

SUMMARY
Diabetic kidney disease (DKD), the most common cause of kidney failure, is a frequent complication of diabetes and obesity, and yet to date, treatments to halt its progression are lacking. We analyze kidney single-cell transcriptomic profiles from DKD patients and two DKD mouse models at multiple time points along disease progression—high-fat diet (HFD)-fed mice aged to 90–100 weeks and BTBR ob/ob mice (a genetic model)—and report an expanding population of macrophages with high expression of triggering receptor expressed on myeloid cells 2 (TREM2) in HFD-fed mice. TREM2high macrophages are enriched in obese and diabetic patients, in contrast to hypertensive patients or healthy controls in an independent validation cohort. Trem2 knockout mice on an HFD have worsening kidney filter damage and increased tubular epithelial cell injury, all signs of worsening DKD. Together, our studies suggest that strategies to enhance kidney TREM2high macrophages may provide therapeutic benefits for DKD.
Graphical abstract
In brief
Subramanian et al. identify a macrophage population expressing a TREM2high transcriptional program in the human adult kidney, matching a homologous population in other tissues (adipose, heart, and liver). Trem2high macrophages expand in diabetic kidney disease in both a high-fat-diet-fed mouse model and an independent cohort of patients. Trem2 deletion results in hastened kidney injury in the presence of a high-fat diet, suggesting a role of these macrophages as responders to local tissue injury in obesity- and diabetes-driven kidney injury.
INTRODUCTION
Diabetic kidney disease (DKD) is a serious complication of diabetes in the kidneys and the leading cause of chronic kidney diseases, affecting 400 million patients worldwide.1,2 Further, the high prevalence of obesity is closely linked to the increasing incidence of chronic diseases such as type 2 diabetes (T2D),3 hypertension,4 and kidney failure,5 and the complex interplay between obesity, T2D, and DKD progression is incompletely understood. Recent interventions that help manage the disease (e.g., SGLT2 inhibitors6,7) are encouraging; however, curative kidney-focused therapies to halt DKD are still urgently needed.
The pathophysiology of DKD involves hemodynamic, metabolic, and immune dysfunction. Increasingly, kidney inflammation is thought to be a key contributor to DKD pathogenesis8,9; however, a detailed understanding of dynamic immune cell changes during the early stages of disease is still missing. While previous studies have focused on metabolic dysfunction and epithelial injury in the onset and progression of DKD,8 chronic low-grade inflammation is increasingly implicated.10 Macrophages are particularly salient to promoting inflammation in disease settings11,12; however, to date, studies of the specific role of macrophages in DKD have been limited by challenges in defining and phenotyping specific macrophage subsets in disease progression. Tissue-specific resident macrophages are of particular interest because they adopt distinct cell states in response to local environmental cues. Single-cell transcriptomics (scRNA-seq) has revealed widespread heterogeneity in macrophage cell states, pointing to protective and immunosuppressive roles in different pathologies.13–16 Hence, understanding the heterogeneity of macrophages in DKD could help define their roles in disease progression and prioritize specific subsets for targeted therapies.
An additional challenge in the DKD field is that, historically, the lack of kidney tissue biopsies from patients with DKD has hindered deep mechanistic insights in human tissue and heightened the need for reliable animal models. Notably, there has been controversy around which mouse models incorporate salient features of human DKD.17 Single-cell genomics has been used to chart rich cellular taxonomies of both the mouse14,18–21 and human kidney,8,22–24 including in the context of various diseases.8,24,25 However, there has not been a detailed comparison of the mouse and human kidney at the earliest stages of DKD that could help identify key contributors to disease progression.
Here, we leveraged single-cell transcriptomics to profile cell-type-specific changes in DKD with the goal of understanding macrophage states. In humans, we found previously unrecognized TREM2high macrophages enriched in the kidneys of obese diabetic patients. Single-cell studies at multiple time points along disease progression in two different mouse models, a high-fat diet (HFD) mouse model followed for 90–100 weeks—a long treatment duration that has not been studied before— and a genetic model of DKD (Black and Tan Brachyuric [BTBR] ob/ob), confirmed the expansion of a Trem2high macrophage population in kidneys of HFD mice. Consistent with the hypothesis that the expansion of Trem2high macrophages is protective, HFD-fed Trem2 knockout mice showed significant kidney filter damage and increased injury markers, consistent with early DKD. Taken together, our work points to Trem2high macrophages as a putative node of intervention for DKD.
RESULTS
The human and mouse diabetic kidney at single-cell resolution
We performed single-cell transcriptomics of human kidney tissue derived from the healthy margins of 12 human nephrectomies (US cohort). Of 12 patient-donors, 6 patient-donors were clinically obese, and a subset of three patient-donors had diabetes and specific histologic evidence of early kidney injury, with diffuse and early nodular diabetic glomerulosclerosis (DKD) on light microscopy, determined by an experienced renal pathologist blinded to our study design (Figure 1A; Table S1). We confirmed podocyte loss in tissue from these DKD patients by in situ hybridization (Figures S1A and S1B). Droplet-based single-cell transcriptomic profiling of kidney cortical sections and analysis captured expected parenchymal, stromal, and immune cell classes (48,154 cells; Figures S2A and S2B).
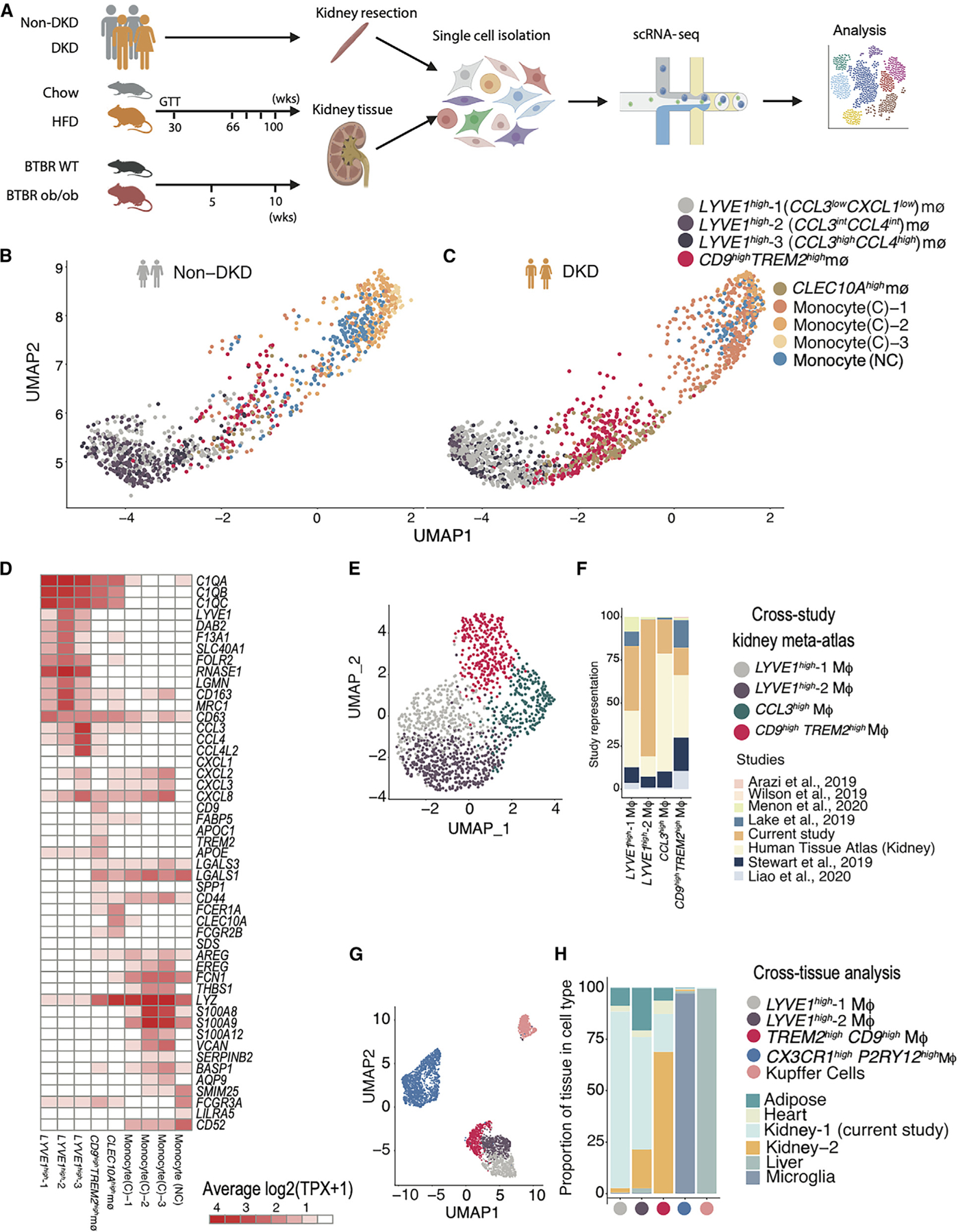
Figure 1. Heterogeneous macrophage subsets in human diabetic kidney at single-cell resolution.
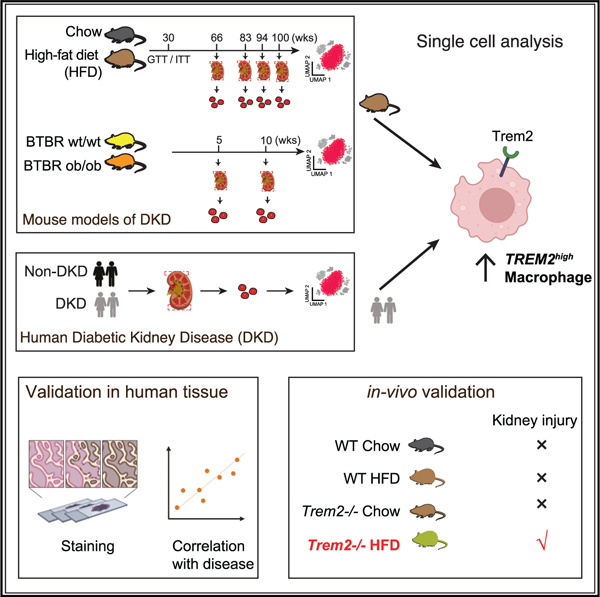
(A) Experimental timeline and sampling outline for kidney tissue from two mouse models and nephrectomy specimens from patients with and without DKD. Kidney cortex obtained at equivalent time points from HFD- and chow-fed mice (aged 66–100 weeks, n = 4 biological replicates in each condition), BTBR ob/ob and BTBR wt/wt mice (aged 5–10 weeks, n = 5 for 5 weeks and N = 3 for 10 weeks), and non-tumor tissue margins of patients undergoing tumor nephrectomy (n = 3 [DKD] and 9 [non-DKD]) was dissociated using the same enzymatic protocol.
(B and C) UMAP visualization of macrophages (MΦ) in the human non-diabetic and diabetic kidney, highlighting macrophage and monocyte populations. Each point represents a cell. Individual populations are represented by distinct colors.
(D) Heatmap visualization of expression programs distinguishing the human macrophage and monocyte populations (columns). Rows are genes that are differentially expressed in each MΦ population. Values (color) represent row-normalized average gene expression in units of log-transformed transcripts per X (log(TPX+1)) (scaling factor, 10,000).
(E) UMAP visualization of MΦs from a human kidney meta-atlas derived from 8 different human kidney scRNA-seq studies (N = 49). Each point represents a cell. Individual populations are represented by distinct colors.
(F) Bar plot of study representation (fraction, y axis) among the human kidney scRNA-seq meta-atlas macrophage subsets (x axis).
(G) UMAP visualization of co-embedding of MΦs from human adipose, heart, liver, microglia, and kidney revealed 5 populations: LYVE1high, CCL3highLYVE1high, CX3CR1highPY2R1high, CD9highTREM2high, and Kupffer cells. Each point represents a cell. Individual populations are represented by distinct colors.
(H) Bar plot of tissue composition (fraction, y axis) among the human cross-tissue macrophage subsets (x axis).
Next, we modeled DKD in mice in (1) a metabolic model of HFD-induced kidney injury and (2) a genetic model of ob/ob leptin deficiency on a BTBR background.26 To best model the long obesity prodrome to DKD in humans, 10-week-old C57BL/6-129 mice were fed an HFD for an extensively long, 100-week period, alongside littermate controls fed a normal chow diet (Figure 1A). We confirmed weight gain, insulin resistance, and kidney dysfunction at 20 and 60 weeks, driven by obesity, dyslipidemia, and endocrine dysregulation27 (Figures S1E–S1N). HFD-fed mice developed progressive kidney disease characterized by glomerular damage, elevated serum creatinine at the terminal time point (Figure S1C, top), and increased urinary albuminuria (Figure S1C, bottom). Histologically, HFD-fed mice developed significant glomerular hypertrophy and nodular sclerotic lesions visible by light microscopy (Figure S1D, bottom left) as well as significant glomerular basement membrane thickening, visualized by electron microscopy (Figure S1D, bottom right), all hallmarks of DKD in humans. In the genetic model, we studied BTBR ob/ob mice, known to exhibit kidney failure by 20 weeks of age.26 Consistent with prior reports,26 and similar to HFD-fed mice, BTBR ob/ob mice at 5 and 10 weeks developed insulin resistance, marked urinary albumin, and glomerular basement membrane thickening as signs of glomerular damage as well as significantly elevated cholesterol (Figures S1O–S1U).
