

Abstract
Individuals with urinary stone disease (USD) exhibit dysbiosis in the urinary tract and the loss of Lactobacillus that promote urinary tract health. However, the microbial metabolic functions that differentiate individuals with USD from healthy individuals are unknown. The objective of the current study was to determine the microbial functions across prokaryotic, viral, fungal, and protozoan domains that are associated with calcium oxalate (CaOx) stone formers through comparative shotgun metagenomics of midstream, voided urine samples for a small number of patients (n = 5 CaOx stone formers, n = 5 healthy controls). Results revealed that CaOx stone formers had reduced levels of genes associated with oxalate metabolism, as well as transmembrane transport, proteolysis, and oxidation–reduction processes. From 17 draft genomes extracted from the data and > 42,000 full length reference genomes, genes enriched in the Control group mapped overwhelming to Lactobacillus crispatus and those associated with CaOx mapped to Pseudomonas aeruginosa and Burkholderia sp. The microbial functions that differentiated the clinical cohorts are associated with known mechanisms of stone formation. While the prokaryotes most differentiated the CaOx and Control groups, a diverse, trans-domain microbiome was apparent. While our sample numbers were small, results corroborate previous studies and suggest specific microbial metabolic pathways in the urinary tract that modulate stone formation. Future studies that target these metabolic pathways as well as the influence of viruses, fungi, and protozoa on urinary tract physiology is warranted.
Keywords: Shotgun metagenomics, Urinary microbiome, Urolithiasis, Oxalate metabolism, Urinary calculi, Calcium oxalate
Introduction
The rising prevalence of urinary stone disease (USD) worldwide over the last fifty years has not only resulted in significant patient morbidity due to high recurrent symptomatic stone events [1], but also considerable burden to healthcare systems, with annual costs of $10 billion in the US alone [2]. The mechanisms that drive this increase in incidence are unknown.
Evidence for a significant role of the human microbiome, which contains all of the microorganisms present in or on the human body, in the onset of urinary stone disease has rapidly accumulated in recent years [3]. Numerous studies have linked the onset of kidney stones to past antibiotic use [4–6]. Additionally, metagenome-wide association studies, which examine the human microbiome through molecular techniques, have found links between USD and the microbiome [3]. There are clear and consistent signatures of a microbiome in non-infection stones themselves, acquired either through molecular analysis and microbial culture techniques [4, 7], or biogeochemical techniques [8]. A recent comparative multi-omic clinical study found that the urinary tract microbiome, but not the gut microbiome, defined cohorts with either USD (excluding infection stone cases) or those with no history of the disease [4]. These results implicate the urinary tract microbiome, previously thought to be sterile [9], as contributing more to the pathogenesis of USD than the gut microbiota.
Metagenome-wide association studies of the urinary tract microbiome have primarily utilized high-throughput 16S rRNA sequencing [4, 7, 10, 11], which provides a taxonomic survey of prokaryotic microorganisms in samples and can be used for comparative analyses to determine if the microbiome is associated with a particular clinical phenotype and quantify which specific taxa are significantly different between clinical cohorts. Few studies have examined the metabolic potential of the urinary tract microbiome through shotgun metagenomics [12, 13], which sequences the entire metagenome of the microbial community in an untargeted manner and allows for broader taxonomic resolution beyond the prokaryotes, as well as a more in depth functional exploration of the microbiome. Therefore, the metabolic potential of the urinary tract microbiome and how it relates to the pathogenesis of urologic disease is largely unknown.
To gain a better understanding of the metabolic potential and broader taxonomic profile of the urinary tract microbiome and how it relates to the pathogenesis of USD, we performed, to our knowledge, the first comparative shotgun metagenomic analysis of the urinary tract microbiome from patients with either an active episode of USD with pure calcium oxalate (CaOx) stones or from patients with no history of the disease. Based on past data, we hypothesized that the microbiome of USD patients would harbor lower metabolic diversity, and lower levels of Lactobacillus species in particular. Results of this analysis will help to elucidate the contribution of the urinary microbiome to USD etiology, with the potential to have broader implications for urologic health.
Methods
Participant recruitment
Following Institutional Review Board approval (IRB # 16–643), patients that either had an active episode of USD, in which patients were clinically diagnosed with kidney stones as confirmed by imaging studies (ultrasound or non-contrast computed tomography; n = 5), or no history of USD (n = 5), were prospectively identified and recruited for study participation after informed consent was obtained by a trained clinical research coordinator. Participants in the USD group presented for care to the Glickman Urological and Kidney Institute, Cleveland Clinic, and were restricted to patients with an active presence of stones, prior to prophylactic antibiotic intervention, to remove potential biases associated with surgical intervention or from being stone-free for a long period of time. Control participants came from the broader population who responded advertisements placed throughout Cleveland Clinic and had imaging to confirm they did not have stones. To balance the potential for the alterations of the microbiome due to antibiotic use, this metric was matched between groups at 30 days and 12 months prior to sample collection. The number of participants that used antibiotics in the last 30 days was low (n = 1 for each group). Stone composition was determined by infrared spectroscopy. For the USD group, only individuals who had confirmed pure CaOx stones were used. Participants were asked to provide information relating to biological sex, prior antibiotic use (past 30 days and past 12 months), age, personal history of USD, family history of USD, co-morbidities, height, weight, and gastrointestinal illness (Table 1). It must be noted, that in a study of this size, non-significant p values in clinical data do not necessarily rule out potential confounders of the clinical indications. Data were collected through participant questionnaires and medical chart review. Exclusion criteria included prior USD history (Control cohort only), known history of chronic gastrointestinal disorders or urinary tract infections, < 18 years of age, urinary tract infection, infected stones (CaOx cohort only), and patients with stones having a composition other than pure CaOx (CaOx cohort only).
Table 1.
Patient demographic data
| Variable | Control | CaOx | p value | Statistic |
|---|---|---|---|---|
|
| ||||
| No. enrolled | 5 | 5 | NA | NA |
| Age | 41 ± 19.59 | 36.6 ± 10.87 | NS | Student’s t test |
| % Female | 40% | 40% | NS | Relative risk, fisher exact test |
| Body mass index | 30.95 ± 4.1 | 24.17 ± 1.65 | NS | Student’s t test |
| % Prior USD | 0% | 40% | NS | Relative risk, fisher exact test |
| % Family history of USD | 40% | 40% | NS | Relative risk, fisher exact test |
| % Diabetic | 0% | 0% | NS | Relative risk, fisher exact test |
| % Hypertension | 0% | 0% | NS | Relative risk, fisher exact test |
| % Gastrointestinal illness | 20% | 0% | NS | Relative risk, fisher exact test |
| % Antibiotics used in past 30 days | 20% | 20% | NS | Relative risk, fisher exact test |
| % Antibiotics used in past 12 months | 40% | 60% | NS | Relative risk, fisher exact test |
Urine collection and DNA extraction
Participants provided a midstream, voided urine sample of at least 25 ml volume in the clinic, prior to any pre- or periprocedural antibiotics and prior to stone removal for the CaOx group, following our IRB-approved protocol. Samples were preserved in boric acid prior to processing within 24 h of receipt. Prior to DNA extraction, urine samples were centrifuged at 15,000 g for five minutes to pellet bacteria.
The DNA from the bacterial pellet was extracted using the Urine DNA Isolation Kit for Exfoliated Cells or Bacteria (Norgen; Thorold, ON, Canada), with modifications from the manufacturer’s protocol, to include lysozyme, proteinase K, and mutanolysin to bacterial pellets to facilitate bacterial lysis, as previously described [4]. Negative controls included preservation and DNA extraction reagents. Only samples exhibiting a clear band on gel electrophoresis and with a DNA concentration greater than 10 ng/μl were sent for shotgun metagenomic sequencing.
DNA sequencing and analysis
Extracted DNA was sent to Argonne National Laboratory (Chicago, IL) for paired-end shotgun metagenomic sequencing on an Illumina HiSeq platform of 151 base pair segments. From the raw data, low quality reads were trimmed and paired ends were merged using default parameters in BBMerge [14]. Additionally, reads mapped to the human reference genome (GRCh38), using BWA mem with default settings [15], were removed. Paired, non-human, sequences were pooled across samples and assembled in MetaSpades with default parameters [16]. Resulting contigs were used in pipelines to extract full-length genes for comparisons between the Control and CaOx groups and to build draft genomes de novo, as described below and presented in Fig. 1.
