

Abstract
The corneocyte lipid envelope (CLE), a monolayer of ω-hydroxyceramides whose function(s) remain(s) uncertain, is absent in patients with autosomal recessive congenital ichthyoses with mutations in enzymes that regulate epidermal lipid synthesis. Secreted lipids fail to transform into lamellar membranes in certain autosomal recessive congenital ichthyosis epidermis, suggesting the CLE provides a scaffold for the extracellular lamellae. However, because cornified envelopes are attenuated in these autosomal recessive congenital ichthyoses, the CLE may also provide a scaffold for subjacent cornified envelope formation, evidenced by restoration of cornified envelopes after CLE rescue. We provide multiple lines of evidence that the CLE originates as lamellar body-limiting membranes fuse with the plasma membrane: (i) ABCA12 patients and Abca12−/− mice display normal CLEs; (ii) CLEs are normal in Netherton syndrome, despite destruction of secreted LB contents; (iii) CLEs are absent in VSP33B-negative patients; (iv) limiting membranes of lamellar bodies are defective in lipid-synthetic autosomal recessive congenital ichthyoses; and (v) lipoxygenases, lipase activity, and LIPN co-localize within putative lamellar bodies.
INTRODUCTION
The corneocyte-bound lipid envelope (CLE), a monolayer enriched in ω-hydroxyceramides, replaces the plasma membrane during epidermal terminal differentiation (Akiyama, 2017; Elias et al., 2014; Gruber et al., 2017; Kihara, 2016; Uchida and Holleran, 2008). The CLE is covalently bound to peptides in the outer surface of the cornified envelope (CE) (Candi et al., 2005; Matsui and Amagai, 2015; Steinert and Marekov, 1999) (Figure 1b). Although its functions remain unknown, it has been speculated that it mediates intercorneocyte cohesion (Wertz et al., 1989), stratum corneum hydration (Elias et al., 2014), and/or permeability barrier homeostasis (Akiyama, 2017; Behne et al., 2000; Elias et al., 2014; Meguro et al., 2000; Takagi et al., 2004).
Figure 1. Dual scaffold functions of CLEs in several ARCIs.
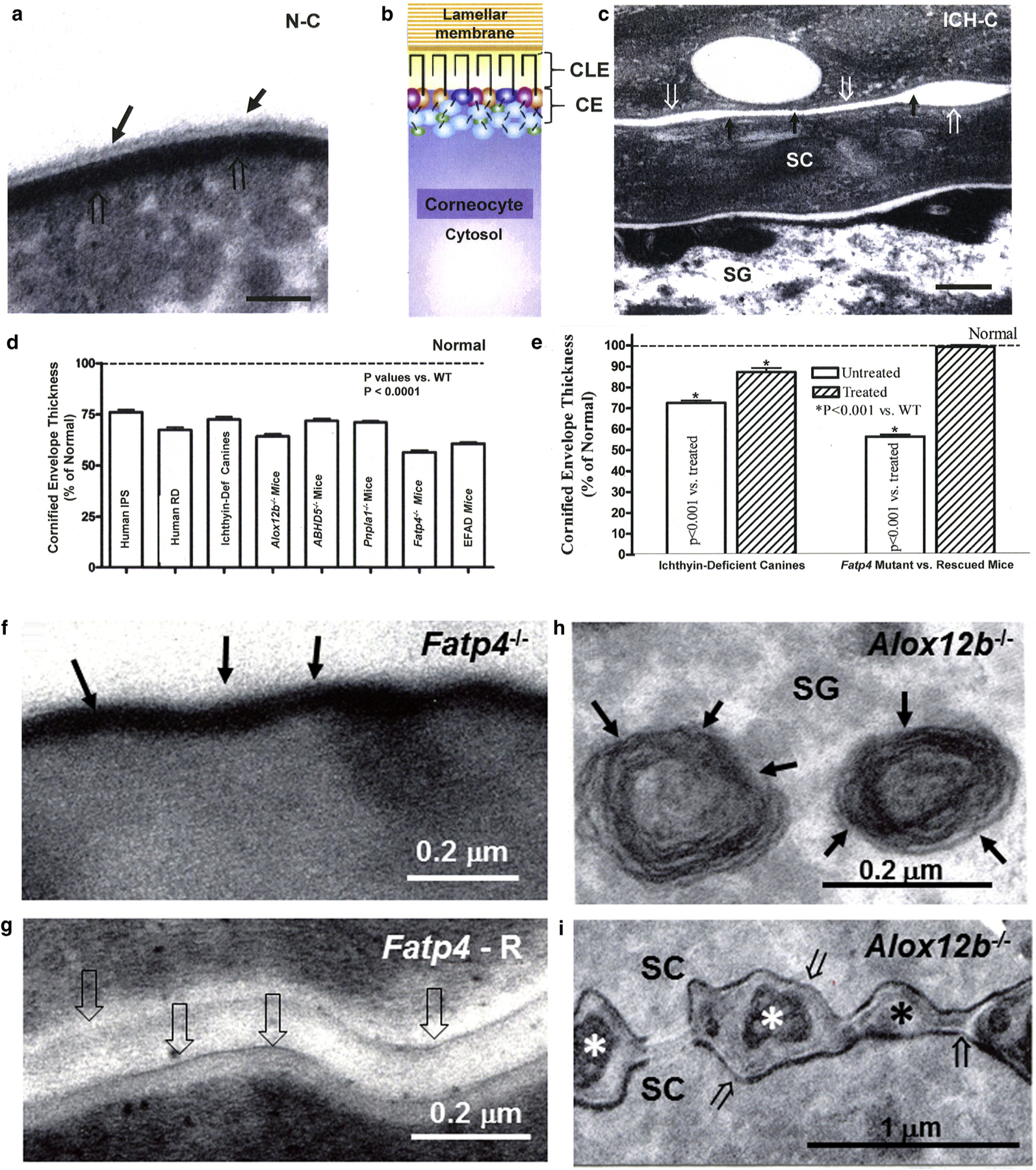
(a, c) Loss of both CLE (solid arrows) and attenuation of cornified envelopes (CE) (open arrows in ichthyin-deficient canines [ICH-C] vs. replete CLEs and CEs in normal littermates [N-C]). (b) Model of the relationship between CLE, CE, lamellar membranes, and corneocyte cytosol in normal stratum corneum. (d, e) Significant decline in CE thickness in several ARCI patients and animal models (mean ± standard error of the mean). (e) Normalization of CE in ichthyin-deficient canines treated with topical ω-O-acylceramide versus vehicle alone. (f) Loss of CLE in Fatp4 knockout (−/−) (solid arrows) and (g) reappearance of CLE (open arrows) after suprabasal transgenic rescue (Fatp4-R) (see Mauldin et al. [2018] for images of CLE in ichthyin-deficient canines]. (h) Despite replete lamellar body contents (solid arrows), (i) secreted contents fail to transform into lamellar membranes in Alox12b−/− mice (asterisk). Note also the loss of CLEs and attenuation of CEs in Alox12b−/− SC (i, open arrows). (c, g, i) osmium tetroxide postfixation. (a, f, m) Ruthenium tetroxide postfixation after pyridine pretreatment. Scale bars = 100 nm in a; 0.2 µm in c, f, g, and h; and 1 µm in i. ARCI, autosomal recessive congenital ichthyosis; CE, cornified envelope; CLE, corneocyte lipid envelope; Def, deficient; IPS, ichthyosis prematurity syndrome; RD, Refsum disease; SC, stratum corneum; SG, stratum granulosum; WT, wild type.