We performed droplet-based single-cell transcriptomics of mouse kidney cortical cells and derived 98,032 cells from the HFD mouse model (Figures S2C and S2D) and 24,484 and 19,643 cells from the BTBR model at 5 (Figures S2E and S2F) and 10 weeks (Figures S2G and S2H), respectively, analyzed independently to capture strain-specific cellular resolution. Cells were annotated with cell type labels at both ‘‘broad cell class’’ and ‘‘granular cell subset’’ resolutions post graph-based clustering and differential gene expression-based marker derivation and based on literature-derived marker genes. At the broad cell class level (e.g., podocyte, proximal convoluted tubule [PCT]), both mouse models captured expected epithelial, stromal, and immune cell types in the kidney. Overall, broad cell classes were consistent between the two mouse models, with 22 shared classes (Table S2). Each strain had its own granular cell subsets (e.g., PCT-1 and PCT-2) most evident among PCT, endothelial, and immune cells. For example, the BTBR model had multiple endothelial cell subsets (e.g., Aplnr+ fenestrated endothelial cells) not found in the HFD model, while the latter had unique PCT transcriptional profiles (e.g., Fgg+ PCTs). Within a strain, broad and granular cell subsets were found under both disease and control conditions.
Human and mouse DKD kidneys had 21 shared cell classes between both mouse models and humans. We looked at transcriptional programs disrupted in DKD by performing a differential gene expression analysis (Figures S2I–S2L) on the broad cell types. We determined genes disrupted in human DKD using a Poisson mixed-effects regression framework that accounted for donor-specific random effects (STAR Methods; Table S3; Figure S2I). Overall, we found more conservation of differentially expressed genes among mouse models than between species (Figures S2J–S2L; Tables S3 and S4), which may be attributed to cross-species differences in the natural history of disease, high variability among human donors, and experimental limitations (smaller numbers of cells recovered and heterogeneity of sample size in human samples). In early human DKD, distal tubular cells and the thick ascending limb displayed the highest numbers of differentially expressed genes, as reported previously (Figure S2I), and also the most cross-species overlap (Figures S2M and S2N). Across both mouse models, podocytes and mesangial cells exhibited significant changes in expression profiles, in line with well-described roles for these cell types in the progression of diabetic kidney injury28 (Figures S2O and S2P). We validated the collagen genes Col4a3 and Col4a2, upregulated in diabetic podocytes (Figures S3A and S3B) and mesangial cells (Figures S3C and S3D), respectively, by in situ hybridization. The podocyte-specific Col4a3 upregulation was reminiscent of the protective COL4A3 allele identified in a genome-wide association study of DKD.29 There were significant numbers of differentially expressed genes in the mouse diabetic PCTs (Figures S2J–S2L), suggesting that these transcriptional changes may reflect early signs of disease that precede histologically detectable damage. Pck1 and Gsta2 emerged as the top upregulated genes in both mouse models (Figures S3E–S3G), validated by immunofluorescence (Figure S3H) and in situ hybridization (Figure S3I). Overall, trends in transcriptional changes were more similar between HFD and 10-week BTBR mice (rather than 5-week mice), with substantial changes in response to injury occurring within the BTBR model from 5 to 10 weeks (Figures S2K and S2L). In summary, while mouse models captured human DKD pathology, trends were largely conserved across strains within species rather than between species, with most cross-species conservation observed in the distal tubules.
Macrophage heterogeneity in the human diabetic kidney
Among immune cells, resident macrophages exhibited the largest number of differentially expressed genes (Figures S2I–S2L). Macrophages are known to play crucial roles in both the progression and resolution of kidney diseases9,30 and offer potential for targeted intervention. We profiled the heterogeneity among human kidney macrophages by iterative clustering and identified five macrophage subsets not previously described in human adult kidney (in addition to classical and non-classical monocytes) and marked by the top upregulated genes: (1) LYVE1high-1 (CCL3lowCXCL1low), (2) LYVE1high-2 (CCL3intCCL4int), (3) LYVE1high-3 (CCL3highCCL4high), (4) a CD9highTREM2high subset, and (5) a CLEC10Ahigh subset (Figures 1B–1D; STAR Methods; Table S5).
The LYVE1high (encoding the protein lymphatic vessel endothelial hyaluronic acid receptor 1) subsets were characterized by high expression of genes associated with homeostatic macrophage populations, including SEPP1, FOLR2, LGMN, CST3, and DAB231,32 (Figure 1D), in line with the LYVE1high CX3CR1low tissue-resident interstitial macrophages recently described in human lung, adipose, and multiple mouse tissues, including heart, fat, and skin.33 LYVE1high-2 kidney macrophages expressed all markers found in LYVE1high heart macrophages34 (Figure S4B). The LYVE1high-2 and LYVE1high-3 subsets were distinguished from the LYVE1high-1 subset by specific chemokine expression (CCL3, CCL4, CCL4L2, and CCL2), suggesting an activated state, similar to previously described senescent-like microglia expressing inflammatory genes.35
The CD9highTREM2high cells showed induction of TREM2 (encoding the protein triggering receptor expressed on myeloid cells 2), SPP1, CD9, FABP5, APOE, APOC1, LGALS3, LGALS1, and cathepsins (Figure 1D), reminiscent of the lipid (or scar)-associated macrophage (LAM/SAM) signature described in human adipose tissue and liver in the setting of inflammation.36,37 CD9highTREM2high cells are suggested to be monocyte derived.36,37 Human kidney macrophages, upon visualization on a two-dimensional uniform manifold approximation and projection (UMAP) manifold, lay along a spectrum spanning monocytes, CD9highTREM2high macrophages, and different subsets of homeostatic LYVE1high macrophages (Figures 1B and 1C). In sum, these kidney CD9highTREM2high cells were transcriptionally situated between monocytes and homeostatic macrophages.
LYVE1high and CD9highTREM2high macrophage subsets were present in tissue from each of 12 human donors (Table S5) in our study (US cohort). To rule out any study bias, we further profiled macrophages from a recently assembled kidney scRNA-seq meta-atlas across 7 additional studies38 spanning 49 human donors in total. In support of kidney-specific macrophage populations, LYVE1high and TREM2high populations were identified in these independent datasets (Figures 1E, 1F, and S4A). Additionally, we analyzed the Kidney Precision Medicine Project (KPMP) scRNA-seq dataset composed of 45 donors and found a prominent population of TREM2high macrophages with prevalence in DKD (Figures S4C–S4E).
We next asked whether the macrophage states we identified in the human kidney are present in other human tissues. Co-embedding our kidney macrophage profiles with macrophages from scRNA-seq studies of adipose tissue, kidney, heart, liver, and brain (microglia)39,40 resulted in five macrophage populations (Figures 1G and 1H; STAR Methods). These included 3 TREM2high populations: CX3CR1highP2RY12high microglia, CD5LhighMARCOhighTREM2high liver Kupffer cells, and the CD9highTREM2high macrophages shared across adipose tissue, heart, and kidney. The LYVE1high and CCL3highLYVE1high populations were present in kidney, liver, as well as heart and adipose tissue.33 We concluded that the distinct LYVE1high and TREM2high macrophage populations we identified in human adult kidney shared signatures across several tissues.
A Trem2high macrophage population tracks with kidney disease progression in obese diabetic mice
To profile the changes in macrophages in mouse DKD, we iteratively clustered the macrophages in each mouse model to identify multiple subsets, broadly characterized as ‘‘resident’’ and ‘‘infiltrating’’ populations, using reported signatures19(Figure S4F) with subset-specific marker genes (Figures 2A–2D and S4G–S4J; Table S6).
Figure 2. Macrophage heterogeneity in the mouse diabetic kidney.
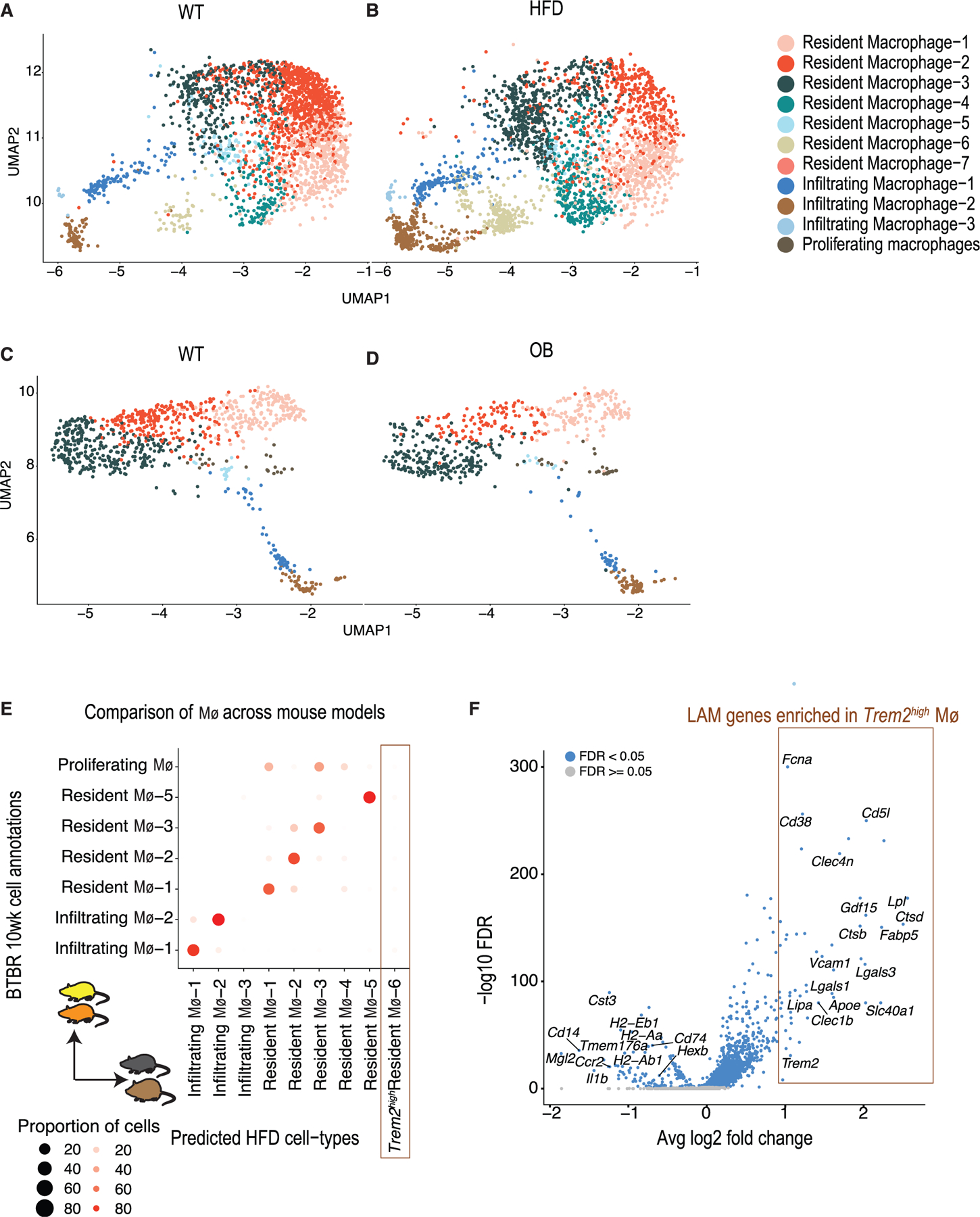
(A and B) UMAP visualization of macrophages recovered from the kidneys of (A) chow-fed and (B) HFD-fed mice (n = 4 biological replicates in each condition). Each point represents a cell. Individual populations are represented by distinct colors.
(C and D) UMAP visualization of macrophages identified in the kidneys of 10-week-old BTBR (C) wt/wt and BTBR (D) ob/ob mice (n = 3 in each condition). Each point represents a cell. Individual populations are represented by distinct colors.
(E) Comparison of macrophage populations between two mouse strains, indicating shared and unique cell populations. Comparisons were performed by training a classifier on the HFD strain/model macrophages (training data) and predicting labels on the 10-week-old BTBR strain/model macrophage data (test data). Each BTBR macrophage is assigned a predicted HFD macrophage label. The plot represents the proportion of each BTBR macrophage subset (y axis) that was assigned an HFD macrophage label (y axis).
(F) Volcano plot showing differentially expressed genes in the Trem2high population (resident Mφ−6) compared to other kidney macrophages of chow- and HFD-fed mice. Each point represents a gene; the y axis represents the −log10(false discovery rate) and the x axis the average log2 fold change.
We cross-referenced macrophage subsets between the two mouse models using a multi-class random forest classifier (Figure 2E; STAR Methods). While most resident and infiltrating macrophage populations corresponded across strains, the Trem2high resident macrophages-6 (M4-6) emerged as a population specific to the HFD-fed mouse model (Figure 2E). When contrasted with other macrophage populations, the resident Trem2high M4-6 population expressed a distinct and specific signature that included the genes Trem2, Lpl, Fabp5, Lipa, Ctsb, Lgals3, Lgals1, Nceh1, Cd63, and Cd36 (Figure 2F), reported previously as important signature genes for LAMs in the adipose tissue of HFD-fed obese mice.36 Intriguingly, this Trem2high population also expressed Cd5l (Figure 2F), a key mediator of lipid synthesis, and its transcriptional regulator Nr1h3, a previously reported biomarker for DKD.41,42 On the other hand, inflammatory genes such as Il1b and antigen-presenting major histocompatibility complex genes were downregulated in these Trem2high macrophages. Macrophages within the BTBR model across time points had high correspondence (Figure S4K).
Visualization by potential of heat diffusion for affinity-based transition embedding (PHATE) analysis70 of the Trem2high macrophages in HFD-fed mice projected them between infiltrating and other resident macrophages (Figure 3A), mirroring the transcriptional trajectory in human kidney. Further, these Trem2high resident Mφ−6 were more prevalent in older and obese HFD-fed diabetic mice compared to chow-fed mice (Figures 3B and 3C). This Trem2high macrophage cluster increased in frequency with disease progression (Poisson regression, Figure 3D). Differentially expressed genes in disease in the Trem2high macrophage cluster were enriched for cholesterol metabolism and, additionally, peroxisome proliferator-activated receptor signaling and lysosomal pathways, as has also been implicated in adipose in the context of obesity (Figure 3E). Two additional macrophage subsets, resident Mφ−3 and Mφ−4, also increased in frequency with disease progression and had a sizable number of cells expressing Trem2 but not the rest of the LAM program. Subsets of resident macrophages (albeit with no detectable Trem2 expression) were also increased in 10-week BTBR ob/ob mice, as shown by in situ hybridization (mean C1qa+/b+ macrophages: 2.7% in BTBR wt/wt mice; 4.8% in BTBR ob/ob, p < 0.01; Figures 3F and 3G).
