

Abstract
INTRODUCTION
The ɛ4 allele of the apolipoprotein E gene (APOE ɛ4) is the strongest genetic risk factor for Alzheimer's disease (AD), but the mechanisms connecting APOE ɛ4 to AD are not clear.
METHODS
Participants (n = 596) were from two clinical‐pathological studies. Tissues from dorsolateral prefrontal cortex were examined to identify 8425 proteins. Post mortem pathological assessment used immunohistochemistry to obtain amyloid beta (Aβ) load and tau tangle density.
RESULTS
In separate models, APOE ɛ4 was associated with 18 proteins, which were associated with Aβ and tau tangles. Examining the proteins in a single model identified Netrin‐1 and secreted frizzled‐related protein 1 (SFRP1) as the two proteins linking APOE ɛ4 with Aβ with the largest effect sizes and Netrin‐1 and testican‐3 linking APOE ɛ4 with tau tangles.
DISCUSSION
We identified Netrin‐1, SFRP1, and testican‐3 as the most promising proteins that link APOE ɛ4 with Aβ and tau tangles.
Highlights
Of 8425 proteins extracted from prefrontal cortex, 18 were related to APOE ɛ4.
The 18 proteins were also related to amyloid beta (Aβ) and tau.
The 18 proteins were more related to APOE ɛ4 than other AD genetic risk variants.
Netrin‐1 and secreted frizzled‐related protein 1 were the two most promising proteins linking APOE ɛ4 with Aβ.
Netrin‐1 and testican‐3 were two most promising proteins linking APOE ɛ4 with tau.
Keywords: Alzheimer's disease, amyloid, apolipoprotein ε4, proteins, tau proteins
1. BACKGROUND
The ɛ4 allele of the apolipoprotein E gene (APOE ɛ4) is the strongest genetic risk factor for Alzheimer's disease (AD). 1 However, the mechanisms connecting APOE ɛ4 to AD are not clear. As APOE ɛ4 is a common allele and present in approximately 20% of Americans, 2 elucidation of the mechanisms connecting APOE ɛ4 to AD will have a large impact in the development of the primary prevention of AD since the allele is sufficiently common to warrant population screening should there be a robust therapeutic.
We and others have shown that AD pathology mediates the association of APOE ɛ4 to Alzheimer's dementia. 3 , 4 Further, we found that amyloid beta (Aβ) mediated the association of APOE ɛ4 with tau tangles. 5 , 6 Finally, we reported that in the absence of Aβ, APOE ɛ4 was not associated with tau tangles. 7 Yet the mechanism underlying the association between APOE ɛ4 and AD remains poorly understood.
As Aβ deposits and tau tangles are aggregates of proteins, few prior studies examined brain proteins in relation to APOE ɛ4, Aβ, and tau tangles to identify molecular pathways connecting APOE ɛ4 with AD. Following the first studies that used targeted proteomics to quantify select proteins including synaptic proteins, 8 recent studies used unbiased hypothesis‐free mass spectrometry‐based proteomic methods to measure hundreds to thousands of proteins with the objective of finding novel druggable molecules and pathways. 9 , 10 , 11 However, the sample sizes were modest and their results inconsistent. Here, we build on prior work in three important ways. First, we employed a far larger sample size, markedly increasing power. Second, we leveraged deep proteomics with more proteins than most prior studies. Finally, we examined, for the first time, a wide range of clinical and pathological phenotypes of AD and AD‐related dementias (ADRD).
2. METHODS
2.1. Participants
Data were collected from community‐dwelling older adults enrolled in one of two longitudinal clinical‐pathological studies of aging conducted at Rush Alzheimer's Disease Center: the Religious Orders Study (ROS) or the Rush Memory and Aging Project (MAP). Both studies enrolled persons without known dementia who agreed to annual clinical evaluations and brain donation at the time of death. ROS participants were nuns, priests, and brothers living across the United States. The study started in 1994. MAP participants were living in northeastern Illinois in retirement facilities, personal accommodations, or subsidized housing. MAP started in 1997. Both studies utilize harmonized data acquisition protocols administered by the same staff, a strategy facilitating joint analysis. Details of the studies are provided elsewhere. 12 Both studies were approved by an Institutional Review Board of Rush University Medical Center.
Through December 2022, of 3750 participants recruited in both studies, 1887 had died and had completed post mortem pathological assessments. However, proteomic analysis of cortical proteins is ongoing and has been completed in 596 participants who composed the analytic sample of the current study.
RESEARCH IN CONTEXT
Systematic review: We reviewed the literature using PubMed. Although APOE ɛ4 is the strongest genetic risk factor for AD, we found no studies that had systematically examined levels of thousands of proteins quantified in human brain in relation to APOE ɛ4 and AD pathological hallmarks, Aβ and tau.
Interpretation: We found 18 out of 8425 examined proteins extracted from the prefrontal cortex that were related to APOE ɛ4. The 18 proteins were also related to Aβ and tau. Of the 18 proteins, Netrin‐1 and secreted frizzled‐related protein 1 were the two most promising proteins linking the association of APOE ɛ4 with Aβ and Netrin‐1 and testican‐3 linking the association of APOE ɛ4 with tau.
Future directions: Future studies should confirm the identified proteins as mediators of the relation between APOE ɛ4 and AD and examine whether targeting these proteins might prevent AD in carriers of APOE ɛ4.
2.2. APOE genotyping
DNA was extracted from peripheral blood or frozen brain tissue. APOE genotyping was performed at Polymorphic DNA Technologies (Alameda, CA, USA) by sequencing rs429358 (codon 112) and rs7412 (codon 158) at exon 4 of the APOE gene. 13 In the current study, the APOE genotype was summarized using a dichotomous variable indicating the presence of at least one APOE ɛ4 allele.