Fig. 1.
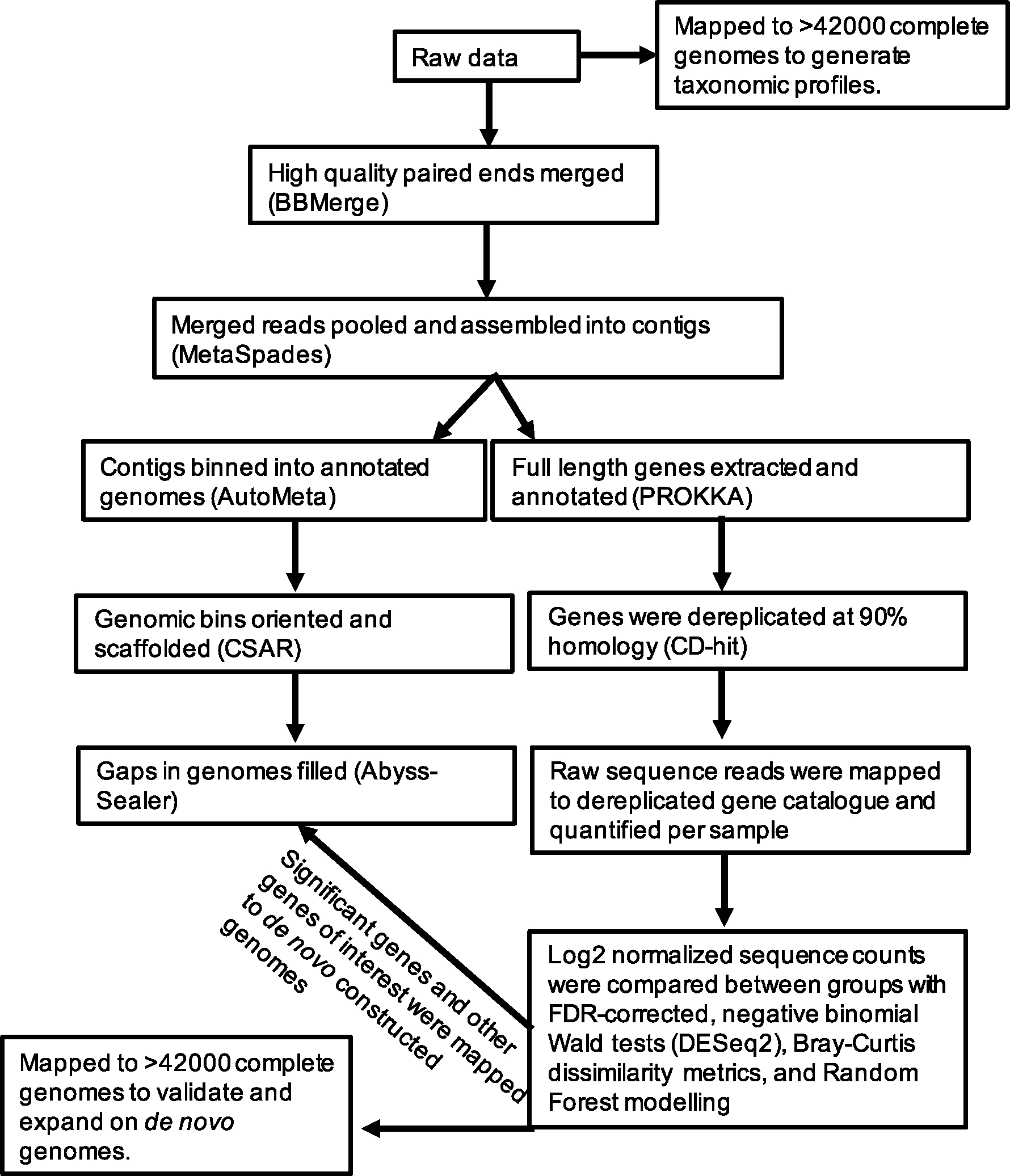
Summary of the bioinformatic pipelines used in the current study
Taxonomic profiling
To generate taxonomic profiles for each sample, non-host sequence reads were mapped to a non-redundant database containing 42,697 complete bacterial, archaeal, viral, fungal, and protozoan genomes downloaded from the NCBI, using BWA mem. Count tables of microbial communities were then reduced to species or lowest assigned taxonomy for profiling and comparative analyses. To establish confidence in taxonomic assignment, sequence homology was calculated for each domain as a whole and for the most abundant taxa by calculating the number of mismatched bases between the sequenced DNA and the reference genomes.
Gene-level profiling
Full-length microbial genes were extracted and annotated from contigs using PROKKA [17]. Gene annotation was achieved by mapping to the UniProt protein database. To dereplicate genes, ensure common nomenclature across samples, and build a reference gene catalog for the quantification of gene counts, full-length genes were clustered at 90% homology in CD-hit [18]. Subsequently, raw reads for each sample were mapped to the high quality, annotated gene catalog to generate gene-level count tables with BWA mem.
Comparative analyses
Raw count tables were normalized using the DESeq2 algorithm [19] and differential abundance analysis was conducted with a negative binomial Wald test [19]. All p values were false discovery rate (FDR) corrected for multiple comparisons with a cut-off at 0.05. Normalized count tables were also used to generate a weighted Bray–Curtis dissimilarity matrix in the Vegan R package, with statistical comparisons with a PERMANOVA at 999 permutations [20] and estimates of taxonomic or genetic richness using the “observed species” function in the Vegan R package, with statistical comparisons generated by a paired t-test. To determine which taxa and genes were differentially abundant between groups, the DESeq2 algorithm was used with FDR correction [19].
de novo genome construction
To determine which microbial taxa harbored the genes that were significantly different between the Control and CaOx groups, and thus help elucidate which taxa are important for maintaining urologic health, we constructed microbial genomes from the shotgun metagenomic data, de novo. For the de novo construction of genomes, assembled contigs were binned to genomes with Autometa [21]. This algorithm takes assembled contigs as input, bins the data to genomes using contig coverage values, GC content, and the Barnes-Hut t-Distributed Stochastic Neighbor Embedding (BH t-SNE) distribution of the contigs. Taxonomy is assigned based on the consensus classification of all contigs in a genomic bin. Completeness and purity calculations are based on the presence of known, unique universal single copy genes in Autometa.
After genomic binning, the contigs were oriented and scaffolded in CSAR [22], which compares the genomic bins to reference genomes of close relatives. Gaps within the genomes were filled with Abyss-Sealer [23] using raw sequencing reads as input. The completeness and purity of genomes were recalculated and taxonomic assignment was validated through phylogenetic analysis of the de novo constructed genomes in comparison with all complete NCBI genomes, achieved through Phylophlan, using the “—accurate” parameter which considers more phylogenetic positions at the cost of computational speed [24]. Phylophlan allows for the phylogenetic analysis of bacteria and archaea using complete genomes rather than gene amplicons, which provides greater resolution on phylogenetic analysis.
Results
Patient characteristics
A total of 10 patients, 5 in each cohort, were recruited for the current study. There were no significant differences in past antibiotic use, age, prior instances of USD, diabetic status, hypertension, body mass index, sex, or gastrointestinal illness between the CaOx and Control group (Table 1).
Shotgun metagenomic analysis and diversity metrics
From 10 samples, over 305 million sequencing reads were generated. After quality control and removal of host-derived reads, 80.6 million reads remained. From urine DNA samples, < 0.75% of reads overlapped with stool samples from the same patients based on targeted sequencing of the 16S rRNA gene [4]. There was a significant difference in the proportion of microbial reads retained, with proportionally more reads retained in the CaOx group compared to Controls (Fig. 2A). After sequence assembly and the extraction of full-length genes, a total of 53,531 microbial genes were defined, of which 27,884 were successfully annotated (52%). Both the CaOx and Control groups were dominated by Prokaryotes (Fig. 2B), with the Portiera and Klebsiella as the dominant Prokaryotic genera (Fig. 2C). The fungal profiles were almost entirely comprised Malassezia restricta in both groups (Fig. 2D), while the BeAn 58,058 virus dominated the viral fraction (Fig. 2E). Protozoan profiles were dominated primarily by Plasmodium vivax and Cyclospora cayetanensis (Fig. 2F). Sequence homology was consistently above 98% for the fungal, prokaryotic, and viral domains as a whole, while the protozoan domain averaged between 93 and 96% homology compared to reference genomes (Fig. 3A). The most abundant prokaryotes and fungi exhibited sequence homology > 97%, while the most abundant viruses and protozoa exhibited ~ 94% sequence homology with reference genomes (Fig. 3B).
