

Abstract
Cytochrome P450 (CYP) 3A4 is an enzyme involved in the metabolism of many drugs that are currently on the market and is therefore a key player in drug–drug interactions (DDIs). ACT‐1004‐1239 is a potent and selective, first‐in‐class ACKR3/CXRC7 antagonist being developed as a treatment for demyelinating diseases including multiple sclerosis. Based on the human absorption, distribution, metabolism, and excretion (ADME) study results, ACT‐1004‐1239 is predominantly metabolized by CYP3A4. This study investigated the effect of the strong CYP3A4 inhibitor, itraconazole, on the pharmacokinetics of single‐dose ACT‐1004‐1239 in healthy male subjects. In the open‐label, fixed‐sequence DDI study, a total of 16 subjects were treated. Each subject received a single dose of 10 mg ACT‐1004‐1239 (Treatment A) in the first period followed by concomitant administration of multiple doses of 200 mg itraconazole and a single dose of 10 mg ACT‐1004‐1239 in the second period. We report a median of difference in t max (90% confidence interval, CI) of 0.5 h (0.0, 1.0) comparing both treatments. The geometric mean ratio (GMR) (90% CI) of C max and AUC0−∞ was 2.16 (1.89, 2.47) and 2.77 (2.55, 3.00), respectively. The GMR (90% CI) of t 1/2 was 1.46 (1.26, 1.70). Both treatments were well‐tolerated with an identical incidence in subjects reporting treatment‐emergent adverse events (TEAE). The most frequently reported TEAEs were headache and nausea. In conclusion, ACT‐1004‐1239 is classified as a moderately sensitive CYP3A4 substrate (i.e., increase of AUC ≥2‐ to <5‐fold), and this should be considered in further clinical studies if CYP3A4 inhibitors are concomitantly administered.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
ACT‐1004‐1239 is a first‐in‐class ACKR3/CXCR7 antagonist with preclinical efficacy in several animal models of multiple sclerosis (MS). Clinically, ACT‐1004‐1239 demonstrated favorable safety and pharmacokinetic (PK) properties in single‐ and multiple‐ascending dose studies. Based on the human absorption, distribution, metabolism, and excretion (ADME) study and preclinical investigations, the pathway contributing ≥25% to ACT‐1004‐1239's elimination is catalyzed by cytochrome P450 (CYP) 3A4.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study investigates the extent of CYP3A4 inhibition using multiple doses of the strong index inhibitor itraconazole on the PK of a single 10 mg dose of ACT‐1004‐1239 in healthy male subjects.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Inhibition of CYP3A4 moderately increased the exposure to ACT‐1004‐1239 (up to 2.9‐fold) indicating that ACT‐1004‐1239 is a moderately sensitive CYP3A4 substrate. Accordingly, other mechanisms are likely to be involved in the elimination of ACT‐1004‐1239.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Combination therapies in patients with MS or other demyelinating diseases are not uncommon. CYP3A4 is a prominent enzyme involved in the metabolism of more than 50% of orally administered pharmaceuticals, and as such it is of importance to understand the impact on substrates such as ACT‐1004‐1239. ACT‐1004‐1239 was characterized to be a moderately sensitive CYP3A4 substrate in this study. Nevertheless, drug–drug interactions in patients may not be ruled out, and thus, attention should be paid on how to implement concomitant use of strong CYP3A4 inhibitors in further clinical development of ACT‐1004‐1239.
INTRODUCTION
Drug metabolism includes processes that transform lipophilic substances into more polar and better excretable molecules. The processes are known as phase I and phase II biotransformation reactions. Phase I metabolic reactions are mainly catalyzed by the cytochrome P450 (CYP) superfamily, of which CYP3A4 is considered the most important enzyme as it is involved in the metabolism of more than 50% of orally administered pharmaceuticals including the herein studied ACT‐1004‐1239. 1 , 2
ACT‐1004‐1239 is an orally available, potent, selective ACKR3/CXRC7 antagonist. 3 ACKR3/CXRC7 is an atypical chemokine receptor, which binds and internalizes its chemokine ligands CXCL11 and CXCL12 leading to their degradation. This scavenging function can facilitate the establishment and maintenance of a concentration gradient for its ligands. Antagonism of ACKR3/CXRC7 by ACT‐1004‐1239 showed in several preclinical models immunomodulatory as well as promyelinating effects. 4 , 5 Specifically, this dual mode of action resulted in reduced disease score in animal models for multiple sclerosis. 5 Therefore, ACT‐1004‐1239 is being developed as treatment for demyelinating diseases including multiple sclerosis. ACT‐1004‐1239 has been clinically investigated in healthy subjects. In the clinical single‐ and multiple‐ascending dose studies ACT‐1004‐1239 showed favorable safety/tolerability, pharmacokinetic (PK), and pharmacodynamic (PD) profiles up to and including a dose of 200 mg in healthy male and female subjects supporting further clinical development. 6 , 7 Briefly, ACT‐1004‐1239 was safe and well‐tolerated up to and including a dose of 200 mg without QTc liability. The PK in healthy subjects were characterized by a quick absorption with an absolute bioavailability of 53%, a high volume of distribution (183 L), and an average terminal half‐life (t 1/2) of 19 h. The exposure to ACT‐1004‐1239 increased in a dose‐proportional manner across the investigated dose range (1–200 mg). In humans, ACT‐1004‐1239 is considered a low‐clearance drug (23 L/h). Target engagement was shown by a dose‐dependent increase in CXCL12 plasma concentration. 