2.3. Post mortem pathological assessment
The median post mortem interval was 6.5 h (IQR: 5.1 to 8.6 h). After brain removal, hemispheres were separated. One hemisphere was frozen for further molecular studies, including proteomic analysis and bulk tissue and single‐nucleus RNA sequencing (snRNA‐seq), and the other hemisphere was fixed in 4% formaldehyde in phosphate buffer. The fixed hemisphere was cut into 1‐cm slabs, and slabs from predetermined regions were further cut into blocks and sections to be used for pathological assessment. The process is explained in more detail elsewhere. 14 , 15
2.3.1. Aβ load
Multiple brain regions were immunohistochemically examined for the presence of Aβ, including anterior cingulate cortex, superior frontal cortex, mid‐frontal cortex, inferior temporal cortex, hippocampus, entorhinal cortex, angular gyrus/supramarginal cortex, and calcarine cortex. Antibodies specific for Aβ (6F/3D [1:50, Dako North America Inc., Carpinteria, CA, USA]; 10D5 [1:600, Elan Pharmaceuticals, San Francisco, CA, USA]; 4G8 [1:9000, Covance Labs, Madison, WI, USA]) were used for staining the 20‐µm sections. The immunostained sections were examined by digital image analysis to calculate percentage of each section occupied by immunohistochemically labeled areas. The percent areas were summarized in and across brain regions to yield brain Aβ load, as described previously. 14 , 16
2.3.2. Tau tangle density
The same brain regions examined for Aβ were also examined for tau tangles using antibodies specific for phosphorylated tau (AT8 [1:1000, Innogenetics, Alpharetta, GA, USA]). The immunostained sections were examined under microscopes equipped with a computer‐aided stereology program to count tau‐labeled tangles, which were summarized in and across brain regions to yield brain tau tangles density, as described previously. 14 , 16
2.3.3. AD pathological diagnosis
Multiple brain regions were stained using a modified Bielschowsky silver stain to identify diffuse and neuritic plaques and neurofibrillary tangles, which were used to adjudicate a pathological diagnosis of AD according to established criteria. 17
2.3.4. Non‐AD brain pathologies
Structured post mortem pathological assessments included collection of indices of eight other brain pathologies, including TAR DNA‐binding protein 43 (TDP‐43), hippocampal sclerosis, Lewy bodies, macroinfarcts, microinfarcts, intracranial atherosclerosis, basal ganglia arteriolosclerosis, and cerebral amyloid angiopathy (CAA). Binary variables were used to summarize the eight brain pathologies, which are described in the eMethods.
2.4. Proteomic analysis
Dorsolateral prefrontal cortex (DLPFC) is a brain region heavily involved in cognitive function and is also vulnerable to the accumulation of AD and other pathologies. Therefore, this brain region was selected for proteomic analysis, described previously in more detail. 18 , 19 In brief, 100 mg DLPFC tissue was homogenized, sonicated, and centrifuged, and protein concentration was determined in the supernatant. Protein digestion was carried out using lysyl endopeptidase and trypsin. The digests were labeled using the tandem mass tag method, which was subsequently fractionated under high pH in a high‐performance liquid chromatography system. The fractions were analyzed by liquid chromatography coupled to mass spectrometry. The spectra were searched against the canonical UniProtKB human proteome database, and, following spectra assignments, peptides were assembled into proteins. Several quality control (QC) measures were in place, including the use of a global internal standard and regressing out effects of protein batch, MS2 versus MS3 quantitation mode, sex, age at death, post mortem interval, and study (ROS vs. MAP), as described previously. 20 The result was a quantification of 8425 proteins in 596 participants.
2.5. Bulk tissue RNA sequencing
Details of RNA‐seq have been published elsewhere. 21 , 22 , 23 After RNA extraction from tissues prepared from DLPFC, standard protocols with minor modifications were used for making libraries and sequencing‐extracted RNA, which were aligned to a human reference genome. The expression levels of transcripts were estimated, aggregated at the gene level, and normalized. Finally, multiple steps were taken to remove major technical and biological factors including batch effect, RNA integrative number, number of aligned reads, study, post mortem interval, age, and sex.
2.6. Single‐nucleus RNA sequencing
The nuclei were isolated from 479 DLPFC samples for snRNA‐seq data acquisition, as previously described. 24 Tissues were processed in 60 batches with eight donors each. The experiment was designed to balance clinical, pathological diagnosis, and sex as much as possible. The libraries were prepared using the 10× Genomics 3 Gene Expression kit (version 3 chemistry), run on the Chromium platform. The libraries were sequenced and aligned to a human reference genome, and the expression levels of transcripts were estimated. Then we used whole genome sequencing data of the participants to identify the original donors of the examined nuclei using genetic demultiplexing approaches. For QC analysis, genotype concordance of RNA and whole genome sequencing, sex check, duplicated individuals, cell doublets, and sequencing depth were assessed. For the cell type annotation step, nuclei were classified into seven major cell types by a weighted ElasticNet‐regularized logistic regression classifier and standard workflows implemented in the R Seurat package. 25 Over 1.64 million nuclei from 424 donors after QC were retained for downstream analysis. Pseudo‐bulk matrices were generated by summing counts per cell type, per donor, and TMM‐voom normalized. 26 Finally, we selected the expression of the targeted genes used in this project.
2.7. Alzheimer's dementia polygenic risk score
Alzheimer's dementia polygenic risk score was calculated using results of a large genome‐wide meta‐analysis 27 and genotyping of the ROS and MAP participants. 28 Based on the association with Alzheimer's dementia and after linkage disequilibrium‐related pruning, sequence variants were selected and weighted by their number and their coefficients of association with Alzheimer's dementia. Then the weights were averaged across the sequence variants to yield Alzheimer's dementia polygenic risk score. As we had aimed to compare other sequence variants of Alzheimer's dementia with APOE ɛ4, we excluded from the polygenic risk score sequence variants that were in the APOE/TOMM40 genetic region. We used two polygenic risk scores by applying two p value thresholds for sequence variants inclusion: <5×10−8 and <1. 29
2.8. Cognition
Global cognition and five cognitive domains were annually assessed using 19 neuropsychological tests. The included tests were as follows: immediate and delayed recall of the East Boston Story and Logical Memory Story A, Word List Memory, Recall, and Recognition (episodic memory); Symbol Digit Modalities Test, Number Comparison, Stroop Color and Word Test (perceptual speed); Boston Naming Test, Verbal Fluency, Word Reading (semantic memory); Digit Span Forward and Backward, and Digit Ordering (working memory); Judgment of Line Orientation and Standard Progressive Matrices (visuospatial ability). The neuropsychological tests’ scores at each annual visit were standardized using means and standard deviations of the scores at baseline and averaged to come up with global cognition and cognitive domain scores. 30 , 31
The neuropsychological tests’ scores were also reviewed by a neuropsychologist and rated. The ratings, along with other clinical data, were reviewed by a clinician with expertise in dementia to adjudicate presence of dementia and its subtypes according to established criteria. 32
2.9. Other covariates
Age at death was calculated using dates of death and birth. In addition, self‐report questions at baseline were used to obtain sex, years of education, and race data.
