

Abstract
Human papilloma virus positive (HPV+) squamous cell carcinoma of the oropharynx (OPSCC) is the most prevalent HPV−associated malignancy in the United States and is primarily caused by HPV16. Favorable treatment outcomes have led to increasing interest in treatment de-escalation to reduce treatment-related morbidity. Prognostic biomarkers are needed to identify appropriately low-risk patients for reduced treatment intensity. Targeted DNA sequencing including all HPV16 open reading frames was performed on tumors from 104 patients with HPV16+ OPSCC treated at a single center. Genotypes closely related to the HPV16-A1 reference were associated with increased numbers of somatic copy-number variants in the human genome and poor recurrence-free survival. Genotypes divergent from HPV16-A1 were associated with favorable recurrence-free survival. These findings were independent of tobacco smoke exposure. Total RNA sequencing was performed on a second independent cohort of 89 HPV16+ OPSCC cases. HPV16 genotypes divergent from HPV16-A1 were again validated in this independent cohort, to be prognostic of improved RFS in patients with moderate (less than 30 pack-years) or low (no more than 10 pack-years) of tobacco smoke exposure. In summary, we show in two independent cohorts that viral sequence divergence from the HPV16-A1 reference is correlated with improved recurrence-free survival in patients with moderate or low tobacco smoke exposure.
Implications:
HPV16 genotype is a potential biomarker that could be easily adopted to guide therapeutic decision-making related to de-escalation therapy.
Introduction:
Human papillomavirus-associated oropharyngeal squamous cell carcinoma (HPV+ OPSCC) has surpassed cervical cancer in incidence and is the most commonly diagnosed malignancy caused by HPV in the USA.1 HPV+ OPSCC has an improved prognosis compared to non-HPV OPSCC, leading to a distinct staging system for these tumors.2,3 The combination of improved oncologic outcomes and significant, lifelong therapeutic toxicity led our center and others and to study de-intensified therapy for patients with HPV+ OPSCC in an effort to limit morbidity while preserving tumor control.4–7 Initial results of these studies have been mixed, but even in those that are promising, there are few available tools to select appropriate patients with favorable prognosis, raising concern that we may inappropriately de-escalate or escalate therapy in a subsets of patients.8,4 Defects in TRAF3 and CYLD, PIK3CA mutations and circulating HPV DNA have been reported to be correlated with patient outcome.6,9,10 Here we explore how the genetic characteristics of the HPV genome itself may be useful to stratify HPV+ OPSCC into subsets with favorable and poor prognosis.
HPV subtype 16 (HPV16) is the most common high-risk HPV causing OPSCC. There are four main variant lineages (A, B, C, D) and ten (or more) sub-lineages (A1, A2, A3, A4, B1, B2, C, D1, D2, D3) of HPV16. Formerly, these sub-lineages were geographically termed European (A1–3), Asian (A4), African-1 (B), African-2 (C), and North-American/Asian-American (D1–3). Lineages are defined by 1–10% differences and sub-lineages defined by 0.5–1% differences in the L1 (capsid protein) sequence.11–13
Along with OPSCC and other anogenital cancers, HPV16 is also the most common high-risk HPV associated with cervical cancer. HPV16 sub-lineage classification of cervical cancer revealed that though lineage A1 is most prevalent, non-A HPV16 lineages (B/C/D) are associated with a higher risk of precancer and cancer.12,13 Further analysis revealed that HPV16 lineage D variants were associated with the highest rate of persistent infection and progression to cervical cancer compared with other variants.13–15
Studies on HPV16 lineages and sub-lineages have led to the exploration of variation in viral oncoproteins E6 and E7, which bind to p53 and pRb respectively and contribute to carcinogenesis and inhibition of apoptosis and entry of cell into the S phase. The well-studied E6 L83V polymorphism has been associated with infection persistence and progression of cervical carcinoma within the A1–3 sub-lineages.16–20 It is currently unknown if this E6 variant has similar biological impact in other HPV lineages.21 As opposed to the E6 viral oncogene, for which many sequence variations have been found in cervical cancer,20,21 multiple studies have shown a high level of conservation of the E7 oncogene.22–24
While exploration of HPV16 genotypic factors which correlate with clinicopathologic features of uterine cervical cancer is relatively robust, few studies have examined HPV16+ OPSCC. Even the TCGA, effort failed to provide comprehensive HPV genotyping; however, RNA sequencing data have been used to determine HPV positivity and viral genome integration.25 HPV integration in OPSCC has been more intensely investigated and has been reported to correlate with genomic methylation26, tumor mutational burden26, genomic instability27, HPV gene expression28, tumor immune landscape29, and survival.29
HPV16 causes the vast majority of OPSCC cases 25,30,31 and HPV Subtype 16 is associated with increased cervical metastasis.30 To begin exploring if HPV16 genotype contribute to the variable clinical and biological behavior of OPSCC we have comprehensively genotyped a cohort of OPSCC patients and correlated our findings with clinical factors and patient outcomes.
Materials and Methods:
DNA sequencing data were collected as a part of the UNCseq tumor sequencing program. The UNCseq targeted sequencing platform involves sequencing exons of a custom list of 650 human genes (covering 3.4 M bases) and 10 pathogen genome segments in fixed or frozen cancer tissue and matched germline DNA from consenting local patients. This custom sequencing platform provided targeted coverage of all HPV16 open reading frames. Tumor sample identifiers and genomic data were derived from the clinical trial LCCC1108: Development of a Tumor Molecular Analyses Program and Its Use to Support Treatment Decisions. This IRB-approved trial opened in 2011. All studies were done with the approval of our Institutional Review Board, patient participation required written informed consent, and all studies were conducted in accordance with recognized ethical guidelines as described in U.S Common Rule. The UNCseq database was queried for all patients with HPV+ oropharyngeal cancer.
Via chart review of electronic medical records, demographic information was obtained for each study subject, including age, gender, race, and smoking history. Clinical stage at presentation according to the AJCC staging system (AJCC 8th edition) was recorded, clinical AJCC8 staging was used, considering that many patients did not receive surgical treatment. Recurrence-free survival was defined from the date of initial diagnosis to the date at which evidence of recurrent disease was first documented after primary treatment. Cases that presented with distant metastasis (n=2) were excluded from recurrence free survival analysis. All survival analysis was performed with the R Survival package. Cox proportional hazards models were implemented with the coxph() function. The validity of the proportional hazards assumption was validated for all cox models using the R cox.zph() function, this assumption was found to be valid for all presented comparisons.
DNA Isolation, Library Preparation, and Sequencing
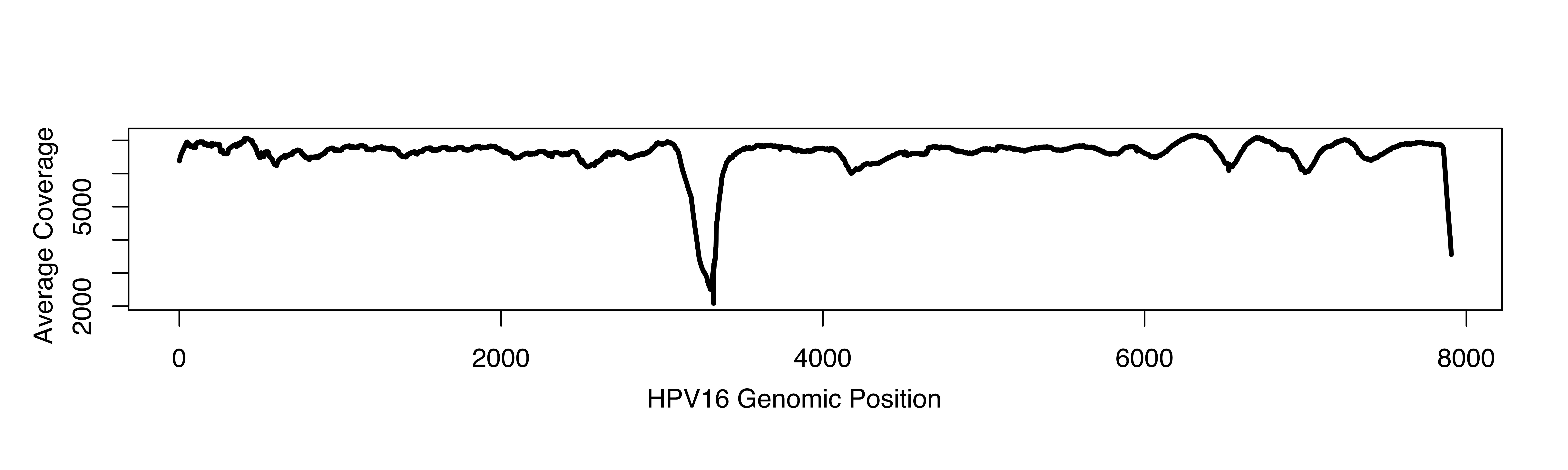
A pathologist examined H&E-stained slides from each case to confirm the diagnosis of squamous cell carcinoma. Automated DNA extraction was from FFPE tissue sections using the Promega Maxwell MDx16™ instruments (Promega) and then fragmented by sonication. Subsequent quality assessments were performed by ultraviolet absorbance and quantity assessments. During DNA isolation and library preparation, DNA concentration was measured by fluorometry and DNA quality was evaluated using the Agilent 2100 Bioanalyzer high sensitivity assay. DNA libraries were pooled for deep sequencing using an Illumina HiSeq2500™ sequencer. For general metrics on human gene sequencing quality, read depth, and coverage statistics please see our recent report which reviewed these features of the data set.32 Excluding the hypervariable region 3150–3351 median coverage of the HPV16 genome was 7904 with an IQR of 6867–7953. The hypervariable region 3150–3351 had median coverage of 3307 with an IQR of 837–7377.