Figure 3. Expansion of a Trem2high macrophage population in kidneys of obese diabetic mice.
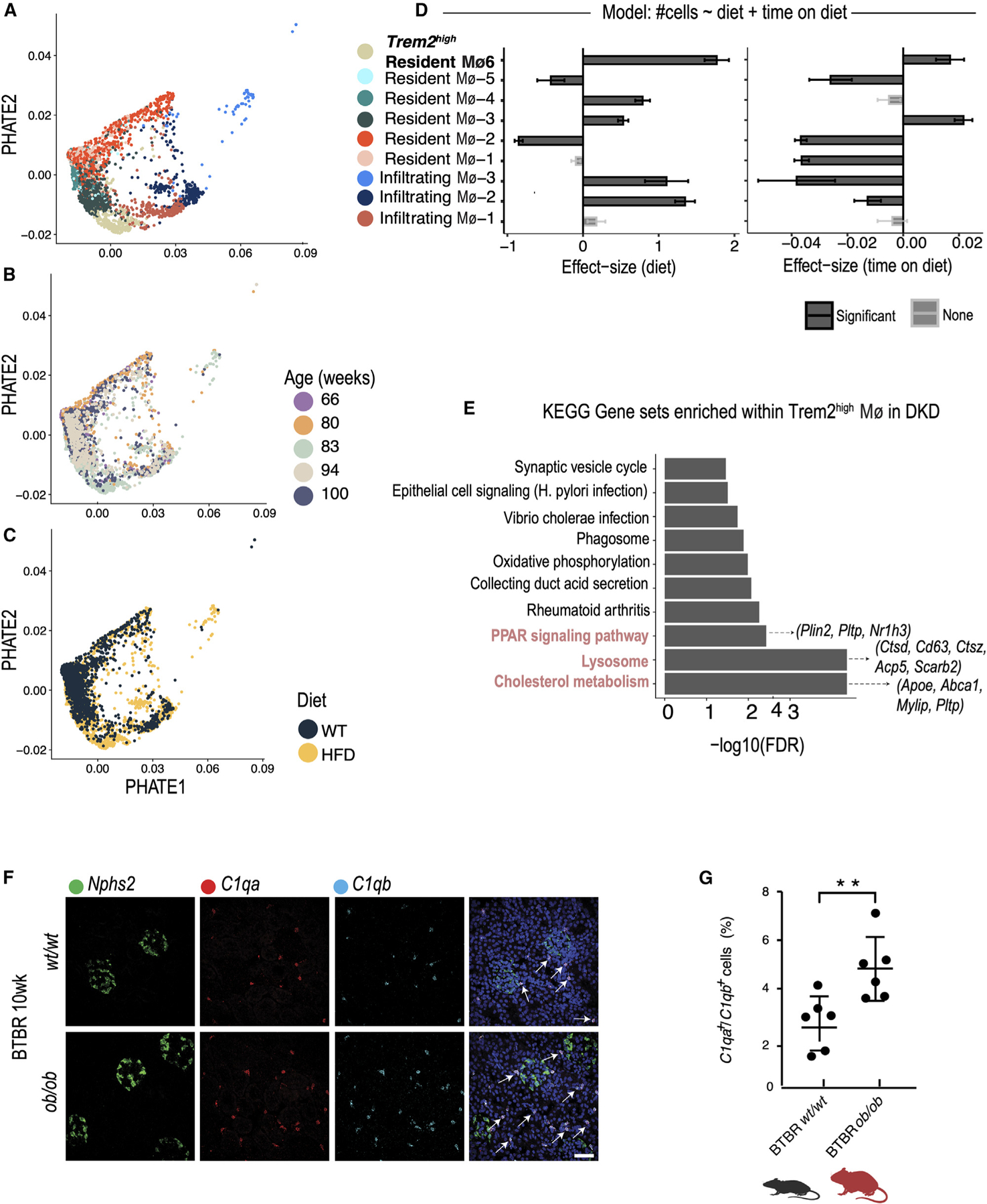
(A–C) Potential of heat diffusion for affinity-based transition embedding (PHATE) visualization of macrophage populations in the kidneys of chow- and HFD-fed mice (n = 4 biological replicates in each condition). Macrophages are colored according to individual cluster (A), age (B), and diet (C).
(D) Results of a Poisson regression fit to estimate the effect of diet (left) and age of mice (right) on the proportion of macrophage subsets between conditions. Standard error bars are shown.
(E) Bar plot of Kyoto Encyclopedia of Genes and Genomes pathway gene sets enriched in genes differentially expressed in DKD in the Trem2high population (resident Mφ−6) in the HFD model.
(F) In situ HCR using probes for C1qa (red) and C1qb (cyan) to identify resident macrophages in kidneys of 10-week-old BTBR wt/wt and ob/ob mice. The Npsh2 probe (green) was used to identify podocytes and provide spatial orientation.
(G) Quantification of C1qa+C1qb+-expressing macrophages, expressed as a percentage of all cells, in kidneys of 10-week-old BTBR wt/wt (mean 2.7% ± 0.4%, n = 6) and BTBR ob/ob (mean 4.8% ± 0.5%, n = 6) mice. p values are derived from an unpaired Student’s t test, **p < 0.01. Arrows indicate individual cells.
TREM2high macrophages are more abundant in kidneys of obese diabetic humans
To further explore TREM2high macrophages in humans and HFD-fed mice with DKD, we compared macrophage composition between mice and humans by using a multi-class random forest classifier (STAR Methods) trained on the subsets of human macrophages and monocytes to classify mouse macrophages in the HFD model. We found that most of the Trem2high mouse macrophage population (Mφ−6) were assigned a corresponding human Mφ prediction of CD9highTREM2high Mφ, suggesting strongest correspondence (Figure 4A). Comparing the average gene expression profiles of the human TREM2high populations with the corresponding mouse HFD Trem2high populations (Figure 4B) showed high correlation (Spearman coefficient = 0.79). Species-specific divergence was noted for APOC1 and CD163 (highly expressed in human TREM2high Mφ), and Lpl and Cd5l (highly expressed in mouse Trem2high macrophages). Overall, this analysis suggested strong cross-species correspondence in TREM2high macrophages.
Figure 4. Expansion of a TREM2high macrophage population in kidneys of obese diabetic humans.
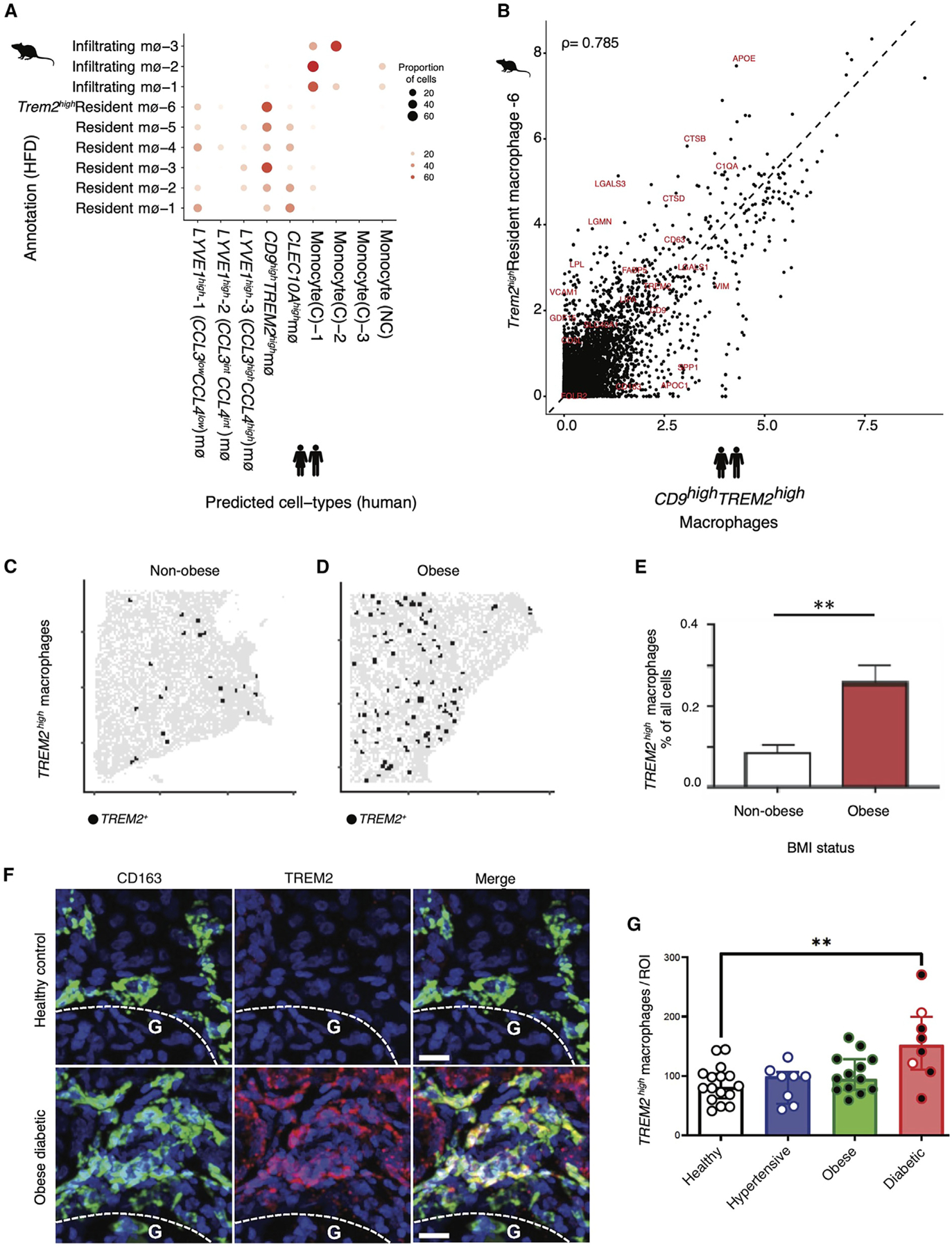
(A) Comparison of macrophage populations between human and mouse. Comparisons were performed by training a classifier on the human macrophages (training data) and predicting labels on the HFD mouse macrophages (test data). Each HFD macrophage is assigned a predicted human macrophage label. The plot represents the proportion of each HFD macrophage subset (y axis) that was assigned a human macrophage label (x axis).
(B) Correlation of TREM2high populations between HFD mouse (y axis) and human kidney (x axis) shows shared and divergent genes. Each data point represents the average normalized and log-transformed expression of the gene in units of log(TPX+1). Spearman correlation is indicated.
(C and D) Digital graphic representation of TREM2high macrophages in nephrectomy tissue from non-obese and obese patients in the US cohort, showing one representative tissue from one non-obese (C) versus an obese (D) patient donor. Red circles denote TREM2high macrophages, with all cells (nuclei) shown in gray. These digital representations make it easier to visualize these otherwise small and hard-to-discern cells in large sections of human kidney tissue.
(E) Quantification of TREM2high macrophage populations in nephrectomy tissue from non-obese (mean 0.09% ± 0.02%, n = 5) and obese (mean 0.26% ± 0.04%, n = 6) patients in the US cohort. p values are derived from an unpaired Student’s t test; **p < 0.01.
(F) Representative images of fluorescence microscopy of kidney tissue from the UK cohort, including a healthy control (top) and diabetic obese patient (bottom), showing staining for the macrophage marker CD163 (green), TREM2 (red) and cell nuclei (Hoechst, blue). Scale bars, 20 μm. G, kidney cortex; indicating that most TREM2 macrophages are located in the medulla.
(G) Quantification of macrophage counts in the UK cohort per region of interest between healthy (n = 16), hypertensive (n = 8), obese (n = 13), and obese/diabetic patients (n = 8). White dots, non-obese; black dots, obese patients (BMI > 30) (see Table S7 for clinical data); p < 0.001.
Next, we quantified TREM2high macrophages in human kidney tissue by an independent method. TREM2high macrophages were detected by immunofluorescence microscopy throughout human kidney nephrectomy tissue sections from 12 patients (US cohort; Figures 4C–4E). When comparing human kidney tissue from patients with high versus normal body mass index (BMI),43 we observed a significant increase in TREM2high macrophages in kidney tissue from obese patients (mean 0.26% ± 0.04%, n = 6) as compared to kidney tissue from patients with a normal BMI (mean 0.09% ± 0.02%, n = 5, p < 0.01; Figures 4C–4E). Most of these patients had preserved kidney function, suggesting that expansion of TREM2high macrophages may coincide with the earliest signs of kidney injury.
We next quantified TREM2high macrophages in an independent validation cohort from the UK. To this end, we stained for the macrophage marker CD163 and for TREM2 in kidney tissue from 45 patient-donors in a UK tissue repository (comprising discarded donor kidney tissue; STAR Methods). In this validation cohort, we found that patients who were obese and diabetic had a significant expansion of TREM2 macrophages in their kidneys as compared to those with normal BMI who were hypertensive or normal controls (Figures 4F and 4G; p < 0.001; healthy, n = 16; hypertensive, n = 8; obese, n = 13) and obese and diabetic patients (n = 6 of 8 total diabetic patients; white dots, non-obese; black dots, obese patients; BMI > 30) (see Table S7 for clinical data). The few obese patients without diabetes showed a trend toward increase in TREM2high macrophages compared to controls, and the abundance of TREM2high macrophages generally correlated with increasing BMI across the UK cohort (Figures S4L and S4M). Taken together, we identified and independently validated a population of TREM2high macrophages in both human and mouse kidneys that tracked with kidney disease progression in mice.
