Birmann BM, Druley TE, Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 7, 12484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, Karger A, Lee S, Chittenden TW, D’Gama AM, Cai X, Luquette LJ, Lee E, Park PJ, Walsh CA, Somatic mutation in single human neurons tracks developmental and transcriptional history. Science. 350, 94–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abyzov A, Tomasini L, Zhou B, Vasmatzis N, Coppola G, Amenduni M, Pattni R, Wilson M, Gerstein M, Weissman S, Urban AE, Vaccarino FM, One thousand somatic SNVs per skin fibroblast cell set baseline of mosaic mutational load with patterns that suggest proliferative origin. Genome Res. 27, 512–523 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, Stebbings L, Menzies A, Widaa S, Stratton MR, Jones PH, Campbell PJ, Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 348, 880–886 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yizhak K, Aguet F, Kim J, Hess JM, Kübler K, Grimsby J, Frazer R, Zhang H, Haradhvala NJ, Rosebrock D, Livitz D, Li X, Arich-Landkof E, Shoresh N, Stewart C, Segrè AV, Branton PA, Polak P, Ardlie KG, Getz G, RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. 364, eaaw0726 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.García-Nieto PE, Morrison AJ, Fraser HB, The somatic mutation landscape of the human body. Genome Biol. 20, 298 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moore L, Cagan A, Coorens THH, Neville MDC, Sanghvi R, Sanders MA, Oliver TRW, Leongamornlert D, Ellis P, Noorani A, Mitchell TJ, Butler TM, Hooks Y, Warren AY, Jorgensen M, Dawson KJ, Menzies A, O’Neill L, Latimer C, Teng M, van Boxtel R, Iacobuzio-Donahue CA, Martincorena I, Heer R, Campbell PJ, Fitzgerald RC, Stratton MR, Rahbari R, The mutational landscape of human somatic and germline cells. Nature. 597, 381–386 (2021). [DOI] [PubMed] [Google Scholar]
- 11.Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, Davies H, Stratton MR, Campbell PJ, Universal patterns of selection in cancer and somatic tissues. Cell. 173, 1823 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, Lamprecht TL, Shen D, Hundal J, Fulton RS, Heath S, Baty JD, Klco JM, Ding L, Mardis ER, Westervelt P, DiPersio JF, Walter MJ, Graubert TA, Ley TJ, Druley T, Link DC, Wilson RK, Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 518, 552–555 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Gole J, Gore A, He Q, Lu M, Min J, Yuan Z, Yang X, Jiang Y, Zhang T, Suo C, Li X, Cheng L, Zhang Z, Niu H, Li Z, Xie Z, Shi H, Zhang X, Fan M, Wang X, Yang Y, Dang J, McConnell C, Zhang J, Wang J, Yu S, Ye W, Gao Y, Zhang K, Liu R, Jin L, Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat. Commun. 11, 3475 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coorens THH, Moore L, Robinson PS, Sanghvi R, Christopher J, Hewinson J, Przybilla MJ, Lawson ARJ, Spencer Chapman M, Cagan A, Oliver TRW, Neville MDC, Hooks Y, Noorani A, Mitchell TJ, Fitzgerald RC, Campbell PJ, Martincorena I, Rahbari R, Stratton MR, Extensive phylogenies of human development inferred from somatic mutations. Nature. 597, 387–392 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Aguet F, Barbeira AN, Bonazzola R, Brown A, Castel SE, Jo B, Kasela S, Kim-Hellmuth S, Liang Y, Oliva M, Parsana PE, Flynn E, Fresard L, Gaamzon ER, Hamel AR, He Y, Hormozdiari F, Mohammadi P, Muñoz-Aguirre M, Park Y, Saha A, Segrć AV, Strober BJ, Wen X, Wucher V, Das S, Garrido-Martín D, Gay NR, Handsaker RE, Hoffman PJ, Kashin S, Kwong A, Li X, MacArthur D, Rouhana JM, Stephens M, Todres E, Viñuela A, Wang G, Zou Y, Brown CD, Cox N, Dermitzakis E, Engelhardt BE, Getz G, Guigo R, Montgomery SB, Stranger BE, Im HK, Battle A, Ardlie KG, Lappalainen T, The GTEx Consortium, The GTEx Consortium atlas of genetic regulatory effects across human tissues,, doi: 10.1101/787903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. See supplementary materials.
- 17.Enge M, Arda HE, Mignardi M, Beausang J, Bottino R, Kim SK, Quake SR, Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell. 171, 321–330.e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.U.S. Cancer Statistics Working Group, U.S. Cancer Statistics Data Visualizations Tool (2021), (available at www.cdc.gov/cancer/dataviz).
