

Summary
Dolichol is a lipid critical for N-glycosylation as a carrier for activated sugars and nascent oligosaccharides. It is commonly thought to be directly produced from polyprenol by the enzyme SRD5A3. Instead, we found that dolichol synthesis requires a three-step detour involving additional metabolites, where SRD5A3 catalyzes only the second reaction. The first and third steps are performed by DHRSX, whose gene resides on the pseudoautosomal regions of the X and Y chromosomes. Accordingly, we report a pseudoautosomal-recessive disease presenting as a congenital disorder of glycosylation in patients with missense variants in DHRSX (DHRSX-CDG). Of note, DHRSX has a unique dual substrate and cofactor specificity, allowing it to act as a NAD+-dependent dehydrogenase and as a NADPH-dependent reductase in two non-consecutive steps. Thus, our work reveals unexpected complexity in the terminal steps of dolichol biosynthesis. Furthermore, we provide insights into the mechanism by which dolichol metabolism defects contribute to disease.
Keywords: dolichol, polyprenol, dolichal, polyprenal, N-glycosylation, pseudoautosomal region, congenital disorders of glycosylation, lipid droplets, polyisoprenoids
Graphical abstract
Highlights
-
•
DHRSX deficiency leads to defective N-glycosylation by disrupting dolichol synthesis
-
•
DHRSX acts both as a reductase and a dehydrogenase in dolichol synthesis
-
•
Dolichol synthesis involves a three-step detour via polyprenal and dolichal
-
•
The function of SRD5A3 is re-assigned as polyprenal reductase
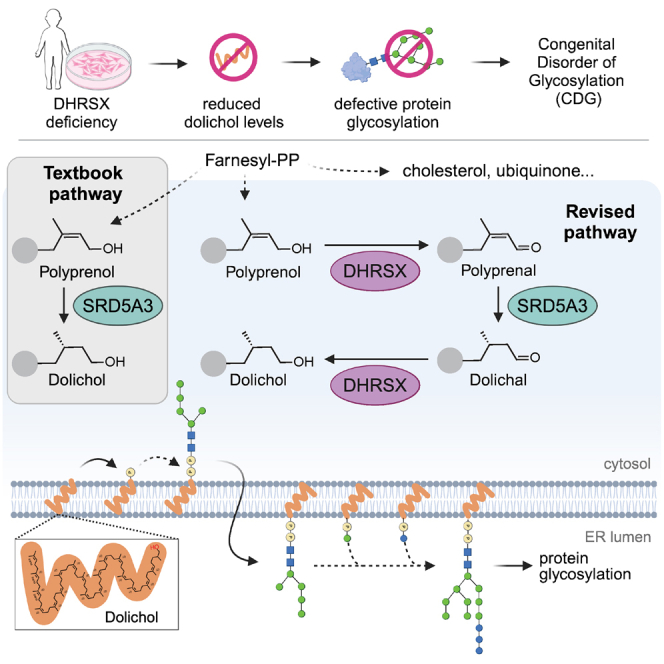
Studying a congenital disorder leads to a revised biosynthetic route for dolichol, a compound that is required for glycosylation.
Introduction
Dolichol is a remarkably long lipid with an essential role in glycosylation pathways that affect the majority of extracellular proteins. In its diphosphate form, it serves as a scaffold for the assembly of the lipid-linked oligosaccharide (LLO) that is transferred en bloc onto nascent glycoproteins during N-glycosylation.1 Dolichol anchors this glycan within the phospholipid bilayer of the endoplasmic reticulum (ER). Furthermore, in its phosphorylated form, it is a carrier for monosaccharides used for N-glycosylation, O-/C-mannosylation, and GPI anchor synthesis. The availability of dolichol-phosphate is rate-limiting for N-glycosylation, since dolichol-phosphate sugars are used in seven steps during the formation of the LLO.2
Dolichol is a polyisoprenoid containing 13–21 five-carbon isoprene units and a terminal hydroxyl group (see graphical abstract). Except for the terminal (α-)isoprene unit, all units contain a double bond between carbons 2 and 3. Dolichol biosynthesis is thought to take place primarily in the membranes of the endoplasmic reticulum (ER).3,4 First, the cis-prenyl transferase complex adds 10–18 isoprene units from isopentenyl diphosphate (IPP) to farnesyl diphosphate (FPP),5 a compound containing three isoprene units that is also used in cholesterol and ubiquinol synthesis (see graphical abstract).6 This process results in polyprenol, which still contains the double bond in its terminal isoprene unit and needs to be converted to dolichol for proper N-glycosylation to occur.7,8 Mammalian SRD5A3 and its yeast ortholog Dfg10 are required for this conversion, and inactivation of the corresponding genes leads to increased polyprenol levels.4 As a consequence, they are widely considered to be polyprenol reductases9 and have been annotated as such in most databases (Uniprot: Q9H8P0 and KEGG: 1.3.1.94). However, a polyprenol reductase activity of SRD5A3 has never been convincingly demonstrated, since impure enzyme preparations with very low enzymatic activity were used in the original studies. In fact, the authors describing the requirement of SRD5A3 in dolichol synthesis did not state that this enzyme is the cellular polyprenol reductase.4 Furthermore, studies on the formation of dolichol in yeast extracts revealed the formation not only of polyprenol and dolichol, but also of dolichal and polyprenal, suggesting that these compounds might be intermediates in the synthesis of dolichol.10 This implied that additional steps in dolichol synthesis remained to be discovered.
Genetic defects in glycosylation processes lead to congenital disorders of glycosylation (CDGs). Affected patients often show severe neurodevelopmental defects and symptoms in a wide range of organs.11 Several CDG due to disrupted dolichol biosynthesis have been described.5 They are caused by pathogenic variants in the two components of the cis-prenyltransferase complex, NUS112 and DHDDS,13 or in the presumptive polyprenol reductase SRD5A3.4,14
Here, we report a CDG caused by biallelic missense variants in DHRSX, a gene located in the pseudoautosomal region 1 of the X and Y chromosomes.15 Extensive studies using patient cells, genetically engineered human and yeast lines, and purified proteins led us to revise the dolichol biosynthesis pathway, discover two unexpected distinct activities of DHRSX, reassign the function of SRD5A3/Dfg10, and provide mechanistic understanding for the glycosylation defect in affected patients.
Results
DHRSX variants in a presumptive pseudoautosomal-recessive congenital disorder of glycosylation
We describe four individuals from three families with distinct facial features (Figure 1A) alongside severe neurological involvement including hypotonia, scoliosis, contractures, profound intellectual disability, epilepsy, and sensorineural hearing loss. These individuals also experienced severe failure to thrive (requiring tube feeding); variable respiratory insufficiency; and involvement of the eyes, the gastrointestinal system, and other organs (Table 1). Since the clinical presentation resembled other congenital disorders of glycosylation (CDGs), we analyzed the glycosylation status of serum transferrin, a glycoprotein that normally carries two N-glycans. This protein is frequently aberrantly glycosylated in disorders affecting N-glycosylation,16 and its analysis is a key step in the diagnostic work-up of patients. Patients 1, 2, and 3 showed changes in transferrin profiles indicative of a defect in the attachment of N-glycans in the ER (i.e., a CDG type I). However, the transferrin glycosylation profile of patient 3 normalized at 17 months of age and was normal in his brother, patient 4.
Figure 1.

DHRSX variants in a presumptive pseudoautosomal-recessive congenital disorder of glycosylation
(A) Patients 1, 3, and 4 carrying DHRSX variants.
(B) The pseudoautosomal regions (PAR) of the X and Y chromosomes.
(C) Pedigree displaying the inheritance of DHRSX variants. Boldface indicates the location of DHRSX variants.
(D) Conservation of DHRSX amino acids affected by variants, and % sequence identity of the entire protein sequences as determined by ClustalW.
(E) 3D models produced by Alphafill optimization of the Alphafold Q8N5I4 model to contain NADP+ at the predicted active site. In pink the adenine nucleoside moiety, in red the phosphate groups, in turquoise the nicotinamide nucleoside moiety.
(F) Alphafold/Alphafill model showing the proximity of the amino acids substituted in patients 1–4 to the predicted active site containing NADP+. Thr49, yellow; Val181, purple; Leu215, blue.
(G) 3D surface model showing the position of the predicted active site with NADP+ bound.
(H) Rotation of the 3D surface model showing the channel presumably allowing access of lipid substrates to the active site.
(I) Localization of Thr49 (yellow), Val181 (purple), and Leu215 (blue) showing proximity to the predicted active site.
(J) Western blot analysis of DHRSX protein levels in EBV-immortalized lymphoblasts from controls, parents, and patients. Bar graphs represent DHRSX protein levels normalized to β-tubulin (mean ± SEM, n = 3).
(K) DHRSX protein levels in dermal fibroblasts from three controls, and patients with DHRSX variants (patients 1 and 3). Data are presented as in (J).
(L) Western blot analysis of LAMP2 mobility and DHRSX in wild-type (“WT”) and DHRSX KO HAP1 cells at baseline, and upon transduction with a lentiviral vector driving expression of DHRSX (“+DHRSX”) or an empty cassette (“EV”).
(M) Immunofluorescence of HAP1 cells expressing DHRSX with a C-terminal FLAG tag, using anti-FLAG for DHRSX, anti-calnexin as ER marker, lipid droplet stain LipidSpot 610, and the nuclear counterstain DAPI. Scale bars, 10 μm.
See also Figure S1.
Table 1.
Summary of clinical features, DHRSX variants, and serum Tf IEF results of the four patients
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | |
|---|---|---|---|---|
| Age | 7 years | 11 months (✝) | 6 years | 4 years |
| Sex | female | female | male | male |
| DHRSX variant (NM_145177.3) | c.541G>T; p.(Val181Phe) | c.146C>T; p.(Thr49Met) | c.541G>T; p.(Val181Phe), c.643C>T; p.(Leu215Phe) | c.541G>T; p.(Val181Phe), c.643C>T; p.(Leu215Phe) |
| Facial dysmorphism | a | a | a | a |
| Neurological findings | ||||
| Developmental/intellectual disability | severe | severe | severe | severe |
| Epilepsy | – | a | a | a |
| Axial hypotonia | a | a | a | a |
| Bilateral sensorineural hearing loss | profound | auditory evoked potentials inconclusive | profound | profound |
| Brain MRI | bilateral hypoplasia of cranial nerves V, VII and VIII | small corpus callosum | mild thinning of corpus callosum; bilateral absence of the cochlear nerves; superior and inferior vestibular nerves bilaterally absent/hypoplastic | bilateral absence of the cochlear nerves, superior and inferior vestibular nerves bilaterally absent/hypoplastic |
| Ophthalmological findings | bilateral neurotrophic keratopathy | normal | bilateral neurotrophic keratopathy | corneal erosion of the right eye |
| Gastrointestinal findings | ||||
| Failure to thrive | a | a | a | a |
| Tube feeding | gastrostomy | gastrostomy | gastrostomy | gastrostomy |
| Hepato(spleno)megaly | hepatosplenomegaly in infancy | cholestasis; hepatomegaly | absent | absent |
| Other | obstipation | gastroesophageal reflux disease | gastroesophageal reflux disease | |
| Dermatological findings | hypertrichosis of arms and legs | ichthyosis | eczema | – |
| Other findings | severe apnea episodes; contractures of elbows and knees | persistent ductus arteriosus | stridor; severe obstructive sleep apnea requiring BiPAP; scoliosis; knee contractures | stridor; respiratory insufficiency requiring BiPAP; scoliosis knee contractures |
| Serum transferrin isoelectrofocusing | type 1 | mild type 1 | type 1 up to 1 year | normal |
present.
The affected individuals were investigated by whole genome or whole exome sequencing after routine investigations had failed to establish a diagnosis. Missense variants were identified in both alleles of DHRSX, a gene in the pseudoautosomal regions (PAR) of the p-arms of the X and Y chromosomes (Figure 1B).17 The PARs are terminal sections of the X and Y chromosomes that are almost perfectly identical.18 Therefore, inheritance of variants in genes in the PARs is pseudoautosomal, and the associated disorders can be recessive or dominant.15,19 Patient 1, a girl, is homozygous for c.541G>T; p.(Val181Phe) in DHRSX (Genbank: NM_145177.3), and Patient 2, a girl, is homozygous for c.146C>T; p.(Thr49Met) in DHRSX. Patients 3 and 4, two brothers, are compound-heterozygous for c.541G>T; p.(Val181Phe) in DHRSX on the X chromosome they inherited from their mother and c.643C>T; p.(Leu215Phe) in DHRSX on the Y chromosome inherited from their father (Figure 1C). The variants were thus inherited in a pseudoautosomal recessive manner.
Variants are predicted to play a role in substrate or cofactor binding, and patient-derived cell lines show strongly reduced DHRSX protein levels
DHRSX is ubiquitously expressed and codes for a putative oxidoreductase of unknown function belonging to the short chain dehydrogenase/reductase family.17 The observed variants are very rare in control populations with allele frequencies of 0.00005 (c.146C>T), 0.000001 (c.541G>T), and 0.00009 (c.643C>T) (December 2023).20 All substituted amino acid residues are located within well-conserved regions of the DHRSX protein (Figure 1D), and substitutions were predicted deleterious by Mutation Taster.21 The 3D structure of DHRSX has not been determined experimentally. However, in a high-confidence model predicted by AlphaFold21,22 the amino acids substituted in patients (Thr49, Val181, and Leu215) are located in the hydrophobic core of the protein (Figures 1E and 1F).
The putative cofactors NAD+ or NADP+ were modeled by the “AlphaFill”23 tool. This revealed an extended channel, reaching from an opening next to a hydrophobic N-terminal alpha helix to the nicotinamide group of NAD(P)+, suggesting that this channel might allow substrate access (Figures 1G and 1H). Using this model, Val181 was predicted to be directly in contact with NAD(P)+ in vicinity of the nicotinamide group (Figures 1E and 1I). The insertion of a large benzyl group due to the Val181Phe substitution is likely to significantly disrupt the structure of the region as well as binding of the cofactor. Furthermore, Val181 is at the interface of the predicted NAD(P)+ binding site and the extended channel that might allow access of the substrate to the active site (Figures 1G–1I). Leu215 is also located along this extended channel and is close to Lys212, predicted to form a hydrogen bond with NAD(P)+ (Figures 1E and 1I). While Thr49 is not predicted to directly interact with NAD(P)+ nor the putative substrate channel, it is an invariable part of a motif that is present in Class IV NAD(P)+ binding domains such as the one in DHRSX (Figures 1E, 1I, and S1A).24 Altogether, the variants observed in our patients were expected to severely impact the putative oxidoreductase function of DHRSX.
Figure S1.

Analysis of the function of DHRSX, related to Figure 1
(A) Conservation of the predicted Class IV NAD or NADP binding site consensus sequence ([AVIC]-[LVIFA]-[VIL]-T-G-[AGSC]-X2-GR-ILF-G-X6-[LFAY]) in the indicated vertebrate species. Between brackets are the % sequence identity of the entire protein-coding sequences as determined by ClustalW alignment. Amino acids indicated under the consensus sequence are those required for either NAD or NADP binding as part of the class IV motif. T49 is obligatory. Amino acid positions indicated above the sequence relate to those in the human DHRSX sequence (Q8N5I4).
(B) Expression of DHRSX mRNA in EBV-immortalized lymphoblasts from controls (C1, C2), DHRSX-CDG patients (P1, P3, P4), the parents of P3/P4, and an SRD5A3-CDG patient as measured by RT-qPCR. Results are normalized to the expression of HPRT1 and then to the mean of controls. Data are represented as the mean of three biological replicates ±SEM.
(C) Sanger sequencing analysis showing a hemizygous 5bp deletion c.467_471 delTCATG; p.(Val156Aspfs∗62) in SRD5A3, confirming gene deletion (KO).
(D) Immunofluorescence analysis of WT HAP1 cells (control), and DHRSX KO HAP1 cells stably transfected with an expression construct for human DHRSX with a C-terminal triple FLAG tag. Labeling with anti-DHRSX (yellow), anti-FLAG DHRSX-FLAG (magenta) and DAPI (cyan) confirms staining in lipid droplet-like structures, and specificity of the anti-FLAG signal. Scale bars = 10 μm.
(E) Western blot analysis shows increased LAMP2 mobility indicative of hypoglycosylation in SRD5A3 KO HAP1 cells. Stable re-expression of WT SRD5A3 led to a migration of LAMP2 comparable to the one seen in WT HAP1 cells.
Immunoblotting revealed substantially lower DHRSX protein levels in patient cells, at an average of 4% of mean control levels in EBV-immortalized lymphoblasts and 5% in fibroblasts (Figures 1J and 1K). The heterozygous parents of patients 3 and 4 had intermediate levels. In contrast, RT-qPCR showed that DHRSX mRNA levels in patients’ lymphoblasts were between 34 and 68% of healthy controls, similar to those observed in samples from the parents of patients 3 and 4 (Figure S1B), suggesting that low protein levels in patient cell lines might be caused by a reduced stability of the variant proteins.
