

Abstract
Previous data indicate that immune mechanisms may be involved in developing capillary leakage during Sin Nombre virus (SNV) infection. Therefore, we investigated production of tumor necrosis factor alpha (TNF-α) by human alveolar macrophages and human umbilical vein endothelial cells (HUVEC) after infection with SNV. In addition, we examined the effect of TNF-α on HUVEC monolayer leakage. Our results reveal that although TNF-α decreases accumulation of viral nucleoproteins, TNF-α levels do not change in SNV-infected cells. In addition, supernatants from SNV-infected human alveolar macrophages did not cause a significant increase in endothelial monolayer permeability.
Hantaviruses are enveloped viruses with a diameter of 120 nm and contain a trisegmented negative-strand RNA genome (32). Hantaan virus and Sin Nombre virus (SNV) are associated with the most severe forms of hantavirus infection in humans, hemorrhagic fever with renal syndrome (HFRS) and hantavirus pulmonary syndrome (HPS), respectively. Autopsy findings of HFRS and HPS victims typically reveal the common feature of increased permeability in microvascular beds, suggesting that the vascular endothelium may be a prime target for virus infection. However, investigations did not reveal endothelial cell necrosis attributable to hantavirus replication (5, 41). These data represent a major paradox of hantavirus infection: development of capillary leakage without visible endothelial cell damage.
A number of observations have suggested that immune mechanisms play a central role in development of HFRS (5, 43). Shortly after onset of disease, activation of the humoral immune response results in formation of specific immunoglobulin M (IgM) and IgG antibodies, which leads to deposition of immune complexes in kidney glomerular capillary basal membranes (41). During the febrile phase, T-cell activation has been demonstrated by flow cytometry (5). This activation is temporally associated with lymphocytosis and the appearance of atypical lymphocytes in the peripheral blood (13). At the same time, a decrease in the CD4/CD8 lymphocyte ratio as a result of CD8 lymphocyte proliferation can be observed in blood samples from HFRS patients (4). With the exception of parameters reflecting renal failure, laboratory findings for HPS patients are basically similar to those for HFRS patients (17).
Recently, an increase in the levels of tumor necrosis factor alpha (TNF-α) in plasma during the acute phase of hantavirus infection was reported (19, 21). Immunohistochemical staining revealed TNF-α-positive cells in lung tissue of patients with HPS who died and in kidney biopsies of HFRS patients (27, 38). TNF-α is a macrophage-derived cytokine first described as a mediator of tumor necrosis in mice (1). Pathological changes following TNF-α infusion include pulmonary inflammation, hemorrhage, microaggregation of leukocytes, and migration of polymorphonuclear leukocytes into the pulmonary microcirculation system (27). Endothelial cells exposed to TNF-α increase procoagulant activity and adhesiveness to lymphocytes and polymorphonuclear cells (29). Also, TNF-α treatment of endothelial cells in vitro results in increased endothelial cell monolayer permeability without visible cytopathic effect. It has been suggested that this effect of TNF-α could be a result of cytoskeleton changes in endothelial cells (35). TNF-α is also a strong activator of macrophages. It stimulates migration, phagocytic activity, cytotoxic activities, respiratory burst, and degranulation of phagocytes (18, 22, 29). Increased levels in serum samples from HPS patients and the presence of TNF-α positive cells in lung biopsies from patients with fatal HPS suggest that it may cause lung capillary leakage. These experiments were initiated to determine if in vitro infection with SNV induced changes in TNF-α levels.
Effect of TNF-α on the accumulation of SNV nucleocapsid protein in Vero E6 cells.
Since Vero E6 cells are known to support the replication of hantaviruses, we first examined the effect of TNF-α on nucleocapsid protein accumulation of SNV (SNV strain Convict Creek 107 [CC107], kindly provided by Connie Schmaljohn). Vero E6 cells were grown in Iscove's medium with 2% fetal bovine serum. Recombinant TNF-α (50 ng/ml) (107 U/ml; Genzyme, Cambridge, Mass.) was added to culture media after infection of Vero E6 cells with SNV. Four days postinfection, fresh medium containing TNF-α was added to the culture medium. At various times postinfection, cells were harvested and the accumulation of virus nucleocapsid protein was examined by Western blotting. Equivalent loading of each lane of sodium dodecyl sulfate-polyacrylamide gels was achieved by quantitation of protein using a modified Lowry protein assay (Pierce, Rockford, Ill.) and was verified by Coomassie blue staining of the gel. Nucleoprotein monoclonal antibody GB04-BF07 (1:1,000; antibody from T. Ksiazek, Centers for Disease Control and Prevention, Atlanta, Ga.) was incubated with membranes at room temperature overnight. Antigen-antibody complexes were identified with goat anti-human horseradish peroxidase (HRP)-conjugated antibodies (Vector Laboratories, Inc., Burlingame, Calif.) and developed under standard HRP substrate conditions (Vector Laboratories, Inc.).