- 19.Goldmann JM, Hampstead JE, Wong WSW, Wilfert AB, Turner TN, Jonker MA, Bernier R, Huynen MA, Eichler EE, Veltman JA, Maxwell GL, Gilissen C, Differences in the number of de novo mutations between individuals are due to small family-specific effects and stochasticity. Genome Res. 31, 1513–1518 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frank SA, Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc. Natl. Acad. Sci. U. S. A. 107 Suppl 1, 1725–1730 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lynch MD, Lynch CNS, Craythorne E, Liakath-Ali K, Mallipeddi R, Barker JN, Watt FM, Spatial constraints govern competition of mutant clones in human epidermis. Nat. Commun. 8, 1119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, Islam SMA, Lopez-Bigas N, Klimczak LJ, McPherson JR, Morganella S, Sabarinathan R, Wheeler DA, Mustonen V, PCAWG Mutational Signatures Working Group, Getz G, Rozen SG, Stratton MR, PCAWG Consortium, The repertoire of mutational signatures in human cancer. Nature. 578, 94–101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao J-J, Pan X-R, Hu J, Ma L, Wu J-M, Shao Y-L, Barton SA, Woodruff RC, Zhang Y-P, Fu Y-X, Highly variable recessive lethal or nearly lethal mutation rates during germ-line development of male Drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A. 108, 15914–15919 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuijk E, Blokzijl F, Jager M, Besselink N, Boymans S, Chuva de Sousa Lopes SM, van Boxtel R, Cuppen E, Early divergence of mutational processes in human fetal tissues. Sci. Adv. 5, eaaw1271 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindsay SJ, Rahbari R, Kaplanis J, Keane T, Hurles ME, Similarities and differences in patterns of germline mutation between mice and humans. Nature Communications. 10 (2019),, doi: 10.1038/s41467-019-12023-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J, A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Genome Aggregation Database Consortium, Neale BM, Daly MJ, MacArthur DG, The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turner TN, Yi Q, Krumm N, Huddleston J, Hoekzema K, F Stessman HA, Doebley A-L, Bernier RA, Nickerson DA, Eichler EE, Denovo-db: A compendium of human de novo variants. Nucleic Acids Res. 45, D804–D811 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium, Pan-cancer analysis of whole genomes. Nature. 578, 82–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldman N, Yang Z, A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 11, 725–736 (1994). [DOI] [PubMed] [Google Scholar]
- 31.Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MR, Fitzgerald RC, Handford PA, Campbell PJ, Saeb-Parsy K, Jones PH, Somatic mutant clones colonize the human esophagus with age. Science. 362, 911–917 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB, The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 17, 225–234 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim K-H, Kohlhammer H, Xu W, Yang Y, Zhao H, Shaffer AL, Romesser P, Wright G, Powell J, Rosenwald A, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Staudt LM, Oncogenically active MYD88 mutations in human lymphoma. Nature. 470, 115–119 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rivas MA, Pirinen M, Conrad DF, Lek M, Tsang EK, Karczewski KJ, Maller JB, Kukurba KR, DeLuca DS, Fromer M, Ferreira PG, Smith KS, Zhang R, Zhao F, Banks E, Poplin R, Ruderfer DM, Purcell SM, Tukiainen T, Minikel EV, Stenson PD, Cooper DN, Huang KH, Sullivan TJ, Nedzel J, Bustamante CD, Li JB, Daly MJ, Guigo R, Donnelly P, Ardlie K, Sammeth M, Dermitzakis ET, McCarthy MI, Montgomery SB, Lappalainen T, MacArthur DG, Segre AV, Young TR, Gelfand ET, Trowbridge CA, Ward LD, Kheradpour P, Iriarte B, Meng Y, Palmer CD, Esko T, Winckler W, Hirschhorn J, Kellis M, Getz G, Shablin AA, Li G, Zhou Y-H, Nobel AB, Rusyn I, Wright FA, Battle A, Mostafavi S, Mele M, Reverter F, Goldmann J, Koller D, Gamazon ER, Im HK, Konkashbaev A, Nicolae DL, Cox NJ, Flutre T, Wen X, Stephens M, Pritchard JK, Tu Z, Zhang B, Huang T, Long Q, Lin L, Yang J, Zhu J, Liu J, Brown A, Mestichelli B, Tidwell D, Lo E, Salvatore M, Shad S, Thomas JA, Lonsdale JT, Choi RC, Karasik E, Ramsey K, Moser MT, Foster BA, Gillard BM, Syron J, Fleming J, Magazine Harold, Hasz R, Walters GD, Bridge JP, Miklos M, Sullivan S, Barker LK, Traino H, Mosavel M, Siminoff LA, Valley DR, Rohrer DC, Jewel S, Branton P, Sobin LH, Barcus M, Qi L, Hariharan P, Wu S, Tabor D, Shive C, Smith AM, Buia SA, Undale AH, Robinson KL, Roche N, Valentino KM, Britton A, Burges R, Bradbury D, Hambright KW, Seleski J, Korzeniewski GE, Erickson K, Marcus Y, Tejada J, Taherian M, Lu C, Robles BE, Basile M, Mash DC, Volpi S, Struewing JP, Temple GF, Boyer J, Colantuoni D, Little R, Koester S, Carithers LJ, Moore HM, Guan P, Compton C, Sawyer SJ, Demchok JP, Vaught JB, Rabiner CA, Lockhart NC, Friedlander MR, ‘t Hoen PAC, Monlong J, Gonzàlez-Porta M, Kurbatova N, Griebel T, Barann M, Wieland T, Greger L, van Iterson M, Almlof J, Ribeca P, Pulyakhina I, Esser D, Giger T, Tikhonov A, Sultan M, Bertier G, Lizano E, Buermans HPJ, Padioleau I, Schwarzmayr T, Karlberg O, Ongen H, Kilpinen H, Beltran S, Gut M, Kahlem K, Amstislavskiy V, Stegle O, Flicek P, Strom TM, Lehrach H, Schreiber S, Sudbrak R, Carracedo A, Antonarakis SE, Hasler R, Syvanen A-C, van Ommen G-J, Brazma A, Meitinger T, Rosenstiel P, Gut IG, Estivill X, The GTEx Consortium, The Geuvadis Consortium, Effect of predicted protein-truncating genetic variants on the human transcriptome. Science. 