Trem2 deletion exacerbates HFD-induced kidney injury in mice
Trem2, encoding the protein triggering receptor expressed on myeloid cells 2, is uniquely expressed by myeloid cells in the mouse kidney (Figure S4N). To explore the functional relevance of Trem2high macrophages in DKD, we studied wild-type (WT) and Trem2-deficient (Trem2−/−) mice (Figures S5A and S5B) with and without DKD. Specifically, we fed WT and Trem2−/− mice either chow or an HFD for a total of 6 months (Figure 5A). Interestingly, HFD treatment could induce Trem2 expression in WT but not in Trem2−/− mice (Figure 5B). Trem2−/− mice exhibited an increased level of glucose intolerance compared with WT mice after the HFD treatment (Figure S5C). Interestingly, HFD-fed Trem2−/− mice exhibited strong structural defects in the kidney filter, as evidenced by increased foot process effacement, visualized by transmission electron microscopy (TEM) (Figure 5C). Indeed, HFD-fed Trem2−/− mice had severe kidney filter dysfunction, as evidenced by the detection of significant proteinuria (the spilling of essential proteins from the blood into the urine) compared to HFD-fed WT mice (Figure 5D). Beyond exhibiting the structural and functional hallmarks of kidney filter damage, the kidney tissues from Trem2-deficient HFD-fed mice was notable for significant upregulation of pro-inflammatory markers such as interleukin-1β (IL-1β) and tumor necrosis factor (TNF)44,45 as well as the kidney tubular injury marker KIM-1 (Havcr1), in contrast to tissues from chow-fed Trem2−/− controls and HFD-fed WT control mice that had no detectable upregulation of these injury markers (Figure 5E). In additional TEM studies, we observed decreased mitochondrial numbers and morphological changes in kidney tubular epithelial cells of HFD-fed Trem2−/− mice (Figure 5F, red arrows). TEM quantification results showed that the total number of mitochondria in HFD-fed Trem2−/− mice was significantly decreased compared to that of chow-fed Trem2−/− mice (Figure 5G). Strikingly, after the HFD treatment, the number of swollen/vacuolated mitochondria was further increased in Trem2−/− mice compared to WT mice (Figure 5H), indicating that Trem2 has a protective effect. In addition to weight gain and increased circulating glucose (Figures S5D and S5E), consistent with early obesity-induced diabetes, Trem2-deficient HFD-fed mice had dysregulated lipid metabolism, as evidenced by elevated serum triglycerides and cholesterol (Figures S5F and S5G). We observed no changes in serum creatinine and blood urea nitrogen (BUN), similar to early DKD in humans who initially present with proteinuria but no elevations in creatinine or BUN.
Figure 5. Trem2 deletion exacerbates HFD-induced kidney damage in mice.
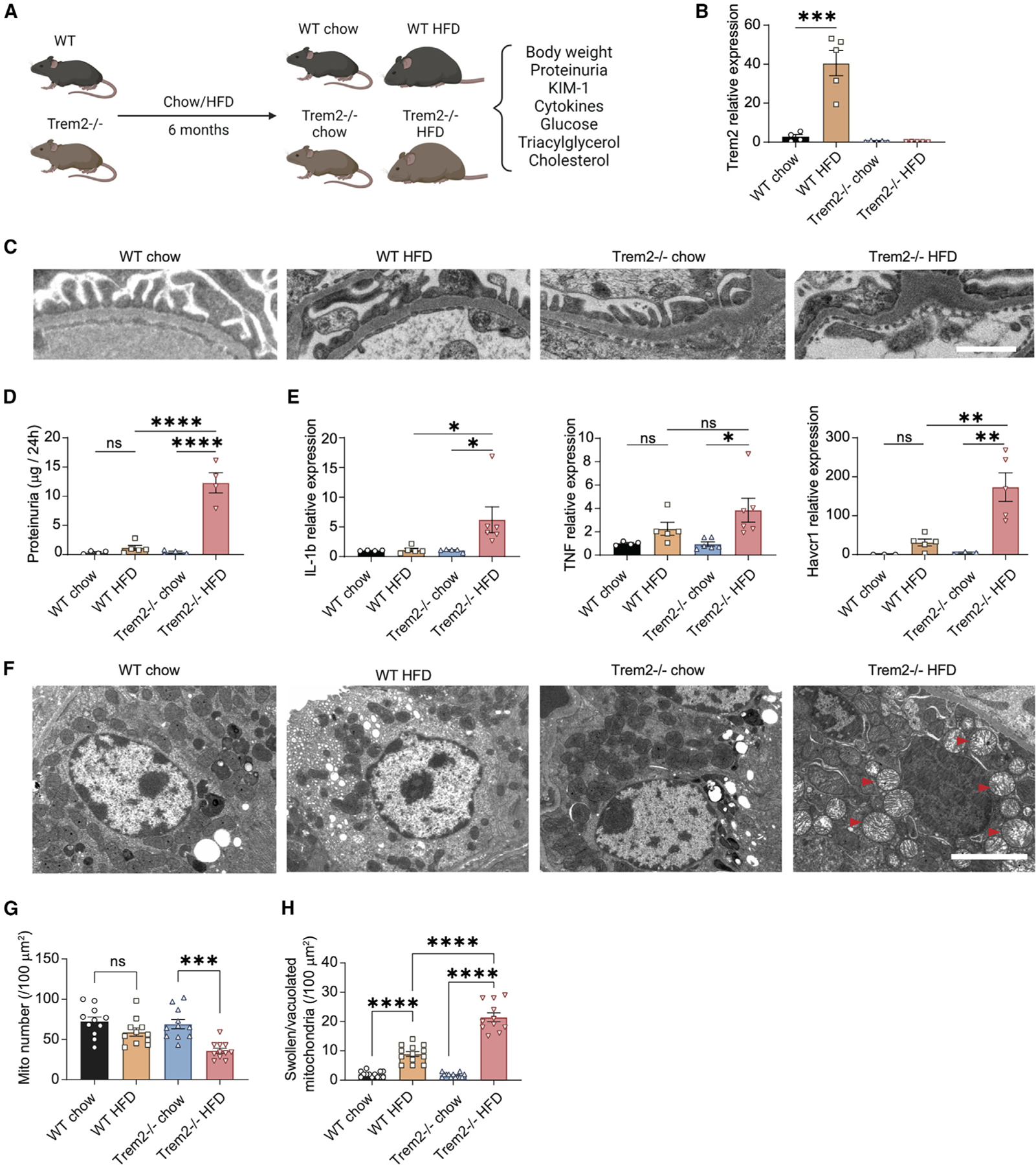
(A) Flow chart of the animal experimental design.
(B) qPCR quantification of the mRNA expression levels of Trem2 in WT chow, WT HFD, Trem2−/− chow, and Trem2−/− HFD mice. ***p < 0.001. n = 5 biological replicates.
(C) Transmission electron microscopy (TEM) image showing foot process effacement in Trem2−/− HFD-treated mice.
(D) Histograms of the proteinuria levels for all four groups. ****p < 0.0001.
(E) qPCR quantification of the mRNA expression levels of Il1b, Tnf, and Havcr1 for all four groups. *p < 0.05, **p < 0.01.
(F) TEM image showing tubular cell mitochondrial injury in Trem2−/− HFD-treated mice. Red arrows indicate mitochondrial swelling.
(G and H) Quantification results of the numbers of total mitochondria (G) and the swollen/vacuolated mitochondria (H) of the renal tubular cells from WT and Trem2−/− mice treated with chow and HFD.
DISCUSSION
We performed a high-resolution, cross-species comparison between mouse and human kidney that revealed the shared transcriptomic architecture and cellular changes driven by obesity and diabetes, with a special focus on the macrophage compartment. A critical feature of our study is that we studied the effects of HFD in mice for 90–100 weeks, a longer period than in most studies to date. Importantly, this approach allowed us to distinguish specific populations of resident and infiltrating macrophages, including a distinct population of Trem2high macrophages in mouse kidney that were subsequently cross-validated in human kidney tissue from obese and diabetic patients.
The significance of resident macrophages, and especially TREM2high cells, in maintaining tissue homeostasis is increasingly being recognized.31,32 These cells play a critical role in fending off inflammatory injury in adipose tissue, liver, and brain.32,36,39 Recent work has revealed the TREM2 receptor as a major pathology-induced immune signaling node that senses tissue damage and activates robust immune remodeling as an adaptive response to injury.46 Trem2-deficient mice have increased susceptibility to inflammation, obesity, and neurodegenerative disease.36,47,48 In this study, we identified TREM2high macrophages in the human adult kidney, and we showed that their absence worsens features of early DKD in mice. Our findings therefore expand our understanding of the role of Trem2high macrophages by showing that they may serve a protective role in the kidney. Specifically, our data suggest that Trem2high macrophages in the kidney may coordinate the local response to injury, leading to a hypothesis that bolstering their expansion may serve to protect kidneys from the harmful sequelae of obesity-driven DKD.
Emerging experimental and epidemiological evidence implicates obesity as an independent risk factor for kidney disease.49 A recent study drawing from 10,547 patients showed that elevated BMI (over 25 kg/m2) accelerates the progression of kidney complications in patients with T2D (ClinicalTrials.gov: NCT00145925).50 A limited number of studies have prospectively examined the relationship between BMI and adverse kidney events among individuals with T2D.51–53 In support of the notion that reversal of obesity benefits kidney health, bariatric surgery improves kidney function.54–56 Our findings encourage follow-up studies focused on preserving and promoting TREM2high macrophages in order to protect kidney function in patients with obesity and diabetes.
In sum, our cross-species comparison of mouse and human kidney cells provides a putative target (TREM2) and a preclinical model (HFD-fed mice) for proof-of-concept studies with direct translatability to human DKD.
Limitations of the study
We note limitations of our study design that must be considered in future work. First, our study evaluated the effects of global deletion of Trem2, a myeloid-specific gene, in mouse kidneys, but we acknowledge that there may be cell crosstalk with myeloid cells from other tissues that could influence kidney disease progression. Second, we did not investigate the detailed mechanism by which Trem2high macrophages protect kidney cells from injury. Many studies have indicated that Trem2 enhances macrophage phagocytosis and reduces inflammation16,19; we therefore speculate that the increased Trem2 macrophages protect kidneys by decreasing inflammation and by removing excess lipids and cell debris via phagocytosis. Future in vitro and in vivo experiments will further explore these detailed mechanisms. Finally, given the focus of our study on the effects of an HFD on the kidneys, we opted to use HFDs and chow diets used in prior published studies.27 Nevertheless, we acknowledge that different diets may influence the gut microbiota, which, in turn, may have effects on kidney injury and disease progression, which our study did not take into account.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Please contact Anna Greka, agreka@broadinstitute.org.
Materials availability
Materials available by contacting lead/corresponding authors.
Data and code availability
We have deposited the mouse scRNA-seq data on NCBI GEO (accession GSE205594).
We have deposited the human scRNA-seq data in the single cell portal (SCP): https://singlecell.broadinstitute.org/single_cell/study/SCP2188/human-and-mouse-dkd-atlas#.
We have deposited all relevant code/scripts in GitHub (https://github.com/ayshwaryas/DKD_paper) with all the analysis described in sufficient detail for others to reproduce.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
All animal experiments were performed in accordance with the guidelines established and approved by the Animal Care and Use Committee at the Broad Institute (Animal Protocol No. 0061-07-15-1) and Brigham and Women’s Hospital, Harvard Medical School (Animal Protocol No. 01538).
HFD model
C57BL/6J mice (JAX stock #000664) were purchased from Jackson Laboratory (Bar Harbor, USA) and backcrossed with 129S1 mice (JAX stock #002448) for at least three generations, resulting in a C57BL/6J-129 mixed background strain. All mice were housed at 25°C with a 12-h light-dark cycle in an AALAC-approved animal facility at Brigham and Women’s Hospital, Harvard Medical School. For the high-fat diet-induced DKD mouse model, adult C57BL/6J-129 mice were fed with a standard chow or high-fat diet ad libitum, starting at 8–10 weeks of age. The high-fat diet was purchased from Envigo (TD.93075 dough), which contains 55% per Kcal fat (23% saturated, 32% trans, 30% monounsaturated, 12% polyunsaturated), and 9.6% sucrose. To confirm the response and diabetic status of the HFD-fed mice, body weights were measured every two weeks after the commencement of the HFD. The intraperitoneal glucose tolerance test (IPGTT) was performed at 30 weeks of age and the intraperitoneal insulin tolerance test (IPITT) one week later. The 24-h urine collection and scRNA-seq experiments were performed at 70, 80, 90, and 100 weeks of age.
Controls for the HFD model
We opted to use as a control the chow diet that has been standard practice in multiple published studies looking at high fat diet-fed mouse models of disease (PMID: 25303528; PMID: 15338127). Our mice developed hyperinsulinemia, hyperglycemia, and glomerular hypertrophy, as described in PMID: 15338127. Therefore, the similarity between our mouse phenotypes and the phenotypes described in PMID: 15338127 confirm that the chow diet used in these studies is an appropriate control diet for studying the injurious effects of a high fat diet on the kidney.
BTBR model
3- and 6-week-old BTBR w/w and ob/ob mice were purchased from the Jackson Laboratory and housed as above at Brigham and Women’s Hospital, Harvard Medical School or the Broad Institute. The intraperitoneal glucose tolerance (IPGTT) and intraperitoneal insulin tolerance (IPITT) tests were performed in both BTBR w/w and ob/ob mice at 4 and 8 weeks of age (along with cholesterol levels), approximately 1 week prior to scRNA-seq experiments. The 24-h urine collection was performed at 5 and 10 weeks of age at the time of scRNA-seq.
Human kidney tissue samples
Kidneys donated for transplantation, but unsuitable for implantation (due to damage to the arterial patch, suspicion of donor malignancy, or adverse retrieval biopsy score) were used. All analysis of human material was performed in the UK; ethical approval was granted by the local ethics committee (REC12/EE/0446) and the study was also approved by NHS Blood and Transplant (NHSBT). All kidneys had a cold ischemic time of less than 30 h prior to processing. Demographic information is shown in Table 4.