RNA Isolation, Library Preparation, and Sequencing
All studies were done with the approval of our Institutional Review Board and were conducted in accordance with recognized ethical guidelines as described in U.S Common Rule. Formalin-fixed paraffin-embedded (FFPE) tissue samples were sent to the UNC Lineberger Comprehensive Cancer Center (LCCC) Translational Genomics Lab (TGL) for RNA isolation using the Maxwell 16 MDx Instrument (Promega AS3000) and the Maxwell 16 LEV RNA FFPE Kit (Promega AS1260) following the manufacturer’s protocol (Promega 9FB167). After a pathology review of a hematoxylin and eosin (H&E) stained slide to identify tumor area, RNA was extracted from unstained slides using macrodissection. Total RNA quality was measured using a NanoDrop spectrophotometer (Thermo Scientific ND-2000C) and a TapeStation 4200 (Agilent G2991AA). Total RNA concentration was quantified using a Qubit 3.0 fluorometer (Life Technologies Q33216). Libraries were prepared with Illumina TruSeq Stranded Total RNA with Ribo-Zero protocol. Libraries were sequenced on an Illumina HiSeq2500 sequencer. Paired end read data, with read lengths of 75 were collected.
Bioinformatics
Viral SNP Calling –
Raw reads were aligned to the human genome plus a comprehensive library of HPV virus sequences, using compiled reference sequences from the ViFi analysis pipeline.33 Human genomic mutations were not investigated as only a subset of samples had matched normal DNA sequencing. Tumors were considered to be HPV16 positive if they had more than 20,000 reads mapping to the HPV16 genome, which was selected based on an obvious bimodal distribution in the data, see supplemental figures from our prior publication justifying this criterion.32 The HPV16 A1 genotype, RefSeq NC_001526.4 was selected as our primary reference sequence for this study. The Varscan pipeline was used for variant/polymorphism calling, using the reference-free approach. The SnpEff pipeline was used to assign and prioritize variant effects. Consensus sequences were derived from the variant calls which represented clonal variants using in-house scrips. SNPs were only analyzed if the sequencing depth was >= 100 and VAF >= 0.5. Variants were considered subclonal if VAF <= 0.9.
Phylogenetics –
The HPV tumor genomes were then aligned against each other and 16 reference HPV16 sub-lineage sequences. Alignments were imported into R for phylogenetic analyses, where maximum parsimony phylogeny was constructed using default parameters provided in the phangorn package. To assign the nearest sub-lineages to the tumor HPV genomes, the R phangorn package was used to construct a sequence distance matrix for each tumor HPV and 16 reference sequences. Each tumor sample was assigned to an HPV16 sub-lineage based on the “closest” reference sequence using the “JC69” distance.
Neo-antigenicity Analysis –
For each patient, targeted genomic sequencing including HLA-A, HLA-B, HLA-C was processed by the Optitype pipeline.34 Comparison with matched normal (blood) sequencing data, where available, demonstrated remarkable consistency of the results between tumor and matched normal blood. Specifically, 93% of cases in which matched normal and tumor HLA typing could be performed were found to have identical results. 4% of cases had apparent loss of heterozygosity at one HLA allele in the tumor sample. 3% of cases had discordant HLA calls at one of 6 loci. Therefore, tumor sequencing data was utilized for HLA typing for the purposes of this study. The netMHCpan pipeline was then applied to generate predicted binding affinities of all possible patient-matched viral peptide and MHC pairs. An empiric threshold of < 325 nM was applied to identify high affinity interaction between MHC and viral peptides.35–37 The log fraction of high-affinity to all possible viral peptide MCH interactions was also investigated as a neo-antigenicity metric, as homozygous haplotypes of HLA loci in some patients made the background number of potential peptide-HLA interactions variable between patients.
APOBEC and Mutational Signature Analysis –
Based on prior reports, mutational contexts represented by COSMIC Version 2 Signature 13 (C>G variants) and Signature 2 (C>T variants) were considered to be potentially APOBEC related.38 Using the trinucleotide contexts most prominent in COSMIC Signatures 2(T[C>T]A, T[C>T]C, T[C>T]T) and 13 (T[C>G]A, T[C>G]C, T[C>G]T), alterations were counted as potentially APOBEC related. Chi-squared test was applied to determine differences in minor proportions of ABOBEC related variants. Nonnegative matrix factorization was used to estimate the contribution of different mutational process influencing the HPV16 genome. This analysis was implemented and visualized with the R package DeconstructSigs39, after generating trinucleotide context SNP matrices with in-house scripts.
Somatic Variant Calling –
Sequencing data were routed through an automated pipeline managed by the Lineberger Bioinformatics Core (LBC). Our current somatic workflow used paired tumor and normal libraries to detect somatic mutations, large and small indels, structural variants, and pathogenic organisms. Raw sequences were aligned using the BWA-mem algorithm and refined using our Assembly Based Realignment process to allow for accurate alignment of complex sequence variation. Only high confidence variants with phred-scaled quality scores greater than 30 were included in the analysis. Average target coverage was ~1000x.
Copy Number Variant Calling –
Copy number calls were generated with the SynthEx algorithm using the tumor sequencing data and a library of 200 un-matched normal samples sequenced with the same technique. We note, the SynthEx pipeline utilizes both on and off target reads to allow large-segment copy number variant calling across the human genome. Other groups have reported genome wide CNV calls with smaller targeted sequencing panels than ours (~250 genes) bolstering the reasonability of this approach.40,41 A conservative approach was taken. Thirty replicates varying the parameter k (number of nearest neighbor) were done per tumor and the model with the fewest deviations from the expected copy number of 2 was selected. Sex chromosomes were excluded.
RNA Data Quantification –
The HPV16 A1 genotype, RefSeq NC_001526.4 was selected as our primary reference sequence for this study. Salmon was used to quantify RNA reads for NC_001526.4 as well as hg38.42 Viral transcripts read counts were transformed into log2 viral read counts per million total mapped reads (human and viral) prior to visualization and analysis. Tumors were considered to be HPV16 positive if there were more than 2000 reads mapping to HPV16 and the log10 ratio of HPV16/Human reads was > −4.5. 24 cases were excluded based on these criteria.
Viral Integration Analysis –
The ViFi pipeline was used to identified discordant (human – viral) read pairs which cluster to potential integration sites in the human and HPV genomes.33 Tumors with more than 25 clustered discordant read pairs were classified as positive for (discordant) split reads. The ratio HPV16 E6 and E7 to HPV16 E5 and E2 was calculated for each tumor based on quantification from Salmon. Above a ratio of −0.304, 88% of tumors also displayed locus specific clusters of human-viral split read pairs. Below this threshold only 26% of tumors had human-viral split read pairs. Therefore, tumors with E6E7/E5E2 ratio above this threshold were considered to have an integrated pattern of viral gene expression.
Viral Genotyping by RNA –
First partial consensus sequences were constructed from viral BAM files output from the ViFi pipeline. With the goal getting an approximate sequence, the Varscan pipeline was used for variant/polymorphism calling, using the reference-free approach, and lax parameters with minimal coverage requirement of 1, VAF minimum of 0.51 and no p-value cut off. These “variants” were used to construct approximate consensus sequences in area of the viral genome covered by the sequencing data. See Supplemental Figure 3, for an illustration of the gross quality of HPV16 genotype data as ascertained by RNAseq. JC69 sequence distances from these partial, approximate viral sequences were then calculated using the R phangorn package. Tumors were classified into viral clades based on the majority voting of the three nearest neighbors from the DNA sequencing cohort. For the 13 tumors with both DNA and RNA sequencing, this method recovered the viral clade in 100% of cases (see Figure 6A).