Mutations in enzymes of epidermal fatty acid or ceramide metabolism account for several of the autosomal recessive congenital ichthyoses (ARCIs), in which the generation of ω-O-acylceramides destined for the CLE (Akiyama, 2017; Elias et al., 2014; Kihara, 2016) is compromised, paralleled by defective extracellular lamellar membranes (Akiyama, 2017; Behne et al., 2000; Elias et al., 2010). Two hypotheses have been advanced to explain the pathogenesis of these ARCIs. One proposes that the dermatoses in these patients reflects a failure to generate oxidized linoleic acid metabolites, that is, hepoxilins, which in turn regulate epidermal homeostasis (Brash, 1999; Brash et al., 2007; Fischer, 2009; Krieg et al., 2013; Munoz-Garcia et al., 2014; Yu et al., 2007). Alternatively, or additionally, absence of the CLE could contribute to disease pathogenesis if it fails to provide a scaffold required to support lamellar membrane formation.
We introduce the term lipid synthetic ARCI (shown in Figure 2m) to distinguish these disorders from other ARCIs such as Netherton syndrome, harlequin ichthyosis, and TG1-deficient or ABCA12-deficient lamellar ichthyosis. These ARCIs represent experiments of nature that could clarify the functions and origin of the CLE. Because the CLE represents a structural end product of epidermal fatty acid and ceramide synthesis, its absence could contribute to the pathogenesis of these disorders (Akiyama, 2017; Elias et al., 2014).
Figure 2. Evidence that CLEs form during LB exocytosis.
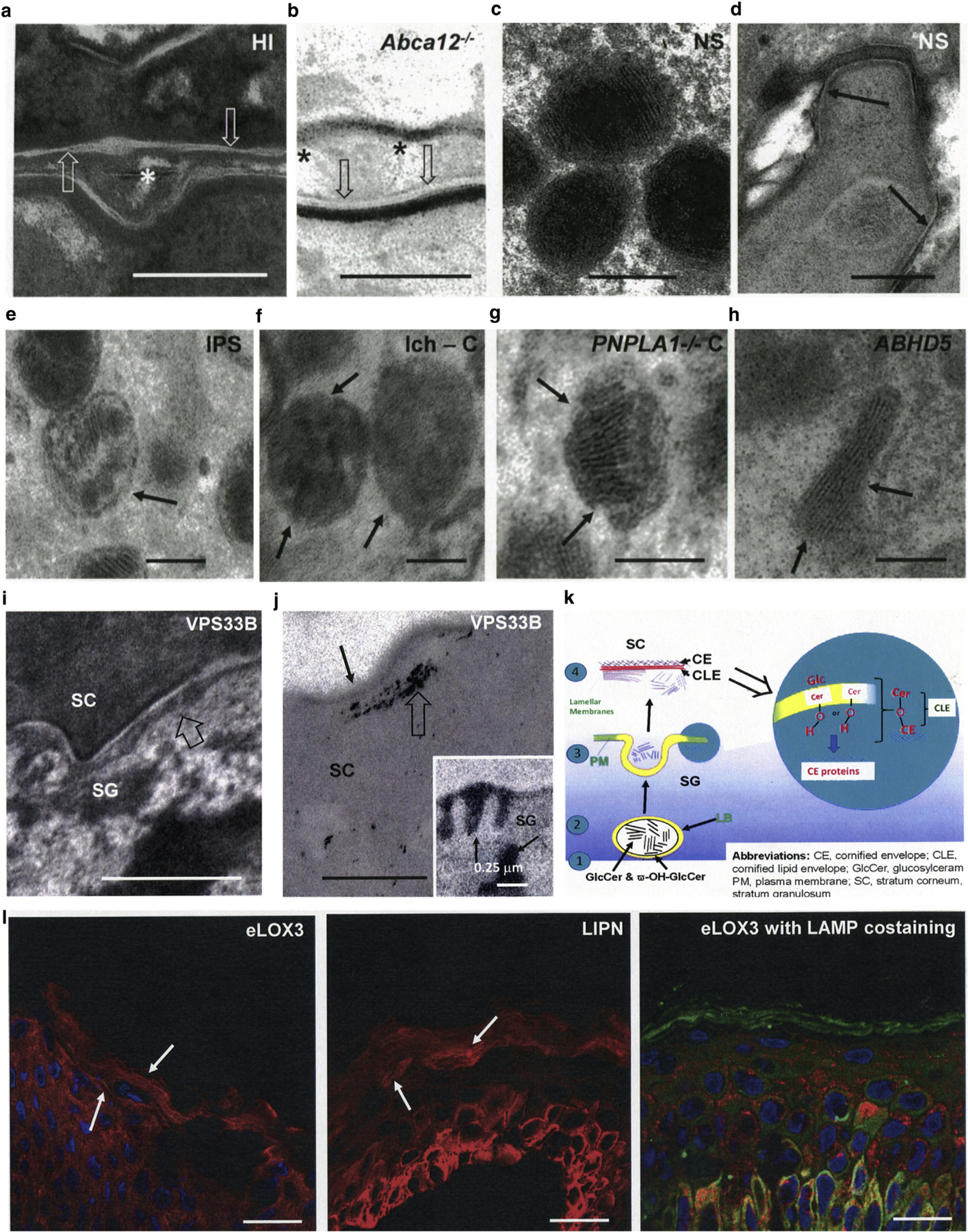
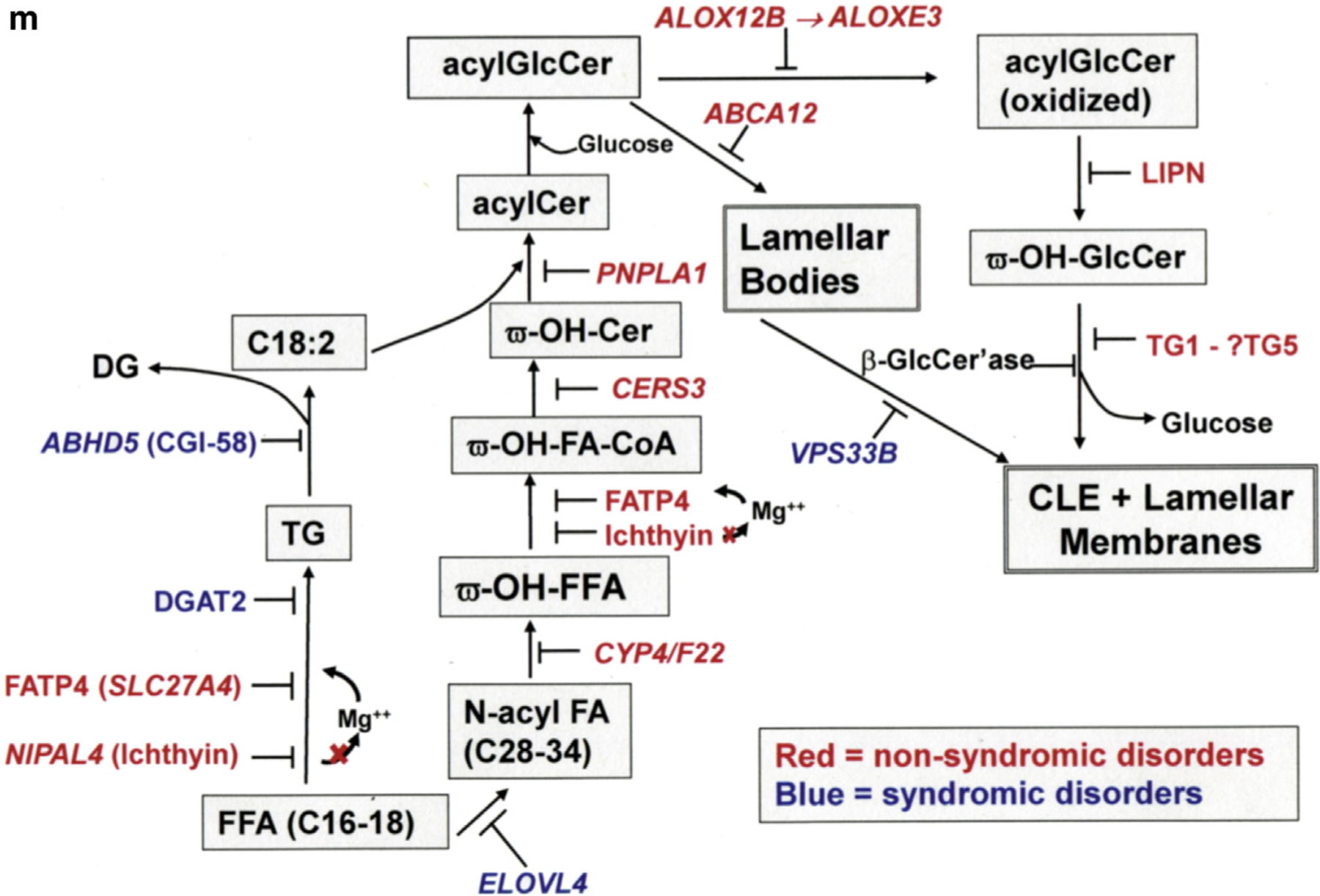
(a, b) Intact CLEs (open arrows), but paucity of extracellular lamellar membranes in harlequin ichthyosis patients and Abca12−/− mice (asterisks). (c, d) Normal LBs and CLEs in Netherton syndrome. (e–h) Abnormal lamellar body-limiting membranes in different ARCI and animal models. Solid arrows point to defects in LB-limiting membranes. (c.f. Figure 1h for another example). (i, j) Paucity of secreted LB contents at the SG-SC interface (i, open arrows) and absence of CLEs (j, solid arrows) in patients with VPS33B mutation (i, open arrow). Note also blockade in secretion results in entombment of lipase activity in the corneocyte cytosol (j, open arrow). (j) (insert) Lipase activity in lamellar bodies and at