348, 666–669 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conrad DF, Keebler JEM, DePristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, Zilversmit M, Cartwright R, Rouleau GA, Daly M, Stone EA, Hurles ME, Awadalla P, 1000 Genomes Project, Variation in genome-wide mutation rates within and between human families. Nat. Genet. 43, 712–714 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roach JC, Glusman G, Smit AFA, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, Shendure J, Drmanac R, Jorde LB, Hood L, Galas DJ, Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 328, 636–639 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rahbari R, UK10K Consortium, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Al Turki S, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, Timing, rates and spectra of human germline mutation. Nat. Genet. 48, 126–133 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, Wong WSW, Sigurdsson G, Walters GB, Steinberg S, Helgason H, Thorleifsson G, Gudbjartsson DF, Helgason A, Magnusson OT, Thorsteinsdottir U, Stefansson K, Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 488, 471–475 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, Wu W, Corominas R, Peoples A, Koren A, Gore A, Kang S, Lin GN, Estabillo J, Gadomski T, Singh B, Zhang K, Akshoomoff N, Corsello C, McCarroll S, Iakoucheva LM, Li Y, Wang J, Sebat J, Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 151, 1431–1442 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang X, Breuss MW, Xu X, Antaki D, James KN, Stanley V, Ball LL, George RD, Wirth SA, Cao B, Nguyen A, McEvoy-Venneri J, Chai G, Nahas S, Van Der Kraan L, Ding Y, Sebat J, Gleeson JG, Developmental and temporal characteristics of clonal sperm mosaicism. Cell. 184, 4772–4783.e15 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hodgkinson A, Idaghdour Y, Gbeha E, Grenier J-C, Hip-Ki E, Bruat V, Goulet J-P, de Malliard T, Awadalla P, High-resolution genomic analysis of human mitochondrial RNA sequence variation. Science. 344, 413–415 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Loh P-R, Genovese G, Handsaker RE, Finucane HK, Reshef YA, Palamara PF, Birmann BM, Talkowski ME, Bakhoum SF, McCarroll SA, Price AL, Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature. 559, 350–355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding J, McConechy MK, Horlings HM, Ha G, Chun Chan F, Funnell T, Mullaly SC, Reimand J, Bashashati A, Bader GD, Huntsman D, Aparicio S, Condon A, Shah SP, Systematic analysis of somatic mutations impacting gene expression in 12 tumour types. Nat. Commun. 6, 8554 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erickson A, He M, Berglund E, Marklund M, Mirzazadeh R, Schultz N, Kvastad L, Andersson A, Bergenstråhle L, Bergenstråhle J, Larsson L, Alonso Galicia L, Shamikh A, Basmaci E, Díaz De Ståhl T, Rajakumar T, Doultsinos D, Thrane K, Ji AL, Khavari PA, Tarish F, Tanoglidi A, Maaskola J, Colling R, Mirtti T, Hamdy FC, Woodcock DJ, Helleday T, Mills IG, Lamb AD, Lundeberg J, Spatially resolved clonal copy number alterations in benign and malignant tissue. Nature. 608, 360–367 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maher GJ, Ralph HK, Ding Z, Koelling N, Mlcochova H, Giannoulatou E, Dhami P, Paul DS, Stricker SH, Beck S, McVean G, Wilkie AOM, Goriely A, Selfish mutations dysregulating RAS-MAPK signaling are pervasive in aged human testes. Genome Res. 28, 1779–1790 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goriely A, Wilkie AOM, Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. Am. J. Hum. Genet. 