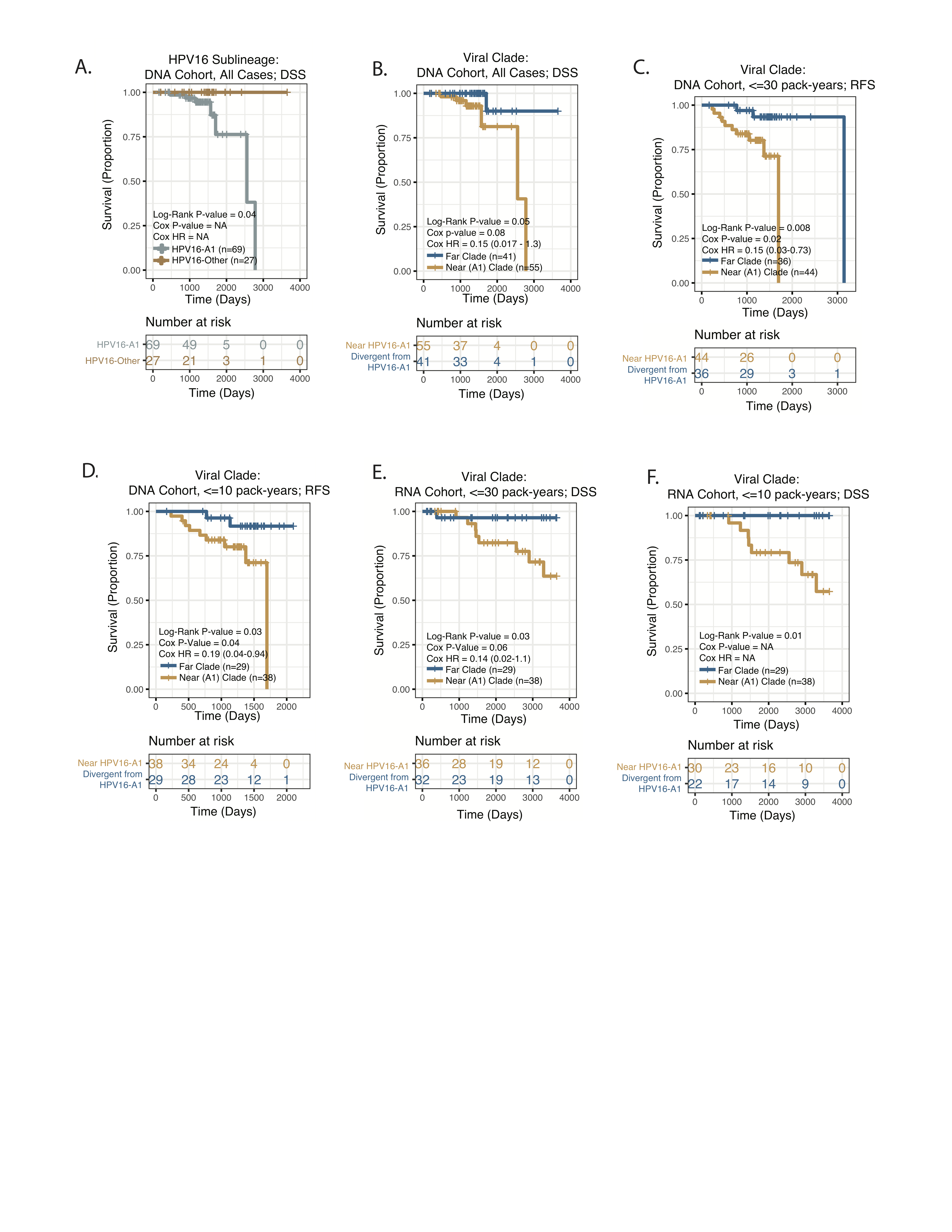
Figure 6. HPV16 Viral Genotyping and Integration Analysis by RNA Sequencing.

A. Maximum Parsimony Phylogeny of HPV16 genomes from the DNA sequencing cohort. Links – Connect known (DNAseq) genotype, with nearest neighbor genotype assigned from RNAseq, for 13 patients with available DNA and RNA sequencing data. Color –Viral clade as assigned in Figure 4. Tip point size – Number of RNAseq cases assigned at a neighbor to the indicated DNAseq case. B-G. Kaplan-Meier Plot of Recurrence-free Survival, comparing viral clades vs. viral integration status, stratified by tobacco smoke exposure. Log-Rank P-value – p-value derived from the Log-Rank test. Cox P-value – the p-value derived from a univariate Cox Model. Cox HR – estimated hazard ratio with 95% confidence interval, from Cox Model. NA – Cox proportional hazard modeling was not possible due to no events (measurable hazard) in one of the two groups. H. Annotated heat map of HPV16 viral gene expression. Columns – tumor samples, organized by E6E7/E5E2 ratio. Viral Clade – as assigned in panel A. Integrated – Assigned integration status based in E6E7/E5E2 ratio. Split Reads – Presence of detectable split read-pairs mapping to both the HPV16 and human genome. I-K. Genomic and clinical features of the <= 10 pack-year smoking exposure sub-group. I. HPV16 Integration status - based in E6E7/E5E2 ratio Significance based on Chi-squared test. J. AJCC8 summary stage. Significance based on Chi-squared test. K. Tobacco Smoke Exposure. Significance based on Wilcoxon Rank-sum test. * P-value < 5*10^−2. NS – not significant. Near – HPV16 viral clade nearest to the HPV16-A1 sub-lineage based on JC69 distance. Far – HPV16 viral clade distal to the HPV16-A1 sub-lineage based on JC69 distance.
Inclusion Criteria
The UNCseq database was queried for p16+ tumors originating from the anatomic oropharynx (tonsil or tongue base) with available tumor sequencing data as well as data on stage, treatment strategy, clinical outcome, and histopathology available. HPV16 positivity was confirmed by DNA sequencing reads which mapped to the HPV16 genome, see above comments.32 For the DNA sequencing cohort, patients were excluded from clinical analyses if tumors were not p16+ or were from atypical oropharyngeal sub-sites (midline soft palate or lateral oropharyngeal wall), see Figure 1. The confirmatory RNA sequencing cohort included cases dating prior to routine p16 IHC was performed at our institution, therefore, p16 status was not considered in this secondary cohort.
Figure1. HPV16 Positivity Assignment and Cohort Composition Flow Diagram.

A. Histogram of sequencing reads mapping to HPV16, normalized to human reads. Dashed line – Threshold for HPV16 positivity applied. B. Normalized reads mapping to HPV16 versus a library of 336 other HPV genomes included in the ViFi package. C. Flow diagram illustrating cohort construction for genotypic and clinical analyses.
Data Availability:
Raw DNA sequencing data has been deposited to dbGaP – accession number phs001713.v1.p1. Data post processing code is available at https://github.com/TravisParkeSchrank/HPV16_genotype.
Results:
Genotypic Variation and Evolutionary Conservation Analysis
To examine the diversity of HPV16 oncogenic genotypes promoting OPSCC in our cohort, we considered polymorphisms with variant allele frequency > 0.9 to be clonal. Based on clonal polymorphisms alone, HPV16 coding genotypes were highly diverse, with 93 distinct protein coding HPV16 genomes amongst the 104 tumors examined. To assess selective pressure for protein sequence conservation, we examined the ratio of coding to synonymous clonal sequence variants. We considered all polymorphisms relative to the HPV16 A1 reference, as well as uncommon polymorphisms, defined as polymorphisms identified in less than 25% of tumors. Based on these metrics, E1^E4 and E2 demonstrated the least conservation amongst all viral genes, see Figure 2 A–B. Consistent with expectations from the uterine cervical carcinoma literature, E7 demonstrated a high degree of conservation with few nonsynonymous polymorphisms, see Figure 2 A–B.22
Figure 2. Sequence Conservation by Viral Gene and Characteristics of Sub-clonal polymorphisms.

A. Conservation of Viral Genes. All clonal polymorphisms relative to the HPV16A1 reference were examined. The ratio of missense to synonymous SNPs were compared between the gene in question and all other viral genes. Significance based on Wilcoxon Rank-sum test. B. Conservation of Viral Genes based on Uncommon Polymorphisms. Clonal polymorphisms were examined if present in less than 25% of cases examined. The ratio of missense to synonymous uncommon SNPs were compared between the gene in question and all other viral genes. Significance based on Wilcoxon Rank-sum test. C. Distribution of non-synonymous polymorphisms amongst HPV16 viral genes. Significance based on chi-squared test. D. Proportion of SNPs by predicted effect. Significance based on chi-squared test. E. Proportion of potentially APOBEC related SNPs in the HPV16 viral genome, compared between clonal and sub-clonal SNPs. Significance based on chi-squared test. *** P-value < 1*10^−3.