METHOD DETAILS
WT and Trem2−/− experiments
Trem2−/− mice were generated by Cyagen (China) using CRISPR/Cas9 and the strategy described in Figure S10A. Genotyping primers were listed in Table S8. The mice were bred and kindly provided by Prof. Yingping Xu at the Institute of Dermatology and Venereology Dermatology Hospital, Southern Medical University. Littermate WT and Trem2−/− mice were fed with a normal chow diet and water ad libitum, and housed at the Animal Facility of Sun Yat-Sen University at a controlled condition (25°C with 60%–65% humidity and a 12-h dark/light cycle). At 6–8 weeks age, WT and Trem2−/− male mice were fed with a normal chow or high-fat diet for 6 months. Mice were then sacrificed and evaluated for proteinuria, serum creatinine, BUN, glucose, triacylglycerol, and cholesterol. Kidney tissues were fresh-frozen for qPCR experiments. qPCR primers were listed in Table S8. All the experimental procedures were in strict agreement with the Guidelines for the Care and Use of Laboratory Animals (NIH Publication, 8th Edition, 2011). All animal protocols were approved by the Institutional Review Boards of the Animal Care and Use Committees of Sun Yat-Sen University (A03LL202303240001).
24-H urine collection and urine albumin assay
Male mice were placed at metabolic cages for 24 h urine collection. After measuring the volumes of mouse urine samples, centrifuge samples at 5,000 rpm, 600 g for 10 min at 4°C. Then extract the supernatants and place them in new 1.5 mL tubes with 5x loading buffer. Denature the urine samples 5 min at 95°C. Urine samples were then run through a Polyacrylamide gel with standards according to our previously published protocol.57 The albumin levels were imaged by CBB staining and quantified by a colorimetric method using ImageJ software.
Glucose tolerance test and insulin tolerance test
Prior to the GTT test, mice were fasted for 16 h and transferred to a procedure room midway through the light phase of the light-dark cycle. Blood was obtained from a tail cut and assessed for baseline glucose levels using a One-touch UltraMini (Lifescan) glucometer. The mice then received 1 g/kg body weight of a 100 mg/mL glucose solution (Sigma, #G8769) in sterile PBS, delivered by i.p. injection. At 15, 30, 60, 90, and 120 min after the administration of glucose, dried blood and tissue were quickly removed from the tail wound and blood was collected again for glucose quantification. Prior to the ITT test, mice were fasted for 5 h and transferred to a procedure room midway through the light phase of the light-dark cycle. The mice received 1 U/kg body weight of humulin N solution (Eli Lily #HI-310) in sterile PBS, delivered by i.p. injection. Blood glucose levels were assessed in the same way as GTT, before and 15, 30, 60, 90, and 120 min after the administration of the insulin solution.
Serum parameters measurement
Whole blood was collected in a 1.5 mL centrifuge tube. After collection, the whole blood was allowed to clot by leaving it undisturbed at room temperature for 15 min. The sample was then centrifuged at 1,000 x g at 4°C for 10 min. Following centrifugation, the supernatant was immediately transferred into a clean polypropylene tube on ice. If the serum was not analyzed immediately, it was divided into 100 μL aliquots and stored at −80°C. The levels of insulin, adiponectin, leptin, total cholesterol, and triglycerides were measured by the Animal Metabolic Physiological Core at Beth Israel Deaconess Medical Center. The levels of BUN and creatinine were measured by the Biochemical Genetics and Metabolic Disease Laboratory at the University of Alabama (Birmingham).
Histology
Mouse kidney tissue was fixed in 4% PFA overnight, embedded in paraffin and sectioned at 5 μm. Samples were deparaffinized and hydrated to water. Sections were oxidized in 0.5% periodic acid solution (Sigma-Aldrich) for 5 min, rinsed with distilled water and then placed in Schiff reagent (Sigma-Aldrich) for 15 min. They were then washed in tap water for 5 min and counterstained with Mayer’s hematoxylin (Sigma-Aldrich) for 1 min. Subsequent washing with tap water for 5 min was followed by dehydration and coverslip mounting using a synthetic mounting medium.
Light microscopy was performed on select formalin-fixed, paraffin-embedded sections of uninvolved renal parenchyma of tumor nephrectomy specimens according to standard clinical pathology operating procedures. 4um sections were stained for periodic acid schiff (PAS), hematoxylin and eosin (H&E), trichrome and Jones methenamine silver. Images were taken using an Olympus BX53 microscope equipped with an Olympus DP73 camera, and processed using Cell Sens Standard software (Olympus).
Immunostaining
kidney sections (6 μm) were prepared using a cryostat. Samples were fixed with 4% PFA at room temperature and permeabilized with 0.1% PBS-Triton X for 10 min and blocked with 3% BSA at room temperature for 1 h. Samples were then incubated with primary antibodies (1:250 dilution for rabbit anti-Pck1 (# 720266, ThermoFisher Scientific, Waltham, USA) and rabbit anti-Gsta2 (# PA5-96757, ThermoFisher Scientific) at room temperature for 1 h and washed 3 times with 0.1% PBS-Tween 20 for 10 min. Appropriate Alexa secondary antibodies (1:250 dilution for goat anti-rabbit IgG 594, # A32740, ThermoFisher Scientific) were used to visualize the proteins. Images were taken using an Olympus FV-1000 confocal microscope (Olympus America Inc, Center Valley, USA) and relative signal intensities were quantified using ImageJ software.
Electron microscopy
Mouse kidney samples were prefixed with 4% PFA and then washed with PBS, prior to/following 2.5% glutaraldehyde (in PBS, pH 7.4) fixation at 4°C overnight. After washing in cacodylate buffer, kidney fragments were then postfixed in 1% osmium tetroxide for 1 h, dehydrated through ascending grades of alcohol, and embedded in Epon resin (Electron Microscopy Science, Hatfield, PA). Ultrathin sections (60–100 nm) were cut on an EM UC7 ultramicrotome (Leica Microsystems, Mannheim, Germany), stained with uranyl acetate and lead citrate, and examined with TEM (Morgagni 268D, Philips, Brno, Czech Republic).
Mitochondria quantification
The morphological scores of mitochondria in renal tubular cells were quantified according to a previous study (PMID: 24361555). For each group, 8–10 TEM sections from 3 animals were collected and the swollen/vacuolated mitochondria were analyzed from at least 5 different microscopic fields. The microscopic fields were chosen and quantified in a double-blind fashion. The average numbers of swollen/vacuolated mitochondria of each group were calculated using ImageJ software.
HCR and tissue collection
PBS perfused kidneys from wild-type mice of a 129/C57BL/6J hybrid background, BTBR w/w, and BTBR ob/ob mice were covered in OCT frozen in liquid nitrogen cooled isopentane. Thin sections of tissue (10μm) were mounted in 24 well glass bottom plates (82050–898, VWR) coated with a 1:50 dilution of APTES (440140, Sigma). All HCR v3 reagents (probes, hairpins, and buffers) were purchased from Molecular Technologies. The following solutions were added to the tissue: 10% formalin (100503–120, VWR) for 15min, 2 washes of 1x PBS (AM9625, ThermoFisher Scientific), ice-cold 70% EtOH at −20°C for 2 h to overnight, 3 washes 5x SSCT (15557044, ThermoFisher Scientific with 0.2% Tween 20), Hybridization buffer (Molecular Technologies) for 10min, probes in Hybridization buffer overnight, 4 15min washes in Wash buffer (Molecular Technologies), 3 washes 5x SSCT, Amplification buffer (Molecular Technologies) for 10min, heat denatured hairpins in Amplification buffer overnight, 3 15min washes in 5x SSCT (1:10,000 DAPI TCA2412-5MG, VWR in the second wash), and storage/imaging in 5x SSCT. Imaging was performed on a spinning disk confocal (Yokogawa W1 on Nikon Eclipse Ti) operating NIS-elements AR software. Image analysis and processing was performed on ImageJ Fiji.
Imaging and analysis of mRNA expression amplified by in situ HCR
All data presented was acquired from multiple, distributed fields of view within each of the probed samples using either a Nikon Plan Fluor 40x 0.75 NA (Col4a3, Col4a2) or a Nikon Plan Apo 10×0.45 NA Objective (Gsta1/2, Pck1). At each region of interest, 15 z series optical sections were acquired with NIS Elements AR 4.51 software (Nikon, Tokyo, Japan) using a step size of 0.5 μm. The gamma, brightness, and contrast for all fluorescence micrographs were adjusted in NIS Elements software identically for compared sets of images.
Image analysis was performed on the maximum z-projections of the images acquired using custom MATLAB scripts (see https://github.com/ssturner-broad/HCRImageAnalysis.git) and the Image Processing Toolbox (MathWorks, Natick, USA). Briefly, the fluorescence signal corresponding to a gene of interest was first isolated and the background was subtracted. Fluorescence intensity was quantified as the integrated mean intensity of pixel values in a given region of interest. To compare fluorescence intensity quantitatively between experimental conditions, the intensity values for each field of view were normalized by the estimated number of nuclei identified in z-projections of paired DAPI channels. Cell density estimations were performed by detecting nuclei using an optimized algorithm for image segmentation and connected components detection. Finally, the resultant relative fluorescence intensity values were uniformly scaled across the compared sets of imaging data and expressed in terms of arbitrary units. For each of the genes visualized, measurements were based on no fewer than three independent fields of view for each sample. To evaluate statistical significance between the conditions compared in image sets, unpaired student’s t tests were performed for each of the gene sets presented.
Immunostaining of macrophages in human nephrectomy samples, US cohort
10mm nephrectomy cryosections were placed into separate wells of a 24-well plate, fixed with 4% PFA for 15 min at 4°C, and then washed twice with PBS. Endogenous peroxidase activity was blocked with 0.03% hydrogen peroxide for 15 min at room temperature, after which sections were washed twice with PBS and blocked (2% fetal calf serum, 2% BSA, 0.2% fish skin gelatin) for 2 h at room temperature. Samples were incubated with mouse monoclonal anti-human SPI1 (clone 2G1; cat# H00006688-M02, ThermoFisher Scientific) and/or rabbit monoclonal anti-human TREM2 (9H4L26; Cat# 702886, ThermoFisher Scientific) antibodies, diluted 1:50 and 1:100 in blocking buffer respectively, overnight at 4°C. Tissue sections were subsequently washed three times with PBS and then incubated with HRP-conjugated donkey anti-mouse IgG (1:1000, diluted in blocking buffer; Cat# A90-337P, Bethyl Laboratories) for 45 min at room temperature. Two subsequent washes were followed by incubation with opal reagent 570 (1:200 (Cat# FP1488001KT) in dilution buffer (Cat# FP1498), Akoya Biosciences) for 10 min at room temperature. Washing and quenching with 0.03% hydrogen peroxide (for 10 min), were followed by incubation with HRP-donkey anti-rabbit IgG (1:200; Cat# A120-208P, Bethyl Laboratories) for 45 min at room temperature. Samples were further washed and incubated with opal reagent 650 (1:200; Cat# FP1496001KT, Akoya Biosciences) for 10 min at room temperature, after which PBS washes were followed by DAPI staining (1:10,000) for 20 min. After final PBS washes, a 1:1 mixture of Gold antifade solution was added to each well and samples were imaged using the Opera Phenix High-Content Screening System (PerkinElmer, Waltham, USA) as described below.
Fluorescence image acquisition and analysis
All fluorescence imaging on human kidney tissue was performed using the Opera Phenix High-Content Screening System (PerkinElmer, Waltham, USA). 10μm cryosections were placed into individual wells of 24-well plates and the entire specimen initially imaged for DAPI at 5X, using the PreciScanTM feature (PerkinElmer, Waltham, USA) to identify tissue. Pre-identified tissue regions were then imaged at higher resolution (20X water immersion objectives, confocal mode). Image analysis was performed using Harmony high-content analysis software (PerkinElmer, Waltham, USA).
Identification of macrophages in human kidney tissue
Each tissue area was identified using Harmony high-content analysis software based on the stitched 20x confocal global image of the entire well using the smoothed DAPI channel. The periphery of the tissue section (30μm) was removed, and fibrotic regions were masked based on a higher smoothed DAPI signal appearance. Within the resulting area, two types of objects were identified: first, all nuclei (total cell number) with their coordinates and second, SPI1-positive objects (i.e., macrophages) with their coordinates. General features used to define the SPI1-positive macrophages included Alexa 568 (SPI1) fluorescence intensity, nuclear Hoechst staining, nuclear area and width: length ratio. Further analysis of the SPI1-positive population was then performed using Tibco Spotfire software (Tibco, Palo Alto, USA). Alexa 488 intensity and distribution within each object were used to determine autofluorescence and enable positive signal to be distinguished from background. Based on negative controls, nuclear roundness and STAR morphology features (nuclear profile and 30% threshold compactness) were additional features used to identify the SPI1-positive population, of which threshold compactness (30%) was the most discriminatory. Using Alexa 647 (TREM2) fluorescence intensity values greater than the upper quartile in control samples to represent positive staining, the number of TREM2high (SPI1-positive) cells within each tissue section was determined and expressed as a percentage of all cells within the section.
Generation of digital graphic images illustrating human kidney TREM2high macrophages
Representative kidney sections from a non-obese and obese patient were chosen to generate the digital graphic images. TREM2high macrophages were identified as described above. Representative regions of each section were then created by first finding the median x and y coordinates of all TREM2high macrophages in a section and setting a target nuclei count of 40,000 total nuclei. The radius of the square, centered around the median that contained the target nuclei count, rounded to the nearest 10, was determined and represented in the graphic image. Graphs were visualized using ggplot2.