Fig. 2.
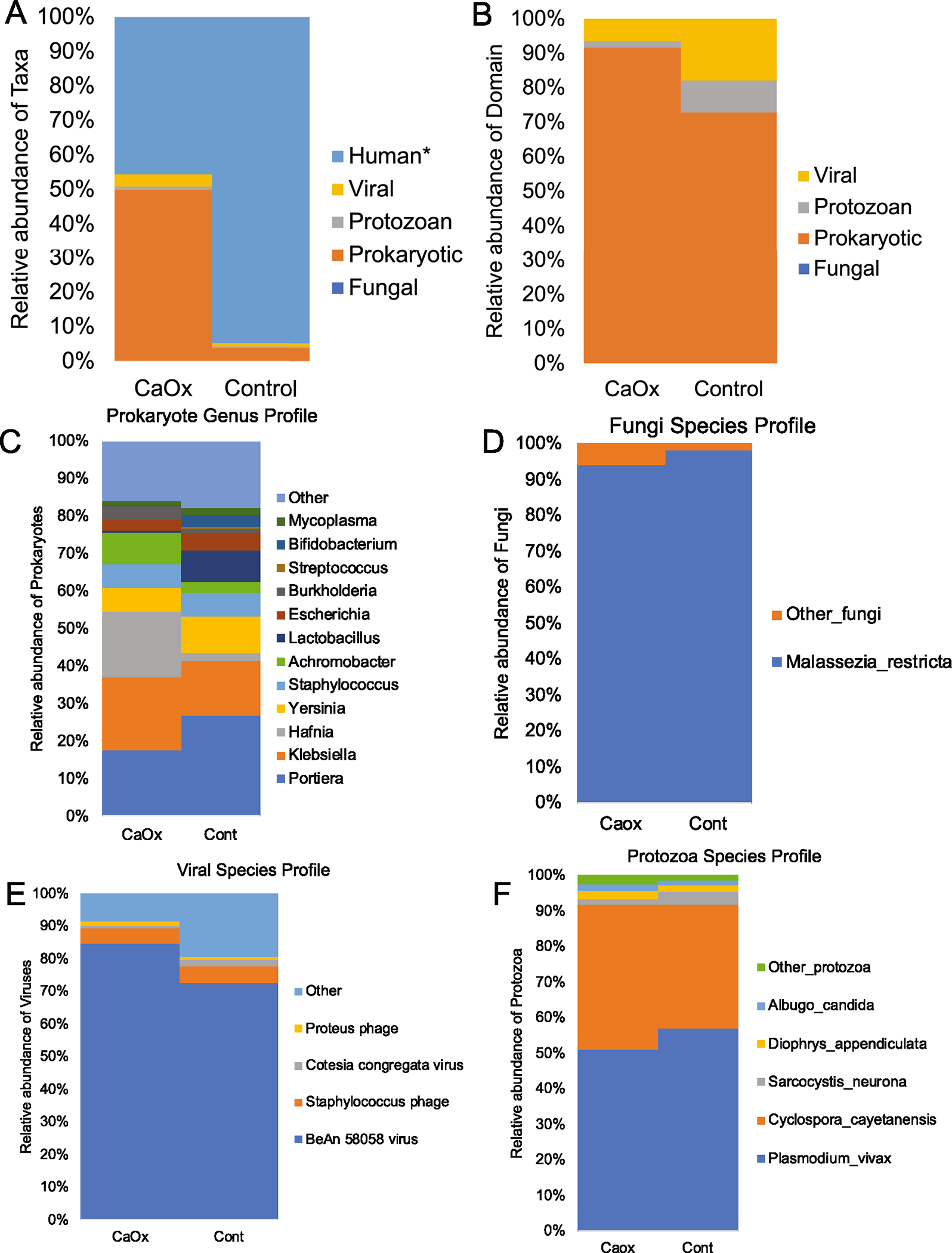
Taxonomic differences in the urobiome between CaOx and Control patients. A All domains; *p value = 0.036; B Microbial domains only; no significant differences were found; C-F Within domain differences at the genus (Prokaryotes) or species level (Fungi, Viral, Protozoa). Statistical differences for within domain comparisons are shown in Fig. 5
Fig. 3.
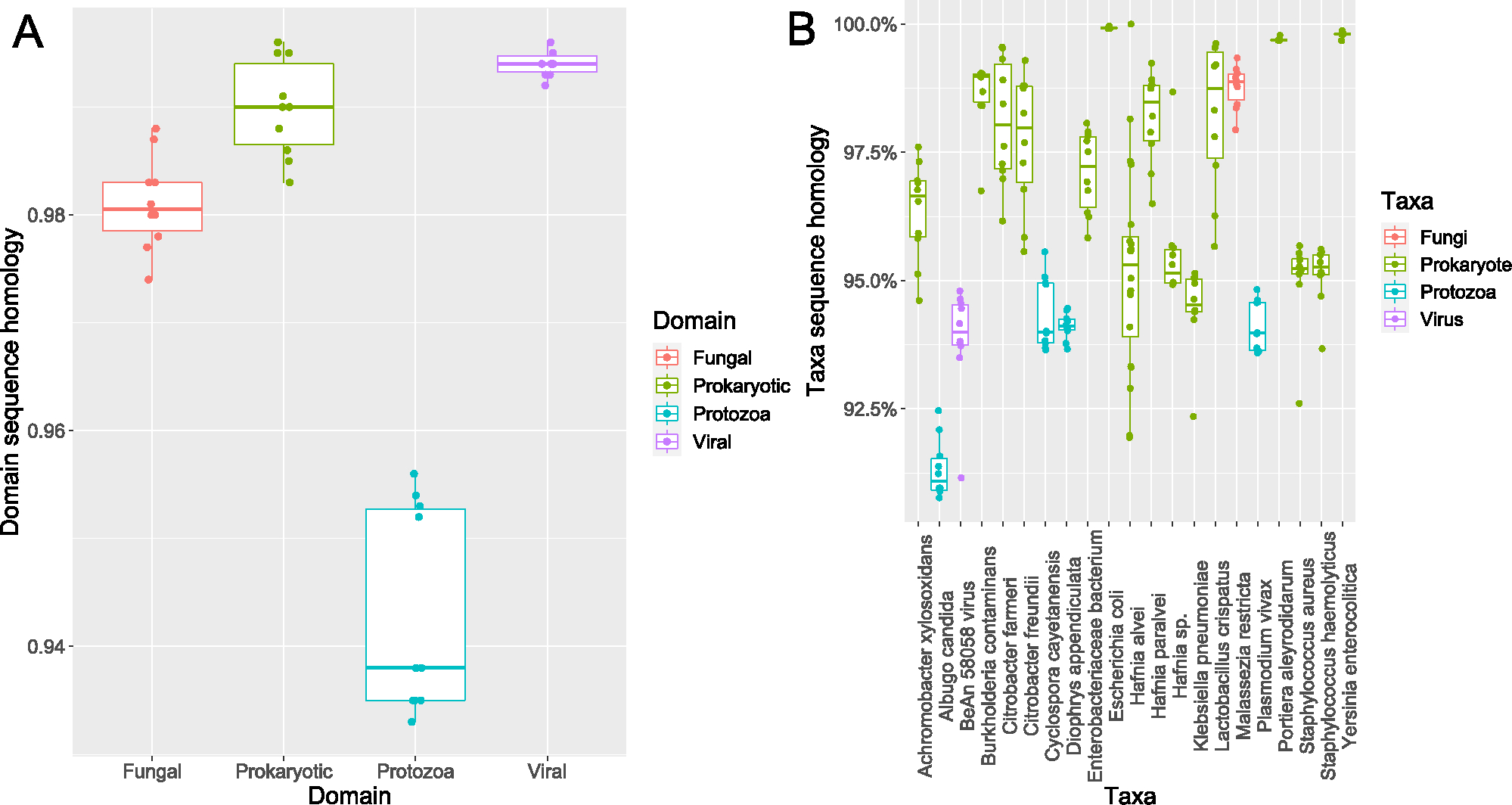
Sequence homology analysis. A Average sequence homology per domain. Each dot represents the average homology per sample. B Sequence homology for specific taxa. Each dot represents the average sequence homology for a taxon within a single sample
The CaOx group exhibited significantly higher levels of genetic diversity overall as determined by Margalef’s index of the normalized counts of full-length genes (Fig. 4A). In contrast, there were no differences in the number of observed species for all taxonomic domains combined, nor for individual domains (Fig. 4B–F).
