

Abstract
Although proteolysis targeting chimeras (PROTACs) have become promising therapeutic modalities, important concerns exist about the potential toxicity of the approach owning to uncontrolled degradation of proteins and undesirable ligase-mediated off-target effects. Precision manipulation of degradation activity of PROTACs could minimize potential toxicity and side effects. As a result, extensive efforts have been devoted to developing cancer biomarker activating prodrugs of PROTACs. In this investigation, we developed a bioorthogonal on-demand prodrug strategy (termed click-release ‘crPROTACs’) that enables on-target activation of PROTAC prodrugs and release of PROTACs in cancer cells selectively. Inactive PROTAC prodrugs TCO-ARV-771 and TCO-DT2216 are rationally designed by conjugating a bioorthogonal trans-cyclooctenes (TCO) group into the ligand of the VHL E3 ubiquitin ligase. The tetrazine (Tz) modified RGD peptide, c(RGDyK)-Tz, which targets integrin αvβ3 biomarker in cancer cells, serves as the activation component for click-release of the PROTAC prodrugs to achieve targeted degradation of proteins of interest (POIs) in cancer cells versus noncancerous normal cells. The results of studies accessing the viability of this strategy show that the PROTAC prodrugs are selectively activated in an integrin αvβ3-dependent manner to produce PROTACs, which degrade POIs in cancer cells. The crPROTAC strategy might be a general, abiotic approach to induce selective cancer cell death through the ubiquitin-proteasome pathway.
Graphical Abstract
INTRODUCTION
Emerging proteolysis-targeting chimaeras (PROTACs) exploit cellular quality control machinery to selectively degrade target proteins including those that are undruggable such as transcriptional factors and scaffold proteins.1–4 PROTACs, which do not rely on occupancy-driven pharmacology, offers unparalleled advantages over traditional small-molecule inhibition strategies because they can be designed to degrade protein targets in a sub-stoichiometric, catalytic fashion and overcome drug resistance.5 However, toxicity arising from degradation of proteins in healthy cells and undesirable ligase-mediated off-target effects could limit clinical applications of PROTACs.6–7 To address these issues, PROTAC prodrugs have been devised to be selectively delivered and/or activated at the tumor sites. These prodrugs rely mainly on tumor biomarkers8, such as tumor microenvironment,9 folate,10 and aptamers.11 However, these approaches are plagued by several issues including activation before the prodrug enters cancer cells. Moreover, protein receptors are rarely tumor cell specific and in many cases the expression level between cancer and normal cells is not significant.12–13 Additionally, mutation of protein receptors that brings about therapeutic resistance enhances the vulnerability of these targeting approaches in treating cancer.14–16 In addition, highly selective antibody-drug conjugates (ADCs) have also been applied in new PROTAC based methodologies.17–22 Unfortunately, the use of antibodies suffers from drawbacks including high production cost, receptor saturation, poor solid tumor penetration and severe immunogenicity.23 In addition, each mAb developed is applicable to only a certain types of cancer because the targeted protein receptors are cancer type selective. Abiotic light-activatable PROTACs have been devised to achieve spatiotemporal regulation of PROTAC activation.24–29 Nevertheless, methods based on this strategy are limited to cancer types that have light accessibility and in some cases, and they sometimes require the use of UV light irradiation that can promote undesired tissue damage.
Bioorthogonal-activatable prodrug-based methods have promise because they enable precise delivery and abiotic activation of drugs.30–33 The high reaction rate, and the excellent bioorthogonality and biocompatibility enable the tetrazine (Tz) and trans-cyclooctene (TCO)-engaged inverse-electron-demand Diels-Alder (IEDDA)34–41 to proceed efficiently in complex biological context. As a result, TCO-caged prodrugs have attracted the attention of medicinal chemists, who have been the first to utilize TCO-caged prodrugs in clinical studies of anticancer drug delivery in human.42–44 However, studies of the prodrug strategy have focused mainly on caging and releasing cytotoxic agents, while applications to the delivery of PROTACs has not been reported.45 Moreover, because the Tz components devised to date lack targeted delivery and activation capacities, they need to be utilized generally in excessively large amounts that can result in side effects.
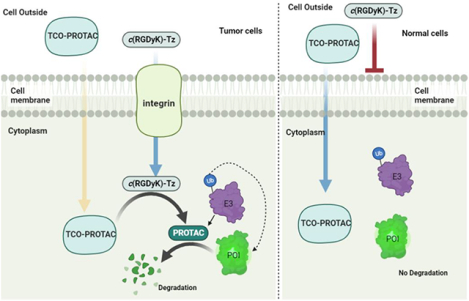
αvβ3 Integrin is one of the most well-studied targets for drug delivery into cancer cells because of its highly expressed on surface of certain cancer cells and the key role it plays in cell invasion and proliferation during tumor vascular remodeling and angiogenesis.46–48 As such, the αvβ3 integrin-targeting strategy has been used for decades for tumor imaging and diagnosis, as well as cancer-targeted drug delivery.49–51 These features stimulated us to incorporate the αvβ3 integrin binding ligand c(RGDyK)52 in the design of the tetrazine containing conjugate c(RGDyK)-Tz for selective activation of TCO-caged PROTAC prodrugs in cancer cells. We believe that this approach would enable controllable targeted degradation of a protein of interest (POI) and minimize potential toxicity to normal tissues (Figure 1A). Toward this end, we designed TCO-PROTACs, TCO-ARV-771 and TCO-DT2216, which possess the TCO moiety on hydroxyl group of the well-studied VHL-based PROATCs, ARV-7717 and DT2216 (Figure 1B).53–55 We anticipated that these prodrugs would be stable and bioorthogonal to biological systems and that they would be selectively activated by IEDDA reaction with c(RGDyK)-Tz. This process would release the corresponding ARV-771 and DT2216 in cancer cells to recruit endogenous VHL E3 ubiquitin ligase to ubiquitinate a POI for subsequent degradation in the proteasome (Figure 1A).
Figure 1:
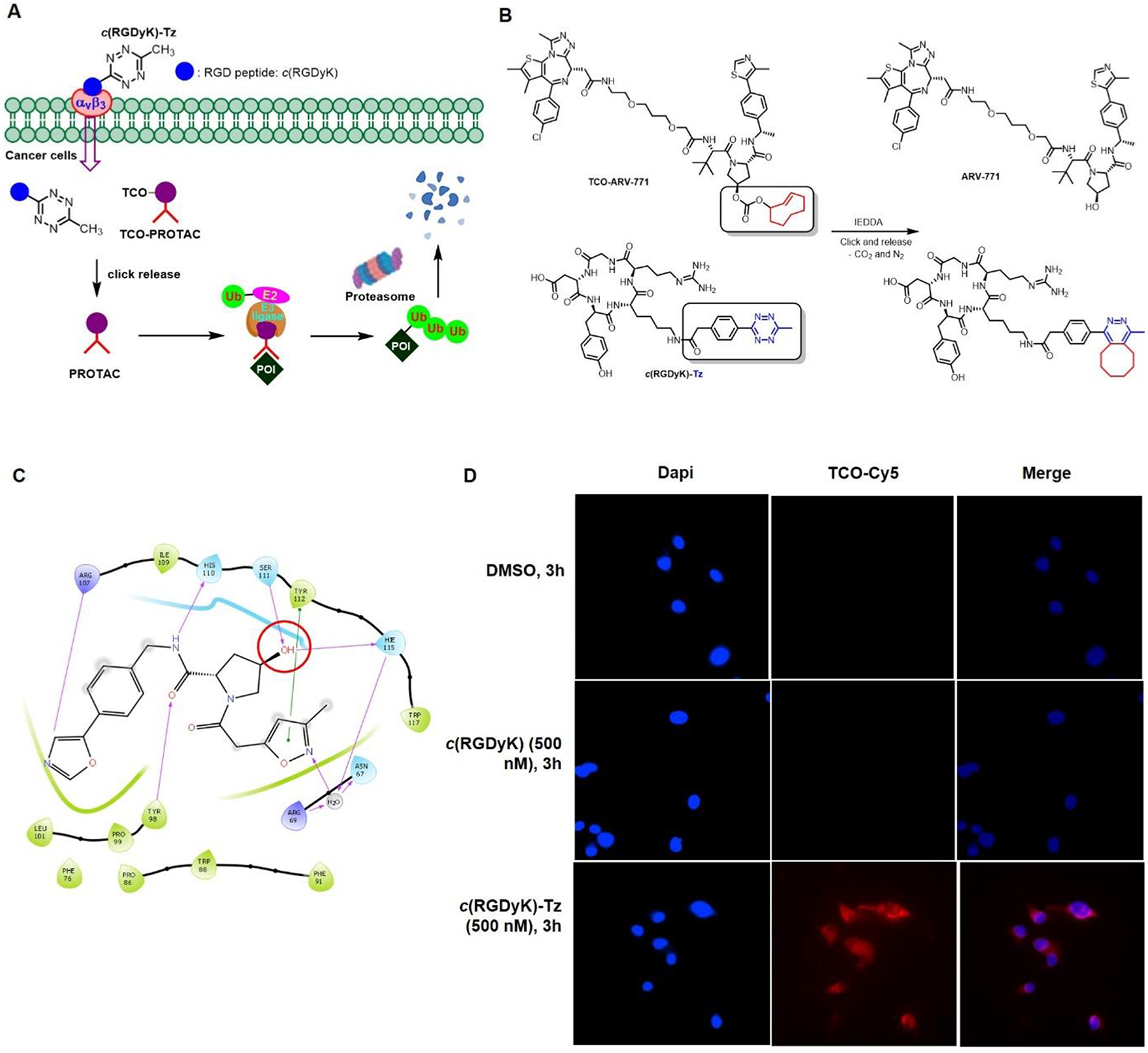
A) Bioorthogonal activation and release of TCO-PROTAC prodrugs by αvβ3 integrin targeted c(RGDyK)-Tz in tumor cells. B) Structures of TCO-ARV-771 and c(RGDyK)-Tz TCO-Tz and The ‘click and release’ reaction between TCO-ARV-771 and c(RGDyK)-Tz releases the activated ARV-771. C) Molecular docking data between VHL protein and VHL ligand. D). HeLa cells imaging with DMSO, c(RGDyK) and c(RGDyK)-Tz. After the 3 h incubation of HeLa cells with DMSO, c(RGDyK) (500 nM) or c(RGDyK)-Tz (500 nM), the cells were washed three times with PBS and then treated with TCO-Cy5 (5.0 μM) for another 15 min. After the cells were washed by PBS three times, the images were taken under the 60 × camera.