DHRSX is localized to lipid droplets, and its deficiency leads to a glycosylation defect in HAP1 cells
Given that the patients were initially identified due to their hypoglycosylation of serum transferrin, we hypothesized that DHRSX had a role in N-glycosylation. First, we investigated whether complete genetic deficiency of DHRSX leads to a glycosylation defect in HAP1 cells, a near haploid human leukemia cell line25 (subsequently called DHRSX KO). The N-glycosylation status was assessed by analyzing the electrophoretic mobility of LAMP2, a heavily glycosylated protein that is hypoglycosylated in CDG.26,27 Increased mobility indicating a glycosylation defect was observed in DHRSX KO cells. Complementation by re-expression of wild-type DHRSX restored the normal migration pattern of LAMP2 (Figure 1L), demonstrating that the loss of DHSRX was indeed responsible for the observed glycosylation defect in DHRSX KO cells.
Next, we explored the subcellular localization of DHRSX. As suitable DHRSX antibodies were not available, we expressed DHRSX carrying an FLAG epitope tag in the corresponding HAP1 KO cell lines. We observed a faint reticular immunofluorescent staining that overlapped with the staining of the ER marker calnexin. Yet, the most intense signal was observed in small (0.2–1 μm diameter) spherical structures that were dispersed throughout the cytoplasm (Figures 1M and S1D). Co-staining with a lipid droplet stain revealed that these structures were lipid droplets, consistent with prior high-throughput studies suggesting enrichment of DHRSX in lipid droplets.28
Metabolite changes in DHRSX and SRD5A3 deficiencies prompt a revision of the dolichol biosynthesis pathway
DHRSX is a putative oxidoreductase and its inactivation leads to a glycosylation defect. Within N-glycosylation, an obvious need for an oxidoreductase only exists in dolichol synthesis, when polyprenol is converted into dolichol. Since this reaction is commonly believed to be catalyzed by SRD5A3, it was not obvious how DHRSX could contribute to dolichol metabolism (Figure 2A).29
Figure 2.

DHRSX or SRD5A3 deficiency leads to metabolic changes prompting a revision of the model of dolichol biosynthesis
(A) Commonly accepted9 model of dolichol biosynthesis, where SRD5A3 directly forms dolichol from polyprenol. Additional related polyisoprenoids detected in our study are shown on the right.
(B) Polyisoprenoid species in wild-type, DHRSX KO, and SRD5A3 KO HAP1 cells and respective complementations. Data represent area under the curve (AUC) normalized to total ion count (TIC) (means ± SEM; n = 4; ∗∗∗∗p < 0.0001). Here and in subsequent figures, one species is shown (“−19” means 19 isoprenoid units), but additional chain lengths and chromatograms are shown in Figures S2A and S2B, and Table S2.
(C) Polyisoprenoid species in EBV-immortalized lymphoblasts from controls, parents and patients. Data are presented as in (B) (means ± SEM; n = 4; §, p < 0.05 compared to every control). See also Table S2.
(D) Working hypothesis of the revised pathway of dolichol biosynthesis. Reaction 1: NAD+-dependent conversion of polyprenol to polyprenal by DHRSX; Reaction 2: NADPH-dependent reduction of polyprenal to dolichal by SRD5A3; Reaction 3: reduction of dolichal to dolichol via an as-yet unknown enzyme.
See also Figure S2.
We explored this by LC-MS in HAP1 cell lines where DHRSX or SRD5A3 had been inactivated by CRISPR/Cas9. As observed in DHRSX KO HAP1 cells, SRD5A3 inactivation also increased mobility of LAMP2, indicating an N-glycosylation defect (Figure S1E).4 We identified polyprenol and dolichol with chain lengths of 18–21 isoprenoid units based on expected m/z values and elution times in comparison to standards (Figure S2A; Table S1). We also detected dolichal and polyprenal, which had been hypothesized to play a role in the synthesis of dolichol in yeast.10 Furthermore, we tentatively identified polyprenoic acids (Figure 2A) that are likely formed via dehydrogenation of polyprenal by an aldehyde dehydrogenase.30
Figure S2.

Polyisoprenoid LCMS chromatograms and changes in DHRSX- and SRD5A3-deficient fibroblasts, related to Figure 2
(A) Representative mass spectra (right) and extracted ion chromatograms (left) for the indicated m/z, obtained from standards for polyprenal (PAL), polyprenol (POL), dolichal (DAL), dolichol (DOL) and a mixture of all four species (bottom).
(B) Representative extracted ion chromatograms of indicated m/z values in WT, DHRSX KO and SRD5A3 KO HAP1 cells showing accumulation of polyprenal (PAL) and polyprenoic acid (POL-COOH) only in SRD5A3 KO cells and polyprenol (POL) in both DHRSX and SRD5A3 KO cells. Dolichol (DOL) levels are reduced in both DHRSX and SRD5A3 KO cells. The [M+2] peak is shown to reduce confounding effects of increased polyprenal M+4 levels on signals for dolichol.
(C) Polyisoprenoids in dermal fibroblasts collected from DHRSX-CDG patients (P1 & P3), compared to those from three controls. Data is the total ion count (TIC)-normalized area under the curve (AUC) (means ± SEM; n = 4). § p < 0.05 compared to every control; # p < 0.05 compared to one of the controls. Only the species with 19 isoprenyl units are shown for clarity. See Table S2 for all isoprenyl chain lengths. Note the up to 100-fold difference in scale between panels.
(D) Polyisoprenoids in dermal fibroblasts collected from six SRD5A3-deficient individuals (SRD5A3-P1-6), compared to those from three controls. Data is TIC-normalized AUC (means ± SEM; n = 4). § p < 0.05 compared to every control; # p < 0.05 compared to one of the controls. Only the isoprenoid species with 19 isoprenyl units are shown for clarity. See Table S2 for all isoprenyl chain lengths.
In DHRSX- and SRD5A3-deficient cells we observed 5-fold and 6-fold reductions in dolichol levels (Figures 2B and S2B; Table S2), respectively. Putative biosynthetic precursors were increased in both cases. Levels of polyprenol, the presumptive substrate for SRD5A3, were increased 30-fold in SRD5A3 KO cells and 70-fold in DHRSX KO cells. Strikingly, polyprenal and polyprenoic acid, which were unchanged in DHRSX KO cells, were massively increased in SRD5A3 KO cells (i.e., 85-fold and 10-fold, respectively). These changes were completely rescued upon re-expression of DHRSX or SRD5A3 in corresponding KO cells (Figure 2B).
Similar results were also obtained in patient-derived cell lines. Immortalized patient lymphoblasts showed a 20- to 30-fold accumulation of polyprenol in DHRSX-deficient cells, alongside a 2- to 3-fold decrease in dolichol levels. Increases in polyprenal (6-fold) and polyprenoic acid (5-fold) were only observed in SRD5A3-deficient cells (Figure 2C; Table S2). In one SRD5A3-deficient lymphoblast line, dolichol levels were unchanged and polyprenol levels only showed a non-significant increase. Polyprenol levels were also increased in SRD5A3 deficient fibroblasts, but to a varying extent, as previously reported.31 In general, changes of these metabolites in patient fibroblasts had the same trends, but fold-changes were much lower (Figures S2C and S2D; Table S2). This context-dependent behavior resembles what has previously been described in SRD5A3-CDG patient cells and might be due to compensatory changes.4,31,32
Taken together, these observations were not consistent with the model where SRD5A3 is directly responsible for polyprenol reduction into dolichol (Figure 2A). They rather suggested that DHRSX was required for the conversion of polyprenol to polyprenal, followed by the reduction of the C2-C3 double bond of polyprenal by SRD5A3. The logical product of this reaction would be dolichal, which could be reduced by an unknown reductase to dolichol (Figure 2D).
DHRSX produces polyprenal from polyprenol using both NAD+ and NADP+ as cofactor
First, we tested the hypothesis that DHRSX may use polyprenol to form polyprenal (reaction 1, Figure 2D). Incubation of recombinant DHRSX with polyprenol led to the production of polyprenal in the presence of NAD+ or NADP+ (Figures 3A, S3A, and S3B), with a KM of 5–10 μM for polyprenol and a kcat of approximately 0.45 s−1 (Figure 3B). Surprisingly, no clear preference for NAD or NADP was apparent, including when the reaction was assessed in reverse direction (Figure S3C) and at different timepoints (Figure S3D).
Figure 3.

DHRSX produces polyprenal from polyprenol using both NAD+ and NADP+ as cofactor (reaction 1)
(A) Formation of polyprenal from polyprenol was measured after incubation of 5 μg/mL polyprenol with 1 mmol/L NADP+ or NAD+ and 0.075 μmol/L recombinant DHRSX (see Figure S3A) for 2 h at 37°C. See also Figures S3B, S3C, and S3D.
(B) Kinetic parameters for DHRSX were determined by measuring polyprenal formation after incubation of the indicated concentrations of polyprenol with 1 mmol/L NADP+ or NAD+ and 0.00375 μmol/L recombinant DHRSX for 5 min at 37°C, or after an identical incubation of 4 μmol/L polyprenol with variable nucleotide concentrations. Data are turnover rates based on formation of polyprenal-18 (means ± SEM; n = 3).
(C) DHRSX is responsible for the polyprenol dehydrogenase activity in HAP1 cells. Polyprenol-18 and polyprenal-18 were monitored after incubation of 1 mg/mL membrane preparations from wild-type (“WT”) and DHRSX KO HAP1 cells with or without polyprenol (5 μg/mL), and NAD+ or NADP+ (5 mmol/L) for 2 h at 37°C. See also Figure S3E.
(D) Endogenous activity in membrane preparations from EBV-immortalized lymphoblast from controls, parents, and patients was assessed in the reverse direction using NADH or NADPH at 5 mmol/L and polyprenal as substrate. See also Figure S3F.
(E) Correlation of polyprenal reductase activity in EBV-immortalized lymphoblast membrane preparations (Figure 3D) with ß-tubulin-normalized DHRSX protein levels (Figure 1J). Figures 3A, 3C, and 3D present TIC-normalized AUC (means ± SEM; n = 3).
See also Figure S3.
Figure S3.

Additional data supporting that DHRSX converts polyprenol to polyprenal, related to Figure 3
(A) Immunoblot using an antibody against the 6xHis epitope of recombinant DHRSX-N-His, alongside Ponceau stain of the same membrane.
(B) Adjunct to Figures 3A and S3C, representative chromatograms of the forward reaction (polyprenol to polyprenal conversion; left side) and reverse reaction (polyprenal to polyprenol conversion; right side) with recombinant DHRSX. Interconversion was measured after incubation of 5 μg/mL of polyprenol or polyprenal with 1 mmol/L of the indicated cofactor, and 0.075 μmol/L recombinant DHRSX for 2 h at 37°C.
(C) Polyprenol formation from polyprenal was measured after incubation of 5 μg/mL polyprenal with 1 mmol/L NADPH or NADH and 0.075 μmol/L recombinant DHRSX for 2 h at 37°C. Measurements are based on the formation of polyprenol with 18 isoprene units.
(D) Time course of polyprenal/polyprenol interconversion by recombinant DHRSX and dependence on NAD(P)(H) concentration measured in both directions. Polyprenol-18 and polyprenal-18 were measured at the indicated time-points after incubation of 5 μg/mL of polyprenol with the indicated concentrations of NADP+ or NAD+, or of 5 μg/mL of polyprenal with the indicated concentrations of NADPH or NADH and 0.075 μmol/L recombinant DHRSX (see Figure S3A) at 37°C.
(E) Adjunct to Figure 3C showing the reverse activity of DHRSX measured in WT and DHRSX KO HAP1 cell membrane extracts. Polyprenol-18 and polyprenal-18 were quantified after incubation of 1 mg/mL HAP1 membrane extracts with/without polyprenal (5 μg/mL) and NADH or NADPH (5 mmol/L), 2h, 37°C.
(F) Measurement of dolichal-18 from the experiment presented in Figure 3D. Isoprenoid species were monitored after incubation of 1 mg/mL EBV-immortalized lymphoblast membrane extracts with/without polyprenal (5 μg/mL) and NADH or NADPH (5 mmol/L), 2h, 37°C. Figures S3C, S3D, S3E and S3F present TIC-normalized AUC (means +/− SEM; n = 3).
This behavior is highly unusual; most oxidoreductases show a marked preference for either NAD+/H or NADP+/H, often with >100-fold differences in KM. In vitro, oxidoreductase reactions are reversible, and the direction is largely determined by the ratio of the reduced to the oxidized cofactor. However, in cells, the ratio of NAD+ to NADH is >100:1, whereas the ratio of NADP+ to NADPH is <1:10.33 Therefore, the preference for NAD or NADP usually determines whether an oxidoreductase preferentially acts as a dehydrogenase or a reductase. In the case of DHRSX, the dual specificity for both cofactors indicated that DHRSX may catalyze both a dehydrogenation reaction (i.e., reaction 1, Figure 2D) and a different, unidentified reduction reaction.
Next, we tested whether DHRSX is responsible for the polyprenol dehydrogenase activity in cells. In membrane preparations from parental HAP1 cells, we observed significant polyprenol dehydrogenase activity both in the forward and reverse direction with either NADP(H) or NAD(H) as cofactors (Figures 3C and S3E). DHRSX KO cells lacked this activity, demonstrating that DHRSX is required for the conversion of polyprenol to polyprenal in HAP1 cells. We also assessed this activity in extracts from patient and control lymphoblasts. We observed activity in cell lines from two controls and the parents of patients 3 and 4, but none in samples from patients 1, 3, or 4 (Figure 3D). Strikingly, the activity in lymphoblasts showed a highly significant positive correlation with levels of DHRSX protein in this cell line (see Figure 1J) (NADH: R2 = 0.9667; p < 0.0001, NADPH: R2 = 0.9910; p < 0.0001; Figure 3E). We concluded that DHRSX is the cellular polyprenol dehydrogenase in HAP1 cells and lymphoblasts.
SRD5A3 and its yeast orthologue Dfg10 produce dolichal from polyprenal (reaction 2), but have undetectable activity on polyprenol
The strong accumulation of polyprenal and polyprenoic acid in SRD5A3-deficient cell lines (see Figures 2B, 2C, S2B, and S2D) led us to hypothesize that SRD5A3 does not act on polyprenol but on polyprenal produced by DHRSX (reaction 2, Figure 2D). Previous efforts to characterize SRD5A3 function encountered difficulties in purifying this integral membrane protein.4,31 To circumvent these problems, we analyzed membrane preparations from HEK293T cells overexpressing SRD5A3 (Figure S4A). We observed the formation of dolichal from polyprenal in SRD5A3-containing extracts in the presence of NADPH, but not in the presence of NADH, nor in extracts that lacked SRD5A3 overexpression (Figures 4A, S4C, S4D, and S4E). We also tested the ability of our SRD5A3 preparation to catalyze the reduction of polyprenol to dolichol, the reaction commonly attributed to SRD5A3 (Figure 2A). We did not detect dolichol formation beyond the endogenous dolichol already present in the enzyme preparation (Figure 4B). This demonstrated that SRD5A3 can catalyze the NADPH-dependent reduction of polyprenal, but cannot act as a polyprenol reductase, explaining why DHRSX and SRD5A3 KO cells both accumulate polyprenol, whereas only SRD5A3 KO cells accumulate polyprenal.
Figure S4.

Additional data supporting that SRD5A3 and Dfg10 convert polyprenal to dolichal, related to Figure 4
(A) Western blot analysis of protein extract from samples used for the preparation of membrane extracts showing expression of SRD5A3-N-His in WT HEK293T cells, detected by His immunoblotting. Overexpression was achieved using pcDNA3.1(+)-N-6His plasmid vector containing H. sapiens SRD5A3 cDNA with an N-terminal 6 x His-tag.
(B) Western blot analysis of protein extract from samples used for the preparation of membrane extracts showing expression of dfg10-N-His in WT HEK293T cells, detected by His immunoblotting. Overexpression was achieved using pcDNA3.1(+)-N-6His plasmid vector containing S. cerevisiae dfg10 cDNA appended to an N-terminal 6 x His-tag.
(C) Polyprenal reductase activity of SRD5A3 is strictly NADPH dependent. NADPH/NADH-dependence of SRD5A3-catalyzed polyprenal reductase activity in SRD5A3-overexpressing HEK293T membrane extracts in the presence of 0.3 mg/mL of membrane protein extract. 5 μg/mL polyprenal was used in the presence of 0, 5, 20, 100, 1000, or 5000 μmol/L of NADPH or NADH, 2h, 37°C. Data are TIC-normalized AUC (mean ± SEM, n = 3).
(D) Representative chromatograms, complementary to Figure 4A and (E), showing that significant dolichal-18 and dolichol-18 are only formed from polyprenal (5 μg/mL) in the presence of NADPH (5 mmol/L), but not NADH (5 mmol/L), after incubation for 2h at 37°C. PAL = polyprenal, POL = polyprenol, DAL = dolichal and DOL = dolichol.
(E and F) SRD5A3-dependent polyprenal and polyprenol reductase activity was assessed in membrane extracts from HEK293T cells (E) or DHRSX KO HAP1 cells (F) overexpressing SRD5A3 or an empty vector. Measurement of polyprenol-18, polyprenal-18, dolichol-18M + 2, and dolichal-18 from 0.3 mg/mL control or SRD5A3-overexpressing cells after incubation in the presence of polyprenal or polyprenol (5 μg/mL) and NADPH or NADH (5 mmol/L), 2h, 37°C. Data are TIC-normalized AUC (mean ± SEM, n = 3). Several metabolites are already shown in Figures 4A and 4B.