6 , 7 Based on the human absorption, distribution, metabolism, and excretion (ADME) study, in which 14C‐labeled ACT‐1004‐1239 was administered orally, ACT‐1004‐1239 is predominately eliminated via feces and is extensively metabolized. Accordingly, only 15% of unchanged drug was recovered in excreta. Elimination of ACT‐1004‐1239 is mainly catalyzed by CYP3A4 with the formation of the primary metabolite M1, which is a pharmacologically inactive amine metabolite formed following oxidative N‐dealkylation of parent drug and found to represent 25.1% of drug‐related material in excreta. 2 Additional metabolite profiling and identification data suggested that M1 can undergo further reactions, namely oxidative defluorination and reduction, which result in the formation of the metabolites A1 and A2, respectively. A1 and A2 represented 4.0% and 15.3% of drug‐related material in excreta, respectively. Therefore, the elimination of ACT‐1004‐1239 via M1 was shown to contribute to approximately 44% of drug's elimination in humans. 2 Other identified elimination pathways included reduction of ACT‐1004‐1239 to metabolite A3, which accounted for 2.7% of total drug only. 2
At that point of time in drug development, the sensitivity of ACT‐1004‐1239 to CYP3A4 was unknown, and following regulatory standards, it was decided to perform a clinical DDI study to investigate this. The Food and Drug Administration (FDA) and European Medicines Agency (EMA) recommend to clinically investigate the impact of possible drug–drug interactions (DDIs) involving all pathways responsible for ≥25% of drug elimination. 8 , 9 Such interactions involve CYP3A4 induction or inhibition, which may result in changes in drug exposure. Consequently, it can inadvertently lead to a lack of efficacy or exacerbation of adverse drug events. 10 , 11 , 12 To evaluate CYP3A4‐related DDIs, clinical studies can be conducted with concomitant drugs recommended as index inhibitors/inducers. For example, itraconazole is a strong CYP3A4 inhibitor used clinically for the treatment of various fungal infections. 13 Previous studies revealed that itraconazole increases the systemic exposure to concomitantly administered sensitive CYP3A4 substrates up to 11‐fold. 14 Itraconazole is a commonly used concomitant drug in DDI studies with acceptable safety profile and clear DDI study design recommendations. 15 , 16 Therefore, it was selected as a perpetrator inhibitor in this DDI study with ACT‐1004‐1239.
The aim of this study was to assess the effect of multiple oral doses of itraconazole on the PK of ACT‐1004‐1239 in healthy male subjects. Results from the clinical study can be further used to provide recommendations on concomitant treatment in successive clinical studies.
MATERIALS AND METHODS
Study design
This open‐label, fixed‐sequence DDI study was conducted at BlueClinical Phase 1 (Porto, Portugal) (ClinicalTrials.gov: NCT05549531) to investigate the effect of itraconazole on the PK of ACT‐1004‐1239 in healthy subjects. The study approvals were obtained from both the National Health Authority of Portugal (Infarmed) and the National Ethics Committee (Comissão de Ética para a Investigação Clínica) before study initiation. The study was performed in accordance with Good Clinical Practice and the Declaration of Helsinki.
Following an eligibility check during a screening period, subjects received in a fixed sequence Treatment A (ACT‐1004‐1239 alone) and thereafter Treatment B (B1: itraconazole alone; B2: ACT‐1004‐1239 + itraconazole). In detail, all subjects received a single oral dose of 10 mg ACT‐1004‐1239 on Day 1 and remained in‐clinic for 24 h. Thereafter, ambulatory visits were scheduled for blood collections and safety check‐ups through Day 6 (Treatment A). From Day 6 to Day 8, a dose of 200 mg itraconazole (available as 100 mg oral capsules, Sporanox®) was administered once‐daily to each subject (Treatment B1). Itraconazole dosing was continued through Day 13, whereas on Day 9, itraconazole was given concomitantly with 10 mg ACT‐1004‐1239 followed by a 24‐h in‐clinic stay and ambulatory visits for blood collections and safety check‐ups through Day 14 (Treatment B2). All treatment administrations were performed under fed conditions after a standardized breakfast served 30 min prior to dosing. End‐of‐study (EOS) assessments were performed on Day 20. Safety follow‐up was performed within 40 days after the EOS visit.
A dose of 10 mg ACT‐1004‐1239 was selected as it was assumed to provide sufficient safety margin to the maximum exposure investigated in humans (achieved with a dose of 200 mg) 6 , 7 in the event itraconazole would increase the exposure to ACT‐1004‐1239 up to 11‐fold as previously observed for other drugs. 14 The dose strength of itraconazole (200 mg) is as per label recommendation as well as best practice guidance on how to conduct DDI studies with itraconazole. 15 , 16 , 17
Study population
In total, 16 nonsmoking healthy male subjects between 18 and 55 years were enrolled in this study. Only male subjects were included since sex was not expected to influence the outcome of the study to a clinically relevant extent 7 while eliminating the need to expose female subjects to itraconazole that is teratogenic. 17 Prior to any study procedure each subject provided informed consent. The study subjects were of good health as assessed by medical history, physical examination, 12‐lead ECG, and clinical laboratory tests (including hematology, clinical chemistry, virus serology, and drug screening). Furthermore, eligible subjects were requested to have a body mass index (BMI) between 18.0 and 30.0 kg/m2 and vital signs within normal ranges defined as 100–140 mmHg for systolic blood pressure, 50–90 mmHg for diastolic blood pressure, and 50–90 bpm for pulse rate.