RESULTS AND DISCUSSION
Design and synthesis of TCO-ARV-771 prodrugs and αvβ3 integrin targeted c(RGDyK)-Tz.
As described above, we envisioned that incorporation of the TCO moiety into PROTACs would generate the bioorthogonal PROATC prodrugs. Critical to the viability of this approach is that introduction of the TCO moiety should abolish the degradation activity of the PROTAC. Moreover, to ensure that the TCO-PROTAC design is generally applicable, we proposed to install the TCO group into a widely used E3 ubiquitin ligase VHL ligand.56 Importantly, the critical role played by the hydroxyl group in the VHL ligand and in recruiting VHL E3 ubiquitin ligase57–58 was demonstrated in previous studies that show that caging the –OH group in this ligand with folate10 and aptamers11 blocks its degradation promoting activity. These observations guided design of a TCO caged PROTAC in which the TCO moiety is linked via a carbonate group to the hydroxyl group of the well-studied VHL-based bromodomain (BRD) degrader, ARV-771 (Figure 1B). The results of molecular docking studies with the TCO-caged ligand suggested that this modification would dramatically decreased the interactions with residues in the binding pocket of the VHL protein (Figure 1C and Figure S1). It is expected that the TCO-ARV-771 should be inactive and stable in biological media, whereas it can be bioorthogonally activated by Tz to trigger release of ARV-771 degrader. In previous described studies, the Tz component of the TCO prodrug activator are not cancer selective. To achieve cancer cell selective activation in the approach, we designed the Tz moiety is conjugated with the αvβ3 integrin binding ligand c(RGDyK) through a PhMeTz linker chosen because of its balanced stability and reactivity and easy preparation (Figure 1A and 1B).41 To explore the viability of the approach described above, c(RGDyK)-Tz and TCO-ARV-771 were synthesized, characterized and employed in the following biological studies (Scheme S1 and S2).
In vitro study of click-release of TCO-ARV-771 and fluorescence imaging of c(RGDyK)-Tz in αvβ3 integrin expressed HeLa cells.
To demonstrate the click-release capability of the newly designed TCO-ARV-771 (1.0 mM), its IEDDA reaction with c(RGDyK)-Tz (5.0 mM) was carried out in a pH 7.4 phosphate buffer. Real-time monitoring using UFLC (Ultra Fast Liquid Chromatography) showed that TCO-ARV-771 was almost completely consumed within 30 min in this process (Figure S2A), and that ca. 62% of consumed substance was converted to ARV-771. The rate of this reaction was determined using the reported UV-Vis spectroscopic method by monitoring changes in the absorption intensity of the 330 nm band with 5.0 μM of TCO-ARV-771 and 50 μM of c(RGDyK)-Tz.59 The IEDDA reaction takes place rapidly in association with kobs = 2.1 × 10−2, t1/2 = 32.78 s, and k2 = 420 M−1s−1 (Figure S2B). This finding is consistent with the results arising from UFLC monitoring of the process. Finally, the stability of TCO-ARV-771 and c(RGDyK)-Tz were determined using UFLC, and separated incubation of each substance in cell culture Dulbecco’s Modified Eagle Medium (DMEM), which contains 10% Fetal bovine serum with UFLC. The results show that TCO-ARV-771 and c(RGDyK)-Tz are stable over 3 h, a period that is more than sufficient to completion of the click-release process (Figure S3).
As indicated above, c(RGDyK)-Tz is designed to selectively deliver the corresponding PROTAC to cancer cell via cancer cell surface integrin. To validate this proposal, the fluorescence imaging studies were conducted in HeLa cells that overexpresses αvβ3 integrin.60 After optimization of pre-incubation time, we chose to pre-incubate c(RGDyK)-Tz (500 nM) for 3 h in HeLa cells, then fluorescence imaging probe TCO-Cy5 was added and incubated for 15 min, washed before being subjected to fluorescent imaging. The results revealed that the c(RGDyK)-Tz treated cells exhibited a red color associated with Cy5 (Figure 1D). In contrast, cells treated with DMSO or the control c(RGDyK) ligand did not exhibit fluorescence. In addition, the cell imaging method was used to probe HS-27 (normal cell, low expression of αvβ3 integrin) and MDA-MB-231 (breast cancer cell, overexpression of αvβ3 integrin). Red colored fluorescence was observed in images of c(RGDyK)-Tz treated MDA-MB-231 cells but very weak emission arose from treated HS-27 (Figure S4). These observations suggest that c(RGDyK)-Tz selectively and efficiently enters αvβ3 integrin overexpressing tumor cells.
BRD4 degradation in HeLa cells and cell viabilities promoted by bioorthogonal activation of TCO-ARV-771 with c(RGDyK)-Tz.
Having demonstrated that c(RGDyK)-Tz capable of αvβ3 integrin dependent entry into HeLa cells, we next tested the capability of TCO-ARV-771 to undergo click-release of the PROTAC, ARV-771. HeLa cells were incubated with c(RGDyK)-Tz for 3 h, then then washed by PBS buffer, followed by the addition of TCO-ARV-771 for a specified time with different concentrations of both reagents. We found that, as predicted, TCO-ARV-771 itself does not possess the ability to degrade BRD4 in HeLa cells at concentrations in the range of 100–400 nM (Figure 2A). However, treatment of the cells containing 0.4 μM TCO-ARV-771 with various concentrations (0.4–2.0 μM) of c(RGDyK)-Tz induced degradation (Figure 2B). At a concentration of 0.4 μM TCO-ARV-771 used, c(RGDyK)-Tz elicited concentration-dependent degradation of BRD4 with complete degradation at 1.0 μM c(RGDyK)-Tz (3 h, pre-treatment) (Figure 2B). Notably, comparable degradation activity was observed at the same concentration of TCO-ARV-771 prodrug (0.4 μM) and parent drug ARV-771 (Figure 2B, C). This implies that the Tz-mediated click-release drug strategy was able to deliver and activate the active drug ARV-771 highly efficiently. We then conducted the experiments by varying the concentration of TCO-ARV-771 with 1.0 μM c(RGDyK)-Tz (3 h, pre-treatment). Again, complete degradation of BRD4 could be achieved at 0.4 μM TCO-ARV-771 (Figure 4D). Finally, assessments of Hela cell viabilities showed that TCO-ARV-771 prodrug and c(RGDyK)-Tz have much lower cell killing propensities with IC50: 4.45 μM and >10 μM, respectively (Figure 2E). However, cells treated by both substances to promote the crPROTAC process have cytotoxicities (IC50: 389 nM) that are similar to those of ARV-771 (IC50: 466 nM). Taken together, these observations validate the proposal that protein degradation is dependent on the presence of both TCO-ARV-771 and c(RGDyK)-Tz, that the Tz-mediated click-release of ARV-771 from TCO-ARV-771 is highly efficient, and that supported that our the crPROTAC strategy enables efficiently activation and release of ARV-771 in the αvβ3 integrin highly expressed HeLa cells.
Figure 2: The degradation ability of TCO-ARV-771 with c(RGDyK)-Tz in HeLa cells.