TNF-α decreased accumulation of virus nucleocapsid protein in Vero E6 cells 1, 3, 4, 5, 6, and 7 days postinfection (Fig. 1) but did not affect the percentage of infected cells or induce a visibly apparent cytopathic effect in treated Vero E6 cells (data not shown). To study the effect of lower concentrations of TNF-α, 50, 10, and 1 ng/ml (5 × 104, 104, and 103 U/ml, respectively) were utilized for treatment of infected Vero E6 cell monolayers. In this experiment, fresh medium containing TNF-α was not added on day 4 of infection. All concentrations of TNF-α had a suppressive effect on virus nucleocapsid accumulation in Vero E6 cells at 1, 3, 4, and 5 days postinfection. No differences in the levels of virus nucleocapsid protein accumulated were detected 6 and 7 days after infection (Fig. 2). This is likely caused by the breakdown of TNF-α after 5 days postinfection.
FIG. 1.
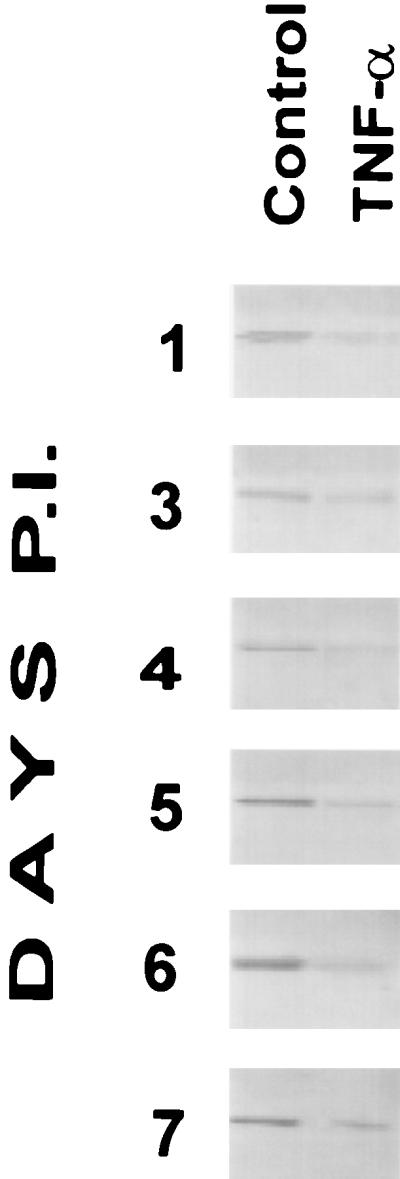
Western blot analysis of the effect of TNF-α on accumulation of SNV nucleocapsid protein in Vero E6 cells. SNV nucleocapsid protein was detected with sera from HPS convalescent patients. Vero E6 cells were treated with 50 ng of TNF-α per ml or not treated with TNF-α as a control. New culture medium was added to the culture medium 4 days postinfection (P.I.), and a new aliquot of cytokine was added.
FIG. 2.

Western blot analysis of the effect of TNF-α on accumulation of SNV nucleocapsid protein in Vero E6 cells. Vero E6 cells were treated with 50, 10, and 1 ng of TNF-α per ml or not treated with TNF-α as a control. There was no addition of new culture medium to the culture medium 4 days postinfection.
TNF-α increases nitric oxide synthase mRNA expression and nitric oxide production in cells (2, 34, 40). Since nitric oxide is a nonspecific antiviral agent (24), an increase in its concentration in infected cells may explain the suppressive effect of this cytokine on SNV nucleocapsid accumulation. To investigate this hypothesis, we utilized the nitric oxide synthase inhibitor N(G)-monomethyl-l-arginine (a kind gift from J. Maciejewski, National Institutes of Health, Bethesda, Md.) alone and in combination with TNF-α. In addition, we used gamma interferon (IFN-γ) (1,000 U/ml; gift from J. Maciejewski), a well-known antiviral cytokine (6), for comparative analysis with TNF-α on suppression of SNV nucleocapsid accumulation in Vero E6 cells (Fig. 3A). In this experiment, TNF-α was added immediately after infection and on day 4 postinfection. TNF-α (1 ng/ml) had a suppressive effect on the accumulation of SNV nucleoprotein in Vero E6 cells at 3, 4, 5, 6, and 7 days postinfection. Nitric oxide synthase inhibitor did not eliminate the suppressive effect of TNF-α on SNV nucleocapsid protein accumulation. IFN-γ (1,000 U/ml) decreased the accumulation of SNV nucleoprotein in infected cells at 3, 4, 5, 6, and 7 days after infection. The effect of IFN-γ exceeded the activity of TNF-α.