(G) Dfg10-dependent polyprenal reductase and polyprenol reductase activity was assessed in membrane-extract from HEK293T cells overexpressing S. cerevisiae dfg10. Measurement of polyprenol-18, polyprenal-18, dolichol-18M + 2, and dolichal-18 from 0.3 mg/mL control or Dfg10-overexpressing HAP1 cells after incubation in the presence of polyprenal or polyprenol (5 μg/mL) and NADPH or NADH (5 mmol/L), 2h, 37°C. Data are TIC-normalized AUC (mean ± SEM, n = 3). Several metabolites are already shown in Figures 4E and 4F.
Figure 4.

SRD5A3 and its yeast orthologue Dfg10 produce dolichal from polyprenal (reaction 2) but have undetectable activity on polyprenol
(A and B) SRD5A3 shows activity on polyprenal but not on polyprenol. Formation of dolichal from polyprenal (reaction 2 of revised model of dolichol synthesis) (A) and formation of dolichol from polyprenol (previously accepted function of SRD5A3 and Dfg10) (B) were assessed using membrane preparations from HEK293T cells overexpressing human SRD5A3 or an empty vector control. Polyisoprenoids were quantified after 2 h incubation at 37°C with polyprenal (A) or polyprenol (B) at 5 μg/mL and NADPH or NADH at 5 mmol/L. Data represent TIC-normalized AUC (means ± SEM, n = 3). See also Figures S4D, S4E, and S4F.
(C) Activity of SRD5A3 determined by measuring dolichal formation after incubation of the indicated concentrations of polyprenal with 1 mmol/L NADPH and 1 μg/mL membrane preparations from DHRSX KO HAP1 cells overexpressing SRD5A3 for 30 min at 37°C (upper) or after an identical incubation of 4 μM of polyprenal with the indicated concentrations of NADPH (lower). Values were calculated based on the formation of dolichal-18 (means ± SEM; n = 3).
(D) Isoprenoid species in WT (BY4741), dfg10 KO, and dfg10 KO + dfg10 S cerevisiae. Data are TIC-normalized AUC (means ± SEM; n = 3; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.01; ∗∗∗∗p < 0.0001). Species with 16 isoprenyl units are shown. See also Table S2.
(E and F) The SRD5A3 yeast orthologue Dfg10 also shows activity on polyprenal but not on polyprenol. Formation of dolichal from polyprenal (reaction 2 of revised model of dolichol synthesis) (E), and formation of dolichol from polyprenol (previously accepted function Dfg10) (F) are presented as described in (A), but using extracts from HEK293T cells overexpressing Dfg10. See also Figure S4G.
Membrane preparations are expected to contain many other enzymes, including DHRSX. Accordingly, we observed formation of polyprenol from polyprenal in all membrane preparations, in the presence of NADH or NADPH (Figures S4D and S4E), likely due to endogenous DHRSX acting in the reverse direction. To avoid confounding effects of DHRSX activity, we determined the kinetic properties of overexpressed SRD5A3 in membrane preparations of DHRSX KO HAP1 cells revealing a KM of 6.5 μM for polyprenal (Figure 4C). Interestingly, SRD5A3-overexpressing membrane fractions formed not only dolichal from polyprenal, but also significant amounts of dolichol (Figures S4D and S4E), indicating they might contain an NADPH-dependent reductase that further metabolizes dolichal to dolichol. Of note, inactivation of DHRSX abolished formation of dolichol, suggesting that it also plays a role in the last step of dolichol synthesis (Figure S4F and DHRSX also catalyzes the final step of dolichol synthesis).
Looking back, the role of SRD5A3 as the cellular polyprenal reductase was also apparent in the experiments where we had analyzed the activity of lymphoblast membrane preparations on polyprenal. Lysates from controls and DHRSX-deficient patients formed significant amounts of dolichal from polyprenal in the presence of NADPH (Figures 3D and S3F). This activity was absent in a SRD5A3-deficient lymphoblast line, indicating that endogenous SRD5A3 was required for the polyprenal reductase activity.
Previous work had demonstrated that Dfg10 is the yeast ortholog of SRD5A3.4 We therefore wondered whether Dfg10 would also act as polyprenal reductase. We first measured polyisoprenoid levels from Dfg10-deficient yeast, and noted a 4-fold decrease in dolichol, accompanied by a 36-fold increase in polyprenol and a 45-fold increase in polyprenal (Figure 4D; Table S2), reminiscent of what we had observed in SRD5A3-deficient mammalian cells (Figure 2B). These changes were suppressed by re-expression of Dfg10. Next, we overexpressed Dfg10 in HEK293T cells (Figure S4B). As for SRD5A3, we observed an NADPH-dependent polyprenal reductase activity in membrane preparations, but no detectable conversion of polyprenol to dolichol (Figures 4E and 4F).
Overall, this demonstrated that SRD5A3 and Dfg10 are responsible for the conversion of polyprenal to dolichal, but cannot directly produce dolichol from polyprenol (Figure 2D).
DHRSX also catalyzes the final step of dolichol synthesis
The revised model of dolichol biosynthesis requires an enzyme to reduce dolichal to dolichol (reaction 3; Figure 2D). While exploring SRD5A3 function, we had noted that membrane preparations contained detectable dolichal reductase activity (Figure S4E), which was absent in DHRSX KO cells (Figure S4F). This indicated that DHRSX was also involved in the last step of dolichol synthesis. We therefore tested whether recombinant DHRSX was able to catalyze this step. Indeed, purified DHRSX had detectable dolichal reductase activity (reaction 3, Figure 2D) using both NADPH or NADH (Figures 5A, S5A, S5B, S5C, and S5D) with a KM of 2 μM for dolichal and a kcat between 1 and 1.4 s−1 (Figure 5B).
Figure 5.

DHRSX also catalyzes the final step in dolichol synthesis
(A) Formation of dolichol from dolichal was assessed after incubation of 5 μg/mL dolichal with 0.075 μmol/L recombinant DHRSX, and 1 mmol/L NAD(P)H, 2 h, 37°C. See also Figures S5A and S5B.
(B) Activity of DHRSX was determined by measuring dolichol formation after incubation of the indicated concentrations of the dolichal mixture with 1 mmol/L NADPH or NADH and 0.00375 μmol/L recombinant DHRSX for 5 min at 37°C (upper), or after an identical incubation of 4 μmol/L of the dolichal mixture with the indicated concentrations of nucleotides (lower). Presented data are turnover rates based on formation of dolichol-18 (means ± SEM; n = 3).
(C) DHRSX KO HAP1 cells lack dolichal reductase activity. Dolichol-18 and dolichal-18 were measured in reactions containing 1 mg/mL HAP1 membrane, 5 μg/mL dolichal-18, and 5 mmol/L NAD(P)H for 2 h at 37°C.
(D) Potential inhibitory interferences arising from the dual lipid and cofactor specificity of DHRSX in the revised model of dolichol synthesis. Red lines indicate potential inhibition of the opposing DHRSX activity by each reciprocal cofactor (NAD+ or NADPH). The members of each cofactor pair in larger font (NAD+ and NADPH) are those proposed to be used in vivo for DHRSX-dependent polyprenol dehydrogenase and dolichal reductase activities.
(E) The NAD+-dependent polyprenal formation from polyprenol is only mildly inhibited by NADPH concentrations found in vivo. Polyprenol-18 and polyprenal-18 were measured after a 15 min, 37°C incubation of 5 μg/mL polyprenol, 0.075 μmol/L recombinant DHRSX, and 1 mmol/L NAD+ with or without 0.1 mmol/L NADPH.
(F) The NADPH-dependent dolichol formation from dolichal is only mildly inhibited by NAD+ concentrations found in vivo. Dolichol-18 and dolichal-18 were measured after a 3 min, 37°C incubation of 5 μg/mL of dolichal, 0.075 μmol/L recombinant DHRSX, and 0.1 mmol/L NADPH with or without 1 mmol/L NAD+. Figures 5A, 5C, 5E, and 5F present TIC-normalized AUC (means ± SEM; n = 3).
See also Figure S5.
Figure S5.

Additional data supporting that DHRSX also converts dolichal to dolichol, related to Figure 5
(A) Representative extracted ion chromatograms of the forward reaction (dolichal to dolichol conversion; left side) and reverse reaction (dolichol to dolichal conversion; right side) of DHRSX presented in Figures 5A and S5B. Metabolites were assessed after incubation of 5 μg/mL dolichal or dolichol with 1 mmol/L of the indicated nucleotides and 0.075 μmol/L recombinant DHRSX for 2 h at 37°C.
(B) Formation of dolichal from dolichol was assessed after incubation of 5 μg/mL dolichol with 0.075 μmol/L recombinant DHRSX, and 1 mmol/L NAD(P)+, 2h, 37°C. Data is TIC-normalized AUC of 18 isoprenoid unit containing lipids (means ± SEM, n = 3). See Figure 5A for forward reaction.
(C and D) Lack of specificity of DHRSX for NAD(H) or NADP(H) in the conversion of polyprenol to polyprenal, as well as in the conversion of dolichal to dolichol in forward and reverse direction. Polyprenol-18 (POL), polyprenal-18 (PAL), dolichol-18 (DOL) or dolichal-18 (DAL) were measured at the indicated timepoints after incubation of 0.075 μmol/L recombinant DHRSX protein with 5 μg/mL of POL, PAL, DOL or DAL and the indicated cofactor concentrations at 37°C. Data are represented as mean TIC-normalized AUC of three replicates ±SEM. Panels showing bidirectional polyprenol to polyprenal conversion have already been shown in Figure S3D and are displayed here to facilitate a comparison. Of note, the progression of these reactions with time indicates that conversion of dolichol to dolichal is much less favorable than the conversion of polyprenol to polyprenal, consistent with prior reports that the presence of a double-bond between C2 and C3 (as present in polyprenol) strongly favors the oxidation of a terminal alcohol group by increasing the equilibrium constant by a factor of more than 100).30.
To confirm that the dolichal reductase reaction in cells was catalyzed by DHRSX, we used DHRSX-deficient HAP1 cells. We detected this activity in control membrane preparations, but not in DHRSX KO (Figure 5C). Similar to the DHRSX-catalyzed polyprenol dehydrogenase activity (reaction 1, Figure 2D), both NADP and NAD were used with comparable efficiency, confirming that DHRSX is responsible for cellular dolichal reductase activity.
The unique dual activity of DHRSX is thermodynamically favorable and not inhibited by the reciprocal cofactor
DHRSX catalyzes the oxidation of an alcohol group to an aldehyde group in reaction 1 and a reduction of an aldehyde group to an alcohol group in reaction 3 (Figure 5D). At first sight, these reactions may seem almost identical, but in opposite directions. Yet, the presence of a double-bond between carbons 2 and 3, as present in polyprenol, favors the oxidation of a terminal alcohol group by increasing the equilibrium constant by a factor of more than 100, facilitating the conversion of polyprenol to polyprenal.30 Consistent with this, we observed that the conversion of polyprenol to polyprenal progressed much more efficiently with time than the conversion of dolichol to dolichal (Figures S5C and S5D).
Since DHRSX can use NAD+/H and NADP+/H with comparable efficiency (Figures S5C and S5D), we speculated that it uses NAD+ in conversion of polyprenol to polyprenal and NADPH for reduction of dolichal to dolichol (Figure 5D). Consequently, conversion of polyprenol to polyprenal would be driven forward by the high cytoplasmic NAD+ to NADH ratio (reaction 1; Figure 5D). In reverse, the reduction of dolichal to dolichol would benefit from the high ratio of NADPH to NADP+ (reaction 3; Figure 5D).
However, the oxidized nucleotide (i.e., NAD+) could inhibit the reduction reaction and vice versa (Figure 5D). We therefore assessed whether the NAD+-dependent polyprenol dehydrogenase activity was inhibited by NADPH at physiological concentrations.34 Polyprenol dehydrogenase activity was only 30% lower when 0.1 mmol/L NADPH was added to a reaction containing 1 mmol/L NAD+ (Figure 5E). Likewise, 1 mmol/L NAD+ inhibited the NADPH-dependent dolichal reductase activity only by 20% (Figure 5F). These findings demonstrated that reduction of dolichal and dehydrogenation of polyprenol could occur simultaneously in vivo, catalyzed by the same enzyme, DHRSX, but using different cofactors. To our knowledge, this is a unique situation in human metabolism.
Phosphorylated and phosphohexose polyprenol accumulate when DHRSX or SRD5A3 are inactivated
When the conversion of polyprenol to polyprenal is defective, dolichol can be replaced by polyprenol as intermediate oligosaccharide acceptor, with polyprenol being much poorer in this respect than dolichol for several of the reactions necessary for N-glycosylation.35,36,37,38,39 We wondered whether an increase of polyprenol-phosphate (Pol-P) or polyprenol-phospho-hexoses (Pol-P-Hex) could cause the glycosylation defect observed in DHRSX-deficient cells (Figure 1L).
Pol-P and Pol-P-Hex were barely detectable in control cells, but DHRSX deficiency caused a marked increase of both metabolites in HAP1 cells (Figures 6A, 6B, S6A, and S6B; Table S2) and lymphoblasts (Figures 6C and 6D; Table S2). Concomitantly, we observed a decrease in dolichol-P (Dol-P) and dolichol-P-hexose (Dol-P-Hex) levels in HAP1 cells and, to a lesser extent, in patient lymphoblasts. Consequently, the ratios of Pol-P to Dol-P and of Pol-P-Hex to Dol-P-Hex were increased by more than 20-fold in HAP1 cells. Similar changes were also seen in the case of SRD5A3 deficiency. Complementation with wild-type genes rescued these changes. These findings indicated that polyprenol can be phosphorylated in cells and that the imbalance between polyprenol-P-hexose and dolichol-P-hexose might be the cause of the glycosylation defect in DHRSX and SRD5A3-deficient cells.35,38
Figure 6.

Accumulation of phospho- and phosphohexose-polyprenol alongside truncated N-linked oligosaccharide species in DHRSX/SRD5A3-deficient cells
(A–D) Dolichol-phosphate or polyprenol-phosphate (A and C), and dolichol-phospho-hexose or polyprenol-phospho-hexose (B and D) were measured in wild-type, DHRSX KO, and SRD5A3 KO HAP1 cells and their respective complementations (A and B), as well as EBV-immortalized lymphoblasts from controls, patients, and parents (C and D). Data are TIC-normalized AUC (means ± SEM, n = 4; ∗p < 0.05; ∗∗∗∗p < 0.0001; §p < 0.05 compared to every control; #p < 0.05 compared to one of the controls). See also Table S2, and Figures S6A and S6B. Dolichol- and polyprenol-phospho-hexose represent mixtures of mannose and glucose derivatives.
(E) Mechanisms underlying the glycosylation defect in DHRSX and SRD5A3 deficient cells integrating published data and our present paper. The inset table presents the ratio of polyprenol to dolichol (see Figure 2B), as well as the ratios of the corresponding phospho and phosphohexose derivatives (A and B) (means ± SEM, n = 4). The percentages alongside red dashed lines indicate the relative activity of the indicated enzyme when using a polyprenol-instead of dolichol-derived substrate (DOLK, dolichol kinase40; DPM1/2/3, Dolichol-phosphate mannosyltransferase41; ALG3, Alpha-1,3-Mannosyltransferase42; DPAGT1, UDP-N-acetylglucosamine—dolichyl-phosphate N-acetylglucosamine-phosphotransferase43,44. Erroneous transfer of an immature glycan (Man5GlcNAc2) to nascent glycoproteins leads to linear Man-5 NLOs (see “abnormal glycosylation” box), that are subsequently trimmed to Man-4 NLOs by the enzyme EDEM3.45 Alternatively, branched Man-5 can be formed during normal N-glycosylation as a consequence of successive glycan trimming (arrows on the right side). Castanospermine allows us to determine the origin of Man-5 glycans, since it inhibits -glucosidases I/II required for this trimming, thereby preventing the formation of branched Man-5.
(F–H) N-linked oligosaccharide (NLO) HPLC profiles obtained from wild-type (F), DHRSX KO (G), and SRD5A3 KO (H) HAP1 cells labeled with 100 μCi [2-3H]-Mannose, showing an accumulation of truncated NLOs, primarily Man-4, Man-5, and Glc1Man5/M6 species upon inactivation of DHRSX or SRD5A3. See also Figures S6D, S6E, and S6F.
(I–K) N-linked oligosaccharide (NLO) HPLC profiles obtained from wild-type (I), DHRSX KO (J), and SRD5A3 KO (K) HAP1 cells labeled with 100 μCi [2-3H]-Mannose and treated with 50 μmol/L castanospermine prior to metabolic labeling. The castanospermine-resistant accumulation of Man4, Man5, and Glc1Man5/M6 species indicates that these are due to the transfer of an incomplete lipid-linked olichosaccharide, rather than trimming of mature NLOs.