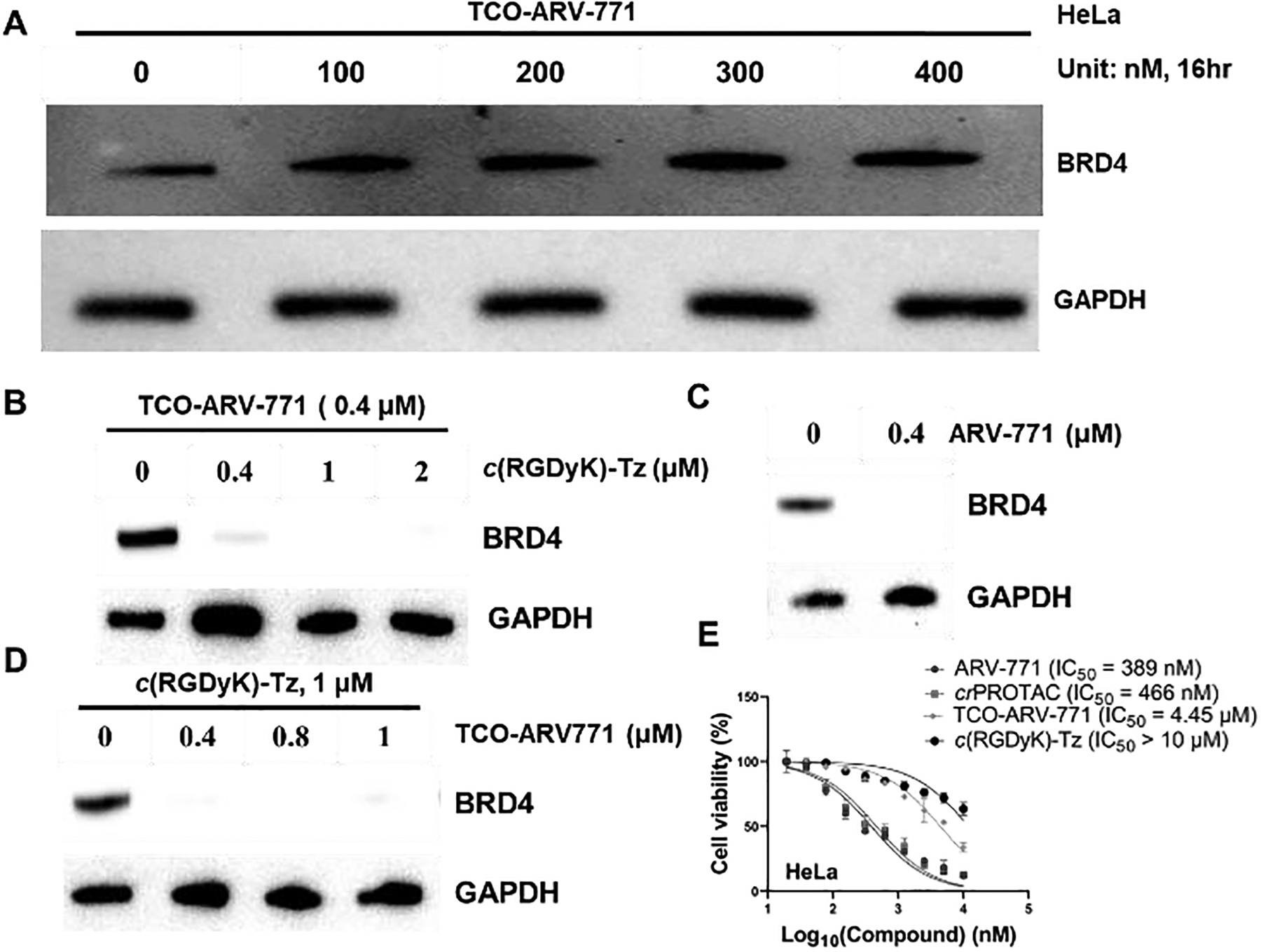
A) Immunoblot analysis of BRD4 levels from HeLa cells treated with TCO-ARV-771 for 16 h. B) Immunoblot analysis of BRD4 levels from HeLa cells treated with 0.4 μM TCO-ARV-771 and various concentration of c(RGDyK)-Tz. HeLa cells were treated with different concentrations of c(RGDyK)-Tz for 3 h followed by treatment with TCO-ARV-771 (0.4 μM) for 16 h. C) Immunoblot analysis of BRD4 levels from HeLa cells treated with positive control ARV-771. D) Immunoblot analysis of BRD4 levels from HeLa cells treated with various concentrations of TCO-ARV-771 and 1.0 μM c(RGDyK)-Tz. HeLa cells were treated with 1.0 μM c(RGDyK)-Tz for 3 h followed by treatment with different concentrations of TCO-ARV-771 for 16 h. E) Cell viability of HeLa cells with ARV-771, TCO-ARV-771, c(RGDyK)-Tz or TCO-ARV-771 and c(RGDyK)-Tz (2.5 equivalent, 3 h pretreatment) for 72 h.
Figure 4: The degradation ability of TCO-ARV-771 with c(RGDyK)-Tz in U87 and HS27 cells.
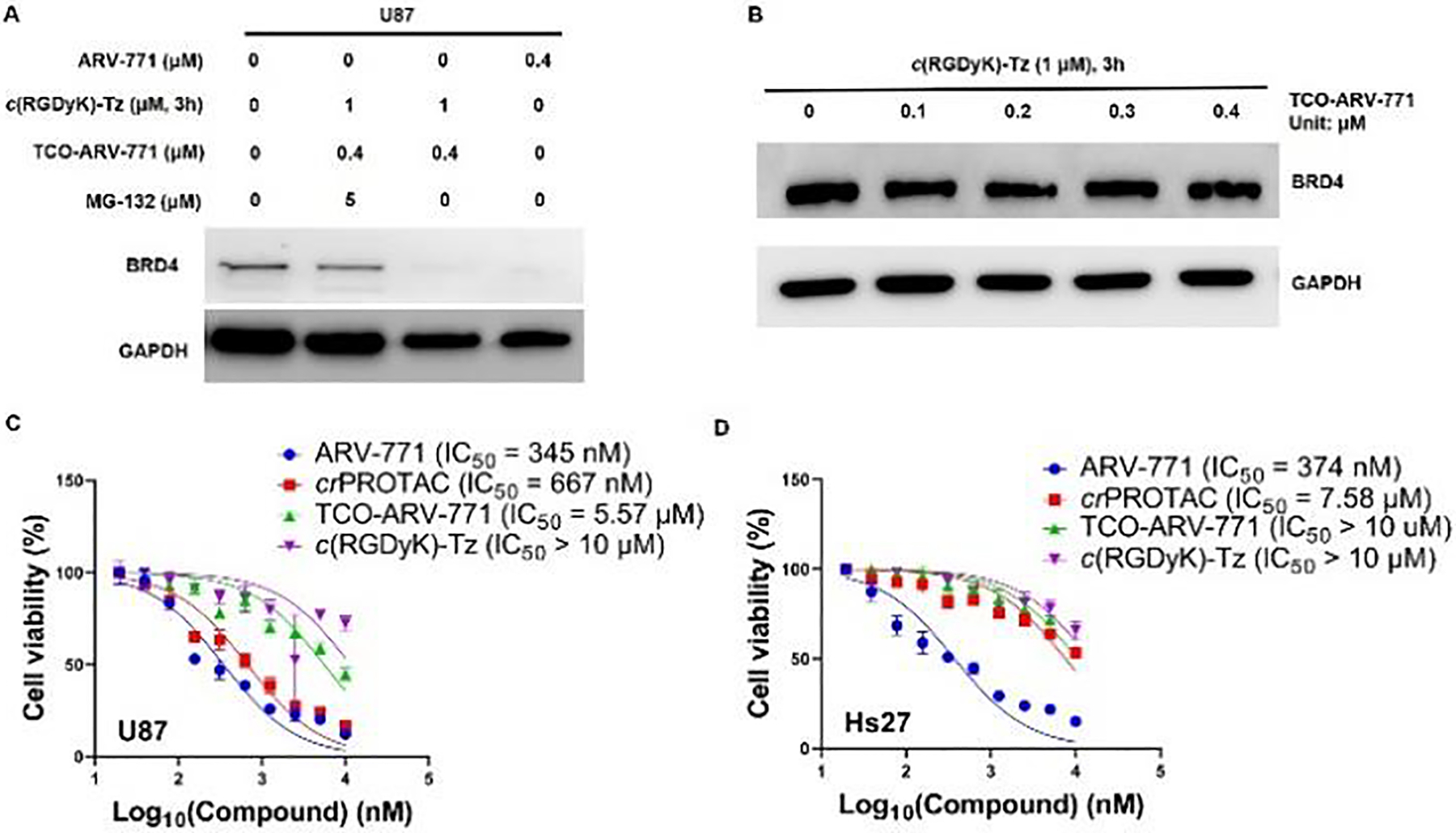
A) Western blot analysis of BRD4 protein levels from U87 cells treated with the indicated doses of treatments for 16 h. B) Western blot analysis of BRD4 and GAPDH levels in HS27 cells with different concentrations of TCO-ARV-771 for 16 h. C, D) Cell viability of U87 or HS27 cells treatment with ARV-771, TCO-ARV-771, c(RGDyK)-Tz or crPROTAC [TCO-ARV-771 + c(RGDyK)-Tz (2.5 equivalent, 3 h pretreatment)] for 72 h.