Consumption of food or beverages that could interfere with CYP3A4 activity (e.g., grapefruit, Seville orange) as well as xanthine‐containing food/beverages, nicotine, and alcohol were forbidden. Concomitant medications were prohibited unless required for the treatment of an adverse event (AE).
Pharmacokinetic analysis
Each administration of ACT‐1004‐1239 was followed by frequent blood collection up to 120 h post dosing to assess its PK. Approximately 3 mL of blood was collected in EDTA‐containing tubes at the following time points: 0 (predose), 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 24, 36, 48, 72, 96, and 120 h of postdose. Following collection, the blood samples were centrifuged (approximately 1800 g for 10 min at 4°C) within 30 min and, subsequently, plasma was aliquoted and stored at −80°C for further analysis.
ACT‐1004‐1239 plasma concentrations were determined using a validated LC–MS/MS assay. 6 The method was monitored using quality control samples, which revealed an inter‐batch precision % coefficient of variation (CV) of ≤7.2%, whereas the inter‐batch accuracy using the relative deviation from the nominal value (% RD) ranged from 2.6% to 6.2%. Concentrations below the limit of quantification (0.5 ng/mL) were set to “0”.
PK parameters (t max, C max, AUC0−t , AUC0−∞, and t 1/2) after each ACT‐1004‐1239 administration were obtained by non‐compartmental analysis using Phoenix WinNonlin version 8.0 (Certara, Princeton, NJ, USA). Measured ACT‐1004‐1239 plasma concentrations were directly used to determine t max and C max, whereas AUC parameters (AUC0−t and AUC0−∞) were calculated based on the linear trapezoidal rule.
Statistical analysis
No formal statistical hypothesis was tested in this study; however, the sample size was based on a precision estimate approach 18 using the CV within subjects for C max and AUC0–∞ of 48.8% and 13.9%, respectively, as observed in a previous study. 6 According to this method, a sample size of 12 subjects would result in lower and upper bounds of the 90% confidence interval (CI) of the geometric mean ratio (GMR) of approximately −29% and +40% and −10% and +11% of the observed ratio for Cmax and AUC0−∞, respectively. Furthermore, accounting for potential subject discontinuations, a sample size of 16 subjects was used in this study. PK parameters were analyzed providing geometric mean and corresponding 95% CI for Cmax, AUC0–t , AUC0–∞, and t 1/2 whereas t max was analyzed with the median and range.
The differences in Cmax, AUC0–t , AUC0–∞, and t 1/2 between Treatment A and Treatment B were explored using the GMR and the 90% CI. Back‐transformed values were analyzed by a mixed‐effects model using log‐transformed PK parameters as dependent variable, treatment as fixed effect, and subject as random effect. Difference for t max between treatments was explored using the nonparametric Hodges‐Lehmann estimate of the median of differences and its 90% CI. 19
Safety and tolerability evaluation
Safety and tolerability were assessed throughout the study based on the nature, frequency, and intensity of AEs. Post‐ACT‐1004‐1239 dosing results of vital signs, 12‐lead ECG, clinical laboratory variables, and body weight were compared with baseline (i.e., prior ACT‐1004‐1239 dosing in Treatment A).
RESULTS
Subjects
Overall, 16 healthy male subjects participated in both treatments and completed the study as per protocol. There was no premature study discontinuation; therefore, all subjects were included in the analysis of PK and safety/tolerability. The mean age was 34.3 years (range: 27–47) and mean BMI 24.2 kg/m2 (range: 19.9–27.6). The majority was White (87.5%), and two subjects were of mixed race (Black or African American and White [12.6%]).
Pharmacokinetics
In the absence of itraconazole, a single oral dose of 10 mg ACT‐1004‐1239 (Treatment A) was quickly absorbed as indicated by a median t max of 2.0 h (Figure 1, Table 1). Following absorption, ACT‐1004‐1239 concentration increased up to a geometric mean Cmax of 25.6 ng/mL and geometric mean AUC0−t and AUC0−∞ were 300 ng * h/mL and 326 ng * h/mL, respectively. ACT‐1004‐1239 was eliminated with a geometric mean t 1/2 of 17.2 h. Corresponding 95% CIs of each PK parameter are provided in Table 1. As for 6 out of 16 subjects, the t 1/2 could not be reliably determined due to insufficient characterization of the elimination phase at time points beyond 48 h after dosing in Treatment A, an exploratory ad hoc sensitivity analysis was performed using AUC0−48h data. Based on the analysis, there was no difference between the GMRs of AUC0−48h and AUC0−∞ (data not shown); therefore, it can be concluded that AUC0−∞ can be used as a measure to determine the effect of itraconazole. The concentration versus time profile of 10 mg ACT‐1004‐1239 was similar in shape to the one observed in a previous study. 6
FIGURE 1.