The viral sequencing data had an average depth of coverage of the HPV16 genome of ~8,000, enabling analysis of sub-clonal genomic variants. Despite the robust detection of sub-clonal variants, the bulk of genomic diversity was clonal with 1295 clonal coding (missense) polymorphisms vs. 144 sub-clonal coding polymorphisms. From 104 tumors, there were 285 unique (only present in one tumor) clonal non-synonymous variants. Sub-clonal variants demonstrated modest but detectable differences in distribution amongst HPV genes and were much more likely to cause protein-coding changes, see Figure 2C–D. Considering prior reports supporting the relative importance of APOBEC-mediated mutagenesis in HPV+ OPSCC, we investigated APOBEC as a potential driver of HPV16 polymorphisms in OPSCC. Interestingly, polymorphisms corresponding to APOBEC deamination targets represented a minority of all clonal HPV polymorphisms identified (9.8%). However, 35.4% of sub-clonal variants (VAF < 90%) were potentially APOBEC related, see Figure 2E. This is consistent with expectations from analysis of cancerous and pre-malignant lesion of the uterine cervix, where APOBEC has been found to induce a minority of the lineage-defining SNPs, but sub-clonal APOBEC related variants are highly associated with viral clearance and failure of lesions to progress to invasive carcinoma.38,43
HPV16 Sub-lineage, Common Polymorphisms, and Viral Copy Loss
HPV16 sub-lineage was also defined by the nearest HPV16 sub-lineage reference sequence in sequence space as determined by the Jukes and Cantor (JC69).44 Sixteen sub-lineages were queried based on contemporary studies, see Figure 3A. As reported for cervical cancer,45 HPV16-A1 genotype accounted the majority of ( 71%) tumors investigated. Eleven other tumors (13%) harbored A2–3 HPV16 and sub-lineages D1–3 were also represented by 11 tumors (13%). Maximum parsimony phylogenetic analysis based on all clonal SNPs grouped HPV16 oncogenic genomes, resulted in groups that were highly related in terms of HPV16 sub-lineage, common non-synonymous polymorphisms, and sequence divergence from HPV16 were all highly correlated, see Figure 3A.
Figure 3. Analysis of HPV16 Genotypes by Sub-lineage.

A. Heatmap of common non-synonymous polymorphisms in the HPV16 genome. Columns – patient tumor samples. Sub-lineage - the nearest HPV16 sub-lineage reference sequence by the JC69 sequence distance. Number of Non-synon. Poly. – Number of non-synonymous polymorphisms relative to the HPV16 A1 reference. Relative Viral Copy Number – Viral copy-number relative to human genomic material. Defined as Log2(HPV16 Reads/Human Reads). Common Polymorphism – The 15 most common non-synonymous polymorphisms relative to HPV16 A1 are listed in order of decreasing prevalence. B. Kaplan-Meier Plot of Recurrence-free Survival, comparing sub-lineage A1 to others. Log-Rank P-value – p-value derived from the Log-Rank test. Cox P-value – the p-value derived from a univariate Cox Model. Cox HR – estimated hazard ratio with 95% confidence interval derived from Cox Model. C. Proportion of Cases Stratified by AJCC8 Clinical Staging. Significance based on chi-squared test. D. Tobacco Smoke Exposure. Significance based on Wilcoxen Rank-sum test. E. Predicted Viral Proteomic Neo-Antigenicity. Neo-Antigenicity was estimated by the number of viral peptides with <350nM affinity for MHC, based on the patients HLA subtype (see methods for further description). Significance based on Wilcoxen Rank-sum test. F. Human Genomic Copy-number Variant Burden. Number of copy number events as identified by the SynthEx pipeline. Significance based on Wilcoxon Rank-sum test. * P-value < 5*10^−2. NS – not significant. A1 – HPV16 genome assigned to HPV16-A1 sub-lineage based on nearest JC69 distance. Other – HPV16 genome assigned to sub-lineage other than HPV16-A1 based on nearest JC69 distance. G. HPV16 Sub-lineages for sequenced HPV16+ tumors from the oropharynx and uterine cervix. H. Common HPV16 Non-synonymous polymorphisms in sequenced HPV16+ tumors from the oropharynx and uterine cervix. I. Proportion of Tumors with HPV16 Deep Copy Loss in E2 or a large loss (involving >10% of the viral genome). Chi-squared test. *** P-value < 5*10^−4.
Review of patient factors identified two patients presented with distant metastases and were treated with palliative intent; therefore 96 patients were available for analysis of recurrence-free-survival (RFS). The A1 sub-lineage was highly associated with poor recurrence-free survival, see Figure 3B. This association persisted in a multivariate regression model including non-HPV16A1 genotype (HR = 0.09, 95%CI = 0.01–0.73, p-value = 0.02), early vs. advanced AJCC8 summary stage (HR = 1.4, 95%CI = 0.38–4.8, p-value = 0.64), and smoking exposure greater than 10 pack-years (HR = 1.8, 95%CI = 0.66–4.8, p-value = 0.25). Overall survival was analyzed, but was not found to be different between patients with HPV16-A1 vs. other genotype, this was likely related to significant non-cancer mortality in the cohort. HPV16-A1 genotype was significantly associated with poor disease specific survival, see Supplemental Figure 1A. Clinical factors related are summarized for patients with HPV16-A1 versus other genotypes in Supplemental Table 1.
Despite known associations of clinical stage and tobacco smoke exposure with outcome, there was no detectable association to viral sub-lineage, see Figure 3C–D. Hypothesizing that some viral proteomes are more immunogenic, we estimated the number of high affinity (< 325 nM) viral peptide / MHC interactions that would be expected given the individual patient’s viral genotype and MHC subtype, see methods above. No difference in antigenicity was identified between peptides encoded by the A1 vs. other sub-lineages, Figure 3E. Based on our recent work associating genomic copy-number variant burden to prognosis in HPV+ OPSCC32, we also examined the total number of copy-altered regions identified in the human genome. Indeed, the A1 sub-lineage correlated with the number of genomic CNVs (p = 0.006, Wilcoxen Rank-sum test), Figure 3F. Follow-up time, race, patient age, patient sex, treatment modality, and anatomic site of tumor origin were also examined, stratified according to HPV16 sub-lineage, see Supplemental Table 1. Notably, there was a trend towards more white patients in the A1, high-risk group. Follow up time was longer in the non-A1, low-risk group.
We compared the prevalence of common coding polymorphisms in HPV+ OPSCC to a population matched (same center and time period) cohort of 44 HPV16+ uterine cervical squamosa cell carcinomas (UCSCC), sequenced with the same technique. Common the prevalence of common (clonal) coding polymorphisms were similar between HPV+ OPSCC and uterine cervical carcinoma (Figure 3H). There as a trend toward Non-A1 HPV16 sub-lineages being more common in the uterine cervical carcinoma (Figure 3G).
In 14/104 tumors, deep copy loss (depth of coverage < 1% of that of tumor matched average E7 coverage) of a portion of the HPV genome was noted (excluding the non-coding hypervariable region 3150–3351). These losses, however, represented only 1.2% of all potential genomic space (tumor HPV bases) interrogated. In only 2 of 104 cases (1.9%), genomic regions with deep loss accounted for more than 10% of the viral genome (75%, 47%). These large genomic losses are likely to be related to (clonal) genomic integration of the HPV16 viral genome. Although integration events can certainly be present as sub-clonal genomic variation, it is interesting to note the deep loss of large segments of the HPV16 genome was quite uncommon in HPV+ OPSCC (Figure 3I). Similarly, only 6 of 104 (5.7%) tumors harbored deep losses of E2 of any size. Fourteen of 44 (32%) HPV16 positive tumors of the uterine cervix sequenced with the same strategy had deep loss involving E2. The difference in proportion of tumors harboring E2 loss between HPV16+ OPSCC (5.7%) and HPV16+ UCSCC (32%) was significant (p < 1*10^−4, Chi squared test), see Figure 3I. Large scale losses (>10% of the viral genome) were also more common in UCSCC (p < 1*10^−4, Chi squared test), see Figure 3I.
Phylogenetic Analysis
Based on the clonal SNPs, relative to the HPV16-A1 reference, full HPV16 genotypes were reconstructed for each tumor. We note that this approach may bias the few (n=2) cases with large areas of deep copy loss as being closer to the A1 reference sequence, as A1 reference is assumed for uncovered bases. To organize the impressive sequence diversity, we implemented a maximum parsimony phylogenetic model. As expected, common non-synonymous polymorphisms were highly correlated with the substructure of the phylogenetic tree. The most common protein-coding polymorphisms are displayed in Figure 3A. Relative viral copy number was estimated based on the ratio of HPV16 to human reads observed. Although relative viral copy number had a wide range (>1000 fold), this was not associated with phylogeny, HPV16 sub-lineage, or recurrence-free survival by any threshold we examined.
We found that the maximum parsimony phylogeny of oncogenic oropharyngeal HPV16 viruses could be reasonably divided into two clades, with membership being highly correlated to the number of non-synonymous polymorphisms (as well as total number of SNPs) relative to the HPV16-A1 reference sequence, see Figure 4A. These de-novo clades were also associated with variable recurrence-free survival. Consistent with the sublineage analysis, the clade more divergent from (far) HPV16-A1 demonstrated relatively favorable recurrence-free survival, see Figure 4B. This association persisted in a multivariate regression model including viral clade (HR = 0.16, 95%CI = 0.04–0.61, p-value = 0.007), early vs. advanced AJCC8 summary stage (HR = 1.6, 95%CI = 0.45–5.9, p-value =0.45), and smoking exposure greater than 10 pack-years (HR =1.6, 95%CI = 0.61–4.4, p-value =0.33). Viral clade remained associated with RFS on subset analysis excluding patients with extensive (> 30 pack-years) tobacco smoke exposure, and also when excluding all patients with greater than 10 pack-years of exposure, see Supplemental Figure 1C–D. Overall survival was examined but, was not significantly different between viral clades. Similar to the sublineage-based analysis, analysis of disease specific survival demonstrated a strong trend towards improved outcomes in the clade more divergent from HPV16-A1, see Supplemental Figure 1B.