2.10. Statistical methods
Separate linear regressions (n = 8425) were used to examine associations of APOE ɛ4 with proteins. The APOE ɛ4‐associated proteins were further examined in association with Aβ load and tau tangle density, which were square‐root‐transformed to have a distribution closer to normality. The APOE ɛ4‐associated proteins were also examined in separate linear mixed‐effects models that used longitudinal measurements of global cognition and cognitive domain scores as the outcome. In these linear mixed‐effects models, the model term of interest was the interaction between APOE ɛ4‐related protein and time, which estimated association of the protein with rate of cognitive decline. In further analyses, linear regressions were used to examine association of APOE ɛ4 with RNA expression levels of the genes of APOE ɛ4‐related proteins and association of AD polygenic risk score with APOE ɛ4‐related proteins. We used backward elimination in the linear regression models that included all APOE ɛ4‐related proteins to identify proteins that were independently associated with Aβ and tau tangles. To estimate effect sizes, we used Cohen's f 2 that indicates relative change in the explained variance of the outcome when we remove the variable of interest. 33 All the linear regressions were controlled for age at death and sex, and linear mixed‐effects models examining cognitive decline were controlled for age at death, sex, education, and their interactions with time. To correct for multiple testing, the p values of all the analyses were adjusted using Bonferroni method.
3. RESULTS
Participant characteristics are summarized in Table 1. On average, participants were 90 years old at death, 70% were women, 30% had Alzheimer's dementia, and 60% had pathologic AD.
TABLE 1.
Characteristics of study participants at the last visit prior to death (n = 596).
| Characteristics | Mean (SD) or n (%) |
|---|---|
| Age at death baseline, years, mean (SD) | 89.6 (6.4) |
| Women, n (%) | 410 (69) |
| Education, years, mean (SD) | 15.4 (3.4) |
| White non‐Hispanic, n (%) | 576 (97) |
| Mini‐Mental State Examination score, mean (SD) | 22.4 (8.3) |
| Global cognition | −0.79 (1.07) |
| Alzheimer's dementia, n (%) | 200 (34) |
| Post mortem pathological assessment | |
| Pathological diagnosis of AD, n (%) | 371 (62) |
| Immunohistochemical assessments | |
| Square root of Aβ load, mean (SD) | 1.6 (0.78) |
| Square root of tau tangle density, mean (SD) | 1.44 (1.11) |
| TDP‐43 | 174 (29) |
| Hippocampal sclerosis | 47 (8) |
| Lewy bodies | 149 (25) |
| Macroinfarcts | 193 (32) |
| Microinfarcts | 164 (28) |
| Moderate to severe atherosclerosis | 189 (32) |
| Moderate to severe arteriolosclerosis | 191 (32) |
| Moderate to severe cerebral amyloid angiopathy | 180 (30) |
3.1. APOE ɛ4 and proteins
One hundred thirty‐three (23%) participants had at least one APOE ɛ4 allele, with 128 of them having one and five having two APOE ɛ4 alleles. Of the 8425 proteins, APOE ɛ4 was associated with the levels of 18. Interestingly, as illustrated in the Chicago plot (Figure 1), all 18 proteins had a positive association with APOE ɛ4 (Table e‐1). The 18 proteins were involved in different cellular functions, including axonal growth and brain development, neurogenesis, angiogenesis, amyloid production and aggregation, and extracellular matrix hemostasis. Of note, the association between APOE ɛ4 and APOE protein was not significant following a conservative Bonferroni correction (estimate = 0.251, SE = 0.099, p = .011).
FIGURE 1.
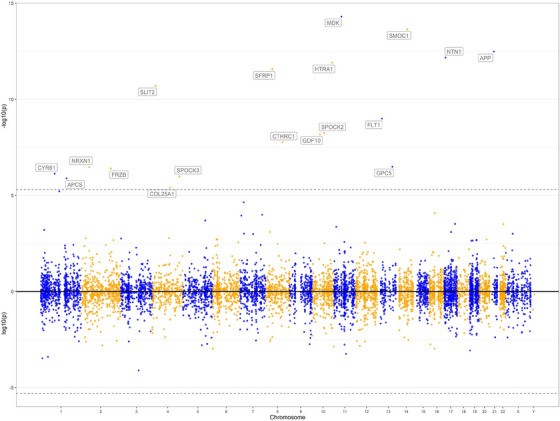
Chicago plot of proteome‐wide association of APOE ɛ4 with proteins in dorsolateral prefrontal cortex.
We next examined whether the 18 proteins were associated with AD pathological indices and cognitive decline. In two series of linear regressions, we separately examined the associations of the 18 APOE ɛ4‐related proteins with Aβ and tau tangles. As expected, due to the associations with APOE ɛ4, 3 , 13 the analyses found that all the proteins were associated with higher levels of Aβ load and tau tangle density (Figure 2A; Table e‐1). Then, in a series of linear mixed‐effects models, we separately examined the associations of the 18 proteins with global cognitive decline and decline in five cognitive domains (Figure 2B; Table e‐2). Higher levels of 16 out of 18 proteins were associated with faster global cognitive decline. Not surprisingly given the relatively selective effect of the allele in our data, 34 the number of proteins associated with faster decline in a cognitive domain was the highest for episodic memory (n = 17), followed by semantic memory (n = 16), working memory (n = 14), processing speed (n = 9), and visuospatial ability (n = 1).
FIGURE 2.
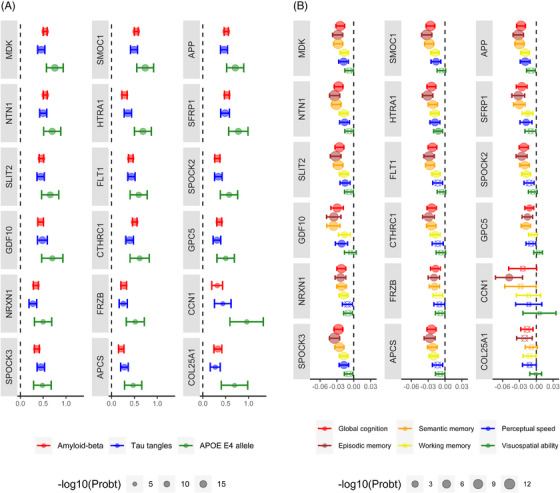
Associations of APOE ɛ4‐related proteins with AD pathological indices and rate of cognitive decline. The circles and bars illustrate estimates and their 95% confidence intervals (CIs) of the associations between the 18 APOE ɛ4‐related proteins and different outcomes. The estimates and 95% CIs are derived from analyzing proteins in separate models. The estimates of the associations with APOE ɛ4 were derived from separate linear regression models where each of the 18 proteins was the outcome and APOE ɛ4 was the model term (A). The associations between the proteins and Aβ or tau tangles were derived from separate linear regression models where Aβ or tau tangle was the outcome and each of the proteins was the model term (A). The association with the rate of cognitive decline is derived from separate mixed‐effects models with repeated measurements of global cognition or the five cognitive domains as the outcome (B). Each mixed‐effects model term was one of the proteins, time (i.e., the years in the study), and interaction of the protein with time. All the linear regression models were controlled for age at death and sex, and the mixed‐effects models were controlled for age at death, sex, education, and their interaction with time. Crossed circles indicate the associations that were not significant after Bonferroni adjustment.
