

Key Points
T cell–specific IFN-γ deficiency abrogates T cell–mediated control of Mtb burden.
IFN-γ−/− T cells are associated with TB lesion granulocyte abundance and pathology.
T cell–specific IFN-γ deficiency polarizes macrophages toward type 2 immune responses.
Visual Abstract

Abstract
T cells producing IFN-γ have long been considered a stalwart for immune protection against Mycobacterium tuberculosis (Mtb), but their relative importance to pulmonary immunity has been challenged by murine studies that achieved protection by adoptively transferred Mtb-specific IFN-γ−/− T cells. Using IFN-γ−/− T cell chimeric mice and adoptive transfer of IFN-γ−/− T cells into TCRβ−/−δ−/− mice, we demonstrate that control of lung Mtb burden is in fact dependent on T cell–derived IFN-γ, and, furthermore, mice selectively deficient in T cell–derived IFN-γ develop exacerbated disease compared with T cell–deficient control animals, despite equivalent lung bacterial burdens. Deficiency in T cell–derived IFN-γ skews infected and bystander monocyte-derived macrophages to an alternative M2 phenotype and promotes neutrophil and eosinophil influx. Our studies support an important role for T cell–derived IFN-γ in pulmonary immunity against tuberculosis.
Introduction
Studies in mice and in human cells have repeatedly demonstrated that immunity to Mycobacterium tuberculosis (Mtb) requires both T cells and IFN-γ. Genetic deficiencies in CD4+ T cells are associated with increased susceptibility to Mtb in mice (1) and humans (2), an outcome mirrored during acquired CD4+ deficiency in advanced HIV (3). Global knockout studies have likewise confirmed the importance of IFN-γ in Mtb restriction in mice (4, 5), and deficits in IFN-γ signaling are associated with human susceptibility to mycobacterial infections, most often with environmentally ubiquitous nontuberculous mycobacteria but also with tuberculosis (TB) (6). Nevertheless, the relative contribution of T cell–derived IFN-γ to protective immunity remains unclear. Currently, vaccine candidates are routinely assessed for their ability to elicit T cell memory responses, particularly the ability of peripheral blood T cells to produce IFN-γ, but this parameter does not consistently correlate with protective immunity to Mtb (7–14). Additionally, IFN-γ-producing T cells can contribute to amelioration (15) or exacerbation (16) of lung pathology in the context of Mtb infection. Studies on the importance of polyfunctional CD4+ T cells, as well as in vivo (15–18) and in vitro (19) studies suggestive of IFN-γ-independent CD4+ T cell functions, have cast doubt on the primacy of T cell–derived IFN-γ. IFN-γ is produced by other cell types in addition to T cells, such as NK cells, during Mtb infection (20), and the relationship between IFN-γ and its source and target cells remains incompletely understood. Understanding the relative role of T cell–derived IFN-γ will inform a rational approach to inducing vaccine-mediated protection against tuberculosis (11, 14).
Prior studies in mice on the C57BL/6 genetic background have challenged the relative importance of T cell–derived IFN-γ in immunity to Mtb. In Mtb-infected Rag2−/− host mice, adoptive transfer of CD4+ T cells from previously Mtb-infected, antibiotic-treated donor mice reduced Mtb lung burden and conferred a survival advantage relative to no-transfer control mice, regardless of whether donor T cells expressed IFN-γ (10). In another study, adoptive transfer of naive CD4+ T cells from uninfected donor mice into Mtb-infected Rag1−/− mice reduced Mtb burden in the lung in a manner only partially dependent on donor T cell IFN-γ (16). In a bone marrow chimera model using Rag1−/− hosts, Mtb burdens were lower in mice with donor-derived IFN-γ−/− lymphocytes than in nonchimeric mice globally deficient in IFN-γ, although protection was greater with wild-type (WT) donor-derived lymphocytes (21). Finally, in WT mice, adoptive transfer of in vitro Th1-polarized CD4+ TCR-transgenic T cells specific for the immunodominant Mtb Ag ESAT6 reduced Mtb burden in a manner partially or, at high doses of transferred cells, completely independently of donor T cell–derived IFN-γ (18). Together, these reports suggest that CD4+ T cell–derived IFN-γ plays a minimal (18) or partial (15, 16, 21) role in protective immunity against Mtb within the lung.
Although these studies highlight potential roles of T cell immunity beyond IFN-γ, the adoptive transfer techniques employed carry limitations that may lead to underestimation of the relative importance of T cell–derived IFN-γ. For example, transfer of a fixed quantity of T cells into lymphopenic mice leads to a limited T cell repertoire (22), potentially skewing the resulting immune response. In this scenario, adoptively transferred T cells also undergo rapid proliferation, differentiation, and activation (22, 23), which would alter their response to subsequent infection and can lead to systemic autoimmunity (24). In addition, RAG-deficient host mice lack not only T cell function but also mature B cells and normal lymphoid structure (25). In TCR-transgenic adoptive transfer studies, a very large number of monoclonal Mtb-specific T cells may amplify the relative contribution of IFN-γ-independent mechanisms of protection that may play a minor role in a more physiologic Mtb-specific cell response. In this study, we used a T cell bone marrow chimera model to address some of the shortcomings of prior adoptive transfer studies and to reassess the hypothesis that T cell–derived IFN-γ plays only a minor role in T cell–dependent immunity to Mtb in the lung. In contrast to methods used in prior studies, we used the more specific T cell–deficient TCRβ−/−δ−/− host strain. This strain lacks TCRαβ and TCRγδ T cells, both of which have been shown to produce IFN-γ in response to Mtb (26). The T cell chimera model allows physiologically relevant T cell development, including generation of a diverse TCR repertoire, thymic selection, and homeostatic regulation, to occur in the host mouse (27). Our findings indicate that T cell–derived IFN-γ is indeed essential for pulmonary immune protection against Mtb, providing a reappraisal of the relative importance of this aspect of T cell–mediated immunity.
Materials and Methods
Mice
TCRβ−/−δ−/− (strain 002122), IFN-γ−/− (strain 002287), and C57BL/6J (WT, strain 000664) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were matched by age (when possible) and sex. Mice across different experimental groups were cohoused in the same cage to minimize confounding differences in environment and microbiota. Mice were sacrificed using cervical dislocation. All animals were housed and maintained in specific pathogen-free conditions at Seattle Children’s Research Institute (SCRI). All animal studies were performed with approval of the SCRI Animal Care and Use Committee.
Bone marrow chimeras
Recipient TCRβ−/−δ−/−mice were sublethally irradiated with 600 rad. Bone marrow was prepared from femurs and tibias of TCRβ−/−δ−/−, WT, and IFN-γ−/− donor mice and depleted of mature T cells using a CD3ε depletion kit (Miltenyi Biotec, 130-094-973). Bone marrow cells (1–2 × 106) were then injected retro-orbitally into each host mouse under isoflurane anesthesia. Mice received enrofloxacin in drinking water for 4 wk after irradiation to prevent neutropenic sepsis. Chimeric mice were rested for 8–10 wk prior to subsequent manipulation to allow T cell development.
Adoptive transfer
Adoptive transfer of total CD4 T cells into immunocompromised host mice was performed 7 d after aerosol Mtb infection of host TCRβ−/−δ−/− or Rag1−/− mice as follows. CD4+ T cells were isolated from spleens and lymph nodes of donor C57BL/6 and IFN-γ−/− mice using the MagniSort CD4+ T cell enrichment kit (Thermo Fisher, 8804-6821-74). Flow cytometry confirmed that >85% of isolated live CD4+ T cells were naive (CD44loCD62Lhi). CD4+ T cells (3 × 106) were injected retro-orbitally into each host mouse under isoflurane anesthesia.