(L) Ratio of the abundance of Man5 N-linked to Man9 N-linked oligosaccharides (NLO) detected in metabolic labeling experiments in wild-type (WT), DHRSX KO, and SRD5A3 KO HAP1 cells and their respective complementations (shown in E–G). Data are presented normalized to WT in a log 2 scale (means ± SEM, n = 2–4; ∗p < 0.05; ∗∗p < 0.01).
See also Figure S6.
Figure S6.

Additional data corroborating effects on polyisoprenoid adducts and glycosylation, supporting Figure 6
(A) Representative mass spectra and extracted ion chromatograms for dimethylated dolichol-phosphate and polyprenol-phosphate (NH4+ adducts in positive mode) acquired in samples from control, DHRSX KO and SRD5A3 KO HAP1 cells.
(B) Representative mass spectra and extracted ion chromatograms (in negative mode) for Dolichol-phosphohexose and polyprenol-phosphohexose in control, DHRSX KO and SRD5A3 KO HAP1 cells. Dolichol-P-hexose and Polyprenol-P-Hexose represent a mixture of mannose and glucose derivatives.
(C) Experimental setup of analysis of newly synthesized N-linked oligosaccharides with radioactive mannose in HAP1 cells. Cells were grown to 90% confluency in a T25 flask, then underwent a glucose deprivation step followed by 1 h of labeling with 100 μCi tritiated 23[H]Mannose. Cells then underwent sequential extraction with chloroform and methanol, then glycoproteins were purified from the resulting protein pellet. After overnight digestion steps with trypsin and PNGase F, N-linked oligosaccharide (NLO) extracts were injected and analyzed by HPLC.
(D) Newly synthesized N-linked oligosaccharides (NLO) were detected by HPLC after incubation of MPDU1 KO HAP1 cells labeled with 100 μCi 23[H]Mannose, showing characteristic accumulation of Man4, Man5 and Glc1Man5/M6 species and deficiency of Man9 species.
(E and F) Newly synthesized NLOs were detected by HPLC after incubation of DHRSX KO HAP1 cells complemented with WT DHRSX (E), or SRD5A3 KO HAP1 cells complemented with WT SRD5A3 (F) with 100 μCi tritiated 23[H]Mannose, showing a restoration of full-length Man9 species.
(G) N-linked Hex4GlcNAc2 (corresponding to Man-4) and Hex8GlcNAc2 (corresponding to Man-8) were identified and quantified by an untargeted proteomics approach in membrane extracts of WT, DHRSX KO and SRD5A3 KO HAP1 cells and their respective complementations. The abundance of the indicated peptides was normalized to the abundance of the corresponding parent proteins. To increase visibility of differences between conditions, data are presented normalized within each modified peptide.
Inactivation of DHRSX and SRD5A3 in HAP1 cells causes defects in the ER N-glycosylation pathway
Four dolichol-P-mannoses and three dolichol-P-glucoses are used by glycosyltransferases to build the dolichol-pyrophosphate-linked (Dol-PP) oligosaccharide, which is then transferred en bloc onto nascent proteins (Figure 6E). Based on our observations and previous work in Chinese hamster ovary (CHO) cells, we hypothesized that the accumulation of polyprenol-P-hexoses could interfere with the activity of the required glycosyltransferases and that glycan assembly on polyprenol-PP might be less efficient.35,36,37,38,39 Consequently, truncated high-mannose lipid linked oligosaccharides might accumulate or even be transferred onto glycoproteins (Figure 6E).
As we were unable to assess the growing Dol/Pol-PP-bound oligosaccharide by LC-MS, we analyzed newly synthesized N-glycans by following the incorporation of radiolabeled mannose (Figure S6C, “metabolic labeling”).46 To this end, we incubated different HAP1 cell lines with radiolabeled [2-3H]-mannose and analyzed N-linked oligosaccharides (NLO) by HPLC (Figure S6C). We focused on (1) the Man9GlcNAc2 NLO (hereafter Man-9), which represents a physiological N-glycan after removal of terminal glucoses by ER -glucosidases I/II (highlighted in gray), and (2) the Man5GlcNAc2 NLO (hereafter Man-5), which can be either the result of further trimming of the Man-9 NLO (giving rise to “branched Man-5” in Figure 6E) or the result of the erroneous transfer of the immature glycan (“linear Man-5” in Figure 6E).
The peak corresponding to the Man-5 NLO was clearly increased in DHRSX KO and in SRD5A3 KO cells (Figures 6F, 6G, and 6H), as well as in MPDU1 deficient cells (Figure S6D) that are defective in dolichol-phosphohexose utilization.47,48 A 3- to 4-fold increase in the ratio of the abundance of the Man-5 to the Man-9 glycan was observed (Figure 6L). Concomitantly, the Man-4 NLO peak was also increased consistent with Man-5 NLOs being partially converted to Man-4 NLO by the enzyme EDEM3.45 Both changes were rescued upon re-expression of the corresponding enzymes (Figures 6L, S6E, and S6F).47,48 We also noted the appearance of several Hex4GlcNAc2-modified glycopeptides (corresponding to the Man-4-NLOs) in DHRSX and SRD5A3 KO cell lines when we analyzed membrane fractions from these cells using an untargeted proteomic approach (Figure S6G). Thus, our data suggested that an immature glycan might be transferred onto proteins in DHRSX or SRD5A3 KO cell lines.
Yet, to allow us to reach this conclusion, we still needed to determine whether the accumulating Man-5 and Man-4 NLOs were indeed the result of an erroneous transfer of an immature glycan (linear Man-5, Figure 6E), or whether it was formed by increased trimming of “normal” NLOs (branched Man-5, Figure 6E). To distinguish between these two possibilities, we treated cells with castanospermine, which prevents removal of terminal glucose residues and subsequent glycan trimming (Figure 6E). We reasoned that this would reduce the production of physiological branched Man-5 and leave the abnormal linear Man-5 unaffected. As expected, castanospermine led to a decrease of Man-5 and Man-4 species in wild-type cells, concomitant with an increase in a glycan retaining a terminal glucose residue (G1M9 in Figure 6I). In contrast, the increase in Man-5 and Man-4 species was fully maintained in castanospermine-treated DHRSX KO and SRD5A3 KO HAP1 cells (Figures 6J and 6K), consistent with a direct transfer from linear Man-5 LLOs to nascent glycoproteins. Taken together, our data indicate that the increased ratio of polyprenol to dolichol affects maturation of the lipid linked oligosaccharide and is associated with the transfer of immature glycans.
Discussion
Dolichol formation from polyprenol involves three steps
Our work reveals the final steps in dolichol synthesis and corrects the misassignment of SRD5A3 function in databases and current literature.9,14,31,49,50 Three steps from polyprenol to dolichol may seem unnecessarily complicated. However, the proposed metabolic “detour” provides a significant advantage due to the presence of a conjugated system in polyprenal, which makes its C2-C3 double bond more amenable to reduction than the one in polyprenol: the C2-C3 double bond adjacent to a carbonyl group enables the formation of a resonance-stabilized intermediate after hydride transfer, a mechanism similar to the reduction of testosterone catalyzed by SRD5A2.51,52 Accordingly, among the ≈100 NAD(P)-dependent enzymes catalyzing the reduction of carbon-carbon double bonds (E.C.1.3.1.x), all but a few act on a conjugated double bond. Notably, metabolic detours involving the transient formation of a conjugated oxo group during the reduction of carbon-carbon double bond are also present in several other pathways, including prostaglandin degradation,53 sex steroid metabolism,54 menthol synthesis,55 and bacterial bile acid dehydroxylation.56 Thus, nature employs such detours as a recurrent strategy to facilitate the reduction of carbon-carbon double bonds.
DHRSX catalyzes two nonconsecutive steps in the same biochemical pathway using different cofactors
DHRSX is unique in catalyzing an oxidation and a reduction reaction on related molecules within the same pathway (reactions 1 and 3 in Figure 5D). This is possible because both steps are thermodynamically favored due to the presence of a double bond conjugated with the aldehyde in polyprenal and the absence of such a bond in dolichal. Based on data obtained with related compounds, the presence of a double bond between C2 and C3 is expected to shift the equilibrium toward the aldehyde by more than 100-fold.57
The quite unique dual cofactor specificity of DHRSX further facilitates its involvement in two reactions33 by allowing it to react with the predominant oxidized nucleotide, NAD+, in the oxidation of polyprenol and with the predominant reduced nucleotide, NADPH, in the dolichal reduction step. Of course, DHRSX might also reduce polyprenal to polyprenol using NADPH (in a reversal of reaction 1). Yet, this seems to be limited in cells due the rapid reduction of polyprenal to dolichal by SRD5A3, given that polyprenal is barely detectable in cells unless SRD5A3 is inactivated. Furthermore, at physiological concentrations, the oxidation of polyprenol with NAD+ is not significantly impeded by NADPH and, reciprocally, NAD+ does not significantly interfere with the reduction of dolichal.
Polyprenol and polyprenal accumulation as pathomechanisms in DHRSX and SRD5A3 deficiencies
The N-linked oligosaccharides obtained from DHRSX and SRD5A3 deficient cells showed an increase in Man5GlcNAc2 species (Man-5) and Man4GlcNAc2 species (Man-4) alongside a reduction in Man9GlcNAc2 species (Man-9). This uncommon pattern had already been observed in two glycosylation-deficient CHO cell lines (Lec5 and Lec9) with an unidentified defect in the conversion of polyprenol to dolichol and a concomitant accumulation of polyprenol.29,58,59 Both DPAGT1, responsible for the transfer of GlcNAc-1-P onto Dol-P; and ALG3, which transfers the sixth mannose residue onto the LLO, are very inefficient when using Pol-P60,44 and Pol-P-Mannose,61 respectively, instead of their dolichol-linked analogues (Figure 6E). Furthermore, the Dol-P-Man synthase (DPM) enzyme has also been shown to be poorly active on Pol-P.41 Therefore, the high Pol-P-Hexose/Dol-P-Hexose ratio, indicative of a high Pol-P-Mannose/Dol-P-Mannose ratio in DHRSX KO and SRD5A3 KO cells, may explain the accumulation of Man-5 species and, due to the activity of the enzyme EDEM3, of Man-4 species.45 Thus, inactivation of DHRSX or SRD5A3 affects maturation of the lipid linked oligosaccharide and is associated with the transfer of immature glycans.
The glycosylation defects in DHRSX- and SRD5A3-deficient cells are similar, but the clinical presentation of DHRSX-CDG and SRD5A3-CDG are different, suggesting that factors beyond the elevated polyprenol to dolichol ratios lead to clinical symptoms. Polyprenal accumulates only in SRD5A3-CDG but not in DHRSX-CDG. This metabolite is highly reactive and might undergo reactions with different nucleophiles similar to the 4-hydroxynonenal.62 The clinical phenotype of SRD5A3-CDG patients includes ocular abnormalities, optic nerve hypoplasia/atrophy, coloboma and retinal dystrophy.61 Given that the terminal part of polyprenal resembles the one of retinaldehyde, it is tempting to speculate that it might interfere with the function of retinaldehyde.
Context-dependent compensation mechanisms in the dolichol biosynthesis pathway
In DHRSX KO or SRD5A3 KO HAP1 cells, dolichol levels were reduced 5- and 6-fold, respectively. In contrast, dolichol levels were only 50% reduced in lymphoblasts from patients with DHRSX-CDG and indistinguishable from controls in lymphoblasts from an SRD5A3-deficient patient. Consistent with these observations, previous studies reported unchanged dolichol levels with an increase in the polyprenol/dolichol ratio in fibroblasts from patients with SRD5A3-CDG.31,32 This indicated that high polyprenol levels allow cells to bypass the requirement for DHRSX and, as previously postulated, for SRD5A3 in a cell-type-dependent manner.4,31 This compensation could explain the relatively mild glycosylation deficiency in certain SRD5A3-CDG patients49,63 and in DHRSX-CDG patients 3 and 4 in our study. Currently, the specifics of the compensation mechanisms remain elusive, but an increased activity of upstream metabolic pathways might be involved.64
DHRSX deficiency is the first pseudoautosomal recessive inborn error of metabolism
Until now, the only published recessive single-gene disorders in the PARs are Langer mesomelic dysplasia, caused by pathogenic variants in the SHOX gene,65 and familial pulmonary alveolar proteinosis caused by mutations in CSF2RA.66,67 Genes in the PAR1, including DHRSX, evade X-inactivation,68,69,70 meaning they might contribute to disorders caused by sex chromosome aneuploidies.71,72 Indeed, two other PAR1 genes, SLC25A6 and CD99, have been linked to cardiac abnormalities and autoimmune disease, respectively, in Klinefelter syndrome (47,XXY).73,74 Thus, a further investigation of DHRSX and dolichol metabolism in these pathologies seems warranted given that sex chromosome aneuploidies are relatively prevalent in the general population (1 in 250–500).75
Limitations of the study
Our study revealed the specific functions of SRD5A3 and DHRSX in dolichol synthesis, but we do not understand how some cell types still can synthesize some dolichol even if DHRSX or SRD5A3 are completely inactivated. Revealing these compensation mechanisms will help us understand patient symptoms and may lead to therapeutic approaches. Our study is also limited regarding the kinetic evaluation of SRD5A3 and DHRSX. On the one hand, this is due to our inability to produce recombinant SRD5A3 protein in a soluble form. On the other hand, this is because we are assessing activities on substrates that are almost insoluble in water, requiring the addition of detergents and phospholipids and preventing us from achieving substrate saturation in kinetics studies. Furthermore, these conditions do not adequately represent the physiological conditions for these enzymes, which likely act on a 2-dimensional phase-border. Lastly, our work revealed dolichal and polyprenal as intermediates in dolichol synthesis, but we do not know anything about specific roles of these metabolites in cell biology. The potential role of these intermediates in health and disease remains to be explored in future studies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Calnexin | StressGen Biotechnologies | SPA-860, RRID: AB_312057 |