Fig. 4.
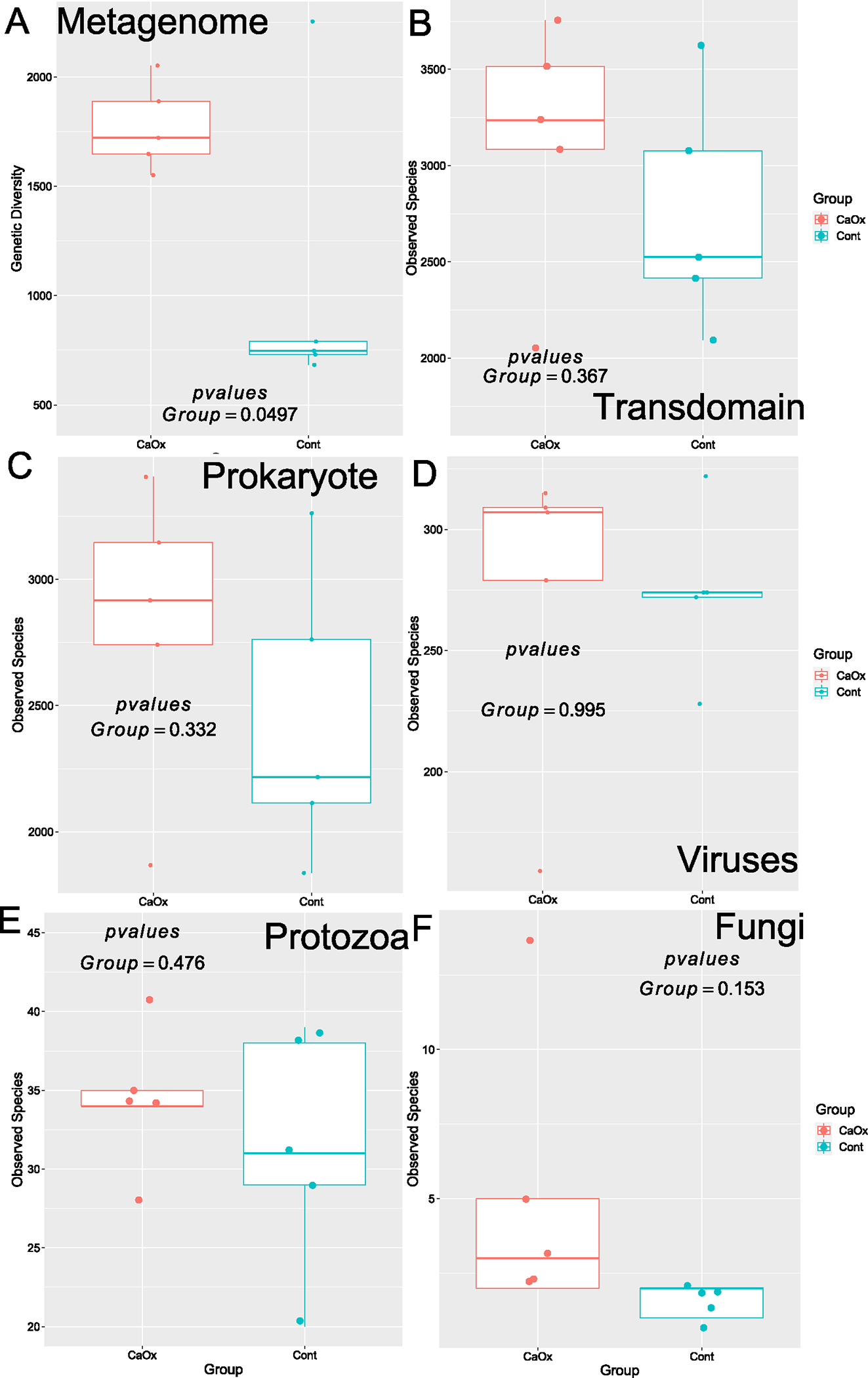
Genetic and species-level alpha-diversity between CaOx stone formers and controls. A Total genetic diversity; B All taxa combined; C Prokaryotes; D Viruses; E Protozoa; F Fungi. Statistical analysis conducted as paired t tests
The dissimilarity of the metagenomes between the CaOx and Control groups was significant by genetic profile (Fig. 5A), with greater within group dissimilarity among the CaOx participants compared to the Controls (Figure S1). The trans-domain profile was also significantly different between groups (Fig. 5B). These results were primarily driven by differences in the Prokaryotic fraction of the microbiome (Fig. 5C) rather than viral, protozoan, or fungal fractions (Fig. 5D–F). Differential abundance analysis of taxa revealed a greater number of taxa enriched in the CaOx group. However, the Lactobacillus enriched in the Control group was the biggest single factor that differentiated the CaOx patients from the Controls (Fig. 6). A total of one fungal species, three protozoans, and 20 viruses differentiated CaOx from Controls (Fig. 6, Table S1).
Fig. 5.
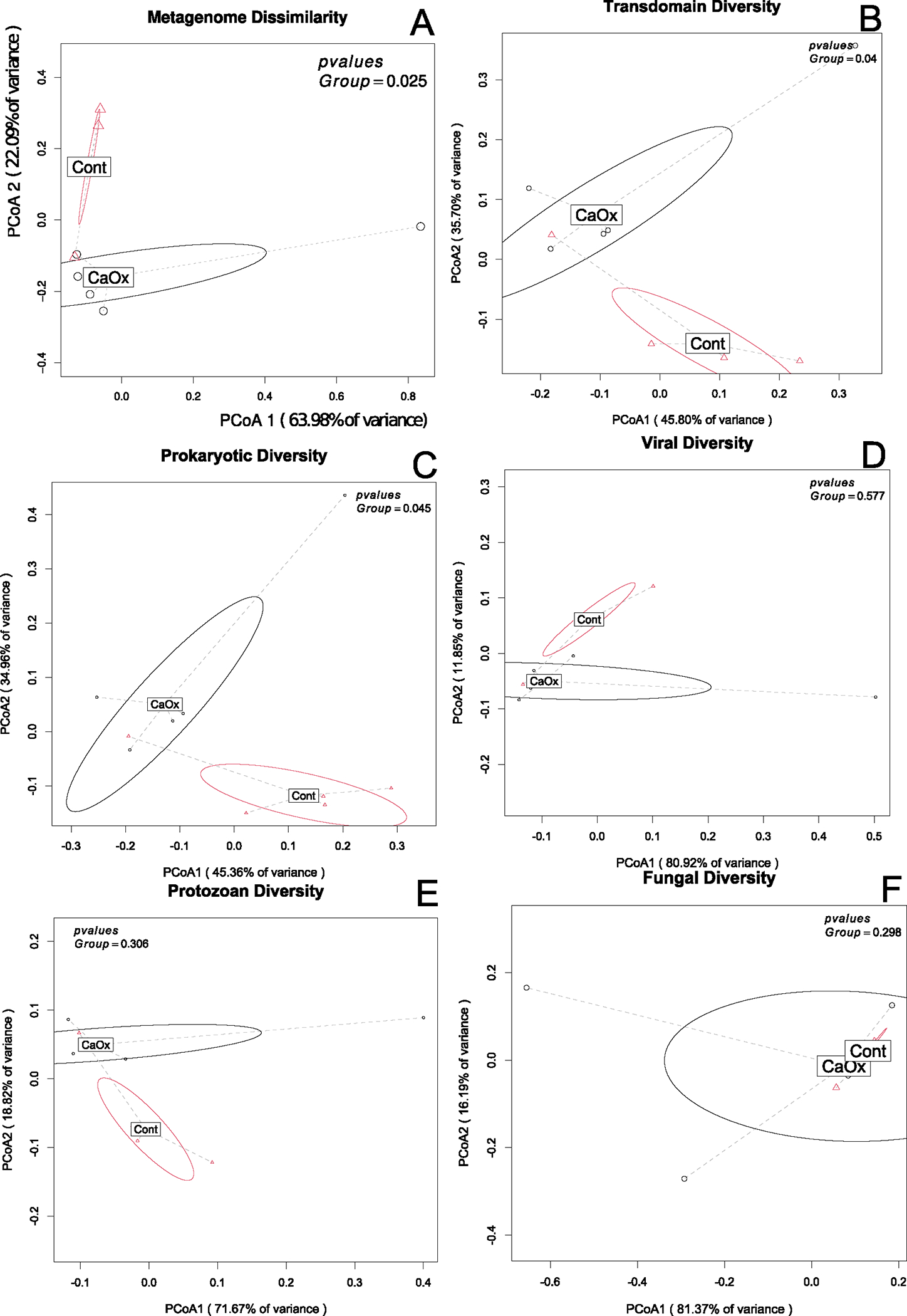
Metagenome and species-level community composition between CaOx stone formers and controls. A Total genetic diversity; B All taxa combined; C Prokaryotes; D Viruses; E Protozoa; F Fungi. Statistical analysis conducted as a PERMANOVA with 999 permutations for a weighted Bray–Curtis dissimilarity matrix
Fig. 6.