Mtb aerosol infections
T cell chimeric mice were infected with 25–100 CFU aerosolized Mtb H37Rv transformed with a reported plasmid bearing the mCherry fluorescent marker constitutively expressed under the pMSP12 promoter sequence (28). Adoptive transfer mice were infected with 25–100 CFU aerosolized Mtb H37Rv. Aerosol infections were performed in a Glas-Col chamber. Two additional mice in each infection were sacrificed directly postinfection to confirm the infectious dose of Mtb CFU per mouse.
CFU plating
Mouse lung lobes and spleens were homogenized in M tubes (Miltenyi Biotec) containing 1 ml PBS + 0.05% Tween 80 (PBS-T) using a GentleMACS tissue dissociator (Miltenyi Biotec). Organ homogenates were then diluted in PBS-T, and aliquots were plated onto 7H10 agar media to quantify Mtb burden. Plates were incubated at for 37°C for at least 21 d prior to CFU counting.
Flow cytometry
CD45.2 Ab (0.2 mg, PE) was injected into mice retro-orbitally 5–10 min prior to sacrifice to label intravascular cells. Lung lobes were excised and dissociated in C tubes (Miltenyi Biotec) in HEPES buffer containing Liberase Blendzyme 3 (70 mg/ml; Roche) and DNase I (30 mg/ml; Sigma-Aldrich) using a GentleMACS dissociator (Miltenyi Biotec). Lung homogenates were incubated for 30 min at 37°C and further processed with the GentleMACS dissociator. Cell suspensions were filtered through a 100-μm cell strainer, treated with RBC lysis buffer (Thermo), and resuspended in FACS buffer (PBS containing 2.5% FBS and 0.1% NaN3). Single-cell suspensions were washed in PBS and then incubated with 50 μl Zombie Aqua viability dye (BioLegend) for 10 min at room temperature in the dark. Cell markers were stained, and viability dye was quenched by the addition of 100 μl of a mixture of fluorophore-conjugated Abs diluted in 50% FACS buffer/50% 24G2 Fc block (Bio X Cell, 2.4G2) and incubated for 20 min at 4°C. Cells were washed once with FACS buffer and fixed with 1% paraformaldehyde for 30 min prior to analysis on an LSRII flow cytometer (BD Biosciences).
Cell sorting
Lungs were processed as described for flow cytometry above, but NaN3 was omitted from FACS buffer. Cells were sorted on a FACSAria cell sorter (BD Biosciences) under BSL3 conditions.
RNA sequencing
Single-cell suspensions from lungs were analyzed and sorted by FACS into Mtb-infected (mCherry+) and bystander (mCherry−) monocyte-derived macrophages (MDMs; dead− Siglec F− Ly6G− CD11b+ CD64+) populations. RNA was isolated using TRIzol and quantified using bulk RNA sequencing (RNA-seq) (Psomagen) after construction of Illumina sequencing libraries using the SMARTer Stranded Total RNA-Seq Kit version 2 Pico Input Mammalian (Takara). Noise from low-expression transcripts was filtered, and analysis of differentially expressed genes across groups was done using the edgeR module in R (29).
Protein quantification
Lung lobes were homogenized in M tubes (Miltenyi Biotec) containing 1 ml ProcartaPlex Cell Lysis Buffer (Invitrogen, EPX-99999-000) with Halt Protease Inhibitor (Invitrogen, 78440) and DNase I (30 mg/ml; Sigma-Aldrich) using a GentleMACS tissue dissociator (Miltenyi Biotec). Homogenates were centrifuged to pellet debris, and supernatants were filtered twice through a 0.22-μm pore size Costar SpinX column (Corning) to exclude mycobacteria, frozen at −80°C, and assayed after a single freeze–thaw cycle. Total protein was measured by bicinchoninic acid assay (Thermo Fisher), and these values were used to normalize individual analyte levels in each sample. IL-4, IL-5, and IL-13 levels in lung homogenates were measured using Cytokine Bead Array Flex Sets (BD); bead fluorescence was measured on an LSRII flow cytometer (BD Biosciences) and analyzed by four-parameter log-logistic curve fitting to the standard curve. IFN-γ levels were quantified using a magnetic Luminex assay (Thermo Fisher Scientific) and analyzed using BioPlex Manager software (Bio-Rad Laboratories).
Histopathologic analysis and confocal imaging
Right inferior lung lobes were dissected and fixed in 20 ml 1:3 dilution of BD Cytofix Buffer (∼1% formaldehyde) for 24 h at 4°C to ensure killing of Mtb, equilibrated in 30% sucrose solution for another 24 h at 4°C, then rapidly frozen in O.C.T. compound in an ethanol dry ice slurry and stored at −80°C. For histopathologic analysis, tissue was embedded in paraffin, 4-mm tissue sections were prepared with a cryostat and mounted on glass slides, stained with H&E by the University of Washington Comparative Pathology Core Facility, then assessed by a trained veterinary pathologist blinded to group assignments. For confocal imaging, 20-mm tissue sections were prepared with a cryostat and mounted on glass slides. Sections were stained with fluorophore-conjugated Abs and Nucspot 750/780 nuclear stain (Biotium) overnight at room temperature and coverslipped with Fluoromount G mounting media (SouthernBiotech). Images were acquired on a Leica Stellaris 8 confocal microscope, compensated for fluorophore spillover using LAS X (Leica), and rendered in Imaris (Bitplane), where ARG1 signal was smoothed using a Gaussian filter with a width of 0.316 μm. Identical settings were applied across experimental groups.
Statistical analysis
Statistical significance was determined using the multcomp and rstatix packages in R using methods indicated in the figure legends. Principal component analysis was done using the stats package in R.
Results
T cell–derived IFN-γ is required to reduce lung Mtb burden and protect from disease
To investigate the role of T cell–derived IFN-γ in TB immunity, we generated T cell chimeric mice (Fig. 1A) in which T cells, but not other cell types, were genetically deficient in their capacity to express IFN-γ. To establish this system, TCRβ−/−δ−/− host mice were partially myeloablated by sublethal irradiation and reconstituted with bone marrow of IFN-γ−/− donor mice or, as controls, bone marrow of TCRβ−/−δ−/− or WT mice. In these chimeras, all T cells are derived from the donor bone marrow (e.g., IFN-γ−/− for the experimental group), whereas >95% of other hematopoietic lineage cells remain WT due to the sublethal dose of radiation (27). After immune reconstitution, T cell chimeric mice were infected with aerosolized Mtb H37Rv, then assessed for bacterial burden in lungs and spleens at 25 d postinfection (dpi). In contrast to prior T cell transfer studies suggesting that T cell–derived IFN-γ may be partially or wholly dispensable for protective T cell responses against murine pulmonary Mtb (16, 18), we found that control of bacterial burden in lungs and spleens of T cell chimeric mice at 25 dpi was indeed dependent on T cell–derived IFN-γ (Fig. 1B, 1C). Furthermore, IFN-γ−/− T cell chimeric mice exhibited clinical deterioration (decreased activity, hunched posture) and lost weight beyond 25 dpi, whereas TCRβ−/−δ−/− chimeric controls did not (Fig. 1D), despite equivalent lung bacterial burdens, suggesting that T cell activity during Mtb infection promotes disease unless countered by T cell–derived IFN-γ.
FIGURE 1.