| DHRSX | Sigma | HPA003035, RRID: AB_1080631 |
| FLAG M2 | Sigma | F3165, RRID: AB_259529 |
| Penta-His | Qiagen | 34660, RRID: AB_2619735 |
| β-tubulin | Thermo Fisher Scientific | MA516308, RRID: AB_2537819 |
| LAMP2 antibody | Santa-Cruz Biotech | SC-18822, RRID: AB_626858 |
| Goat-anti-Mouse antibody | DAKO | P0447, RRID: AB_2617137 |
| Goat-anti-Rabbit antibody | DAKO | P0448, RRID:AB_2617138 |
| Alexa 488-labeled goat anti-rabbit IgG antibody | Thermo Fisher Scientific | A-11034, RRID: AB_2576217 |
| Alexa 568-labeled goat anti-mouse IgG antibody | Thermo Fisher Scientific | A-11019, RRID: AB_143162 |
| Alexa 488-labeled goat anti-mouse IgG antibody | Thermo Fisher Scientific | A-11001, RRID: AB_2534069 |
| Alexa 568-labeled goat anti-rabbit IgG antibody | Thermo Fisher Scientific | A-11011, RRID: AB_143157 |
| Chemicals, reagents, and recombinant proteins | ||
| Bovine Serum Albumin (BSA) Fraction V | Sigma | 10735086001 |
| Digitonin | Invitrogen (Thermo Fisher Scientific) | BN2006 |
| DBPS | Cytiva | SH30264.01 |
| Fetal Bovine Serum | Dutcher | S1810 |
| Fetal Bovine Serum | BioWest | S1400 |
| TurboFectin 8.0 | Origene | TF81005 |
| Lipofectamine3000 | Thermo Fisher Scientific | 15292465 |
| Opti-MEM™ | Gibco | 11058021 |
| DMEM/F-12 | Gibco | 21041–025 |
| DMEM/F-12, HEPES | Gibco | 31330038 |
| IMDM | Gibco | 12440–053 |
| DMEM | Gibco | 41966052 |
| Cytiva HyClone™ Fetal Clone III Serum | Cytiva | 12319862 |
| 0.5 mm glass beads | Sigma Aldrich | Z250465 |
| DAPI Solution (1 mg/mL) | Thermo Fisher Scientific | 62248 |
| Saponin | Sigma-Aldrich | 84510 |
| Triton™ X-100 | Sigma-Aldrich | T8787 |
| beta-mercaptoethanol | Sigma-Aldrich | M3148 |
| Pageruler Plus Prestained Protein Ladder | Thermo Fisher Scientific | 11832124 |
| DMSO | Sigma-Aldrich | 317275 |
| Para-formaldehyde | Thermo Fisher Scientific | 28908 |
| Mowiol® 4-88 | Sigma-Aldrich | 81381 |
| RIPA buffer | Custom | N/A |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | Thermo Fisher Scientific | 34579 |
| Puromycin Dihydrochloride | Gibco | A1113802 |
| Difco™ Casamino Acids, Vitamin Assay | Gibco | 16219861 |
| Bacto™ Yeast Extract | Gibco | 212750 |
| Bacto™ Peptone | Gibco | 16299741 |
| Geneticin™ Selective Antibiotic | Gibco | 10131035 |
| Hygromycin B | Gibco | 10687010 |
| GeneRuler | Thermo Fisher Scientific | SM1331 |
| PBS | Thermo Fisher Scientific | 10010023 |
| TBS 10X | Euromedex | ET220 |
| Versene | Gibco | 15040033 |
| Methanol | Thermo Fisher Scientific | 10606652 |
| Methanol (LC-MS grade) | Biosolve | 136878 |
| Chloroform | VWR | 22711290 |
| Chloroform (LC-MS grade) | Biosolve | 34806 |
| trimethylsilyl diazomethane (TMSD) | Sigma-Aldrich | 362832 |
| NAD+ free acid grade II | ROCHE | 10127990001 |
| NADH disodium salt | ROCHE | 10128023001 |
| NADP+ sodium salt hydrate | Sigma-Aldrich | N0505-1G |
| NADPH tetrasodium salt | ROCHE | 10102824001 |
| Dolichol | Aventi polar lipids | 9002000 |
| Polyprenol | Aventi polar lipids | 9002100 |
| Dolichal | This study | N/A |
| Polyprenal | Aventi polar lipids | 9002200 |
| Phosphatidylcholine | Sigma-Aldrich | P-0378 |
| Phosphatidylethanolamine | Sigma-Aldrich | P-0503 |
| Glucose | Sigma-Aldrich | G7021 |
| Magnesium Sulfate | Thermo Fisher Scientific | A14491.0I |
| Asahipak NH2P-50 4E column | Shodex | N/A |
| 7-AAD | Thermo Fisher Scientific | A1310 |
| Carbenicillin | VWR | J67159.AD |
| Dithiothreitol | Thermo Fisher Scientific | R0861 |
| Gelred nucleic acid stain | Sigma-Aldrich | SCT123 |
| T7 DNA Ligase | New England Biolabs | M0318S |
| CutSmart® Buffer | New England Biolabs | B7204 |
| Agilent 6546 ion funnel mass spectrometer | Agilent | N/A |
| Agilent 1290 HPLC System | Agilent | N/A |
| Accucore C30 150 × 2.1 mm column | Thermo Fisher Scientific | 27826–152130 |
| Isopropopanol | Biosolve | 162678 |
| Acetonitrile | Biosolve | 12078 |
| Ammonium formate | Biosolve | 19878 |
| Formic acid | Biosolve | 232478 |
| iBlot® 2 NC mini Stacks | Invitrogen | IB23002 |
| ECL WB substrate | Thermo Fisher Scientific | PIER32106 |
| First-strand cDNA Synthesis Kit | Sigma Aldrich | GE27-9261-01 |
| DNAse I | Sigma Aldrich | 10104159001 |
| Trypsin | Gibco | 25300096 |
| IPTG | Thermo Fisher Scientific | 15529019 |
| Bolt® 4–12% Bis-Tris Plus Gels, 12-well | Thermo Fisher Scientific | 15324604 |
| Bolt Transfer Buffer (20X)-1 L | Thermo Fisher Scientific | 15256066 |
| 20X Bolt® MES SDS Running Buffer (500 mL) | Thermo Fisher Scientific | 13266499 |
| LDS Sample buffer | Thermo Fisher Scientific | 11549166 |
| cOmplete(TM), Mini, EDTA-free Protease | Thermo Fisher Scientific | 11836170001 |
| Trypsin | Sigma Aldrich | T8003-500MG |
| Trypsin-EDTA 1X in PBS w/o Calcium w/o Magnesium | Dominique Dutscher | L0940-100 |
| Tween 20 | Thermo Fisher Scientific | P1379 |
| N-Glycosidase F | Roche | 11365193001 |
| 6X DNA Loading Dye | Thermo Fisher Scientific | R0611 |
| Cell Disruption Media | Scientific Industries, Inc. | SI-BG05 |
| LipidSpot™ 610 | Biotium | 70069 |
| Polybrene | Merck life science N.V (ex Sigma Aldrich) | TR-1003 |
| Phusion™ DNA polymerase | Thermo Fisher Scientific | F630S |
| 2-Chloroacetamide (CAM) | Sigma-Aldrich | C0267 |
| Trizma® base | Merck Life Science BV (ex Sigma Aldrich) | T1503-100g |
| S-Trap™ micro units | Protifi LLC | N/A |
| Axygen™ MaxyClear Snaplock Microtubes, 1.5 mL (MCT-150-L-C) | Thermo Fisher Scientific | 11311984 |
| CaCl2 | Sigma-Aldrich | 793639 |
| Water ULC/MS - CC/SFC | Biosolve | 232141 |
| Sequencing Grade Modified Trypsin (1x100μg) | Promega | V5117 |
| Lys-C endopeptidase | Sopachem NV | 125–02543 |
| Phosphoric acid | Merck Life Science BV (ex Sigma Aldrich) | 695017-100ML |
| Sodium dodecyl sulfate SDS | Merck Life Science BV (ex Sigma Aldrich) | 62862-1KG |
| Critical commercial assays | ||
| Micro BCA™ Protein Assay Kit | Thermo Fisher Scientific | Cat# 23235 |
| DNeasy Blood & Tissue Kit | Qiagen | 69504 |
| Ni-NTA Fast Start Kit | Qiagen | 30600 |
| Ni-NTA spin kit | Qiagen | 31314 |
| RNeasy Mini KIT | Qiagen | 74104 |
| NucleoSpin Plasmid Mini kit | Macherey-Nagel | 740588.5 |
| Plasmid mini kit | Qiagen | 12123 |
| Yeast Transformation Kit | Sigma Aldrich | YEAST1-1KT |
| NEBridge® Golden Gate Assembly Kit | New England Biolabs | R3733S |
| Experimental models: Cell lines | ||
| HAP1 Parental cells | Horizon Discovery | C631 |
| HAP1 DHRSX KO cells | Horizon Discovery | HZGHC007899c011 |
| HAP1 SRD5A3 KO Cells | This paper | N/A |
| HAP1 MPDU1 KO cells | Horizon Discovery | HZGHC004060c012 |
| HEK293T | Gift from Reid Gilmore, Uni. Of Mass. | This paper |
| EBV-immortalized lymphoblasts (Controls and affected DHRSX and SRD5A3-deficient individuals) | This paper | N/A |
| Dermal fibroblasts (Controls and affected DHRSX and SRD5A3-deficient individuals) | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Yeast BY4741 | Euroscarf | Y00000 |
| Yeast BY4741 dfg10 KO | This paper | N/A |
| BL21(DE3) E. Coli | Thermo Fisher Scientific | 10749734 |
| One Shot® TOP10 E. coli | Thermo Fisher Scientific | C404003 |
| Oligonucleotides | ||
| DHRSX_gDNA_PCR_F TCTCCACTGTCTTCCCCAGT | This paper | N/A |
| DHRSX_gDNA_PCR_R CGCACAGACAGAGGGAGATG | This paper | N/A |
| SRD5A3_gDNA1_PCR_F GCCTTTGATGTCCCCAAGAGATATT | This paper | N/A |
| SRD5A3_gDNA1_PCR_R CATGGCACTTATACTGATGGGC | This paper | N/A |
| SRD5A3_gDNA2_PCR_F TCAGAATTCTCGGGGCGGC | This paper | N/A |
| SRD5A3_gDNA2_PCR_R AGTGAATGACCACTCCTGCTTTA | This paper | N/A |
| Ori-Amp_CW_F GAGGTACCGGTCTCTGGG GCGGTAATACGGTTATCCACAG |
This paper | N/A |
| Ori-PmeI_CCW_F GAGGTACCGGTCTCAGTT TAAACTCATATATACTTTAGATTGATTTAAAAC |
This paper | N/A |
| AmpR_CW_F GAGGTACCGGTCTCTAAACTT GGTCTGACAGTTACCAATGCTTAATC |
This paper | N/A |
| Ori-Amp_CCW_F GAGGTACCGGTCTCATC ACTGGCACTTTTCGGGGAAATGTGC |
This paper | N/A |
| DFG10-L_CW_F GAGGTACCGGTCTCTGT GAGTTTAAACTTTTTGCCCAAATAAATCATAATC |
This paper | N/A |
| DFG10-L_CCW_F GAGGTACCGGTCTCTC GCAGCAATTGTTCTTCATCAAAGTACAT |
This paper | N/A |
| pUG74_NatK7b_CW-F GAGGTACCGGTC TCGTGCGGTACGCTGCAGGTCGACAACC |
This paper | N/A |
| pUG74_NatK7_CCW_F GAGGTACCGGTCT CTACGACACTAGTGGATCTGATATCACC |
This paper | N/A |
| DFG10-R_CW_F GAGGTACCGGTCTCTTCG TTAATGTGCACACTAGGGCATTCATATAC |
This paper | N/A |
| DFG10-R_CCW_F GAGGTACCGGTCTCT CCCCGTTTAAACAATGCCTAAAATATT AAATAAAAAATAGG |
This paper | N/A |
| AmpR-Mut_CCW_F GAGGTACCGGTCTC GTCACGCGGTATCATTGCAGCACTG |
This paper | N/A |
| pEF-1_CCW_F GAGGTACCGGTCTCTCG GTTGCTAGTGAACACAGTTGTGTCAG |
This paper | N/A |
| DHRSX_CW_F GAGGTACCGGTCTCTACCG ATGTCGCCATTGTCTGCGGC |
This paper | N/A |
| DHRSX-mut_CCW_F GAGGTACCGGTCTCTCT TCGTCTCTTTCTCGTTGTATAGGTAAC |
This paper | N/A |
| DHRSX-mut_CW_F GAGGTACCGGTCTCTG AAGTCCCTCCACGTCACCTACAAC |
This paper | N/A |
| DHRSX-stop_CCW_F GAGGTACCGGTCTCT GTTGTCACAGGGTCACATCAAGGAC |
This paper | N/A |
| hBGt-R-CCW-F GAGGTACCGGTCTCTCA ACATCGATTAGACTAGTCTAGAAATTCACC |
This paper | N/A |
| AmpR_Mut_CW_F GAGGTACCGGTCTCC GTGACCCACGCTCACCGGCTCCAG |
This paper | N/A |
| pEF-2_CCW_F GAGGTACCGGTCTCTCGGTT CACGACACCTGAAATGGAAG |
This paper | N/A |
| DHRSX-Ctag_CCW_F GAGGTACCGGT CTCTCCAGCAGGGTCACATCAAGGACCC |
This paper | N/A |
| C3xFlag_CW_F GAGGTACCGGTCTCAC TGGGTCACCATCACCATCACCATGAC TACAAAGACCATGACGGTG |
This paper | N/A |
| C3xFlag_CCW_F GAGGTACCGGTCTCG GTTGTCAGGCCGCAAGCTTGTCATCGT |
This paper | N/A |
| hBGt-2-R-CW-F GAGGTACCGGTCTCT CAACCGACTGTGCCTTCTAGTTGCC |
This paper | N/A |
| CeGFP_CW_F GAGGTACCGGTCTCA CTGGGTGTGAGCAAGGGCGAGGAGCTG |
This paper | N/A |
| CeGFP_CCW_F GAGGTACCGGTCTCGG TTGTTACTTGTACAGCTCGTCCATGCCG |
This paper | N/A |
| SRD5A3_CW_F GAGGTACCGGTCTCTA CCGATGGCTCCCTGGGCGGAGG |
This paper | N/A |
| SRD5A3-Ctag_CCW_F GAGGTACCGGT CTCTCCAGAAACAAAAATGGTA GGAAAGCTTTCCTATG |
This paper | N/A |
| Cen4Ars1_CW_F GAGGTACCGGTCTCTG TGACCGCATATATCGCTGGGCCATTC |
This paper | N/A |
| Cen4Ars1_CCW_F GAGGTACCGGTCTCA CGCAGTTATTACTGAGTAGTATTTATT TAAGTATTGTTTG |
This paper | N/A |
| pRS_DrugK7_CW_F GAGGTACCGGTCTCGTG CGGCATCAGAGCAGATTGTACTGAGAGTG |
This paper | N/A |
| pRS_DrugK7_CCW_F GAGGTACCGGTCTC TACGATGCGGTATTTTCTCCTTACGCATCTGTG |
This paper | N/A |
| DFG10-L2_CW_F GAGGTACCGGTCTCTTCGTG TTTAAACTTTTTGCCCAAATAAATCATAATC |
This paper | N/A |
| DFG10-A_CW_F ACCAGTAGAATGCTGTCCTGC | This paper | N/A |
| DFG10-B_CW_F CTATGAAAAGTGAGCAAATGCCTAC | This paper | N/A |
| DFG10-C_CW_F CATGTTATTCTGGCCAATCTG | This paper | N/A |
| DFG10-D_CW_F GTTAGGCATTTGTCTCTGAAG | This paper | N/A |
| kanB CTGCAGCGAGGAGCCGTAAT | This paper | N/A |
| kanC TGATTTTGATGACGAGCGTAAT | This paper | N/A |
| SRD5A3sense_BglII_HIS TTATATAGATCTTGCCACCATGCATCATCAC |
This paper | N/A |
| SRD5A3_rev_bsrGI ATACAATGTACACTAGAAGGCACAGTCGAGGC |
This paper | N/A |
| SRD5A3_sense_BglII TTATATAGATCTCCACCATGGCTCCCTGGGCGGAG |
This paper | N/A |
| hDHRSX_rev_bsrGI ATACCATGTACATCACAGGGTCACATCAAGGAC |
This paper | N/A |
| hDHRSX_ sense_BglII TTATATAGATCTCCACCATGTCGCCATTGTCTGCGG |
This paper | N/A |
| Deposited data | ||
| Proteomics data | This paper | ProteomeXchange: PXD049174 |
| Recombinant DNA | ||
| pET15b DHRSX-N-His NM_145177.3 | This paper | N/A |
| pcDNA3.1(+) SRD5A3-N-His NM_024592.5 | This paper | N/A |
| pcDNA3.1(+) DFG10-N-His (Human optimized cDNA) | This paper | N/A |
| pSpCas9(BB)-2A-GFP (PX458) gRNA_1 sequence CACGACGTGAATCATGACAT | This paper | N/A |
| pSpCas9(BB)-2A-GFP (PX458) gRNA_2 sequence CTAGCCACCAAGTTAAGTTG | This paper | N/A |
| pDel-DFG10 | This paper | N/A |
| pEF-DHRSX-3xFlag | This paper | N/A |
| pYCH-DFG10 | This paper | N/A |
| psPAX2 | Addgene #12260 | |
| pMD2.G | Addgene #12259 | |
| pUB83 SRD5A3 | This paper | N/A |
| pUB83 SRD5A3-N-His | This paper | N/A |
| Software and algorithms | ||
| ImageJ | NIH | https://www.imagej.net/ij/ |
| Fiji | NIH | https://doi.org/10.1038/nmeth.2019 |
| Adobe Illustrator | Adobe | https://www.adobe.com |
| LAURA 6 | LabLogic, UK | https://lablogic.com/life-sciences/software/laura |
| Alissa Interpret | Agilent | https://www.agilent.com/en/product/next-generation-sequencing/clinical-informatics-platform/alissa-interpret-930086 |
| Interactive Genomics Viewer | Broad Institute | https://software.broadinstitute.org/software/igv/ |
| Ugene | Unipro | https://www.ugene.net/ |
| Graphpad Prism | Dotmatics | https://www.graphpad.com |
| Mass Hunter Quantitative Analysis | Agilent | https://www.agilent.com/en/product/software-informatics/mass-spectrometry-software/data-analysis/quantitative-analysis |
| Mass Hunter Qualitative Analysis | Agilent | https://www.agilent.com/en/product/software-informatics/mass-spectrometry-software/data-analysis/quantitative-analysis |
| Proteome Discoverer | Thermo Fisher Scientific | https://www.thermofisher.com/be/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis |
| Biorender | Science Suite Inc. | https://www.biorender.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact: Guido T Bommer, Metabolic Research Group, de Duve Institute & WELRI, Université Catholique de Louvain, 1200 Brussels, Belgium; guido.bommer@uclouvain.be.
Materials availability
All unique/stable reagents generated in this study are available from the lead contact without restriction.
Data and code availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD049174 and 10.6019/PXD049174. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
HAP1 cells
This adherent cell line (#C631, Horizon Discovery) and derived knockouts were cultured at 37°C, 5% CO2 in IMDM (Gibco) supplemented with 10% FBS (Dominique Dutscher). At 70–90% confluency, cells were passaged approximately once every 2 days, 1:3, by addition of Versene (Gibco) followed by 0.05% Trypsin (Gibco), centrifugation at 200 x g for 5 min, and resuspension in fresh medium.
HEK293T cells
This adherent cell line was cultured at 37°C, 5% CO2 in DMEM (Gibco) supplemented with 10% FBS (Dominique Dutscher). At 70–90% confluency, cells were passaged approximately once every 2 days, 1:3, by addition of Versene (Gibco) followed by 0.05% Trypsin (Gibco), centrifugation at 200 x g for 5 min, and resuspension in fresh medium.
EBV-immortalized lymphoblasts
These patient-derived cell lines were cultured in suspension at 37°C, 5% CO2 in DMEM/F-12 w/HEPES (Gibco) and passaged approximately 1:3 every 2 days, by centrifugation at 200 x g for 5 min, and resuspension in fresh medium. For details on the profiles of affected patients and controls from whom these cells were derived, see Tables 1 and S3.