the SG-SC interface in normal human epidermis (solid arrows). (k) Proposed model of CLE formation with fusion of LB-limiting membranes to plasma membrane in outer SG. (l) Immunolocalization of eLOX3 (EI303 antibody) and LAMP1 in vesicular structures and lamellar membranes in frozen sections of normal human epidermis (solid arrows). (m) Epidermal lipid synthetic pathway leading to formation of the CLE and extracellular lamellar membranes, as well as sites of enzyme blockade in the lipid synthetic ARCI. Scale bars in a, b, d, e, and j = 0.2 µm; in c, f, g, and h = 0.1 µm; in i = 0.5 µm; in j = 0.25 µm; and in j, insert = l–10 µm. ARCI, autosomal recessive congenital ichthyosis; C, canine; CE, cornified envelop; CLE, corneocyte lipid envelope; HI, harlequin ichthyosis; ICH, ichthyin; IPS, ichthyosis prematurity syndrome; LB, lamellar body; M, transgenic mice; NS, Netherton syndrome; SC, stratum corneum; SG, stratum granulosum.
Absent CLEs in lipid synthetic ARCI
CLEs reportedly are defective, and bound ceramides are largely absent, in patients with several lipid synthetic ARCIs and in essential fatty acid-deficient mice (Akiyama, 2017; Elias et al., 2014; Grond et al., 2017a, 2017b; Hirabayashi et al., 2017; Jennemann et al., 2012; Kihara, 2016). To further clarify whether the mutated enzymes in this pathway universally compromise CLE formation, we assessed its status in several additional ARCIs and their canine/mouse analogues. Extracellular lamellar membranes again were defective in all of these additional diseases/animal models (Figure 1c, f, and i vs. 1a) (see also Elias et al., 2010, 2014; Grond et al., 2017a, 2017b; Zhang et al., 2016), expanded to now include (i) ichthyosis prematurity syndrome, (ii) transgenic mice with epidermal targeted deletion of Fatp4, (iii) NIPAL4 (ichthyin)-deficient canines (Mauldin et al., 2018), and (iv) PNPLA1-deficient canines (Grond et al., 2017a; Hirabayashi et al., 2017). Moreover, in many of these ARCI, biochemical studies have shown parallel reductions in bound ω-hydroxyceramide, often with concurrent elevations in unesterified free fatty acids (Grond et al., 2017a, 2017b; Jennemann et al., 2012; Uchida et al., 2010; Zheng et al., 2011). Thus, epidermal fatty acid and ceramide metabolism generate the ω-hydroxyceramide required to form the CLE.
