

Abstract
BACKGROUND
Variants in ABCA7, a member of the ABC transporter superfamily, have been associated with increased risk for developing late onset Alzheimer's disease (LOAD).
METHODS
CRISPR‐Cas9 was used to generate an Abca7V1613M variant in mice, modeling the homologous human ABCA7V1599M variant, and extensive characterization was performed.
RESULTS
Abca7V1613M microglia show differential gene expression profiles upon lipopolysaccharide challenge and increased phagocytic capacity. Homozygous Abca7V1613M mice display elevated circulating cholesterol and altered brain lipid composition. When crossed with 5xFAD mice, homozygous Abca7V1613M mice display fewer Thioflavin S‐positive plaques, decreased amyloid beta (Aβ) peptides, and altered amyloid precursor protein processing and trafficking. They also exhibit reduced Aβ‐associated inflammation, gliosis, and neuronal damage.
DISCUSSION
Overall, homozygosity for the Abca7V1613M variant influences phagocytosis, response to inflammation, lipid metabolism, Aβ pathology, and neuronal damage in mice. This variant may confer a gain of function and offer a protective effect against Alzheimer's disease‐related pathology.
Highlights
ABCA7 recognized as a top 10 risk gene for developing Alzheimer's disease.
Loss of function mutations result in increased risk for LOAD.
V1613M variant reduces amyloid beta plaque burden in 5xFAD mice.
V1613M variant modulates APP processing and trafficking in 5xFAD mice.
V1613M variant reduces amyloid beta‐associated damage in 5xFAD mice.
Keywords: ABCA7, Alzheimer's disease, Aβ, lipids, neuroinflammation
1. BACKGROUND
Alzheimer's disease (AD) is a progressive neurodegenerative disease characterized by the presence of extracellular amyloid beta (Aβ) plaques and intracellular neurofibrillary tau tangles. 1 Causative mutations in genes related to amyloid precursor protein (APP) processing have been identified for autosomal dominant AD. In contrast, the disease etiology for sporadic or late‐onset AD (LOAD) remains unclear. To better understand this, genome‐wide association studies (GWAS) have identified genetic variations associated with risk of developing LOAD, 2 , 3 , 4 including variants in ABCA7. 2 , 3 , 5
ABCA7 encodes ATP‐binding cassette, subfamily A, member 7 (ABCA7), a multidomain transmembrane protein member of the conserved superfamily of ATP‐binding cassette (ABC) transporters that transfer various molecules across membranes in an ATP‐dependent manner. 6 ABCA7 mediates efflux of phospholipids and to a much lesser extent cholesterol across the plasma membrane. 7 , 8 , 9 , 10 In Abca7 knockout (KO) mice, cholesterol metabolism, characterized by reduced plasma high‐density lipoprotein (HDL) and cholesterol levels, brain lipid profiles, and lipid rafts in antigen‐presenting cells, are all disrupted compared to wildtype (WT) littermates. 11 , 12 , 13 , 14 ABCA7 deficiency also reduced phagocytosis of apoptotic cells, Aβ oligomers, and diminished proinflammatory response in macrophages. 15 , 16 , 17 In transgenic AD mouse models, complete or haploinsufficiency of Abca7 results in increased Aβ accumulation in the brain and reduced uptake of Aβ in macrophages and microglia. 16 , 18 ABCA7 loss of function also increases Aβ via altered APP processing. 19
Consistent with these findings, rare premature termination codon and/or loss of function mutations in ABCA7 have been associated with increased risk of AD and are enriched in AD patients, 20 whereas a low‐frequency coding variant (p.G215S) has been identified as potentially protective against AD. 21 Additional study is needed to understand the biological implications of specific ABCA7 polymorphisms in AD.
Despite being a major risk factor for AD, there is limited knowledge on the role of ABCA7 in the development and progression of AD, particularly regarding the impact of specific variants on AD risk and disease pathology. One specific ABCA7 single nucleotide polymorphism (SNP), rs117187003, produces the rare V1599M variant in humans. Several exome sequencing studies predicted this variant, which is located in a region of ABCA7 that is conserved between humans and mouse, as being deleterious and potentially damaging. 4 , 21 To test the hypothesis that this variant is potentially damaging we used CRISPR‐CAS9 to introduce the corresponding coding variant (V1613M) in the mouse genome, which we refer to as Abca7V1613M hereafter. Homozygous Abca7V1613M microglia exhibit a more inflammatory response to acute lipopolysaccharide (LPS) stimulation and have increased phagocytic capacity for Aβ1‐42 and beads. To examine the impact of this genetic variation on AD pathology, we analyzed homozygous Abca7V1613M mice that were hemizygous for the 5xFAD transgene array. 5xFAD/Abca7V1613M mice have altered brain and plasma lipids, reduced Aβ plaques, neuroinflammation, dystrophic neurites, and neurofilament light chain (NfL) levels. Finally, we used both in vivo and in vitro approaches to show that the Abca7V1613M variant induces alterations in APP processing and trafficking. These results suggest that the effects of the Abca7V1613M variant are consistent with a gain of function, rather than a loss of function, and may offer protection against the development of LOAD.
2. METHODS
2.1. Animals
All experiments involving mice were approved by the UC Irvine (UCI) Institutional Animal Care and Use Committee (IACUC) and were conducted in compliance with all relevant ethical regulations for animal testing and research. All experiments involving mice comply with the Animal Research: Reporting of in Vivo Experiments (ARRIVE‐10) guidelines.
2.2. Mouse generation and breeding
CRISPR/Cas9 was used to generate a V1613M missense variant allele of Abca7. This variant models a SNP (rs117187003) in human ABCA7 that encodes a missense variant (V1599M). Alt‐R Crispr RNA (TMF1268—cttggtggcagtgtgcatag) and tracrRNA plus CAS9 protein (HiFi Cas9 nuclease V3, Integrated DNA Technologies [IDT), Coralville, IA) as a ribonucleoprotein (RNP) were electroporated into C57BL/6J zygotes (Jackson Lab Stock # 000664) along with a ssODN sequence (TMF1276—sequence available upon request) to introduce the V1613M missense variant. G0 founder animals containing the desired DNA sequence changes were backcrossed with C57BL/6J mice, and N1 Abca7V1613M heterozygous mice were sequenced to determine the variant allele. N1 Abca7V1613M heterozygous mice were again backcrossed with C57BL/6J mice to produce N2F1 Abca7V1613M heterozygotes, which were subsequently crossed with 5xFAD hemizygous congenic B6J (B6.CgTg(APPSwFlLon,PSEN1*M146L*L286V)6799Vas/Mmjax, Jackson Lab Stock # 34848, MMRRC) mice to produce N3F1 animals that were heterozygous or wildtype for Abca7V1613M and hemizygous or nontransgenic for 5xFAD. These N3F1 animals were used to produce N3Fx experimental and control animals by natural mating or in vitro fertilization procedures (Figure S1). After weaning, they were housed together by sex with littermates and aged until the harvest dates. All animals in this study were on a co‐isogenic or congenic C57BL/6J strain background. All animals were bred by the Transgenic Mouse Facility at UCI. Abca7V1613M mice (B6‐ Abca7 em1Aduci /J) are available from The Jackson Laboratory (Bar Harbor, ME), stock number #035316.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (eg, PubMed) sources and meeting abstracts and presentations. While the role of ABCA7, and its variants, in Alzheimer's disease (AD) pathogenesis is not yet as widely studied as other aspects of AD biology, there have been several publications using Abca7 knockout models which have highlighted a key role for ABCA7 in amyloid beta (Aβ) production and processing. These relevant works are appropriately cited.
Interpretation: Our findings show introduction of Abca7V1613M variant modulates Aβ production, processing, and ultimately results in reduced Aβ plaque formation and the variant protects against Aβ‐induced damage in a 5xFAD mouse model.
Future directions: This study highlights the effect of the Abca7V1613M variant on Aβ pathology. Further work is required to understand the underlying molecular mechanisms of ABCA7 and variant function and whether promoting ABCA7 function may be beneficial to treatment of AD.
2.3. Genotyping
Oligonucleotides for polymerase chain reaction (PCR)‐based genotyping were purchased from IDT. Abca7V1613M genotyping was performed using a common primer set to amplify both Abca7 wildtype allele and Abca7V1613M allele (For 5′‐ TGGATACAGTGTAACTACTTGG −3′ and Rev 5′‐ ATAGGCTCTCTGCTGAAAG −3′). Two fluorophore labeled‐hydrolysis probes which hybridized specific to mouse Abca7 wildtype amplicon (5′‐ TGAACACCACTATGCACACTGCCA −3′+HEX) and Abca7V1613M variant (5′‐ TGA ACATGACGATGCACACTGCCA −3′‐FAM) were used to detect the allelic ratio in the amplicon. The relative fluorescence from each probe was quantified at the end point of PCR cycles to call the genotype using the allelic discrimination function in Bio‐Rad CFX Maestro software (Bio‐Rad, Hercules, CA). For 5xFAD genotyping, a hydrolysis probe which hybridizes to APP(Swe) mutation amplicon was used (For 5′‐TGGGTTCAAACAAAGGTGCAA‐3′ and Rev 5′‐GATGACGATCACTGTCGCTATGAC‐3′: APP(Swe) probe 5′‐CATTGGACTCATGGTGGGCGGTG‐3′) to detect transgenes. We used endogenous ApoB allele (For 5′‐CACGTGGGCTCCAGCATT‐3′ and Rev 5′‐TCACCAGTCATTTCTGCCTTTG‐3′: ApoB probe 5′‐CCAATGGTCGGGCACTGCTCAA‐3′) to normalize the cycle threshold values.
2.4. Off‐target analysis
Genomic DNA was extracted from mouse tail biopsies using DirectPCR Lysis Reagent (Viagen Biotech, Los Angeles, CA) and Proteinase K (Roche, Indianapolis, IN). Amplification was performed using a Bio‐Rad CFX‐96 instrument. For each amplicon, a single PCR product was confirmed by capillary electrophoresis (Fragment Analyzer, AATI / Agilent, Santa Clara, CA) then subjected to Sanger sequencing (Retrogen, San Diego, CA) and analyzed using SeqMan Pro 17.2 (DNASTAR, Madison, WI). Potential off‐target sites are listed in Table S1 while primers for PCR amplification and sequencing of potential off‐target sites are listed in Table S2.
2.5. Bulk RNA‐sequencing
Frozen hippocampi were pulverized, and total RNA was extracted according to manufacturer's instructions using RNAeasy Plus mini kit (Qiagen, 74134). The RNA integrity number (RIN) was measured, and samples with RIN ≥ 7.0 were kept for library construction. cDNA synthesis, amplification, library construction, and sequencing were performed by Novogene (Sacramento, CA) using Illumina NovaSeq and HiSeq platforms with paired‐end 150 bp (PE 150) sequencing strategy. Fastqs were aligned to the mouse genome (mm10) and annotation was done using GENCODE v21. Pair‐end RNA‐seq reads were mapped with STAR (2.5.1b‐static) and RSEM (1.2.22) was used for quantification of gene expression.
2.5.1. Differential gene expression analysis
Differential gene expression analysis was performed using edgeR per timepoint and genotype or treatment on polyA genes. Top significant genes are displayed as a volcano plot constructed using GLimma, ggplot2, and EnhancedVolcano (false discovery rate [FDR] < 0.05, LogFC > 1). Based on the differentially expressed gene (DEG) comparisons, gene transcripts per million (TPMs) of interest were plotted as a heatmap or graphically using Prism (GraphPad, Boston, MA).