Figure 4. Tumor Genomic and Patient Factors Stratified by Maximum Parsimony Phylogeny of OPSCC HPV16 Viral Genomes.

A. Maximum Parsimony Phylogeny of HPV16 genomes. Color – Above or below the median number of non-synonymous clonal polymorphisms in the HPV16 genome relative to the A1 reference sequence. Tip point size/color – Total number of non-synonymous clonal polymorphisms in the HPV16 genome relative to the A1 reference sequence. B. Kaplan-Meier Plot of Recurrence-free Survival, comparing viral clades indicated in Panel A. Log-Rank P-value – p-value derived from the Log-Rank test. Cox P-value – the p-value derived from a univariate Cox Model. Cox HR – estimated hazard ratio with 95% confidence interval derived from Cox Model. C. Proportion of Cases Stratified by AJCC8 Clinical Staging in near and far clades. Significance based on chi-squared test. D. Tobacco Smoke Exposure for near and far clades. Significance based on Wilcoxon Rank-sum test. E. Predicted Viral Proteomic Neo-Antigenicity for near and far clades. Neo-Antigenicity was estimated by the number viral peptides with <350nM affinity for MHC, based on the patients HLA subtype (see methods for further description). Significance based on Wilcoxon Rank-sum test. F. Human Genomic Copy-number Variant Burden. Number of copy number events as identified by the SynthEx pipeline. Significance based on Wilcoxon Rank-sum test. * P-value < 5*10^−2.
Because clinical stage and tobacco exposure history are accepted clinical prognostic markers for HPV+ OPSCC, we directly queried their correlation with viral clade and detected no association, see Figure 4C–D. As was found in our sub-lineage analysis, no difference in antigenicity was identified, Figure 4E. A subset of tumors (n=37) also had available matched normal DNA sequencing which allowed somatic variant calling (Supplemental Figure 2). Mutations in PIK3CA have been reported as a poor prognosticator,46 but did the frequency of these mutations was similar between the near and far clades (See Supplemental Figure 2). One TP53 variant was identified in a patient with a low-risk, divergent clade HPV16 genotype. Similar to the above sub-lineage analysis, an increased number of genomic CNV was associated with the near A1 clade (p = 0.015), see Figure 4F. Follow-up time, race, patient age, patient sex, treatment modality, and anatomic site of tumor origin were also examined, stratified according to HPV16 viral clade, see Supplemental Table 2. There was a trend towards more white patients in the near A1, high-risk clade. Follow up time was longer in the divergent (from HPV16-A1) low-risk clade.
Origins of Genomic Diversity in Oncogenic Oropharyngeal HPV16 – Environmental vs. Intra-tumoral.
Considering that tumors harboring HPV16 genomes more distantly related to HPV16-A1 had distinct biological characteristics, we considered whether the origins of the clonal (environmental) HPV genomic diversity of these groups were also distinct. SNPs and their related trinucleotide contexts were generated for identified non-synonymous polymorphisms (in the HPV16 genome). To focus our analysis on more evolutionarily recent, we limited our analysis to those polymorphisms identified in < 25% of all tumors investigated (limiting analysis to even more uncommon polymorphisms did not change the qualitative results). Tumors groups were then stratified by viral clade as in Figure 4, and non-negative matrix factorization for the COSMIC Signatures (V2)47 was performed, see Figure 5A–B. Diversity amongst HPV16 genomes more related to HPV16-A1 (Figure 5A) was dominated by Signature 9 (NMF weight of 0.61) which is thought to be related to DNA Polymerase Eta related mutagenesis. Alternatively, diversity amongst HPV16 genomes more distantly related to HPV16-A1 (Figure 5B) was dominated by Signature 3 (NMF weight of 0.50) which is thought to be related to defective homologous recombination repair of double-strand breaks. Although it is impossible to know the origins of the investigated SNPs the trinucleotide context-dependent base substitutions appear to be grossly dissimilar between these two groups of tumors bolstering the concept that they are biologically distinct groups of tumor viruses.
Figure 5. Origins of Environmental and Intra-tumoral Genomic Diversity of Oncogenic HPV16 in HNSCC.

A-B. Mutational Signature Analysis of Uncommon HPV16 Non-synonymous Clonal (Environmental) Polymorphisms. All non-synonymous polymorphisms identified in less than 25% of tumors were included. Non-negative matrix decomposition of all included SNPs was performed based on the COSMIC signatures V2, as implemented by the DeconstructSigs R package. A. Tumors in the viral clade near the HPV16-A1 reference sequence. B. Viral genomes distal to relative to HPV16-A1. C-E. Estimating HPV16 Sub-clonality with Binomial Mixture Clustering Analysis. C. Scatter plot of Sub-clone Defining Viral SNPs. VAF – variant allele frequency. D. Bar Plot of frequency of tumors by number of HPV16 sub-clonal populations. Populations defined by VAF Groups defined by binomial mixture clustering with BIC analysis as implemented in the R Canopy package.
Sub-clonal viral polymorphisms, which likely arose during or after oncogenesis also demonstrated a distinct pattern of trinucleotide contexts, compared to clonal polymorphisms (Figure 2E) suggesting a prominent role for APOBEC mediated mutagenesis in defining viral subclones in HNSCC tumor cells. The number of distinct viral subclones were estimated by the Bayesian information criteria (BIC) after clustering of viral sub-clonal VAFs. 31% of tumors had discernable viral genomic sub-clonal populations with an allele fraction > 5% (Figure 5C). Sub-clonal populations were often defined by SNPs (in addition to similar VAFs), had similar trinucleotide contexts, and were close in genomic space, suggestive of multi-mutational kataegis events, which have been previously reported to be the result of APOBEC mediated mutagenesis (Figure 5D).48 Neither the frequency of sub-clonal polymorphisms or sub-clonal populations were associated with patient outcome by any metric investigated.
Validation of HPV16 genotyping by RNA sequencing
To determine if RNA sequencing data was sufficient to assign the HPV16 genotype of a given tumor to the viral clades investigated in Figure 4, 13 patients from the DNA sequencing cohort were also subject to RNA sequencing. Gross agreement of SNPs as determined by DNA and RNA sequencing were seen in all 13 cases, see Supplemental Figure 3 for representative data. Excluding areas with zero coverage in the RNA data, we assigned clade membership according to the nearest neighbors in sequence space (excluding the parent sample) based on JC69 sequence distance. In 100% (13/13) of cases the viral clade assigned by DNA sequencing was recovered by analysis of the RNA sequencing data. With a single exception, nearest neighbors assigned by RNA were quite close in phylogenetic space to the parent tumor based on DNA, see Figure 6A. Based on prevalence of tumors in the two clade groups, of the probability of 13/13 correct classifications by random chance would be ~3.8*10^−5. These data confirming that RNA sequencing correctly classified HPV to near or far clades encouraged us to perform further validation studies using tumor with RNA sequencing data alone.
Validation RNA sequencing Cohort
Considering the strong relationship of HPV16 viral genotype to recurrence free survival in the cohort presented above, we endeavored to validate our finding in a secondary, independent set of patients. Inclusion criteria were the same as the DNA sequencing cohort, with the exception that p16 status was not examined, because many patients with older archival tissue from OPSCC were not subject to routine p16 testing. 120 patients with archival FFPE from OPSCC were identified and processed for next generation RNA sequencing. Of these, 89 patients were found to express HPV16 genes and had sufficient clinical data for inclusion.
Validation of HPV16 genotype by RNA sequencing as predictor of RFS
Prior studies have demonstrated a relationship between viral genomic integration and survival in HPV+ OPSCC. To compare the prognostic values of HPV16 genotype and viral integration, we assigned integration status using a combination of human-viral split read pair identification, as well as the ratio of expression of HPV16 genes E6/E7 to E5/E2, see Figure 6H.29 A trend towards improved RFSl for HPV16 genotypes divergent from the A1 clade was noted in the RNA-only cohort, see Figure 6B. Patients with high degrees of tobacco smoke exposure were somewhat over represented in this new data set, with 20/89 (22%) patients having >= 30 pack-years of tobacco smoke exposure. Subset analysis of patients with <30 pack years of tobacco smoke exposure revealed a strong relationship to RFS, with improved survival in patients with more divergent HPV16 sequences, see Figure 6C. This relationship persisted in the subset of patients with <=10 pack-years of smoking exposure, see Figure 6D. Consistent with prior reports, viral integration was prognostic of poor RFS when all patients were examined, see Figure 6E.29 However, this association lost significance for patients with less than 30 pack-years or 10 pack-years of smoking exposure, see Figure 6F–G. Within the subset of patients with <= 10 pack-years of smoking exposure, HPV16 genotypes closely related to HPV16-A1 were more likely to be integrated (p = 0.02, Chi-squared test), see Figure 6I. No significant differences in AJCC8 stage or tobacco smoke exposure were noted in the <= 10 pack-year sub-group, but the far clade (favorable prognosis group) trended toward higher stage and fewer pack years (p=0.09), Figure 6J–K.