CLEs provide a bidirectional scaffold also necessary for normal CE formation
To assess the potential scaffold role(s) of the CLE, we examined tissue samples from several patients and animal models with lipid synthetic ARCI (Table 1). Not only were lamellar membranes defective, but CEs also were attenuated in these ARCI (Figure 1c and d), suggesting that the CLE could provide a template for the formation of underlying replete CEs. To solidify this possible function, we assessed (and showed that) topical ω-O-acylceramide treatment normalized CE dimensions in ichthyin-deficient canines (Mauldin et al., 2018) (Figure 1e) and that transgenic rescue of the CLE in Fatp4-deficient mice normalized CEs (Figure 1g vs. f). Hence, the CLE provides a bidirectional scaffold that supports both the lamellar membranes and the formation of replete CEs.
Table 1.
Samples assessed in these studies
| Disease Categories (OMIM) | Mutation | Available Material CLE Human Canine Murine Repletion | |||
|---|---|---|---|---|---|
| Fatty acid availability | |||||
| Essential fatty acid deficiency | Not applicable | − | − | + | − |
| Refsum disease (266500) | PHYH | + | − | − | − |
| Fatty acid CoA esterification | |||||
| Ichthyosis prematurity syndrome (608649) | SLC27A4 (Fatp4) | + | − | + | + |
| Ichthyin deficiency (609383) | NIPAL4 | − | + | − | + |
| Acylceramide synthesis | |||||
| Neutral lipid storage disease (275630) | ABHD5 | + | − | + | + |
| ω-hydroxy-transacylation (615024) | PNPLA1 | − | + | + | + |
| Acylceramide metabolism | |||||
| ALOX mutations (607206 and 603741) | Alox12b, Aloxe3 | − | − | + | + |
| Glucosylceramide loading into lamellar bodies | |||||
| Harlequin ichthyosis (242500) | ABCA12 | + | − | + | − |
| Destruction of lamellar body contents | |||||
| Netherton syndrome (256500) | SPINK5 | + | − | − | − |
| Failure of lamellar body exocytosis | |||||
| ARKID syndrome (208085) | VPS33B | + | − | − | − |
Abbreviations: ARKID, autosomal recessive keratoderma-ichthyosis-deafness syndrome; CLE, corneocyte lipid envelope; CoA, coenzyme A.
The CLE, rather than the CE, is required for the transformation of secreted lipids into lamellar membranes
To address more specifically how the CLE supports the generation of the extracellular lamellar membranes, we next examined lamellar membranes in mice with transgenic knockout of 12RLox, one of the two lipoxygenase enzymes. In the ALOXs, the defect in the CLE synthetic pathway occurs distal to loading of glucosylceramide and acyl glucosylceramide into lamellar bodies (LBs) (Figure 2m). Here again, CLEs were absent and CEs attenuated (Figure 1i), although LB production appeared unimpaired (Figure 1h). However, newly secreted LB contents failed to transform into lamellar membranes (Figure 1i).
In contrast, the CE is not required for the generation of lamellar membranes, because CLEs and bound ω-hydroxyceramide (Paige et al., 1994) are normal in TG1-negative lamellar ichthyosis and in loricrin keratoderma, despite markedly abnormal CEs (Elias et al., 2002; Schmuth et al., 2004). However, secreted lamellar contents subsequently become disorganized in both of these disorders, suggesting that the CE mediates a separate scaffold function, that is, maintenance of the supramolecular architecture of the extracellular lamellar membranes.
The CLE originates with exocytosis of LBs
The ω-hydroxyceramides that form the CLE could originate from either a pool of secreted acylglucosylceramides (Kalinin et al., 2001) or with insertion of LBs into the plasma membrane during their exocytosis late in epidermal terminal differentiation. We had long suspected the latter, because normal-appearing CLEs were present in biopsy samples from patients with clinical features of harlequin ichthyosis, obtained during the pre-genotyping era (Elias et al., 2000). To address more definitively whether the CLE originates from a secreted pool of acylglucosylceramides or with insertion of organelle-limiting membranes during terminal differentiation, we next examined a cohort of genetically characterized harlequin ichthyosis patients and Abca12−/− mice (Zhang et al., 2016). In both genotyped humans and Abca12−/− mice, CLEs were present, although extracellular lamellar membranes were largely absent (Figure 2a and b), despite unabated secretion of myriad forme fruste LBs (Chan et al., 2015; Zhang et al., 2016), likely accounting for the ongoing generation of CLEs in both harlequin ichthyosis patients and Abca12−/− mice.
Further evidence for the role of LB exocytosis in the formation of the CLE came from patients with autosomal recessive keratoderma-ichthyosis-deafness syndrome (i.e., ARKID) syndrome due to VPS33B mutations, in which LB exocytosis is impeded (Gissen et al., 2004; Gruber et al., 2017; Hershkovitz et al., 2008) and CLEs again appeared to be absent (Figure 2i and j).
To further ascertain whether the CLE originates from a pool of secreted acylglucosylceramides or during fusion of LB-limiting membranes during exocytosis, we examined CE/CLE structure in Netherton syndrome, in which LB contents form normally and secretion accelerates (Figure 2c), but secreted LB contents are rapidly degraded (Fartasch et al., 1999; Hachem et al., 2006). Accordingly, both CE and CLE structure and dimensions appeared unaltered in Netherton syndrome (Figure 2d).
If fusion of LBs generates the CLE, the limiting membranes of LBs should be defective in the lipid synthetic ARCI. Indeed, LB-limiting membranes appeared defective in all lipid-synthetic ARCIs examined to date (note discontinuities in membranes in Figure 2e–h). Finally, should fusion of LB-limiting membranes generate CLEs, the lipoxygenase enzymes necessary to generate the requisite oxidized ω-hydroxyceramides should localize to LBs. Accordingly, using polyclonal antibodies with specificity against either murine eLOX3 or arachidonate 12-lipoxygenase displayed a prominent, vesicular immunostaining pattern, comparable to the immunolocalization of a standard LB marker, LAMP1 (Figure 2l).
The penultimate step in CLE formation requires lipolysis of the oxidized linoleate moiety from acylglucosylcer-amide before the transesterification of the newly liberated ω-hydroxyglucosylceramides to the CE. Accordingly, LBs contain abundant acidic lipase activity, which remains segregated within the stratum corneum interstices after organelle secretion (Figure 2j, insert) (Menon et al., 1986, 1992). Although properly positioned to remove oxidized, ω-esterified linoleic acid residues from acylglucosylceramide, the responsible protein is not known. Because a small subset of ARCI patients who show mutations in LIPN (Israeli et al., 2011), this epidermal-localized member of the acidic lipase superfamily could account for the observed acidic lipase activity in normal epidermis. Like the lipoxygenases, LIPN displayed a vesicular distribution pattern that mirrored the localization of LBs in the outer epidermis (Figure 2l). Together, these studies suggest that the CLE originates during fusion of LBs with plasma membranes during epidermal terminal differentiation.