2.5.2. Weighted correlation gene network analysis
Weighted gene correlation network analysis (WGCNA) was performed using PyWGCNA 22 on the Abca7V1613M dataset with matching controls at two different timepoints (4 months and 12 months old) in the hippocampus for both sexes, using genes with more than 1 TPM. One sample was removed based on hierarchical clustering at the sample level. Based on our datasets, we used power 19 as a soft threshold. PyWGCNA were also applied on the LPS dataset (Figure 1) at all three timepoints (0 hours, 6 and 24 hours) for both treatment groups using genes with more than 1 TPM. The other parameters were the same for both including min. module size = 50 and MEDissThres = 0.2. Significant modules were identified by calculating the correlation with the traits, then plotting the behavior per sample of the genes in the inflammatory module by doing a GO term analysis using PyWGCNA.
FIGURE 1.
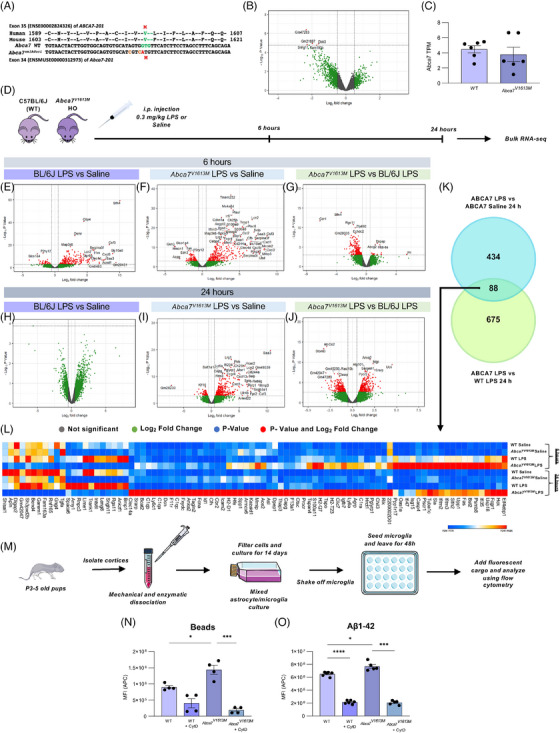
The Abca7V1613M allele in mice models the human single nucleotide polymorphism rs117187003 (ABCA7 V1599M ) and induces distinct responses to lipopolysaccharide (LPS) challenge and increased phagocytosis. (A) Human and mouse amino acid sequence alignment indicating position of valine (V) to methionine (M) missense variant in each species (in green/red). Two silent mutations (tan) were co‐introduced into mouse Abca7 to prevent recutting of the edited allele by CRISPR‐Cas9. (B) Volcano plot of differentially expressed genes (DEGs) between C57BL6/J (wildtype [WT]) and Abca7V1613M homozygous mice at 6 to 8 months of age. Only five DEGs were deemed significant (denoted by red dots). (C) Transcripts per million (TPMs) of Abca7 in WT and Abca7V1613M homozygotes (unpaired Student t‐test). (D) Experimental paradigm used for the LPS experiment. I Volcano plot of DEGs between WT mice treated with 0.3 mg/kg LPS versus saline 6 hours postinjection. (F) Volcano plot of DEGs between Abca7V1613M mice treated with 0.3 mg/kg LPS versus saline 6 hours postinjection. (G) Volcano plot of DEGs between Abca7V1613M and WT B6/J mice treated with 0.3 mg/kg LPS 6 hours postinjection. (H) Volcano plot of DEGs between Abca7V1613M mice treated with 0.3 mg/kg LPS versus saline 24 hours postinjection. (I) Volcano plot of DEGs between Abca7V1613M mice treated with 0.3 mg/kg LPS versus saline 24 hours postinjection. (J) Volcano plot of DEGs between Abca7V1613M and WT mice treated with 0.3 mg/kg LPS 24 hours post injection. (K) Venn diagram highlighting the 88 overlapping genes between Abca7V1613M LPS versus Abca7V1613M saline and Abca7V1613M LPS versus WT LPS DEGs at 24 hours. (L) Heatmap of 88 overlapping genes between Abca7V1613M LPS versus saline mice and Abca7V1613M and WT LPS treated mice 24 hours postinjection highlighting increased expression of inflammatory genes in Abca7 V1613M mice compared to WT mice at both 6 and 24 hours postinjection. N = 3 to 4 mice per genotype/treatment/timepoint. Mice from each genotype/treatment/timepoint were pooled and the mean TPM value was plotted as the heatmap. A log2 fold change of 0.5 was used for all volcano plots; however, the ‐log10 p‐value changed according to different comparisons analyzed. (M) Schematic of primary microglial extraction from neonatal pups. (N,O) Uptake assays of primary microglia extracted from WT and Abca7V1613M mice for (N) carboxylated beads, N = 4 independent experiments, and (O) Aβ 1‐42 peptide, N = 5 independent experiments; one‐way analysis of variance was used to examine group differences. MFI (APC) indicates the mean fluorescence intensity of allophycocyanin (APC) fluorophore inside microglia; 10 μM cytochalasin D was used as an inhibitor for phagocytosis. Data are represented as mean ± SEM. *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001.
2.6. LPS treatment
Eight‐month‐old WT and homozygous Abca7V1613M mice of both sexes were injected intraperitoneally with either 0.3 mg/kg LPS or 1× phosphate‐buffered saline (PBS). At 6 or 24 hours post administration, mice were euthanized via CO2 inhalation and transcardially perfused with ice‐cold 1× PBS. Brains were removed and hemispheres separated along the midline. Brain halves were flash frozen for RNA‐sequencing analysis.
2.7. Generation of mouse mixed primary microglia‐astrocyte cultures
Primary mixed microglia‐astrocyte cultures were generated as described. 23 Whole brains were extracted from neonatal 3‐ to 5‐day‐old mice and cortical tissue was cut into small pieces before digestion with trypsin. Trypsin was quenched using glia media (DMEM supplemented with 10% performance + heat inactivated serum (10082147; Thermo Fisher Scientific) and 1% penicillin/streptomycin (P4333‐100ML; Sigma‐Aldrich) and tissue was dissociated by pipetting up and down 20 times with a 1000‐μL tip. Following digestion, the tissue was dissociated and homogenized through pipetting and then centrifuged at 150 × g for 7 minutes with slow start‐stop at room temperature. Cells were resuspended in fresh glia media and filtered using 100‐μm (352360; Falcon), followed by 40‐μm strainers (352340; Falcon). Finally, the cells were reconstituted with 10 mL of glia media and placed in T‐75 cm2 flasks. Flasks were precoated with 0.002% poly‐lysine (P4707‐50ML; Sigma‐Aldrich) for at least 30 minutes, at room temperature. After 24 hours, 20 mL of fresh media was added to the cell cultures. After 7 to 14 days in vitro, mixed microglia‐astrocyte cultures were used for experiments.
2.8. Phagocytosis assays
Primary microglia were removed from mixed microglia‐astrocyte culture by gentle shaking as described. 23 To investigate the ability of cells to engage in general phagocytosis, 0.005% w/v 5‐μm carboxylated beads (CFP‐5070‐2; Spherotech) were incubated for 2 hours with 50,000 primary microglia (seeded in 2:1 fresh:conditioned DMEM in 24‐well plates in triplicate 24 hours before adding cargo). For Alexa Fluor 647‐labelled Aβ1‐42 (AS‐64161; AnaSpec) a final concentration of 2 μM was added for 1 hour to a total of 20,000 primary microglia (seeded in 2:1 fresh:conditioned DMEM in 96‐well plates in triplicate 24 hours before adding cargo). For both experiments, microglia were pretreated +/‐ 10 μM cytochalasin D (CytD) for 1 hour as a negative control for phagocytic uptake. After incubation, cells were washed twice with PBS to remove non‐phagocytosed cargo and then harvested using trypsin, pelleted, and finally resuspended in PBS for flow cytometry (NovoCyte, Agilent, Santa Clara, CA).
2.9. Flow cytometry
For flow cytometry, ACEA Quanteon (NovoCyte) analyzer was used. At least 5000 events were analyzed for each treatment replicate. Fc blocking antibodies were not used. Forward and side scatter was used to distinguish cells from unphagocytosed targets, that is, beads and Aβ, by gating on cells in the forward and side scatter plots. Within this scatter gate, a fluorescence gate was set to identify cells that were over a threshold of fluorescence. For phagocytic targets, this fluorescence gate was set so that for cells incubated in the absence of any fluorescent targets (ie, absence of fluorescent beads or Aβ), 99% of the cells were below this gated fluorescence and 1% were above this gate. For cells incubated in the presence of fluorescent targets, the percentage of cells with fluorescence greater than the gated fluorescence was used as the measure of mean fluorescence intensity of cells that had phagocytosed the targets. For detection of 5‐μm sky blue fluorescent beads and Aβ− Alexa Fluor 647, a 640‐nm excitation laser and allophycocyanin (APC) detector were used.
2.10. Lipid/cholesterol Piccolo analysis
Blood plasma was collected and analyzed using a Piccolo blood chemistry analyzer (Abaxis, Union City, CA) according to the manufacturer's instructions. Briefly, plasma was diluted 1:1 with ddH20 and 100 μL was loaded onto the Piccolo lipid plus panel plate (#07P0212, Abaxis). For lipid analysis, total cholesterol (CHOL), HDL, non‐HDL cholesterol (nHDLc), triglycerides, low‐density lipoprotein (LDL), and very LDL (vLDL) were analyzed and plotted. Lipid and general chemistry controls (#07P0401, Abaxis) were used.
2.11. Bulk lipidomics
Lipid analysis was conducted by UC Davis West Coast Metabolomics Center as described. 24 Briefly, 1 mL of ice‐cold solvent mixture (N2‐purged 3:10 methanol:methyl‐tertiary butyl ether) was added to 2 mg (dry weight) of pulverized cortical tissue, followed by lipophilic/hydrophilic phase separation. The upper organic phase was transferred into two separate tubes and was analyzed via a Charged Surface Hybrid column. The lower aqueous phase was transferred into two separate tubes and was analyzed by hydrophilic interaction liquid chromatography. The abundance of 552 lipids were quantified for the WT, 5xFAD hemizygous, Abca7V1613M homozygous, and 5xFAD hemizygous/Abca7V1613M homozygous (ie, 5xFAD/Abca7V1613M ) mice. Raw lipid abundances were log10 transformed and pareto scaled prior to analysis. Metaboanalyst 5.0 (http://Metaboanalyst.ca) was used for all lipidomic analyses. Analysis of variance (ANOVA) with Tukey's post hoc test was used to examine group differences in the lipidomic profiles. Differential abundance of the lipids was calculated using a fold change of >1.5 or an FDR p‐value of <0.05 for significance.
2.12. Histology
Mice were euthanized at 4 and 12 months old and brain hemispheres were drop‐fixed in 4% paraformaldehyde (PFA) for immunohistochemical analysis. Fixed half brains were cryopreserved with 0.05% sodium azide and 30% sucrose and stored at 4°C before being sliced at 40 μm using a Leica SM2000R freezing microtome (between −2.78 mm posterior and −3.38 mm posterior to Bregma according to the Allen Mouse Brain Atlas, Reference Atlas version 1, 2008). Sliced brains were placed in 30% ethyl glycerol and 30% glycerol at −20°C for long‐term storage. One representative brain slice from each mouse of the same experimental group (ie, same genotype, age, and sex) was stained. Free‐floating sections were washed three times with 1× PBS (1 × 10 minutes and 2 × 5 minutes) and for Thioflavin S (ThioS) staining, incubated for 10 minutes in 0.5% ThioS (T1892; Sigma‐Aldrich) diluted in 50% ethanol. Sections were washed 2 × 5 minutes each in 50% ethanol and 1 × 10 minutes in 1× PBS. For Amylo‐Glo staining, after PBS washes, brain slices were washed in 70% ethanol for 5 minutes and rinsed in ddH2O for 2 minutes before being immersed for 10 minutes in Amylo‐Glo RTD Amyloid Plaque Staining Reagent (1:100; TR‐200‐AG; Biosensis, Temecula, CA) diluted in 0.9% saline solution. Afterwards, sections were washed in 0.9% saline solution for 5 minutes, then rinsed in deionized water for 15 seconds before proceeding with a standard indirect immunohistochemical protocol. After staining with ThioS or Amylo‐Glo, sections were protected from light. Sections were immersed in blocking solution (5% normal goat serum with 0.2% Triton X‐100 in 1× PBS) for 1 hour before overnight incubation at 4°C with primary antibodies diluted in normal blocking serum solution.