Discussion:
In this study, we analyzed two independent cohorts of HPV16+ OPSCC tumors for a total of 187 cases, representing one of the largest reported series of HPV+ OPSCC with viral sequencing. For reference, TCGA included 56 HPV+ OPSCC cases, if non-oropharyngeal HNSCC cases are excluded.25 Since HPV16 accounts for greater than 90% of OPSCC, and HPV sub-type may correlate with outcome30, we limited our study to HPV16 positive tumors as confirmed by DNA or RNA sequencing.
This study has uncovered a surprising degree of genomic diversity in oncogenic HPV16 viral genomes, with 93 distinct protein coding HPV16 genomes amongst the 104 tumors examined. This rich diversity has been previously under investigated in OPSCC. Previous reports have attempted to with limited success to correlate coarser HPV sub-types (strains) to clinical and biological features of OPSCC. We demonstrate that subtler genotypic variations within the HPV16 genome may have important relationships to clinical outcomes in OPSCC. To our knowledge, no prior study has provided comprehensive HPV16 genotyping for a clinically annotated cohort of HPV16+ OPSCC.
Clinical and Translational Value
HPV+ and HPV−negative OPSCC respond quite differently to treatment, with much better survival for patients with HPV+ tumors.49 Bolstering the evidence that HPV+ and HPV−negative tumors are distinct biological diseases, genomic analyses have revealed exclusive occurrence of TRAF3 and CYLD mutations in HPV+ HNSCC, which have been associated with decreased innate immune response and an excess of APOBEC-driven mutations in PIK3CA.1,9,50,51
Excellent rates of 5-year disease control at the cost of relatively high rates of unfortunate treatment-related morbidities such as gastrostomy tube dependence and osteoradionecrosis of the mandible, have driven many to investigate reduced-intensity treatment regiments for HPV+ OPSCC, with the goal of maintaining excellent cure rates while limiting morbidity.4,5 Although there is tremendous interest in de-intensification of therapy for HPV+ OPSCC, inaccuracy of current prognostic markers such as smoking history, stage or radiologic characteristics in predicting long-term disease control are of concern to clinicians who fear undertreating a patient who may otherwise be cured with standard regimes.52 However, long-term analysis of patients treated with high-intensity chemoradiation protocols has revealed increased non-cancer mortality that may negate the survival advantage of aggressive therapy.53,54 Hoping to improve prognostic predictions through bio-marker development, our group has explored molecular characteristics of HPV+ OPSCC55–57 finding that circulating HPV DNA and defects in TRAF3 or CYLD correlate with survival.9,10
Here, we demonstrate that targeted HPV16 sequencing or total RNA sequencing from FFPE from routine clinical specimens is sufficient to acquire clinically relevant and risk-stratifying information that could inform treatment decisions. We have confirmed the prognostic value of HPV16 genotype classification within two independent cohorts for the subgroup of patients with no more than 10 pack-years of tobacco smoke exposure (the eligibility threshold for reduced treatment intensity at our center and others).58 Although sequencing of the relatively small ~7900 base HPV genome from FFPE would be easily performed in many modern clinical laboratories, other studies have also demonstrated the circulating cell-free HPV DNA shed from OPSCC cancer cells can be robustly detected,10 quantified6 and even genotyped59 from routine clinical blood draw specimens. Therefore, it is also may be possible to similarly acquire prognostic HPV16 genotypic data from clinical blood samples; making HPV16 genotyping a relatively ideal biomarker for clinical application.
Biological Value
We have previously reported that genomic instability determined by copy number variant burden is also prognostic in HPV16+ OPSCC.32 Herein we have shown that HPV16-A1 and closely related genotypes are also associated with increased CNV burden. Genomic CNVs identified were primarily numerical chromosomal aberrations or large-scale sub-chromosomal amplifications or losses.32 This correlation could be explained by the fact that high-risk HPV16 infection and expression of HPV16 E6 and E7 have been demonstrated to directly result in numerical and structural chromosomal instability.60
Many studies have established that HPV infection both induces replication stress,61 causes DNA damage, and disrupts cellular DNA double-strand break (DSB) repair via multiple mechanisms.62 Together with other groups, we have recently reported that HPV−positive head and neck cancer cells are deficient in homologous recombination repair of double-strand breaks.63 Double-strand break repair dysfunction has been broadly linked to genomic instability in human cancers.64,65 Therefore, it is quite possible that functional polymorphisms in the HPV16 genome result in differential modulation of the host of human DDR factors known to be influenced by high risk HPV infection.62 And indeed low-risk HPVs (HPV6, HPV11) are reported to have relatively diminished ability to disrupt the human DNA damage response (DDR).62
Although somewhat counterintuitive that double-strand break mediated viral mutagenesis was correlated with the low-risk group of tumors (divergent from A1) with relatively stable human genomes, see Figure 5B. This may reflect relatively ineffectual recruitment of human DDR factors to viral replications centers62,66 rather than heightened disruption of the DDR in general. Indeed, experimental depletion of host factors involved in the replication of the HPV genome has been demonstrated to primarily decrease the fidelity (increased viral mutagenesis) without greatly altering the throughput of the replicative process.67 Tumors with HPV16 genomes more similar to the A1 sub-lineage were associated with Polymerase Eta (POLH)-related variants. Interestingly, HPV16 E6 expression has been recently found to modulate the activity Polymerase Eta.68 Although our analysis of the origin of variants is merely correlative, it is notable that the two dominant mutagenetic processes identified (deficiency of DSB repair by homologous recombination deficiency and Pol. Eta mutagenesis) both have experimentally validated relationships to high-risk alpha-HPV biology.66,68 It is also interesting that E1^E4, E2, and to some degree E5 ––the HPV proteins known to be involved in HPV replication, HPV gene expression, and recently shown to drive alternative episome-based carcinogenesis-– were found to be the least conserved amongst HPV16 viral genes (Figure 2A–B).69
Prior studies of uterine cervical cancer have demonstrated that HPV18 is more likely to integrated as compared to HPV16, suggesting that HPV genotype can influence the likelihood of genomic integration during carcinogenesis.70 In the present work, the RNA sequencing cohort which allowed viral integration analysis, demonstrated a relationship between viral integration and HPV16 genotype in patients with <= 10 pack-years of smoking. These data suggest that even more subtle genotypic variations in the HPV genome (amongst HPV16 viruses) may influence the chance of genomic integration. Future investigation of the variable biomolecular characteristics of the HPV16 polymorphisms identified in this work, may help determine what biomolecular processes are key for viral integration.
Conclusions:
Striking genomic diversity is present among oncogenic HPV16 viruses associated with squamous cell carcinomas of the oropharynx. Our data from two independent patient cohorts suggests that molecular risk-stratification based on HPV16 genotype is a promising genomic tool for the identification of very low-risk patients which may enable safe treatment de-escalation and limit long-term, treatment-related toxicity which is a key goal in the field.5,8,71
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments and Conflict of Interest Statement:
This research was supported by NIH NIDCR KO8-DE029241-01A (PI: TS), an American Academy of Otolaryngology Head and Neck Surgery, Translational Innovator Award (PI: TS), and NIH NIDCR R01DE027942 (PI: NI and WY). TS has submitted a patent related to this work UNC Ref. 21-0132; MB Ref. 5470.905PR. We have no other relevant disclosures.
Funding Sources:
American Academy of Otolaryngology Head and Neck Surgery, Translational Innovator Award to T.S.
NIH NIDCR KO8-DE029241-01A, Awarded to T.S.
NIH NIDCR R01DE027942, Awarded to N.I. and W.G.Y.
Footnotes
Conflict of Interest Statement: T.S. has submitted a patent related to this work UNC Ref. 21-0132; MB Ref. 5470.905PR. We have no other relevant disclosures.