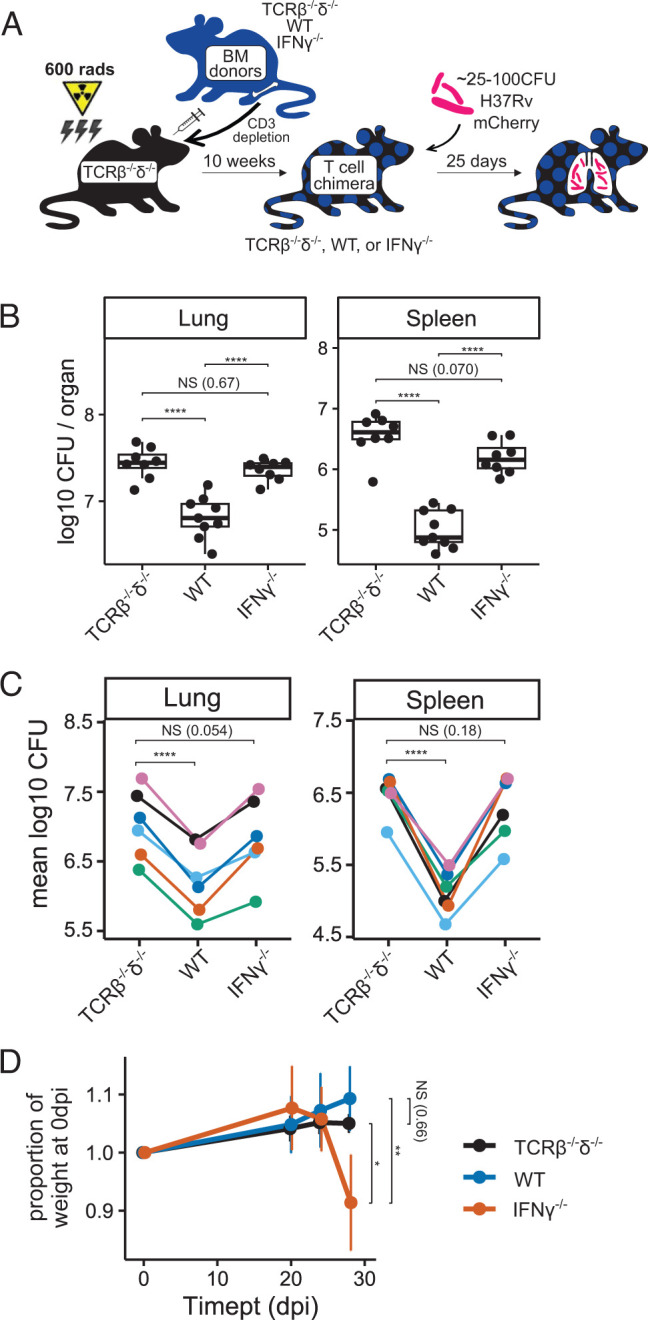
IFN-γ−/− T cells do not reduce Mtb bacterial burden, and they exacerbate disease in T cell chimeric mice. (A) Schematic of the preparation of TCRβ−/−δ−/−, WT, and IFN-γ−/− T cell chimeric mice, followed by infection with aerosolized Mtb. (B and C) Bacterial burden in lungs and spleens of Mtb-infected T cell chimeric mice at 25 dpi in one representative experiment (B), as well as group means from six independent experiments (C). Total n = 35 TCRβ−/−δ−/−, n = 34 WT, and n = 33 IFN-γ−/− T cell chimeric mice. (D) Weight trends of Mtb-infected T cell chimeric mice through 29 dpi for n = 45 TCRβ−/−δ−/−, n = 3 WT, and n = 6 IFN-γ−/− T cell chimeric mice in one representative experiment. Statistical significance is shown for weights at 29 dpi. Statistical significance was determined by Tukey’s range test (B and D) and paired t test (C). *p ≤ 0.05, **p ≤ 0.01, ****p ≤ 0.0001. BM, bone marrow.
In contrast to our results in the T cell chimera model, prior studies have shown that adoptive transfer of IFN-γ−/− T cells into T cell–deficient (Rag1−/−) host mice decreases lung and spleen Mtb CFU relative to no transfer, although the protective effect was smaller than that observed after adoptive transfer of WT T cells (16). We used a similar adoptive transfer strategy (Fig. 2A) to reconcile those findings with our results. Instead of RAG-deficient host mice as in Sakai et al., however, we used TCRβ−/−δ−/− host mice (as we had used in the T cell chimera experiments; Fig. 1A). TCRβ−/−δ−/− host mice were infected with aerosolized Mtb, and CD4+ T cells (3 × 106/mouse) isolated from WT or IFN-γ−/− donor mice were administered i.v. at 7 dpi. To investigate whether IFN-γ−/− T cells mediate pathologic effects and whether WT T cells can counteract those effects in this model, an additional group of mice received a 50%/50% mix of WT and IFN-γ−/− T cells. Similar to our observations in T cell chimeric animals, WT T cells significantly decreased Mtb burden in both the lungs and spleens of TCRβ−/−δ−/− host mice at 36 dpi, whereas IFN-γ−/− T cells did not (Fig. 2B), again supporting a requirement for T cell–derived IFN-γ for control of bacterial burden in murine Mtb infection. Furthermore, mice receiving adoptively transferred IFN-γ−/− CD4+ T cells exhibited significantly more weight loss over the course of infection than either no-transfer control animals or WT CD4+ T cell transfer mice (Fig. 2C), despite having mycobacterial burdens equivalent to no-transfer control mice. This is again consistent with a pathologic effect mediated by T cells that are unable to produce IFN-γ, as observed in T cell chimeric mice. Interestingly, coadministration of WT and IFN-γ−/− donor CD4+ T cells restored the ability of recipient mice to control mycobacterial burden in lungs and spleens (Fig. 2B, “mix”) and decreased the rate of weight loss postinfection compared with mice receiving IFN-γ−/− T cells alone. This suggests that IFN-γ−/− deficient T cells are not inherently pathogenic and can be complemented in trans by the presence of other T cells that can produce IFN-γ, but not by other IFN-γ-producing cell types (such as NK or NKT cells), during Mtb infection.
FIGURE 2.
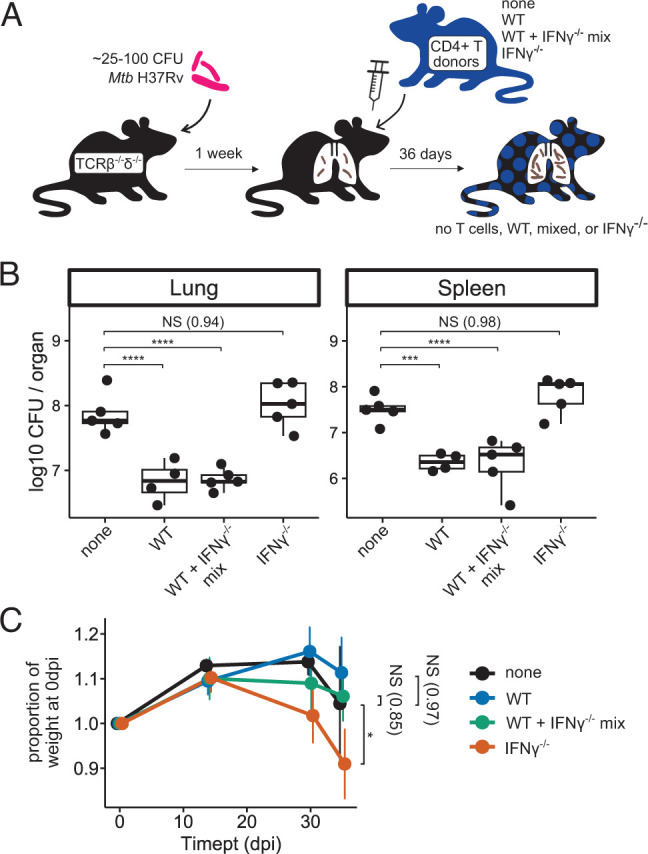
Adoptively transferred IFN-γ−/− CD4+ T cells do not reduce Mtb burden, and they exacerbate disease in TCRβ−/−δ−/− mice. (A) Schematic of adoptive transfer of zero (none) or 3 × 106 WT, 50%/50% mixed WT+IFN-γ−/−, or IFN-γ−/− CD4+ T cells to T cell–deficient host mice postinfection with aerosolized Mtb. (B) Bacterial burden in lungs and spleens of Mtb-infected adoptive transfer mice at 36 dpi. Results are representative of two independent experiments. (C) Weight trends of Mtb-infected adoptive transfer mice through 36 dpi. Statistical significance is shown for weights at 36 dpi. n = 5 none, n = 4 WT, n = 5 WT+IFN-γ−/−, and n = 5 IFN-γ−/− adoptive transfer mice. Results are representative of two independent experiments. Statistical significance was determined by Dunnett’s test using “none” as the control group. *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001.