90, 175–200 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aiello NM, Stanger BZ, Echoes of the embryo: using the developmental biology toolkit to study cancer. Dis. Model. Mech 9, 105–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gong T, Zhang C, Ni X, Li X, Li J, Liu M, Zhan D, Xia X, Song L, Zhou Q, Ding C, Qin J, Wang Y, A time-resolved multi-omic atlas of the developing mouse liver. Genome Res. 30, 263–275 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin Y-T, Capel B, Cell fate commitment during mammalian sex determination. Curr. Opin. Genet. Dev. 32, 144–152 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sasaki K, Yokobayashi S, Nakamura T, Okamoto I, Yabuta Y, Kurimoto K, Ohta H, Moritoki Y, Iwatani C, Tsuchiya H, Nakamura S, Sekiguchi K, Sakuma T, Yamamoto T, Mori T, Woltjen K, Nakagawa M, Yamamoto T, Takahashi K, Yamanaka S, Saitou M, Robust in vitro induction of human germ cell fate from pluripotent stem cells. Cell Stem Cell. 17, 178–194 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Kobayashi T, Zhang H, Tang WWC, Irie N, Withey S, Klisch D, Sybirna A, Dietmann S, Contreras DA, Webb R, Allegrucci C, Alberio R, Surani MA, Principles of early human development and germ cell program from conserved model systems. Nature. 546, 416–420 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tyser RCV, Mahammadov E, Nakanoh S, Vallier L, Scialdone A, Srinivas S, Single-cell transcriptomic characterization of a gastrulating human embryo. Nature. 600, 285–289 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB, Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 9, e1003709 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eilbeck K, Quinlan A, Yandell M, Settling the score: variant prioritization and Mendelian disease. Nat. Rev. Genet. 18, 599–612 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup, The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Costello M, Pugh TJ, Fennell TJ, Stewart C, Lichtenstein L, Meldrim JC, Fostel JL, Friedrich DC, Perrin D, Dionne D, Kim S, Gabriel SB, Lander ES, Fisher S, Getz G, Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 41, e67 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen L, Liu P, Evans TC Jr, Ettwiller LM, DNA damage is a pervasive cause of sequencing errors, directly confounding variant identification. Science. 355, 752–756 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Carrot-Zhang J, Majewski J, LoLoPicker: detecting low allelic-fraction variants from low-quality cancer samples. Oncotarget. 8, 37032–37040 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, Johnson R, Segrè AV, Djebali S, Niarchou A, GTEx Consortium, Wright FA, Lappalainen T, Calvo M, Getz G, Dermitzakis ET, Ardlie KG, Guigó R, Human genomics. The human transcriptome across tissues and individuals. Science. 348, 660–665 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.The GTEx Consortium, Ardlie KG, Deluca DS, Segrè AV, Sullivan TJ, Young TR, Gelfand ET, Trowbridge CA, Maller JB, Tukiainen T, Lek M, Ward LD, Kheradpour P, Iriarte B, Meng Y, Palmer CD, Esko T, Winckler W, Hirschhorn JN, Kellis M, MacArthur DG, Getz G, Shabalin AA, Li G, Zhou Y-H, Nobel AB, Rusyn I, Wright FA, Lappalainen T, Ferreira PG, Ongen H, Rivas MA, Battle A, Mostafavi S, Monlong J, Sammeth M, Mele M, Reverter F, Goldmann JM, Koller D, Guigó R, McCarthy MI, Dermitzakis ET, Gamazon ER, Im HK, Konkashbaev A, Nicolae DL, Cox NJ, Flutre T, Wen X, Stephens M, Pritchard JK, Tu Z, Zhang B, Huang T, Long Q, Lin L, Yang J, Zhu J, Liu J, Brown A, Mestichelli B, Tidwell D, Lo E, Salvatore M, Shad S, Thomas JA, Lonsdale JT, Moser MT, Gillard BM, Karasik E, Ramsey K, Choi C, Foster BA, Syron J, Fleming J, Magazine Harold, Hasz R, Walters GD, Bridge JP, Miklos M, Sullivan S, Barker LK, Traino HM, Mosavel M, Siminoff LA, Valley DR, Rohrer DC, Jewell SD, Branton PA, Sobin LH, Barcus M, Qi L, McLean J, Hariharan P, Um KS, Wu S, Tabor D, Shive C, Smith AM, Buia SA, Undale AH, Robinson KL, Roche N, Valentino KM, Britton A, Burges R, Bradbury D, Hambright KW, Seleski J, Korzeniewski GE, Erickson K, Marcus Y, Tejada J, Taherian M, Lu C, Basile M, Mash DC, Volpi S, Struewing JP, Temple GF, Boyer J, Colantuoni D, Little R, Koester S, Carithers LJ, Moore HM, Guan P, Compton C, Sawyer SJ, Demchok JP, Vaught JB, Rabiner CA, Lockhart NC, Ardlie KG, Getz G, Wright FA, Kellis M, Volpi S, Dermitzakis ET, The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science. 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Engström PG, The RGASP Consortium, Steijger T, Sipos B, Grant GR, Kahles A, Rätsch G, Goldman N, Hubbard TJ, Harrow J, Guigó R, Bertone P, Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods. 10, 1185–1191 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nishikura K, Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 79, 321–349 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li S, Mason CE, The pivotal regulatory landscape of RNA modifications. Annu. Rev. Genomics Hum. Genet. 