Brain sections were stained following a standard indirect technique as described 25 , 26 , 27 with the following primary antibodies against ionized calcium‐binding adapter molecule 1 (IBA1; 1:2000; 019–19741; Wako or IBA1, 1:1000, 234 009, Synaptic systems), cluster of differentiation 68 (CD68; 1:500; AB125212; ABCAM), Aβ1‐16 (6E10; 1:2000; 8030001; BioLegend), glial fibrillary acidic protein (GFAP; 1:1000; AB134436; Abcam), S100 calcium binding protein β (S100β; 1:200; AB41548; Abcam), lysosome‐associated membrane protein 1 (LAMP1; 1:200; AB25245, Abcam), BASSOON (1:250, 75‐491, NeuroMab), HOMER1 (1:250; 160003, SYSY), OC (1:1000; AB2286; Sigma‐Aldrich), and CD11c (1:100, 14‐0114‐82, Invitrogen). Brain sections were mounted on poly‐lysine microscope slides using polyvinylpyrrolidone (AC227541000; Thermo Fisher Scientific) then cover slipped (48393‐106, Avantor) using Fluoromount‐G (0100‐01, Southern Biotech).
Images of whole hemispheres were acquired with a Zeiss Axio Scan Z1 Slidescanner (Carl Zeiss), using a 10 × 0.45 NA Plan‐Apo objective. High‐resolution fluorescence images were obtained using a Leica TCS SPE‐II confocal microscope and LAS‐X software. For confocal imaging, one field of view per brain region was acquired per mouse.
For super‐resolution imaging of synaptic puncta, images of the visual cortex, subiculum, and CA1 regions were acquired using Super–Resolution Lattice Structured Illumination Microscopy (Lattice‐SIM) with an Elyra 7 microscope system (Carl Zeiss, White Plains, NY). Samples were imaged using a 63 × 1.4 NA Plan‐Apo objective lens and Immersol 518 F (23°C) immersion oil. Images were collected as z‐stacks (110 nm step interval, within a depth of 3 to 8 μm, covering an area of 64 × 64 μm) and for each focal plane, nine phase images were acquired. Images were then processed using ZEN SIM2 with ZEN software (black edition, Zeiss). Two images per brain region/mouse/genotype/age/sex were acquired.
2.13. Imaris quantitative analysis
Image analysis was performed and quantified using Imaris 9.7 Software (Bitplane, South Windsor, CT). Confocal images of Aβ plaques, LAMP1 halos, microglia, and astrocytes were quantified using the spots module. Similarly, volumetric measurements (ie, ThioS+ plaque volume, IBA1+ microglia volume, etc.) were acquired utilizing the surfaces module. Quantitative comparisons between experimental groups were carried out in sections stained simultaneously. For synaptic quantification, the total number of BASSOON or HOMER1 puncta was quantified using the spots function on Imaris. Results were normalized to the total volume of each image, to correct for any difference in the depth of imaging.
2.14. Quantification of soluble and insoluble fraction Aβ and NfL
Preparation of samples and quantification of Aβ was performed as described. 25 , 26 , 27 Microdissected hippocampal and cortical regions of each mouse were flash‐frozen and processed for biochemical analysis. Samples were pulverized using a Bessman Tissue Pulverizer. Pulverized hippocampal tissue was homogenized in 150 μL of Tissue Protein Extraction Reagent (TPER; Life Technologies, Carlsbad, CA), while cortical tissue was homogenized in 1000 μL/150 mg of TPER. This composition of TPER includes 25 mM bicine and 150 mM sodium chloride (pH 7.6) to efficiently solubilize proteins within brain tissue following homogenization. Together with protease (11836170001, Roche) and phosphatase inhibitors (78426, Thermo Fisher Scientific), the homogenized samples were centrifuged at 100,000 × g for 1 h at 4°C to generate TPER‐soluble fractions. For formic acid‐fractions, TPER‐insoluble pellets were homogenized in 70% formic acid. Afterwards, samples were again centrifuged at 100,000 × g for 1 hour at 4°C. Protein in the insoluble fraction of microdissected hippocampal and cortical tissue was normalized to its respective brain region weight, while protein in soluble fractions was normalized to the protein concentration determined via Bradford Protein Assay. Formic acid neutralization buffer (1 M Tris base, 0.5 M Na2HPO4, 10% NaN3) was used to adjust pH prior to running Meso Scale Discovery (MSD) assays.
Human Aβ in soluble and insoluble fractions was measured using the V‐PLEX Aβ Peptide Panel 1 (6E10) (K15200G‐1; Meso Scale Discovery, Rockville, MD). Plasma NfL was measured using the R‐Plex Human Neurofilament L Assay (K1517XR‐2; Meso Scale Discovery). Mouse Aβ levels in N2a cell lines were measured using V‐PLEX Aβ peptide Panel 1 (4G8) (K15199G‐2; Meso Scale Discovery) and mouse plasma proinflammatory cytokines were measured using V‐PLEX mouse proinflammatory cytokine Panel 1 (K15048D‐1; Meso Scale Discovery)
2.15. Western blotting
The cortical soluble protein fraction extracted for Aβ MSD assay was also used for western blotting. Protein concentrations were determined by the Pierce protein assay (22660; Thermo Fisher Scientific, Waltham, MA). Proteins were separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) through a 4% to 12% Bis/Tris gel (Life Technologies) for 2.5 hours at 80 V for APP blots and 1.5 hour at 100 V for all other blots. Proteins were transferred to 0.22 μM nitrocellulose membranes (926‐31092, LI‐COR, Lincoln, NE) and stained with REVERT 700 total protein stain (TPS; 926‐11016, LI‐COR) for 5 minutes at room temperature. After a brief rinse, the TPS was quantified using Image Studio software and an Odyssey CLx with TPS values used for subsequent normalization. Membranes were then blocked for 1 hour in Odyssey block solution (927‐50000; LI‐COR). Primary antibodies and dilutions used in this study include the following: 6E10 for full‐length APP (1:1000, 803002, BioLegend), C‐terminal (751‐770) APP for C99 and C83 fragments (1:1000, 17161050UL, MilliporeSigma), BACE1 (1:1000, AB2077, ABCAM), ADAM10 (1:1000, AB1997, ABCAM), PSEN1 (1:1000, AB76083, ABCAM), and ABCA7 (1:1000, 32942S, Cell Signaling Technology). Membranes were incubated with primary antibodies in tris‐buffered saline (TBS) with 0.1% Tween‐20 overnight at 4°C with rocking. The following day, membranes were washed in PBS then incubated with secondary antibodies for 1 hour at room temperature. Secondary antibodies and dilutions used were antimouse IRDye 800CW (1:15,000; 926‐32210; LI‐COR) and antirabbit IRDye 800CW (1:15,000; 926‐32211; LI‐COR). After three washes of 5 minutes with TBS‐Tween, membranes were imaged using an Odyssey CLx. Quantitative analyses were performed with Empiria Studio Software v2.3 (LI‐COR).
2.16. Generation of ABCA7 overexpressing N2a cells
Mouse N2a neuroblastoma cells were provided by Dr. Masashi Kitazawa (UCI). An empty vector (contains EGFP only, Prp[Exp]‐EGFP/neo‐CAG > ORF_stuffer), mouse WT ABCA7 (Prp[Exp]‐EGFP/neo‐CAG > NM_001346081.2), and V1613M ABCA7 (Prp[Exp]‐EGFP/neo‐CAG > NM_001346081.2*V1613M) were designed and cloned using VectorBuilder (Chicago, IL). Transformed E. coli glycerol stocks were propagated, and plasmids purified using Monarch plasmid miniprep kit (T1010S, New England Biolabs, Ipswich, MA). Plasmids were linearized by digestion with Asc I, verified by agarose gel electrophoresis, the protein precipitated using 2.5 M ammonium acetate, and plasmid DNA precipitated and sterilized using ethanol. N2a cells were plated into a six‐well plate (100,000/well) 24 hours before transfection. Then 2.5 μg plasmid DNA was transfected into the N2a cells using lipofectamine 3000 transfection kit (L3000008, Invitrogen). P3000 was added to increase transfection efficiency. Plasmid DNA and lipofectamine complexes were made in reduced serum media, OptiMEM (31985070, Invitrogen). Complexes were added to cells in 10% fetal bovine serum (FBS) containing DMEM and left for 48 hours before analyzing transfection efficiency via EGFP expression. Selection of positive clones was conducted by selection in G418 (500 μg/mL) for 14 days. Cells were further sorted via FACS buffer to increase the percentage of successfully transfected cells. Transfected cells were maintained in 10% FBS containing DMEM supplemented with 100 μg/mL G418 which was replaced every 3 days.
2.17. Immunocytochemistry of ABCA7 overexpressing N2a cells
Cells were seeded into 12‐well plates (200,000 cells/well) on coverslips in reduced serum media, OptiMEM (31985070, Invitrogen) 24 hours before staining. Cells were washed in 1× PBS (2 × 5 minutes) then fixed in 4% PFA for 20 minutes at room temperature. Following fixation, cells were washed with 1× PBS (3 × 5 minutes) before blocking in 5% normal goat serum supplemented with 0.2% Triton‐X, for 1 hour at room temperature. Cells were then incubated with primary antibodies, mouse antimouse APP (1:100; MAB348, MilliporeSigma) and rat antimouse LAMP1 (1:100; AB25245, ABCAM), overnight at 4°C. Cells were subsequently washed with 1× PBS (1 × 10 minutes, 2 × 5 minutes) and incubated with secondary antibodies, antimouse Alexa Fluor 647 (1:200, A‐21235, Thermo Fisher Scientific) and antirat Alexa Fluor 555 (1:200, A‐21434, Thermo Fisher Scientific), for 1 hour at room temperature. Finally, cells were washed with 1× PBS (1 × 10 minutes, 2 × 5 minutes) and coverslips mounted using Fluoromount‐G (0100‐01, Southern Biotech). Cells were imaged using LSM 900 (Carl Zeiss) with Airyscan processing (Zen 3.6, Blue edition), 63 × 1.3NA immersion oil objective and 5× digital zoom. Five images were acquired per cell type/stain and APP+ intracellular puncta were counted using ImageJ (Version 1.53t).
2.18. Statistical analysis
The number of independent biological replicates are indicated by N in Figures 1 through 6. The sample sizes are similar to those found in prior studies conducted by MODEL‐AD. 25 , 26 , 27 Immunohistochemical and biochemical data were analyzed using a Student t‐test or two‐way ANOVA via Prism v.9 (GraphPad). Tukey's post hoc tests were utilized to examine biologically relevant interactions from the two‐way ANOVA. Where sex‐differences are apparent, a Student t‐test was used within the genotype group. Outlier tests were performed via Prism v.9 where relevant and any datapoint removed from the analyses is indicated in the relevant figure legend. Unless otherwise stated, data are presented as raw means and standard error of the mean (SEM).