In a parallel experiment, we also tested whether the difference between our results and those reported by Sakai et al. could be explained by differences in the host mice used, as ours were specifically deficient in T cells (TCRβ−/−δ−/−), whereas Sakai et al. used mice deficient in both B and T cells (Rag1−/−) (Supplemental Fig. 1A). Although Sakai et al. reported 60-fold and 5-fold decreases, respectively, in lung and spleen Mtb burden in Rag1−/− mice that had received IFN-γ−/− T cells compared with no-transfer control mice at 42 dpi (16), we observed similar lung and spleen mycobacterial burdens in these groups (Supplemental Fig. 1B). However, we had to assess mycobacterial burden in Rag1−/− host mice at the earlier 34 dpi time point because, in our laboratory, Mtb-infected Rag1−/− host mice started to lose weight after 23 d of infection, regardless of transferred T cell genotype (Supplemental Fig. 1C), and, in a separate experiment, many in fact required euthanasia prior to 42 dpi. In addition, within-group variance among lung Mtb CFU in Rag1−/− host mice in our hands was high, which limited our ability to observe statistically significant intragroup effects in lung Mtb burden (Supplemental Fig. 1B, left) and thus may have also contributed to discordance between our findings and those of Sakai et al. Although our results do not fully explain the discrepancy between our findings and those previously published, they suggest that using Rag1−/− recipients that lack both T cells and B cells and that have aberrant lymph nodes may lead to confounding factors that increase variability in some settings. Taken together, our data using both T cell chimeric mice and adoptive transfer into T cell–deficient mice suggest that T cell–derived IFN-γ is required for pulmonary immunity against Mtb and that a T cell–specific incapability to produce IFN-γ can in fact promote detrimental pathologic effects.
TB lesions in IFN-γ−/− T cell chimeric mice exhibit increased neutrophilic and eosinophilic infiltration
To investigate the immune landscape associated in mice with T cell intrinsic IFN-γ deficiency, we analyzed the cellular composition of lung tissue of T cell chimeric mice at 25 dpi using flow cytometry (Fig. 3A, gating as in Supplemental Fig. 2A). Strikingly, the number of both neutrophils and eosinophils in IFN-γ−/− T cell chimeric mice was approximately 1 log higher than in either TCRβ−/−δ−/− (lacking T cells) or WT T cell chimeras (Fig. 3A), although T cell–dependent recruitment of MDMs was preserved in both IFN-γ−/− and WT T cell chimeras. Consistent with these findings, confocal microscopy revealed robust neutrophil and eosinophil infiltration into pulmonary TB lesions in IFN-γ−/− chimeric mice (Fig. 3B).
FIGURE 3.
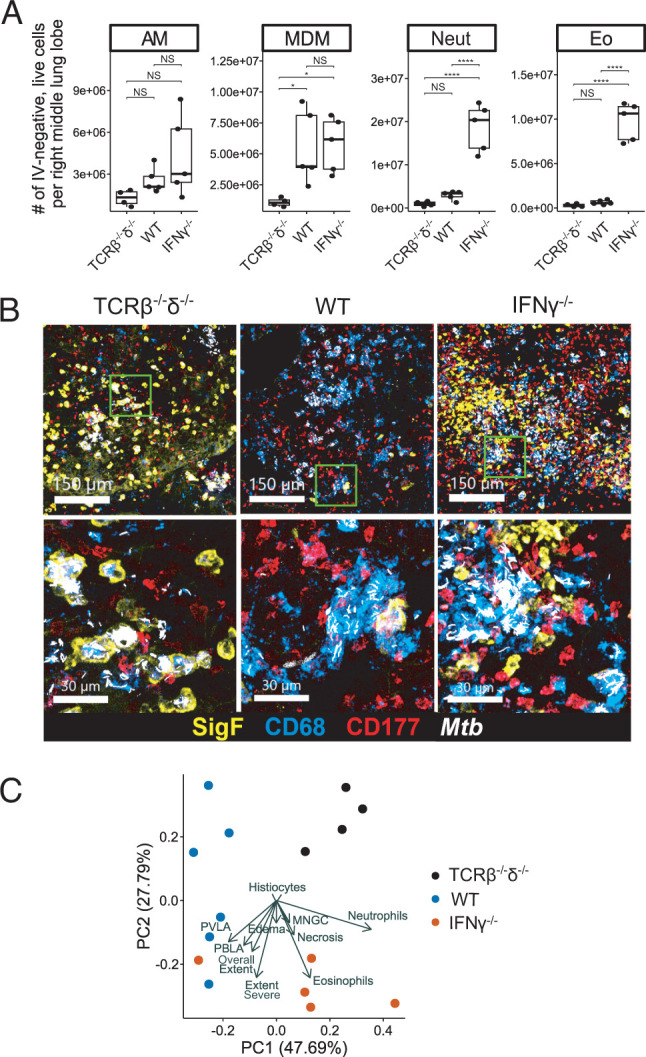
IFN-γ−/− T cells promote neutrophil and eosinophil recruitment to pulmonary lesions in T cell chimeric mice infected with Mtb. (A) Absolute number of each indicated cell type among live, parenchymal (IV-) cells in the right lung of T cell chimeric mice at 25 dpi. Results are representative of six independent experiments. Statistical significance was determined by Tukey’s range test. *p ≤ 0.05, ****p ≤ 0.0001. AM, alveolar macrophage; Eo, eosinophil; Neut, neutrophil. (B) Representative confocal microscopy demonstrating AMs (Siglec F+, CD68+), MDMs (Siglec F−, CD68+), neutrophils (CD177+), and eosinophils (Siglec F+, CD68−) in TB lesions in T cell chimeric mice. (C) Principal component analysis of 15 histopathologic features assessed in representative sections of fixed and H&E-stained lung of T cell chimeric mice at 25 dpi. MNGC, multinucleated giant cells; PBLA, peribronchial lymphoid aggregates; PVLA, perivascular lymphoid aggregates.
We next asked whether histopathologic tissue analysis might help give insight into how increased neutrophil and eosinophil responses (Fig. 3A, 3B) may be linked with clinical decline in IFN-γ−/− T cell chimeric mice relative to TCRβ−/−δ−/− and WT T cell chimeras (Fig. 1D). H&E-stained lung sections were scored in a blinded fashion across 10 standardized histopathologic features (Supplemental Fig. 3A), confirming that Mtb lesions in IFN-γ−/− T cell chimeric mouse lungs were marked by abundant neutrophil and eosinophil infiltration (Supplemental Fig. 3B; 800×). Absence of T cells correlated with fewer and more poorly organized TB lesions in TCRβ−/−δ−/− T cell chimeric mice, whereas both IFN-γ−/− and WT T cell chimeric mice were able to form dense, organized TB lesions (Supplemental Fig. 3B; 25×). Although the differences in histopathology across groups were individually subtle, combined assessment using principal component analysis of the blinded histopathology feature scores clustered samples within each genotype together, indicating similar pathology; this was driven mainly by lesion neutrophils and eosinophils in IFN-γ−/− T cell chimeric mice and decreased extent and severity of lung involvement, as well as absence of lymphoid aggregates, in TCRβ−/−δ−/− chimeric mice (Fig. 3C). In summary, T cells were able to promote organized TB lesions in lung tissue of T cell chimeric mice during Mtb infection, regardless of their ability to produce IFN-γ, but T cell–derived IFN-γ was required to restrict neutrophil and eosinophil infiltration of these lesions.