COMMENTS
Although abnormalities in permeability barrier homeostasis shape clinical phenotypes in the ichthyoses (Elias et al., 2010; Schmuth et al., 2007; Williams and Elias, 2000), how specific epidermal structures dictate these disease phenotypes is not always clear. Likewise, the function of these structures in normal epidermis can also remain uncertain. One distinctive structure, with still-obscure function(s) in normal epidermis, is the CLE, a monolayer enriched in ω-hydroxyceramides that coats the outer surface of the CE (Akiyama, 2017; Elias et al., 2014; Kihara, 2016; Wertz and Downing, 1986). The CLE replaces the plasma membrane during epidermal terminal differentiation, forming a broad lipid raft that envelopes nascent corneocytes. Although this structure physically bridges the CE to the extracellular lamellar membranes, its precise function(s) remain(s) speculative.
We hypothesized that the lipid synthetic ARCIs comprise experiments of nature, much like knockout mice, that could pinpoint both the role(s) and the origin of the CLE. After assessing the structural consequences of mutations in multiple lipid synthetic ARCI and their animal analogues (Table 1), it seems clear that this pathway converges upon the CLE (Figure 2m) and that its absence (although not always confirmed by lipid biochemistry) compromises its function in these ichthyoses (Akiyama, 2017; Elias et al., 2014). However, we did not examine an alternate or additional possibility, that is, that failure to generate hepoxilin metabolites from oxidized linoleate could also contribute to disease pathogenesis (Brash et al., 2007; Fischer, 2009; Krieg and Furstenberger, 2014; Munoz-Garcia et al., 2014; Yu et al., 2007). Failure to generate these metabolites could compromise the generation of eicosanoid ligands of PPARs, which regulate epidermal homeostasis (Schmuth et al., 2008).
Although loss of the CLE likely contributes to the pathogenesis and clinical phenotype of the lipid synthetic ARCI, the question then becomes How? Based on the lack of identifiable extracellular lamellar membranes in Alox12b−/− mice, despite a demonstration of the ongoing bioavailability of secreted ceramides, the CLE likely supports the initial transformation of secreted lipids into the lamellar membranes that mediate the permeability barrier. In contrast, normal CLEs (and bound ω-hydroxyceramides) (Paige et al., 1994) are generated in both TG1-negative lamellar ichthyosis and loricrin keratoderma, despite prominent abnormalities in CE structure (Elias et al., 2002; Schmuth et al., 2004). In contrast to ALOX deficiency, however, extracellular lamellar membranes form initially in both lamellar ichthyosis and loricrin keratoderma, but they subsequently become disorganized, accounting for their associated permeability barrier abnormalities. Thus, the CE, independent of the CLE, apparently supports a separate function, that is, maintenance of the supramolecular architecture of the extracellular lamellar membranes.
Unexpectedly, mutations in the lipid synthetic ARCI result not only in an absence of the CLE but also in attenuation of underlying CEs. To determine whether the CLE supports the formation of replete CEs, we assessed whether either topical replenishment of acylceramide or transgenic rescue reconstituted CLEs, while also normalizing underlying CEs. These unrelated approaches both restored CLEs and/or bound ω-hydroxyceramides in parallel with normalization of CE dimensions in (i) NIPAL4-deficient canines (Mauldin et al., 2018), (ii) Fatp4−/− mice (Moulson et al., 2007), and (iii) Pnpla1−/− mice (Grond et al., 2017a). Hence, the CLE provides a bidirectional scaffold required for both lamellar membrane formation and generation of a replete, underlying CE. However, additional replenishment or genetic rescue studies in other lipid-synthetic ARCI would help to solidify this paradigm.
Because a normal-appearing CLE is formed (Elias et al., 2002; Schmuth et al., 2004) and covalently bound ω-hydroxyceramides remain normal in TG1-deficient skin (Paige et al., 1994), the transesterification of ω-hydroxyglucosylceramides to involucrin residues in the CE cannot be catalyzed by TG1 (as proposed by Nemes and Steinert, 1999). Instead, another transglutaminase isozyme (e.g., TG5) likely is up-regulated in these patients. The localization of lipoxygenase enzymes, acidic lipase, and LIPN protein to LBs shows that this cluster is positioned to potentially remove oxidized linoleate moieties from acyl glucosylceramide, and this lipid still remains anchored to the LB-limiting membranes. Lipolysis of the oxidized linoleate then would be followed by cross-linking of the newly liberated ω-hydroxyceramides to the CE by the putative alternate transglutaminase isomer.
Finally, we provide multiple lines of evidence that the CLE forms as the limiting membranes of LBs fuse with the apical plasma membrane of stratum granulosum cells (Figure 2k) rather than from a pool of secreted acyl glucosylceramide (Kalinin et al., 2001). This evidence includes our demonstration (i) that normal-appearing CLEs are present in both harlequin ichthyosis and Abca12−/− mice, where forme fruste LBs continue to be formed and secreted (Chan et al., 2015; Zhang et al., 2016); (ii) the presence of defective limiting membranes of LBs in lipid-synthetic ARCI; (iii) the persistence of normal CLEs (and CEs) in Netherton syndrome, where LB secretion accelerates but secreted LB contents are destroyed by unopposed kallikrein activity (Fartasch et al., 1999; Hachem et al., 2006); (iv) the absence of CLEs in autosomal recessive keratoderma-ichthyosis-deafness syndrome, where mutations in VPS33B compromise LB trafficking and secretion (Gissen et al., 2004; Gruber et al., 2017); and (v) the immunolocalization of lipoxygenase enzymes, LIPN protein. and acidic lipase activity to vesicular structures likely representing LBs. Although the lipoxygenases co-localize to vesicular organelles that also express LAMP1, a standard LB marker, LIPN apparently inhabits a separate pool of LAMP1-negative organelles (Figure 2l). Although LIPN’s function is unknown, it is an epidermal-localized member of the acidic lipase superfamily (Hide et al., 1992; Holmes et al., 2010; Wong and Schotz, 2002). Because this protein immunolocalizes to LBs and because abundant enzyme activity is present in LBs (Menon et al., 1986, 1992), LIPN could account for this acidic lipase activity. Although these disparate observations provide robust evidence that the CLE forms during the exocytosis of LBs, definitive proof of this hypothesis will require the lipid biochemical characterization of LB-limiting membranes.