3.2. APOE ɛ4 and RNA expressions of the 18 proteins related to APOE ɛ4
To test whether the associations between APOE ɛ4 and the 18 proteins were also seen at the RNA level, we examined bulk tissue RNA expression levels of genes corresponding to the proteins. RNA expression level of APCS did not pass QC. Of the remaining 17 proteins, three had modest correlations with their RNA expression levels (Table e‐3). We found no associations of APOE ɛ4 with the 17 RNAs (Table e‐4). RNA expression of the 17 genes had also been measured in 600 other participants whose proteomic data were not available. Examining the association of APOE ɛ4 with RNA expression of the 17 genes in all participants with available RNA data (n = 1196) did not change the finding that APOE ɛ4 was not related to RNA expression (Table e‐5).
In 218 participants, we had snRNA‐seq pseudo‐bulked data of the 17 genes (similarly to bulk tissue RNA data, APCS was not expressed in snRNA‐seq data). Seven genes were expressed in all cell types (Figure 3). However, replacing bulk tissue with snRNA expression levels did not change the finding that APOE ɛ4 was not associated with RNA expression of any of the examined genes (Table e‐6). snRNA‐seq data were also available in 206 other participants whose proteomic data were not available. Examining the association of APOE ɛ4 with snRNA expression of the 17 genes in all participants with available snRNA‐Seq data (n = 424) indicated that only the association between APOE ɛ4 and snRNA expression of FLT1 (vascular endothelial growth factor receptor 1) in microglia was significant after Bonferroni correction (Table e‐7).
FIGURE 3.
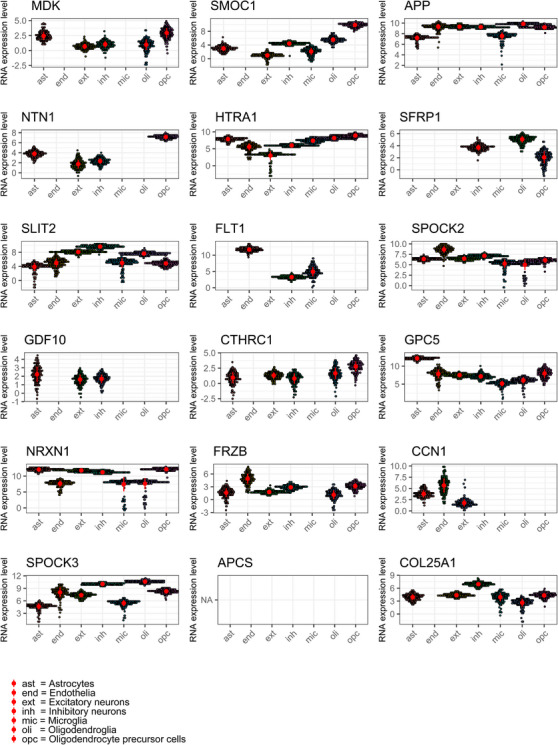
Pseudo‐bulk snRNA‐seq gene expression data for the 18 proteins related to APOE ɛ4.
FIGURE 4.
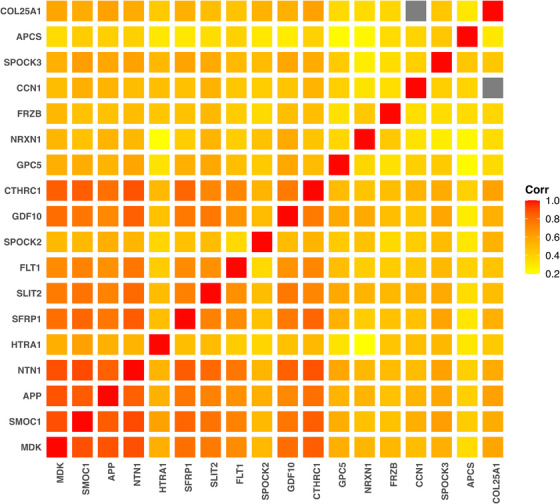
A heatmap plot illustrating correlations of the 18 proteins associated with APOE ɛ4. The gray squares denote missing in the levels of Cellular Communication Network family member 1 (O00622) and collagen alpha‐1(XXV) chain (Q9BXS0) as there were no participants having non‐missing levels of both proteins.
3.3. Alzheimer's dementia polygenic risk scores and the 18 APOE ɛ4‐related proteins
Next, we examined whether the 18 proteins were exclusively related to APOE ɛ4 or whether they were also related to other Alzheimer's dementia genetic risk factors. We used an Alzheimer's dementia polygenic risk score developed by applying a p value threshold of <5×10−8 and by excluding the APOE region. Examination of the 18 proteins in separate linear regression models indicated that the polygenic risk score was associated with only two proteins (SPOCK2 and NRXN1; Table e‐8). However, these associations were not robust as two other proteins (secreted frizzled‐related protein 1 [SFRP1] and SPOCK3) were associated with Alzheimer's dementia polygenic risk score when we changed the p value threshold to <1 and included more sequence variants in the risk score (Table e‐8). The aforementioned findings indicated that the 18 proteins were mostly related to APOE ɛ4 as opposed to other Alzheimer's dementia genetic risk variants.
3.4. Proteins linking associations of APOE ɛ4 with Aβ and tau tangles
The 18 APOE ɛ4‐related proteins were moderately to strongly correlated (Figure 4). Only seven proteins were present in all participants (Table e‐9), and no participant had non‐missing levels of all 18 proteins. Therefore, we excluded three proteins with the most missing (230, 343, 450 missing, Table e‐9) and examined 302 participants with non‐missing data of the remaining 15 proteins to examine the 15 proteins together in a single model. These 302 participants were not different from the other 294 participants in sex, race, education, frequency of APOE ɛ4, and levels of Aβ, tau tangle, and the 18 proteins, but on average they were 1 year older in age at death (Table e‐10).