Degradation of BRD4 by ARV-771 produced from the bioorthogonal reaction of TCO-ARV-771 with c(RGDyK)-Tz is believed to take place via the ubiquitin-proteasome pathway involving ubiquitination and subsequent proteasome degradation. Experiments were performed to confirm this proposal. Specifically, HeLa cells treated with 1.0 μM c(RGDyK)-Tz, incubated for 3 h, washed with PBS, and then incubated for 16 h with either TCO-ARV-771 at various concentrations, ARV-771 and DMSO as control. As described above, 0.4 μM TCO-ARV-771 promoted a level of BRD4 degradation which matches that caused by ARV-771. The results revealed that when the concentration of TCO-ARV-771 was 0.4 μM, the BRD4 protein was degraded efficiently at a similar level to that with ARV-771, and no degradation was observed with DMSO (Figure 3A). However, when the proteasome inhibitor MG-132 was present, BRD4 protein was not degraded (Figure 3B). Furthermore, degradation was reduced significantly when the HeLa cells was first treated with the free VHL ligand, because the VHL ligand competed with generated ARV-771 for ubiquitination (Figure 3C). In a control experiment, co-treatment c(RGDyK) peptide, which does not contain the tetrazine group, with TCO-ARV-771 did not induce BRD4 degradation (Figure 3D). The studies show that ARV-771 formed by crPROTAC process promotes protein degradation via the ubiquitin-proteasome pathway.
Figure 3: The degradation ability of TCO-ARV-771 with c(RGDyK)-Tz in HeLa cells.
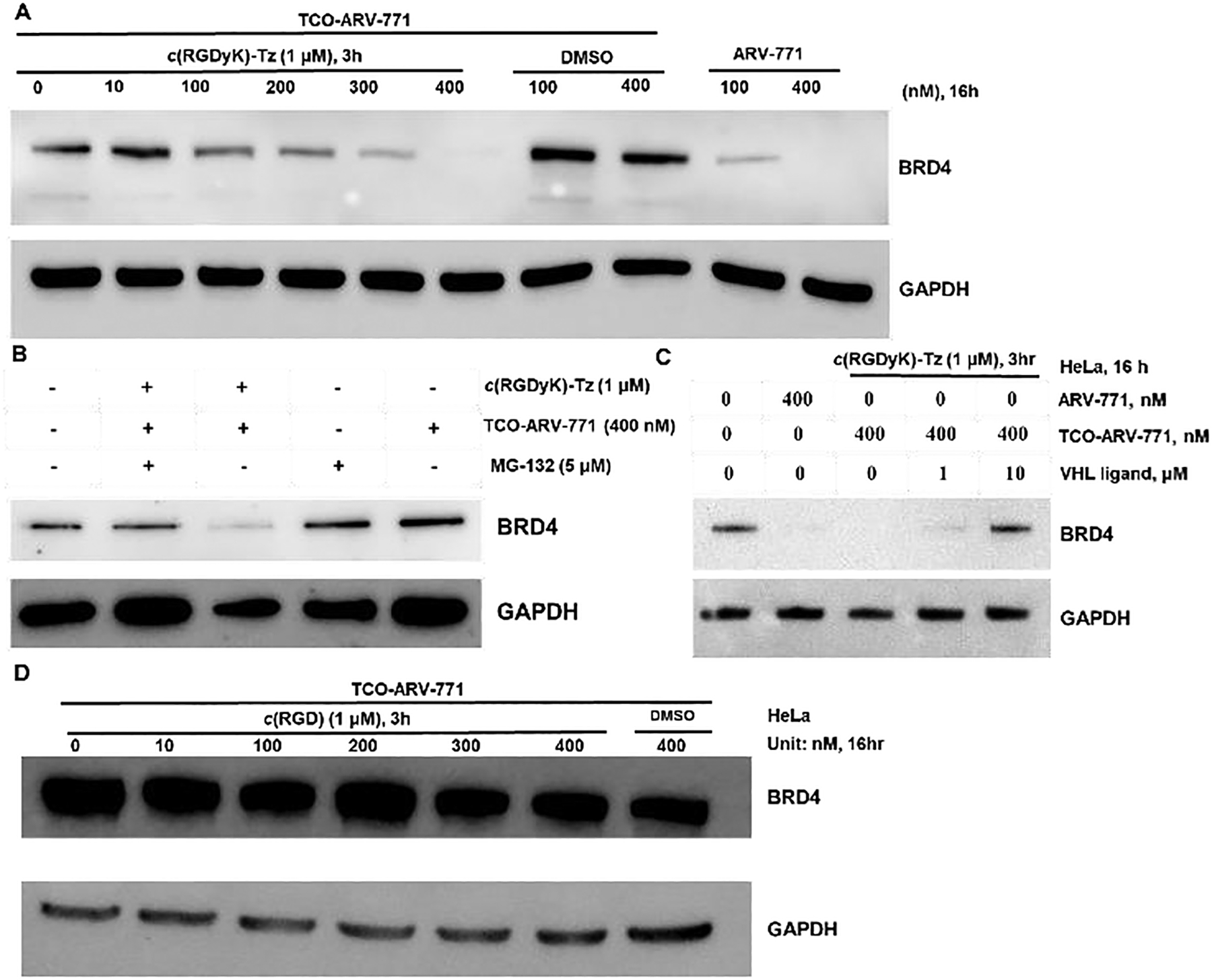
A) Western blot analysis of BRD4 protein levels from HeLa cells treated for 16 h. B) Western blot analysis of BRD4 and GAPDH levels in HeLa cells after the co-treatment with 5 μM MG-132 and crPROTAC (TCO-ARV-771 + c(RGDyK)-Tz, 3h) for 16 h. C) Western blot analysis of BRD4 and GAPDH levels in HeLa cells after the co-treatment with VHL ligand and crPROTAC (TCO-ARV-771 + c(RGDyK)-Tz, 3h) for 16 h. D) Western blot analysis of BRD4 and GAPDH levels in HeLa cells after the co-treatment with c(RGDyK) (3 h) and TCO-ARV-771 for 16 h.
Selective degradation of BRD4 in other αvβ3 integrin highly expressed cancer cells.
We have shown that the bioorthogonal on-demand and on-target strategy described above selectively activates TCO-ARV-771 prodrugs in αvβ3 integrin overexpressed HeLa cells to release the active PROATC ARV-771 for degradation of BRD4 proteins. To demonstrate the generality of this selective delivery and activation of approach, our studies focused on U87 and MDA-MB-231 cancer cell lines, which overexpress αvβ3 integrin as well (Figure S7).61–63 In this effort, low αvβ3 integrin expressing normal fibroblast HS-27 and WI-38 cells were used as controls (Figure S7).64–65 The same protocol used in HeLa cells was employed in both tumor and normal cell lines. Incubation of U87 cells with 0.4 μM TCO-ARV-771 and 1.0 μM c(RGDyK)-Tz gave rise to a BRD4 degradation activity level that is comparable to that induced by treatment of U87 cells with 0.4 μM ARV-771, and when HeLa cells were treated in the same manner (Figure 4A). In the absence of TCO-ARV-771 or 1.0 μM c(RGDyK)-Tz, no BRD4 degradation occurred. Similar observations were made in studies using MDA-MB-231 cells (Figure S5). In addition, blocking αvβ3 integrin by its inhibitor c(RGDyK) affected the efficiency of c(RGDyK)-Tz uptake, subsequent click-release event and ultimately degradation activity. Specifically, compared to MDA-MB-231 cells directly treated with c(RGDyK)-Tz, those pretreated with c(RGDyK) and then incubated c(RGDyK)-Tz and TCO-ARV-771 displayed a significantly decreased BRD4 degradation propensity (Figure S5C). In contrast, protein degradation activity was not promoted in normal cells (HS-27 and WI-38) when they were treated with 0.4 μM TCO-ARV-771 and 1.0 μM c(RGDyK)-Tz. These findings suggest that c(RGDyK)-Tz was not selectively delivered into HS27 or WI-38 cells due to the lack of αvβ3 integrin (Figure S8), which is believed to play the roles in targeting and drug transport (Figure 4B and Figure S6). The effective targeted delivery of ARV-771 from TCO-ARV-771 in U87 cells enabled inducing potent toxicity (IC50 667 nM), which is comparable to those of cells, treated directly with ARV-771 (IC50 345 nM, Figure 4C) and similar to those of HeLa cells treated in the same manner. In contrast, a significantly lower cytotoxicity (IC50: 7.58 μM) was displayed by HS27 cells (Figure 4D). Taken together, the findings demonstrate that the TCO-tetrazine prodrug strategy can be employed to bioorthogonally and selectively target and activate prodrugs in the αvβ3 integrin-positive cancer cells.
Bioorthogonal reaction of TCO-ARV-771 and c(RGDyK)-Tz induces apoptosis in HeLa cells.
Flow cytometry experiments were performed to determine if the bioorthogonal c(RGDyK)-Tz promoted release of ARV-771 from TCO-ARV-771, which induces BRD4 degradation, causes cell apoptosis. While treatment with only 0.4 μM TCO-ARV-771 showed a low toxic effect on HeLa cells (Fig. 5A), 13.58% apoptosis of these cells was induced when the cells were incubated with both TCO-ARV-771 (0.4 μM) and 1.0 μM c(RGDyK)-Tz group (3 h). A similar high apoptosis level was reached when Hela cells were treated with ARV-771 alone (Figure 5A and 5B).
Figure 5: Flow cytometry of apoptosis assays.