IFN-γ−/− T cell chimeric mice exhibit a type 2 cytokine milieu and alternative activation of MDMs
To further investigate the possible immune effector mechanisms associated with the maladaptive response to Mtb infection in IFN-γ−/− T cell chimeric mice, we assessed gene expression in lung MDMs, the primary infected cell type in pulmonary TB. In FACS-sorted Mtb-mCherry–infected and bystander MDMs at 25 dpi, we noted prominent suppression of multiple hallmark M1 genes commonly associated with antimycobacterial responses, including Nos2, IL12a, and IL12b, and concurrent upregulation of a subset of hallmark alternative activation (M2) genes, including Arg1, Chil3 (Ym1), Mrc1 (CD206), Fn1, Retnla (Fizz1), and Ccl22 (Fig. 4A, gating as in Supplemental Fig. 2B). Cytokine quantification in whole-lung lysates correlated the observed M2-related gene expression in IFN-γ−/− T cell chimera MDMs (Fig. 4A) with the presence of the canonical type 2 cytokines IL-4, IL-5, and IL-13 (Fig. 4B), along with nearly complete absence of IFN-γ (Fig. 4C). Confocal microscopy confirmed that, consistent with transcriptional data, expression of NOS2 was dramatically reduced in lung MDMs of IFN-γ−/− T cell chimeric mice at 25 dpi, whereas the M2 marker ARG1 was expressed on a much greater proportion of MDMs (Fig. 4D). We asked whether the type I IFN response, which has been associated with an ineffective and pathogenic response to Mtb infection (30–33), including neutrophil-associated pathology (34), may be responsible for the shutdown of type II IFN responses in IFN-γ−/− T cell chimeras. However, transcription of both type I and type II IFN responses was suppressed in IFN-γ−/− T cell chimera mouse lung MDMs, suggesting that type I IFNs do not play a major role in suppressing type II IFN-induced responses in these mice (Supplemental Fig. 4).
FIGURE 4.

IFN-γ−/− T cells drive a type 2 cytokine milieu and alternative activation of MDMs in T cell chimeric mice infected with Mtb. (A) Relative expression of classical (M1) and alternative (M2) genes in FACS-sorted bystander and Mtb-infected MDMs in lungs of T cell chimeric mice at 25 dpi. Results are representative of two independent experiments. (B) Concentration of type 2 cytokines in lung lysates from Mtb-infected T cell chimeric mice at 25 dpi, as measured by cytokine bead array. n = 4 TCRβ−/−δ−/−, n = 5 WT, and n = 5 IFN-γ−/− T cell chimeric mice. Results are representative of three independent experiments. (C) Concentration of IFN-γ in lung lysates from Mtb-infected T cell chimeric mice at 25 dpi, as measured by Luminex assay. n = 5 TCRβ−/−δ−/−, n = 5 WT, and n = 6 IFN-γ−/− T cell chimeric mice. Results are representative of three independent experiments. (D) Representative confocal microscopy demonstrating expression of iNOS and ARG1 in lungs of Mtb-infected T cell chimeric mice at 25 dpi.
Discussion
Despite prior reports that T cell–derived IFN-γ plays a minimal role in Mtb restriction in the C57BL/6 mouse model, our studies confirm it is indeed essential to control lung Mtb burden and disease in this setting. Furthermore, our results indicate a detrimental role for T cell signals unopposed by concomitant T cell–derived IFN-γ in pulmonary Mtb infection. Unopposed IFN-γ-independent T cell signaling correlates with clinical decline and recruitment of neutrophils and eosinophils to Mtb lung lesions. These effects did not involve a type I IFN response but correlated with a type 2 cytokine signature and skewing of MDMs to an M2 phenotype. Pathology was worse in IFN-γ−/− than in TCRβ−/−δ−/− T cell chimeric mice, despite no statistical difference in lung Mtb CFU burden, suggesting that T cell–derived IFN-γ regulates tolerance of the host to manifestations of disease from Mtb infection, such as tissue damage and weight loss. An effective vaccine strategy against TB disease will likely require both IFN-γ-dependent and -independent T cell effects, and, as is frequently the case for immune responses in vivo, a balance between these two arms of the T cell response is required for effective protection while minimizing immunopathology.
Pulmonary lesions in TB disease–susceptible IFN-γ−/− T cell chimeric mice were marked by neutrophil and eosinophil infiltration. Whether the recruitment of these cell types is a major driver of lung pathology and clinical deterioration remains to be determined. Although neutrophils may play a host-protective role in mycobacterial clearance early in infection through phagocytosis and reactive oxygen species production, adverse outcomes in Mtb infection are usually associated with a dysregulated neutrophil response that plays a major role in driving detrimental pathology (35). Prior studies have also shown that IFN-γR−/− neutrophils accumulate in the lungs of Mtb-infected WT/IFN-γR−/− mixed bone marrow chimeric mice and that these mice exhibit accelerated weight loss (15), consistent with a direct role for IFN-γ in suppressing harmful neutrophil accumulation in the lung during Mtb infection. Our results build on this model by suggesting that T cells may provide an essential source of IFN-γ that inhibits the pathogenic effects of neutrophils. Whether the abundant eosinophils we observed in pulmonary TB lesions of IFN-γ−/− T cell chimeric mice also drive detrimental pathology or potentially mediate a host-beneficial response to severe disease deserves further study. Prior work using two independent genetic models of global eosinophil deficiency demonstrated a role for these cells in restriction of mycobacterial burden (36). However, the contribution of eosinophils to the host–pathogen balance may be context-specific.
Lung lysates of Mtb-infected IFN-γ−/− T cell chimeric mice were marked by a significant type 2 cytokine profile, with abundant IL-4, IL-5, and IL-13. In prior murine studies, depletion of IL-4 led to improved control of Mtb burden in BALB/c mice (37). In C57BL/6 mice, known to have a strong type 1–skewed immune response to Mtb infection, pulmonary Mtb burden was not affected by global deficiency in IL-4 or IL-13 (38). However, overexpression of IL-13 in C57BL/6 mice led to formation of necrotizing granulomas (39) in an IL-4Rα–dependent manner (40). Furthermore, alternatively activated M2 macrophages expressing ARG1, associated with the presence of type 2 cytokines, were abundant in this model (39). Accordingly, we observed suppression of the type 1 cytokine-driven NOS2 and induction of the type 2 cytokine–driven M2 marker ARG1 in macrophages in Mtb lesions of IFN-γ−/− T cell chimeric mice. Consistent with these findings, type 2 responses have been shown to be responsible for more severe pulmonary inflammation and TB disease in mice exposed to Schistosoma mansoni parasites or Ag than in untreated mice across a range of murine genotypes (41). Together, this evidence again suggests a context-specific effect that may depend on factors including genetic background, helminth coinfection, and environment.