3. RESULTS
3.1. Mouse Abca7V1613M allele models human ABCA7V1599M allele with no off‐target effects
We developed an Abca7V1613M mouse model on an inbred C57BL/6J mouse background utilizing previously reported strategies. 27 Sequencing confirmed the desired change on Exon 35 of methionine (M) to valine (V) at amino acid 1613 (Figure 1A). Abca7 em1 Aduci/J mice are available free of restrictions through The Jackson Laboratory (Stock No. #035316). Bulk RNA sequencing (RNA‐seq) analysis of 8‐month‐old mice indicated relatively few differences in gene expression between WT C57BL/6J mice and Abca7V1613M mice, with five genes above the threshold for significance, including reduced expression of Dok3 which encodes a negative regulator of LPS‐induced Toll‐like receptor signaling (PMID 22761938) (Figure 1B). 28 Bulk RNA‐seq also supports the view that the Abca7V1613M allele is expressed at a similar level to the WT Abca7 allele without evidence of any cryptic splicing products from the modified allele (Figure 1C and data not shown). We confirmed that ABCA7 protein expression is similar in WT and Abca7V1613M mice at 4 months of age via western blotting (Figure S2). To further validate the Abca7V1613M model we conducted off‐target analyses of other putative cut sites within genes or in conserved intergenic regions that might have been targeted by CRISPR‐Cas9 during generation of the Abca7V1613M allele. Abca7V1613M founder mice were backcrossed with WT C57BL/6J animals for three generations before use to generate animals for this study, making it unlikely that a mutation caused by an off‐target effect of CRISPR/Cas9 would be present on a chromosome other than chromosome 10, that is, the location of Abca7 (C57BL/6J; Chr10; 79.8 Mb, GRCm39, Ensembl release 110) (Figure S3). Potential CRISPR/Cas9 off‐target sites with up to four mismatches using crRNA TMF1268 were screened for using Cas‐OFFinder (http://www.rgenome.net/cas‐offinder/;). Nineteen potential off‐target sites on mouse chromosome 10 were identified (Table S1). Five potential off‐target sites were within introns and three were located within conserved intergenic regions while the remaining eleven were within nonconserved intergenic regions. To screen for evidence of CRISPR/Cas9 RNP activity at the intronic and conserved intergenic sites, DNA from a WT and two homozygous Abca7V1613M mice were amplified by PCR using the primers listed (Table S2), then sequenced across the potential off‐target region at each locus. None of the eight potential off‐target sites analyzed showed a difference in sequence between WT and homozygous Abca7V1613M mice (Figure S3). These data suggest that selective mutagenesis of mouse Abca7 at amino acid position 1613 was successful with no unwanted off‐target effects. Furthermore, introduction of this missense variant does not change expression of ABCA7 at the RNA or protein level, suggesting that any phenotypic changes seen in animals with this variant are due to differences in ABCA7 function, not expression.
3.2. Homozygous Abca7V1613M mice display a differential response to LPS challenge and increased microglial phagocytosis
Previous studies implicate a role for ABCA7 in inflammation and microglial function, 15 , 17 but the effects of the V1599M/V1613M variant are unknown, including whether it behaves as a loss of function variant. Abca7 haploinsufficient mice display diminished responses to LPS stimulation. 17 To investigate the impact of the V1613M variant on the brain's ability to mount an immune response to immune challenge, we injected 8‐month‐old C57BL/6J (WT) and homozygous Abca7V1613M mice intraperitoneally with saline or 0.3 mg/kg LPS and collected brains for bulk tissue RNA‐seq analysis at 6 and 24 hours postinjection. LPS administration induces a robust inflammatory response in both in WT (Figure 1E) and Abca7V1613M (Figure 1F) animals at 6 hours postinjection, as indicated by increased transcription of inflammatory genes such as Ccl2, Cxcl1, Il1b, and Tnfa, accompanied by downregulation of the homeostatic microglial gene, P2ry12. Specifically, there are 273 upregulated and 101 downregulated genes in WT LPS versus saline at 6 hours and 325 upregulated and 51 downregulated genes in Abca7V1613M LPS versus saline at 6 hours (Figure 1E,F). To identify LPS‐dependent DEGs in Abca7V1613M mice, we compared Abca7V1613M LPS‐treated versus Abca7V1613M saline‐treated, and Abca7V1613M LPS‐treated versus WT LPS‐treated mice at 24 hours postinjection and found 88 overlapping genes (Figure 1K). Of these 88 genes, the majority are upregulated in Abca7V1613M mice compared to WT at both 6 and 24 hours postinjection (Figure 1L). Of note, Abca7V1613M mice do not fully resolve LPS‐induced gene expression changes by 24 hours postinjection and have upregulation of a large number of inflammatory‐associated genes such as C1ra, C1qa, C1qb, Casp4, and Capg compared to their WT counterparts (Figure 1H,I). Pathway analyses of these 88 genes reveal a high association with interferon signaling and classical complement/inflammatory signaling pathways. Given these changes in the central nervous system, we next explored the plasma response to LPS. Abca7V1613M mice had similar or lower levels of most cytokines analyzed, including IFN‐γ, IL‐6, and TNFα, when compared to WT animals 24 hours postinjection (Figure S4). These data indicate that homozygous Abca7V1613M mice evoke a differential response to LPS stimulation compared to WT animals, which appears to be specific in the brain and not due to augmentation of LPS‐mediated peripheral cytokine levels. Together, these findings indicate that Abca7V1613M mice are less able to resolve LPS‐induced brain‐associated inflammation and/or remain more activated after challenge. These data are in contrast to previous studies using Abca7 haploinsufficient mice, in which inflammation was suppressed.
We analyzed the in vitro phagocytic capacity of microglia isolated from cortices of Abca7V1613M homozygous and WT control p3‐ to 5‐day‐old mice. Primary microglia were incubated with either carboxylated fluorescent beads, which mimics uptake of dead/dying cells, or fluorescently labelled Aβ1‐42 peptide for 1 hour before uptake was analyzed via flow cytometry (Figure 1M). Microglia from Abca7V1613M mice have increased phagocytic capacity of both beads and Aβ1‐42 peptide compared to WT controls (Figure 1N,O). These data also contrast with previous studies that report reduced phagocytosis of apoptotic cells and Aβ oligomers in ABCA7 KO macrophages and microglia. 15 , 16 , 17 Together, these data suggest that the V1613M variant in murine Abca7 produces a gain of function (hypermorphic or neomorphic) effect with respect to inflammation and microglial phagocytosis, and not a loss of function.
3.3. Abca7V1613M mediates distinct changes in the lipid profile of 5xFAD mice
To explore the impact of the Abca7V1613M variant in the context of plaque development, and its effects on the brain we crossed Abca7V1613M mice with the 5xFAD mouse model of amyloidosis to generate four experimental genotypes, C57BL/6J (WT), Abca7V1613M homozygous (Abca7V1613M ), 5xFAD hemizygous (5xFAD HEMI), and 5xFAD/Abca7V1613M . All mice were inbred or co‐isogenic C57BL/6J background. Cohorts of mice of both sexes and each genotype were aged to 4 and 12 months of age to reflect early and late stages of pathology, respectively (Figure 2A).
FIGURE 2.
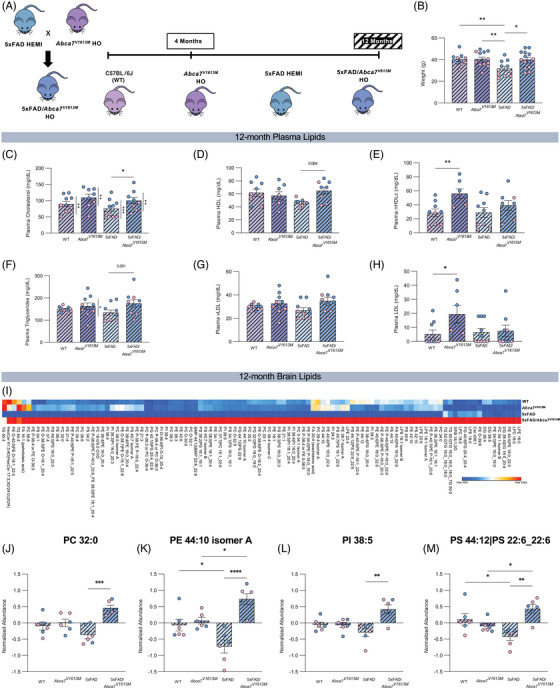
Altered plasma and brain lipids in 12‐month‐old 5xFAD/Abca7V1613M mice. (A) Schematic demonstrating the initial cross used to generate the desired genotypes and the timepoints used in this study. All data in this figure are from the 12‐month timepoint. (B) Weight of mice from all four experimental groups at 12 months of age. (C‐H) Analysis of the effect of the Abca7V1613M variant on circulating lipids in plasma in 12‐month‐old mice. Quantification of (C) total cholesterol, (D) high‐density lipoprotein (HDL), (E) non‐ HDL cholesterol (nHDLc), (F) triglycerides, (G) very low‐density lipoprotein (vLDL), (H) LDL. N = 3 to 6 per sex/genotype/age. Statistically significant differences between sexes are denoted by vertical bars and associated significance markers. (I‐M) Liquid chromatography/mass spectrometry analysis of cortical brain lipids in 12‐month‐old mice. (I) Heatmap of significantly altered lipids identified by analysis of variance (ANOVA). (J‐M) Normalized abundance of (J) phosphatidylcholine (PC) 32:0, (K) phosphatidylethanolamine (PE) 44:10 isomer A, (L) phosphatidylinositol (PI) 38:5, (M) phosphatidylserine (PS) 44:12|PS 22:6_22:6. N = 6 per genotype. Data are represented as mean ± SEM. Statistics (B‐H, J‐M) by two‐way ANOVA. WT, wildtype. *p ≤ 0.05, **p ≤ 0.01 ***p ≤ 0.001, ****p ≤ 0.0001.
ABCA7 is a lipid transporter predominantly involved in cholesterol/phospholipid efflux and Abca7 KO mice have altered lipid profiles. 9 , 11 , 12 , 14 , 29 Because dyslipidemia can be related to weight 30 we measured animal total body weight. Twelve‐month‐old WT and Abca7 V1613M animals have similar mass (Figure 2B). In contrast, 12‐month‐old 5XFAD mice have reduced body weight compared to 5XFAD/Abca7V1613M , WT, and homozygous Abca7V1613M animals (Figure 2B), indicating a protective effect of the Abca7V1613M variant against 5xFAD‐mediated weight loss.
We next examined the impact of the Abca7V1613M variant on lipid homeostasis by analyzing plasma and cortical tissue from WT, Abca7V1613M , 5xFAD, and 5xFAD/Abca7V1613M mice of both sexes and 12 months of age. We quantified circulating plasma lipids including CHOL, HDL, and triglycerides. In plasma, 5xFAD/Abca7V1613M mice have increased CHOL compared to 5xFAD mice (Figure 2C). Male mice have higher CHOL levels compared to females in all genotypes (Figure 2C). Abca7V1613M mice trended to have increased cholesterol compared to WT mice (Figure 2C). We report a statistically significant effect of genotype on cholesterol levels (F = 6.336 and p = 0.0026), suggesting the V1613M variant has an effect on levels of circulating cholesterol. 5xFAD/Abca7V1613M have higher levels of plasma HDL (Figure 2D) and triglycerides (Figure 2F) compared to 5xFAD mice, with differences close to statistical significance. In the absence of the 5xFAD transgene, Abca7V1613M mice show increased plasma nHDLc (Figure 2E) and LDL (Figure 2H) compared to WT controls. No difference was detected in plasma vLDL across genotypes (Figure 2G). These data indicate ABCA7‐dependent roles in circulating cholesterol homeostasis, and a differential role with the introduction of the Abca7V1613M variant.
To assess lipid changes in the brain, we performed unbiased bulk lipidomics on cortical tissue using liquid chromatography‐mass spectrometry. ANOVA revealed significant difference in lipid abundance between the groups for 106 of 552 identified lipids (p < 0.05) comprising a range of species, mostly phosphatidylcholines (PCs), phosphatidylethanolamines (PEs), phosphatidylinositols (PIs), and phosphatidylserines (PSs), shown as a heatmap (Figure 2I). Of these 106 lipids we plotted several key metabolites, PC 32:0, PE 44:10 isomer A, PI 38:5, and PS 44:12|PS 22:6_22:6 (Figure 2J–M), to highlight differences between all four groups. There is a consistent increase in the levels of lipids in 5xFAD/Abca7V1613M mice compared to 5xFAD controls.