Dermal fibroblasts
These adherent cell lines were cultured at 37°C, 5% CO2 in DMEM/F-12 (Gibco) supplemented with 10% FBS (Dominique Dutscher). At 70–90% confluency, cells were passaged approximately 1:3, by addition of Versene (Gibco) followed by 0.05% Trypsin (Gibco), centrifugation at 200 x g for 5 min, and resuspension in fresh medium. For details on the profiles of affected patients and controls from whom these cells were derived, please see Tables 1 and S3.
Yeast BY4741
Saccharomyces cerevisiae BY4741 control cells were acquired from Euroscarf, cultured on YPD (Bacto Yeast Extract (Gibco); Bacto Peptone (Gibco) and 2% glucose (Sigma-Aldrich)) plates or medium at 30°C. Medium was agitated at 200 RPM during growth.
Ethics statement
Written informed consent was obtained from the individuals involved in this study or their guardians. Research on human material was approved by the ethical committee of the University Hospital Leuven (Study number: S58358) or the ethical committee of the General University Hospital in Prague (18/21 GRANT AZV VES 2022 1. LF UK, 20.5.2021).
Method details
Genetic analyses
Patients 1 and 4 were investigated via exome and/or genome sequencing as part of a cohort of individuals with biomarkers indicative of CDG-I. Filtering and variant curation were prioritized on the basis of the analysis of known or predicted CDG-I associated genes. Patients 3 and 4 underwent quad genome sequencing as part of a research study of children with unexplained medical complexity.76 Sequencing methods, variant calling, and variant filtering/prioritization strategies were described previously.76 In all individuals, no other pathogenic or probably pathogenic candidate variants were identified. Inheritance of variants were also confirmed when DNA from family members was available. For RNA analysis, the RNeasy Mini kit (Qiagen) was used to isolate Total RNA prior to DNase treatment (Roche Diagnostics) to remove genomic DNA. Reverse transcription was performed using 2 μg purified total RNA with the First-Strand cDNA synthesis kit (Cytiva).
Clinical biochemical analyses
Transferrin N-glycosylation was analyzed by isoelectric focusing, capillary zone electrophoresis, immunoblotting or HPLC (Bruneel et al., 2020).
Human cell culture
Human fibroblasts were cultured in DMEM/F12 (Gibco, 21041-025) supplemented with 10% FBS (Clone III, HyClones) and antibiotics streptomycin (100 μg/mL), penicillin (100 U/mL), and amphotericin (0.5 μg/mL) at 37°C under 5% CO2. EBV-immortalized lymphoblasts, HEK293T cells and HAP1 cells were cultured identically but with DMEM/F12 w/HEPES (Gibco 11039-021), DMEM (Gibco, 41966-029) and IMDM (Gibco, 12440-053), respectively. Cells were passaged using versene (Gibco, 15040033) and trypsin (Gibco, 25300096).
CRISPR/Cas9 editing
HAP1 cells deficient in DHRSX were purchased from Horizon Discovery, United Kingdom. This line was produced from a wild-type HAP1 line (Horizon catalog number: C631) and contains a hemizygous 32bp deletion in exon 5 of DHRSX which was confirmed by Sanger sequencing (Horizon ref no. HZGHC007899c011). Lack of DHRSX protein was confirmed by immunoblotting. HAP1 cells deficient in SRD5A3 were produced by CRISPR/Cas9-editing. Two batches of wild-type (Horizon catalog number: C631) HAP1 cells were seeded in a 6-well plate to allow a confluency of 60–80% after 24 h, at which time they were transfected according to the lipofectamine 3000 (ThermoFisher) manufacturer’s instructions with two pSpCas9(BB)-2A-GFP (PX458) plasmids containing two different gRNA sequences targeted to alternate regions of the SRD5A3 gene, while minimizing possible off-target effects (gRNA_1: CACGACGTGAATCATGACAT; gRNA_2: CTAGCCACCAAGTTAAGTTG). After 24 h, cells were dissociated enzymatically and subjected to fluorescence-activated cell sorting (FACS) into 5 x 96-well plates using a BD Biosciences Influx flow cytometer, gated according to GFP signal and a 7AAD stain, in order to exclude dead cells. Several were selected and Sanger sequenced in order to confirm successful alteration of the wild-type sequence. The clone selected for further analysis contained a hemizygous 5bp deletion in SRD5A3 (c.467_471delTCATG; p.(Val156Aspfs∗62)).
Plasmid constructions
The plasmids used in this work were generated through NEBridge Golden Gate Assembly of PCR products. PCR products necessary for each respective plasmid construction (see key resources table) were used in a reaction containing 0.1 pmol of each fragment, 10 mmol/L of DTT, 1 mmol/L of ATP, 0.5μL (10U) of BsaIHF (NEB), 1μL of T7-DNA-Ligase(3000 U) and 1X Cutsmart buffer (NEB), in a final volume of 20μL. The assembly was carried out in a thermal cycler using the following program: 3 min at 37°C, 35 cycles of incubation for 2 min at 37°C and for 3 min at 25°C, 5 min of incubation at 50°C and 5 min of incubation at 80°C. Five microliters of the assembly reaction were transformed in `NEB 5α competent cells subcloning efficiency’ cells according to the manufacturer’s protocol. Colonies growing on selective plates were selected. Plasmid purification was performed using the Nucleospin plasmid kit (Macherey Nagel) following the manufacturer’s protocol. DNA concentrations were quantified with a nanospectrophotometer ND 1000 (NanoDrop). The constructs were checked by restriction enzyme digestion and separation by electrophoresis on 0.8% agarose gels in TBE 1X buffer using GelRed as a dye. Sanger sequencing of parts of the constructs was performed by Eurofins Genomics using specific oligonucleotides found in the key resources table. The recombinant plasmids pET15b DHRSX-N-His (NM_145177.3), pcDNA3.1(+) SRD5A3-N-His (NM_024592.5) and pcDNA3.1(+) DFG10-N-His (Human optimized cDNA) were purchased from GenScript Biotech (Rijswijk, Netherlands) and sequences were checked by Sanger sequencing.
Transfection of HEK293T cells
Transfection of wild-type HEK293T cells was performed using the Lipofectamine 3000 transfection reagent (Invitrogen, ref: L30000001). Briefly, cells were seeded in a 6-well plate to allow a confluency of 60–80% after 24 h, at which time they were transfected according to the manufacturer’s instructions with the pcDNA3.1(+)-N-6His plasmid vector containing respective cDNA sequences appended to an N-terminal 6 x His-tag (see key resources table). After a further 24 h, cells from one well were processed for immunoblotting to confirm successful overexpression of the target protein and the other five were processed for membrane preparation as described below.
Transfection of HAP1 cells
HAP1 cells were grown in 6 well plates to reach 70–80% confluence on the day of transfection in IMDM (Gibco) supplemented with 10% FBS (Dominique Dutscher). Cells were transfected using 0.5 μg of respective plasmid DNA, including a puromycin resistance gene, with 1.5 μL of TurboFectin (OriGene) in Opti-MEM (Gibco) for 24 h. The next day, medium was changed to IMDM (Gibco) supplemented with 10% FBS (Dominique Dutscher) and 1 μg/mL puromycin selective antibiotic to obtain and maintain a stable transfection state.
Recombinant lentiviruses production and infection of HAP1 cells
Recombinant lentiviruses were produced in HEK293T cells as previously described77 using the vectors pUB83 SRD5A3, pUB83 SRD5A3-N-His or pUB83 DHRSX. The primer sets SRD5A3_sense_BglII/SRD5A3_rev_bsrGI, SRD5A3_sense_BglII_HIS/SRD5A3_rev_bsrGI, or hDHRSX_sense_BglII_HIS/hDHRSX_rev_BsrGI, respectively, were used to PCR-amplify SRD5A3 or DHRSX from pcDNA3.1(+) SRD5A3-N-His or pET15b DHRSX-N-His using Phusion DNA polymerase (ThermoFisher scientific). The restriction enzymes BgIII and BsrGI were used for cloning into pUB83, a lentiviral expression vector based on the pLVX-PURO (Clontech) plasmid containing a CMV promoter, and sequences were verified by Sanger sequencing. Viruses were then used to infect control and SRD5A3 KO HAP1 cells. HAP1 cells were grown in 6 well plates to reach 60% confluence on the day of infection in IMDM. The medium was replaced by IMDM containing diluted recombinant lentiviruses (lentivirus:IMDM 1:4 V/V) in the presence of 4μg/mL polybrene. After 24 h of growth the medium was replaced by virus free IMDM containing 2 μg/mL puromycin.
Yeast culture
S. cerevisiae WT (BY4741) purchased from Euroscarf and dfg10 knockout yeast were pre-cultured in complete YPD medium (Bacto Yeast Extract, 1X Bacto Peptone, 2% glucose) overnight. Cultures of dfg10 knockout yeast were supplemented with the selection antibiotic Geneticin at 200 μg/mL. Yeast strains were cultured at a starting OD600 of 0.2 at 30°C while shaking at 200 rpm for 5–6 h until mid-log phase and a final OD600 of 0.8–1.0.
Yeast gene inactivation
All yeast strains were constructed from the WT BY4741 strain (Euroscarf). In order to inactivate dfg10, a deletion cassette was generated and cloned to obtain plasmid pDel-dfg10. This plasmid contains a 224bp fragment corresponding to bp −198 to +24 of dfg10 start codon, linked to a G418 selection marker and to a 306bp fragment corresponding to the sequence immediately downstream of dfg10 ORF. From pDel-dfg10, a 2129 bp fragment generated through digestion with PmeI containing the deletion cassette has been purified and used to inactivate dfg10 by yeast transformation of WT strains. Correct integration of the deletion cassette for selected clones was checked by PCR at both ends, using oligonucleotides DFG10-A located at −394bp from the start and DFG10-D located 523bp after the stop codon of dfg10 as well as divergent oligonucleotides kanB and KanC located inside the G418 marker.
Yeast competent cell preparation
The S. cerevisiae strain to be transformed was seeded in 10 mL of YPD media (BactoYeast Extract, 1X Bacto Peptone, 2% glucose) overnight at 30°C while shaking at 200 rpm. On the day after, pre-cultured yeast were seeded in 50 mL of fresh YPD media at a starting OD600 of 0.2 for 5 h or until an OD600 of 0.5–0.8 was reached. Cells were collected by a 5-min centrifugation at 3000 rpm at room temperature. Cells were washed once with 25 mL of sterile water, then centrifuged again for 5 min at 3000 rpm at room temperature. The supernatant was discarded and cells were washed with 10 mL of sterile SORB buffer (1 M Li-acetate, 1 M Tris-HCl pH 7.8, 0.5 M EDTA pH 8.0, 1 M sorbitol in water) followed by another 5-min centrifugation step at 3000 rpm at room temperature. The supernatant was discarded and cells were resuspended in 360 μL of sterile SORB buffer with 40 μL of SS-DNA (heat-denatured salmon sperm DNA at 10 mg/mL in water) and stored at −80°C.
Yeast transformation
Competent S. cerevisiae cells were thawed on ice and added to sterile 1.5 mL tubes containing 2 μg of plasmid DNA and six volumes of sterile PEG buffer (100 mmol/L LiOAc, 10 mmol/L Tris-HCl pH 8.0, 1 mmol/L EDTA/NaOH pH 8.0, 40% PEG 3350). The suspension was mixed without vortexing and incubated for 30 min at room temperature. 10 μL of DMSO were added and the cell suspension was incubated in a 42°C water bath for 15 min, followed by a 3-min centrifugation step at 2000 rpm at room temperature. The supernatant was discarded, and cells resuspended in 3 mL of YPD media (BactoYeast Extract, 1X Bacto Peptone, 2% glucose) for a 5-h incubation at 30°C at 200 rpm. The cell suspension was centrifuged at 3000 rpm for 5 min, and 2.8 mL of supernatant discarded while the remaining yeast cell suspension was spread on YPD selective (G418 (100 μg/mL) for selection of knockout strains; additional hygromycin B (600 μg/mL) resistance for subsequent transformation) agar plates to grow at 30°C for up to 3 days.
Immunoblotting of protein lysates from human cell lines
Protein lysates from fibroblasts, EBV-immortalized lymphoblasts, HAP1 cells and HEK293T cells were prepared by the addition of RIPA buffer (10 mmol/L Tris-HCl [pH 7.4], 150 mmol/L NaCl, 0.5% sodium deoxycholate, 0.1% SDS, and 1X cOmplete protease inhibitor cocktail [Sigma Aldrich]) at 4°C, mechanical lysis by passing 5 times through a sterile 27G needle and incubation for 30 min followed by centrifugation at 4°C (15,000 g, 30 min). The total protein concentration in the supernatant was calculated with the Pierce BCA protein assay kit (Thermo Fisher Scientific). Protein lysates (10–20 μg) were analyzed by SDS/PAGE and blotted onto a nitrocellulose membrane (Invitrogen, LC2000). Blocking was performed in 2–5% milk or bovine serum albumin (98022, Sigma-aldrich) with the relevant primary and secondary antibodies. Washing was performed with 1 x Tris-buffered saline solution (12498S, Bioké) with 0.1% Tween 20 (P1379, Sigma-Aldrich). Signal detection was performed by chemiluminescence using an Amersham ImageQuant 800 imager (Cytiva), and quantification was performed using ImageJ. All antibodies used for immunoblotting can be found in the key resources table.
Overexpression and Ni-NTA agarose purification of DHRSX in E. coli
pET15b plasmids containing the cDNA sequence of human DHRSX with a 6 x His tag appended to the N-terminus were initially transformed into One Shot TOP10 Chemically Competent E. coli (Life technologies, ref: C404003). Clones were selected and grown to prepare minipreps. Vectors were then transformed into BL21(DE3) E. coli, colonies were selected according to carbenicillin resistance and grown in 10mL LB medium (100 μg/mL carbenicillin) overnight at 37°C before dilution in 250mL LB medium (100 μg/mL carbenicillin) and further growth until an OD600 of 0.6 was reached. Isopropylthio-β-galactoside (IPTG) was then added to a final concentration of 1 mmol/L and the culture was grown for a further 5 h at 37°C before centrifugation at 4000 x g for 20 min to harvest cells. Native purification of 6 x His-tagged DHRSX was performed using the Ni-NTA fast start kit (Qiagen ref. 30600) according to manufacturer’s instructions.
Immunofluorescent labeling
HAP1 DHRSX KO, SRD5A3 KO and control cells were grown on glass coverslips in 12-well plates in IMDM (Gibco) supplemented with 10% FBS (Clone III, HyClone) under 5% CO2 atmosphere at 37°C until 80–90% confluency. The coverslips were then washed with DPBS (+/+) (Cytiva) and fixed with 4% paraformaldehyde in PBS pH 7.3 for 20 min at room temperature. Coverslips were washed 3 x with PBS and mounted in a wet chamber covered with DPBS, all steps were performed at room temperature. Cells were permeabilized with 0.5% Triton X-100 (Sigma-Aldrich) in DPBS for 15 min. Cells were incubated in a blocking buffer (DPBS with 2% FBS, 2% BSA and 0.2% gelatine) for 1 h. The primary antibody was diluted 1:100 in blocking buffer and cover slips incubated for 1 h, followed by three DPBS washing steps. The fluorescent secondary antibody (AlexaFluor, Invitrogen) was diluted 1:600 in blocking buffer and cover slips incubated for 1 h, followed by three DPBS washing steps. All antibodies used for immunofluorescence can be found in the key resources table. Cell nuclei were stained using DAPI (Thermo Scientific) at 5 μg/mL in DPBS for 15 min. Coverslips were mounted on glass slides with Mowiol and DABCO (Sigma-Aldrich). Images were acquired using an inverted Zeiss LSM780 confocal microscope. Contrast/brightness of individual channels were adjusted where necessary to show localization more clearly.
HAP1 cell metabolic radiolabeling
HAP1 cells at 90% confluency were pre-incubated in DMEM containing 10% of dialyzed FBS (BioWest) and 0.5 mmol/L of glucose for 45 min. Cells were then labeled with 100 μCi of [2-3H] mannose (American Radiolabeled Chemicals, Inc (ARC), Saint Louis, USA/Isobio, Fleurus, Belgium) for 1 h. After labeling, the cells were washed three times with DPBS and underwent biphasic sequential Folch extraction and purification of oligosaccharide moieties from the extracted glycoproteins, as previously published and shown in detail in Figure S6C.46 The purified oligosaccharides were analyzed with high-performance liquid chromatography (HPLC) using an amino-derived Asahipak NH2P-50 4E column (250 × 4.6 mm; Shodex, Showa Denko K.K. [SDK], Tokyo, Japan) with a solvent system of acetonitrile/water from 70:30 (v/v) to 50:50 (v/v) at a flow rate of 1 mL/min over 80 min. Radioactivity was detected by a B-RAM 5B (LabLogic, UK) on-line detector and the data were processed using the radiochromatography software LAURA 6 (LabLogic, UK).