FIG. 3.

Western blot analysis of the effects of TNF-α, N(G)-monomethyl-l-arginine (MMA), IFN-γ, and pentoxyfilline on accumulation of SNV nucleocapsid protein in Vero E6 cells. (A) Vero E6 cells were treated with TNF-α (1 ng/ml), TNF-α and MMA (5 μM), or IFN-γ (1,000 U/ml) or not treated as a control. P.I., postinfection. (B) Vero E6 cells were treated with TNF-α (1 ng/ml) or pentoxyfilline (PTF) (5 mM) alone or combined; some cells were not treated as a control. Four days postinfection, new culture medium was added to the culture medium, and new aliquots of cytokine and PTF were added.
Investigations of the last decade revealed that pentoxifylline, a potential antiinflammatory drug, abolished the effects of TNF-α (7, 28, 37). The accumulation of viral nucleoprotein in the presence of pentoxyfilline (5 mM; Sigma, St. Louis, Mo.) alone or in combination with TNF-α (10 ng/ml) was investigated in SNV-infected Vero E6 cells. TNF-α decreased accumulation of viral nucleoprotein in Vero E6 cells at 7 days post- infection. Pentoxifylline alone did not affect the accumulation of SNV nucleoprotein 7 days after infection. The combination of cytokine and pentoxifylline reversed the effect of TNF-α and restored the level of viral nucleocapsid protein to that of the control (Fig. 3B). TNF-α-treated Vero E6 cells produced slightly fewer plaques (data not shown) than the control (uninfected) cells.
Treatment of human umbilical vein endothelial cells (HUVEC) with TNF-α (10 ng/ml) decreased the accumulation of SN virus nucleocapsid protein in HUVEC 1, 3, 4, 5, 6, and 7 days postinfection, and no cytopathic effect was observed in infected-cell monolayers (data not shown).
Effect of SNV infection and TNF-α treatment on permeability of HUVEC monolayer.
Modified Boyden chamber systems were used to determine if SNV infection could alter endothelial cell monolayer permeability. HUVEC were isolated from umbilical cords by the method of Jaffe and colleagues (16) and used between passages 2 and 4. Cells were plated on membrane inserts and placed in 24-well plates to comprise upper and lower compartments. Transmembrane diffusion of HRP was used to detect changes in permeability of endothelial monolayers, as described by Feldmann and colleagues (9). When the HUVEC monolayers reached confluence (2 or 3 days), cells were infected with SNV at a multiplicity of infection of 0.01. Immunostaining of infected-cell monolayers revealed that about 90% of HUVEC expressed SNV proteins 10 days after infection (data not shown). SNV infection of HUVEC monolayer did not cause statistically significant leakage (Fig. 4). However, during the observation period, the concentration of HRP in the lower compartment of infected-cell monolayers was always higher than in the uninfected control. Although not statistically significant, this tendency may reflect activation of endothelial cells after SNV infection. There was no detectable cytopathic effect caused by SNV replication that could explain this phenomenon.
FIG. 4.

Effect of SNV infection on the leakage of endothelial cell monolayer. Changes in optical density, using light with a wavelength of 470 nm, over time are shown for SNV-infected (stippled squares) and control (uninfected) (solid diamonds) HUVEC monolayers. Each point is the mean of three separate experiments (two samples per experiment). The means were compared by an unpaired t test, and a P value of <0.05 was considered statistically significant.
To determine the effect of TNF-α on vascular endothelial cell monolayer leakage, cytokine (50 ng/ml) was added to the upper compartment and the permeability of HUVEC monolayers was examined. The choice of cytokine concentration was based on data from Feldmann and colleagues (9) and Ishii and colleagues (14), who reported that no increase of endothelial cell permeability occurred at concentrations of TNF-α below 4 ng/ml. However, TNF-α (10 ng/ml) caused a stable suppressive effect on the accumulation of SNV nucleoprotein in previous experiments. In our experiments, TNF-α showed statistically significant (P < 0.05, P < 0.001) increases in HUVEC monolayer permeability 5 and 24 h after initiation of the experiment (Fig. 5).
FIG. 5.

Effect of TNF-α on endothelial cell monolayer leakage. Changes in optical density, using light with a wavelength of 470 nm, over time are shown for untreated endothelial cell monolayer (open bars) and endothelial cell monolayer after treatment with TNF-α (50 ng/ml) (solid bars). Values that are significantly different from the control values are indicated by asterisks (∗, P < 0.05; ∗∗, P < 0.001).
Infection of human alveolar macrophages with SNV.