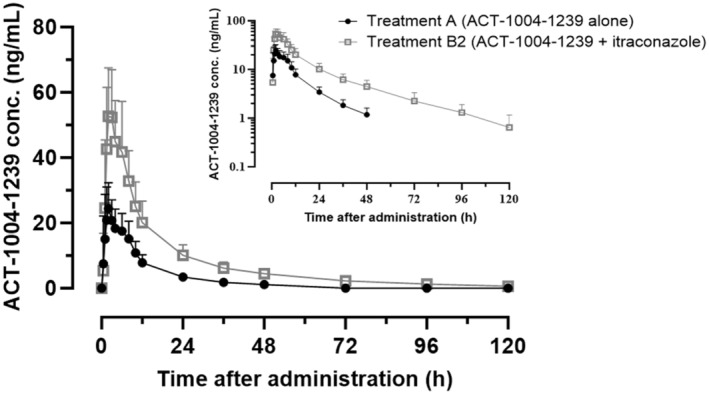
ACT‐1004‐1239 plasma concentration‐time profile of 10 mg ACT‐1004‐1239 (Treatment A) and 10 mg ACT‐1004‐1239 concomitantly to 200 mg itraconazole (Treatment B) in humans. Arithmetic mean plasma concentrations (+SD) of ACT‐1004‐1239 by treatment on linear and semilogarithmic (inset) scales (n = 16 per treatment).
TABLE 1.
Pharmacokinetic parameters of single‐dose ACT‐1004‐1239 in humans.
| Parameter (unit) | Treatment A (ACT‐1004‐1239) alone, N = 16 | Treatment B2 (ACT‐1004‐1239 + itraconazole), N = 16 |
|---|---|---|
| Cmax (ng/mL) | 25.6 (21.1, 31.0) | 55.3 (47.0, 65.2) |
| AUC0−t (ng * h/mL) | 300 (257, 352) | 862 (734, 1012) |
| AUC0−inf (ng * h/mL) | 326 (279, 380) a | 729 (627, 849) |
| t max (h) | 2.00 (0.5, 6.0) | 2.00 (1.0, 6.0) |
| t ½ (h) a | 17.2 (13.5, 21.9) a | 25.2 (22.2, 28.5) |
Note: Data are displayed as geometric means (95% CI) except for t max that is provided as median (range).
Abbreviations: AUC0−inf, area under the plasma concentration‐time curve from 0 h to infinity; AUC0−t , area under the plasma concentration‐time curve from 0 h to time t of the last measured concentration above the limit of quantification; CI, confidence interval; C max, maximum plasma concentration; n, number of subjects; N, number of subjects in the population; t ½, terminal half‐life; t max, time to reach maximum plasma concentration.
n = 10 for t 1/2 as for 6 subjects the t 1/2 could not be reliably determined due to insufficient characterization of the elimination phase at time points beyond 48 h after dosing in Treatment A.
In the presence of multiple doses of 200 mg itraconazole (Treatment B2), the concentration versus time profile of ACT‐1004‐1239 was consistent in shape with that in Treatment A (Figure 1). There was almost no difference in t max indicated by the median of differences in t max (90% CI) of 0.5 h (0.0, 1.0). The exposure to ACT‐1004‐1239 was moderately higher when concomitantly administered with itraconazole as shown by the GMR (90% CI) of Cmax, AUC0−t , and AUC0−∞ which were 2.16 (1.89, 2.47), 2.87 (2.64, 3.11), and 2.77 (2.55, 3.00), respectively. The t 1/2 of ACT‐1004‐1239 was slightly longer following concomitant administration with itraconazole based on the GMR (90% CI) of 1.46 (1.26, 1.70).
Safety and tolerability
Overall, all treatments were well‐tolerated. There were neither serious AEs nor AEs leading to premature discontinuation in this study. A total of eight AEs were reported as treatment‐emergent. All treatment‐emergent AEs (TEAEs) were of mild intensity and resolved at EOS without sequelae. The overall incidence of subjects with TEAEs was identical across treatments with 18.8% in each treatment. The most frequent TEAEs were headache and nausea (each reported by 2 subjects; Table 2). Other TEAEs were abdominal pain, diarrhea, and somnolence (each reported by one subject; Table 2). Diarrhea and headache (in Treatment B2) were considered to be related to itraconazole only, whereas abdominal pain was assessed to be related to ACT‐1004‐1239 and itraconazole. No clinically relevant treatment‐related mean changes from baseline in vital signs, body weight, clinical laboratory, or ECG variables were detected in the corresponding period after administration of ACT‐1004‐1239 in Treatment A or Treatment B2.
TABLE 2.
Incidence and number of treatment‐emergent adverse events by treatment.
| Adverse events (Preferred term) | Treatment A (N = 16) | Treatment B2 (N = 16) | ||
|---|---|---|---|---|
| nAEs | n (%) | nAEs | n (%) | |
| Number of subjects with at least one TEAE | – | 3 (18.8) | – | 3 (18.8) |
| Number of different TEAEs | 3 | – | 3 | – |
| Total number of TEAEs | 5 | – | 3 | – |
| Headache | 2 | 1 (6.3) | 1 | 1 (6.3) |
| Nausea | 2 | 2 (12.5) | 0 | 0 |
| Abdominal pain | 0 | 0 | 1 | 1 (6.3) |
| Diarrhea | 0 | 0 | 1 | 1 (6.3) |
| Somnolence | 1 | 1 (6.3) | 0 | 0 |
Abbreviations: N, number of subjects in the population; n, number of subjects; nAEs, number of adverse events; TEAE, treatment‐emergent adverse event.