3.4. Abca7V1613M reduces Aβ plaque pathology in 5xFAD mice
Loss of ABCA7 exacerbates Aβ plaque pathology in APP‐overexpressing mice. 12 , 19 To investigate the effect of the Abca7V1613M variant on development of plaque pathology, coronal brain sections from 5xFAD and 5xFAD/Abca7V1613M mice were stained with ThioS and a conformation‐specific antibody (Aβ fibrils OC) to evaluate amyloid plaque and fibrillar Aβ deposition, respectively. 5xFAD mice homozygous for the Abca7V1613M variant have fewer ThioS+ plaques in the cortex and hippocampus at both 4 and 12 months of age (Figure 3C–H, K–Q). OC+ staining is also reduced in both the cortex and subiculum at 4 months and in the cortex at 12 months (Figure 3E–J, N–S). Hence, the Abca7V1613M variant appears to have the opposite effect on development of Aβ plaque compared to mice with a complete loss of function of Abca7.
FIGURE 3.
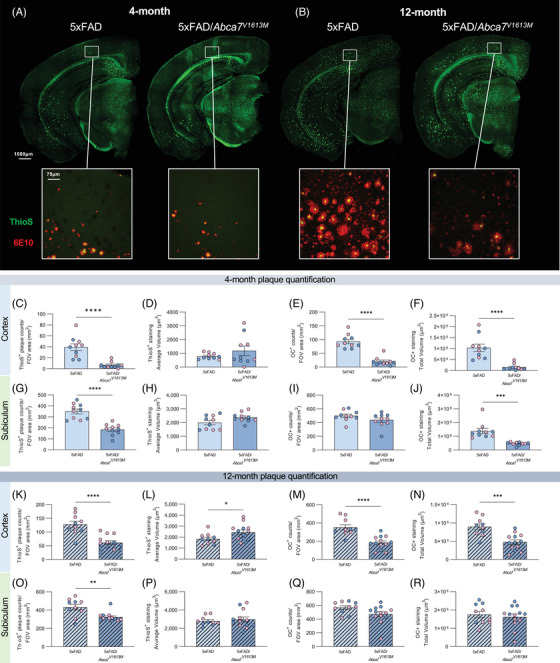
Reduced amyloid beta (Aβ) plaque burden in 5xFAD/Abca7V1613M mice. Plaque burden in 4‐ and 12‐month‐old 5xFAD hemizygous (5xFAD), and 5xFAD hemizygous /Abca7V1613M homozygous (5xFAD/Abca7V1613M ) animals was assessed with Thioflavin S (ThioS) and OC staining. (A‐B) Representative hemispheric brain and higher magnification cortical images from 5xFAD and 5xFAD/Abca7V1613M females showing ThioS+ dense core plaques and more fibrillar OC staining at (A) 4 months and (B) 12 months. (C‐R) Quantification of (C) ThioS+ plaques in cortex at 4 months, (D) average volume of ThioS+ plaques in cortex at 4 months, (E) OC+ plaques in cortex at 4 months, (F) total volume of OC+ staining in cortex at 4 months, (G) ThioS+ plaques in subiculum at 4 months, (H) average volume of ThioS+ plaques in subiculum at 4 months, (I) OC+ plaques in subiculum at 4 months, (J) Total volume of OC+ staining in subiculum at 4 months, (K) ThioS+ plaques in cortex at 12 months, (L) average volume of ThioS+ plaques in cortex at 12 months, (M) OC+ plaques in cortex at 12 months, (N) total volume of OC+ staining in cortex at 12 months, (O) ThioS+ plaques in subiculum at 12 months, (P) average volume of ThioS+ plaques in subiculum at 12 months, (Q) OC+ plaques in subiculum at 12 months, (R) total volume of OC+ plaques in subiculum at 12 months. N = 4 to 6 per sex/genotype/age. Data are represented as mean ± SEM. Statistics (C‐S) by unpaired Student t‐test. FOV, field of view. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
3.5. Abca7V1613M reduces Aβ‐associated neuronal damage but does not affect synapse loss in 5xFAD mouse model
Aβ deposition is known to be associated with neuronal damage and neurodegeneration, as seen by the formation of dystrophic neurites, elevated NfL in plasma and cerebral spinal fluid, as well as loss of synapses. 31 , 32 , 33 To assess whether the reduction in Aβ in Abca7V1613M mice was associated with reduced damage and neurodegeneration in the 5xFAD brain, we examined dystrophic neurites in 5xFAD and 5xFAD/Abca7V1613M mice, as well as synapses via immunohistochemistry and NfL via MSD assay in all four groups. Brain sections were stained with ThioS and LAMP1, a marker for dystrophic neurites. At 4 and 12 months of age there is a large reduction in total LAMP1 staining in the cortex, although this reduction is only significant at 4 months in the subiculum between 5xFAD and 5xFAD/Abca7V1613M mice (Figure 4A–F). The reduced LAMP1 correlates well with the reduced Aβ plaques in the brain. Similarly, a significant and sustained reduction in NfL was observed in both plasma and cortex in 5xFAD/Abca7V1613M mice compared to 5xFAD controls at both ages analyzed (Figure 4G–J).
FIGURE 4.
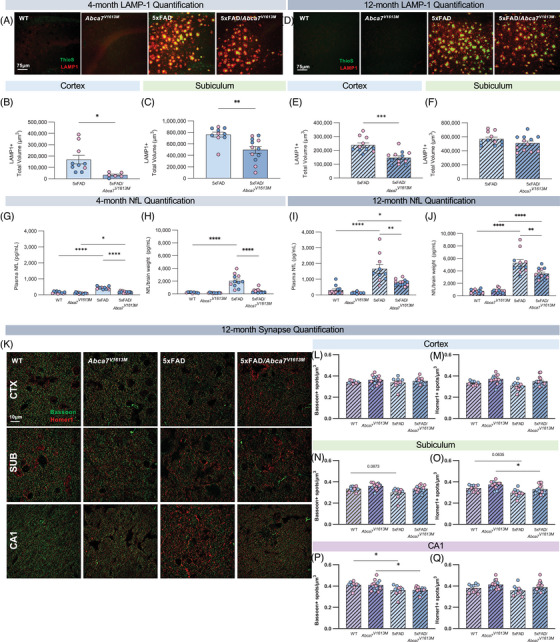
5xFAD/Abca7V1613M mice have reduced amyloid beta (Aβ)‐associated neuronal damage. (A) Representative confocal images of subiculum in 4‐month‐old wildtype (WT), Abca7V1613M , 5Xfad, and 5xFAD/Abca7V1613M mice stained with Thioflavin S (ThioS; green) and LAMP1 (red). Scale bar = 75 microns. (B‐C) Quantification of total LAMP1+ staining in (B) cortex and (C) subiculum at 4 months of age. (D) Representative confocal images of subiculum in 12‐month‐old WT, Abca7V1613M , 5xFAD, and 5xFAD/Abca7V1613M mice stained with ThioS (green) and LAMP1 (red). Scale bar = 75 microns. (E‐F) Quantification of total LAMP1+ staining in (E) cortex and (F) subiculum at 12 months of age. (G‐J) Compared with 5xFAD mice, 5xFAD/Abca7V1613M animals display lower neurofilament light chain (NfL) in plasma and cortex at 4 and 12 months of age. (K) Representative super resolution images of synaptic densities in different sites in 12‐month‐old WT, Abca7V1613M , 5xFAD, and 5xFAD/Abca7V1613M mice. Scale bar = 10 microns. (L‐Q) Quantification of (L) presynaptic BASSOON spots and (M) postsynaptic HOMER1 spots normalized to volume of z stack in cortex; (N) presynaptic BASSOON spots and (O) postsynaptic HOMER1 spots normalized to volume of z stack in subiculum; and (P) presynaptic BASSOON spots and (Q) postsynaptic HOMER1 spots normalized to volume of z stack in CA1. N = 4 to 6 per sex/genotype/age. Data are represented as mean ± SEM. Statistics (B‐F) by unpaired Student t‐test, (G‐Q) by two‐way analysis of variance. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, **** ≤ 0.0001.
We investigated whether the Abca7V1613M variant affected synaptic density by quantifying the presynaptic marker BASSOON, and postsynaptic marker HOMER1 in the 12‐month cohort. There were fewer BASSOON spots in the CA1 region of hippocampus in 5xFAD mice compared to WT controls (Figure 4P). However, no difference was found between 5xFAD and 5xFAD/Abca7V1613M animals (Figure 4P). In cortex and subiculum no difference was found in the number of BASSOON spots between all groups (Figure 4L–O). There was no significant difference in HOMER1 spots in any of the brain region images, except for a minor reduction in HOMER1 spots in the subiculum between Abca7V1613M homozygous mice with pathology versus those without (Figure 4O).
Together, these data indicate that the 5xFAD/Abca7V1613M mice have reduced neuron‐associated damage, consistent with less Aβ pathology. Additionally, the V1613M variant does not influence plaque induced synaptic loss, or synapse densities under homeostatic conditions.
3.6. Abca7V1613M reduces Aβ‐associated gliosis in 4‐month‐old 5xFAD mice
Microglia are implicated in the pathology of AD and are found in close proximity and actively react to Aβ plaques, inducing distinct transcriptional changes. 34 To analyze the effect of the Abca7V1613M variant on the plaque–microglial association in 5xFAD mice, we stained for microglia with IBA1, and Aβ plaques with ThioS, in both brain regions at 4 and 12 months. 5xFAD/ Abca7V1613M mice have reduced microglial density in the cortex and subiculum at both 4 and 12 months of age (Figure 5B, D, G, I) compared to 5xFAD, although no difference was observed in the number of microglia associated per Aβ plaque in both regions and timepoints (Figure 5C, E, H, J).
FIGURE 5.
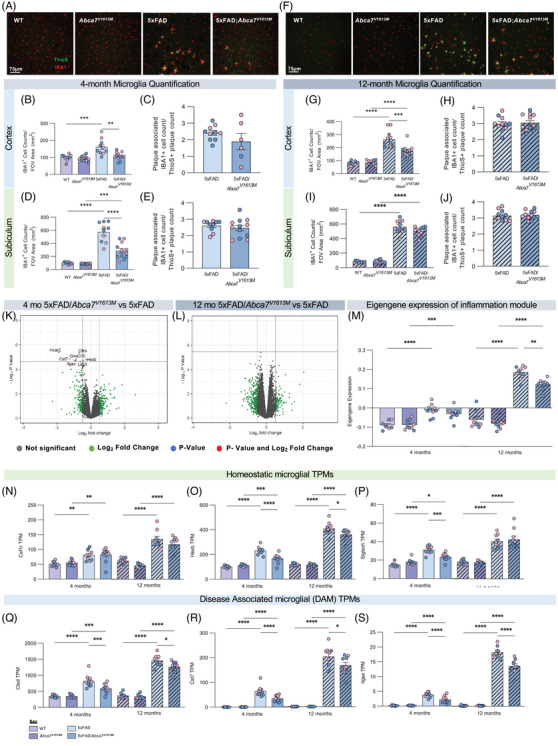
5xFAD/Abca7V1613M mice display reduced microgliosis at 4 and 12 months. Microglia number and size were assessed by staining coronal sections with IBA1 at 4 and 12 months. (A) Representative confocal images from cortex of wildtype (WT), Abca7V1613M homozygous, 5Xfad, and 5XFAD/Abca7V1613M mice displaying reduced number of microglia at 4 months. Scale bar = 75 microns. (B) IBA1+ cell density in cortex at 4 months. (C) Quantification of IBA1+ cells associated with Thioflavin S (ThioS)+ plaque in cortex at 4 months. (D) IBA1+ cell density in subiculum at 4 months. (E) Quantification of IBA1+ cells associated with ThioS+ plaque in subiculum at 4 months. (F) Representative confocal images from cortex of WT, Abca7V1613M homozygous, 5xFAD, and 5XFAD/Abca7V1613M mice displaying reduced number and size of microglia at 12 months. Scale bar = 75 microns. (G) IBA1+ cell density in cortex at 12 months. (H) Quantification of IBA1+ cells associated with ThioS+ plaque in cortex at 12 months. (I) IBA1+ cell density in subiculum at 12 months. (J) Quantification of IBA1+ cells associated with ThioS+ plaque in subiculum at 12 months. (K,L) Volcano plots of bulk RNA‐seq displaying differentially expressed genes (DEGs) between 5xFAD/Abca7V1613M versus 5xFAD at (K) 4 and (L) 12 months of age. (M) inflammatory module eigengene values in WT, Abca7V1613M homozygous, 5Xfad, and 5XFAD/Abca7V1613M mice at 4 and 12 months of age. Results indicate reduced expression of inflammatory genes in 5xFAD/Abca7V1613M mice compared to 5xFAD. (N‐S) Transcripts per million (TPMs) of homeostatic markers (N) Csf1r, (O) Hexb, and (P) Siglech; and of disease‐associated microglia (DAM) markers (Q) Ctsd, (R) Cst7, and (S) Itgax. A Log2 fold change of 0.5 was used for all volcano plots; however, ‐Log10 p‐value changed according to different comparisons analyzed. N = 4 to 6 per sex/genotype/age. Data are represented as mean ± SEM. Statistics (B,D,G,I) by two‐way analysis of variance (ANOVA), (C,H,E,J) by unpaired Student t‐test, (M‐S) by two‐way ANOVA on 4 or 12 months separately. FOV, field of view. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, **** ≤ 0.0001.