An earlier study (43) reported alveolar macrophages expressing SNV antigens in the lungs of patients with HPS. To determine whether macrophages can be infected with SNV, the following experiment was done. Human alveolar macrophages were isolated from bronchoalveolar lavage (BAL) following routine diagnostic procedures at the Veterans Affairs Sierra Nevada Health Care System, Reno, Nev. The University of Nevada Biomedical Research Committee approved this project, and patients gave informed consent. BAL was performed using an Olympus B4 bronchoscope wedged in distal airways and four 50-ml aliquots of normal saline. Alveolar macrophages were separated from human BAL contents by Ficoll density gradient centrifugation, and cells were plated into six-well tissue culture plates. To avoid contamination, an antibiotic-antimycotic mixture (Sigma) was used. On average, 2 to 5 million cells were recovered from each lavage sample. Cells were divided equally to form two groups: control and SNV infected. After 1 h of adsorption, unattached virus was removed by extensive washing and new medium was added. Supernatant (200-μl) aliquots were collected at 4 and 7 days postinfection to determine the amount of infectious virus present. At 7 days postinfection, cells were washed, fixed, and stained with immune convalescent HPS sera to reveal SNV-positive cells. Human alveolar macrophages are permissive to SNV infection, as illustrated by the punctate staining pattern (approximately 65% cells were positive) and localization of viral antigens in the cytoplasm of infected cells (Fig. 6A and B). No viral antigens were observed in uninfected control cells (Fig. 6C). To detect virus production by human alveolar macrophages, Vero E6 monolayers were incubated with macrophage culture supernatants for 1 h at 37°C. After infection, Vero E6 cells were washed, overlaid with 0.6% agarose, and incubated for 14 days. Agarose was removed, and cells were washed, fixed, and stained. Human alveolar macrophages produced relatively low levels of SNV (<0.1 PFU/ml) (data not shown).
FIG. 6.

SNV infection of human lung alveolar macrophages isolated from BAL. Cells were infected with SNV stock (multiplicity of infection of 0.01) and cultured for 7 days. SNV proteins were detected by immunostaining with convalescent HPS serum. Immunostaining results were visualized with a Nikon ES 800 microscope using differential interference contrast microscopy. Images were captured with an integrating charge-coupled device camera (Photonic Science, Millham, England) and Image Pro Plus image analysis software (Media Cybernetics, Silver Springs, Md.). Images were sharpened with Micro-tome version 4.0 (Vay-Tek, Inc., Fairfield, Iowa). (A) Human alveolar macrophages infected with SNV. The presence of SNV antigens is revealed by red alkaline phosphatase staining. (B) SNV-infected alveolar macrophages illustrating the characteristic punctate staining pattern and cytoplasmic localization of viral antigens. (C) Uninfected human lung alveolar macrophages. Magnifications, ×200 (A and C) and ×400 (B).
Detection of TNF-α production by SNV-infected and LPS-treated human alveolar macrophages.
Isolated human alveolar macrophages were counted and added to six-well plates at a density of 106 cells/well. After 24 h of incubation, cells were infected with SNV or treated with lipopolysaccharide (LPS) (1μg/ml; Sigma). Four days after treatment, supernatants were collected for quantitative enzyme-linked immunosorbent assay analysis using a Quantikine ELISA Kit (R&D System, Minneapolis, Minn.). SNV-infected human alveolar macrophages produced significantly less TNF-α than alveolar macrophages treated with LPS (Fig. 7). Interestingly, alveolar macrophages from some donors did not produce any detectable TNF-α in response to virus infection, whereas the same cells developed a strong cytokine response upon LPS treatment.
FIG. 7.

Detection of TNF-α in supernatant from SNV-infected and LPS-treated human alveolar macrophages. Human alveolar macrophages were not treated (bar 1), infected with SNV (bar 2), or treated with LPS (bar 3). Values that are significantly different from the control values are indicated by asterisks (∗∗, P < 0.001).
Effect of supernatant from SNV-infected and LPS-treated alveolar macrophages on HUVEC monolayer leakage.
The supernatant culture fluids from infected and LPS-treated human alveolar macrophages were added to the upper compartment of 24-well plates of confluent HUVEC monolayers. Supernatant from SNV-infected human alveolar macrophages did not induce statistically significant changes in HUVEC monolayer permeability. However, a statistically significant increase of HUVEC monolayer leakage was observed 24 h after the addition of supernatant from LPS-treated alveolar macrophages (Fig. 8). It is well-known that inflammatory cytokines may regulate virus gene expression and virus protein production. For example, TNF-α inhibits hepatitis B virus gene expression in transgenic mice (11) and human immunodeficiency virus type 1 replication in peripheral blood monocytes and alveolar macrophages (20). TNF-α and IFN-γ act synergistically to inhibit murine cytomegalovirus and herpes simplex virus replication (8, 23). TNF-α markedly inhibits respiratory syncytial virus replication in a dose and time-dependent manner (26). Also, treating MDCK cells with TNF-α inhibits influenza virus replication and protein synthesis (39). Our data revealed that TNF-α inhibits SNV nucleoprotein accumulation in infected Vero E6 cells. TNF-α exhibits suppressive effects at concentrations as low as 1 ng/ml which is close to that produced by infected lung macrophages in our experiments. The effect of TNF-α was reversible, and periodic addition of fresh medium containing TNF-α was required to maintain the suppressive effect.