Isoprenoid species and preparation of dolichal
Dolichol (# 9002000), polyprenol (# 9002100) and polyprenal (# 9002200) were purchased from Avanti Polar Lipid. Dolichal was synthesized by oxidizing dolichol in the presence of pyridinium dichromate78 (# 214698-100G, Merck life science BV): 300 mL of 1 mg/mL dolichol in chloroform was dried down and resuspended in 200 mL of dichloromethane containing 10 mg/mL of pyridinium dichromate. After shaking at 1000 rpm for 15 min, the preparation was incubated overnight at 20°C. The mixture was centrifuged and the supernatant was transferred to a new tube. Several cycles of back-extractions were performed by using 200 mL methanol/water (5:3) (MS-grade, Biosolve) to remove pyridinium dichromate until the preparation became translucent. The quality of dolichal synthesis was assessed by LC-MS, showing a high yield of conversion (>95%). Dolichal was stored at −80°C for future enzymatic assays. All isoprenoid solutions represent mixtures with 13–21 isoprene units (with 18 being the most abundant species). Molar concentrations were calculated based on the distribution of chain lengths. Thus, a 5 μg/mL solution corresponds to approximately 4 μM.
Sample preparation and extraction of metabolites from HAP1, fibroblast and EBV-lymphoblast cells
For adherent cells (i.e., HAP1 and fibroblasts), the medium was removed, plates were rapidly washed with ice-cold water and plunged in liquid nitrogen to quench metabolic activity. The frozen dishes were placed on dry ice, and 500 μL ice-cold methanol was immediately added, followed by 300 μL of ice-cold water. The cells were scraped and collected in 2 mL tubes containing 1 mL of chloroform (MS-grade, Biosolve). For isoprenoid extraction from EBV-lymphoblasts, ∼3 × 106 cells were used for the extraction of metabolites. First, cells were incubated on ice for 5 min to cool down and slow down the metabolism, followed by centrifugation at 1200 rpm for 5 min at 4°C. The medium was removed and cells were washed by centrifugation with ice-cold PBS. After a second centrifugation, 800 μL of methanol/water (5:3) was added to the cells followed by a vigorous shaking for 2 min, then transferred in 2 mL tubes containing 1 mL of chloroform. The 2 mL tubes containing lysates of HAP1 cells, fibroblasts or EBV-lymphoblasts were vigorously mixed at 2000 rpm for 20 min at 4°C, followed by a centrifugation at 16000 x g for 30 min at 4°C. The lower layer, containing hydrophobic metabolites, was dried down under a gentle stream of nitrogen and resuspended in 100 μL of methanol/isopropanol (1:1) before LC-MS analysis.
Sample preparation and extraction of metabolites from yeast
S. cerevisiae WT, dfg10 KO, and dfg10 KO + WT dfg10 strains were cultured overnight in YPD medium at 30°C and 200 rpm. OD600 was measured and the cultures diluted to an OD of 1.2 mL were then centrifuged at 13200 rpm at 4°C. The pellet was washed with ice-cold PBS, centrifuged, resuspended in ice-cold methanol/water (5:3) and homogenized using ceramic beads (ten 1.4 mm and five 2.8 mm beads per tube; PerkinElmer) at 6000 rpm using Precellys Evolution homogenizer (Bertin Technologies). The program used consisted of six cycles of homogenization (20 s ON and 10 s OFF for each cycle). 1 mL of chloroform was added to the yeast homogenate, followed by a vigorous shaking for 20 min at 2000 rpm and at 4°C, and by a centrifugation at 13200 rpm for 30 min at 4°C. The lower organic layer was dried down under a gentle stream of nitrogen and resuspended in 100 μL of methanol/isopropanol (1:1) for LC-MS analysis.
Dimethylation of isoprenoid phosphates using trimethylsilyl diazomethane (TMSD)
The dimethylation of dolichol phosphate and polyprenol phosphate was performed as described in Kale et al.32 The organic fraction containing isoprenoid species was completely dried and resuspended in 200 μL of dichloromethane:methanol (6.5:5.2, v/v). 10 μL of 2 mol/L TMSD (in hexane) was added and incubated for 40 min at room temperature. The incubation was halted by adding 1 μL of acetic acid. Samples were then dried and dissolved in methanol:isopropanol 1:1 (V/V) for LC-MS analysis.
Extraction of membrane proteins from HAP1, EBV-lymphoblasts or HEK293 cells
For adherent cells, confluent 10 cm plates were washed with ice-cold PBS and cells were collected in 0.5 mL/plate of lysis buffer (25 mmol/L HEPES pH 7.5, 2 μg/mL antipain and leupeptin, 0.5 mmol/L PMSF and 10% glycerol) by scraping. For EBV-lymphoblasts growing in suspension, 3 million cells were centrifuged at 500 g for 5 min, and the supernatant was removed before proceeding. Cells were then lysed in 500 μL of lysis buffer, subjected to 2 cycles of freezing-thawing in liquid nitrogen, treated with DNAse I (0.1 mg/mL in 10 mmol/L MgSO4) for 15 min and centrifuged (16000 x g for 15 min at 4°C) to recover a membrane fraction in the pellet. The resulting pellet was washed twice by centrifugation as above with the same volume of lysis buffer and the resulting pellet was resuspended in the initial volume of lysis buffer. This preparation was aliquoted and kept at −80°C.
Measurement of DHRSX activity in membrane protein extracts of HAP1 and EBV-transformed lymphoblastoid cells
Enzymatic activity measurements were performed in buffer containing 10 mmol/L HEPES, pH 8.0, 10 mmol/L KCl, 0.3% Triton X-100, 0.5 mmol/L β-mercaptoethanol, 1% phosphatidylcholine (PC), and 0.2% phosphatidylethanolamine (PE). To assess dehydrogenase or reductase activities, 5 μg/mL of polyprenol, polyprenal or dolichal were used in the presence of 5 mmol/L of NAD(H)+ or NADP(H)+. First, 5 μL of a 50 μg/mL solution of the respective polyisoprenoid mixtures in chloroform was added into an empty tube, and dried under a stream of nitrogen. After that, 5 μL of 10X buffer [100 mmol/L HEPES, pH 8.0, 100 mmol/L KCl, 3% Triton X-100, 0.5 mmol/L β-mercaptoethanol, 10% phosphatidylcholine (PC), and 2% phosphatidylethanolamine (PE)] were added to the tube. 5 μL of NAD(P)+ or NAD(P)H (50 mM) was then added. The incubations were started by adding protein extracts at a final concentration of 1 mg/mL in a total volume of 50 μL. Incubation was performed at 37°C for 2h. The assays with EBV-transformed lymphoblast membrane protein preparations (1 mg/mL) were carried out in the same conditions but only incubations with polyprenal and 5 mmol/L NADH or NADPH were performed. Samples were analyzed by LC-MS after methanol-chloroform extraction of analytes.
Analysis of recombinant DHRSX activity on polyprenal, dolichal, polyprenol or dolichol
DHRSX activity time course measurements were performed in a buffer containing 10 mmol/L HEPES pH 8.0, 10 mmol/L KCl, 0.5 mmol/L β-mercaptoethanol, 1% PC, and 0.2% PE. 5 μg/mL of lipid substrates were used in the presence of 0, 5, 20, 100, or 1000 μmol/L of NAD(P)++ or NAD(P)H. The assay was started by addition of 0.075 μmol/L recombinant DHRSX and carried out at 37°C in a total volume of 300 μL. Subsequently, five aliquots of 50 μL were taken from this reaction at different timepoints, and immediately quenched by methanol-chloroform extraction. To determine kinetic properties of recombinant DHRSX, the incubation time was reduced to 5 min in the presence of 0.00375 μmol/L enzyme. Non-linear curve fitting with GraphPad prism was used assuming a Michaelis Menten kinetic.
To assess the inhibitory effect of NADPH on the Polyprenol dehydrogenase, or NAD+ on the Dolichal reductase activities of DHRSX, time points were chosen based on the results from the time-course experiments. Thus, polyprenol dehydrogenase activity was assessed in 15 min incubations, whereas 3 min incubations were used to assess dolichal reductase activity.
Measurement of SRD5A3/DFG10 activity in HEK293T membrane protein extracts
Enzymatic activity measurements were performed in a buffer containing 10 mmol/L HEPES pH 8.0, 10 mmol/L KCl, 0.3% Triton X-100, 0.5 mmol/L β-mercaptoethanol, 1% PC, and 0.2% PE. 5 μg/mL of polyprenal or polyprenol were used in the presence of 5 mmol/L of NADH or NADPH. The incubation was started by addition of 0.3 mg/mL of membrane protein preparations in a final volume of 50 μL. The incubation was performed at 37°C for 2h. Samples were analyzed by LC-MS after methanol-chloroform extraction. To determine kinetic properties of SRD5A3, the incubation time was reduced to 30 min using 1 mg/mL membrane preparations from SRD5A3-overexpressing DHRSX KO HAP1 cells. Cellular concentrations of SRD5A3 were determined by comparing the signal of His-tagged SRD5A3 with the signal of six different purified His-tagged proteins in western blot. Non-linear curve fitting with Graphpad prism was used assuming a Michaelis Menten kinetic model.
Sample preparation and extraction of metabolites from enzymatic assays
After the end of each respective incubation, 450 μL of ice-cold methanol/water (5:3) was added into the tube followed by 550 μL of ice-cold chloroform. The mixture was vigorously mixed at 2000 rpm for 20 min and at 4°C, followed by a centrifugation at 13200 rpm for 10 min at 4°C. The lower layer containing hydrophobic metabolites was dried down under a gentle stream of nitrogen and resuspended in 50 μL methanol/isopropanol (1:1) before LC-MS analysis.
LC-MS analysis of metabolites
LC-MS analysis of organic fractions obtained from cells (or enzymatic assays) was carried out using a method adapted from that used by Dewulf et al. 2019.79 Briefly, 5 μL of sample was injected and subjected to reverse phase chromatography with an Accucore C30 150 × 2.1 mm column (ref. 27826–152130, ThermoFisher), operated at 45°C on an Agilent 1290 HPLC system. The flow rate was constant at 0.2 mL/min using mobile phase A (60% acetonitrile, and 40% water, 10 mmol/L ammonium formate, and 0.1% formic acid) and B (90% isopropanol, 10% acetonitrile, 10 mmol/L ammonium formate, and 0.1% formic acid (Biosolve)). An Agilent 6546 ion funnel mass spectrometer was used in the positive or negative ionization modes with an electrospray ionization (ESI) (voltage 3500 V, Nozzle voltage 1000V, sheath gas 350 °C at 11 L/min, nebulizer pressure 35 psi and drying gas 300 °C at 8 L/min). Starting from 5 min onwards, one spectrum encompassing a range of 69–1700 m/z was acquired per second, generated from 10772 transients. The mass spectrometer was operated in positive mode for the detection of dolichal, polyprenal, dolichol, polyprenol, dimethylated dolichol-P, and dimethylated polyprenol-P. For the elution, the solvent gradient was: 0–5 min at 90% B; 5–33 min from 90 to 97% B; 33–34 min from 97 to 99% B; 34–35 min from 99 to 90% B. The negative mode was used to detect and measure polyprenoic acid, dolichol-P-hexose, and polyprenol-P-hexose. The elution gradient consisted of: 0–3 min at 30% B; 3–8 min from 30 to 43% B; 8–9 min from 43 to 50% B; 9–18 min from 50 to 90% B; 18–26 min from 90 to 99% B; 26–30 min at 99% B; 30–30.1 min from 99 to 30% B; 30.1–35 min at 30% B. The different theoretical m/z values of [M + NH4+] and [M - H+] ions are given in Table S1. To avoid confounding effects of high levels of polyprenal M+4, the M+2 isotopologue of dolichol was used for quantification in Figures S4E, S4F, and S4G. The resulting data were analyzed and processed by the software Agilent MassHunter Qualitative Analysis 10.0 for the identification and the visualization of peaks/metabolites. The quantification was performed by Agilent MassHunter Quantitative Analysis software (Agilent Technologies, CA, USA).
Proteomic analysis
100 μg of membrane preparations were resuspended in 23 μL of a solution containing 5% SDS, 50 mmol/L Tris pH 8.5 and 5 mmol/L DTT. After 15 min at 55°C, chloroacetamide was added to a final concentration of 20 mmol/L followed by another 10 min at 55°C. Samples where then acidified by addition of phosphoric acid to a final concentration of 2.5%, and transferred onto a micro S-Trap column (Protifi LLC, USA). Digestion was performed at 37°C overnight by adding 1:100 of trypsin and LysC.
Digested peptides were eluted in three steps using 40 μL each of 50 mmol/L Tris pH 8.5, 0.2% formic acid and 50% acetonitrile. The eluted peptides were dried down in a vacuum concentrator (SpeedVac, Thermo Scientific) and resuspended in 2% acetonitrile and 0.2% formic acid. Peptide concentration was determined by Pierce Quantitative Peptide Assay (Thermo Scientific). Peptides were directly loaded onto reversed-phase trap-column (Acclaim PepMap 100, Thermo Scientific) and eluted in backflush mode. Peptide separation was performed using a reversed-phase analytical column (EasySpray, 0.075 × 250 mm,Thermo Scientific) with solvent A (0.1% formic acid) and a linear gradient of 4%–27.5% solvent B (0.1% formic acid in 98% acetonitrile) for 100 min, 27.5%–40% solvent B for 10 min, 40%–95% solvent B for 10 min at a constant flow rate of 300 nL/min on a Vanquish Neo HPLC system. The peptides were analyzed by an Orbitrap Fusion Lumos tribrid mass spectrometer with an ESI source (ThermoFisher Scientific) in positive mode coupled online to the nano-LC. Peptides were detected in the Orbitrap at a resolution of 120,000. Peptides were selected for MS/MS using the HCD setting at 30; ion fragments were detected in the Orbitrap at a resolution of 30 000. A data-dependent procedure was used with a cycle time of 3s alternating between one MS scan and multiple MS/MS scans for ions above a threshold ion count of 3.8E4 in the MS survey scan with 30.0s dynamic exclusion. The electrospray voltage applied was 2.1 kV MS1 spectra were obtained with an AGC target of 400000 and a maximum injection time set to custom, and MS2 spectra were acquired with an AGC target of 10000 and a maximum injection time set to custom. For MS scans, the m/z scan range was 350–1800.
The resulting MS/MS data were processed using the Sequest HT search engine within Proteome Discoverer 2.5 SP1 against a Homo sapiens protein database obtained from Uniprot (sp_canonical TaxID = 9606). Trypsin was specified as cleavage enzyme allowing up to 2 missed cleavages, 4 modifications per peptide and up to 7 charges. The mass error was set to 20 ppm for precursor ions and 0.05 Da for fragment ions. Oxidation on Met (+15.995 Da), Hex(1)HexNAc(2) (+568.212 Da), Hex(2)HexNAc(2) (+730.264 Da), Hex(3)HexNAc(2) (+892.317 Da), Hex(4)HexNAc(2) (+1054.370 Da), Hex(5)HexNAc(2) (+1216.423) Da Hex(6)HexNAc(2) (+1378.476 Da), Hex(7)HexNAc(2) (+1540.529 Da), Hex(8)HexNAc(2) (+1702.581 Da), Hex(9)HexNAc(2) (+1864.634 Da), Hex(10)HexNAc(2) (+2026.687 Da) on Asparagine, and Methionine loss (−131.040 Da) on the N-terminus of the protein and peptides were considered as variable modifications, whereas carbamidomethylation of cysteine was considered as a fixed modification. The False discovery rate (FDR) was assessed using Percolator and thresholds for the identification of proteins, peptides and modification sites were specified at 1%. Label-free quantification of peptides is based on the precursor ion intensity. Signals were normalized to the sum of all signals within each individual sample. Protein abundances were calculated as the sum of the abundances of unmodified peptides. Signals obtained for modified peptides were normalized to the abundance of the parent protein. To facilitate visualization in a heatmap, data were subsequently normalized to the median of abundance across samples.
Quantification and statistical analysis
All analyses were carried out via GraphPad Prism version 9.20 for Mac (GraphPad Software, La Jolla, CA). Statistical analysis was performed by a student’s T-test or one-way ANOVA followed by Dunnett’s multiple comparisons test. The graphical abstract and Figure S6C were generated with Biorender.
Acknowledgments
We thank the families, patients, and healthcare providers, specifically all staff affiliated with the Division of Clinical & Metabolic Genetics, the SickKids Complex Care Program, and The Center for Applied Genomics. We thank Marie-Line Jacquemont and Marta Spodenkiewicz of CHU La Réunion for referring samples of patient 2 and clinical reports; Wim Huybrechts and the cell culture facility at the Center for Human Genetics, Leuven, Belgium; Julie Graff, UCLouvain, for technical assistance; Ramon Duran-Romaña, Switch Laboratory, VIB-KU Leuven, for assistance with 3D-modelling; Liesbeth Keldermans and Erika Souche, Center for Human Genetics, Leuven, for assistance with whole genome analysis of patient 1; Paul Van Veldhoven, KU Leuven, for comments on the manuscript, and Didier Vertommen, UCLouvain, for help with proteomics data analysis. Flow cytometry was carried out at the KU Leuven Flow and Mass Cytometry Facility.