To identify changes in gene expression associated with homozygosity for the Abca7V1613M variant in 5xFAD mice we conducted bulk tissue RNA‐seq on hippocampi. Consistent with the reduced plaque load in 4‐month‐old 5xFAD/Abca7V1613M mice, we found reduced transcripts for inflammatory/microglial expressed genes such as Cst7, Trem2, Itgax, and Hexb in 5xFAD/Abca7V1613M mice compared with 5xFAD animals (Figure 5K). By contrast, in 12‐month‐old animals, despite lower plaque loads, we did not identify any differentially expressed genes between 5xFAD/Abca7V1613M and 5xFAD mice (Figure 5L). However, there were sustained reductions in TPMs of disease‐associated microglia genes when considered individually: Ctsd, Cst7, and Itgax (Figure 5Q–S). We confirmed the reduction in Itgax at the protein level by analyzing CD11c via immunohistochemistry in hippocampi of 12‐month‐old animals (Figure S5). To further explore gene expression changes across groups, we analyzed functional networks of correlated genes (WGCNA) and identified one module which contains inflammation/immune‐related genes that are highly upregulated in 5xFAD mice but significantly reduced in 5xFAD/Abca7V1613M mice. We plotted eigengene values of these inflammation module for all groups (Figure 5M), revealing a reduction in 5xFAD/Abca7V1613M mice compared to 5xFAD at both 4 and 12 months of age.
We also explored astrocytic responses to plaques through immunohistochemistry for GFAP, a general marker of activation, and S100β, which stains most astrocytes regardless of activation state, in both regions and timepoints. At the 4‐ and 12‐month timepoints there is reduced total volume of GFAP+ astrocytes in the cortex and subiculum in 5xFAD/Abca7V1613M mice compared to 5xFAD controls (Figure S6B, D, G, I), with no change in the number of S100β + astrocytes in the cortex (Figure S6C, H). Immunohistology GFAP data in the subiculum are consistent with reduced GFAP TPM values in 5xFAD/Abca7V1613M mice compared to 5xFAD at both 4 and 12 months of age from bulk RNA‐seq analysis of hippocampi (Figure S6K). In the subiculum at 4 months there is a small but significant reduction in the number of S100β+ astrocytes between 5xFAD/Abca7V1613M and 5xFAD mice, which is no longer significant at 12 months of age (Figure S6E, J).
Collectively, these data indicate that 5xFAD/Abca7V1613M mice exhibit reduced microgliosis and inflammation, which is likely due to lower Aβ plaque burden compared to 5xFAD controls rather than distinct changes to microglial gene expression and function.
3.7. Abca7V1613M results in altered APP processing and trafficking but does not affect cholesterol efflux in vitro
Prior studies suggested that ABCA7 KO could mediate increased plaque burden through impairment in microglial clearance of Aβ. 12 , 17 However, we found no overt difference in microglial response to plaques in 5xFAD/Abca7V1613M mice when normalized for the reduction in plaque load. We postulated that the effect of the Abca7V1613M variant on plaques might instead be due to altered Aβ production. To test this theory, we quantified Aβ40 and Aβ42 in both the detergent insoluble and soluble fractions representing Aβ sequestered in plaques versus that being actively produced, respectively, from hippocampal and cortical tissue. Consistent with plaque analysis, we observed reduced Aβ40 and Aβ42 in the insoluble and soluble fractions of the cortex and hippocampus at 4 months in 5xFAD/Abca7V1613M mice compared to 5xFAD controls (Figure 6A–H). Broadly, Aβ levels are also reduced at 12 months in both the soluble and insoluble fractions in 5xFAD/Abca7V1613M compared to 5xFAD mice (Figure 6I–P). Together, these data suggest that Abca7V1613M reduces Aβ production, and that this results in less Aβ sequestered in plaques.
FIGURE 6.
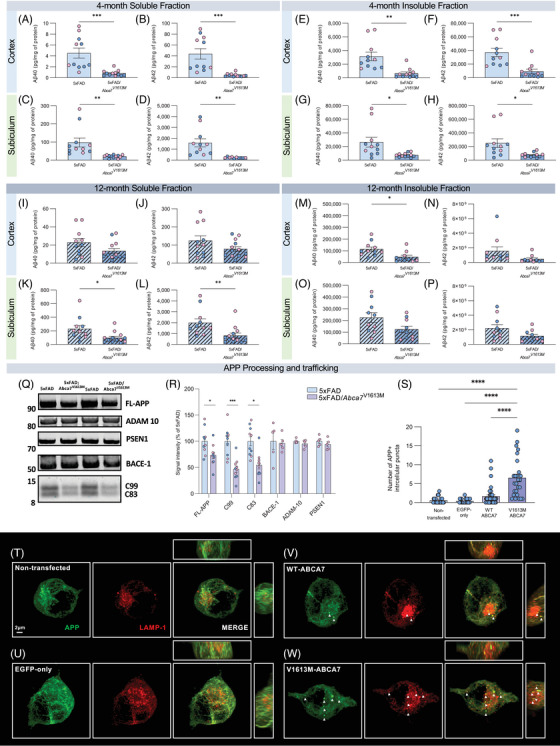
Abca7V1613M variant reduces amyloid beta (Aβ) production and processing in vivo and alters amyloid precursor protein (APP) trafficking in vitro. (A‐P) Quantification of Aβ40 and Aβ42 in detergent insoluble and soluble fractions in 5xFAD and 5XFAD/Abca7V1613M hippocampus and cortex in (A‐H) 4 month‐old and (I‐P) 12‐month‐old mice. In 4‐month‐old mice, both Aβ40 and Aβ42 are reduced in all samples. At 12‐months‐old, soluble Aβ40 and Aβ42 are reduced in subiculum and insoluble Aβ40 is reduced in cortex, with both soluble and insoluble Aβ40 and Aβ42 trending lower in other samples from 5xFAD/Abca7V1613M animals compared to 5xFAD mice. (Q) Western blot of soluble cortical protein from representative female 5xFAD and 5XFAD/Abca7V1613M mice at 4 months for full‐length APP (FL‐APP), APP C‐terminal fragments (C99 and C83), BACE‐1, ADAM‐10, PSEN1, and C‐terminal PSEN1 fragment. (R) Quantification of FL‐APP, C99 and C83, BACE‐1, ADAM‐10, and PSEN1 normalized to total protein stain (TPS) as a percentage of 5xFAD signal intensity. (S) Quantification of intracellular APP+ puncta in transfected N2a cells. (T‐W) Representative confocal images showing 22C11 (APP) and LAMP1 staining in (T) nontransfected control, (U) EGFP‐only (empty vector) control, (V) Wildtype (WT)‐ABCA7 overexpressing, and (W) V1613M‐ABCA7 overexpressing N2a cells. Scale bar = 2 microns. N = 4 to 6 per sex/genotype/age (A‐R); N = 26 to 36 cells per group (S). Data are represented as mean ± SEM. Statistics (A‐P) by unpaired Student t‐test, (R) by individual unpaired Student t‐test of each target protein, (S) by one‐way analysis of variance. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, **** p ≤ 0.0001.
Aβ is sequentially cleaved from its parent protein, APP, by BACE1 followed by the γ‐secretase complex. We conducted western blot analysis of key enzymes and products of the APP processing pathway: full‐length APP, C‐terminal APP fragments C83 and C99, BACE‐1 (β‐secretase), PSEN1 (component of the γ‐secretase complex), and ADAM‐10 (α‐secretase). 5xFAD/Abca7V1613M mice have reduced full‐length APP and corresponding C terminal fragments (C83 and C99) (Figure 6Q, R). However, there was no difference in BACE1, PSEN1, or ADAM10 between 5xFAD and 5xFAD/Abca7V1613M animals (Figure 6Q, R). Uncropped versions of these blots can be found in Figure S7. These data suggest that reduced Aβ generation and subsequent plaque development are due to changes in the proteolytic cleavage of both α‐ and β‐secretases (ADAM10 and BACE1, respectively), possibly reflecting a change in the availability of the substrate (ie, APP).
We hypothesized that the Abca7V1613M variant was altering intracellular APP trafficking, such that it was less available for α‐ or β‐secretase cleavage. To test this theory, we generated N2a cell lines overexpressing either WT ABCA7 or V1613M‐ABCA7 and evaluated the subcellular localization of APP via super resolution microscopy. First, to assess whether Aβ levels were reduced in N2a cells overexpressing WT ABCA7 or V1613M‐ABCA7, we measured Aβ40 and Aβ42 in both the supernatant and cell lysate of all four cell lines. We found a reduction of both Aβ40 and Aβ42 in the supernatant and lysate of N2a cells overexpressing WT and V1613M‐ABCA7 compared to nontransfected and EGFP‐only (empty vector) control lines (Figure S8D–G). These data are consistent with our in vivo findings that show a reduction in Aβ40 and Aβ42 in 5xFAD/Abca7V1613M mice compared to 5xFAD controls. N2a cells overexpressing mouse WT ABCA7 exhibited a small number of APP‐positive intracellular puncta (average of 3 per cell) (Figure 6S, V). We also see these puncta in nontransfected and EGFP‐only controls but at a much lower frequency (average of one punctum per cell) (Figure 6S, T, U). N2a cells overexpressing V1613M‐ABCA7 display significantly increased intracellular APP puncta (average seven puncta per cell) compared to WT ABCA7 overexpressing and control cells (Figure 6S, W). To assess whether overexpression of WT or V1613M‐ABCA7 could be associated with changes in APP trafficking we stained N2a cells with LAMP1, a marker for both lysosomes and late endosomes, and RAB7A, a marker for late endosomes and SXN4, a marker for sorting/recycling endosomes. We found that APP puncta colocalized with LAMP1+ and RAB7A+ structures (Figure 6W, Figure S8H) but not with SNX4 (Figure S8I). These data suggest overexpression of ABCA7 alters APP trafficking by promoting intracellular accumulation in LAMP1+ and RAB7+ structures, and that the V1613M variant enhances this effect. This intracellular accumulation suggests APP is being retained/degraded in the lysosomal pathway rather than being recycled back to the cell membrane, thereby leading to a reduction in Aβ production.
Given differences in circulating cholesterol levels with introduction of the V1613M variant, we conducted a cholesterol efflux assay on our WT and V1613M overexpressing N2a cells, along with their controls. We found no statistical significance in cholesterol efflux between control cells and WT or V1613M ABCA7 overexpressing N2a cells (Figure S8K).