FIG. 8.

Increased permeability of endothelial cell monolayer after the addition of culture supernatants. The cultures were untreated endothelial cells (open bars) and human alveolar macrophages that were treated with LPS (shaded bars) or infected with SNV (solid bars). Changes in optical density, using light with a wavelength of 470 nm, over time are shown. Values that are significantly different from the control values are indicated by asterisks (∗∗, P < 0.001).
The suppressive effect of TNF-α on the accumulation of SNV nucleoprotein was not mediated by induction of endogenous production of nitric oxide by infected cells. This finding is supported by Rosenkranz-Weiss and colleagues (31), who showed that neither TNF-α, IFN-γ, or IL-1 alone is able to activate nitric oxide synthase and increase production of nitric oxide in cells; however, a combination of cytokines was effective. The suppression of SNV nucleocapsid accumulation in Vero E6 cells after TNF-α treatment was not due to activation of the IFN-γ antiviral pathway, because SNV nucleocapsid accumulation in Vero E6 cells was almost completely suppressed by IFN-γ, whereas suppression induced by TNF-α was not that pronounced.
To further investigate the mechanism behind TNF-α-related suppression of SNV nucleoprotein accumulation in infected cells, we tested the influence of pentoxifylline on the level of virus nucleocapsid in infected cells. Investigations over the last decade revealed the ability of this drug to abolish the effects of TNF-α. Thus, pentoxifylline protected the L929 cell line against TNF-α-mediated cytotoxicity and cytostasis (37). Ohdama and colleagues (28) reported prevention of TNF-α-induced suppression of endothelial cell surface thrombomodulin expression after incubation of cells with pentoxifylline. In our experiments, pentoxifylline alone did not change the level of SNV nucleocapsid. However, in combination with TNF-α, it reversed the suppressive effect of the cytokine. The mechanism of anti-TNF-α activity of pentoxifylline is not fully understood, but investigators proposed that the effect might be associated with its selective inhibition of postreceptor signaling. For example, pentoxifylline abrogates TNF-α-induced actin filament polymerization, which has been reported to participate in receptor cycling (7).
The lowest concentration of TNF-α used in our experiments was 1 ng/ml. Of concern was whether this concentration of cytokine could be produced by infected alveolar macrophages in humans infected with SNV. Experiments with infected human alveolar macrophages revealed that cells from at least 3 of 10 (33%) donors produced approximately 1 ng of TNF-α per ml after SNV infection. Surprisingly, we were not able to detect release of TNF-α by alveolar macrophages from seven donors following infection with SNV in vitro. In contrast, LPS treatment of alveolar macrophages collected from the same donors resulted in dramatic increases of TNF-α release in supernatants of cells from all donors. The macrophage cultures from some individuals responded with up to 8 ng of cytokine released per ml after activation with LPS. These data indicate that productive SNV infection of human alveolar macrophages is not always associated with an increase in TNF-α release and that cytokine production appears to depend on individual patient reactivity.
Histological observations of tissue samples from people with HPS who died reveal that hantaviruses do not cause detectable cytopathic effects on blood vessel endothelial cells, despite the presence of hantavirus antigens in these cells (17, 42, 43). However, recent investigations (10, 15) found that vascular leakage could occur without endothelial cell damage leading to cell death, through destruction of the adherens-type junctions between endothelial cells and degradation of the fibrin layer underneath that supports growth and viability of the endothelium. Productive infection of endothelial cells by SNV did not result in a significant increase of endothelium monolayer leakage. However, our study with infected HUVEC monolayer permeability shows an increased tendency of monolayers to leak after SNV infection. We propose that this statistically insignificant but stable increase in permeability of HUVEC is a sign of structural changes in interendothelial junctions or fibrin production in response to SN infection.