METHODS AND MATERIALS
Patient material and animal models
We examined biopsy material from our library of genotyped patient samples and animal models, which included patient biopsy samples submitted to us from outside centers under a protocol approved by the UCSF Committee for Human Research no. 10–00944 (Table 1). FATP4-knockout mice and rescued mice (FATP4 transgene under keratinocyte-specific involucrin promoter) have been described (Moulson et al., 2007). Abdominal skin of 5-month-old sibling female canines with NIPAL4 mutations were treated daily with ω-esterified C18:2, as described (Mauldin et al., 2018).
Light microscopy and immunohistochemistry
Next, 6-µm skin punch biopsies were performed under local anesthesia, divided into flash-frozen or formation-fixed samples, and processed for hematoxylin and eosin staining and immunohistochemistry. Paraffin-embedded sections from affected and wild-type control animals were incubated with either rabbit polyclonal anti-ichthyin (sc-133280; Santa Cruz Biotechnology, Dallas, TX), or rabbit polyclonal FATP4 antibody (Abcam, Cambridge, UK) (0.865 mg/ml). For immunodetection of ichthyin and FATP4, a LSAB2 kit (Dako, Carpinteria, CA) was used with 3,3′-diaminobenzidine and hematoxylin counterstaining.
For immunofluorescence, neonatal foreskins were fixed in 4% formalin, embedded in optimal cutting temperature compound and snap-frozen in liquid nitrogen. Next, 5-µm cryosections were postfixed in pre-chilled acetone at −20 °C for 10 minutes and washed in phosphate buffered saline/0.5% TritonX-100 (Millipore Sigma, St. Louis, MO). After blocking for 30 minutes in 1% bovine serum albumin/phosphate buffered saline plus 5% cold-water fish gelatin, sections were incubated overnight at 4 °C with primary antibodies: anti-mouse arachidonate 12-lipoxygenase (Y14334), 1:100; anti-mouse el3.3 (mouse epidermal lipoxygenase-3) (Y14885), 1:200, anti-human LIPN (Thermo Fisher Scientific, Waltham, MA), 1:50; and anti-human LAMP1 (Abcam), 1:25. Slides were washed and incubated with appropriate secondary antibodies (1:1,000) for 1.5 hours: anti-rabbit Alexa 555, anti-mouse Alexa 488, anti-guinea pig Alexa 594 (Thermo Fisher Scientific). Slides were mounted with Fluoroshield with DAPI (Sigma-Aldrich, St. Louis, MO), and imaged on a Zeiss (Oberkochen, Germany) confocal microscope using a 63X Plan-Apochromat lens; 5-µm Z stacks were obtained every 0.272 um, processed with Fiji (National Institutes of Health, Bethesda, MD), and displayed at maximum intensities.
Additional frozen human skin samples were cryoprotected in 30% sucrose, embedded in optimal cutting temperature compound, cryosectioned (5 µm), air-dried, washed with 0.15 mol/L phosphate buffered saline and incubated for 18 hours with either (i) rabbit anti-ichthyin (Santa Cruz Biotechnology) (1:300) in Antibody Diluent Reagent Solution (Invitrogen, Waltham, MA), (ii) anti-LAMP1 (Alexa 488), (iii) anti-eLOX3 (guinea pig, Alexa 597), (iv) or anti-LIPN antibodies (Alexa 597/568) (1:500), followed by incubation with Alexa 568 goat anti-rabbit IgG secondary antibody (Thermo Fisher Scientific). After being washed with phosphate buffered saline, slides were mounted in Vectashield with 4′,6-dia-midino-2-phenylindole (DAPI; Vector Labs, Burlingame, CA).
Ultrastructural methods
For standard electron microscopy, skin biopsy samples were minced to smaller than 0.5-mm3 fragments, rinsed three times in 0.1 mol/L cacodylate buffer, and prefixed in half-strength Karnovsky’s fixative, followed by postfixation in (i) reduced osmium tetroxide and ruthenium tetroxide (Polysciences, Warrington, PA), followed by epoxy-Epon (Hexion, Columbus, OH) embedding (Hou et al., 1991). To quantify CE dimensions, 2-mm punch biopsy samples were flash frozen and thawed in absolute pyridine for 2 hours at room temperature (Lin et al., 2012), rinsed in 0.1 mol/L cacodylate buffer, and postfixed with reduced osmium tetroxide before imaging on a JEOL (Peabody, MA) 100CX electron microscope at 60 V, using a Gatan (Pleasanton, CA) Bioscan camera (model 792).
Ultrastructural quantitation
CE measurements were performed directly on the JEOL microscope at 29,000 with the Gatan camera by using a measuring tool in the camera’s software, calibrated using catalase crystals or a diffraction grating replica for higher and lower magnifications, respectively. Five images were taken of each sample, with 15 measurements obtained at the thinnest (most perpendicular) portions of CEs from each image (two or three different tissue samples).
Lipase ultrastructural cytochemistry
The delivery and secretion of LB contents was assessed by lipase ultracytochemistry, as previously described (Menon et al., 1986, 1992; Rassner et al., 1997). Tissue samples then were immersed in 0.1% lead nitrate for 10 minutes and postfixed as described for 1 hour in the dark, followed by dehydration and embedding.
Lipid extraction, fractionation, and quantitation of epidermal ceramides
Scale was acquired by surface scrapings from several ARCI patients and affected animal models (Table 1). Scale samples were homogenized in water, followed by extraction and quantitation of bound and unbound ceramides by gas liquid chromatography, coupled to electrospray ionization tandem mass spectrometry (i.e., LC-ESI-MS/MS), monitored in the positive ion mode (Eckl et al., 2013; Uchida, et al, 2010). Ceramide species were identified using internal ceramide standards (d17:1/C18:0) and were normalized to sample dry weight.
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institute of Arthritis, Musculoskeletal and Skin Diseases of the National Institutes of Health (NIH) (R01 AR061106), administered by the Northern California Institute for Research and Education, with additional resources provided by the Veterans Affairs Medical Center, San Francisco, CA; NIH grants P40 OD010939 and R01 AR068392; and the Commonwealth of Pennsylvania. We gratefully acknowledge our patient with ichthyin deficiency and the Foundation for Ichthyosis and Related Skin Types (FIRST). This study was supported in part by Helen Diller Family Comprehensive Cancer Center (HDFCCC) Laboratory for Cell Analysis Shared Resource Facility through a grant from NIH (P30 CA082103) and through NIH grant R01 AR51968 (to A.R.B.). This content is solely the responsibility of the authors and does not necessarily represent the official views of either the National Institutes of Health or the Department of Veterans Affairs.