In a single linear regression model, we examined the associations of the 15 proteins with Aβ, controlling for age at death and sex using backward elimination. Only five proteins remained associated with Aβ (Figure 5A‐E) and explained 58% of the variance of Aβ. Examination of the effect sizes of the five proteins indicated that two proteins (netrin‐1 and SFRP1) had the largest effect sizes, twice or more compared with the other three proteins (Table e‐11). Moreover, in a model that included the two proteins and APOE ɛ4, the association of APOE ɛ4 with Aβ was attenuated and no longer significant, indicating that the two proteins linked APOE ɛ4 with Aβ (Figure 6, Table e‐12).
FIGURE 5.
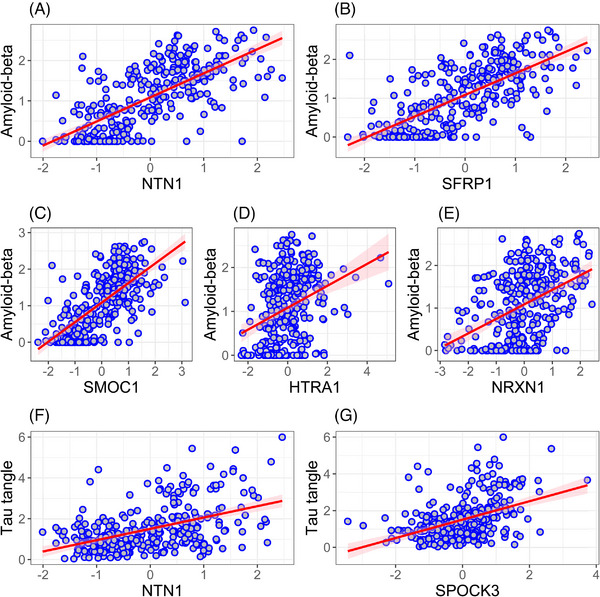
Associations of five and two APOE ɛ4‐related proteins independently associated with Aβ and tau tangles, respectively. In two separate models including 15 APOE ɛ4‐related proteins, only five proteins (A–E) remained associated with Aβ and two proteins (F–G) with tau tangles.
FIGURE 6.
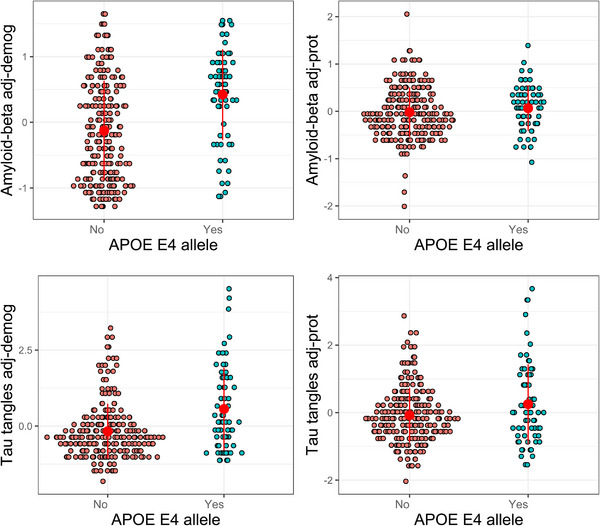
Associations of APOE ɛ4 with Aβ and tau tangles with and without adjustment for APOE ɛ4‐related proteins. The left panel illustrates associations of APOE ɛ4 with Aβ (upper panel) and tau tangles (lower panel) where levels of Aβ and tau tangles were adjusted for age at death and sex. The right panels illustrate the same associations where levels of Aβ (upper panel) and tau tangles (lower panel) were further adjusted for the related proteins, which were Netrin‐1 and secreted frizzled‐related protein 1 for Aβ and netrin‐1 and testican‐3 for tau tangles. The figure illustrates attenuation of the association between APOE ɛ4 and Aβ or tau tangles after adjustment for the proteins.
Similarly, in a separate linear regression model, we examined the associations of the 15 proteins with tau tangles as the outcome, controlled for age at death and sex using a backward elimination method. Only two proteins (netrin‐1 and testican‐3) remained associated with tau tangles (Figure 5F‐G) and explained 32% of the variance of tau tangles. The effect size of netrin‐1 was three times larger than the effect size of testican‐3 (Table e‐11). In a model that included the two proteins and APOE ɛ4, the association of APOE ɛ4 with tau tangles was attenuated by 50% but still significant (Figure 6, Table e‐12). Inclusion of Aβ in the model did not change the finding (Tables e‐12).
3.5. Pleiotropy of the 18 APOE ɛ4‐related proteins
We examined whether the 18 proteins were related exclusively to AD pathological hallmarks or to other brain pathologies as well. In eight series of logistic regression models, we separately examined associations of the 18 APOE ɛ4‐related proteins with eight other brain pathologies. As binary indices were used to summarize the other pathologies, we also examined the association of the 18 proteins with the binary variable of pathological diagnosis of AD to make comparable comparisons. While all 18 proteins were associated with higher odds of pathological diagnosis of AD, 16, 11, and eight proteins were related to CAA, TDP‐43, and Lewy bodies, which are also correlated with AD, no protein was related to hippocampal sclerosis, macroinfarcts, microinfarct, or arteriolosclerosis, and one protein was inversely related to atherosclerosis (Table e‐13). Moreover, the proteins that were related to both AD and other brain pathologies had stronger associations (larger odds ratios) with AD than with other pathologies. These findings suggest that the 18 APOE ɛ4‐related proteins had relative specificity in association with AD.
4. DISCUSSION
Clinical, pathological, and omics data from nearly 600 older adults were examined to identify proteins that link APOE ɛ4 with AD hallmarks, Aβ and tau tangles. The proteome‐wide association analysis identified 18 proteins that had higher levels in carriers of APOE ɛ4. Moreover, higher levels of the 18 proteins were associated with higher levels of Aβ and tau tangles, and faster cognitive decline was observed in higher levels of 16 of the proteins. Examining the proteins in a unified model identified two proteins (netrin‐1 and SFRP1) linking APOE ɛ4 with Aβ and two proteins (netrin‐1 and testican‐3) linking APOE ɛ4 with tau tangles. Replication of these findings in other studies with different methodologies will support targeting these proteins for drug development against AD in general and among those with APOE ɛ4 in particular.