Single cell isolation from mouse kidney
Mice were anesthetized using isoflurane and perfused with PBS (1x) via the left ventricle. The kidneys were then removed and immediately placed in PBS on ice. For coronal sectioning, the renal capsule was removed, the kidney mounted and 300μm-thick sections cut using a vibrating blade microtome (Leica Biosystems Inc., Buffalo Grove, USA). The coronal sections were then returned to ice-cold PBS ready for dissection. For the regional sectioning, the renal capsule was removed from the other kidney and the hilum sampled first. The kidney was then cut in half along its coronal axis and cortical and medullary tissue was sequentially sampled. As each region was sampled, the tissue was chopped into 1 mm × 1 mm cubes and placed in 0.25 mg/ml liberase TH (Roche Diagnostics, Indianapolis, USA) dissociation medium. Following further dissection, the samples were incubated at 37°C for 1 h at 600rpm. Samples were regularly triturated during the incubation period using a 1mL pipette, after which 10% heat-inactivated FBS RPMI was added to stop the digestion. Centrifugation at 500g for 5 min at room temperature with the removal of the supernatant, was followed by the addition of ACK lysing buffer to remove erythrocytes (Thermo Fisher Scientific, Waltham, USA). Following centrifugation, the resulting cell pellet was incubated with Accumax at 37°C for 3 min (Innovative Cell Technologies Inc, San Diego, USA). 10% FBS RPMI was again used to neutralize the Accumax and centrifugation was followed by resuspension of the cell pellet in 0.4% BSA/PBS. The single cell suspension was filtered using a 30um CellTrics filter (Sysmex America Inc, Lincolnshire, USA) with cell viability and concentration determined using trypan blue and the Auto T4 cellometer (Nexcelom Bioscience LLC, Lawrence, USA). According to the manufacturer’s guidelines, 17,500 cells were loaded into the 10x Genomics microfluidic system/onto the 10x Genomics platform to achieve a targeted recovery of 10,000 cells (10x Genomics, Pleasanton, USA).
Single cell isolation from human kidney tissue, US cohort
Samples of macroscopically normal cortex, medulla and hilum were obtained from tumor nephrectomy specimens, distant from the tumor site and after appropriate patient consent, in accordance with IRB and institutional guidelines. Following transfer in 2% FBS RPMI, tissue was cut into 1 mm × 1 mm cubes and placed in 0.25 mg/ml liberase TH (Roche Diagnostics, Indianapolis, USA) dissociation medium. Following further dissection, the tissue was incubated at 37°C for 1 h at 600rpm as described previously. Samples were again regularly triturated during the incubation period using a 1mL pipette, after which the digestion was stopped by the addition of 10% heat-inactivated FBS RPMI. The addition of ACK lysing buffer (Thermo Fisher Scientific, Waltham, USA) following centrifugation at 500g for 5 min at room temperature, was performed twice in light of the lack of perfusion prior to nephrectomy. After centrifugation, the cell pellet was incubated with Accumax at 37°C for 3 min (Innovative Cell Technologies Inc, San Diego, USA), with 10% FBS RPMI again used for its subsequent neutralization. The resulting cell pellet was resuspended in 0.4% BSA/PBS and filtered using a 30um CellTrics filter (Sysmex America Inc, Lincolnshire, USA). Cell viability and concentration were determined as before, with 10,000 cells loaded into the 10x Genomics microfluidic system according to the manufacturer’s guidelines (10x Genomics, Pleasanton, USA).
Droplet-based scRNA-seq
Single cells were partitioned into gel bead-in-emulsions (GEMs) and incubated to generate barcoded cDNA by reverse transcription. Barcoded cDNA was then amplified by PCR prior to library construction. Fragmentation, sample index and adaptor ligation, and PCR were used to generate libraries of paired-end constructs according to the manufacturer’s recommendations (10x Genomics, Pleasanton, USA). Libraries were pooled and sequenced using the Illumina HiSeq X system (San Diego, USA). Whenever feasible, we pooled 10x libraries on sequencing lanes to ensure that any individual sample was not confounded by batch (kidney section, day of sample collection, condition, timepoint) and were randomly distributed across lanes.
Human kidney samples, UK cohort
Kidneys donated for transplantation, but unsuitable for implantation (due to damage to the arterial patch, suspicion of donor malignancy, or adverse retrieval biopsy score) were used. All analysis of human material was performed in the UK; ethical approval was granted by the local ethics committee (REC12/EE/0446) and the study was also approved by NHS Blood and Transplant (NHSBT). All kidneys had a cold ischemic time of less than 30 h prior to processing. Demographic information is shown in Table 4).
Confocal microscopy
Samples were fixed with AntigenFix (DiaPath) for 2 h at 4°C, dehydrated in 30% sucrose solution in PBS and embedded in OCT. 30μm sections were permeabilized and blocked in 0.1M Tris, containing 0.1% Triton (Sigma-Aldrich), 1% normal mouse serum, 1% normal donkey serum and 1% BSA (R&D systems). Samples were stained for 2 h at room temperature with Hoechst 33258 (1/5000 dilution) and primary antibodies (CD163-AF647, 1/100; goat anti-LYVE1 1/250; rabbit anti-TREM2, 1/100) in a humid chamber and washed 3 times in PBS. Secondary staining was performed for 2 h at room temperature (donkey anti-rabbit-AF555 and donkey anti-goat AF488 1/250 both; see the Table below). Slides were then washed as previously and mounted in Fluoromount-G (Southern Biotech).
Images were acquired using a TCS SP8 (Leica microsystems) inverted confocal microscope, using a 40 × 1.1N/A water objective. Raw imaging data were processed using Imaris v9.7.0 (Bitplane). The spot function was used to define total macrophage numbers based on CD163 and nuclear staining. TREM2 and LYVE1 expression was then attributed to each macrophage manually. Two areas 1.25 mm2 from each sample were imaged and counted. The data presented represents the average of the two areas quantified for each sample.
The mean values were compared between groups with differing donor comorbidities, including arterial hypertension, obesity and type 2 diabetes mellitus.
* = p < 0.05, ** = p < 0.01, ns = non significant. Mann-Whitney test (for between group comparisons)
* = p < 0.05. Spearman test (for correlation analysis)
| Marker | Conjugate | Clone | Cat# | Vendor | Dilution |
|---|---|---|---|---|---|
| Hoechst 33258 | x | x | 40044 | Biotum | 1/5 000 |
| CD163 | AF647 | GH1/61 | 333619 | BioLegend | 1/100 |
| TREM2 | x | Polyclonal | 13483–1 - AP | Proteintech | 1/100 |
| LYVE1 | x | Polyclonal | AF2089 | R&D Systems | 1/250 |
| Donkey anti-rabbit | AF555 | Polyclonal | A32794 | Invitrogen | 1/250 |
| Donkey anti-goat | AF488 | Polyclonal | A32814 | Invitrogen | 1/250 |
QUANTIFICATION AND STATISTICAL ANALYSIS
Computational methods for data analysis
Study design and DKD cell atlases
-
i
DKD Mouse Study Design:
For the BTBR ob/ob genetic mouse model, we included n = 5 and n = 3 biological replicates for the 5- and 10-week time-point respectively for both ob/ob and wildtype strains. For the HFD model, we combined n = 4 biological replicates into a pooled ‘‘aged’’ group, in each of the chow and HFD conditions. The ‘‘aged’’ group ranged from 66 to 100 weeks old. For 3 biological replicates in each condition (chow ages: 94,100; HFD: ages 83,100), there were 2 technical replicates or 10x channels run from the same cortical sample leading to a total of 7 libraries per condition.
| Mouse model | Biological replicates per condition | Technical replicates |
|---|---|---|
| Chow/HFD | 4 | 2 each for 3 biological replicates |
| BTBR wt and ob/ob (5 weeks) | 5 | 0 |
| BTBR wt and ob/ob (10 weeks) | 3 | 0 |
-
ii
Human kidney samples:
Human kidney samples arrived as per the nephrectomy surgery schedule, with no selection on the age or sex of the patients. In case of 3 patients (Nx12, Nx10 and Nx8, the kidney sections were split to allow CD45 enrichment of one-half. Though libraries were prepared soon after sample collection, we randomized pooling of samples during sequencing.
-
iii
Sequencing Design:
We pooled the 10x libraries on sequencing lanes using a randomized design to ensure that replicates from an individual batch (mouse, replicate, kidney section, day of sample collection, condition, timepoint) were distributed across lanes. Despite a deliberate random sequencing design to mitigate batch effects, donor effects were prominent in human data. Within a mouse strain, batch effects (separation by mouse) was not observed.
Generation of mouse and human diabetic cell atlases
Preprocessing of 10x droplet-based sequencing outputs
The HFD/Chow mouse, BTBR ob/ob mouse and human DKD scRNAseq data were generated via 10X Genomics Chromium 3′ droplet-based sequencing. Specifically, the 10X Genomics kit used was v2. The sequencing platform was Novaseq at the Genomics platform. Final libraries (4nM) were sequenced on a NovaSeq S2 or S4 with 100–200 million reads per sample at the Genomics Platform at the Broad Institute. The Cellranger toolkit (v6.0.2)57,58 by 10X Genomics was used to (1) de-multiplex the sequencing outputs using the command cellranger mkfastq with default options, (2) align the sequencing reads to the reference transcriptomes (mm10 for mouse, GrCh38 for human), and (3) quantify gene expression, resulting in gene-by-cell UMI count matrices. (2) and (3) were done using the command cellranger count with default options. We used Cumulus to perform preprocessing on Google cloud using the Terra scientific computing workflow.
Quality control (QC) of scRNAseq datasets
Ambient RNA correction: We ran Cellbender59 (v0.2.0) to correct for contamination by ambient RNA using the FPR cutoff of “0.01” using the Terra pipelines.
Across all datasets, as an initial QC, we only retained cells that had reads mapping to a minimum of 1000 UMIs and 200 genes.
Because the kidney has the second highest mitochondrial metabolic activity among organs in the human body,60 we first imposed a permissive threshold, allowing all cells with less than 80% mitochondrial reads to be included in the downstream analysis. We plotted boxplots showing the number of UMIs, genes and percent mitochondrial content after these filters.We hypothesized that each cell type will have its own profile of metabolic activity and filtering must proceed in a cell type specific manner, leveraging exploratory data analysis. We proceeded to perform downstream steps and cluster cells.
First, we examined if there were any clusters that could be distinguished by a majority of cells expressing high-percentages of mitochondrial reads, and by examining the top differentially expressed genes for each cluster. In every (btbr 4 week, btbr 10 week, HFD mouse models, human kidney data) model, we did not observe such a ‘‘technical’’ cluster. Because of the absence of technical clusters, we proceeded to assign high-level cell-type assignments to the clusters. On plotting percent mitochondrial reads across cells, we saw cells with higher percentages in cell types with high expected metabolic activity like the proximal tubule, thick ascending limb and the distal convoluted tubule. For any downstream analysis involving sub clustering or assessment of differentially expressed genes in disease, we discarded cells with over 20% mitochondrial reads except the proximal tubules which is explained in more detail below.
Besides ambient correction using CellBender, we found ambient contamination during differential expression analysis in the mouse data. Specifically, genes highly expressed in the predominant parenchymal cell types proximal tubule and distal nephron, and some endothelial genes were frequently found to be ambient, i.e., differentially expressed in non-native cell types. We generated a list of such genes (“Pck1”, “Pecam1”, “Gsta2”, “Klk1”, “Spink1”, “Wfdc2”, “Miox”, “Slc34a1”, “Kap”, “Aldob”, “Cubn”, “Ttc36”, “Umod”, “Fxyd2”) and flagged them during differential expression analysis except in the native cell types where they are expected.
Doublet detection
The scDblFinder61 (v1.4.0) package within the R statistical computing environment (v4.0.3) was used to detect and remove doublets for scRNA-Seq samples. Each sample was processed individually and converted to a SingleCellExperiment62 (v1.12.0) object before running scDblFinder. Samples were converted to Seurat (v4.1.1) objects after processing them, and then merged into a single object using the merge function within R. UMAP projections were used to visualize single-cell doublet detection using the DimPlot function within Seurat, as well as stacked bar plots using the ggplot2 package to visualize the singlets-doublets proportions for both within samples and clusters.
Inferring cell-types from mouse and human kidney cells
Normalization, scaling and dimensionality reduction.
All single cell data analysis was performed in the R statistical computing environment (v4.0.3) using package Seurat (v4.1.1).63 First, we normalized the gene UMI counts per cell using total sum scaling followed by multiplication by a factor of 1e+5 (TP10k or TPX, here we will refer to TPX), and log-transformation using a pseudocount of 1 using the NormalizeData function to obtain log(TPX+1) units for each gene. Next, we identified high varying features of gene expression using the FindVariableGenes function using default parameters. We computed the top 100 principal components (PCs) using the RunPCA function on the expression matrix composed of only the most highly variable genes after mean centering and scaling. We were more permissive in the number of PCs to enable discovery of rare cell types and states, as it is easier to post hoc merge together redundant states.
Unsupervised determination of putative cell types or clusters.
For identification of cell types, we ran unsupervised clustering using the FindClusters function on all the computed PCs. In general, we used domain knowledge and exploratory data analyses to aid choice of parameters for the unsupervised analysis (resolution of clustering, annotation of cell types and number of PCs) FindClusters builds a shared k-nearest neighbor graph64 (k = 30) followed by Louvain community detection65 to determine clusters. We first used a resolution of 0.4, followed by iterative clustering on major cell types and as needed. For visualization, we embedded cells in the Uniform manifold approximation and projection (UMAP) space using the RunUMAP function using all computed PCs, and with default parameters.
Joint analyses of human cells from multiple donors.
Batch effects are often detected by segregation of clusters by technical (e.g., donor origin, replicate, day of sequencing etc) rather than expected biological identity. While there were no explicit clusters from a technical replicate in the mice data, the human data separated by donor. We used the RunHarmony function of the Harmony66 R package setting the number of dimensions to 20, to co-embed human scRNAseq data prior to clustering and visualization. Each individual was used as a separate batch.
Assignment of cell-identity.
We ran differential gene expression (DGE) tests using the FindAllMarkers function to compute highly differentially expressed (DE) genes distinguishing each cluster from all other cells using the Wilcoxon Rank-Sum test with False Discovery Rate (FDR) adjustment using the method of Benjamini-Hochberg.67 We performed the test only on genes expressed in at least 25% of the cells using the min.pct argument. We annotated clusters by checking the presence of literature derived cell-type specific genes among the top DE genes when possible. Where applicable, Seurat’s AddModuleScore and CellCycleScoring functions were used to score signatures of gene sets and cell-cycle genes respectively.