Abbreviations:
- ARCI
autosomal recessive congenital ichthyosis
- CE
cornified envelope
- CLE
corneocyte lipid envelope
- LB
lamellar body
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
REFERENCES
- Akiyama M Corneocyte lipid envelope (CLE), the key structure for skin barrier function and ichthyosis pathogenesis. J Dermatol Sci 2017;88: 3–9. [DOI] [PubMed] [Google Scholar]
- Behne M, Uchida Y, Seki T, de Montellano PO, Elias PM, Holleran WM. Omega-hydroxyceramides are required for corneocyte lipid envelope (CLE) formation and normal epidermal permeability barrier function. J Invest Dermatol 2000;114:185–92. [DOI] [PubMed] [Google Scholar]
- Brash AR. Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem 1999;274:23679–82. [DOI] [PubMed] [Google Scholar]
- Brash AR, Yu Z, Boeglin WE, Schneider C. The hepoxilin connection in the epidermis. FEBS J 2007;274:3494–502. [DOI] [PubMed] [Google Scholar]
- Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol 2005;6:328–40. [DOI] [PubMed] [Google Scholar]
- Chan A, Godoy-Gijon E, Nuno-Gonzalez A, Crumrine D, Hupe M, Choi EH, et al. Cellular basis of secondary infections and impaired desquamation in certain inherited ichthyoses. JAMA Dermatol 2015;151:285–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckl KM, Tidhar R, Thiele H, Oji V, Hausser I, Brodesser S, et al. Impaired epidermal ceramide synthesis causes autosomal recessive congenital ichthyosis and reveals the importance of ceramide acyl chain length. J Invest Dermatol 2013;133:2202–11. [DOI] [PubMed] [Google Scholar]
- Elias PM, Fartasch M, Crumrine D, Behne M, Uchida Y, Holleran WM. Origin of the corneocyte lipid envelope (CLE): observations in harlequin ichthyosis and cultured human keratinocytes. J Invest Dermatol 2000;115:765–9. [DOI] [PubMed] [Google Scholar]
- Elias PM, Gruber R, Crumrine D, Menon G, Williams ML, Wakefield JS, et al. Formation and functions of the corneocyte lipid envelope (CLE). Biochim Biophys Acta 2014;1841: 314–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias PM, Schmuth M, Uchida Y, Rice RH, Behne M, Crumrine D, et al. Basis for the permeability barrier abnormality in lamellar ichthyosis. Exp Dermatol 2002;11:248–56. [DOI] [PubMed] [Google Scholar]
- Elias PM, Williams ML, Crumrine D, Schmuth M. Ichthyoses—clinical, biochemical, pathogenic, and diagnostic assessment. In: Itin P, editor. Current problems in dermatology, vol. 39. Basel, Switzerland: S. Kargar AG; 2010. [Google Scholar]
- Fartasch M, Williams ML, Elias PM. Altered lamellar body secretion and stratum corneum membrane structure in Netherton syndrome: differentiation from other infantile erythrodermas and pathogenic implications. Arch Dermatol 1999;135:823–32. [DOI] [PubMed] [Google Scholar]
- Fischer J Autosomal recessive congenital ichthyosis. J Invest Dermatol 2009;129:1319–21. [DOI] [PubMed] [Google Scholar]
- Gissen P, Johnson CA, Morgan NV, Stapelbroek JM, Forshew T, Cooper WN, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet 2004;36:400–4. [DOI] [PubMed] [Google Scholar]
- Grond S, Eichmann TO, Dubrac S, Kolb D, Schmuth M, Fischer J, et al. PNPLA1 deficiency in mice and humans leads to a defect in the synthesis of omega-O-acylceramides. J Invest Dermatol 2017a;137:394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grond S, Radner FP, Eichmann TO, Kolb D, Grabner GF, Wolinski H, et al. Skin barrier development depends on CGI-58 protein expression during late-stage keratinocyte differentiation. J Invest Dermatol 2017b;137: 403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber R, Rogerson C, Windpassinger C, Banushi B, Straatman-Iwanowska A, Hanley J, et al. Autosomal recessive keratoderma-ichthyosis-deafness (ARKID) syndrome is caused by VPS33B mutations affecting Rab protein interaction and collagen modification. J Invest Dermatol 2017;137:845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachem JP, Wagberg F, Schmuth M, Crumrine D, Lissens W, Jayakumar A, et al. Serine protease activity and residual LEKTI expression determine phenotype in Netherton syndrome. J Invest Dermatol 2006;126:1609–21. [DOI] [PubMed] [Google Scholar]
- Hershkovitz D, Mandel H, Ishida-Yamamoto A, Chefetz I, Hino B, Luder A, et al. Defective lamellar granule secretion in arthrogryposis, renal dysfunction, and cholestasis syndrome caused by a mutation in VPS33B. Arch Dermatol 2008;144:334–40. [DOI] [PubMed] [Google Scholar]
- Hide WA, Chan L, Li WH. Structure and evolution of the lipase superfamily. J Lipid Res 1992;33: 167–78. [PubMed] [Google Scholar]
- Hirabayashi T, Anjo T, Kaneko A, Senoo Y, Shibata A, Takama H, et al. PNPLA1 has a crucial role in skin barrier function by directing acylceramide biosynthesis. Nat Commun 2017;8:14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes RS, Cox LA, VandeBerg JL. Comparative studies of mammalian acid lipases: evidence for a new gene family in mouse and rat (Lipo). Comp Biochem Physiol Part D Genomics Proteomics 2010;5:217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou SY, Mitra AK, White SH, Menon GK, Ghadially R, Elias PM. Membrane structures in normal and essential fatty acid-deficient stratum corneum: characterization by ruthenium tetroxide staining and x-ray diffraction. J Invest Dermatol 1991;96:215–23. [DOI] [PubMed] [Google Scholar]
- Israeli S, Khamaysi Z, Fuchs-Telem D, Nousbeck J, Bergman R, Sarig O, et al. A mutation in LIPN, encoding epidermal lipase N, causes a late-onset form of autosomal-recessive congenital ichthyosis. Am J Hum Genet 2011;88:482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennemann R, Rabionet M, Gorgas K, Epstein S, Dalpke A, Rothermel U, et al. Loss of ceramide synthase 3 causes lethal skin barrier disruption. Hum Mol Genet 2012;21:586–608. [DOI] [PubMed] [Google Scholar]
- Kalinin A, Marekov LN, Steinert PM. Assembly of the epidermal cornified cell envelope. J Cell Sci 2001;114:3069–70. [DOI] [PubMed] [Google Scholar]