There is also mounting evidence from human clinical and experimental studies supporting a detrimental role for type 2 cytokine signaling in TB pathology (42). A study of 1971 HIV-negative patients with sputum culture–positive pulmonary TB in Ghana revealed that a variant of IL-4Rα associated with increased signal transduction was associated with increased cavity size (40). In addition, a significantly higher IL-4/IFN-γ ratio was observed in stimulated lung lymphocytes from bronchoalveolar lavage in patients with miliary (43) or cavitary (44) rather than pleural (43) or noncavitary (44) TB. The same pattern was seen among peripheral blood lymphocytes (45), although the proportion of circulating IL-4–expressing T cells was much smaller than in bronchoalveolar lavage when measured concurrently in the same patients (43), implying an important role for tissue-specific responses. Indeed, in situ hybridization using resected lung tissue from patients with severe pulmonary TB demonstrated IFN-γ and IL-4 mRNA-producing cells within the same granulomas, as well as coexistence of these mixed granulomas alongside granulomas expressing only IFN-γ within the same patient (46); each lesion may therefore represent a unique cytokine microenvironment. Thus, although comorbidities associated with type 2 immunity, such as helminth infection or allergy/atopy, have not consistently correlated with severity of TB disease (47–49), studies have repeatedly demonstrated an association between the ratio of Th2 versus Th1 cells and the severity of Mtb infection outcomes. It has been suggested that a vaccine that would protect against Mtb in areas where both TB and helminthic infections are endemic should both support the Th1 response and block the Th2 response (42, 50). Our findings correlating type 2 cytokines and M2 macrophage responses with severe TB disease support this framework.
Although T cell–derived IFN-γ is most often thought of in terms of its effect on macrophages, it is not known which cellular target is primarily responsible for the differences in Mtb infection outcome between WT and IFN-γ−/− T cell chimeric mice. T cell–derived IFN-γ can be sensed by T cells themselves and plays a role in subsequent Th1 polarization (51). Furthermore, effects of IFN-γ on Mtb immunity have been correlated with induction of gene expression in epithelial cells (52) and by shaping T cell compartments by inducing apoptosis of activated CD4+ T cells (53). In addition, our studies do not identify the relative contribution of IFN-γ-producing T cell subsets (e.g., CD4+, CD8+, or γδ) (26) to the phenotypes observed. Future studies to characterize cell compartments, apoptosis, and activation status in lungs of Mtb-infected IFN-γ−/− T cell chimeric mice will help address these questions.
In prior studies, depletion of CD4+ T cells only led to a transient decrease in total IFN-γ levels in the lung (1, 20). In vivo and in vitro studies using RAG-deficient mice demonstrated significant IFN-γ expression in response to Mtb by cell types other than T cells, primarily by NK cells, which expand in a compensatory manner in this T cell–deficient setting (54). Notably, IFN-γ produced by non-T cells in Rag−/− mice was required for induction of Nos2 and concomitant suppression of type 2 markers (Arg1, Chil3) in lung macrophages after Mtb infection (54), consistent with a redundant role between T cell– and NK cell–derived IFN-γ. In our studies, lung IFN-γ levels and expression of NOS2 by lung MDMs were significantly lower in IFN-γ−/− T cell chimeric mice than in T cell chimeras that lacked T cells completely. This observation suggests that NK cells may be suppressed or unable to meaningfully increase IFN-γ production when T cells lack IFN-γ. It is plausible that mice in the CD4+ T cell depletion studies were previously exposed to T cell–derived IFN-γ and may have therefore achieved a baseline tonic state driven by type 1 immunity that could support the production of IFN-γ by NK and/or other cell types when needed. Our studies with T cell chimeric mice support a more nuanced model in which T cell–derived IFN-γ is required to suppress T cell–dependent type 2 immunity that inhibits compensatory NK cell functions.
Prior studies showed that IFN-γ produced by adoptively transferred Th1-polarized, Mtb Ag-specific CD4+ T cells is dispensable to control of pulmonary Mtb burden in WT host mice (6) when transferred at a dose of 1 × 107 cells/host and partially dispensable at a dose of 1 × 106 cells/host. Together with our results, these data indicate that high numbers of Mtb Ag-specific, Th1-polarized IFN-γ−/− CD4+ T cells likely amplify the importance of protective IFN-γ-independent T cell effects and overcome a requirement for T cell–derived IFN-γ to reduce Mtb burden in the lung, whereas dependence on T cell–derived IFN-γ is unmasked at more physiologic numbers of Ag-specific T cells that are more closely aligned with an expected vaccine response.
One factor that limits the interpretation of our studies is that C57BL/6 mice, the strain used in our work, is known to elicit type 1–skewed immune responses, whereas other genetic backgrounds may be less dependent on IFN-γ for protection against TB (55, 56). Furthermore, type 1 immune-driven mechanisms in C57BL/6 mice may differ from those in humans. Although the role of IFN-γ-induced NOS2 in Mtb restriction is well established in mice, Mtb-infected human peripheral blood–derived monocytes produce only small amounts of NO in response to IFN-γ signaling in vitro (57). Whether NOS2 is induced at sites of Mtb infection in human lungs is controversial, with studies showing different results (58–60). Nevertheless, the most frequently cited publications arguing for a minimal role of T cell–derived IFN-γ in pulmonary immunity against TB are studies in C57BL/6 mice, which are inconsistent with our results.
Our study was performed in unvaccinated mice, and it remains possible that vaccination could boost other mechanisms of immunity independently of T cell–derived IFN-γ. For example, although prior studies have definitively proved the requirement for IFN-γ in control of Mtb, IFN-γR−/− mice vaccinated with bacillus Calmette-Guérin still have a survival advantage over unvaccinated IFN-γR−/− mice (17); although T cell–independent effects such as trained immunity could be responsible, IFN-γ-independent T cell activity may also play a role. Nevertheless, our results indicate that T cell–derived IFN-γ can be critical for immunity within the Mtb-infected lung, and vaccination or host-directed therapy strategies that restore or augment the ability of Mtb-specific T cells to produce IFN-γ should continue to be explored.
Supplementary Material
Acknowledgments
We thank Daniel Kim, Lindsay Engels, Kaitlin Durga, and the SCRI Animal Care staff for technical assistance; Alan Diercks for RNA-seq data alignment; SCRI Research Scientific Computing for HPC resources; and members of the Urdahl Lab for helpful discussions.
This article is featured in Top Reads, p. 245
Footnotes
This work was supported by National Institutes of Health Grants U19AI135976 (to K.B.U.), 75N93019C00070 (to K.B.U.), T32AI007044 (to K.M.), and 5K08AI166072 (to B.H.G.); Firland Foundation Grant 20230026C (to K.M.); American Lung Association Grant CAALA2023 (to K.M.); and NIH-funded Seattle TB Research Advancement Center (SEATRAC) Grant 1P30AI168034-01 (to K.M.). The sponsors had no role in the design, conduct, analysis, or interpretation of the study or in the preparation, review, or approval of the manuscript.
The online version of this article contains supplemental material.
- dpi
- days postinfection
- MDM
- monocyte-derived macrophage
- Mtb
- Mycobacterium tuberculosis
- TB
- tuberculosis
- WT
- wild type
Disclosures
The authors have no financial conflicts of interest.