Curation of gene signatures.
We used literature derived genes for kidney parenchymal, stromal and immune cell types.22–24,68
Identification of cell type markers.
We derived cluster specific DE genes using the FindAllMarkers function to distinguish cell-types at the higher and subcluster levels. For visualization on the heatmap, we selected the both data-driven markers obtained by DGE (at least top 3 in case of immune cells) in addition to literature-derived canonical markers.
Plotting and visualization.
We used the ggplot269 R package for generation of boxplots (geom_box), violin plots (geom_vln), proportional bar plots (geom_col), UMAP visualization and dotplots (geom_point).
In the dot plot representation, each dot size represented the percentage of cells expressing the gene, and the color represented the average nonzero gene expression in log2 scale.
Cell proportions are computed as the ratio of the number of cells in a certain cell type divided by the total number of cells in the unit of interest (e.g., condition, region, individual, Figures G,L,Q, Supp Fig.1P–R).
We visualized heat maps using the pheatmap70 package. Units of expression are log transformed transcript counts per 10,0000 (TPX). A pseudocount of 1 was added during the log transformation.
Characterization of macrophages in human adult kidney
We iteratively clustered C1Q + positive macrophages to derive subsets, followed by DGE analysis using the FindAllMarkers functions to determine subset specific signatures.
Identification of DKD-associated genes and pathways
Derivation of differentially expressed (DE) genes in DKD
In the mouse data, after assignment of cell-identity as described previously, we performed differential expression analyses between control and DKD cells using the Wilcoxon Rank-sum test using the ‘‘wilcox.test’’ base R function on genes present in at least 25% of cells in either condition, and a Benjamini-Hochberg FDR cutoff of 0.05 using the ‘‘p.adjust’’ base R function to prioritize for downstream interpretation. We did the analysis for each cell type independently. We analyzed five mice in each of the HFD- and chow-fed groups, pooled across 66–100 weeks of age into a single ‘‘aged mice’’ group for DGE analysis.
For human data, we used a poisson mixed-effects model with the donor as the random covariate, disease as covariate being tested and the average number of UMIs per cell as the offset. We used the glmer command from the lme4 package in R using the formula ‘.’ The model was run for each cell type independently. We adjusted for multiple hypotheses by the method of Benjamini and Hochberg. We prioritized genes by setting fdr <0.1.
Characterization of macrophages in mouse diabetic kidney
Annotation of immune cells and macrophages
For both the HFD and BTBR mouse models (two time points), we subset the Cd45+ cells and iteratively clustered them to derive myeloid and lymphoid lineages. We used the previously characterized resident and infiltrating kidney macrophage signatures to mark macrophages as resident vs. infiltrating.19 For the heatmap visualizations, we used the top 3 data-driven DE genes marking the subset, alongside selected canonical and literature driven markers. On all heatmaps, gene expression in units of log(TPX+1) was averaged across all cells in a subset, followed by scaling across all subsets.
As macrophage subsets are in a transcriptional continuum, we used the Potential of Heat-diffusion for Affinity-based Trajectory Embedding (PHATE,71) embedding to visualize the cells across age, condition and subset. The macrophage genexcell data matrices were first converted to the anndata object format using the anndata Python library(https://anndata.readthedocs.io/en/stable/#), and PHATE was run using the phate Python library, and the commands ‘‘phate.PHATE()’’ and ‘‘phate_op.fit_transform(anndata).’’
The Lipid associated macrophage (LAM) signature was derived from.36 We performed DGE using the Wilcoxon rank-sum test using the function call wilcox.test in R, between the HFD LAMSs and other HFD macrophages. The results were visualized as a volcano plot, where genes that passed the FDR alpha threshold of 0.05 were colored blue, while the rest were gray. Top DE genes were annotated on the plot with gene names.
Comparison of macrophage populations across strains
We trained an RF classifier on the HFD macrophage subset labels using either the intersection of HVG with BTBR week 10 (Figure 5A) or week 5 macrophages as features and then predicted on the BTBR data to determine correspondence.
Association of cell type proportions to age and disease
We used a Poisson regression model to determine the association between macrophage proportions (using counts) and covariates (age, disease condition). The model was defined as follows using the glm command from the stats package in R and formula “” for the HFD model and “” for the BTBR models.
Characterization of macrophages in human diabetic kidney
Derivation of human DKD macrophage subsets
We subsetted and clustered all the CD45+ immune cells across DKD and non-DKD kidney data, including the CD45+ enriched samples, and subsetted the clusters differentially enriched in the macrophage marker genes C1QA and C1QB into the human kidney macrophage subset. We then iteratively clustered the human kidney macrophage subset to derive 4 distinct subsets (MΦ 1–4), after excluding monocytes and DCs, and performed DGE using the FindAllMarkers Seurat command with the default Wilcoxon rank-sum test to get subset-specific marker genes.
For the heatmap, each marker gene was assigned the average gene expression across all cells in the subset in units of log(TPX+1), followed by row normalization across the subsets.
Comparison of mouse and human macrophages in DKD
To map the mouse macrophages with human macrophages, a multi-class random forest (RF) was trained on the human DKD macrophages as described above on the feature set of orthologous HVG shared with the HFD model mice. The trained RF was used to predict labels for HFD mouse macrophages. The resulting confusion matrix was visualized as a dot plot as described above.
Analysis of KPMP scRNA-seq data
The Kidney Precision Medicine Project (KPMP) scRNA-seq Seurat data object was shared by the authors of the study (PMID: 37468583). Myeloid cells were subset by selecting for the author provided cell annotations: “MON”,”ncMON”,”MDC”,”cDC”, “MAC-M2”,”cycMNP”,”MAST.” The subset was further analyzed by finding highly variable genes, principal component analysis (default number of dimensions), correction for donor effects by Harmony, and clustered using graph-based clustering in the Harmony embedding (10 dimensions) at a resolution 0.5 using functions in the Seurat package. Cluster specific markers were computed using the FindAllMarkers function. The ‘‘SampleType’’ variable in the metadata column was used for stratification by disease.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Rabbit anti-Pck1 | ThermoFisher Scientific | #720266; RRID: AB_2633227 |
| Rabbit anti-Gsta2 | ThermoFisher Scientific | #PA5-96757; RRID: AB_2808559 |
| Alexa Goat anti-rabbit IgG 594 | ThermoFisher Scientific | #A32740; RRID: AB_2762824 |
| Mouse monoclonal anti-human SPI1 | ThermoFisher Scientific | #H00006688-M02; RRID: AB_913827 |
| Rabbit monoclonal anti-human TREM2 | ThermoFisher Scientific | #702886; RRID: AB_2762383 |
| Rabbit anti-TREM2 | Proteintech | #13483-1 - AP; RRID: AB_2605803 |
| HRP-conjugated donkey anti-mouse IgG | Bethyl Laboratories | #A90-337P; RRID: AB_10631367 |
| HRP- conjugated donkey anti-rabbit IgG | Bethyl Laboratories | #A120-208P ; RRID: AB_66770 |
| CD163-AF647 | BioLegend | #333619; RRID: AB_2563474 |
| Goat anti-LYVE1 | R&D Systems | # AF2089; RRID: AB_355144 |
| Donkey anti-rabbit-AF555 | ThermoFisher Scientific | # A32794; RRID: AB_162543 |
| Donkey anti-goat AF488 | ThermoFisher Scientific | # A32814; RRID: AB_2534102 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| PBS | ThermoFisher Scientific | #AM9625 |
| BSA | Sigma | # A1595 |
| 4% PFA | ThermoFisher Scientific | #101176-014 |
| 10% formalin | VWR | #100503-120 |
| 0.5% periodic acid solution | Sigma | #1.00482 |
| Schiff reagent | Sigma | # 1.09033 |
| Mayer’s hematoxylin | Sigma | # 51275 |
| 5x SSCT | ThermoFisher Scientific | # 15557044 |
| Opal reagent 570 | Akoya Bio | # FP1488001KT |
| Opal reagent 650 | Akoya Bio | # FP1496001KT |
| Liberase TH | Roche Diagnostics | # 5401119001 |
| RPMI-1640 | ThermoFisher Scientific | # 11875119 |
| ACK lysing buffer | Thermo Fisher Scientific | # A1049201 |
| CellTrics filter | Sysmex America Inc | # 04-0042-2316 |
| Humulin N solution | Eli Lilly | #HI-310 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Chromium Single Cell | 10x Genomics | https://www.10xgenomics.com/platforms/chromium |
|
| ||
| Deposited data | ||
|
| ||
| Mouse single cell data | Current study | GEO: GSE205594 |
| Human single cell data | Current study | Accession: SCP2188 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6 | The Jackson Lab | IMSR_JAX:000664 |
| 129S1 | The Jackson Lab | IMSR_JAX:002448 |
| BTBR.Cg-Lepob/WiscJ | The Jackson Lab | IMSR_JAX:004824 |
| C57BL/6N-Trem2em1cyagen | Cyagen | KOCMP-83433-Trem2-B6NVA |
|
| ||
| Software and algorithms | ||
|
| ||
| Graphpad Prism | GraphPad Software | https://www.graphpad.com/ |
| ImageJ | ImageJ | https://imagej.net/software/imagej/ |
| Adobe Illustrator | Adobe | https://www.adobe.com/ |
| Preprocessing of single-cell RNAseq sequencing files | Cellranger toolkit (v6.0.2), Cellbender (v0.2.0) | N/A |
| Genome references | mm10 for mouse, GrCh38 for human | N/A |
| Analysis of single-cell RNAseq data using R packages | R (v4.0.3), Seurat (v4.1.1), ggplot2, cowplot, pheatmap, UpsetR, scDblFinder (v1.4.0), Harmony | N/A |
|
| ||
| Other | ||
|
| ||
| High-fat diet | Envigo | TD.93075 dough |
| Standard chow | LabDiet | PicoLab Rodent Diet 20 pellets |
| Glucose meter One-touch UltraMini | Lifescan | https://www.onetouch.com/ |
Highlights.
TREM2high macrophages reside in the human adult kidney
Trem2high macrophages increase in frequency in a high-fat-diet mouse model of diabetic kidney disease
Human adult diabetic and obese kidney tissue has increased frequency of TREM2high macrophages
Trem2 deletion promotes kidney injury after exposure to a high-fat diet but not a regular diet
ACKNOWLEDGMENTS
We thank our colleagues in the BWH Department of Urology for generous assistance with recovering tissue from tumor nephrectomies under IRB protocol 2011P002692. This work was funded by NIH grants DK095045 and DK099465 (to A. Greka) and the Chan Zuckerberg Foundation (CZF019-002447) (to A. Greka). We would like to thank Katie Liguori for exceptional graphic design work.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114253.
DECLARATION OF INTERESTS
A.Greka has a financial interest in Sail Bio, which was reviewed and is managed by Brigham and Women’s Hospital, Mass General Brigham (MGB), and the Broad Institute of MIT and Harvard in accordance with their conflict of interest policies. K.A.V. is an employee and shareholder of Q32 Bio, Inc. J.L.S. is an equity holder of Magnetic Ventures. A.R. is a cofounder and equity holder of Celsius Therapeutics and equity holder of Immunitas and, until August 2020, was an SAB member of Thermo Fisher Scientific, Syros Pharmaceuticals, Neogene Therapeutics, and Asimov. A.R. is an employee of Genentech, Inc. O.R.-R is an employee of Genentech, Inc. O.R.-R. is a coinventor on patent applications filed by the Broad Institute related to single-cell genomics.