15, 127–150 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Ramaswami G, Li JB, RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 42, D109–13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haeussler M, Zweig AS, Tyner C, Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Hinrichs AS, Gonzalez JN, Gibson D, Diekhans M, Clawson H, Casper J, Barber GP, Haussler D, Kuhn RM, Kent WJ, The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 47, D853–D858 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, Levanon EY, A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 24, 365–376 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA, The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ju YS, Martincorena I, Gerstung M, Petljak M, Alexandrov LB, Rahbari R, Wedge DC, Davies HR, Ramakrishna M, Fullam A, Martin S, Alder C, Patel N, Gamble S, O’Meara S, Giri DD, Sauer T, Pinder SE, Purdie CA, Borg Å, Stunnenberg H, van de Vijver M, Tan BKT, Caldas C, Tutt A, Ueno NT, van ‘t Veer LJ, Martens JWM, Sotiriou C, Knappskog S, Span PN, Lakhani SR, Eyfjörd JE, Børresen-Dale A-L, Richardson A, Thompson AM, Viari A, Hurles ME, Nik-Zainal S, Campbell PJ, Stratton MR, Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature. 543, 714–718 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Freed D, Pevsner J, The contribution of mosaic variants to autism spectrum disorder. PLoS Genet. 12, e1006245 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dou Y, Gold HD, Luquette LJ, Park PJ, Detecting somatic mutations in normal cells. Trends Genet. 34, 545–557 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Luo Y, Hitz BC, Gabdank I, Hilton JA, Kagda MS, Lam B, Myers Z, Sud P, Jou J, Lin K, Baymuradov UK, Graham K, Litton C, Miyasato SR, Strattan JS, Jolanki O, Lee J-W, Tanaka FY, Adenekan P, O’Neill E, Cherry JM, New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 48, D882–D889 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.ENCODE Project Consortium, An integrated encyclopedia of DNA elements in the human genome. Nature. 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ramu A, Conrad DF, Arnav: Site specific error models to identify variants in RNA. bioRxiv (2018),, doi: 10.1101/397539. [DOI] [Google Scholar]
- 75.Bolger AM, Lohse M, Usadel B, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li H, Durbin R, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fox J, Weisberg S, An R companion to applied regression (SAGE Publications, Thousand Oaks, CA, ed. 3, 2018). [Google Scholar]
- 78.Stoffel MA, Nakagawa S, Schielzeth H, rptR: repeatability estimation and variance decomposition by generalized linear mixed-effects models. Methods Ecol. Evol. 8, 1639–1644 (2017). [Google Scholar]
- 79.Oliva M, Muñoz-Aguirre M, Kim-Hellmuth S, Wucher V, Gewirtz ADH, Cotter DJ, Parsana P, Kasela S, Balliu B, Viñuela A, Castel SE, Mohammadi P, Aguet F, Zou Y, Khramtsova EA, Skol AD, Garrido-Martín D, Reverter F, Brown A, Evans P, Gamazon ER, Payne A, Bonazzola R, Barbeira AN, Hamel AR, Martinez-Perez A, Soria JM, Pierce BL, Stephens M, Eskin E, Dermitzakis ET, Segrè AV, Im HK, Engelhardt BE, Ardlie KG, Montgomery SB, Battle AJ, Lappalainen T, Guigó R, Stranger BE, Aguet F, Anand S, Ardlie KG, Gabriel S, Getz GA, Graubert A, Hadley K, Handsaker RE, Huang KH, Kashin S, Li X, MacArthur DG, Meier SR, Nedzel JL, Nguyen DT, Segrè AV, Todres E, Balliu B, Barbeira AN, Battle A, Bonazzola R, Brown A, Brown CD, Castel SE, Conrad DF, Cotter DJ, Cox N, Das S, de Goede OM, Dermitzakis ET, Einson J, Engelhardt BE, Eskin E, Eulalio TY, Ferraro NM, Flynn ED, Fresard L, Gamazon ER, Garrido-Martín D, Gay NR, Gloudemans MJ, Guigó R, Hame AR, He Y, Hoffman PJ, Hormozdiari F, Hou L, Im HK, Jo B, Kasela S, Kellis M, Kim-Hellmuth S, Kwong A, Lappalainen T, Li X, Liang Y, Mangul S, Mohammadi P, Montgomery SB, Muñoz-Aguirre M, Nachun DC, Nobel AB, Oliva M, Park Y, Park Y, Parsana P, Rao AS, Reverter F, Rouhana JM, Sabatti C, Saha A, Stephens M, Stranger BE, Strober BJ, Teran NA, Viñuela A, Wang G, Wen X, Wright F, Wucher V, Zou Y, Ferreira PG, Li G, Melé M, Yeger-Lotem E, Barcus ME, Bradbury D, Krubit T, McLean JA, Qi L, Robinson K, Roche NV, Smith AM, Sobin L, Tabor DE, Undale A, Bridge J, Brigham LE, Foster BA, Gillard BM, Hasz R, Hunter M, Johns C, Johnson M, Karasik E, Kopen G, Leinweber WF, McDonald A, Moser MT, Myer K, Ramsey KD, Roe B, Shad S, Thomas JA, Walters G, Washington M, Wheeler J, Jewell SD, Rohrer DC, Valley DR, Davis DA, Mash DC, Branton PA, Barker LK, Gardiner HM, Mosavel M, Siminoff LA, Flicek P, Haeussler M, Juettemann T, Kent WJ, Lee CM, Powell CC, Rosenbloom KR, Ruffier M, Sheppard D, Taylor K, Trevanion SJ, Zerbino DR, Abell NS, Akey J, Chen L, Demanelis K, Doherty JA, Feinberg AP, Hansen KD, Hickey PF, Jasmine F, Jiang L, Kaul R, Kibriya MG, Li JB, Li Q, Lin S, Linder SE, Pierce BL, Rizzardi LF, Skol AD, Smith KS, Snyder M, Stamatoyannopoulos J, Tang H, Wang M, Carithers LJ, Guan P, Koester SE, Little AR, Moore HM, Nierras CR, Rao AK, Vaught JB, Volpi S, GTEx Consortium, The impact of sex on gene expression across human tissues. Science. 369, eaba3066 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.PreAnalytiX, “PAXgene Tissue FIX Container (50 ml) Handbook” (2019), (available at https://www.qiagen.com/us/resources/download.aspx?id=9e333add-b31e-4d1e-a4a1-).