4. DISCUSSION
GWAS of AD and control individuals identified variants associated with the ABCA7 locus as having increased risk of development of LOAD. 5 , 35 More recently, exome sequencing of ABCA7 identified nonsense and missense coding variants that are predicted to affect protein structure and function. Many of these identified changes appear likely to result in loss of function mutations and have been found to be enriched in AD individuals. 4 , 20 , 21 , 36 , 37 , 38 , 39 , 40 , 41 , 42 Consistent with this finding, results of expression studies that indicate that AD brains with low levels of ABCA7 develop AD at a younger age than those with higher expression of ABCA7, while individuals who expressed ABCA7 at similar levels to healthy controls developed AD at a very late age. 43 , 44 To date, only one protective low frequency coding missense variant has been identified (rs72973581, p.G215S) and has been hypothesized to induce a small gain of function or increase in ABCA7 expression, although this has not yet been confirmed. 21 Together, these findings suggest that reduced function of ABCA7 may be an important risk factor in the development of AD. 44
Investigating the ability of disease‐associated SNPs to modulate brain function and aging to promote or protect against the development of AD is important to build testable models to understand mechanisms of the disease. We evaluated the V1599M (V1613M in mouse) coding variant for potential inclusion into new LOAD models due to its location within a relatively well conserved region of homology between mouse and human ABCA7 and due to its prediction to be a deleterious mutation, thereby potentially acting as a loss of function mutation, which has been linked to worsening of AD‐relevant pathologies. 4 , 21 , 36 , 39 , 40 Notably, our results here are inconsistent with the actions of a variant that would increase risk for the development of AD, with profound reductions in both Aβ and plaques seen with Abca7V1613M . Consistent with this, recent findings suggest the V1599M variant does not confer risk for AD. 4 , 21 , 45 , 46 , 47 , 48 It is important to note that the bar for proving protection against AD risk is higher, and it is established that the variant is present in centenarians, and therefore does not preclude extreme aging. 47 Larger studies incorporating more V1599M carriers may help to clarify whether this variant confers protection against AD risk.
Single‐cell transcriptional and western analysis has shown that Abca7 is expressed at comparable levels in all cell types of the murine brain. 19 , 49 In the human brain, ABCA7 expression is relatively low under physiological conditions 50 and like mice, is also expressed at similar levels in all cell types. 51 , 52 , 53 , 54 ABCA7 is a member of the ABC superfamily and is thought to be a key regulator of lipid efflux, phagocytosis, and APP processing. 9 , 11 , 12 , 15 , 16 , 19 , 55 , 56 Human ABCA7 can promote phospholipid and, to a certain degree, cholesterol efflux to apolipoproteins in vitro. 7 , 9 , 11 , 56 More recently, an in vitro model of the blood‐brain barrier was used to demonstrate an effect of ABCA7 on regulation of cholesterol and Aβ efflux. 57 Consistent with these findings, we find that homozygosity for the V1613M variant in mice influences brain lipids in a relatively minor manner compared to WT control mice, and that ABCA7, along with the V1613M variant, did not strongly affect cholesterol efflux in neuronal cells in vitro. However, despite this, circulating cholesterol levels are increased in Abca7V1613M mice suggesting that other cell types in the body may be having greater effects on cholesterol metabolism or transport. Further, when combined with an amyloidogenic insult produced by the 5xFAD transgene array, the V1613M variant produces an increase in a wide range of brain lipids in addition to increasing circulating cholesterol and triglycerides. Most significantly, we demonstrate that the V1613M variant has a profound effect on APP processing and Aβ plaque deposition in hemizygous 5xFAD mice.
Loss of ABCA7 function has been associated with increased amyloid deposition in humans via neuroimaging studies. 58 , 59 Aβ is cleaved from its precursor, APP, via the amylogenic pathway which is mediated first by β‐secretase (BACE1), producing soluble APPβ (sAPPβ) and C99 fragments, followed by γ‐secretase to release Aβ and AICD. Studies using distinct human APP transgenic mouse models show that KO of Abca7 in both TgCRND8 and APP/PS1 mice results in increased Aβ pathology as well as increased levels of sAPPβ and soluble Aβ. 12 , 19 In contrast, here we find that the Abca7V1613M variant robustly decreases plaque load and Aβ generation in 5xFAD mice, across multiple timepoints, suggesting that this variant is not a loss of function, but rather promotes ABCA7 functions by increasing normal functions (ie, hypermorphic) or developing new functions (ie, neomorphic), or a combination of both. We also observed a reduction in downstream reactions to plaques, including in astrocyte reactivity, accumulation of dystrophic neurites and presence of NfL in the periphery, but no differences in synapse loss compared to 5xFAD mice. Whether these reductions are a consequence of the reduced plaque load in 5xFAD/Abca7V1613M mice or due to an ABCA7‐driven mechanism remains to be determined.
Our study demonstrates that the Abca7V1613M variant robustly decreases steady‐state levels of both C99 and C83, along with a decrease in full‐length APP in the brain of hemizygous 5xFAD mice. Overexpression of ABCA7 can produce a similar phenotype in both APPwt and APPswe overexpressing CHO cells, also resulting in decreases in SAPPβ and Aβ and localization of APP in perinuclear and other subcellular structures. 56 The reduction in both C99 and C83 is not consistent with inhibition or promotion of either the α‐ or β‐secretase pathways as they compete for substrate (APP), but instead suggests that APP is located in a manner that reduces its availability to either pathway equally. To assess whether ABCA7 also influenced the subcellular localization of APP, and therefore its availability to be processed, we overexpressed WT or V1613M mouse ABCA7 in N2a cells and stained for APP and markers of the endo‐lysosomal pathway. The increase in intracellular APP puncta in N2a cells overexpressing the ABCA7 V1613M variant compared to WT ABCA7 and their colocalization with LAMP1 and RAB7A structures is consistent with a model of increased accumulation of APP in endo‐lysosomal structures, which could result in increased degradation accounting for the equal reductions in both C99 and C83, as well as full‐length APP.
In addition to effects on Aβ generation and plaque pathology, KO of ABCA7 can modulate microglia and macrophage function. 15 , 16 , 17 Microglia are key mediators of AD pathology and by constantly sensing their surroundings, they can facilitate responses to different stimuli. 60 , 61 , 62 To assess whether the V1613M variant affects microglial function we assessed microglial response to acute stimulation via LPS injection and chronic stimulation, by their reaction to plaques. The results indicate that homozygosity for the V1613M variant extends the response to LPS stimulation, characterized by prolonged increase in classical proinflammatory genes such as C1qa, C1qb, C1qc, C1ra, and Hcar2. One possible explanation for this prolonged response in Abca7V1613M mice could be the downregulation of Dok3 in Abca7V1613M homozygous mice compared to WT mice under homeostatic conditions (Figure 1B). Dok3 is a negative regulator of LPS‐mediated inflammation and reduced expression of Dok3 results in increased LPS signaling in macrophages. 28 To explore if Abca7V1613M was modulating the phagocytic abilities of microglia we incubated primary microglia from WT and Abca7V1613M mice with beads, which represent dead/dying cells, and Aβ1‐42 peptide. Microglia from homozygous Abca7V1613M have increased phagocytic capacity for both beads and Aβ1‐42. In contrast to our findings in vitro and in response to acute stimulation, we did not find striking differences in microglial responses to chronic stimulation (Aβ plaques) in 5xFAD mice with the Abca7V1613M variant. Microglial numbers and associated inflammatory response reflected reduced Aβ plaque numbers in 5xFAD/Abca7V1613M mice. Thus, despite high expression of ABCA7 in microglia, this variant does not appear to grossly alter microglial function, in disease context, but rather its effect may be more prominent in neurons on Aβ generation and deposition in the aging brain rather than the reaction to the plaques driving damage and clinical symptoms. These different responses to different stimuli, acute versus chronic, may be due to the distinct recognition and signaling mechanisms of microglial activation associated with these different insults, further highlighting the complex nature of microglial function in health and disease.
This study is the first to explore functions of the V1599M (humans)/V1613M (mice) AD‐associated ABCA7 variant. 5xFAD/Abca7V1613M mice display persistent reductions in Aβ and plaque numbers. This reduction is mirrored by reduced inflammation, associated microgliosis, astrogliosis, and pathology‐induced damage. 5xFAD/Abca7V1613M mice also exhibit altered APP processing. Our data suggest that the Abca7V1613M variant is not a loss of function mutation, counter to the original predictions, but rather may act as a neomorph. These data raise the interesting possibility that the rs117187003 ABCA7 V1599M variant might be protective and potentially larger cohorts of individuals harboring ABCA7V1599M are required to elucidate this.
CONFLICT OF INTEREST STATEMENT
KNG is a member of the advisory board of Ashvattha Therapeutics. Author disclosures are available in the supporting information. This study was supported by the Model Organism Development and Evaluation for Late‐onset Alzheimer's Disease (MODEL‐AD) consortium funded by the National Institute on Aging (U54 AG054349).
CONSENT STATEMENT
No human subjects were used for the present study. Therefore, consent was not necessary.
Supporting information
Supporting Information
Table S1
Table S2
ICMJE Disclosure Form
ACKNOWLEDGMENTS
The authors thank Dr. Masashi Kitazawa (UC Irvine) for generously providing mouse N2a neuroblastoma cells and the UC Davis West Coast Metabolomics Center for bulk lipidomics analyses. This study was made possible in part through access to the Optical Biology Core Facility of the Developmental Biology Center, and the UCI Transgenic Mouse Facility (TMF), shared resources supported by the Cancer Center Support Grant (CA‐62203) and Center for Complex Biological Systems Support Grant (GM‐076516) at the University of California, Irvine.
Butler CA, Mendoza Arvilla A, Milinkeviciute G, et al. The Abca7V1613M variant reduces Aβ generation, plaque load, and neuronal damage. Alzheimer's Dement. 2024;20:4914–4934. 10.1002/alz.13783
Contributor Information
Grant R. MacGregor, Email: gmacg@uci.edu.
Kim N. Green, Email: kngreen@uci.edu.
DATA AVAILABILITY STATEMENT
Protocols, data, and results are available via the AD Knowledge Portal (https://adknowledgeportal.synapse.org). The AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the Accelerating Medicines Partnership (AMP‐AD) Target Discovery Program and other National Institute on Aging (NIA)‐supported programs to enable open‐science practices and accelerate translational learning. The data, analyses, and tools are shared early in the research cycle without a publication embargo on secondary use. Data are available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.org/DataAccess/Instructions). Data can be accessed in an interactive matter at UCI Mouse Mind Explorer (admodelexplorer.org).
The Abca7V1613M model is available from The Jackson Laboratory (Stock #035316) without restrictions on its use by both academic and commercial users. The content is solely the responsibility of the authors and does not necessarily represent the official view of the National Institutes of Health.