DISCUSSION
This open‐label, fixed‐sequence DDI study investigated the effect of multiple doses of itraconazole on the PK of a single dose of 10 mg ACT‐1004‐1239. The latter is a potent, selective, and orally available first‐in‐class ACKR3/CXRC7 antagonist. Previous clinical investigations revealed that ≥25% of ACT‐1004‐1239 elimination goes through the primary metabolite M1, which is predominately formed by CYP3A4. 2 Therefore, a DDI study with ACT‐1004‐1239 and a strong CYP3A4 inhibitor is recommended by health authorities to investigate the extent of an increase in ACT‐1004‐1239 exposure associated with CYP3A4 interactions. There are several strong CYP3A4 inhibitors that have been previously used as perpetrator drugs in DDI studies such as ketoconazole, ritonavir, or voriconazole. The use of ketoconazole is discouraged due to the high risk of severe liver injury. 20 , 21 Furthermore, none of these CYP3A4 inhibitors are listed as index inhibitors, 22 which are nowadays preferred concomitant drugs for prospective DDI studies due to their known strength of inhibition and selectivity. 23
Strong index inhibitors are drugs that can elevate the AUC of a sensitive substrate by ≥5‐fold, 24 , 25 examples being clarithromycin and itraconazole in the context of CYP3A4. 22 Clarithromycin was not selected for this study as it is an antibiotic associated with AEs related to, amongst others, the gastrointestinal tract and QTc interval prolongation. 26 In addition, clarithromycin must be administered twice‐daily 26 and this would have posed operational challenges as ACT‐1004‐1239 is dosed once‐daily. Thus, itraconazole, approved for the treatment of various fungal infections, was chosen for this DDI study with ACT‐1004‐1239.
Thus far, two oral formulations of itraconazole are available on the market, namely a capsule and a solution. Recently, Hoch et al. reported a DDI study that showed that cyclodextrin, an excipient that is available in excess in the itraconazole oral solution, can interact with victim drugs, and lead to unreliable study outcomes. 27 In short, in this study contrary to expectation, asciminib (a substrate of CYP3A4) exposure decreased approximately 50% when given concomitantly with an itraconazole solution. This observation was attributed to the sequestration phenomenon of β‐cyclodextrin as previously also observed with other drugs. 28 , 29 Based on the physicochemical properties of ACT‐1004‐1239, 3 sequestration of ACT‐1004‐1239 by β‐cyclodextrin could not be excluded. Therefore, the itraconazole capsule formulation was preferred as a perpetrator in this DDI study. According to the dosing recommendation of itraconazole given as a capsule formulation, higher exposures to itraconazole can be reached when it is given with food. Furthermore, as no effect of food on ACT‐1004‐1239 exposure had been observed, 6 all treatment administrations in this study were performed under fed conditions.
In Treatment A of the herein reported study, the exposure to ACT‐1004‐1239 when administered alone under fed conditions was slightly higher than the one observed in the previous single‐ascending dose study at the same dose level in which the drug was given without food. However, the CIs of the PK exposures of both studies were overlapping. This may be explained by the small sample size and the high variability in ACT‐1004‐1239 exposure in the single‐ascending dose study. 6
A dose of 10 mg ACT‐1004‐1239 was selected for this study to ensure that the maximum ACT‐1004‐1239 exposure investigated in humans would not be exceeded if itraconazole increases ACT‐1004‐1239 exposure by up to 11‐fold. 6 , 14 , 30 In general, adopting a conservative approach when conducting DDI studies with itraconazole is advisable to mitigate the risk of unexpected safety events. Moreover, if the PK properties of the investigated drug permit, particularly when dose‐proportionality is observed, as exemplified by ACT‐1004‐1239, the results obtained from a DDI study utilizing a lower dose of the victim drug can be extrapolated to higher therapeutic doses. This extrapolation strategy, supported by robust PK data and an exposure‐safety relationship, can streamline the assessment of potential DDIs. The exposure to ACT‐1004‐1239 concomitantly with itraconazole (Treatment B2) was up to 2.9‐fold that of ACT‐1004‐1239 alone. An increase in ACT‐1004‐1239 exposure was expected given that itraconazole and its metabolites inhibit the CYP3A4‐dependent metabolism of ACT‐1004‐1239 in the intestine and liver. 31 , 32 , 33 Although itraconazole concentrations were not measured in this study, the multiple dosing regimen of 200 mg itraconazole is routinely used in investigations of CYP3A4 inhibition and therefore, the study results are considered valid. 15 , 16 , 22 Elimination of ACT‐1004‐1239 was slower following concomitant administration of itraconazole compared to ACT‐1004‐1239 alone. This may be linked to the blockade of one ACT‐1004‐1239 elimination route (i.e., through CYP3A4) 31 , 32 that can consequently lead to a general delay in drug elimination if no compensatory alternative routes are in place. Such alternative routes may include the elimination through CYP2C19, CYP2C8, and CYP1A1, which were shown to also catalyze formation of M1 in vitro or other elimination pathways such as reduction of ACT‐1004‐1239 to the metabolite A3 as reported previously. 2
In accordance with the definition given by the health authorities, ACT‐1004‐1239 is a moderately sensitive CYP3A4 substrate (i.e., increase of AUC ≥2‐ to <5‐fold). 22 Moreover, using the formula for fraction of metabolism (i.e., 1‐AUCcontrol/AUCinhibited) (control: AUC of victim drug, inhibited: AUC of victim drug given concomitantly with CYP inhibitor), which is a known approach to determine the involvement of CYPs in the metabolism of drugs, 34 , 35 , 36 , 37 , 38 approximately 55% of ACT‐1004‐1239 elimination is dependent on CYP3A4 in humans. This estimation on CYP3A4 dependency differs from the preclinical observation that revealed a 94% dependency of ACT‐1004‐1239 metabolism on CYP3A4 in vitro. 2 Given that the increase of ACT‐1004‐1239 exposure was less than fivefold and that the dependency on CYP3A4 is moderate, it is likely that other mechanisms are involved in the elimination of ACT‐1004‐1239. As reported in the human ADME study, alternative elimination pathways of ACT‐1004‐1239 encompass the excretion of unchanged parent drug, as well as biotransformation to the metabolite A3 (reduction of parent drug). 2 These findings additionally reinforce the observation that ACT‐1004‐1239 elimination is not exclusively reliant on CYP3A4. Other tools, such as a physiologically‐based PK (PBPK) model, can be further leveraged in drug development to provide a greater understanding to CYP dependencies in support of clinical data. 39 , 40 Such a mechanistic tool can be utilized to make predictions that are physiologically driven, as for example it can account for enzyme abundance and function, blood flow, PK variability across patients, and other biological processes. 39 In addition, the use of PBPK modeling can be advantageous in designing and optimizing clinical DDI studies. 40 Ultimately, it may serve as a replacement for additional clinical DDI studies. For ACT‐1004‐1239 development, modeling may be useful to predict other scenarios such as concomitant use of ACT‐1004‐1239 and moderate CYP3A4 inhibitors. Although no relevant increase in ACT‐1004‐1239 exposure is expected with a moderate CYP3A4 inhibitor, PBPK modeling would give more certainty on this assumption. Furthermore, PBPK modeling can be leveraged to provide more insight on the impact of different ACT‐1004‐1239 concentration and enzyme kinetics as the fraction metabolized by CYP3A4 may theoretically vary depending on the drug's concentration.