Prior studies suggested different mechanisms underlying the association of APOE ɛ4 with AD, including impaired Aβ removal through less expression of amyloid degrading proteases, less Aβ degradation by astrocytes and microglia, and less efficient perivascular drainage of Aβ. 35 However, these mechanisms are based on findings from experimental studies including cell cultures and animal models, with limitations of such findings for generalization to the human body. Moreover, as the expression of genes and proteins may vary in different tissues, it is essential to examine omics data of human brain tissue rather than other tissues, including plasma, to uncover molecular mechanisms linking APOE ɛ4 to AD. Although a handful of studies used mass spectrometry methods to quantify brain tissue‐derived proteins and examine their relation to AD, 36 , 37 , 38 , 39 , 40 , 41 fewer studies specifically investigated the quantified proteins in relation to APOE ɛ4. 9 , 10 , 11 Compared with the prior studies, the current study quantified proteins in a much larger sample of approximately 600 participants. Further, we linked the proteins to a wide range of AD/ADRD clinical and pathologic traits and to other AD‐related genetic risk variants. Thus, the current study builds on prior work in important ways.
We found 18 proteins that showed higher expression in APOE ɛ4 carriers and were related to greater Aβ load and more tau tangles. The proteins were involved in diverse cellular functions, just as proteins that were found related to Aβ and tau tangles in proteome‐wide association studies of AD. 42 Moreover, 11 of the 18 proteins were among 58 proteins that were related to AD and validated across three cohort studies. 40 Interestingly, the 18 proteins were not among the top proteins related to APOE ɛ4 in prior studies 9 , 10 , 11 possibly due to different sample sizes, participant age, number of quantified proteins, and examined brain regions. Of note, there was almost no association between APOE ɛ4 and RNA expression of these proteins, which indicates that the pathways linking APOE ɛ4 and the 18 proteins are at the posttranslational stages of the protein synthesis, not at the RNA transcription level.
By examining the APOE ɛ4‐related proteins together in one model, we identified netrin‐1 as the protein linking APOE ɛ4 with Aβ and tau tangles with the largest effect sizes. Netrin‐1 is a matrix protein contributing to axonal guidance during brain development. 43 Prior studies showed that netrin‐1 was enriched in Aβ deposits but not tau tangles 41 and also bound to Aβ. 40 , 43 Moreover, a variant of a netrin‐1 receptor, UNC5C, is a rare genetic risk factor for late‐onset AD 44 and is related to CAA 45 and cognition. 46 As both netrin‐1 and APOE protein are present in the brain matrix and APOE may contribute to Aβ clearance with less efficient clearance by APOE E4 protein compared with E2 and E3 isoforms, 35 we hypothesized that the contribution of netrin‐1 to Aβ clearance pathways facilitated by APOE was the mechanism through which netrin‐1 mediated the association between APOE ɛ4 and AD. This hypothesis needs to be tested in future studies.
Fewer data are available to suggest how SFRP1 and testican‐3 link APOE ɛ4 with Aβ and tau tangles, respectively. SFRP1 is an inhibitor of disintegrin and metalloproteinase domain‐containing protein 10 (ADAM10), 47 , 48 which is a part of α‐secretase, which provides an alternative processing pathway for amyloid precursor proteins yielding less toxic soluble amyloid precursor protein α rather than Aβ. In addition, lower levels of SFRP1 were observed in brain organelles derived from APOE‐deficient stem cells. 49 We hypothesize that higher levels of SFRP1 associated with APOE ɛ4 result in the production of more Aβ as a result of inhibiting α‐secretase. If our hypotheses are correct, netrin‐1 and SFRP1 link APOE ɛ4 with more Aβ in two separate pathways; one yields less Aβ clearance and the other more Aβ production. Testican‐3 is a secreted proteoglycan that is involved in the regulation of extracellular protease cascade and neuronal function. 50 We could not find a plausible mechanism explaining the relation between testican‐3, APOE ɛ4, and tau tangles.
Some of the 18 proteins were also related to TDP‐43, Lewy bodies, and CAA, which were found to be correlated with AD. 51 However, the proteins were not related to cerebrovascular disease pathologies that were not correlated with AD. 51 The latter, together with the stronger associations of the proteins with AD compared with their associations with TDP‐43 and Lewy bodies, suggest that the proteins are specifically involved in the molecular biology of AD. However, the proteins may be representative of the molecular biology of neurodegeneration and neuronal loss, that is, the end stage of all the neurodegenerative diseases of the brain. 52 These possibilities should be addressed in future studies.
The current study has several strengths. Proteome data of frontal cortex of approximately 600 older adults were examined with AD pathological markers and APOE ɛ4. A common structured harmonized data collection system reduced the variability of measurements and increased the reliability of the findings. The availability of both proteome and transcriptome data provided the opportunity to examine whether the findings of proteomes linking APOE ɛ4 with AD were because of changes in gene expression or because of posttranslational changes. However, limitations must be noted when drawing inferences from the findings. The findings come from analyzing observational cross‐sectional study data and are more indicative of correlations rather than cause and effect. While more than 15,000 genes are expressed in the human brain, we could quantify only 8500 proteins. Future studies with advanced proteomic techniques may quantify more proteins and more pathways linking APOE ɛ4 with AD. The study participants were volunteers, mostly Whites and with high educational level; further studies with more diverse participants are required for the generalizability of the findings. As the association of APOE ɛ4 with AD varies across race and ethnicity, 53 the findings of this study may not be generalizable to more diverse populations. Less than 1% of the participants had an APOE ɛ4ɛ4 genotype, which is much less than expected. 54 Because individuals with two APOE ɛ4 alleles have a much higher risk of AD compared with one APOE ɛ4 allele, 54 our findings need replication in samples with more persons with APOE ɛ4ɛ4 genotype.
CONFLICT OF INTEREST STATEMENT
The authors report no disclosures relevant to the manuscript. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All participants signed informed and repository consents and an Anatomic Gift Act.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors are truly thankful to the ROSMAP participants who generously donated their time, in life by participation in the long‐lasting clinical examinations, and their brain for post mortem pathological and biological examinations. We also appreciate the staff of Rush Alzheimer's Disease Center; this study would not have been possible without their work. ROSMAP is supported by P30AG10161, P30AG72975, R01AG15819, R01AG17917, U01AG46152, and U01AG61356. ROSMAP resources can be requested at https://www.radc.rush.edu and https://www.synapse.org. The study was supported by National Institute on Aging grants P30AG010161, P30AG072975, R01AG015819, R01AG017917, U01AG046152, and U01AG061356.