- 81.Hara K, Watanabe A, Matsumoto S, Matsuda Y, Kuwata T, Kan H, Yamada T, Koizumi M, Shinji S, Yamagishi A, Ishiwata T, Naito Z, Shimada T, Uchida E, Surgical specimens of colorectal cancer fixed with PAXgene Tissue system preserve high-quality RNA. Biopreserv. Biobank. 13, 325–334 (2015). [DOI] [PubMed] [Google Scholar]
- 82.Mathieson W, Marcon N, Antunes L, Ashford DA, Betsou F, Frasquilho SG, Kofanova OA, McKay SC, Pericleous S, Smith C, Unger KM, Zeller C, Thomas GA, A critical evaluation of the PAXgene tissue fixation system: Morphology, immunohistochemistry, molecular biology, and proteomics. Am. J. Clin. Pathol. 146, 25–40 (2016). [DOI] [PubMed] [Google Scholar]
- 83.Högnäs G, Kivinummi K, Kallio HML, Hieta R, Ruusuvuori P, Koskenalho A, Kesseli J, Tammela TLJ, Riikonen J, Ilvesaro J, Kares S, Hirvikoski PP, Laurila M, Mirtti T, Nykter M, Kujala PM, Visakorpi T, Tolonen T, Bova GS, Feasibility of prostate PAXgene fixation for molecular research and diagnostic surgical pathology. Am. J. Surg. Pathol. 42, 103–115 (2018). [DOI] [PubMed] [Google Scholar]
- 84.Bates D, Mächler M, Bolker B, Walker S, Fitting Linear Mixed-Effects Models using lme4. arXiv [stat.CO] (2014), (available at http://arxiv.org/abs/1406.5823). [Google Scholar]
- 85.Kimura M, The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics. 61, 893–903 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.The National Cancer Institute, The Biospecimen Research Database, (available at http://biospecimens.cancer.gov/brd).
- 87.Sulston JE, Schierenberg E, White JG, Thomson JN, The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64–119 (1983). [DOI] [PubMed] [Google Scholar]
- 88.Edgar R, Mazor Y, Rinon A, Blumenthal J, Golan Y, Buzhor E, Livnat I, Ben-Ari S, Lieder I, Shitrit A, Gilboa Y, Ben-Yehudah A, Edri O, Shraga N, Bogoch Y, Leshansky L, Aharoni S, West MD, Warshawsky D, Shtrichman R, LifeMap Discovery™: the embryonic development, stem cells, and regenerative medicine research portal. PLoS One. 8, e66629 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Editorial office Of journal “Morphologia,” Langman’s Medical Embryology. 14th Edition, 2018 Author: T.W. Sadler. Morphologia. 13, 90–96 (2019). [Google Scholar]
- 90.Teshima THN, Ianez RF, Coutinho-Camillo CM, Buim ME, Soares FA, Lourenço SV, Development of human minor salivary glands: expression of mucins according to stage of morphogenesis. J. Anat. 219, 410–417 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.GI Motility online, (available at http://www.nature.com/gimo/).
- 92.Hill M, Embryology (2020), (available at https://embryology.med.unsw.edu.au/embryology/index.php/Main_Page).
- 93.Forsberg LA, Gisselsson D, Dumanski JP, Mosaicism in health and disease - clones picking up speed. Nat. Rev. Genet. 18, 128–142 (2017). [DOI] [PubMed] [Google Scholar]
- 94.Behjati S, Huch M, van Boxtel R, Karthaus W, Wedge DC, Tamuri AU, Martincorena I, Petljak M, Alexandrov LB, Gundem G, Tarpey PS, Roerink S, Blokker J, Maddison M, Mudie L, Robinson B, Nik-Zainal S, Campbell P, Goldman N, van de Wetering M, Cuppen E, Clevers H, Stratton MR, Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature. 513, 422–425 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hodgkinson A, Eyre-Walker A, Variation in the mutation rate across mammalian genomes. Nat. Rev. Genet. 12, 756–766 (2011). [DOI] [PubMed] [Google Scholar]
- 96.Zhang Z, Gerstein M, Patterns of nucleotide substitution, insertion and deletion in the human genome inferred from pseudogenes. Nucleic Acids Res. 31, 5338–5348 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Buil A, Brown AA, Lappalainen T, Viñuela A, Davies MN, Zheng H-F, Richards JB, Glass D, Small KS, Durbin R, Spector TD, Dermitzakis ET, Gene-gene and gene-environment interactions detected by transcriptome sequence analysis in twins. Nat. Genet. 47, 88–91 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.The UK10K Consortium, Walter K, Min JL, Huang J, Crooks L, Memari Y, McCarthy S, Perry JRB, Xu C, Futema M, Lawson D, Iotchkova V, Schiffels S, Hendricks AE, Danecek P, Li R, Floyd J, Wain LV, Barroso I, Humphries SE, Hurles ME, Zeggini E, Barrett JC, Plagnol V, Brent Richards J, Greenwood CMT, Timpson NJ, Durbin R, Soranzo N, Bala S, Clapham P, Coates G, Cox T, Daly A, Danecek P, Du Y, Durbin R, Edkins S, Ellis P, Flicek P, Guo X, Guo X, Huang L, Jackson DK, Joyce C, Keane T, Kolb-Kokocinski A, Langford C, Li Y, Liang J, Lin H, Liu R, Maslen J, McCarthy S, Muddyman D, Quail MA, Stalker J, Sun J, Tian J, Wang G, Wang J, Wang Y, Wong K, Zhang P, Barroso I, Birney E, Boustred C, Chen L, Clement G, Cocca M, Danecek