Results of vitro experiments showed that TNF-α alone could cause endothelial monolayer leakage (3). However, the concentrations of cytokine used in those experiments were significantly higher than that (1.8 ng/ml) produced in vitro by SNV-infected cells (our data). Our data are supported by work done by Ishii and colleagues (14), who found that endothelial monolayer permeability did not increase in response to TNF-α concentrations below 4 ng/ml. TNF-α concentrations in serum samples from patients with hemorragic fever caused by different agents (Puumala virus, Hantaan virus, Junin virus, etc.) (12, 19) never exceeded 100 pg/ml. We believe that because of the low production of TNF-α by alveolar macrophages after SNV infection, this cytokine alone could not be considered the sole causative agent of lung edema.
In conclusion, in this study we demonstrated the following. (i) TNF-α has a suppressive effect on the accumulation of SNV nuclecapsid protein in infected cells. (ii) Human alveolar macrophages are permissive for SNV infection and react with low production of TNF-α. (iii) Supernatant from infected human alveolar macrophages fails to induce endothelial monolayer leakage. (iv) SNV infection of endothelial cells results in an insignificant increase of permeability, which could be a sign of structural changes in interendothelial junctions.
Acknowledgments
This study was supported in part by NIH grants AI36418, AI39808, and AI45059 and by the Medical Service and Research Service of the Veteran Affairs Sierra Nevada Health Care System.
We thank M. Hall for helpful discussions and critical reading of the manuscript.
REFERENCES
- 1.Beutler B, Greenwald D, Hulmes J D, Chang M, Pan Y-C E, Mathison J, Ulevitch R, Cerami A. Identity of tumor necrosis factor and the macrophage-secreted factor cachectin. Nature. 1985;316:552–553. doi: 10.1038/316552a0. [DOI] [PubMed] [Google Scholar]
- 2.Beyaert R, Fiers W. Tumor necrosis factor and lymphotoxin. In: Mire-Sluis A R, Thorpe R, editors. Cytokines. San Diego, Calif: Academic Press; 1998. pp. 335–361. [Google Scholar]
- 3.Brett J, Gerlach H, Nawroth P, Steinberg S, Godman G, Stern D. Tumor necrosis factor/cachectin increases permeability of endothelial cell monolayers by a mechanism involving regulatory G proteins. J Exp Med. 1989;169:1977–1991. doi: 10.1084/jem.169.6.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, Yang W. Abnormalities of T cell immunoregulation in hemorrhagic fever with renal syndrome. J Infect Dis. 1990;161:1016–1019. doi: 10.1093/infdis/161.5.1016. [DOI] [PubMed] [Google Scholar]
- 5.Cosgriff T M. Mechanisms of disease in hantavirus infection: pathophysiology of haemorrhagic fever with renal syndrome. Rev Infect Dis. 1991;13:97–107. doi: 10.1093/clinids/13.1.97. [DOI] [PubMed] [Google Scholar]
- 6.De Maeyer E, De Maeyer-Guignard J. Interferon-gamma. In: Mire-Suis A R, Thorpe R, editors. Cytokines. San Diego, Calif: Academic Press; 1998. pp. 391–401. [Google Scholar]
- 7.Elbim C, Lefebvre M, Hakim J, Gougerot-Pocidalo M A. Effects of pentoxifylline on human polymorphonuclear neutrophil responses to TNF in whole blood. Eur Cytokine Netw. 1995;6:113–120. [PubMed] [Google Scholar]
- 8.Feduchi E, Alonso M A, Carrasco L. Human gamma interferon and tumor necrosis factor exert a synergistic blockade on the replication of herpes simplex virus. J Virol. 1989;63:1354–1359. doi: 10.1128/jvi.63.3.1354-1359.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feldmann H, Bugany H, Mahner F, Klenk H-D, Drenckhahn D, Schnittler H-J. Filovirus-induced endothelial leakage triggered by infected monocytes/macrophages. J Virol. 1996;70:2208–2214. doi: 10.1128/jvi.70.4.2208-2214.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franke W W, Kaaprell H-P, Cowin P. Immunolocalization of plakoglobin in endothelial junctions: identification as a special type of zonulae adherents. Biol Cell. 1987;59:205–218. doi: 10.1111/j.1768-322x.1987.tb00532.x. [DOI] [PubMed] [Google Scholar]