References
- 1. Caruso, A. M., Serbina N., Klein E., Triebold K., Bloom B. R., Flynn J. L.. 1999. Mice deficient in CD4 T cells have only transiently diminished levels of IFN-gamma, yet succumb to tuberculosis. J. Immunol. 162: 5407–5416. [PubMed] [Google Scholar]
- 2. Casanova, J.-L., Abel L.. 2002. Genetic dissection of immunity to mycobacteria: the human model. Annu. Rev. Immunol. 20: 581–620. [DOI] [PubMed] [Google Scholar]
- 3. Esmail, H., Riou C., Du Bruyn E., Lai R. P.-J., Harley Y. X. R., Meintjes G., Wilkinson K. A., Wilkinson R. J.. 2018. The immune response to Mycobacterium tuberculosis in HIV-1-coinfected persons. Annu. Rev. Immunol. 36: 603–638. [DOI] [PubMed] [Google Scholar]
- 4. Flynn, J., Chan J., Triebold K., Dalton D., Stewart T., Bloom B.. 1993. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 178: 2249–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pearl, J. E., Saunders B., Ehlers S., Orme I. M., Cooper A. M.. 2001. Inflammation and lymphocyte activation during mycobacterial infection in the interferon-γ-deficient mouse. Cell. Immunol. 211: 43–50. [DOI] [PubMed] [Google Scholar]
- 6. Boisson-Dupuis, S., Bustamante J., El-Baghdadi J., Camcioglu Y., Parvaneh N., El Azbaoui S., Agader A., Hassani A., El Hafidi N., Mrani N. A., et al. 2015. Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol. Rev. 264: 103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Majlessi, L., Simsova M., Jarvis Z., Brodin P., Rojas M.-J., Bauche C., Nouzé C., Ladant D., Cole S. T., Sebo P., Leclerc C.. 2006. An increase in antimycobacterial Th1-cell responses by prime-boost protocols of immunization does not enhance protection against tuberculosis. Infect. Immun. 74: 2128–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mittrücker, H.-W., Steinhoff U., Köhler A., Krause M., Lazar D., Mex P., Miekley D., Kaufmann S. H. E.. 2007. Poor correlation between BCG vaccination-induced T cell responses and protection against tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 104: 12434–12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jeevan, A., Bonilla D. L., David M. N.. 2009. Expression of interferon-gamma and tumour necrosis factor-alpha messenger RNA does not correlate with protection in guinea pigs challenged with virulent Mycobacterium tuberculosis by the respiratory route. Immunology 128: e296–e305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kagina, B. M. N., Abel B., Scriba T. J., Hughes E. J., Keyser A., Soares A., Gamieldien H., Sidibana M., Hatherill M., Gelderbloem S., et al. South African Tuberculosis Vaccine Initiative . 2010. Specific T cell frequency and cytokine expression profile do not correlate with protection against tuberculosis after bacillus Calmette-Guérin vaccination of newborns. Am. J. Respir. Crit. Care Med. 182: 1073–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abebe, F. 2012. Is interferon-gamma the right marker for bacille Calmette–Guérin-induced immune protection? The missing link in our understanding of tuberculosis immunology. Clin. Exp. Immunol. 169: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Billeskov, R., Christensen J. P., Aagaard C., Andersen P., Dietrich J.. 2013. Comparing adjuvanted H28 and modified vaccinia virus ankara expressingH28 in a mouse and a non-human primate tuberculosis model. PLoS One 8: e72185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tameris, M., Geldenhuys H., Luabeya A. K., Smit E., Hughes J. E., Vermaak S., Hanekom W. A., Hatherill M., Mahomed H., McShane H., Scriba T. J.. 2014. The candidate TB vaccine, MVA85A, induces highly durable Th1 responses. PLoS One 9: e87340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rodo, M. J., Rozot V., Nemes E., Dintwe O., Hatherill M., Little F., Scriba T. J.. 2019. A comparison of antigen-specific T cell responses induced by six novel tuberculosis vaccine candidates. PLoS Pathog. 15: e1007643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nandi, B., Behar S. M.. 2011. Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J. Exp. Med. 208: 2251–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sakai, S., Kauffman K. D., Sallin M. A., Sharpe A. H., Young H. A., Ganusov V. V., Barber D. L.. 2016. CD4 T cell-derived IFN-γ plays a minimal role in control of pulmonary Mycobacterium tuberculosis infection and must be actively repressed by PD-1 to prevent lethal disease. PLoS Pathog. 12: e1005667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cowley, S. C., Elkins K. L.. 2003. CD4+ T cells mediate IFN-gamma-independent control of Mycobacterium tuberculosis infection both in vitro and in vivo. J. Immunol. 171: 4689–4699. [DOI] [PubMed] [Google Scholar]
- 18. Gallegos, A. M., van Heijst J. W. J., Samstein M., Su X., Pamer E. G., Glickman M. S.. 2011. A gamma interferon independent mechanism of CD4 T cell mediated control of M. tuberculosis infection in vivo. PLoS Pathog. 7: e1002052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dis, E. V., Fox D. M., Morrison H. M., Fines D. M., Babirye J. P., McCann L. H., Rawal S., Cox J. S., Stanley S. A.. 2022. IFN-γ-independent control of M. tuberculosis requires CD4 T cell-derived GM-CSF and activation of HIF-1α. PLoS Pathog. 18: e1010721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scanga, C. A., Mohan V. P., Yu K., Joseph H., Tanaka K., Chan J., Flynn J. L.. 2000. Depletion of CD4+ T cells causes reactivation of murine persistent tuberculosis despite continued expression of interferon gamma and nitric oxide synthase 2. J. Exp. Med. 192: 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miller, H. E., Robinson R. T.. 2012. Early control of Mycobacterium tuberculosis infection requires il12rb1 expression by rag1-dependent lineages. Infect. Immun. 80: 3828–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Surh, C. D., Sprent J.. 2000. Homeostatic T cell proliferation: how far can T cells be activated to self-ligands? J. Exp. Med. 192: F9–F14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim, J., Lee J. Y., Cho K., Hong S.-W., Kim K. S., Sprent J., Im S.-H., Surh C. D., Cho J.-H.. 2018. Spontaneous proliferation of CD4+ T cells in RAG-deficient hosts promotes antigen-independent but IL-2-dependent strong proliferative response of naïve CD8+ T cells. Front. Immunol. 9: 1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ancelet, L., Rich F. J., Delahunt B., Kirman J. R.. 2012. Dissecting memory T cell responses to TB: concerns using adoptive transfer into immunodeficient mice. Tuberculosis (Edinb.) 92: 422–433. [DOI] [PubMed] [Google Scholar]
- 25. Falk, I., Potocnik A. J., Barthlott T., Levelt C. N., Eichmann K.. 1996. Immature T cells in peripheral lymphoid organs of recombinase-activating gene-1/-2-deficient mice. Thymus dependence and responsiveness to anti-CD3 epsilon antibody. J. Immunol. 156: 1362–1368. [PubMed] [Google Scholar]
- 26. Tsukaguchi, K., Balaji K., Boom W.. 1995. CD4+ alpha beta T cell and gamma delta T cell responses to Mycobacterium tuberculosis. Similarities and differences in Ag recognition, cytotoxic effector function, and cytokine production. J. Immunol. 154: 1786–1796. [PubMed] [Google Scholar]
- 27. Moguche, A. O., Shafiani S., Clemons C., Larson R. P., Dinh C., Higdon L. E., Cambier C. J., Sissons J. R., Gallegos A. M., Fink P. J., Urdahl K. B.. 2015. ICOS and Bcl6-dependent pathways maintain a CD4 T cell population with memory-like properties during tuberculosis. J. Exp. Med. 212: 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cosma, C. L., Humbert O., Ramakrishnan L.. 2004. Superinfecting mycobacteria home to established tuberculous granulomas. Nat. Immunol. 5: 828–835. [DOI] [PubMed] [Google Scholar]