A single dose of 10 mg ACT‐1004‐1239 in absence and presence of itraconazole was well‐tolerated and the incidence of subjects with TEAEs was identical between both treatments. Nevertheless, this type of study is not suitable to draw firm conclusions on the safety profiles of these treatments as it was an open‐label study without a placebo control group.
The study was designed, taking into account various factors such as the selection of the concomitant drug, its formulation, and dose, to provide insights into the DDI potential of ACT‐1004‐1239 when co‐administered with a strong CYP3A4 inhibitor. Although this study design is widely accepted and implemented, measurement of itraconazole concentrations at selected time points could further strengthen the confidence in the study results by confirming that the desired exposure levels, at which CYP3A4 inhibitory efficacy is postulated, were indeed achieved. Furthermore, as the study results suggested the involvement of other elimination routes, the application of PBPK modeling is encouraged to provide a better understanding of the role of alternative elimination routes and to estimate CYP3A4 dependency. This additional analysis may provide clarity to the unexpected moderate increase in ACT‐1004‐1239 exposure following concomitant dosing with itraconazole. Moreover, PBPK modeling and/or in vitro studies could provide valuable insights into the potential for non‐linear metabolism of ACT‐1004‐1239 by CYP3A4, despite the enzyme's broad substrate specificity and high catalytic capacity, aiding in the interpretation of the DDI interaction results presented in this study.
In conclusion, in the presence of itraconazole, a strong CYP3A4 inhibitor, the exposure to ACT‐1004‐1239 was moderately higher (up to 2.9‐fold) than in the absence of the concomitant drug. Therefore, ACT‐1004‐1239 is a moderately sensitive substrate of CYP3A4. At this early stage in clinical development of ACT‐1004‐1239, the findings of this study should be taken into consideration when planning further studies in which strong CYP3A4 inhibitors may be used concomitantly.
AUTHOR CONTRIBUTIONS
C.H, J.D., H.E.M.Z.S., M.F., and P.N.S. wrote the manuscript. C.H., J.D., H.E.M.Z.S., and P.N.S. designed the research. C.H. and M.F. performed the research. C.H., J.D., and P.N.S. analyzed the data.
FUNDING INFORMATION
This study was sponsored by Idorsia Pharmaceuticals Ltd, Allschwil, Switzerland.
CONFLICT OF INTEREST STATEMENT
C.H. and J.D. were employees of Idorsia Pharmaceuticals Ltd at the time of the study. P.N.S. is an employee of Idorsia Pharmaceuticals Ltd. M.F. is the principal investigator who is employed by BlueClinical Phase 1. H.E.M.Z.S. has no conflict of interest.
ACKNOWLEDGMENTS
The authors would like to thank the team members from Idorsia Pharmaceuticals Ltd as well as from BlueClinical Phase 1 for their dedicated support in terms of data generation and/or discussion including Denis Boutin, Susanne Globig, Carmela Gnerre, Beya Khouildi, Vincent Lemoine, Roberta Renai, and Giancarlo Sabattini.