- 11.Gilles P N, Fey G, Chisari F V. Tumor necrosis factor alpha negatively regulates hepatitis B virus gene expression in transgenic mice. J Virol. 1992;66:3955–3960. doi: 10.1128/jvi.66.6.3955-3960.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heller M V, Saavedra M C, Falcoff R, Maiztegui J I, Molinas F C. Increased tumor necrosis factor-α levels in Argentine hemorrhagic fever. J Infect Dis. 1992;166:1203–1204. doi: 10.1093/infdis/166.5.1203. [DOI] [PubMed] [Google Scholar]
- 13.Huang C, Jin B, Wang M, Li E, Sun C. Hemorrhagic fever with renal syndrome: relationship between pathogenesis and cellular immunity. J Infect Dis. 1994;169:868–870. doi: 10.1093/infdis/169.4.868. [DOI] [PubMed] [Google Scholar]
- 14.Ishii Y, Partridge C A, Del Vecchio P J, Malik A B. Tumor necrosis factor-α-mediated decrease in glutathione increases the sensitivity of pulmonary vascular endothelial cells to H2O2. J Clin Invest. 1992;89:794–802. doi: 10.1172/JCI115658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaffe E A, Mosher D F. Synthesis of fibronectin by cultured human endothelial cells. J Exp Med. 1978;147:1779–1791. doi: 10.1084/jem.147.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaffe E A, Nachman R L, Becker C G, Minick C R. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karneva M, Mustonen J, Vaheri A. Pathogenesis of Puumala and other hantavirus infections. Rev Med Virol. 1998;8:67–86. doi: 10.1002/(sici)1099-1654(199804/06)8:2<67::aid-rmv217>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 18.Khwaja A, Carver J E, Linch D C. Interactions of granulocyte macrophage colony stimulating factor (CSF), granulocyte CSF, and tumor necrosis factor-α in the priming of the neutrophil respiratory burst. Blood. 1992;79:745–753. [PubMed] [Google Scholar]
- 19.Krakauer T, Leduc J W, Krakauer H. Serum levels of tumor necrosis factor-α, interleukin-1, and interleukin-6 in hemorrhagic fever with renal syndrome. Viral Immunol. 1995;8:75–79. doi: 10.1089/vim.1995.8.75. [DOI] [PubMed] [Google Scholar]
- 20.Lane B R, Markovitz D M, Woodford N L, Rochford R, Strieter R M, Coffey M J. TNF-alpha inhibits HIV-1 replication in peripheral blood monocytes and alveolar macrophages by inducing the production of RANTES and decreasing C-C chemokine receptor 5 (CCR5) expression. J Immunol. 1999;163:3653–3661. [PubMed] [Google Scholar]
- 21.Linderholm M, Ahlm C, Settergren B, Waage A, Tarnvik A. Elevated plasma levels of tumor necrosis factor (TNF)-α, soluble TNF receptors, interleukin (IL)-6, and IL-10 in patients with hemorrhagic fever with renal syndrome. J Infect Dis. 1996;173:38–43. doi: 10.1093/infdis/173.1.38. [DOI] [PubMed] [Google Scholar]
- 22.Lo S K, Everitt J, Gu J, Malik A B. Tumor necrosis factor mediates experimental pulmonary edema by ICAM-1 and CD 18-dependent mechanisms. J Clin Invest. 1992;89:981–988. doi: 10.1172/JCI115681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lucin P, Jonjic S, Messerle M, Polic B, Hengel H, Koszinovski U H. Late phase inhibition of murine cytomegalovirus replication by synergistic action of interferon-gamma and tumor necrosis factor. J Gen Virol. 1994;75(Pt. 1):101–110. doi: 10.1099/0022-1317-75-1-101. [DOI] [PubMed] [Google Scholar]
- 24.Mannick J B. The antiviral role of nitric oxide. Res Immunol. 1995;146:693–697. doi: 10.1016/0923-2494(96)84920-0. [DOI] [PubMed] [Google Scholar]
- 25.Martinet I, Yamauchi K, Crystal R G. Differential expression of the tumor necrosis factor/cachectin gene by blood and lung mononuclear phagocytes. Am Rev Respir Dis. 1988;138:659–665. doi: 10.1164/ajrccm/138.3.659. [DOI] [PubMed] [Google Scholar]
- 26.Merolla R, Rebert N A, Tsivite P T, Hoffmann S P, Panuska J R. Respiratory syncytial virus replication in human lung epithelial cells: inhibition by tumor necrosis factor alpha and interferon beta. Am J Respir Crit Care Med. 1995;152:1358–1366. doi: 10.1164/ajrccm.152.4.7551395. [DOI] [PubMed] [Google Scholar]
- 27.Mori M, Rothman A L, Kurane I, Montoya J M, Nolte K B, Norman J E, Waite D C, Koster F T, Ennis F A. High levels of cytokine-producing cells in the lung tissue of patients with fatal hantavirus pulmonary syndrome. J Infect Dis. 1999;179:295–302. doi: 10.1086/314597. [DOI] [PubMed] [Google Scholar]