- 29. McCarthy, D. J., Chen Y., Smyth G. K.. 2012. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40: 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ji, D. X., Yamashiro L. H., Chen K. J., Mukaida N., Kramnik I., Darwin K. H., Vance R. E.. 2019. Type I interferon-driven susceptibility to Mycobacterium tuberculosis is mediated by IL-1Ra. Nat. Microbiol. 4: 2128–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moreira-Teixeira, L., Mayer-Barber K., Sher A., O’Garra A.. 2018. Type I interferons in tuberculosis: foe and occasionally friend. J. Exp. Med. 215: 1273–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang, L., Jiang X., Pfau D., Ling Y., Nathan C. F.. 2021. Type I interferon signaling mediates Mycobacterium tuberculosis-induced macrophage death. J. Exp. Med. 218: e20200887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kotov, D. I., Lee O. V., Fattinger S. A., Langner C. A., Guillen J. V., Peters J. M., Moon A., Burd E. M., Witt K. C., Stetson D. B., et al. 2023. Early cellular mechanisms of type I interferon-driven susceptibility to tuberculosis. Cell 186: 5536–5553.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Berry, M. P., Graham C. M., Finlay M. W., Xu Z., Bloch S. A. A., Oni T., Wilkinson K. A., Banchereau R., Skinner J., Wilkinson R. J., et al. 2010. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466: 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gaffney, E., Murphy D., Walsh A., Connolly S., Basdeo S. A., Keane J., Phelan J. J.. 2022. Defining the role of neutrophils in the lung during infection: implications for tuberculosis disease. Front. Immunol. 13: 984293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bohrer, A. C., Castro E., Hu Z., Queiroz A. T. L., Tocheny C. E., Assmann M., Sakai S., Nelson C., Baker P. J., Ma H., et al. Tuberculosis Imaging Program . 2021. Eosinophils are part of the granulocyte response in tuberculosis and promote host resistance in mice. J. Exp. Med. 218: e20210469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Buccheri, S., Reljic R., Caccamo N., Ivanyi J., Singh M., Salerno A., Dieli F.. 2007. IL-4 depletion enhances host resistance and passive IgA protection against tuberculosis infection in BALB/c mice. Eur. J. Immunol. 37: 729–737. [DOI] [PubMed] [Google Scholar]
- 38. Jung, Y.-J., LaCourse R., Ryan L., North R. J.. 2002. Evidence inconsistent with a negative influence of T helper 2 cells on protection afforded by a dominant T helper 1 response against Mycobacterium tuberculosis lung infection in mice. Infect. Immun. 70: 6436–6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heitmann, L., Abad Dar M., Schreiber T., Erdmann H., Behrends J., Mckenzie A. N. J., Brombacher F., Ehlers S., Hölscher C.. 2014. The IL-13/IL-4Rα axis is involved in tuberculosis-associated pathology. J. Pathol. 234: 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hölscher, C., Heitmann L., Owusu-Dabo E., Horstmann R. D., Meyer C. G., Ehlers S., Thye T.. 2016. A mutation in IL4RA is associated with the degree of pathology in human TB patients. Mediators Inflamm. 2016: 4245028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Monin, L., Griffiths K. L., Lam W. Y., Gopal R., Kang D. D., Ahmed M., Rajamanickam A., Cruz-Lagunas A., Zúñiga J., Babu S., et al. 2015. Helminth-induced arginase-1 exacerbates lung inflammation and disease severity in tuberculosis. J. Clin. Invest. 125: 4699–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rook, G. A. W. 2007. Th2 cytokines in susceptibility to tuberculosis. Curr. Mol. Med. 7: 327–337. [DOI] [PubMed] [Google Scholar]
- 43. Sharma, S. K., Mitra D. K., Balamurugan A., Pandey R. M., Mehra N. K.. 2002. Cytokine polarization in miliary and pleural tuberculosis. J. Clin. Immunol. 22: 345–352. [DOI] [PubMed] [Google Scholar]
- 44. Mazzarella, G., Bianco A., Perna F., D’Auria D., Grella E., Moscariello E., Sanduzzi A.. 2003. T lymphocyte phenotypic profile in lung segments affected by cavitary and non-cavitary tuberculosis. Clin. Exp. Immunol. 132: 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. van Crevel, R., Karyadi E., Preyers F., Leenders M., Kullberg B. J., Nelwan R. H., van der Meer J. W.. 2000. Increased production of interleukin 4 by CD4+ and CD8+ T cells from patients with tuberculosis is related to the presence of pulmonary cavities. J. Infect. Dis. 181: 1194–1197. [DOI] [PubMed] [Google Scholar]
- 46. Fenhalls, G., Wong A., Bezuidenhout J., van Helden P., Bardin P., Lukey P. T.. 2000. In situ production of gamma interferon, interleukin-4, and tumor necrosis factor alpha mRNA in human lung tuberculous granulomas. Infect. Immun. 68: 2827–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lienhardt, C., Azzurri A., Amedei A., Fielding K., Sillah J., Sow O. Y., Bah B., Benagiano M., Diallo A., Manetti R., et al. 2002. Active tuberculosis in Africa is associated with reduced Th1 and increased Th2 activity in vivo. Eur. J. Immunol. 32: 1605–1613. [DOI] [PubMed] [Google Scholar]
- 48. Aira, N., Andersson A.-M., Singh S. K., McKay D. M., Blomgran R.. 2017. Species dependent impact of helminth-derived antigens on human macrophages infected with Mycobacterium tuberculosis: direct effect on the innate anti-mycobacterial response. PLoS Negl. Trop. Dis. 11: e0005390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cozmei, C., Constantinescu D., Carasevici E., Anisie E., Ungureanu D., Sorete-Arbore A., Gramadă D., Mihăescu T., Croitoru C., Popa D.. 2007. Th1 and Th2 cytokine response in patients with pulmonary tuberculosis and health care workers occupationally exposed to M. tuberculosis. Rev. Med. Chir. Soc. Med. Nat. Iasi. 111: 702–709. [PubMed] [Google Scholar]
- 50. Rook, G. A. W., Hernandez-Pando R., Dheda K., Teng Seah G.. 2004. IL-4 in tuberculosis: implications for vaccine design. Trends Immunol. 25: 483–488. [DOI] [PubMed] [Google Scholar]
- 51. Das, G., Sheridan S., Janeway C. A.. 2001. The source of early IFN-gamma that plays a role in Th1 priming. J. Immunol. 167: 2004–2010. [DOI] [PubMed] [Google Scholar]
- 52. Desvignes, L., Ernst J. D.. 2009. Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity 31: 974–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dalton, D. K., Haynes L., Chu C. Q., Swain S. L., Wittmer S.. 2000. Interferon gamma eliminates responding CD4 T cells during mycobacterial infection by inducing apoptosis of activated CD4 T cells. J. Exp. Med. 192: 117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Feng, C. G., Kaviratne M., Rothfuchs A. G., Cheever A., Hieny S., Young H. A., Wynn T. A., Sher A.. 2006. NK cell-derived IFN-gamma differentially regulates innate resistance and neutrophil response in T cell-deficient hosts infected with Mycobacterium tuberculosis. J. Immunol. 177: 7086–7093. [DOI] [PubMed] [Google Scholar]
- 55. Kurtz, S. L., Rossi A. P., Beamer G. L., Gatti D. M., Kramnik I., Elkins K. L.. 2020. The diversity outbred mouse population is an improved animal model of vaccination against tuberculosis that reflects heterogeneity of protection. mSphere 5: e00097-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Smith, C. M., Baker R. E., Proulx M. K., Mishra B. B., Long J. E., Park S. W., Lee H.-N., Kiritsy M. C., Bellerose M. M., Olive A. J., et al. 2022. Host-pathogen genetic interactions underlie tuberculosis susceptibility in genetically diverse mice. Elife 11: e74419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jagannath, C., Actor J. K., Hunter R. L.. 1998. Induction of nitric oxide in human monocytes and monocyte cell lines by Mycobacterium tuberculosis. Nitric Oxide 2: 174–186. [DOI] [PubMed] [Google Scholar]
- 58. MacMicking, J., Xie Q. W., Nathan C.. 1997. Nitric oxide and macrophage function. Annu. Rev. Immunol. 15: 323–350. [DOI] [PubMed] [Google Scholar]
- 59. Choi, H.-S., Rai P. R., Chu H. W., Cool C., Chan E. D.. 2002. Analysis of nitric oxide synthase and nitrotyrosine expression in human pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 166: 178–186. [DOI] [PubMed] [Google Scholar]
- 60. Mattila, J. T., Ojo O. O., Kepka-Lenhart D., Marino S., Kim J. H., Eum S. Y., Via L. E., Barry C. E., Klein E., Kirschner D. E., et al. 2013. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J. Immunol. 191: 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.