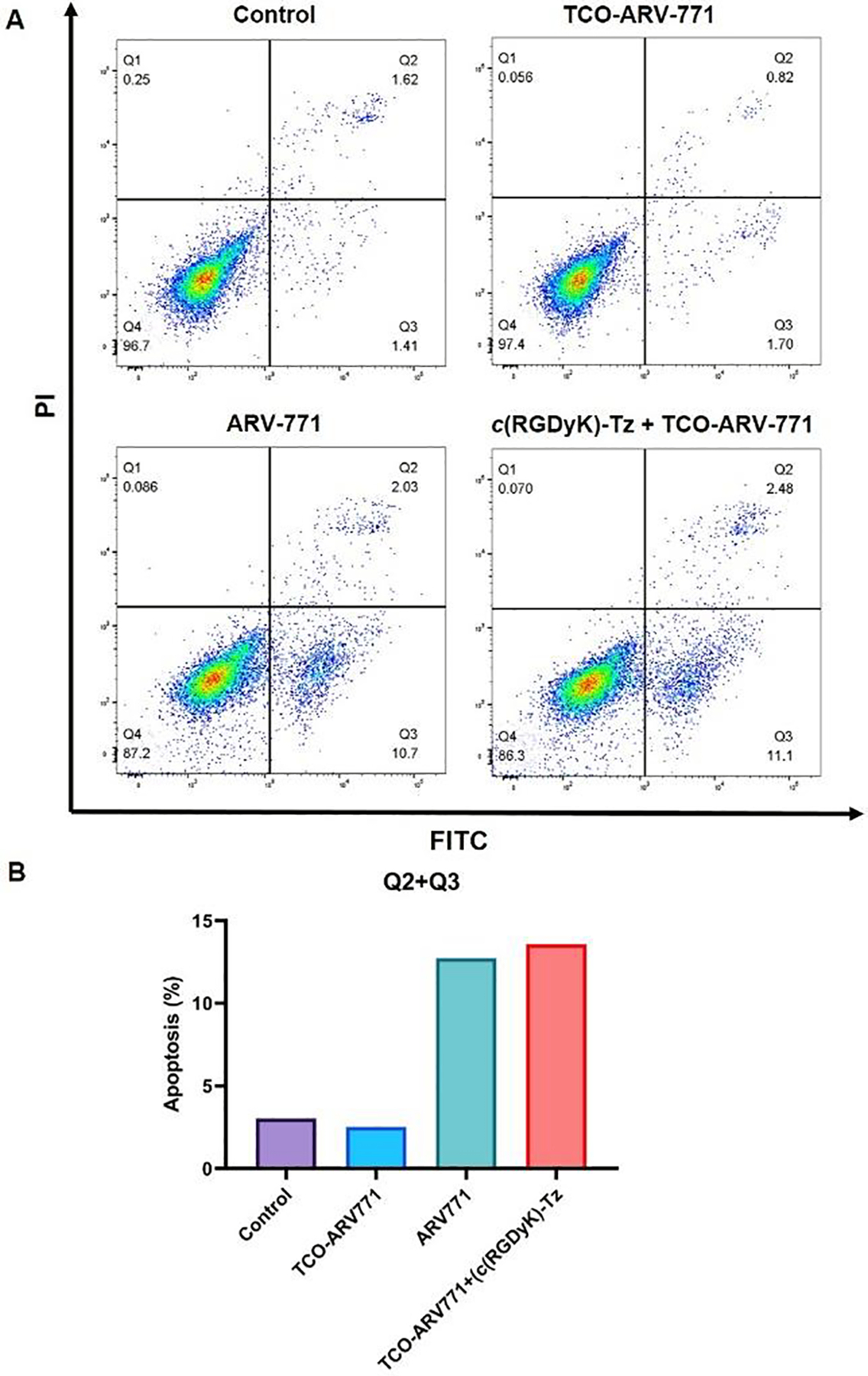
A) Effects of compounds TCO-ARV-771, crPROTAC (TCO-ARV-771 + c(RGDyK)-Tz, 3 h) and ARV-771 on the induction of apoptosis in HeLa cells. HeLa cells were cultured with or without TCO-ARV-771 and c(RGDyK)-Tz and ARV-771 for 16 h, and Annexin V and 7-AAD staining for flow cytometry was performed. Representative dot-plot graphs of each group. B) Apoptosis comparison.
Proteomic analysis demonstrating that crPROTAC selectively degrades BRD4 protein.
A quantitative multiplexed approach was employed to evaluate the selectivity of BRD4 degradation promoted by the bioorthogonal crPROTAC. For this purpose, the levels of cellular protein in the proteome of HeLa cells treated with TCO-ARV-771 or crPROTAC (TCO-ARV-771 + c(RGDyK)-Tz, 3 h) (Figure 6). In contrast to the TCO-ARV-771 treated cells, those in the crPROTAC treated group displayed an increase in the levels of 120 proteins and a decrease in the levels of 328 proteins (Figure S8). Moreover, the results (Figure 6A) showed that the level of BRD4, decreased significantly in the crPROTAC treated group. In addition, changes occurred in the levels of up/down-stream proteins, which might be associated with BRD4. For example, we found a decrease took place in expression level of the BCOR protein, which is an interacting corepressor of BCL-6 that enhances BCL-6-mediated transcriptional repression in cancers.66–68 Furthermore, the level of thioredoxin-interacting protein (TXNIP) increased the crPROTAC treated group. TXNIP, a member of the alpha-arrestin family that prevents proliferation by inducing the apoptosis signal-regulating kinase 1 enzyme (ASK1).69–72 The changes detected in these tumor-related proteins suggest that a potential correlation exists between BRD4 and BCOR or TXNIP proteins. The significant changes in the protein levels are listed in Figure 6B and Figure S7. It should be noted that the information gained from this analysis could be used to better understand the mechanism for degradation of BRD4.
Figure 6: The proteomic analysis of TCO-ARV-771 and crPROTAC.
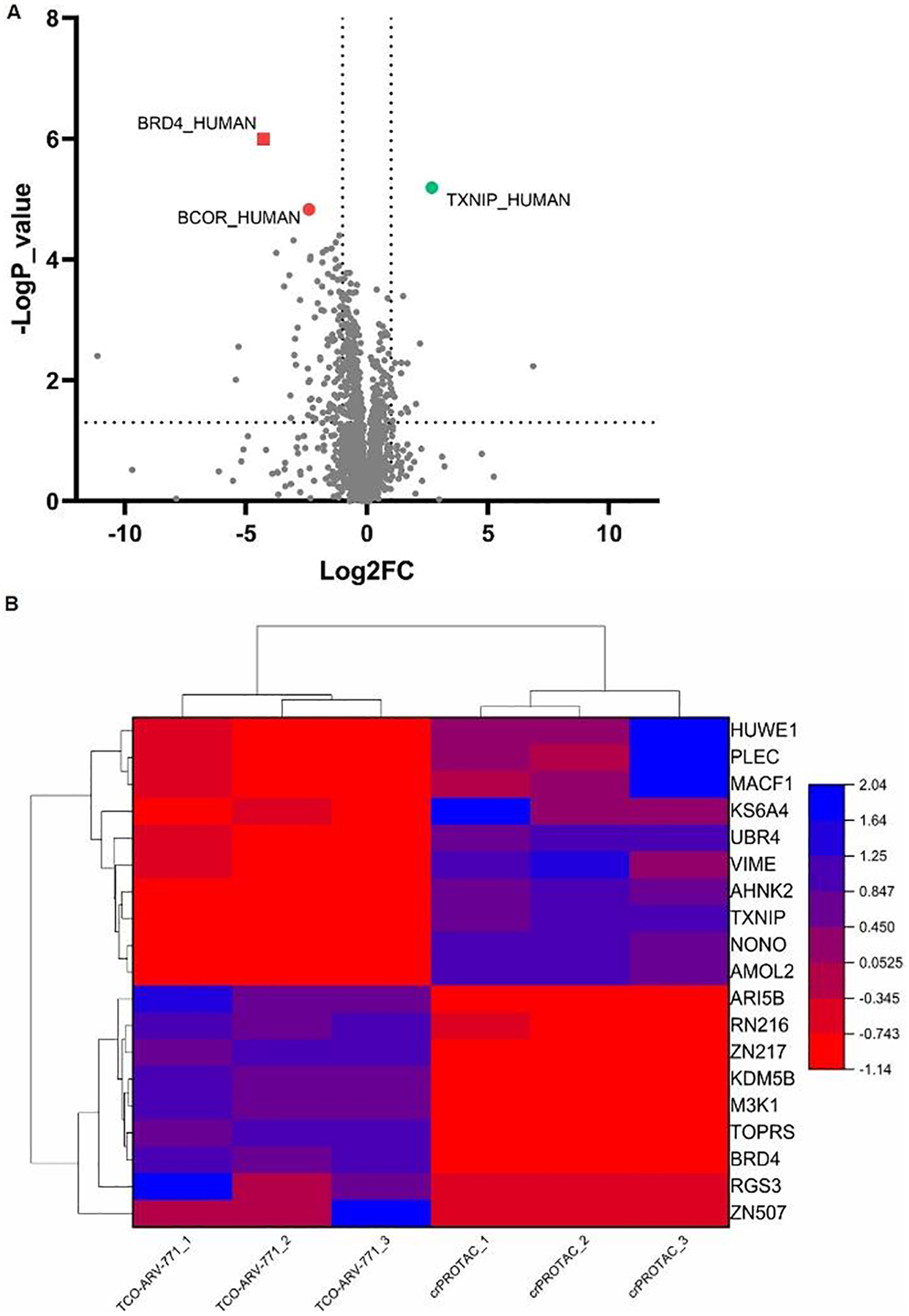
HeLa cells were treated with either compound 0.4 μM TCO-ARV-771, or 1 μM c(RGDyK)-Tz (3 h, pretreatment) + 0.4 μM TCO-ARV-771 for 16 h. Lysates were subjected to mass spec-based proteomics analysis. A) Volcano plot shows protein abundance (log2) as a function of significance level (-log10). B) Heat map analysis to screen between TCO-ARV-771 and crPROTAC groups (TCO-ARV-771 + c(RGDyK)-Tz, 3 h).
crPROTAC of TCO-DT2216 and c(RGDyK)-Tz induces Bcl-XL degradation in HeLa cells.