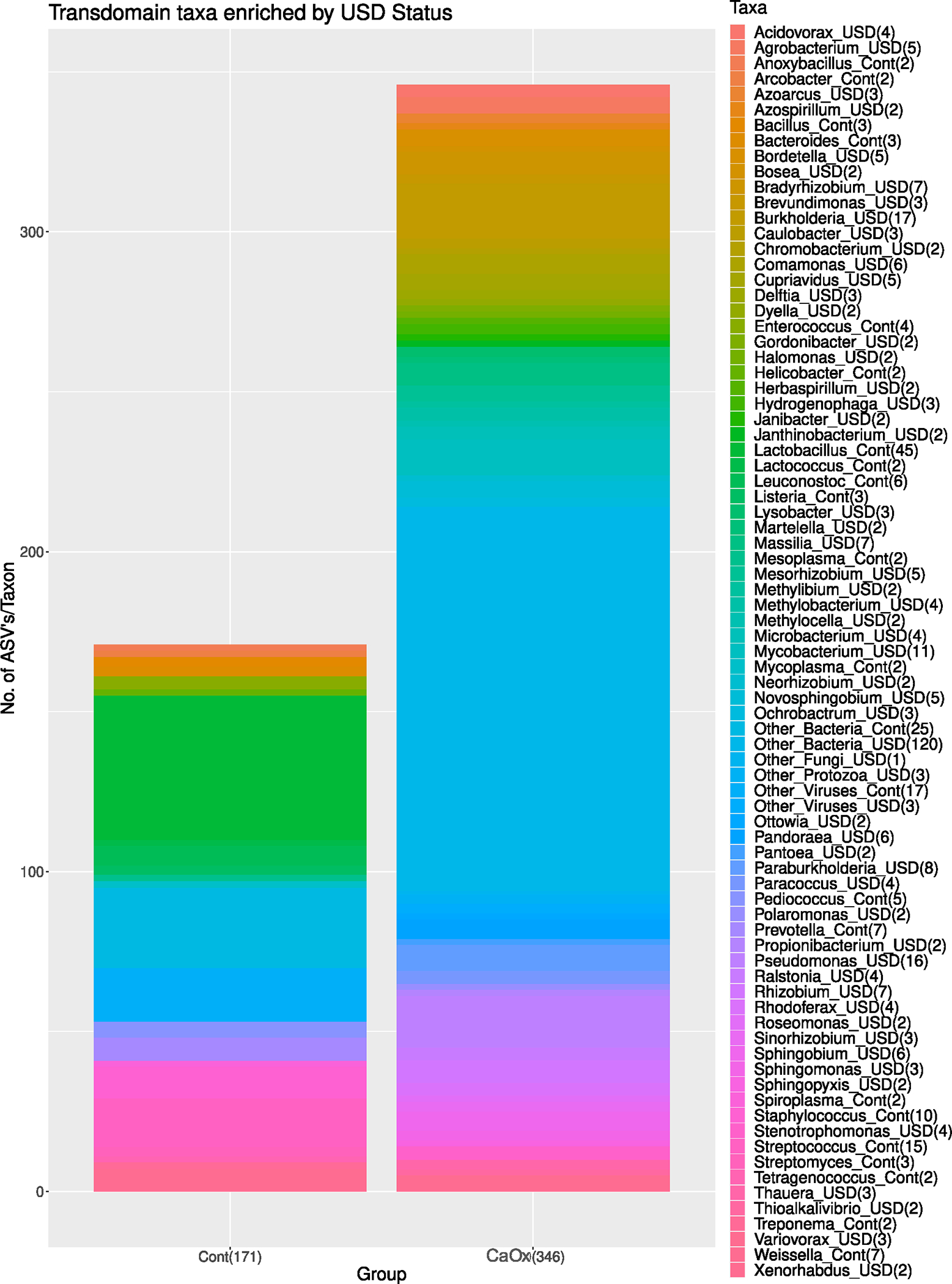
Differential abundance analysis across all domains, between Caox and Control groups. Total number of taxa differing between groups are listed in the x-axis in parentheses. Number of species within each taxa enriched in either the CaOx or Control groups are listed in the legend
Functional differences between Controls and CaOx
To determine the functional differences in the urinary tract microbiome between the Control and CaOx groups, the differential abundance of full-length genes was quantified. From a total of 53,531 microbial genes, 3581 were enriched in the Control group while only 303 were enriched in the CaOx group (Fig. 7A). All significantly different, fully annotated, microbial genes are presented in Table S2. The primary metabolic pathways responsible for these differences include transmembrane transport, metal ion binding, carbohydrate synthesis/metabolism, amino acid metabolism, zinc ion binding/transport, and oxidoreductase activity, all of which were higher in the Controls (Fig. 7B). In addition to these pathways, we also identified the oxalate-degrading genes Oxalyl-CoA decarboxylase, Formyl-CoA transferase, and Acetyl-CoA:Oxalate-CoA transferase in the data. One copy each of the Oxalyl-CoA decarboxylase and Formyl-CoA transferase was significantly higher in the Control group compared to the CaOx group (Fig. 7B, Table S2). Other pathways related to the synthesis, degradation, or transport of oxalate were also differentially abundant between the two groups (Fig. 7B, Table S2).
Fig. 7.