REFERENCES
- 1. Knopman DS, Amieva H, Petersen RC, et al. Alzheimer disease. Nat Rev Dis Primers. 2021;7(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45(12):1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reitz C, Jun G, Naj A, et al. Variants in the ATP‐binding cassette transporter (ABCA7), apolipoprotein E ϵ4,and the risk of late‐onset Alzheimer disease in African Americans. JAMA. 2013;309(14):1483‐1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vardarajan BN, Ghani M, Kahn A, et al. Rare coding mutations identified by sequencing of Alzheimer disease genome‐wide association studies loci. Ann Neurol. 2015;78(3):487‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. 2011;43(5):429‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim WS, Weickert CS, Garner B. Role of ATP‐binding cassette transporters in brain lipid transport and neurological disease. J Neurochem. 2008;104(5):1145‐1166. [DOI] [PubMed] [Google Scholar]
- 7. Picataggi A, Rodrigues A, Cromley DA, et al. Specificity of ABCA7‐mediated cell lipid efflux. Biochim Biophys Acta Mol Cell Biol Lipids. 2022;1867(7):159157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wiener JP, Desire S, Garliyev V, Lyssenko Iii N, Praticò D, Lyssenko NN. Down‐regulation of ABCA7 in human microglia, astrocyte and THP‐1 cell lines by cholesterol depletion, IL‐1β and TNFα, or PMA. Cells. 2023;12(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang N, Lan D, Gerbod‐Giannone M, et al. ATP‐binding cassette transporter A7 (ABCA7) binds apolipoprotein A‐I and mediates cellular phospholipid but not cholesterol efflux. J Biol Chem. 2003;278(44):42906‐42912. [DOI] [PubMed] [Google Scholar]
- 10. Abe‐Dohmae S, Yokoyama S. ABCA7 links sterol metabolism to the host defense system: molecular background for potential management measure of Alzheimer's disease. Gene. 2021;768:145316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abe‐Dohmae S, Ikeda Y, Matsuo M, et al. Human ABCA7 supports apolipoprotein‐mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J Biol Chem. 2004;279(1):604‐611. [DOI] [PubMed] [Google Scholar]
- 12. Sakae N, Liu CC, Shinohara M, et al. ABCA7 deficiency accelerates amyloid‐beta generation and Alzheimer's neuronal pathology. J Neurosci. 2016;36(13):3848‐3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nowyhed HN, Chandra S, Kiosses W, et al. ATP binding cassette transporter ABCA7 regulates NKT cell development and function by controlling CD1d expression and lipid raft content. Sci Rep. 2017;7:40273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fu Y, He Y, Phan K, et al. Sex‐specific lipid dysregulation in the Abca7 knockout mouse brain. Brain Commun. 2022;4(3):fcac120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jehle AW, Gardai SJ, Li S, et al. ATP‐binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J Cell Biol. 2006;174(4):547‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim WS, Li H, Ruberu K, et al. Deletion of Abca7 increases cerebral amyloid‐β accumulation in the J20 mouse model of Alzheimer's disease. J Neurosci. 2013;33(10):4387‐4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aikawa T, Ren Y, Yamazaki Y, et al. ABCA7 haplodeficiency disturbs microglial immune responses in the mouse brain. Proc Natl Acad Sci U S A. 2019;116(47):23790‐23796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fu Y, Hsiao JH, Paxinos G, Halliday GM, Kim WS. ABCA7 mediates phagocytic clearance of amyloid‐beta in the brain. J Alzheimers Dis. 2016;54(2):569‐584. [DOI] [PubMed] [Google Scholar]
- 19. Satoh K, Abe‐Dohmae S, Yokoyama S, St George‐Hyslop P, Fraser PE. ATP‐binding cassette transporter A7 (ABCA7) loss of function alters Alzheimer amyloid processing. J Biol Chem. 2015;290(40):24152‐24165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bossaerts L, Hendrickx Van de Craen E, Cacace R, Asselbergh B, Van Broeckhoven C. Rare missense mutations in ABCA7 might increase Alzheimer's disease risk by plasma membrane exclusion. Acta Neuropathol Commun. 2022;10(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sassi C, Nalls MA, Ridge PG, et al. ABCA7 p.G215S as potential protective factor for Alzheimer's disease. Neurobiol Aging. 2016;46:235. e1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rezaie N, Reese F, Mortazavi A. PyWGCNA: a Python package for weighted gene co‐expression network analysis. bioRxiv. 2022. 2022.08.22.504852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carrillo‐Jimenez A, Puigdellívol M, Vilalta A, et al. Effective knockdown of gene expression in primary microglia with siRNA and magnetic nanoparticles without cell death or inflammation. Front Cell Neurosci. 2018;12:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cajka T, Smilowitz JT, Fiehn O. Validating quantitative untargeted lipidomics across nine liquid chromatography‐high‐resolution mass spectrometry platforms. Anal Chem. 2017;89(22):12360‐12368. [DOI] [PubMed] [Google Scholar]
- 25. Forner S, Kawauchi S, Balderrama‐Gutierrez G, et al. Systematic phenotyping and characterization of the 5xFAD mouse model of Alzheimer's disease. Sci Data. 2021;8(1):270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Javonillo DI, Tran KM, Phan J, et al. Systematic phenotyping and characterization of the 3xTg‐AD mouse model of Alzheimer's Disease. Front Neurosci. 2021;15:785276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tran KM, Kawauchi S, Kramár EA, et al. A Trem2(R47H) mouse model without cryptic splicing drives age‐ and disease‐dependent tissue damage and synaptic loss in response to plaques. Mol Neurodegener. 2023;18(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peng Q, O'Loughlin JL, Humphrey MB. DOK3 negatively regulates LPS responses and endotoxin tolerance. PLoS One. 2012;7(6):e39967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ikeda Y, Abe‐Dohmae S, Munehira Y, et al. Posttranscriptional regulation of human ABCA7 and its function for the apoA‐I‐dependent lipid release. Biochem Biophys Res Commun. 2003;311(2):313‐318. [DOI] [PubMed] [Google Scholar]
- 30. Truesdale KP, Stevens J, Cai J. The effect of weight history on glucose and lipids: the Atherosclerosis Risk in Communities Study. Am J Epidemiol. 2005;161(12):1133‐1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Serrano‐Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Spires‐Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron. 2014;82(4):756‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kang MS, Aliaga AA, Shin M, et al. Amyloid‐beta modulates the association between neurofilament light chain and brain atrophy in Alzheimer's disease. Mol Psychiatry. 2021;26(10):5989‐6001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Keren‐Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell. 2017;169(7):1276‐1290. e17. [DOI] [PubMed] [Google Scholar]
- 35. Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late‐onset Alzheimer's disease. Nat Genet. 2011;43(5):436‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cuyvers E, De Roeck A, Van den Bossche T, et al. Mutations in ABCA7 in a Belgian cohort of Alzheimer's disease patients: a targeted resequencing study. Lancet Neurol. 2015;14(8):814‐822. [DOI] [PubMed] [Google Scholar]
- 37. Kunkle BW, Carney RM, Kohli MA, et al. Targeted sequencing of ABCA7 identifies splicing, stop‐gain and intronic risk variants for Alzheimer disease. Neurosci Letters. 2017;649:124‐129. [DOI] [PubMed] [Google Scholar]
- 38. Cukier HN, Kunkle BW, Vardarajan BN, et al. ABCA7 frameshift deletion associated with Alzheimer disease in African Americans. Neurol Genet. 2016;2(3):e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Steinberg S, Stefansson H, Jonsson T, et al. Loss‐of‐function variants in ABCA7 confer risk of Alzheimer's disease. Nat Genet. 2015;47(5):445‐447. [DOI] [PubMed] [Google Scholar]
- 40. Allen M, Lincoln SJ, Corda M, et al. ABCA7 loss‐of‐function variants, expression, and neurologic disease risk. Neurol Genet. 2017;3(1):e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bellenguez C, Charbonnier C, Grenier‐Boley B, et al. Contribution to Alzheimer's disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol Aging. 2017;59:220. e1‐ e9. [DOI] [PubMed] [Google Scholar]
- 42. De Roeck A, Van den Bossche T, van der Zee J, et al. Deleterious ABCA7 mutations and transcript rescue mechanisms in early onset Alzheimer's disease. Acta Neuropathol. 2017;134(3):475‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lyssenko NN, Shi X, Praticò D. The Alzheimer's disease GWAS risk alleles in the ABCA7 promoter and 5' region reduce ABCA7 expression. Acta Neuropathol. 2022;144(3):585‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lyssenko NN, Praticò D. ABCA7 and the altered lipidostasis hypothesis of Alzheimer's disease. Alzheimers Dement. 2021;17(2):164‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Holstege H, Hulsman M, Charbonnier C, et al. Exome sequencing identifies rare damaging variants in ATP8B4 and ABCA1 as risk factors for Alzheimer's disease. Nat Genet. 2022;54(12):1786‐1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. N'Songo A, Carrasquillo MM, Wang X, et al. African American exome sequencing identifies potential risk variants at Alzheimer disease loci. Neurol Genet. 2017;3(2):e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nygaard HB, Erson‐Omay EZ, Wu X, et al. Whole‐exome sequencing of an exceptional longevity cohort. J Gerontol A Biol Sci Med Sci. 2019;74(9):1386‐1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee WP, Choi SH, Shea MG, et al. Association of common and rare variants with Alzheimer's Disease in over 13,000 diverse individuals with whole‐genome sequencing from the Alzheimer's disease sequencing project. medRxiv. 2023. [Google Scholar]
- 49. Zhang Y, Chen K, Sloan SA, et al. An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929‐11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. De Roeck A, Van Broeckhoven C, Sleegers K. The role of ABCA7 in Alzheimer's disease: evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 2019;138(2):201‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mathys H, Davila‐Velderrain J, Peng Z, et al. Single‐cell transcriptomic analysis of Alzheimer's disease. Nature. 2019;570(7761):332‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Morabito S, Miyoshi E, Michael N, et al. Single‐nucleus chromatin accessibility and transcriptomic characterization of Alzheimer's disease. Nat Genet. 2021;53(8):1143‐1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Morabito S, Miyoshi E, Michael N, Swarup V. Integrative genomics approach identifies conserved transcriptomic networks in Alzheimer's disease. Hum Mol Genet. 2020;29(17):2899‐2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Leng K, Li E, Eser R, et al. Molecular characterization of selectively vulnerable neurons in Alzheimer's disease. Nat Neurosci. 2021;24(2):276‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim WS, Fitzgerald ML, Kang K, et al. Abca7 null mice retain normal macrophage phosphatidylcholine and cholesterol efflux activity despite alterations in adipose mass and serum cholesterol levels. J Biol Chem. 2005;280(5):3989‐3995. [DOI] [PubMed] [Google Scholar]
- 56. Chan SL, Kim WS, Kwok JB, et al. ATP‐binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J Neurochem. 2008;106(2):793‐804. [DOI] [PubMed] [Google Scholar]
- 57. Lamartinière Y, Boucau MC, Dehouck L, et al. ABCA7 downregulation modifies cellular cholesterol homeostasis and decreases amyloid‐β peptide efflux in an in vitro model of the blood‐brain barrier. J Alzheimers Dis. 2018;64(4):1195‐1211. [DOI] [PubMed] [Google Scholar]
- 58. Hughes TM, Lopez OL, Evans RW, et al. Markers of cholesterol transport are associated with amyloid deposition in the brain. Neurobiol Aging. 2014;35(4):802‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Apostolova LG, Risacher SL, Duran T, et al. Associations of the Top 20 Alzheimer disease risk variants with brain amyloidosis. JAMA Neurol. 2018;75(3):328‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wolf SA, Boddeke HW. Kettenmann H. microglia in physiology and disease. Annu Rev Physiol. 2017;79:619‐643. [DOI] [PubMed] [Google Scholar]
- 61. Vilalta A, Brown GC. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. Febs j. 2018;285(19):3566‐3575. [DOI] [PubMed] [Google Scholar]
- 62. Galloway DA, Phillips AEM, Owen DRJ, Moore CS. Phagocytosis in the Brain: homeostasis and Disease. Front Immunol. 2019;10:790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Table S1
Table S2
ICMJE Disclosure Form
Data Availability Statement
Protocols, data, and results are available via the AD Knowledge Portal (https://adknowledgeportal.synapse.org). The AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the Accelerating Medicines Partnership (AMP‐AD) Target Discovery Program and other National Institute on Aging (NIA)‐supported programs to enable open‐science practices and accelerate translational learning. The data, analyses, and tools are shared early in the research cycle without a publication embargo on secondary use. Data are available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.org/DataAccess/Instructions). Data can be accessed in an interactive matter at UCI Mouse Mind Explorer (admodelexplorer.org).
The Abca7V1613M model is available from The Jackson Laboratory (Stock #035316) without restrictions on its use by both academic and commercial users. The content is solely the responsibility of the authors and does not necessarily represent the official view of the National Institutes of Health.