P, Davey Smith G, Day INM, Day-Williams A, Down T, Dunham I, Durbin R, Evans DM, Gaunt TR, Geihs M, Greenwood CMT, Hart D, Hendricks AE, Howie B, Huang J, Hubbard T, Hysi P, Iotchkova V, Jamshidi Y, Karczewski KJ, Kemp JP, Lachance G, Lawson D, Lek M, Lopes M, MacArthur DG, Marchini J, Mangino M, Mathieson I, McCarthy S, Memari Y, Metrustry S, Min JL, Moayyeri A, Muddyman D, Northstone K, Panoutsopoulou K, Paternoster L, Perry JRB, Quaye L, Brent Richards J, Ring S, Ritchie GRS, Schiffels S, Shihab HA, Shin S-Y, Small KS, Soler Artigas M, Soranzo N, Southam L, Spector TD, St Pourcain B, Surdulescu G, Tachmazidou I, Timpson NJ, Tobin MD, Valdes AM, Visscher PM, Wain LV, Walter K, Ward K, Wilson SG, Wong K, Yang J, Zeggini E, Zhang F, Zheng H-F, Anney R, Ayub M, Barrett JC, Blackwood D, Bolton PF, Breen G, Collier DA, Craddock N, Crooks L, Curran S, Curtis D, Durbin R, Gallagher L, Geschwind D, Gurling H, Holmans P, Lee I, Lönnqvist J, McCarthy S, McGuffin P, McIntosh AM, McKechanie AG, McQuillin A, Morris J, Muddyman D, O’Donovan MC, Owen MJ, Palotie A, Parr JR, Paunio T, Pietilainen O, Rehnström K, Sharp SI, Skuse D, St Clair D, Suvisaari J, Walters JTR, Williams HJ, Barroso I, Bochukova E, Bounds R, Dominiczak A, Durbin R, Farooqi IS, Hendricks AE, Keogh J, Marenne G, McCarthy S, Morris A, Muddyman D, O’Rahilly S, Porteous DJ, Smith BH, Tachmazidou I, Wheeler E, Zeggini E, Al Turki S, Anderson CA, Antony D, Barroso I, Beales P, Bentham J, Bhattacharya S, Calissano M, Carss K, Chatterjee K, Cirak S, Cosgrove C, Durbin R, Fitzpatrick DR, Floyd J, Reghan Foley A, Franklin CS, Futema M, Grozeva D, Humphries SE, Hurles ME, McCarthy S, Mitchison HM, Muddyman D, Muntoni F, O’Rahilly S, Onoufriadis A, Parker V, Payne F, Plagnol V, Lucy Raymond F, Roberts N, Savage DB, Scambler P, Schmidts M, Schoenmakers N, Semple RK, Serra E, Spasic-Boskovic O, Stevens E, van Kogelenberg M, Vijayarangakannan P, Walter K, Williamson KA, Wilson C, Whyte T, Ciampi A, Greenwood CMT, Hendricks AE, Li R, Metrustry S, Oualkacha K, Tachmazidou I, Xu C, Zeggini E, Bobrow M, Bolton PF, Durbin R, Fitzpatrick DR, Griffin H, Hurles ME, Kaye J, Kennedy K, Kent A, Muddyman D, Muntoni F, Lucy Raymond F, Semple RK, Smee C, Spector TD, Timpson NJ, Charlton R, Ekong R, Futema M, Humphries SE, Khawaja F, Lopes LR, Migone N, Payne SJ, Plagnol V, Pollitt RC, Povey S, Ridout CK, Robinson RL, Scott RH, Shaw A, Syrris P, Taylor R, Vandersteen AM, Barrett JC, Barroso I, Davey Smith G, Durbin R, Farooqi IS, Fitzpatrick DR, Hurles ME, Kaye J, Kennedy K, Langford C, McCarthy S, Muddyman D, Owen MJ, Palotie A, Brent Richards J, Soranzo N, Spector TD, Stalker J, Timpson NJ, Zeggini E, Amuzu A, Pablo Casas J, Chambers JC, Cocca M, Dedoussis G, Gambaro G, Gasparini P, Gaunt TR, Huang J, Iotchkova V, Isaacs A, Johnson J, Kleber ME, Kooner JS, Langenberg C, Luan J, Malerba G, März W, Matchan A, Min JL, Morris R, Nordestgaard BG, Benn M, Ring S, Scott RA, Soranzo N, Southam L, Timpson NJ, Toniolo D, Traglia M, Tybjaerg-Hansen A, van Duijn CM, van Leeuwen EM, Varbo A, Whincup P, Zaza G, Zeggini E, Zhang W, Writing group, Production group, Cohorts group, Neurodevelopmental disorders group, Obesity group, Rare disease group, Statistics group, Ethics group, Incidental findings group, Management committee, Lipid meta-analysis group, The UCLEB Consortium, The UK10K project identifies rare variants in health and disease. Nature. 526, 82–90 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Verdi S, Abbasian G, Bowyer RCE, Lachance G, Yarand D, Christofidou P, Mangino M, Menni C, Bell JT, Falchi M, Small KS, Williams FMK, Hammond CJ, Hart DJ, Spector TD, Steves CJ, TwinsUK: The UK adult twin registry update. Twin Res. Hum. Genet. 22, 523–529 (2019). [DOI] [PubMed] [Google Scholar]
- 100.Long T, Hicks M, Yu H-C, Biggs WH, Kirkness EF, Menni C, Zierer J, Small KS, Mangino M, Messier H, Brewerton S, Turpaz Y, Perkins BA, Evans AM, Miller LAD, Guo L, Caskey CT, Schork NJ, Garner C, Spector TD, Venter JC, Telenti A, Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites. Nat. Genet. 49, 568–578 (2017). [DOI] [PubMed] [Google Scholar]