Huynh C, Dingemanse J, Meyer zu Schwabedissen HE, Fonseca M, Sidharta PN. The effect of itraconazole, a strong CYP3A4 inhibitor, on the pharmacokinetics of the first‐in‐class ACKR3/CXCR7 antagonist, ACT‐1004‐1239. Clin Transl Sci. 2024;17:e13883. doi: 10.1111/cts.13883
REFERENCES
- 1. Yu J, Zhou Z, Tay‐Sontheimer J, Levy RH, Ragueneau‐Majlessi I. Risk of clinically relevant pharmacokinetic‐based drug‐drug interactions with drugs approved by the U.S. Food and Drug Administration between 2013 and 2016. Drug Metab Dispos. 2018;46(6):835‐845. doi: 10.1124/dmd.117.078691 [DOI] [PubMed] [Google Scholar]
- 2. Huynh C, Seeland S, Segrestaa J, et al. Absorption, metabolism, and excretion of ACT‐1004‐1239, a first‐in‐class CXCR7 antagonist: in vitro, preclinical, and clinical data. Front Pharmacol. 2022;13(March):1‐15. doi: 10.3389/fphar.2022.812065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Richard‐Bildstein S, Aissaoui H, Pothier J, et al. Discovery of the potent, selective, orally available CXCR7 antagonist ACT‐1004‐1239. J Med Chem. 2020;63(24):15864‐15882. doi: 10.1021/acs.jmedchem.0c01588 [DOI] [PubMed] [Google Scholar]
- 4. Pouzol L, Sassi A, Baumlin N, et al. CXCR7 antagonism reduces acute lung injury pathogenesis. Front Pharmacol. 2021;12:12. doi: 10.3389/fphar.2021.748740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pouzol L, Baumlin N, Sassi A, et al. ACT‐1004‐1239, a first‐in‐class CXCR7 antagonist with both immunomodulatory and promyelinating effects for the treatment of inflammatory demyelinating diseases. FASEB J. 2021;35(3):1‐17. doi: 10.1096/fj.202002465R [DOI] [PubMed] [Google Scholar]
- 6. Huynh C, Henrich A, Strasser DS, et al. A multipurpose first‐in‐human study with the novel CXCR7 antagonist ACT‐1004‐1239 using CXCL12 plasma concentrations as target engagement biomarker. Clin Pharmacol Ther. 2021;109(6):1648‐1659. doi: 10.1002/cpt.2154 [DOI] [PubMed] [Google Scholar]
- 7. Huynh C, Brussee JM, Pouzol L, et al. Target engagement of the first‐in‐class CXCR7 antagonist ACT‐1004‐1239 following multiple‐dose administration in mice and humans. Biomed Pharmacother. 2021;144(September):112363. doi: 10.1016/j.biopha.2021.112363 [DOI] [PubMed] [Google Scholar]
- 8. United States Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research . In Vitro Drug Interaction Studies – Cytochrome P450 Enzyme and Transporter Mediated Drug Interactions, Guidance for Industry. 2020. Accessed September 1, 2021. https://www.fda.gov/media/134582/download
- 9. European Medicines Agency Evaluation of Medicines for Human Use . Guideline on the investigation of drug interactions. 2013. Accessed July 28, 2022. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐investigation‐drug‐interactions‐revision‐1_en.pdf
- 10. Pal D, Mitra AK. MDR‐ and CYP3A4‐mediated drug–drug interactions. J Neuroimmune Pharmacol. 2006;1(3):323‐339. doi: 10.1007/s11481-006-9034-2 [DOI] [PubMed] [Google Scholar]
- 11. Bellosta S, Corsini A. Statin drug interactions and related adverse reactions: an update. Expert Opin Drug Saf. 2018;17(1):25‐37. doi: 10.1080/14740338.2018.1394455 [DOI] [PubMed] [Google Scholar]
- 12. Patsalos PN, Perucca E. Clinically important drug interactions in epilepsy: general features and interactions between antiepileptic drugs. Lancet Neurol. 2003;2(6):347‐356. doi: 10.1016/S1474-4422(03)00409-5 [DOI] [PubMed] [Google Scholar]
- 13. Isoherranen N, Kunze KL, Allen KE, Nelson WL, Thummel KE. Role of itraconazole metabolites in CYP3A4 inhibition. Drug Metab Dispos. 2004;32(10):1121‐1131. doi: 10.1124/dmd.104.000315 [DOI] [PubMed] [Google Scholar]
- 14. Czyrski A, Resztak M, Świderski P, Brylak J, Główka FK. The overview on the pharmacokinetic and pharmacodynamic interactions of triazoles. Pharmaceutics. 2021;13(11):1‐27. doi: 10.3390/pharmaceutics13111961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen Y, Cabalu TD, Callegari E, et al. Recommendations for the design of clinical drug–drug interaction studies with itraconazole using a mechanistic physiologically‐based pharmacokinetic model. CPT Pharmacometrics Syst Pharmacol. 2019;8(9):685‐695. doi: 10.1002/psp4.12449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu L, Bello A, Dresser MJ, et al. Best practices for the use of itraconazole as a replacement for ketoconazole in drug‐drug interaction studies. J Clin Pharmacol. 2016;56(2):143‐151. doi: 10.1002/jcph.562 [DOI] [PubMed] [Google Scholar]
- 17. SPORANOX® (itraconazole) Capsules. Accessed October 5, 2022. https://www.janssenlabels.com/package‐insert/product‐monograph/prescribing‐information/SPORANOX‐Capsules‐pi.pdf
- 18. Patterson SD, Jones B. Bioequivalence and Statistics in Clinical Pharmacology. 2nd ed. Taylor and Francis Group LLC; 2006. [Google Scholar]
- 19. Han L. Calculating the point estimate and confidence interval of Hodges‐Lehmann's median using SAS® software. 2008. Accessed April 1, 2022. https://analytics.ncsu.edu/sesug/2008/ST‐154.pdf
- 20. European medicines agency evaluation of medicines for human use . European medicines agency recommends suspension of marketing authorisations for oral ketoconazole. 2013. Accessed October 5, 2022. https://www.ema.europa.eu/en/news/european‐medicines‐agency‐recommends‐suspension‐marketing‐authorisations‐oral‐ketoconazole