References:
- 1.Pan C, Issaeva N, Yarbrough WG. HPV−driven oropharyngeal cancer: current knowledge of molecular biology and mechanisms of carcinogenesis. Cancers Head Neck. 2018;3. doi: 10.1186/s41199-018-0039-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doescher J, Veit JA, Hoffmann TK. [The 8th edition of the AJCC Cancer Staging Manual : Updates in otorhinolaryngology, head and neck surgery]. HNO. 2017;65(12):956–961. doi: 10.1007/s00106-017-0391-3 [DOI] [PubMed] [Google Scholar]
- 3.Zhan KY, Eskander A, Kang SY, et al. Appraisal of the AJCC 8th edition pathologic staging modifications for HPV−positive oropharyngeal cancer, a study of the National Cancer Data Base. Oral Oncol. 2017;73:152–159. doi: 10.1016/j.oraloncology.2017.08.020 [DOI] [PubMed] [Google Scholar]
- 4.Cheraghlou S, Yu PK, Otremba MD, et al. Treatment deintensification in human papillomavirus-positive oropharynx cancer: Outcomes from the National Cancer Data Base. Cancer. 2018;124(4):717–726. doi: 10.1002/cncr.31104 [DOI] [PubMed] [Google Scholar]
- 5.Chera BS, Amdur RJ, Tepper JE, et al. Mature results of a prospective study of deintensified chemoradiotherapy for low-risk human papillomavirus-associated oropharyngeal squamous cell carcinoma. Cancer. 2018;124(11):2347–2354. doi: 10.1002/cncr.31338 [DOI] [PubMed] [Google Scholar]
- 6.Chera BS, Kumar S, Beaty BT, et al. Rapid Clearance Profile of Plasma Circulating Tumor HPV Type 16 DNA during Chemoradiotherapy Correlates with Disease Control in HPV−Associated Oropharyngeal Cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2019;25(15):4682–4690. doi: 10.1158/1078-0432.CCR-19-0211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marur S, Li S, Cmelak AJ, et al. E1308: Phase II Trial of Induction Chemotherapy Followed by Reduced-Dose Radiation and Weekly Cetuximab in Patients With HPV−Associated Resectable Squamous Cell Carcinoma of the Oropharynx- ECOG-ACRIN Cancer Research Group. J Clin Oncol Off J Am Soc Clin Oncol. 2017;35(5):490–497. doi: 10.1200/JCO.2016.68.3300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pearlstein KA, Wang K, Amdur RJ, et al. Quality of Life for Patients With Favorable-Risk HPV−Associated Oropharyngeal Cancer After De-intensified Chemoradiotherapy. Int J Radiat Oncol Biol Phys. 2019;103(3):646–653. doi: 10.1016/j.ijrobp.2018.10.033 [DOI] [PubMed] [Google Scholar]
- 9.Hajek M, Sewell A, Kaech S, Burtness B, Yarbrough WG, Issaeva N. TRAF3/CYLD mutations identify a distinct subset of human papillomavirus-associated head and neck squamous cell carcinoma. Cancer. 2017;123(10):1778–1790. doi: 10.1002/cncr.30570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chera BS, Kumar S, Shen C, et al. Plasma Circulating Tumor HPV DNA for the Surveillance of Cancer Recurrence in HPV−Associated Oropharyngeal Cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2020;38(10):1050–1058. doi: 10.1200/JCO.19.02444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith B, Chen Z, Reimers L, et al. Sequence Imputation of HPV16 Genomes for Genetic Association Studies. PLOS ONE. 2011;6(6):e21375. doi: 10.1371/journal.pone.0021375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burk RD, Harari A, Chen Z. Human papillomavirus genome variants. Virology. 2013;445(1–2):232–243. doi: 10.1016/j.virol.2013.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirabello L, Yeager M, Cullen M, et al. HPV16 Sublineage Associations With Histology-Specific Cancer Risk Using HPV Whole-Genome Sequences in 3200 Women. J Natl Cancer Inst. 2016;108(9). doi: 10.1093/jnci/djw100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clifford GM, Tenet V, Georges D, et al. Human papillomavirus 16 sub-lineage dispersal and cervical cancer risk worldwide: Whole viral genome sequences from 7116 HPV16-positive women. Papillomavirus Res Amst Neth. 2019;7:67–74. doi: 10.1016/j.pvr.2019.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lou H, Boland JF, Torres-Gonzalez E, et al. The D2 and D3 Sublineages of Human Papilloma Virus 16-Positive Cervical Cancer in Guatemala Differ in Integration Rate and Age of Diagnosis. Cancer Res. 2020;80(18):3803–3809. doi: 10.1158/0008-5472.CAN-20-0029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Londesborough P, Ho L, Terry G, Cuzick J, Wheeler C, Singer A. Human papillomavirus genotype as a predictor of persistence and development of high-grade lesions in women with minor cervical abnormalities. Int J Cancer. 1996;69(5):364–368. doi: [DOI] [PubMed] [Google Scholar]
- 17.Gheit T, Cornet I, Clifford GM, et al. Risks for persistence and progression by human papillomavirus type 16 variant lineages among a population-based sample of Danish women. Cancer Epidemiol Biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol. 2011;20(7):1315–1321. doi: 10.1158/1055-9965.EPI-10-1187 [DOI] [PubMed] [Google Scholar]
- 18.Grodzki M, Besson G, Clavel C, et al. Increased risk for cervical disease progression of French women infected with the human papillomavirus type 16 E6–350G variant. Cancer Epidemiol Biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol. 2006;15(4):820–822. doi: 10.1158/1055-9965.EPI-05-0864 [DOI] [PubMed] [Google Scholar]
- 19.Zehbe I, Tachezy R, Mytilineos J, et al. Human papillomavirus 16 E6 polymorphisms in cervical lesions from different European populations and their correlation with human leukocyte antigen class II haplotypes. Int J Cancer. 2001;94(5):711–716. doi: 10.1002/ijc.1520 [DOI] [PubMed] [Google Scholar]
- 20.Zacapala-Gómez AE, Del Moral-Hernández O, Villegas-Sepúlveda N, et al. Changes in global gene expression profiles induced by HPV 16 E6 oncoprotein variants in cervical carcinoma C33-A cells. Virology. 2016;488:187–195. doi: 10.1016/j.virol.2015.11.017 [DOI] [PubMed] [Google Scholar]
- 21.Tan G, Duan M, Li Y, et al. Distribution of HPV 16 E6 gene variants in screening women and its associations with cervical lesions progression. Virus Res. 2019;273:197740. doi: 10.1016/j.virusres.2019.197740 [DOI] [PubMed] [Google Scholar]
- 22.Mirabello L, Yeager M, Yu K, et al. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell. 2017;170(6):1164–1174.e6. doi: 10.1016/j.cell.2017.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhe X, Xin H, Pan Z, et al. Genetic variations in E6, E7 and the long control region of human papillomavirus type 16 among patients with cervical lesions in Xinjiang, China. Cancer Cell Int. 2019;19:65. doi: 10.1186/s12935-019-0774-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research Network, Albert Einstein College of Medicine, Analytical Biological Services, et al. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543(7645):378–384. doi: 10.1038/nature21386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–582. doi: 10.1038/nature14129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parfenov M, Pedamallu CS, Gehlenborg N, et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc Natl Acad Sci U S A. 2014;111(43):15544–15549. doi: 10.1073/pnas.1416074111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akagi K, Li J, Broutian TR, et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014;24(2):185–199. doi: 10.1101/gr.164806.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walline HM, Komarck CM, McHugh JB, et al. Genomic Integration of High-Risk HPV Alters Gene Expression in Oropharyngeal Squamous Cell Carcinoma. Mol Cancer Res MCR. 2016;14(10):941–952. doi: 10.1158/1541-7786.MCR-16-0105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koneva LA, Zhang Y, Virani S, et al. HPV Integration in HNSCC Correlates with Survival Outcomes, Immune Response Signatures, and Candidate Drivers. Mol Cancer Res MCR. 2018;16(1):90–102. doi: 10.1158/1541-7786.MCR-17-0153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nichols AC, Dhaliwal SS, Palma DA, et al. Does HPV type affect outcome in oropharyngeal cancer? J Otolaryngol - Head Neck Surg. 2013;42(1):9. doi: 10.1186/1916-0216-42-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis JS, Mirabello L, Liu P, et al. Oropharyngeal Squamous Cell Carcinoma Morphology and Subtypes by Human Papillomavirus Type and by 16 Lineages and Sublineages. Head Neck Pathol. Published online April 2, 2021. doi: 10.1007/s12105-021-01318-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schrank TP, Lenze N, Landess LP, et al. Genomic heterogeneity and copy number variant burden are associated with poor recurrence-free survival and 11q loss in human papillomavirus-positive squamous cell carcinoma of the oropharynx. Cancer. Published online April 5, 2021. doi: 10.1002/cncr.33504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen NPD, Deshpande V, Luebeck J, Mischel PS, Bafna V. ViFi: accurate detection of viral integration and mRNA fusion reveals indiscriminate and unregulated transcription in proximal genomic regions in cervical cancer. Nucleic Acids Res. 2018;46(7):3309–3325. doi: 10.1093/nar/gky180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szolek A. HLA Typing from Short-Read Sequencing Data with OptiType. Methods Mol Biol Clifton NJ. 2018;1802:215–223. doi: 10.1007/978-1-4939-8546-3_15 [DOI] [PubMed] [Google Scholar]