- Kihara A. Synthesis and degradation pathways, functions, and pathology of ceramides and epidermal acylceramides. Prog Lipid Res 2016;63:50–69. [DOI] [PubMed] [Google Scholar]
- Krieg P, Furstenberger G. The role of lipoxygenases in epidermis. Biochim Biophys Acta 2014;1841:390–400. [DOI] [PubMed] [Google Scholar]
- Krieg P, Rosenberger S, de Juanes S, Latzko S, Hou J, Dick A, et al. Aloxe3 knockout mice reveal a function of epidermal lipoxygenase-3 as hepoxilin synthase and its pivotal role in barrier formation. J Invest Dermatol 2013;133: 172–80. [DOI] [PubMed] [Google Scholar]
- Lin TK, Crumrine D, Ackerman LD, Santiago JL, Roelandt T, Uchida Y, et al. Cellular changes that accompany shedding of human corneocytes. J Invest Dermatol 2012;132:2430–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Amagai M. Dissecting the formation, structure and barrier function of the stratum corneum. Int Immunol 2015;27:269–80. [DOI] [PubMed] [Google Scholar]
- Mauldin EA, Crumrine D, Casal ML, Jeong S, Opalka L, Vavrova K, et al. Cellular and metabolic basis for the Ichthyotic phenotype in NIPAL4 (Ichthyin)-deficient canines. Am J Pathol 2018;188:1419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meguro S, Arai Y, Masukawa Y, Uie K, Tokimitsu I. Relationship between covalently bound ceramides and transepidermal water loss (TEWL). Arch Dermatol Res 2000;292:463–8. [DOI] [PubMed] [Google Scholar]
- Menon GK, Ghadially R, Williams ML, Elias PM. Lamellar bodies as delivery systems of hydrolytic enzymes: implications for normal and abnormal desquamation. Br J Dermatol 1992;126:337–45. [DOI] [PubMed] [Google Scholar]
- Menon GK, Grayson S, Elias PM. Cytochemical and biochemical localization of lipase and sphingomyelinase activity in mammalian epidermis. J Invest Dermatol 1986;86:591–7. [DOI] [PubMed] [Google Scholar]
- Moulson CL, Lin MH, White JM, Newberry EP, Davidson NO, Miner JH. Keratinocyte-specific expression of fatty acid transport protein 4 rescues the wrinkle-free phenotype in Slc27a4/Fatp4 mutant mice. J Biol Chem 2007;282: 15912–20. [DOI] [PubMed] [Google Scholar]
- Munoz-Garcia A, Thomas CP, Keeney DS, Zheng Y, Brash AR. The importance of the lipoxygenase-hepoxilin pathway in the mammalian epidermal barrier. Biochim Biophys Acta 2014;1841:401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemes Z, Steinert PM. Bricks and mortar of the epidermal barrier. Exp Mol Med 1999;31: 5–19. [DOI] [PubMed] [Google Scholar]
- Paige DG, Morse-Fisher N, Harper JI. Quantification of stratum corneum ceramides and lipid envelope ceramides in the hereditary ichthyoses. Br J Dermatol 1994;131:23–7. [DOI] [PubMed] [Google Scholar]
- Rassner UA, Crumrine DA, Nau P, Elias PM. Microwave incubation improves lipolytic enzyme preservation for ultrastructural cytochemistry. Histochem J 1997;29:387–92. [DOI] [PubMed] [Google Scholar]
- Schmuth M, Fluhr JW, Crumrine DC, Uchida Y, Hachem JP, Behne M, et al. Structural and functional consequences of loricrin mutations in human loricrin keratoderma (Vohwinkel syndrome with ichthyosis). J Invest Dermatol 2004;122:909–22. [DOI] [PubMed] [Google Scholar]
- Schmuth M, Gruber R, Elias P, Williams M. Ichthyosis update: towards a function-driven model of pathogenesis of the disorders of cornification and the role of corneocyte proteins in these disorders. Adv Dermatol 2007;23: 231–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmuth M, Jiang YJ, Dubrac S, Elias PM, Feingold KR. Thematic review series: skin lipids. Peroxisome proliferator-activated receptors and liver X receptors in epidermal biology. J Lipid Res 2008;49:499–509. [DOI] [PubMed] [Google Scholar]
- Steinert PM, Marekov LN. Initiation of assembly of the cell envelope barrier structure of stratified squamous epithelia. Mol Biol Cell 1999;10: 4247–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi Y, Nakagawa H, Kondo H, Takema Y, Imokawa G. Decreased levels of covalently bound ceramide are associated with ultraviolet B-induced perturbation of the skin barrier. J Invest Dermatol 2004;123:1102–9. [DOI] [PubMed] [Google Scholar]
- Uchida Y, Cho Y, Moradian S, Kim J, Nakajima K, Crumrine D, et al. Neutral lipid storage leads to acylceramide deficiency, likely contributing to the pathogenesis of Dorfman-Chanarin syndrome. J Invest Dermatol 2010;130:2497–9. [DOI] [PubMed] [Google Scholar]
- Uchida Y, Holleran WM. Omega-O-acylceramide, a lipid essential for mammalian survival. J Dermatol Sci 2008;51:77–87. [DOI] [PubMed] [Google Scholar]
- Wertz PW, Downing DT. Covalent attachment of ω-hydroxyacid derivatives to epidermal macromolecules: a preliminary characterization. Biochem Biophys Res Commun 1986;137: 992–7. [DOI] [PubMed] [Google Scholar]
- Wertz PW, Swartzendruber DC, Kitko DJ, Madison KC, Downing DT. The role of the corneocyte lipid envelope in cohesion of the stratum corneum. J Invest Dermatol 1989;93: 169–72. [DOI] [PubMed] [Google Scholar]
- Williams M, Elias P. Ichthyosis: where we have been. Disorders of cornification: where we are going. Curr Probl Dermatol 2000;12: 170–6. [Google Scholar]
- Wong H, Schotz MC. The lipase gene family. J Lipid Res 2002;43:993–9. [DOI] [PubMed] [Google Scholar]
- Yu Z, Schneider C, Boeglin WE, Brash AR. Epidermal lipoxygenase products of the hepoxilin pathway selectively activate the nuclear receptor PPARa. Lipids 2007;42:491–7. [DOI] [PubMed] [Google Scholar]
- Zhang L, Ferreyros M, Feng W, Hupe M, Crumrine DA, Chen J, et al. Defects in stratum corneum desquamation are the predominant effect of impaired Abca12 function in a novel mouse model of harlequin ichthyosis. PLoS One 2016;11:e0161465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Yin H, Boeglin WE, Elias PM, Crumrine D, Beier DR, et al. Lipoxygenases mediate the effect of essential fatty acid in skin barrier formation: a proposed role in releasing omega-hydroxyceramide for construction of the corneocyte lipid envelope. J Biol Chem 2011;286:24046–56. [DOI] [PMC free article] [PubMed] [Google Scholar]