Oveisgharan S, Yu L, de Paiva Lopes K, et al. Proteins linking APOE ɛ4 with Alzheimer's disease. Alzheimer's Dement. 2024;20:4499–4511. 10.1002/alz.13867
Statistical analyses: The statistical analyses were supervised by Dr. Lei Yu, PhD at Rush Alzheimer's Disease Center.
REFERENCES
- 1. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349‐1356. [PubMed] [Google Scholar]
- 2. Rajan KB, Barnes LL, Wilson RS, et al. Racial differences in the association between apolipoprotein E risk alleles and overall and total cardiovascular mortality over 18 years. J Am Geriatr Soc. 2017;65:2425‐2430. doi: 10.1111/jgs.15059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bennett DA, Wilson RS, Schneider JA, et al. Apolipoprotein E epsilon4 allele, AD pathology, and the clinical expression of Alzheimer's disease. Neurology. 2003;60:246‐252. doi: 10.1212/01.wnl.0000042478.08543.f7 [DOI] [PubMed] [Google Scholar]
- 4. Mortimer JA, Snowdon DA, Markesbery WR. The effect of APOE‐epsilon4 on dementia is mediated by Alzheimer neuropathology. Alzheimer Dis Assoc Disord. 2009;23:152‐157. doi: 10.1097/wad.0b013e318190a855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bennett DA, Schneider JA, Wilson RS, Bienias JL, Berry‐Kravis E, Arnold SE. Amyloid mediates the association of apolipoprotein E e4 allele to cognitive function in older people. J Neurol Neurosurg Psychiatry. 2005;76:1194‐1199. doi: 10.1136/jnnp.2004.054445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu L, Boyle PA, Leurgans S, Schneider JA, Bennett DA. Disentangling the effects of age and APOE on neuropathology and late life cognitive decline. Neurobiol Aging. 2014;35:819‐826. doi: 10.1016/j.neurobiolaging.2013.10.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Farfel JM, Yu L, De Jager PL, Schneider JA, Bennett DA. Association of APOE with tau‐tangle pathology with and without β‐amyloid. Neurobiol Aging. 2016;37:19‐25. doi: 10.1016/j.neurobiolaging.2015.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sweet RA, MacDonald ML, Kirkwood CM, et al. Apolipoprotein E*4 (APOE*4) genotype is associated with altered levels of glutamate signaling proteins and synaptic coexpression networks in the prefrontal cortex in mild to moderate Alzheimer disease. Mol Cell Proteomics. 2016;15:2252‐2262. doi: 10.1074/mcp.M115.056580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dai J, Johnson ECB, Dammer EB, et al. Effects of APOE genotype on brain proteomic network and cell type changes in Alzheimer's disease. Front Mol Neurosci. 2018;11:454. doi: 10.3389/fnmol.2018.00454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roberts JA, Varma VR, An Y, et al. A brain proteomic signature of incipient Alzheimer's disease in young APOE ε4 carriers identifies novel drug targets. Sci Adv. 2021;7:eabi8178. doi: 10.1126/sciadv.abi8178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ojo JO, Reed JM, Crynen G, et al. APOE genotype dependent molecular abnormalities in the cerebrovasculature of Alzheimer's disease and age‐matched non‐demented brains. Mol Brain. 2021;14:110. doi: 10.1186/s13041-021-00803-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious orders study and rush memory and aging project. J Alzheimers Dis. 2018;64:S161‐S189. doi: 10.3233/JAD-179939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oveisgharan S, Buchman AS, Yu L, et al. APOE epsilon2epsilon4 genotype, incident AD and MCI, cognitive decline, and AD pathology in older adults. Neurology. 2018;90:e2127‐e2134. doi: 10.1212/WNL.0000000000005677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oveisgharan S, Arvanitakis Z, Yu L, Farfel J, Schneider JA, Bennett DA. Sex differences in Alzheimer's disease and common neuropathologies of aging. Acta Neuropathol. 2018;136:887‐900. doi: 10.1007/s00401-018-1920-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the Rush Memory and Aging Project. Curr Alzheimer Res. 2012;9:646‐663. doi: 10.2174/156720512801322663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61:378‐384. doi: 10.1001/archneur.61.3.378 [DOI] [PubMed] [Google Scholar]
- 17. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8:1‐13. doi: 10.1016/j.jalz.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wingo AP, Fan W, Duong DM, et al. Shared proteomic effects of cerebral atherosclerosis and Alzheimer's disease on the human brain. Nat Neurosci. 2020;23:696‐700. doi: 10.1038/s41593-020-0635-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Higginbotham L, Carter EK, Dammer EB, et al. Unbiased classification of the elderly human brain proteome resolves distinct clinical and pathophysiological subtypes of cognitive impairment. Neurobiol Dis. 2023;186:106286. doi: 10.1016/j.nbd.2023.106286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wingo AP, Liu Y, Gerasimov ES, et al. Sex differences in brain protein expression and disease. Nat Med. 2023;29:2224‐2232. doi: 10.1038/s41591-023-02509-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mostafavi S, Gaiteri C, Sullivan SE, et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer's disease. Nature Neuroscience. 2018;21:811‐819. doi: 10.1038/s41593-018-0154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oveisgharan S, Yang J, Yu L, et al. Estrogen receptor genes, cognitive decline, and Alzheimer disease. Neurology. 2023;100:e1474‐e1487. doi: 10.1212/WNL.0000000000206833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tasaki S, Xu J, Avey DR, et al. Inferring protein expression changes from mRNA in Alzheimer's dementia using deep neural networks. Nat Commun. 2022;13:655. doi: 10.1038/s41467-022-28280-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Green GS, Fujita M, Yang H‐S, et al. Cellular dynamics across aged human brains uncover a multicellular cascade leading to Alzheimer's disease. BioRxiv 2023:2023.03.07.531493. doi: 10.1101/2023.03.07.531493 bioRxiv [DOI]