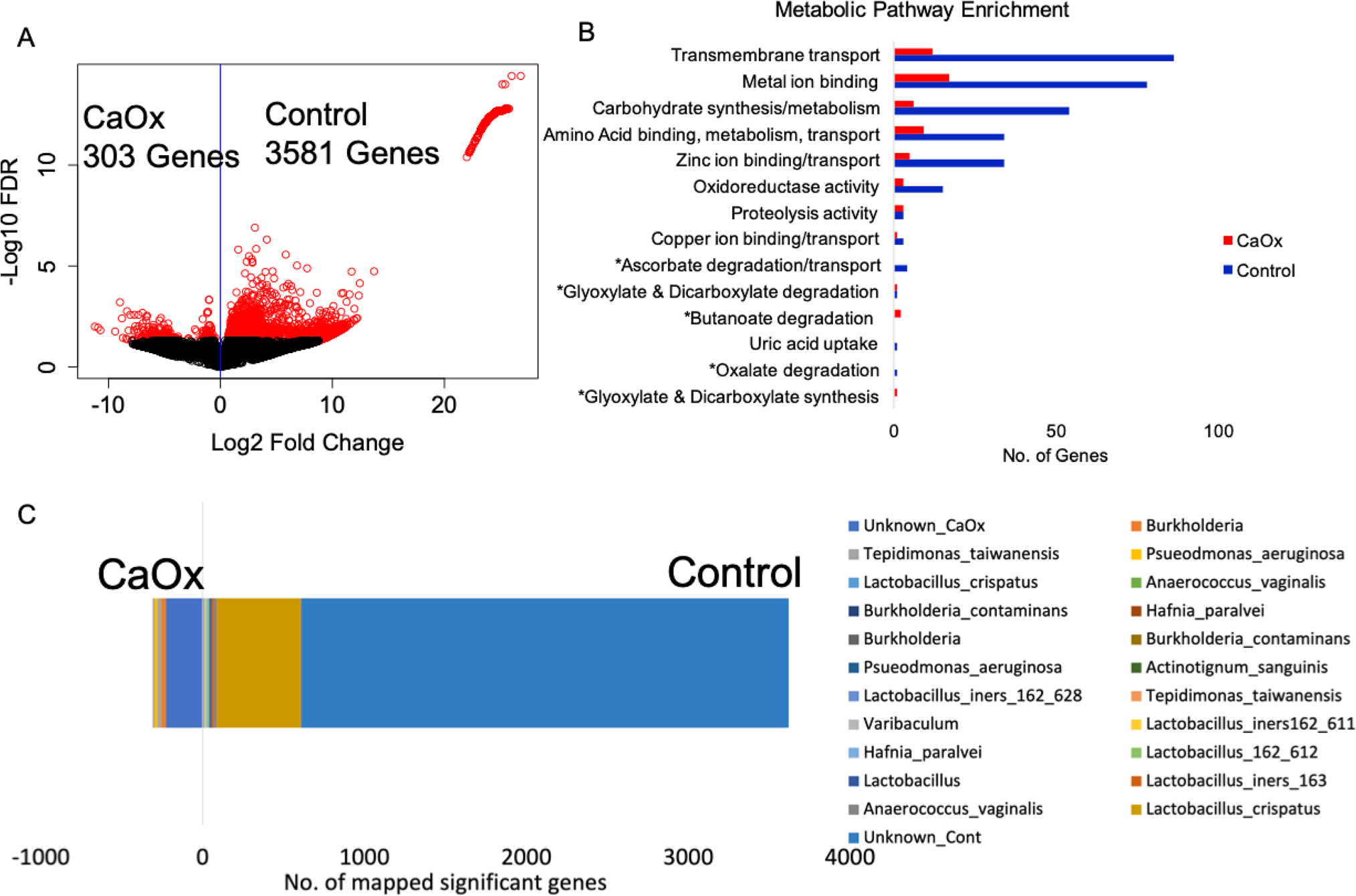
A Differential abundance gene analysis; B metabolic pathway enrichment in terms of the number of microbial genes significantly enriched in either the CaOx or Control group. Pathways were chosen for inclusion if either a large number of significantly different genes fit the pathway or the pathway had been previously associated with urolithiasis. *Indicates pathways associated with oxalate metabolism, synthesis, or transport; C Significantly difference genes mapped to de novo constructed genomes
A total of 17 genomes were constructed from the shotgun metagenomic data with completeness ranging from 17 to 98% and purity ranging from 89 to 100% (Fig. 8, Table 2). Genomes are provided in Supplemental files 1–17, with a summary of genome characteristics in Table S3. Genomes were submitted to the NCBI under BioProject #PRJNA717274. The taxa of the genomes were dominated by Lactobacillus, with other taxa such as Burkholderia, Pseudomonas, and Propionimicrobium known to comprise the urinary tract microbiome, also present (Table 2, Fig. 8) [4, 7]. For the Controls, 687 of the 3581 significantly higher genes (19%) mapped to L. crispatus de novo genome (Table 2, Fig. 7C). However, the L. crispatus genome was only 35% complete. When the significantly different genes are mapped to full length reference genomes, the number of Control genes mapped to L. crispatus increased to 1018 or 28.4% of the total (Figure S2, Table S2). The genes that mapped to L. crispatus included the oxalate-degrading genes that were higher in the Controls. In contrast, of the 303 genes enriched in the CaOx cohort, most mapped primarily to an unspecified Burkholderia (32 genes or 10.5%) and Pseudomonas aeruginosa (43 genes or 14.2%) based on the de novo genomes. From reference genomes, 60 genes (19.8%) mapped to Burkholderia contaminans and 40 (13.2%) mapped to Portiera aleyrodidarum (Figure S2, Table S2). Collectively, mapping significantly different genes to the de novo constructed genomes accounted for 21% the genes enriched in the Controls and 36% of the genes enriched in the CaOx group. Interestingly, the P. aeruginosa also harbored 4 copies of an Acetyl-CoA:Oxalate-CoA transferase gene, although these were not significantly higher in the CaOx group.
Fig. 8.
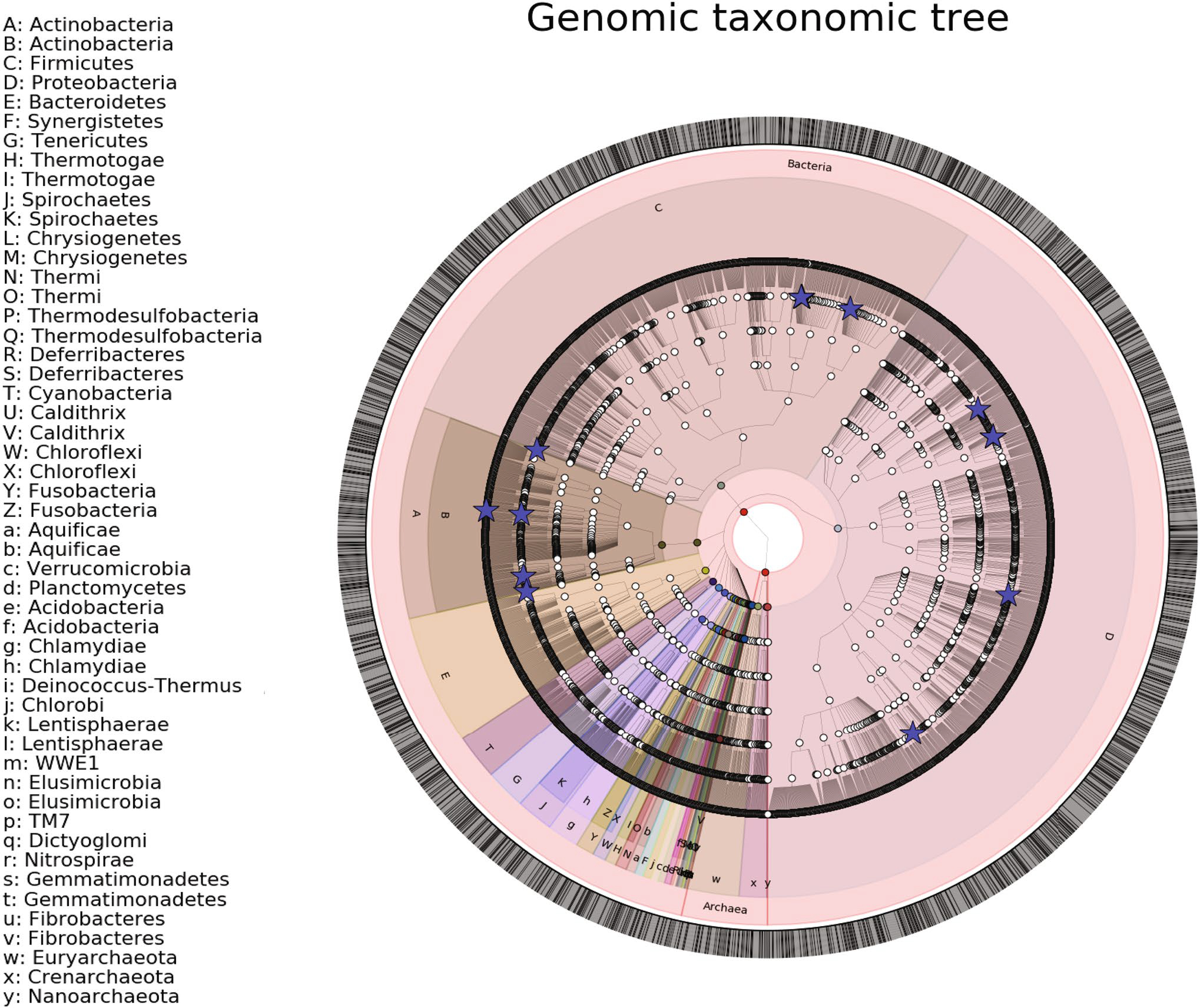
Phylogenetic tree of genomes constructed from shotgun metagenomic data of the urinary tract. Blue stars represent constructed genomes, while the remaining datapoints were extracted from the microbial tree of life (Phylophlan)
Table 2.