- 28.Ohdama S, Takano S, Ohashi K, Miyake S, Aoki N. Pentoxifylline prevents tumor necrosis factor-induced suppression of endothelial cell surface thrombomodulin. Thromb Res. 1991;62:745–755. doi: 10.1016/0049-3848(91)90378-a. [DOI] [PubMed] [Google Scholar]
- 29.Pober J S, Cotran R S. Cytokines and endothelial cell biology. Pathol Rev. 1990;70:427–451. doi: 10.1152/physrev.1990.70.2.427. [DOI] [PubMed] [Google Scholar]
- 30.Ravkov E V, Nichol S T, Peters C J, Compans R W. Role of actin filaments in Black Creek Canal virus morphogenesis. J Virol. 1998;72:2865–2870. doi: 10.1128/jvi.72.4.2865-2870.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rozenkranz-Weiss P, Sessa W C, Milstein S, Kaufman S, Watson C A, Pober J S. Regulation of nitric oxide synthesis by proinflammatory cytokines in human umbilical vein endothelial cells. J Clin Invest. 1994;93:2236–2243. doi: 10.1172/JCI117221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmaljohn C S. Bunyaviridae: the viruses and their replication. In: Fields B N, Knipe D M, Howley P M, editors. Fundamental virology. 3th ed. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 649–675. [Google Scholar]
- 33.Shope R E. Bunyaviruses. In: Fields B N, Knipe D M, Chanock R M, Melnick J L, Roizman B, Shope R E, editors. Virology. New York, N.Y: Raven Press; 1985. pp. 1055–1082. [Google Scholar]
- 34.Song W, Lu X, Feng Q. Tumor necrosis factor-alpha induces apoptosis via inducible nitric oxide synthase in neonatal mouse cardiomyocytes. Cardiovasc Res. 2000;45:595–602. doi: 10.1016/s0008-6363(99)00395-8. [DOI] [PubMed] [Google Scholar]
- 35.Stolpen A H, Guinan E C, Fiers W, Pober J S. Tumor necrosis factor and immune interferon act singly and in combination to reorganize human vascular endothelial cell monolayers. Am J Pathol. 1986;123:16–24. [PMC free article] [PubMed] [Google Scholar]
- 36.Tabibzadeh S, Kong Q F, Kapur S, Satyaswaroop P G, Aktories K. Tumor necrosis factor-alpha-mediated dyscohesion of epithelial cells is associated with disordered expression of cadherin/beta-catenin and disassembly of actin filaments. Hum Reprod. 1995;10:994–1004. doi: 10.1093/oxfordjournals.humrep.a136084. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi G W, Montgomery R B, Stahl W L, Crittenden C A, Valentine M A, Thorning D R, Andrews III D F, Lilly M B. Pentoxifylline inhibits tumor necrosis factor-alpha-mediated cytotoxity and cytostatis in L929 murine fibrosarcoma cells. Int J Immunopharmacol. 1994;16:723–736. doi: 10.1016/0192-0561(94)90092-2. [DOI] [PubMed] [Google Scholar]
- 38.Temonen M, Mustonen J, Helin H, Pasternack A, Vaheri A, Holthoffer H. Cytokines, adhesion molecules, and cellular infiltration in nephropathia epidemica kidneys: an immunohistochemical study. Clin Immunol Immunopathol. 1996;78:47–55. doi: 10.1006/clin.1996.0007. [DOI] [PubMed] [Google Scholar]
- 39.Van Campen H. Influenza A virus replication is inhibited by tumor necrosis factor-alpha in vitro. Arch Virol. 1994;136:439–446. doi: 10.1007/BF01321073. [DOI] [PubMed] [Google Scholar]
- 40.Yamaoka J, Kume T, Akaike A, Miyachi Y. Suppressive effect of zinc ion on iNOS expression induced by interferon-gamma or tumor necrosis factor-alpha in murine keratinocytes. J Dermatol Sci. 2000;23:27–35. doi: 10.1016/s0923-1811(99)00062-6. [DOI] [PubMed] [Google Scholar]
- 41.Yan D, Gu X, Wang D, Yang S. Studies on immunopathogenesis in epidemic hemorrhagic fever: sequential observation on activation of the first component complement in serum from patients with epidemic hemorrhagic fever. J Immunol. 1981;127:1064–1067. [PubMed] [Google Scholar]
- 42.Yanagihara R, Silverman D J. Experimental infection of human vascular endothelial cells by pathogenic and non-pathogenic hantaviruses. Arch Virol. 1990;111:281–286. doi: 10.1007/BF01311063. [DOI] [PubMed] [Google Scholar]
- 43.Zaki S R, Greer P W, Coffield L M, Goldsmith C S, Nolte K B, Foucar K, Feddersen R M, Zumwalt R E, Miller G L, Khan A S, Rollin P E, Ksiazek T G, Nichol S T, Mahy B W J, Peters C J. Hantavirus pulmonary syndrome: pathogenesis of an emerging infectious disease. Am J Pathol. 1995;146:552–579. [PMC free article] [PubMed] [Google Scholar]