In the final phase of this effort, we demonstrated that the crPROTAC mediated bioorthogonal strategy is applicable to other VHL-based PROTACs. For this purpose, we designed and synthesized TCO-DT2216 prodrug (Figure 7A), which could release DT2216 that is known to degrade the Bcl-XL protein in HeLa cells.53–55 TCO-DT2216 was synthesized in a similar manner that one employed to prepare TCO-ARV-771 (Scheme S3). The results of experiments conducted to examine the prodrug strategy, we found that DT2216, formed by in cell reaction of TCO-DT2216 with c(RGDyK)-Tz, degraded Bcl-XL as efficiently as DT2216 (Figure 7B). No degradation activity was observed when the HeLa cells were treated with c(RGDyK) alone. We also confirmed that the produced DT2216 degraded the Bcl-XL protein through the ubiquitin-proteasome pathway. Finally, we found that the presence of the VHL ligand (Figure 7C) and proteasome inhibitor MG-132 (Figure 7D) significantly blocked the Bcl-XL degradation activity promoted by crPROTAC [TCO-DT2216 + c(RGDyK)-Tz].
Figure 7: The degradation assay of TCO-DT2216 with c(RGDyK)-Tz.
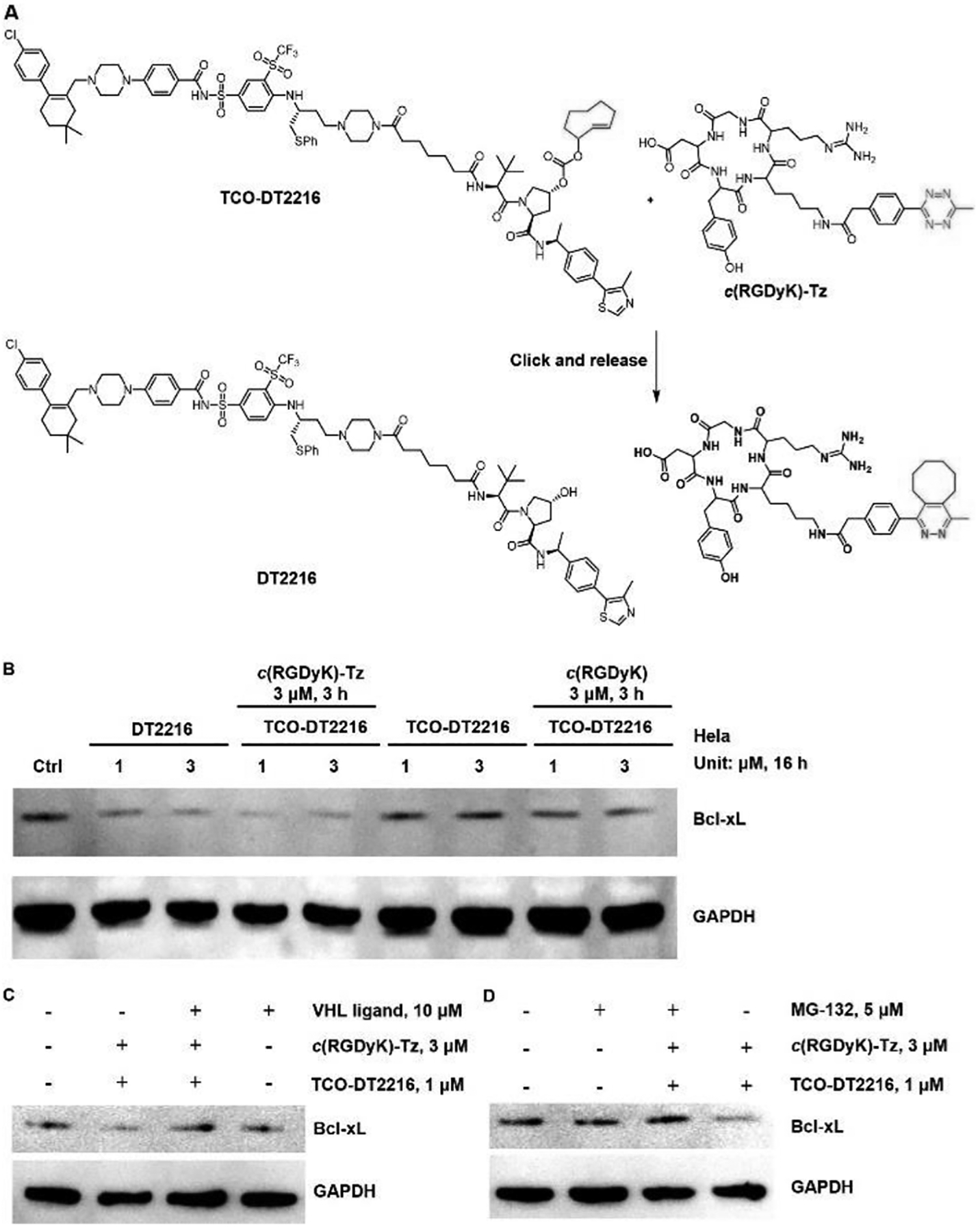
A) The ‘click’ reaction between TCO-DT2216 and c(RGDyK)-Tz releases the activated DT2216. B) Western blot analysis of Bcl-XL protein levels from HeLa cells treated with the indicated doses of treatments for 16 h. C) Western blot analysis of Bcl-XL and GAPDH levels in HeLa cells after the co-treatment with 10 μM VHL ligand and crPROTAC (TCO-DT2216 + c(RGDyK)-Tz, 3 h) for 16 h. D) Western blot analysis of BRD4 and GAPDH levels in HeLa cells after the co-treatment with 5 μM MG-132 and crPROTAC (TCO-DT2216 + c(RGDyK)-Tz, 3 h) for 16 h.
CONCLUSION
In the investigation described above, we have developed a new bioorthogonal on-demand prodrug strategy for selective control of PROTAC’s on-target degradation activity in a cancer cell selective manner. Differing from conventional prodrugs for PROTACs, which rely on cancer cell biomarkers or tumor microenvironment for drug delivery and activation, the new bioorthogonal on-demand approach enables on-target activation and release of PROTACs at tumor sites and consequently the approach minimizes premature drug activation. We have also demonstrated that inactive PROTAC prodrugs TCO-ARV-771 and TCO-DT2216 are selectively activated in cancer cells, not in noncancerous normal cells owing to the presence of the αvβ3 integrin binding ligand c(RGDyK) in activating agent c(RGDyK)-Tz. The results of the effort also show that the crPROTAC process produces ARV-771 and DT2216 in cancer cells where they promote protein degradation through the ubiquitin-proteasome pathway. Further studies of crPROTACs will be pursued to evaluate their in vivo efficiency of this protocol. We believe that the novel approach developed in this investigation will be generally applicable to all VHL-recruiting PROTACs that degrade POIs in cancer cells selectively.
Supplementary Material
ACKNOWLEDGMENT
We gratefully acknowledge financial support from NIH 5R01GM130772 and Arizona Center for Drug Discovery at the University of Arizona.
Footnotes
The authors claim there is no competing interest.
Supporting Information
The Supporting Information is available free of charge via the Internet at http://pubs.acs.org.