- 25. Hao Y, Hao S, Andersen‐Nissen E, et al. Integrated analysis of multimodal single‐cell data. Cell. 2021;184:3573‐3587. doi: 10.1016/j.cell.2021.04.048. e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA‐seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51:404‐413. doi: 10.1038/s41588-018-0311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shulman JM, Chen K, Keenan BT, et al. Genetic susceptibility for Alzheimer disease neuritic plaque pathology. JAMA Neurol. 2013;70:1150‐1157. doi: 10.1001/jamaneurol.2013.2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tasaki S, Gaiteri C, Petyuk VA, et al. Genetic risk for Alzheimer's dementia predicts motor deficits through multi‐omic systems in older adults. Transl Psychiatry. 2019;9:241. doi: 10.1038/s41398-019-0577-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oveisgharan S, Wilson RS, Yu L, Schneider JA, Bennett DA. Association of early‐life cognitive enrichment with Alzheimer disease pathological changes and cognitive decline. JAMA Neurol. 2020;77:1217. doi: 10.1001/jamaneurol.2020.1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oveisgharan S, Dawe RJ, Yu L, et al. Frequency and underlying pathology of pure vascular cognitive impairment. JAMA Neurol. 2022;79:1277‐1286. doi: 10.1001/jamaneurol.2022.3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263‐269. doi: 10.1016/j.jalz.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ialongo C. Understanding the effect size and its measures. Biochem Med (Zagreb). 2016;26:150‐163. doi: 10.11613/BM.2016.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilson RS, Bienias JL, Berry‐Kravis E, Evans DA, Bennett DA. The apolipoprotein E epsilon 2 allele and decline in episodic memory. J Neurol Neurosurg Psychiatry. 2002;73:672‐677. doi: 10.1136/jnnp.73.6.672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yamazaki Y, Zhao N, Caulfield TR, Liu C‐C, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15:501‐518. doi: 10.1038/s41582-019-0228-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seyfried NT, Dammer EB, Swarup V, et al. A multi‐network approach identifies protein‐specific co‐expression in asymptomatic and symptomatic Alzheimer's disease. Cell Syst. 2017;4:60‐72. doi: 10.1016/j.cels.2016.11.006. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Andreev VP, Petyuk VA, Brewer HM, et al. Label‐free quantitative LC‐MS proteomics of Alzheimer's disease and normally aged human brains. J Proteome Res. 2012;11:3053‐3067. doi: 10.1021/pr3001546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hondius DC, van Nierop P, Li KW, et al. Profiling the human hippocampal proteome at all pathologic stages of Alzheimer's disease. Alzheimers Dement. 2016;12:654‐668. doi: 10.1016/j.jalz.2015.11.002 [DOI] [PubMed] [Google Scholar]
- 39. Ho Kim J, Franck J, Kang T, et al. Proteome‐wide characterization of signalling interactions in the hippocampal CA4/DG subfield of patients with Alzheimer's disease. Sci Rep. 2015;5:11138. doi: 10.1038/srep11138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bai B, Wang X, Li Y, et al. Deep multilayer brain proteomics identifies molecular networks in Alzheimer's disease progression. Neuron. 2020;105:975‐991. doi: 10.1016/j.neuron.2019.12.015. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Askenazi M, Kavanagh T, Pires G, Ueberheide B, Wisniewski T, Drummond E. Compilation of reported protein changes in the brain in Alzheimer's disease. Nat Commun. 2023;14:4466. doi: 10.1038/s41467-023-40208-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johnson ECB, Carter EK, Dammer EB, et al. Large‐scale deep multi‐layer analysis of Alzheimer's disease brain reveals strong proteomic disease‐related changes not observed at the RNA level. Nat Neurosci. 2022;25:213‐225. doi: 10.1038/s41593-021-00999-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Borel F, Marzocca F, Delcros J‐G, Rama N, Mehlen P, Ferrer J‐L. Molecular characterization of Netrin‐1 and APP receptor binding: new leads to block the progression of senile plaques in Alzheimer's disease. Biochem Biophys Res Commun. 2017;488:466‐470. doi: 10.1016/j.bbrc.2017.05.056 [DOI] [PubMed] [Google Scholar]
- 44. Wetzel‐Smith MK, Hunkapiller J, Bhangale TR, et al. A rare mutation in UNC5C predisposes to late‐onset Alzheimer's disease and increases neuronal cell death. Nat Med. 2014;20:1452‐1457. doi: 10.1038/nm.3736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang H‐S, White CC, Chibnik LB, et al. UNC5C variants are associated with cerebral amyloid angiopathy. Neurol Genet. 2017;3:e176. doi: 10.1212/NXG.0000000000000176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. White CC, Yang H‐S, Yu L, et al. Identification of genes associated with dissociation of cognitive performance and neuropathological burden: multistep analysis of genetic, epigenetic, and transcriptional data. PLoS Med. 2017;14:e1002287. doi: 10.1371/journal.pmed.1002287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Esteve P, Rueda‐Carrasco J, Inés Mateo M, et al. Elevated levels of Secreted‐Frizzled‐Related‐Protein 1 contribute to Alzheimer's disease pathogenesis. Nat Neurosci. 2019;22:1258‐1268. doi: 10.1038/s41593-019-0432-1 [DOI] [PubMed] [Google Scholar]
- 48. Ramamurthy E, Welch G, Cheng J, et al. Cell type‐specific histone acetylation profiling of Alzheimer's disease subjects and integration with genetics. Front Mol Neurosci. 2022;15:948456. doi: 10.3389/fnmol.2022.948456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao J, Ikezu TC, Lu W, et al. APOE deficiency impacts neural differentiation and cholesterol biosynthesis in human iPSC‐derived cerebral organoids. Stem Cell Res Ther. 2023;14:214. doi: 10.1186/s13287-023-03444-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hartmann U, Hülsmann H, Seul J, et al. Testican‐3: a brain‐specific proteoglycan member of the BM‐40/SPARC/osteonectin family. J Neurochem. 2013;125:399‐409. doi: 10.1111/jnc.12212 [DOI] [PubMed] [Google Scholar]
- 51. Boyle PA, Wang T, Yu L, et al. To what degree is late life cognitive decline driven by age‐related neuropathologies? Brain. 2021;144:2166‐2175. doi: 10.1093/brain/awab092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Simons M, Levin J, Dichgans M. Tipping points in neurodegeneration. Neuron. 2023;111:2954‐2968. doi: 10.1016/j.neuron.2023.05.031 [DOI] [PubMed] [Google Scholar]
- 53. Belloy ME, Andrews SJ, Le Guen Y, et al. APOE genotype and Alzheimer disease risk across age, sex, and population ancestry. JAMA Neurol. 2023;80:1284‐1294. doi: 10.1001/jamaneurol.2023.3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sando SB, Melquist S, Cannon A, et al. APOE epsilon 4 lowers age at onset and is a high risk factor for Alzheimer's disease; a case control study from central Norway. BMC Neurol. 2008;8:9. doi: 10.1186/1471-2377-8-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information