Summary of genome construction from shotgun metagenomic data. Included is the size of constructed genome, completeness and purity of genome after the autometa and abyss-sealer steps, as well as the number of oxalate-degrading genes and genes enriched in either the control of CaOx groups that map to the genomes
| Taxonomy | Size (bp) | Completeness | Purity | NCBI taxid (closest relative) | Oxalyl-CoA decarboxy lase | Formyl-CoA:oxalate CoA-transferase | Acetyl-CoA:oxalate CoA-transferase | Total no. of Control genes | Total no. of CaOx genes |
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Actinotignum sanguinis | 711,869 | 36.43% | 100% | 1,445,614 | 0 | 0 | 0 | 6 | 0 |
| Anaerococcus vaginalis | 648,911 | 52.14% | 100% | 33,037 | 0 | 0 | 0 | 16 | 2 |
| Bifidobacterium breve | 2,246,848 | 97.86% | 97.86% | 1685 | 0 | 0 | 0 | 0 | 0 |
| Burkholderia | 44,847 | 41.43% | 100% | 32,008 | 0 | 0 | 0 | 0 | 32 |
| Burkholderia contaminans | 3,990,717 | 34.23% | 97.96% | 488,447 | 0 | 0 | 0 | 0 | 2 |
| Hafnia paralvei | 4,293,986 | 97.86% | 99.27% | 546,367 | 0 | 0 | 0 | 8 | 2 |
| Lactobacillus crispatus | 1,505,152 | 35% | 100% | 47,770 | 2 | 2 | 1 | 687 | 3 |
| Lactobacillus iners | 427,908 | 36.43% | 100% | 147,802 | 0 | 0 | 0 | 6 | 0 |
| Lactobacillus iners | 259,786 | 33.57% | 100% | 147,802 | 0 | 0 | 0 | 9 | 0 |
| Lactobacillus iners | 366,732 | 31.43% | 100% | 147,802 | 0 | 0 | 0 | 2 | 0 |
| Lactobacillus iners | 876,541 | 71.43% | 100% | 147,802 | 0 | 0 | 0 | 13 | 0 |
| Propionimicrobium lymphophilum | 959,140 | 42.14% | 96.70% | 33,012 | 0 | 0 | 0 | 0 | 0 |
| Pseudomonas aeruginosa | 4,258,017 | 17.86% | 89.29% | 287 | 0 | 0 | 4 | 0 | 43 |
| Streptococcus mitis | 1,957,560 | 97.58% | 98.56% | 28,037 | 0 | 0 | 0 | 0 | 0 |
| Tepidimonas taiwanensis | 617,127 | 19.29% | 100% | 307,486 | 0 | 0 | 0 | 1 | 25 |
| Varibaculum sp | 1,324,554 | 59.29% | 100% | 1,852,372 | 0 | 0 | 0 | 3 | 0 |
Discussion
Recent metagenome-wide association studies focused on urolithiasis have revealed links between this disease and the human microbiome [3]. In the current study, we found that CaOx stone formers had a significantly higher proportion of microbial reads and overall genetic diversity than the Controls (Figs. 2A, 4A). These data suggest that there was greater microbial load in the urinary tract of the CaOx stone formers, even though none of the participants had a history of urinary tract infections. Additionally, there was a significant difference in metagenome, trans-domain, and prokaryotic composition between the two groups overall (Fig. 5A–C), which further elucidates the distinct microbial communities present within these groups. In contrast to data showing greater microbial abundance in CaOx stone formers, the Control group harbored a far greater number of microbial genes enriched in the urine (Fig. 4A). These data suggest that individuals with no history of urologic disease harbor a core set of diverse bacteria that are consistently present in the urinary tract compared to CaOx patients, which is corroborated by the fact that the CaOx had significantly greater, within group variance in the metagenome dissimilarity (Figure S1). When a perturbation event occurs, such as through the use of antibiotics, the urinary tract microbiome can destabilize, leading to a loss of core bacteria, followed by an overgrowth of a subset of the bacteria that are resistant to the perturbation, which can eventually result in the onset of urologic disorders. Numerous studies have independently linked past antibiotic use to the onset of USD, with the urinary tract microbiome exhibiting a greater impact of antibiotics than the gut [4–6]. Thus, antibiotic use could provide the mechanism of perturbance that leads to the urinary tract dysbiosis.
Metabolic pathway analysis of the shotgun metagenomic data reveal several potential mechanisms about how the urinary tract microbiome may contribute to the onset of USD. Importantly, our study is the first to show that not only are oxalate-degrading bacteria present in the urinary tract (Table 2), but that oxalate-degrading genes are significantly lower in the CaOx group (Fig. 7B). Additionally, pathways associated with the metabolism of oxalate precursors, such as ascorbic acid and glyoxylate, are also altered and suggests that higher levels of these precursors could be present in CaOx stone formers (Fig. 7B). Considerable attention has focused on oxalate-degrading bacteria in the gut and their role in USD [3]. However, our data indicate that the mitigation of oxalate by urinary tract bacteria may also play an important role in USD pathogenesis.
Beyond oxalate, our data point toward the reduction of microbial functions associated with other key pathways known to be important mechanisms for stone formation (Fig. 7B). These include transmembrane transport [25], metal-ion binding and zinc binding in particular [26, 27], carbohydrate synthesis/metabolism [28], and oxidoreductase activity [29, 30]. Thus, many metabolic processes that have been linked to stone formation may in part be influenced by the urinary tract microbiome in a physiologically relevant manner.
Taxonomic surveys, based on 16S rRNA surveys of the urinary tract microbiome, have pointed toward bacteria such as L. crispatus [4], Prevotella copri [4, 11], and Finegoldia spp [10] as important species that differentiate healthy individuals from those with stones. In the current study, we constructed multiple genomes, de novo, from the metagenomic data (Table 2). These genomes were taxonomically annotated based on the consensus classification of multiple, large contigs. The average size of our genomes was > 1.5 million base pairs (Table S3), which makes the taxonomic classification far more robust than taxonomic assignment from 300 base pairs or less. Confidence in the taxonomic classification of our de novo constructed genomes and of sequence reads can be assessed through our sequence homology analysis compared to reference genomes of the same taxa (Fig. 3). By mapping genes associated with the Control or CaOx cohorts to our de novo genomes, we found that 19% of the Control-associated genes were attributed directly to L. crispatus, which included the oxalate-degrading genes enriched in the Control group. This number increased to 28.4% when the significantly different genes were mapped to full length reference genomes. Additionally, a total of 24.7% of the CaOx-associated genes were attributed to a Burkholderia sp. and P. aeruginosa based on our de novo genomes. Reference genome analysis shows that (19.8%) of CaOx genes mapped to Burkholderia contaminans and 40 (13.2%) mapped to Portiera aleyrodidarum (Figure S2, Table S2). The L. crispatus species is known to be uroprotective and has successfully been used in clinical trials to prevent recurrent urinary tract infections [31]. Given the breadth of metabolic pathways associated to L. crispatus in the current study, there are multiple potential mechanisms that may contribute to this species’ uroprotective role. In contrast, P. aeruginosa and Burkholderia spp. (formerly Pseudomonas) are known uropathogens that commonly cause urinary tract infections [32]. Though none of the patients in the current study had a clinical urinary tract infection on standard urine culture, sub-clinical colonization by pathogenic bacteria may promote stone growth. This hypothesis is corroborated by the documented presence of other known uropathogens directly in non-infection kidney stones, in the absence of clinical infections [4, 7].
The current study is the first to provide a trans-domain microbial profile for the urinary tract. While non-Prokaryotic domains did not contribute much to the differentiation of the Control and CaOx groups, some important results must be considered. First, the fungal fractions were almost entirely dominated by Malassezia restricta, while the viral fractions were almost entirely dominated by a virus called BeAn 58,058. The M. restricta is a common skin-associated fungus and has also been found to be prevalent on the glans of the penis [33]. Therefore, this dominant fungus is likely a skin-contaminant of the microflora. In contrast, the BeAn 58,058 virus is a poxvirus originally isolated from a wild rodent in Brazil [34] that has been found to be abundant in the lungs [35], groundwater samples [36], and ocular lymphomas [37]. However, this particular virus has high sequence homology with the human genome and previous studies have attributed the finding of the BeAn 58,058 virus to be an artifact [35]. Finally, there were several protozoan species found in the urine samples, which were predominantly attributed to the Albugo candida, Cyclospora cayetanensis, Diophrys appendiculata, and Plasmodium vivax species (Figs. 2F, 3B). These protists include plant pathogens, intestinal parasites, marine ciliates, and blood parasites, respectively. Sequence homology analysis reveals that the actual taxonomic assignment at the species level is incorrect and instead the DNA of microorganisms found in voided urine may actually represent more benign, closely related species not well represented in public databases. These related organisms may have been either ingested or perhaps injected into the bloodstream by mosquitoes prior to arriving in the urinary tract. Indeed, RNA and DNA is well known to circulate in the bloodstream in exomes and extracellular vesicles [38]. Though, the concept of “dietary DNA” is controversial.