Materials and experimental procedures including compound synthesis, western blotting analysis, proteomic analysis, cell culture and imaging, and cell viability; characterization data for novel compounds; docking studies; click release kinetic study data; uncropped immunoblots (PDF)
REFERENCES
- 1.Lai AC; Crews CM, Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov 2017, 16, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schapira M; Calabrese MF; Bullock AN; Crews CM, Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discov 2019, 18, 949–963. [DOI] [PubMed] [Google Scholar]
- 3.Sun X; Gao H; Yang Y; He M; Wu Y; Song Y; Tong Y; Rao Y, PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther 2019, 4, Article number: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burslem GM; Crews CM, Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 2020, 181, 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burslem GM; Smith BE; Lai AC; Jaime-Figueroa S; McQuaid DC; Bondeson DP; Toure M; Dong H; Qian Y; Wang J; Crew AP; Hines J; Crews CM, The Advantages of targeted protein degradation over inhibition: An RTK case study. Cell. Chem. Biol 2018, 25, 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bondeson DP; Smith BE; Burslem GM; Buhimschi AD; Hines J; Jaime-Figueroa S; Wang J; Hamman BD; Ishchenko A; Crews CM, Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol 2018, 25, 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raina K; Lu J; Qian Y; Altieri M; Gordon D; Rossi AMK; Wang J; Chen X; Dong H; Siu K; Winkler JD; Crew AP; Crews CM; Coleman KG, PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong Y; Chi F; Wu H; Liu Y; Xie Z; Huang W; Shi W; Qian H, Emerging targeted protein degradation tools for innovative drug discovery: From classical PROTACs to the novel and beyond. Eur. J. Med. Chem 2022, 231, 114142. [DOI] [PubMed] [Google Scholar]
- 9.Zhang C; He S; Zeng Z; Cheng P; Pu K, Smart nano-PROTACs reprogram tumor microenvironment for activatable photo-metabolic cancer immunotherapy. Angew. Chem. Int. Ed 2022, 61, e202114957. [DOI] [PubMed] [Google Scholar]
- 10.Liu J; Chen H; Liu Y; Shen Y; Meng F; Kaniskan HU; Jin J; Wei W, Cancer selective target degradation by folate-caged PROTACs. J. Am. Chem. Soc 2021, 143, 7380–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He S; Gao F; Ma J; Ma H; Dong G; Sheng C, Aptamer-PROTAC conjugates (APCs) for tumor-specific targeting in breast cancer. Angew. Chem., Int. Ed 2021, 60, 23299–23305. [DOI] [PubMed] [Google Scholar]
- 12.Manzari MT; Shamay Y; Kiguchi H; Rosen N; Scaltriti M; Heller DA, Targeted drug delivery strategies for precision medicines. Nat. Rev. Mater 2021, 6, 351–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Z; Ukidve A; Kim J; Mitragotri S, Targeting strategies for Tissue-specific drug delivery. Cell 2020, 181, 151–167. [DOI] [PubMed] [Google Scholar]
- 14.Mombaerts P; Mizoguchi E; Grusby MJ; Glimcher LH; Bhan AK; Tonegawa S, Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell 1993, 75, 275–282. [DOI] [PubMed] [Google Scholar]
- 15.Puffenberger EG; Hosoda K; Washington SS; Nakao K; deWit D; Yanagisawa M; Chakravarti A, A missense mutation of the endothelin-B receptor gene in multigenic hirschsprung’s disease. Cell 1994, 79, 1257–1266. [DOI] [PubMed] [Google Scholar]
- 16.Liaw D; Marsh DJ; Li J; Dahia PLM; Wang SI; Zheng Z; Bose S; Call KM; Tsou HC; Peacoke M; Eng C; Parsons R, Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nature Genetics 1997, 16, 64–67. [DOI] [PubMed] [Google Scholar]
- 17.Cotton AD; Nguyen DP; Gramespacher JA; Seiple IB; Wells JA, Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J. Am. Chem. Soc 2021, 143, 395–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pillow TH; Adhikari P; Blake RA; Chen J; Del Rosario G; Deshmukh G; Figueroa I; Gascoigne KE; Kamath AV; Kaufman S; Kleinheinz T; Kozak KR; Latifi B; Leipold DD; Sing Li C; Li R; Mulvihill MM; O’Donohue A; Rowntree RK; Sadowsky JD; Wai J; Wang X; Wu C; Xu Z; Yao H; Yu SF; Zhang D; Zang R; Zhang H; Zhou H; Zhu X; Dragovich PS, Antibody conjugation of a chimeric BET degrader enables in vivo activity. ChemMedChem 2020, 5, 17–25. [DOI] [PubMed] [Google Scholar]
- 19.Banik SM; Pedram K; Wisnovsky S; Ahn G; Riley NM; Bertozzi CR, Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maneiro MA; Forte N; Shchepinova MM; Kounde CS; Chudasama V; Baker JR; Tate EW, Antibody-PROTAC conjugates enable HER2-dependent targeted protein degradation of BRD4. ACS Chem. Biol 2020, 15, 1306–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dragovich PS; Adhikari P; Blake RA; Blaquiere N; Chen J; Cheng YX; den Besten W; Han J; Hartman SJ; He J; He M; Rei Ingalla E; Kamath AV; Kleinheinz T; Lai T; Leipold DD; Li CS; Liu Q; Lu J; Lu Y; Meng F; Meng L; Ng C; Peng K; Lewis Phillips G; Pillow TH; Rowntree RK; Sadowsky JD; Sampath D; Staben L; Staben ST; Wai J; Wan K; Wang X; Wei B; Wertz IE; Xin J; Xu K; Yao H; Zang R; Zhang D; Zhou H; Zhao Y, Antibody-mediated delivery of chimeric protein degraders which target estrogen receptor alpha (ERalpha). Bioorg. Med. Chem. Lett 2020, 30, 126907. [DOI] [PubMed] [Google Scholar]
- 22.Dragovich PS; Pillow TH; Blake RA; Sadowsky JD; Adaligil E; Adhikari P; Bhakta S; Blaquiere N; Chen J; Dela Cruz-Chuh J; Gascoigne KE; Hartman SJ; He M; Kaufman S; Kleinheinz T; Kozak KR; Liu L; Liu Q; Lu Y; Meng F; Mulvihill MM; O’Donohue A; Rowntree RK; Staben LR; Staben ST; Wai J; Wang J; Wei B; Wilson C; Xin J; Xu Z; Yao H; Zhang D; Zhang H; Zhou H; Zhu X, Antibody-mediated delivery of chimeric BRD4 degraders. Part 1: exploration of antibody linker, payload loading, and payload molecular properties. J. Med. Chem 2021, 64, 2534–2575. [DOI] [PubMed] [Google Scholar]
- 23.Beck A; Goetsch L; Dumontet C; Corvaïa N, Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov 2017, 16, 351–337. [DOI] [PubMed] [Google Scholar]
- 24.Teichmann E; H. S, Shining a light on proteolysis targeting chimeras. ACS Cent. Sci 2019, 5, 1645–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfaff P; Samarasinghe KTG; Crews CM; Carreira EM, Reversible spatiotemporal control of induced protein degradation by bistable photoPROTACs. ACS Cent. Sci 2019, 5, 1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xue G; Wang K; Zhou D; Zhong H; Pan Z, Light-induced protein degradation with photocaged PROTACs. J. Am. Chem. Soc 2019, 141, 18370–18374. [DOI] [PubMed] [Google Scholar]
- 27.Liu J; Chen H; Ma L; He Z; Wang D; Liu Y; Lin Q; Zhang T; Gray N; Kaniskan HU; Jin J; Wei W, Light-induced control of protein destruction by opto-PROTAC. Sci. Adv 2020, 6, eaay5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naro Y; Darrah K; Deiters A, Optical control of small molecule-induced protein degradation. J. Am. Chem. Soc 2020, 142, 2193–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reynders M; Matsuura BS; Bérouti M; Simoneschi D; Marzio A; Pagano M; Trauner D, PHOTACs enable optical control of protein degradation. Sci. Adv 2020, 6, eaay5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.J. L; Chen PR, Development and application of bond cleavage reactions in bioorthogonal chemistry. Nat. Chem. Biol 2016, 12, 129–137. [DOI] [PubMed] [Google Scholar]
- 31.Ji X; Pan Z; Yu B; De La Cruz LK; Zheng Y; Ke B; Binghe Wang B, Click and release: bioorthogonal approaches to “on-demand” activation of prodrugs. Chem. Soc. Rev 2019, 48, 1077–1094. [DOI] [PubMed] [Google Scholar]
- 32.Wang J; Wang X; Fan X; Chen PR, Unleashing the power of bond cleavage chemistry in living systems. ACS Cent. Sci 2021, 7, 929–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Devaraj NK; Finn MG, Introduction: Click chemistry. Chem. Rev 2021, 121, 6697–6698. [DOI] [PubMed] [Google Scholar]
- 34.Oliveira BL; Guo Z; Bernardes GJL, Inverse electron demand Diels–Alder reactions in chemical biology. Chem. Soc. Rev 2017, 46, 4895–4950. [DOI] [PubMed] [Google Scholar]
- 35.Blackman ML; Royzen M; Fox JM, Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J. Am. Chem. Soc 2008, 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knall A-C; Slugovc C, Inverse electron demand Diels–Alder (iEDDA)-initiated conjugation: a (high) potential click chemistry scheme. Chem. Soc. Rev 2013, 42, 5131–5142. [DOI] [PubMed] [Google Scholar]
- 37.Taylor MT; Blackman ML; Dmitrenko O; Fox JM, Design and synthesis of highly reactive dienophiles for the tetrazine–trans-cyclooctene ligation. J. Am. Chem. Soc 2011, 133, 9646–9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan X; Ge Y; Lin F; Yang Y; Zhang G; Ngai W; Lin Z; Zheng S; Wang J; Zhao J; Li J; Chen P, Optimized tetrazine derivatives for rapid bioorthogonal decaging in living cells. Angew. Chem., Int. Ed 2016, 55, 14046–14050. [DOI] [PubMed] [Google Scholar]