REFERENCES
- 1.Thomas MC, Brownlee M, Susztak K, Sharma K, Jandeleit-Dahm KAM, Zoungas S, Rossing P, Groop P-H, and Cooper ME (2015). Diabetic kidney disease. Nat. Rev. Dis. Prim. 1, 15018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alicic RZ, Rooney MT, and Tuttle KR (2017). Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 12, 2032–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lingvay I, Sumithran P, Cohen RV, and le Roux CW (2022). Obesity management as a primary treatment goal for type 2 diabetes: time to reframe the conversation. Lancet 399, 394–405. 10.1016/S0140-6736(21)01919-X. [DOI] [PubMed] [Google Scholar]
- 4.Hall ME, Cohen JB, Ard JD, Egan BM, Hall JE, Lavie CJ, Ma J, Ndumele CE, Schauer PR, and Shimbo D; American Heart Association Council on Hypertension; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Lifestyle and Cardiometabolic Health; and Stroke Council (2021). Weight-Loss Strategies for Prevention and Treatment of Hypertension: A Scientific Statement From the American Heart Association. Hypertension 78, e38–e50. [DOI] [PubMed] [Google Scholar]
- 5.Stenvinkel P, Zoccali C, and Ikizler TA (2013). Obesity in CKD—What Should Nephrologists Know? J. Am. Soc. Nephrol. 24, 1727–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ingelfinger JR, and Rosen CJ (2019). Clinical Credence — SGLT2 Inhibitors, Diabetes, and Chronic Kidney Disease. N. Engl. J. Med. 380, 2371–2373. 10.1056/nejme1904740. [DOI] [PubMed] [Google Scholar]
- 7.Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, Edwards R, Agarwal R, Bakris G, Bull S, et al. (2019). Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 380, 2295–2306. [DOI] [PubMed] [Google Scholar]
- 8.Wilson PC, Wu H, Kirita Y, Uchimura K, Ledru N, Rennke HG, Welling PA, Waikar SS, and Humphreys BD (2019). The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc. Natl. Acad. Sci. USA 116, 19619–19625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu J, Sun Z, Wang X, Zhang T, Yuan W, Salem F, Yu SM-W, Zhang W, Lee K, and He JC (2022). The single-cell landscape of kidney immune cells reveals transcriptional heterogeneity in early diabetic kidney disease. Kidney Int. 102, 1291–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wada J, and Makino H (2016). Innate immunity in diabetes and diabetic nephropathy. Nat. Rev. Nephrol. 12, 13–26. [DOI] [PubMed] [Google Scholar]
- 11.Klessens CQF, Zandbergen M, Wolterbeek R, Bruijn JA, Rabelink TJ, Bajema IM, and IJpelaar DHT (2017). Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol. Dial. Transplant. 32, 1322–1329. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Yang Y, and Zhao Y (2019). Macrophage phenotype and its relationship with renal function in human diabetic nephropathy. PLoS One 14, e0221991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mould KJ, Jackson ND, Henson PM, Seibold M, and Janssen WJ (2019). Single cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI Insight 4, e126556. 10.1172/jci.insight.126556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conway BR, O’Sullivan ED, Cairns C, O’Sullivan J, Simpson DJ, Salzano A, Connor K, Ding P, Humphries D, Stewart K, et al. (2020). Kidney Single-Cell Atlas Reveals Myeloid Heterogeneity in Progression and Regression of Kidney Disease. J. Am. Soc. Nephrol. 31, 2833–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lantz C, Radmanesh B, Liu E, Thorp EB, and Lin J (2020). Single-cell RNA sequencing uncovers heterogenous transcriptional signatures in macrophages during efferocytosis. Sci. Rep. 10, 14333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park MD, Silvin A, Ginhoux F, and Merad M (2022). Macrophages in health and disease. Cell 185, 4259–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Betz B, and Conway BR (2014). Recent advances in animal models of diabetic nephropathy. Nephron Exp. Nephrol. 126, 191–195. [DOI] [PubMed] [Google Scholar]
- 18.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, Li M, Barasch J, and Suszták K (2018). Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360, 758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimmerman KA, Bentley MR, Lever JM, Li Z, Crossman DK, Song CJ, Liu S, Crowley MR, George JF, Mrug M, and Yoder BK (2019). Single-Cell RNA Sequencing Identifies Candidate Renal Resident Macrophage Gene Expression Signatures across Species. J. Am. Soc. Nephrol. 30, 767–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ransick A, Lindström NO, Liu J, Zhu Q, Guo J-J, Alvarado GF, Kim AD, Black HG, Kim J, and McMahon AP (2019). Single-Cell Profiling Reveals Sex, Lineage, and Regional Diversity in the Mouse Kidney. Dev. Cell 51, 399–413.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu J, Akat KM, Sun Z, Zhang W, Schlondorff D, Liu Z, Tuschl T, Lee K, and He JC (2019). Single-Cell RNA Profiling of Glomerular Cells Shows Dynamic Changes in Experimental Diabetic Kidney Disease. J. Am. Soc. Nephrol. 30, 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart BJ, Ferdinand JR, Young MD, Mitchell TJ, Loudon KW, Riding AM, Richoz N, Frazer GL, Staniforth JUL, Vieira Braga FA, et al. (2019). Spatiotemporal immune zonation of the human kidney. Science 365, 1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sivakamasundari V, Bolisetty M, Sivajothi S, Bessonett S, Ruan D, and Robson P (2017). Comprehensive Cell Type Specific Transcriptomics of the Human Kidney. Preprint at bioRxiv. 238063. 10.1101/238063. [DOI] [Google Scholar]
- 24.Young MD, Mitchell TJ, Vieira Braga FA, Tran MGB, Stewart BJ, Ferdinand JR, Collord G, Botting RA, Popescu D-M, Loudon KW, et al. (2018). Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 361, 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirita Y, Wu H, Uchimura K, Wilson PC, and Humphreys BD (2020). Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc. Natl. Acad. Sci. USA 117, 15874–15883. 10.1073/pnas.2005477117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hudkins KL, Pichaiwong W, Wietecha T, Kowalewska J, Banas MC, Spencer MW, Mühlfeld A, Koelling M, Pippin JW, Shankland SJ, et al. (2010). BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J. Am. Soc. Nephrol. 21, 1533–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yore MM, Syed I, Moraes-Vieira PM, Zhang T, Herman MA, Homan EA, Patel RT, Lee J, Chen S, Peroni OD, et al. (2014). Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell 159, 318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brunskill EW, and Potter SS (2012). Changes in the gene expression programs of renal mesangial cells during diabetic nephropathy. BMC Nephrol. 13, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salem RM, Todd JN, Sandholm N, Cole JB, Chen W-M, Andrews D, Pezzolesi MG, McKeigue PM, Hiraki LT, Qiu C, et al. (2019). Genome-Wide Association Study of Diabetic Kidney Disease Highlights Biology Involved in Glomerular Basement Membrane Collagen. J. Am. Soc. Nephrol. 30, 2000–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, and Harris DC (2011). Macrophages in renal disease. J. Am. Soc. Nephrol. 22, 21–27. [DOI] [PubMed] [Google Scholar]
- 31.Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, Bilbao-Cortes D, Godwin JW, and Rosenthal NA (2012). An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS One 7, e36814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba A-E, and Zernecke A (2018). Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res. 122, 1661–1674. [DOI] [PubMed] [Google Scholar]
- 33.Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, Zhang X-M, Foo S, Nakamizo S, Duan K, et al. (2019). Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 363, eaau0964. 10.1126/science.aau0964. [DOI] [PubMed] [Google Scholar]
- 34.Litviňuková M, Talavera-López C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, et al. (2020). Cells of the adult human heart. Nature 588, 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geirsdottir L, David E, Keren-Shaul H, Weiner A, Bohlen SC, Neuber J, Balic A, Giladi A, Sheban F, Dutertre C-A, et al. (2020). Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell 181, 746. [DOI] [PubMed] [Google Scholar]
- 36.Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, et al. (2019). Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 178, 686–698.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice M, Marwick JA, et al. (2019). Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subramanian A, Vernon K, Slyper M, and Waldman J (2020). RAAS blockade, kidney disease, and expression of ACE2, the entry receptor for SARS-CoV-2, in kidney epithelial and endothelial cells. Preprint at bio-Rxiv. 10.1101/2020.06.23.167098. [DOI] [Google Scholar]
- 39.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- 40.Han X, Zhou Z, Fei L, Sun H, Wang R, Chen Y, Chen H, Wang J, Tang H, Ge W, et al. (2020). Construction of a human cell landscape at single-cell level. Nature 581, 303–309. 10.1038/s41586-020-2157-4. [DOI] [PubMed] [Google Scholar]
- 41.Peters KE, Davis WA, Ito J, Winfield K, Stoll T, Bringans SD, Lipscombe RJ, and Davis TME (2017). Identification of Novel Circulating Biomarkers Predicting Rapid Decline in Renal Function in Type 2 Diabetes: The Fremantle Diabetes Study Phase II. Diabetes Care 40, 1548–1555. [DOI] [PubMed] [Google Scholar]
- 42.Noelia A, Guillen JA, Gallardo G, Diaz M, De La Rosa JV, Hernandez IH, Casanova-Acebes M, Lopez F, Tabraue C, Beceiro S, et al. (2013). The nuclear receptor LXRa controls the functional specialization of splenic macrophages. Nat. Immunol. 14, 831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kato M, Shimazu M, Moriguchi S, and Kishino Y (1996). Body mass index (BMI) is a reliable index to estimate obesity as a risk factor for deteriorating health. Tokushima J. Exp. Med. 43, 1–6. [PubMed] [Google Scholar]
- 44.Milas O, Gadalean F, Vlad A, Dumitrascu V, Velciov S, Gluhovschi C, Bob F, Popescu R, Ursoniu S, Jianu DC, et al. (2020). Pro-inflammatory cytokines are associated with podocyte damage and proximal tubular dysfunction in the early stage of diabetic kidney disease in type 2 diabetes mellitus patients. J. Diabet. Complicat. 34, 107479. [DOI] [PubMed] [Google Scholar]
- 45.Donate-Correa J, Ferri CM, Sánchez-Quintana F, Pérez-Castro A, González-Luis A, Martín-Núñez E, Mora-Fernández C, and Navarro-González JF (2020). Inflammatory Cytokines in Diabetic Kidney Disease: Pathophysiologic and Therapeutic Implications. Front. Med. 7, 628289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, and Amit I (2018). Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 173, 1073–1081. [DOI] [PubMed] [Google Scholar]
- 47.Deczkowska A, Weiner A, and Amit I (2020). The Physiology, Pathology, and Potential Therapeutic Applications of the TREM2 Signaling Pathway. Cell 181, 1207–1217. [DOI] [PubMed] [Google Scholar]
- 48.Hammond TR, Marsh SE, and Stevens B (2019). Immune Signaling in Neurodegeneration. Immunity 50, 955–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Henegar JR, Bigler SA, Henegar LK, Tyagi SC, and Hall JE (2001). Functional and structural changes in the kidney in the early stages of obesity. J. Am. Soc. Nephrol. 12, 1211–1217. [DOI] [PubMed] [Google Scholar]
- 50.Mohammedi K, Chalmers J, Herrington W, Li Q, Mancia G, Marre M, Poulter N, Rodgers A, Williams B, Perkovic V, et al. (2018). Associations between body mass index and the risk of renal events in patients with type 2 diabetes. Nutr. Diabetes 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang W-H, Chen C-Y, Lin J-L, Lin-Tan D-T, Hsu C-W, and Yen T-H (2014). High body mass index reduces glomerular filtration rate decline in type II diabetes mellitus patients with stage 3 or 4 chronic kidney disease. Medicine 93, e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luk AOY, So W-Y, Ma RCW, Kong APS, Ozaki R, Ng VSW, Yu LWL, Lau WWY, Yang X, Chow FCC, et al. (2008). Metabolic syndrome predicts new onset of chronic kidney disease in 5,829 patients with type 2 diabetes: a 5-year prospective analysis of the Hong Kong Diabetes Registry. Diabetes Care 31, 2357–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Belhatem N, Mohammedi K, Rouzet F, Matallah N, Al Baloshi A, Travert F, Velho G, Roussel R, Le Guludec D, Marre M, and Hansel B (2015). Impact of morbid obesity on the kidney function of patients with type 2 diabetes. Diabetes Res. Clin. Pract. 108, 143–149. [DOI] [PubMed] [Google Scholar]
- 54.Chang AR, Chen Y, Still C, Wood GC, Kirchner HL, Lewis M, Kramer H, Hartle JE, Carey D, Appel LJ, and Grams ME (2016). Bariatric surgery is associated with improvement in kidney outcomes. Kidney Int. 90, 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hou C-C, Shyu R-S, Lee W-J, Ser K-H, Lee Y-C, and Chen S-C (2013). Improved renal function 12 months after bariatric surgery. Surg. Obes. Relat. Dis. 9, 202–206. [DOI] [PubMed] [Google Scholar]
- 56.Navarro-Díaz M, Serra A, Romero R, Bonet J, Bayés B, Homs M, Pérez N, and Bonal J (2006). Effect of drastic weight loss after bariatric surgery on renal parameters in extremely obese patients: long-term follow-up. J. Am. Soc. Nephrol. 17, S213–S217. [DOI] [PubMed] [Google Scholar]
- 57.10x Genomics (2024). Cell Ranger (10x Genomics). [Google Scholar]
- 58.Fleming SJ, Marioni JC, and Babadi M (2019). CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. Preprint at bioRxiv. 10.1101/791699. [DOI] [Google Scholar]
- 59.Fleming SJ, Chaffin MD, Arduini A, Akkad A-D, Banks E, Marioni JC, Philippakis AA, Ellinor PT, and Babadi M (2019). Unsupervised removal of systematic background noise from droplet-based single-cell experiments using CellBender. Preprint at bioRxiv. 10.1101/791699. [DOI] [PubMed] [Google Scholar]
- 60.Mercer TR, Neph S, Dinger ME, Crawford J, Smith MA, Shearwood A-MJ, Haugen E, Bracken CP, Rackham O, Stamatoyannopoulos JA, et al. (2011). The human mitochondrial transcriptome. Cell 146, 645–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Germain P-L, Lun A, Garcia Meixide C, Macnair W, and Robinson MD (2021). Doublet identification in single-cell sequencing data using scDblFinder. F1000Res. 10, 979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Amezquita RA, Lun ATL, Becht E, Carey VJ, Carpp LN, Geistlinger L, Marini F, Rue-Albrecht K, Risso D, Soneson C, et al. (2020). Orchestrating single-cell analysis with Bioconductor. Nat. Methods 17, 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jarvis RA, and Patrick EA (1973). Clustering Using a Similarity Measure Based on Shared Near Neighbors. IEEE Trans. Comput. C-22, 1025–1034. [Google Scholar]
- 65.Blondel VD, Guillaume J-L, Lambiotte R, and Lefebvre E (2008). Fast unfolding of communities in large networks. Preprint at arXiv. 10.48550/arXiv.0803.0476. [DOI] [Google Scholar]
- 66.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P-R, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296. 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Benjamani Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B 57, 289–300. [Google Scholar]
- 68.Subramanian A, Sidhom E-H, Emani M, Vernon K, Sahakian N, Zhou Y, Kost-Alimova M, Slyper M, Waldman J, Dionne D, et al. (2019). Single cell census of human kidney organoids shows reproducibility and diminished off-target cells after transplantation. Nat. Commun. 10, 5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag; ). [Google Scholar]
- 70.Kolde R (2012). Pheatmap: pretty heatmaps. [Google Scholar]
- 71.Moon KR, van Dijk D, Wang Z, Gigante S, Burkhardt DB, Chen WS, Yim K, Elzen A.v.d. Hirn MJ Coifman RR et al. (2019). Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 37, 1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We have deposited the mouse scRNA-seq data on NCBI GEO (accession GSE205594).
We have deposited the human scRNA-seq data in the single cell portal (SCP): https://singlecell.broadinstitute.org/single_cell/study/SCP2188/human-and-mouse-dkd-atlas#.
We have deposited all relevant code/scripts in GitHub (https://github.com/ayshwaryas/DKD_paper) with all the analysis described in sufficient detail for others to reproduce.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.