- 101.Brazhnik K, Sun S, Alani O, Kinkhabwala M, Wolkoff AW, Maslov AY, Dong X, Vijg J, Single-cell analysis reveals different age-related somatic mutation profiles between stem and differentiated cells in human liver. Sci. Adv. 6, eaax2659 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sun S, Wang Y, Maslov AY, Dong X, Vijg J, SomaMutDB: a database of somatic mutations in normal human tissues. Nucleic Acids Res. 50, D1100–D1108 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. (1986), doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 104.Hernando B, Dietzen M, Parra G, Gil-Barrachina M, Pitarch G, Mahiques L, Valcuende-Cavero F, McGranahan N, Martinez-Cadenas C, The effect of age on the acquisition and selection of cancer driver mutations in sun-exposed normal skin. Ann. Oncol. 32, 412–421 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Deglincerti A, Croft GF, Pietila LN, Zernicka-Goetz M, Siggia ED, Brivanlou AH, Self-organization of the in vitro attached human embryo. Nature. 533, 251–254 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Behringer R, Gertsenstein M, Nagy K, Nagy A, Manipulating the mouse embryo: A laboratory manual, fourth edition (Cold Spring Harbor Laboratory Press, New York, NY, 2013). [Google Scholar]
- 107.Rubin H, The disparity between human cell senescence in vitro and lifelong replication in vivo. Nat. Biotechnol. 20, 675–681 (2002). [DOI] [PubMed] [Google Scholar]
- 108.Jaiswal S, Ebert BL, Clonal hematopoiesis in human aging and disease. Science. 366, eaan4673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G, Correa A, Banahan BF, Gabriel S, Kathiresan S, Stringham HM, McCarthy MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, Wilson JG, Neuberg D, Altshuler D, Ebert BL, Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, Fish P, Harsha B, Hathaway C, Jupe SC, Kok CY, Noble K, Ponting L, Ramshaw CC, Rye CE, Speedy HE, Stefancsik R, Thompson SL, Wang S, Ward S, Campbell PJ, Forbes SA, COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 47, D941–D947 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dang Y, Yan L, Hu B, Fan X, Ren Y, Li R, Lian Y, Yan J, Li Q, Zhang Y, Li M, Ren X, Huang J, Wu Y, Liu P, Wen L, Zhang C, Huang Y, Tang F, Qiao J, Tracing the expression of circular RNAs in human pre-implantation embryos. Genome Biol. 17 (2016), doi: 10.1186/s13059-016-0991-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mahyari E, Guo J, Lima AC, Lewinsohn DP, Stendahl AM, Vigh-Conrad KA, Nie X, Nagirnaja L, Rockweiler NB, Carrell DT, Hotaling JM, Aston KI, Conrad DF, Comparative single-cell analysis of biopsies clarifies pathogenic mechanisms in Klinefelter syndrome. Am. J. Hum. Genet. 108, 1924–1945 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weyrich A, Preparation of genomic DNA from mammalian sperm. Curr. Protoc. Mol. Biol. 2 (2012), doi: 10.1002/0471142727.mb0213s98. [DOI] [PubMed] [Google Scholar]
- 114.Zong C, Lu S, Chapman AR, Xie XS, Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 338, 1622–1626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G, Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L, VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 25, 2283–2285 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Solano F, Photoprotection and skin pigmentation: Melanin-related molecules and some other new agents obtained from natural sources. Molecules. 25, 1537 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pfeifer GP, You Y-H, Besaratinia A, Mutations induced by ultraviolet light. Mutat. Res. 571, 19–31 (2005). [DOI] [PubMed] [Google Scholar]
- 119.Kryazhimskiy S, Plotkin JB, The population genetics of dN/dS. PLoS Genet. 4, e1000304 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Williams MJ, Zapata L, Werner B, Barnes CP, Sottoriva A, Graham TA, Measuring the distribution of fitness effects in somatic evolution by combining clonal dynamics with dN/dS ratios. Elife. 9 (2020), doi: 10.7554/eLife.48714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pérez-Figueroa A, Posada D, Interpreting dN/dS under different selective regimes in cancer evolution. bioRxiv (2021),, doi: 10.1101/2021.11.30.470556. [DOI] [Google Scholar]
- 122.Reeves G, Specific stroma in the cortex and medulla of the ovary. Cell types and vascular supply in relation to follicular apparatus and ovulation. Obstet. Gynecol. 37, 832–844 (1971). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All GTEx protected data are available through the database of Genotypes and Phenotypes (dbGaP) (accession no. phs000424.v8). Access to the raw sequence data is provided through the AnVIL platform (https://gtexportal.org/home/protectedDataAccess).
The cancer results are based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. de novo mutations were obtained from denovo-db (https://denovo-db.gs.washington.edu/denovo-db/).
Data generated on GTEx tissues by the ENCODE project are available from http://www.encodeproject.org
Source code used in this study is available from https://github.com/conradlab/RockweilerEtAl.Archived source code at time of publication: doi:10.5281/zenodo.7378922