- 39.Devaraj NK; Weissleder R; Hilderbrand SA, Tetrazine-based cycloadditions: application to pretargeted live cell imaging. Bioconju. Chem 2008, 19, 2297–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carlson JCT; Mikula H; Weissleder R, Unraveling tetrazine-triggered bioorthogonal elimination enables chemical tools for ultrafast release and universal cleavage. J. Am. Chem. Soc 2018, 140, 3603–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carlson JC; Meimetis LG; Hilderbrand SA; Weissleder R, BODIPY-tetrazine derivatives as superbright bioorthogonal turn-on probes. Angew. Chem., Int. Ed 2013, 52, 6917–6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khan I; Seebald LM; Robertson NM; Yigit MV; Royzen M, Controlled in-cell Activation of RNA Therapeutics Using Bond-cleaving Bio-orthogonal Chemistry. Chem. Sci 2017, 8, 5705–5712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Versteegen RM; Rossin R; ten Hoeve W; Janssen HM; Robillard MS, Click to release: instantaneous doxorubicin elimination upon tetrazine ligation. Angew. Chem., Int. Ed 2013, 52, 14112–14116. [DOI] [PubMed] [Google Scholar]
- 44.Zheng Y; Ji X; Yu B; Ji K; Gallo D; Csizmadia E; Zhu M; De La Cruz LK; Choudhary MR; Chittavong V; Pan Z; Yuan Z; Otterbein L; Wang B, Enrichment-triggered Prodrug Activation Demonstrated through Mitochondria-targeted Delivery of Doxorubicin and Carbon Monoxide. Nat. Chem 2018, 10, 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lebraud H; Wright DJ; Johnson CN; Heightman TD, Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Cen. Sci 2016, 2, 927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brooks PC; Montgomery AM; Rosenfeld M; Reisfeld RA; Hu T; Klier G; Cheresh DA, Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 1994, 79, 1157–1164. [DOI] [PubMed] [Google Scholar]
- 47.Albeda SM; Motto SA; Elder DE; Stewart RM; Damjanovich L; Herlyn M; Buck CA, Integrin distribution in malignant melanoma: Association of the β3 subunit with tumor progression. Cancer Res. 1990, 50, 6757–6764. [PubMed] [Google Scholar]
- 48.Gladson CL; Cheresh DA, Glioblastoma expression of vitronectin and the alpha v beta 3 integrin. Adhesion mechanism for transformed glial cells. J. Clin. Invest 1991, 88, 1924–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaertner FC; Kessler H; Wester H-J; Schwaiger M; Beer AJ, Radiolabelled RGD peptides for imaging and therapy. Eur. J. Nucl. Med. Mol. Imaging 2012, 39 (Suppl 1), S126–S13. [DOI] [PubMed] [Google Scholar]
- 50.Wang F; Li Y; Shen Y; Wang A; Wang S; Xie T, The functions and applications of RGD in tumor therapy and tissue engineering. Int. J. Mol. Sci 2013, 14, 13447–13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alipour M; Baneshi M; Hosseinkhani S; Mahmoudi R; Arabzadeh AJ; Akrami M; Mehrzad J; Bardania H, Recent progress in biomedical applications of RGD-based ligand: From precise cancer theranostics to biomaterial engineering: A systematic review. J. Biomed. Mater. Res 2020, 108, 839–850. [DOI] [PubMed] [Google Scholar]
- 52.Haubner R; Gratias R; Diefenbach B; Goodman SL; Jonczyk A; Kessler H, Structural and functional aspects of RGD-containing cyclic pentapeptides as highly potent and selective integrin αVβ3 antagonists. J. Am. Chem. Soc 1996, 118, 7461–7472. [Google Scholar]
- 53.He Y; Koch R; Budamagunta V; Zhang P; Zhang X; Khan S; Thummuri D; Ortiz YT; Zhang X; Lv D; Wiegand JS; Li W; Palmer AC; Zheng G; Weinstock DM; Zhou D, DT2216-a Bcl-xL-specific degrader is highly active against Bcl-xL-dependent T cell lymphomas. J. Hematol. Oncol 2020, 13, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lv D; Pal P; Liu X; Jia Y; Thummuri D; Zhang P; Hu W; Pei J; Zhang Q; Zhou S; Khan S; Zhang X; Hua N; Yang Q; Arango S; Zhang W; Nayak D; Olsen SK; Weintraub ST; Hromas R; Konopleva M; Yuan Y; Zheng G; Zhou D, Development of a BCL-xL and BCL-2 dual degrader with improved anti-leukemic activity. Nat. Commun 2021, 12, 6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khan S; Zhang X; Lv D; Zhang Q; He Y; Zhang P; Liu X; Thummuri D; Yuan Y; Wiegand JS; Pei J; Zhang W; Sharma A; McCurdy CR; Kuruvilla VM; Baran N; Ferrando AA; Kim YM; Rogojina A; Houghton PJ; Huang G; Hromas R; Konopleva M; Zheng G; Zhou D, A selective BCL-X(L) PROTAC degrader achieves safe and potent antitumor activity. Nat. Med 2019, 25, 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buckley DL; Van Molle I; Gareiss PC; Tae HS; Michel J; Noblin DJ; Jorgensen WL; Ciulli A; Crews CM, Targeting the von Hippel–Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J. Am. Chem. Soc 2012, 134, 4465–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Min JH; Yang H; Ivan M; Gertler F; Kaelin WG Jr.; Pavletich NP, Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science 2002, 296, 1886–1889. [DOI] [PubMed] [Google Scholar]
- 58.Hon WC; Wilson MI; Harlos K; Claridge TD; Schofield CJ; Pugh CW; Maxwell PH; Ratcliffe PJ; Stuart DI; Jones EY, Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 2002, 417, 975–978. [DOI] [PubMed] [Google Scholar]
- 59.Darko A; Wallace S; Dmitrenko O; Machovina MM; Mehl RA; Chin JW; Fox JM, Conformationally strained trans-cyclooctene with improved stability and excellent reactivity in tetrazine ligation. Chem. Sci 2014, 5, 3770–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rizvi SFA; Mu S; Wang Y; Li S; Zhang H, Fluorescent RGD-based pro-apoptotic peptide conjugates as mitochondria-targeting probes for enhanced anticancer activities. Biomed. Pharmacother 2020, 127, 110179. [DOI] [PubMed] [Google Scholar]
- 61.Cui Y; Song X; Li S; He B; Yuan L; Dai W; Zhang H; Wang X; Yang B; Zhang Q, The impact of receptor recycling on the exocytosis of alphavbeta3 integrin targeted gold nanoparticles. Oncotarget 2017, 8, 38618–38630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nestic D; Uil TG; Ma J; Roy S; Vellinga J; Baker AH; Custers J; Majhen D, alphavbeta3 Integrin Is Required for Efficient Infection of Epithelial Cells with Human Adenovirus Type 26. J. Virol 2019, 93, e01474–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen R; Ni S; Chen W; Liu M; Feng J; Hu K, Improved Anti-Triple Negative Breast Cancer Effects of Docetaxel by RGD-Modified Lipid-Core Micelles. Int. J. Nanomedicine 2021, 16, 5265–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Poitz DM; Stolzel F; Arabanian L; Friedrichs J; Docheva D; Schieker M; Fierro FA; Platzbecker U; Ordemann R; Werner C; Bornhauser M; Strasser RH; Ehninger G; Illmer T, MiR-134-mediated beta1 integrin expression and function in mesenchymal stem cells. Biochim. Biophys. Acta 2013, 1833, 3396–3404. [DOI] [PubMed] [Google Scholar]
- 65.E EME; A SA; Koosha S; A AA; Azam F; I MT; Khalilullah H; Sadiq Al-Qubaisi M; M AA, Zerumbone Induces Apoptosis in Breast Cancer Cells by Targeting alphavbeta3 Integrin upon Co-Administration with TP5-iRGD Peptide. Molecules 2019, 24, 2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Astolfi A; Fiore M; Melchionda F; Indio V; Bertuccio SN; Pession A, BCOR involvement in cancer. Epigenomics 2019, 11, 835–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huynh KD; Fischle W; Verdin E; Bardwell VJ, BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev. 2000, 14, 1810–1823. [PMC free article] [PubMed] [Google Scholar]
- 68.Kang JH; Lee SH; Lee J; Choi M; Cho J; Kim SJ; Kim WS; Ko YH; Yoo HY, The mutation of BCOR is highly recurrent and oncogenic in mature T-cell lymphoma. BMC Cancer 2021, 21, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patwari P; Higgins LJ; Chutkow WA; Yoshioka J; Lee RT, The interaction of thioredoxin with Txnip. Evidence for formation of a mixed disulfide by disulfide exchange. J. Biol. Chem 2006, 281, 21884–21891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aitken CJ; Hodge JM; Nishinaka Y; Vaughan T; Yodoi J; Day CJ; Morrison NA; Nicholson GC, Regulation of human osteoclast differentiation by thioredoxin binding protein-2 and redox-sensitive signaling. J. Bone Miner. Res 2004, 19, 2057–2064. [DOI] [PubMed] [Google Scholar]
- 71.Goldberg SF; Miele ME; Hatta N; Takata M; Paquette-Straub C; Freedman LP; Welch DR, Melanoma metastasis suppression by chromosome 6: evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Res. 2003, 63, 432–40. [PubMed] [Google Scholar]
- 72.Nagaraj K; Lapkina-Gendler L; Sarfstein R; Gurwitz D; Pasmanik-Chor M; Laron Z; Yakar S; Werner H, Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 1045–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.