- 21. United States Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research . FDA drug safety communication: FDA limits usage of Nizoral (ketoconazole) oral tablets due to potentially fatal liver injury and risk of drug interactions and adrenal gland problems. 2017. Accessed October 5, 2022. https://www.fda.gov/drugs/drug‐safety‐and‐availability/fda‐drug‐safety‐communication‐fda‐limits‐usage‐nizoral‐ketoconazole‐oral‐tablets‐due‐potentially
- 22. United States Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research . Drug development and drug interactions | table of substrates, inhibitors and inducers. 2022. Accessed October 5, 2022. https://www.fda.gov/drugs/drug‐interactions‐labeling/drug‐development‐and‐drug‐interactions‐table‐substrates‐inhibitors‐and‐inducers#table2‐2
- 23. Tornio A, Filppula AM, Niemi M, Backman JT. Clinical studies on drug–drug interactions involving metabolism and transport: methodology, pitfalls, and interpretation. Clin Pharmacol Ther. 2019;105(6):1345‐1361. doi: 10.1002/cpt.1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. United States Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research . Clinical drug interaction studies—cytochrome P450 enzyme‐ and transporter‐mediated drug interactions. 2020. Accessed October 5, 2022. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/clinical‐drug‐interaction‐studies‐cytochrome‐p450‐enzyme‐and‐transporter‐mediated‐drug‐interactions
- 25. European Medicines Agency . Guideline on the investigation of drug interactions. 2013. doi: 10.1093/deafed/ens058 [DOI]
- 26. BIAXIN®, clarithromycin USP . Accessed October 5, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/050662s042,050698s024,050775s013lbl.pdf
- 27. Hoch M, Huth F, Sato M, et al. Pharmacokinetics of asciminib in the presence of CYP3A or P‐gp inhibitors, CYP3A inducers, and acid‐reducing agents. Clin Transl Sci. 2022;15(7):1698‐1712. doi: 10.1111/cts.13285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Al‐Dubaili N, Saleh N. Sequestration effect on the open‐cyclic switchable property of warfarin induced by cyclodextrin: time‐resolved fluorescence study. Molecules. 2017;22(8):1326. doi: 10.3390/molecules22081326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Durk MR, Jones NS, Liu J, et al. Understanding the effect of hydroxypropyl‐β‐cyclodextrin on fenebrutinib absorption in an itraconazole–fenebrutinib drug–drug interaction study. Clin Pharmacol Ther. 2020;108(6):1224‐1232. doi: 10.1002/cpt.1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Olkkola KT, Backman JT, Neuvonen PJ. Midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;55(5):481‐485. doi: 10.1038/clpt.1994.60 [DOI] [PubMed] [Google Scholar]
- 31. Kolars JC, Watkins PB, Merion RM, Awni WM. First‐pass metabolism of cyclosporin by the gut. Lancet. 1991;338(8781):1488‐1490. doi: 10.1016/0140-6736(91)92302-I [DOI] [PubMed] [Google Scholar]
- 32. Wrighton SA, Stevens JC. The human hepatic cytochromes P450 involved in drug metabolism. Crit Rev Toxicol. 1992;22(1):1‐21. doi: 10.3109/10408449209145319 [DOI] [PubMed] [Google Scholar]
- 33. Wang H, LeCluyse EL. Role of orphan nuclear receptors in the regulation of drug‐Metabolising enzymes. Clin Pharmacokinet. 2003;42(15):1331‐1357. doi: 10.2165/00003088-200342150-00003 [DOI] [PubMed] [Google Scholar]
- 34. Ito K, Hallifax D, Obach RS, Houston JB. Impact of parallel pathways of drug elimination and multiple cytochrome P450 involvement on drug‐drug interactions: CYP2D6 paradigm. Drug Metab Dispos. 2005;33(6):837‐844. doi: 10.1124/dmd.105.003715 [DOI] [PubMed] [Google Scholar]
- 35. Zhou X, Hui YH, Hudson L, et al. In vitro and in vivo correlation of hepatic fraction of metabolism by P450 in dogs. J Pharm Sci. 2019;108(2):1017‐1026. doi: 10.1016/j.xphs.2018.09.012 [DOI] [PubMed] [Google Scholar]
- 36. Yeung C, Yoshida K, Kusama M, et al. Organ impairment—drug–drug interaction database: a tool for evaluating the impact of renal or hepatic impairment and pharmacologic inhibition on the systemic exposure of drugs. CPT Pharmacometrics Syst Pharmacol. 2015;4(8):489‐494. doi: 10.1002/psp4.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hua L, Chiang C, Cong W, et al. The cancer drug fraction of metabolism database. CPT Pharmacometrics Syst Pharmacol. 2019;8(7):511‐519. doi: 10.1002/psp4.12417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Klammers F, Goetschi A, Ekiciler A, et al. Estimation of fraction metabolized by cytochrome P450 enzymes using long‐term cocultured human hepatocytes. Drug Metab Dispos. 2022;50(5):566‐575. doi: 10.1124/dmd.121.000765 [DOI] [PubMed] [Google Scholar]
- 39. Hanke N, Frechen S, Moj D, et al. PBPK models for CYP3A4 and P‐gp DDI prediction: a modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacometrics Syst Pharmacol. 2018;7(10):647‐659. doi: 10.1002/psp4.12343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. European Medicines Agency Evaluation of Medicines for Human Use . Guideline on the reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation. 2019. Accessed December 12, 2023. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐reporting‐physiologically‐based‐pharmacokinetic‐pbpk‐modelling‐and‐simulation_en.pdf