In conclusion, the present study provides additional evidence that the urinary tract microbiome influences the pathogenesis of USD, specifically driven by the loss of bacteria such as L. crispatus that may provide uroprotective functions. Several metabolic pathways previously linked to USD pathogenesis were attributed to the urinary tract microbiome here and were significantly altered between the Control and CaOx groups. Limitations of this initial exploratory study include small sample sizes and the fact that the midstream voided urine samples reflect the lower urinary tract rather than the upper urinary tract where stones form. However, the lower urinary tract microbiome does not differ from the lower urinary tract [7]. Additionally, given the clear differences in the metagenome between CaOx stone formers and healthy controls, along with the strong overlap between differentiated pathways and those known to be involved with stone formation, our results warrant further investigations into the mechanistic roles that L. crispatus and the uropathogens Pseudomonas aeruginosa and Burkholderia play in the pathogenesis of USD.
Supplementary Material
Funding
NK was supported in part by the Urology Care Foundation Research Scholar Award Program and Endourological Society/Raju Thomas M.D. Award. Funding was also provided by NIH/NIDDK Grant 1R01DK121689-01A1 to AM.
Abbreviations
- USD
Urinary stone disease
- CaOx
Calcium oxalate
Footnotes
Conflict of interest The authors have no conflicts of interests to report.
Ethics approval The questionnaire and methodology for this study was approved by the Institutional Review Board of Cleveland Clinic (IRB# 16–643).
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s00240-022-01314-5.
Data availability
Raw sequence reads and metadata are available at the Sequence Read Archive under BioProject # PRJNA718167. Constructed genomes are available under BioProject #PRJNA717274.
References
- 1.Ziemba JB, Matlaga BR (2017) Epidemiology and economics of nephrolithiasis. Investigative and clinical urology 58:299–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saigal CS, Joyce G, Timilsina AR, Project UDiA (2005) Direct and indirect costs of nephrolithiasis in an employed population: opportunity for disease management? Kidney Int 68:1808–1814 [DOI] [PubMed] [Google Scholar]
- 3.Batagello CM, Manoj Miller, Aaron W. (2018) Urolithiasis: a case of missing microbes? J Endourol 32(11):995–1005 [DOI] [PubMed] [Google Scholar]
- 4.Zampini A, Nguyen AH, Rose E, Monga M, Miller AW (2019) Defining dysbiosis in patients with urolithiasis. Sci Rep 9:5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tasian GE, Thomas J, Goldfarb David S, Copelovitch L, Gerber Jeffrey S, Qufei Wu, Denburg Michelle R (2018) Oral antibiotic exposure and kidney stone disease. J Am Soc Nephrol 29(6):1731–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferraro PM, Curhan GC, Gambaro G, Taylor EN (2019) Antibiotic use and risk of incident kidney stones in female nurses. Am J Kidney Dis 74:736–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dornbier RA, Bajic P, Van Kuiken M et al. (2019) The microbiome of calcium-based urinary stones. Urolithiasis. 48(3):191–199 [DOI] [PubMed] [Google Scholar]
- 8.Sivaguru M, Saw JJ, Williams JC et al. (2018) Geobiology reveals how human kidney stones dissolve in vivo. Sci Rep 8:13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolfe AJ, Brubaker L (2015) “Sterile urine” and the presence of bacteria. Eur Urol 68:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu F, Zhang N, Jiang P et al. (2020) Characteristics of the urinary microbiome in kidney stone patients with hypertension. J Transl Med 18:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie J, Huang J-s, Huang X-j et al. (2020) Profiling the urinary microbiome in men with calcium-based kidney stones. BMC Microbiol 20:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rani A, Ranjan R, McGee HS et al. (2017) Urinary microbiome of kidney transplant patients reveals dysbiosis with potential for antibiotic resistance. Transl Res 181:59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garretto A, Thomas-White K, Wolfe AJ, Putonti C (2018) Detecting viral genomes in the female urinary microbiome. J Gen Virol 99:1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bushnell B, Rood J, Singer E (2017) BBMerge–accurate paired shotgun read merging via overlap. PLoS ONE 12:e0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 1–3. arXiv preprint arXiv:1303.3997. [Google Scholar]
- 16.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA (2017) metaSPAdes: a new versatile metagenomic assembler. Genome Res 27:824–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–206924642063 [Google Scholar]
- 18.Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oksanen J, Blanchet FG, Kindt R, et al. (2010) Vegan: community ecology package. R package version 1.17–4. http://cran.r-project.org. Acesso em.23:2010. [Google Scholar]
- 21.Miller IJ, Rees ER, Ross J et al. (2019) Autometa: automated extraction of microbial genomes from individual shotgun metagenomes. Nucleic Acids Res 47:e57–e57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen K-T, Liu C-L, Huang S-H et al. (2018) CSAR: a contig scaffolding tool using algebraic rearrangements. Bioinformatics 34:109–111 [DOI] [PubMed] [Google Scholar]
- 23.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I (2009) ABySS: a parallel assembler for short read sequence data. Genome Res 19:1117–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asnicar F, Thomas AM, Beghini F et al. (2020) Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat Commun 11:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holmes RP, Assimos DG (2004) The impact of dietary oxalate on kidney stone formation. Urol Res 32:311–316 [DOI] [PubMed] [Google Scholar]
- 26.Tang J, McFann K, Chonchol M (2012) Dietary zinc intake and kidney stone formation: evaluation of NHANES III. Am J Nephrol 36:549–553 [DOI] [PubMed] [Google Scholar]
- 27.Chi T, Kim MS, Lang S et al. (2015) A Drosophila model identifies a critical role for zinc in mineralization for kidney stone disease. PLoS ONE 10:e0124150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reddy ST, Wang C-Y, Sakhaee K, Brinkley L, Pak CY (2002) Effect of low-carbohydrate high-protein diets on acid-base balance, stone-forming propensity, and calcium metabolism. Am J Kidney Dis 40:265–274 [DOI] [PubMed] [Google Scholar]
- 29.Khan SR (2013) Reactive oxygen species as the molecular modulators of calcium oxalate kidney stone formation: evidence from clinical and experimental investigations. J Urol 189:803–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khan SR (2014) Reactive oxygen species, inflammation and calcium oxalate nephrolithiasis. Transl Androl Urol 3:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grin PM, Kowalewska PM, Alhazzan W, Fox-Robichaud AE (2013) Lactobacillus for preventing recurrent urinary tract infections in women: meta-analysis. Can J Urol 20:6607–6614 [PubMed] [Google Scholar]
- 32.Nerurkar A, Solanky P, Naik SS (2012) Bacterial pathogens in urinary tract infection and antibiotic susceptibility pattern. J Pharm Biomed Sci 21:1–3. [Google Scholar]
- 33.Atilla Aridoğan I, Ilkit M, Izol V, Ates A (2005) Malassezia and Candida colonisation on glans penis of circumcised men. Mycoses 48:352–356 [DOI] [PubMed] [Google Scholar]
- 34.Fonseca F, Lanna M, Campos M et al. (1998) Morphological and molecular characterization of the poxvirus BeAn 58058. Adv Virol 143:1171–1186 [DOI] [PubMed] [Google Scholar]
- 35.Goolam Mahomed T, Peters R, Allam M et al. (2021) Lung microbiome of stable and exacerbated COPD patients in Tshwane South Africa. Sci Rep 11:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van de Vossenberg J, Hoiting Y, Kamara AK et al. (2021) Double-Stranded DNA virus assemblages in groundwater in three informal urban settlements in sub-Saharan Africa differ from each other. ACS ES&T Water 1:1992–2000 [Google Scholar]
- 37.Mollerup S, Mikkelsen LH, Hansen AJ, Heegaard S (2019) High-throughput sequencing reveals no viral pathogens in eight cases of ocular adnexal extranodal marginal zone B-cell lymphoma. Exp Eye Res 185:107677. [DOI] [PubMed] [Google Scholar]
- 38.Isaac R, Reis FCG, Ying W, Olefsky JM (2021) Exosomes as mediators of intercellular crosstalk in metabolism. Cell Metab 33:1744–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequence reads and metadata are available at the Sequence Read Archive under BioProject # PRJNA718167. Constructed genomes are available under BioProject #PRJNA717274.