- 35.Rajasagi M, Shukla SA, Fritsch EF, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. 2014;124(3):453–462. doi: 10.1182/blood-2014-04-567933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wells DK, van Buuren MM, Dang KK, et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell. 2020;183(3):818–834.e13. doi: 10.1016/j.cell.2020.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith CC, Entwistle S, Willis C, et al. Landscape and Selection of Vaccine Epitopes in SARS-CoV-2. BioRxiv Prepr Serv Biol. Published online June 4, 2020. doi: 10.1101/2020.06.04.135004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Revathidevi S, Murugan AK, Nakaoka H, Inoue I, Munirajan AK. APOBEC: A molecular driver in cervical cancer pathogenesis. Cancer Lett. 2021;496:104–116. doi: 10.1016/j.canlet.2020.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenthal R. DeconstructSigs: Identifies Signatures Present in a Tumor Sample.; 2016. https://CRAN.R-project.org/package=deconstructSigs
- 40.Laver TW, Franco ED, Johnson MB, et al. SavvyCNV: Genome-Wide CNV Calling from off-Target Reads.; 2019:617605. doi: 10.1101/617605 [DOI] [Google Scholar]
- 41.Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLOS Comput Biol. 2016;12(4):e1004873. doi: 10.1371/journal.pcbi.1004873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14(4):417–419. doi: 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu B, Xiao Y, Yeager M, et al. Mutations in the HPV16 genome induced by APOBEC3 are associated with viral clearance. Nat Commun. 2020;11(1):886. doi: 10.1038/s41467-020-14730-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jukes TH, Cantor CR. CHAPTER 24 - Evolution of Protein Molecules. In: Munro HN, ed. Mammalian Protein Metabolism. Academic Press; 1969:21–132. doi: 10.1016/B978-1-4832-3211-9.50009-7 [DOI] [Google Scholar]
- 45.Arroyo-Mühr LS, Lagheden C, Hultin E, et al. Human papillomavirus type 16 genomic variation in women with subsequent in situ or invasive cervical cancer: prospective population-based study. Br J Cancer. 2018;119(9):1163–1168. doi: 10.1038/s41416-018-0311-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beaty BT, Moon DH, Shen CJ, et al. PIK3CA mutation in HPV−associated OPSCC patients receiving deintensified chemoradiation. J Natl Cancer Inst. Published online November 20, 2019. doi: 10.1093/jnci/djz224 [DOI] [PubMed] [Google Scholar]
- 47.Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3(1):246–259. doi: 10.1016/j.celrep.2012.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lada AG, Dhar A, Boissy RJ, et al. AID/APOBEC cytosine deaminase induces genome-wide kataegis. Biol Direct. 2012;7:47; discussion 47. doi: 10.1186/1745-6150-7-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ben-David U, Siranosian B, Ha G, et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature. 2018;560(7718):325–330. doi: 10.1038/s41586-018-0409-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J, Chen T, Yang X, et al. Attenuated TRAF3 Fosters Activation of Alternative NF-κB and Reduced Expression of Antiviral Interferon, TP53, and RB to Promote HPV−Positive Head and Neck Cancers. Cancer Res. 2018;78(16):4613–4626. doi: 10.1158/0008-5472.CAN-17-0642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cannataro VL, Gaffney SG, Sasaki T, et al. APOBEC-induced mutations and their cancer effect size in head and neck squamous cell carcinoma. Oncogene. 2019;38(18):3475–3487. doi: 10.1038/s41388-018-0657-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheraghlou S, Yu PK, Otremba MD, et al. Treatment deintensification in human papillomavirus-positive oropharynx cancer: Outcomes from the National Cancer Data Base. Cancer. 2018;124(4):717–726. doi: 10.1002/cncr.31104 [DOI] [PubMed] [Google Scholar]
- 53.Forastiere AA, Zhang Q, Weber RS, et al. Long-term results of RTOG 91–11: a comparison of three nonsurgical treatment strategies to preserve the larynx in patients with locally advanced larynx cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31(7):845–852. doi: 10.1200/JCO.2012.43.6097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tasoulas J, Lenze NR, Farquhar D, et al. The addition of chemotherapy to adjuvant radiation is associated with inferior survival outcomes in intermediate-risk HPV−negative HNSCC. Cancer Med. 2021;10(10):3231–3239. doi: 10.1002/cam4.3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Slebos RJC, Li M, Evjen AN, Coffa J, Shyr Y, Yarbrough WG. Mutagenic effect of cadmium on tetranucleotide repeats in human cells. Mutat Res. 2006;602(1–2):92–99. doi: 10.1016/j.mrfmmm.2006.08.003 [DOI] [PubMed] [Google Scholar]
- 56.Slebos RJC, Jehmlich N, Brown B, et al. Proteomic analysis of oropharyngeal carcinomas reveals novel HPV−associated biological pathways. Int J Cancer. 2013;132(3):568–579. doi: 10.1002/ijc.27699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sewell A, Brown B, Biktasova A, et al. Reverse-phase protein array profiling of oropharyngeal cancer and significance of PIK3CA mutations in HPV−associated head and neck cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2014;20(9):2300–2311. doi: 10.1158/1078-0432.CCR-13-2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chera BS, Amdur RJ, Tepper JE, et al. Mature results of a prospective study of deintensified chemoradiotherapy for low-risk human papillomavirus-associated oropharyngeal squamous cell carcinoma. Cancer. 2018;124(11):2347–2354. doi: 10.1002/cncr.31338 [DOI] [PubMed] [Google Scholar]
- 59.Lopez EM, Tanner AM, Du E, et al. Decline in circulating viral and human tumor markers after resection of head and neck carcinoma. Head Neck. 2021;43(1):27–34. doi: 10.1002/hed.26444 [DOI] [PubMed] [Google Scholar]
- 60.Duensing S, Münger K. The Human Papillomavirus Type 16 E6 and E7 Oncoproteins Independently Induce Numerical and Structural Chromosome Instability. Cancer Res. 2002;62(23):7075–7082. [PubMed] [Google Scholar]
- 61.Moody CA. Impact of Replication Stress in Human Papillomavirus Pathogenesis. J Virol. 2019;93(2). doi: 10.1128/JVI.01012-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wallace NA. Catching HPV in the Homologous Recombination Cookie Jar. Trends Microbiol. 2020;28(3):191–201. doi: 10.1016/j.tim.2019.10.008 [DOI] [PubMed] [Google Scholar]
- 63.Hajek M, Biktasova A, Sewell A, et al. Global Genome Demethylation Causes Transcription-Associated DNA Double Strand Breaks in HPV−Associated Head and Neck Cancer Cells. Cancers. 2020;13(1). doi: 10.3390/cancers13010021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu X, Li F, Huang Q, et al. Self-inflicted DNA double-strand breaks sustain tumorigenicity and stemness of cancer cells. Cell Res. 2017;27(6):764–783. doi: 10.1038/cr.2017.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.So A, Guen TL, Lopez BS, Guirouilh-Barbat J. Genomic rearrangements induced by unscheduled DNA double strand breaks in somatic mammalian cells. FEBS J. 2017;284(15):2324–2344. doi: 10.1111/febs.14053 [DOI] [PubMed] [Google Scholar]
- 66.Mehta K, Laimins L. Human Papillomaviruses Preferentially Recruit DNA Repair Factors to Viral Genomes for Rapid Repair and Amplification. mBio. 2018;9(1). doi: 10.1128/mBio.00064-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bristol ML, Wang X, Smith NW, Son MP, Evans MR, Morgan IM. DNA Damage Reduces the Quality, but Not the Quantity of Human Papillomavirus 16 E1 and E2 DNA Replication. Viruses. 2016;8(6). doi: 10.3390/v8060175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wendel SO, Snow JA, Bastian T, et al. High Risk α-HPV E6 Impairs Translesion Synthesis by Blocking POLη Induction. Cancers. 2020;13(1). doi: 10.3390/cancers13010028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ren S, Gaykalova DA, Guo T, et al. HPV E2, E4, E5 drive alternative carcinogenic pathways in HPV positive cancers. Oncogene. 2020;39(40):6327–6339. doi: 10.1038/s41388-020-01431-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McBride AA, Warburton A. The role of integration in oncogenic progression of HPV−associated cancers. PLoS Pathog. 2017;13(4):e1006211. doi: 10.1371/journal.ppat.1006211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mavroidis P, Price A, Fried D, et al. Dose-volume toxicity modeling for de-intensified chemo-radiation therapy for HPV−positive oropharynx cancer. Radiother Oncol J Eur Soc Ther Radiol Oncol. 2017;124(2):240–247. doi: 10.1016/j.radonc.2017.06.020 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw DNA sequencing data has been deposited to dbGaP – accession number phs001713.v1.p1. Data post processing code is available at https://github.com/TravisParkeSchrank/HPV16_genotype.