Funding: ERA-Net for Research on Rare Diseases (ERARE118-117 - EUROGLYCAN-omics) to G.M., F.F., E.V.S., H.H.); the French National Agency (ENIGMncA project, ANR-21-CE14-0049-01) to F.F.; the CNRS IRP GLYCOCDG project to F.F; an FWO senior postdoctoral fellowship (Project ID: 1289023N to M.P.W.); Marie Curie Individual Fellowship (H2020- MSCA-IF-2019; project ID: 894669 to M.P.W.); the Jaeken-Theunissen CDG Fund; Mizutani Foundation for Glycoscience (Grant 210119 to G.M. and 240097 to G.T.B.); CELSA fund grant (CELSA/21/027) to G.M. and H.H., KU Leuven Global PhD Partnership (to C.R.A., F.F., and G.M. 3M200250). WELBIO 2019, (to G.T.B.), Fondation Médicale Reine Elisabeth, FNRS equipment grant UN06220F and research credit J.0016.23, ARC UCLouvain ARC17/22-079 and ERC consolidator grant #771704 (all to G.T.B.); FWO-FNRS WEAVE program (G061524N to G.T.B., M.P.W., and G.M.), Fonds Baillet Latour (to G.T.B. and E.V.S.), AZV MZ ČR NU22-07-00474 (H.H.)], SickKids Research Institute and University of Toronto McLaughlin Centre (G.C.).
Author contributions
E.V.S., G.M., F.F. and G.T.B. conceived and supervised this study. M.P.W., T.K., C.R.A., G.d.B., H.H., J.J., V.C., E.V.S., G.M., F.F. and G.T.B. designed experiments and analyses. M.P.W., T.K., C.R.A., C.S., G.d.B., D.C., G.M.C., L.R., F.C., M.B., and J.P.D. performed experimental work, in silico analyses, or method optimizations. L.C., B.B., G.Y., G.C., S.V.-B., D.R., and J.J. provided clinical information on affected patients. M.P.W., G.Y., G.C., and S.V.-B. conducted next generation sequencing analysis. M.P.W., T.K., C.R.A., J.J., E.V.S., G.M., F.F., and G.T.B. wrote and prepared the manuscript. All authors proofread and corrected the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 30, 2024; Corrected online: June 10, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2024.04.041.
Contributor Information
Emile Van Schaftingen, Email: emile.vanschaftingen@uclouvain.be.
Gert Matthijs, Email: gert.matthijs@kuleuven.be.
François Foulquier, Email: francois.foulquier@univ-lille.fr.
Guido T. Bommer, Email: guido.bommer@uclouvain.be.
Supplemental information
References
- 1.Aebi M. N-linked protein glycosylation in the ER. Biochim. Biophys. Acta. 2013;1833:2430–2437. doi: 10.1016/j.bbamcr.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Denecke J., Kranz C. Hypoglycosylation due to dolichol metabolism defects. Biochim. Biophys. Acta. 2009;1792:888–895. doi: 10.1016/J.BBADIS.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 3.Shridas P., Rush J.S., Waechter C.J. Identification and characterization of a cDNA encoding a long-chain cis-isoprenyltranferase involved in dolichyl monophosphate biosynthesis in the ER of brain cells. Biochem. Biophys. Res. Commun. 2003;312:1349–1356. doi: 10.1016/j.bbrc.2003.11.065. [DOI] [PubMed] [Google Scholar]
- 4.Cantagrel V., Lefeber D.J., Ng B.G., Guan Z., Silhavy J.L., Bielas S.L., Lehle L., Hombauer H., Adamowicz M., Swiezewska E., et al. SRD5A3 Is Required for Converting Polyprenol to Dolichol and Is Mutated in a Congenital Glycosylation Disorder. Cell. 2010;142:203–217. doi: 10.1016/j.cell.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cantagrel V., Lefeber D.J. From glycosylation disorders to dolichol biosynthesis defects: a new class of metabolic diseases. J. Inherit. Metab. Dis. 2011;34:859–867. doi: 10.1007/S10545-011-9301-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grabińska K.A., Park E.J., Sessa W.C. cis-Prenyltransferase: New Insights into Protein Glycosylation, Rubber Synthesis, and Human Diseases. J. Biol. Chem. 2016;291:18582–18590. doi: 10.1074/JBC.R116.739490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burda P., Aebi M. The dolichol pathway of N-linked glycosylation. Biochim. Biophys. Acta. 1999;1426:239–257. doi: 10.1016/S0304-4165(98)00127-5. [DOI] [PubMed] [Google Scholar]
- 8.Lehle L., Strahl S., Tanner W. Protein glycosylation, conserved from yeast to man: a model organism helps elucidate congenital human diseases. Angew. Chem. Int. Ed. Engl. 2006;45:6802–6818. doi: 10.1002/ANIE.200601645. [DOI] [PubMed] [Google Scholar]
- 9.Stiles A.R., Russell D.W. SRD5A3: A surprising role in glycosylation. Cell. 2010;142:196–198. doi: 10.1016/J.CELL.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sagami H., Igarashi Y., Tateyama S., Ogura K., Roos J., Lennarz W.J. Enzymatic formation of dehydrodolichal and dolichal, new products related to yeast dolichol biosynthesis. J. Biol. Chem. 1996;271:9560–9566. doi: 10.1074/jbc.271.16.9560. [DOI] [PubMed] [Google Scholar]
- 11.Ondruskova N., Cechova A., Hansikova H., Honzik T., Jaeken J. Congenital Disorders of Glycosylation: Still “Hot” in 2020. Biochim Biophys Act Gen Subj. 2021;1865:129751. doi: 10.1016/j.bbagen.2020.129751. [DOI] [PubMed] [Google Scholar]
- 12.Park E.J., Grabińska K.A., Guan Z., Stránecký V., Hartmannová H., Hodaňová K., Barešová V., Sovová J., Jozsef L., Ondrušková N., et al. Mutation of Nogo-B receptor, a subunit of cis-prenyltransferase, causes a congenital disorder of glycosylation. Cell Metab. 2014;20:448–457. doi: 10.1016/j.cmet.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabry S., Vuillaumier-Barrot S., Mintet E., Fasseu M., Valayannopoulos V., Héron D., Dorison N., Mignot C., Seta N., Chantret I., et al. A case of fatal Type I congenital disorders of glycosylation (CDG I) associated with low dehydrodolichol diphosphate synthase (DHDDS) activity. Orphanet J. Rare Dis. 2016;11 doi: 10.1186/S13023-016-0468-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morava E., Wevers R.A., Cantagrel V., Hoefsloot L.H., Al-Gazali L., Schoots J., Van Rooij A., Huijben K., Van Ravenswaaij-Arts C.M.A., Jongmans M.C.J., et al. A novel cerebello-ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism. Brain. 2010;133:3210–3220. doi: 10.1093/BRAIN/AWQ261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rappold G.A. The pseudoautosomal regions of the human sex chromosomes. Hum. Genet. 1993;92:315–324. doi: 10.1007/BF01247327. [DOI] [PubMed] [Google Scholar]
- 16.Bruneel A., Cholet S., Tran N.T., Mai T.D., Fenaille F. CDG biochemical screening: Where do we stand? Biochim. Biophys. Acta. Gen. Subj. 2020;1864 doi: 10.1016/j.bbagen.2020.129652. [DOI] [PubMed] [Google Scholar]
- 17.Zhang G., Luo Y., Li G., Wang L., Na D., Wu X., Zhang Y., Mo X., Wang L. DHRSX, a novel non-classical secretory protein associated with starvation induced autophagy. Int. J. Med. Sci. 2014;11:962–970. doi: 10.7150/ijms.9529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hinch A.G., Altemose N., Noor N., Donnelly P., Myers S.R. Recombination in the human Pseudoautosomal region PAR1. PLoS Genet. 2014;10 doi: 10.1371/JOURNAL.PGEN.1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellis N., Goodfellow P.N. The mammalian pseudoautosomal region. Trends Genet. 1989;5:406–410. doi: 10.1016/0168-9525(89)90199-6. [DOI] [PubMed] [Google Scholar]
- 20.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varadi M., Anyango S., Deshpande M., Nair S., Natassia C., Yordanova G., Yuan D., Stroe O., Wood G., Laydon A., et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022;50:D439–D444. doi: 10.1093/NAR/GKAB1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akdel M., Pires D.E.V., Pardo E.P., Jänes J., Zalevsky A.O., Mészáros B., Bryant P., Good L.L., Laskowski R.A., Pozzati G., et al. A structural biology community assessment of AlphaFold2 applications. Nat. Struct. Mol. Biol. 2022;29:1056–1067. doi: 10.1038/s41594-022-00849-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hekkelman M.L., de Vries I., Joosten R.P., Perrakis A. AlphaFill: enriching AlphaFold models with ligands and cofactors. Nat. Methods. 2023:205–213. doi: 10.1038/s41592-022-01685-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hua Y.H., Wu C.Y., Sargsyan K., Lim C. Sequence-motif Detection of NAD(P)-binding Proteins: Discovery of a Unique Antibacterial Drug Target. Sci. Rep. 2014:6471–6477. doi: 10.1038/srep06471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Llargués-Sistac G., Bonjoch L., Castellvi-Bel S. HAP1, a new revolutionary cell model for gene editing using CRISPR-Cas9. Front. Cell Dev. Biol. 2023;11 doi: 10.3389/FCELL.2023.1111488/FULL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foulquier F., Amyere M., Jaeken J., Zeevaert R., Schollen E., Race V., Bammens R., Morelle W., Rosnoblet C., Legrand D., et al. TMEM165 deficiency causes a congenital disorder of glycosylation. Am. J. Hum. Genet. 2012;91:15–26. doi: 10.1016/j.ajhg.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morelle W., Potelle S., Witters P., Wong S., Climer L., Lupashin V., Matthijs G., Gadomski T., Jaeken J., Cassiman D., et al. Galactose supplementation in patients with TMEM165-CDG rescues the glycosylation defects. J. Clin. Endocrinol. Metab. 2017;102:1375–1386. doi: 10.1210/jc.2016-3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mejhert N., Gabriel K.R., Frendo-Cumbo S., Krahmer N., Song J., Kuruvilla L., Chitraju C., Boland S., Jang D.K., von Grotthuss M., et al. The Lipid Droplet Knowledge Portal: A resource for systematic analyses of lipid droplet biology. Dev. Cell. 2022;57:387–397.e4. doi: 10.1016/J.DEVCEL.2022.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenwald A.G., Krag S.S. Lec9 CHO glycosylation mutants are defective in the synthesis of dolichol. J. Lipid Res. 1990;31:523–533. doi: 10.1016/s0022-2275(20)43174-8. [DOI] [PubMed] [Google Scholar]
- 30.Könning D., Olbrisch T., Sypaseuth F.D., Tzschucke C.C., Christmann M. Oxidation of allylic and benzylic alcohols to aldehydes and carboxylic acids. Chem. Commun. 2014;50:5014–5016. doi: 10.1039/C4CC01305K. [DOI] [PubMed] [Google Scholar]
- 31.Gründahl J.E.H., Guan Z., Rust S., Reunert J., Müller B., Du Chesne I., Zerres K., Rudnik-Schöneborn S., Ortiz-Brüchle N., Häusler M.G., et al. Life with too much polyprenol: polyprenol reductase deficiency. Mol. Genet. Metab. 2012;105:642–651. doi: 10.1016/J.YMGME.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kale D., Kikul F., Phapale P., Beedgen L., Thiel C., Brügger B. Quantification of Dolichyl Phosphates Using Phosphate Methylation and Reverse-Phase Liquid Chromatography-High Resolution Mass Spectrometry. Anal. Chem. 2023;95:3210–3217. doi: 10.1021/ACS.ANALCHEM.2C03623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veech R.L., Eggleston L.V., Krebs H.A. The redox state of free nicotinamide-adenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem. J. 1969;115:609–619. doi: 10.1042/BJ1150609A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cantó C., Menzies K.J., Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/J.CMET.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hall C.W., McLachlan K.R., Krag S.S., Robbins A.R. Reduced utilization of Man5GlcNAc2-P-P-lipid in a Lec9 mutant of Chinese hamster ovary cells: analysis of the steps in oligosaccharide-lipid assembly. J. Cell. Biochem. 1997;67:201–215. [PubMed] [Google Scholar]
- 36.Lu H., Sathe A.A., Xing C., Lehrman M.A. The Lec5 glycosylation mutant links homeobox genes with cholesterol and lipid-linked oligosaccharides. Glycobiology. 2019;29:106–109. doi: 10.1093/GLYCOB/CWY103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaiden A., Rosenwald A.G., Cacan R., Verbert A., Krag S.S. Transfer of two oligosaccharides to protein in a Chinese hamster ovary cell B211 which utilizes polyprenol for its N-linked glycosylation intermediates. Arch. Biochem. Biophys. 1998;358:303–312. doi: 10.1006/ABBI.1998.0839. [DOI] [PubMed] [Google Scholar]
- 38.Beck P.J., Gething M.J., Sambrook J., Lehrman M.A. Complementing mutant alleles define three loci involved in mannosylation of Man5-GlcNAc2-P-P-dolichol in Chinese hamster ovary cells. Somat. Cell Mol. Genet. 1990;16:539–548. doi: 10.1007/BF01233094. [DOI] [PubMed] [Google Scholar]
- 39.Hall C.W., Robbins A.R., Krag S.S. Preliminary characterization of a Chinese hamster ovary cell glycosylation mutant isolated by screening for low intracellular lysosomal enzyme activity. Mol. Cell. Biochem. 1986;72:35–45. doi: 10.1007/BF00230634. [DOI] [PubMed] [Google Scholar]
- 40.Keller R.K., Rottler G.D., Cafmeyer N., Adair W.L., Jr. Subcellular localization and substrate specificity of dolichol kinase from rat liver. Biochim. Biophys. Acta. 1982;719:118–125. doi: 10.1016/0304-4165(82)90315-4. [DOI] [PubMed] [Google Scholar]
- 41.McLachlan K.R., Krag S.S. Three enzymes involved in oligosaccharide-lipid assembly in Chinese hamster ovary cells differ in lipid substrate preference. J. Lipid Res. 1994;35:1861–1868. [PubMed] [Google Scholar]
- 42.D’Souza-Schorey C., McLachlan K.R., Krag S.S., Elbein A.D. Mammalian glycosyltransferases prefer glycosyl phosphoryl dolichols rather than glycosyl phosphoryl polyprenols as substrates for oligosaccharyl synthesis. Arch. Biochem. Biophys. 1994;308:497–503. doi: 10.1006/abbi.1994.1070. [DOI] [PubMed] [Google Scholar]
- 43.Palamarczyk G., Lehle L., Mankowski T., Chojnacki T., Tanner W. Specificity of solubilized yeast glycosyl transferases for polyprenyl derivatives. Eur. J. Biochem. 1980;105:517–523. doi: 10.1111/J.1432-1033.1980.TB04527.X. [DOI] [PubMed] [Google Scholar]
- 44.McLachlan K.R., Krag S.S. Substrate specificity of N-acetylglucosamine 1-phosphate transferase activity in Chinese hamster ovary cells. Glycobiology. 1992;2:313–319. doi: 10.1093/GLYCOB/2.4.313. [DOI] [PubMed] [Google Scholar]
- 45.Polla D.L., Edmondson A.C., Duvet S., March M.E., Sousa A.B., Lehman A., CAUSES Study. Niyazov D., van Dijk F., Demirdas S., et al. Bi-allelic variants in the ER quality-control mannosidase gene EDEM3 cause a congenital disorder of glycosylation. Am. J. Hum. Genet. 2021;108:1342–1349. doi: 10.1016/J.AJHG.2021.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Péanne R., Vanbeselaere J., Vicogne D., Mir A.M., Biot C., Matthijs G., Guérardel Y., Foulquier F. Assessing ER and Golgi N-glycosylation process using metabolic labeling in mammalian cultured cells. Methods Cell Biol. 2013;118:157–176. doi: 10.1016/B978-0-12-417164-0.00010-0. [DOI] [PubMed] [Google Scholar]
- 47.Schenk B., Imbach T., Frank C.G., Grubenmann C.E., Raymond G.V., Hurvitz H., Korn-Lubetzki I., Revel-Vik S., Raas-Rotschild A., Luder A.S., et al. MPDU1 mutations underlie a novel human congenital disorder of glycosylation, designated type If. J. Clin. Invest. 2001;108:1687–1695. doi: 10.1172/JCI13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kranz C., Denecke J., Lehrman M.A., Ray S., Kienz P., Kreissel G., Sagi D., Peter-Katalinic J., Freeze H.H., Schmid T., et al. A mutation in the human MPDU1 gene causes congenital disorder of glycosylation type If (CDG-If) J. Clin. Invest. 2001;108:1613–1619. doi: 10.1172/JCI13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wheeler P.G., Ng B.G., Sanford L., Sutton V.R., Bartholomew D.W., Pastore M.T., Bamshad M.J., Kircher M., Buckingham K.J., Nickerson D.A., et al. SRD5A3-CDG: Expanding the phenotype of a congenital disorder of glycosylation with emphasis on adult onset features. Am. J. Med. Genet. 2016;170:3165–3171. doi: 10.1002/AJMG.A.37875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buczkowska A., Swiezewska E., Lefeber D.J. Genetic defects in dolichol metabolism. J. Inherit. Metab. Dis. 2015;38:157–169. doi: 10.1007/s10545-014-9760-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao Q., Wang L., Supekar S., Shen T., Liu H., Ye F., Huang J., Fan H., Wei Z., Zhang C. Structure of human steroid 5α-reductase 2 with the anti-androgen drug finasteride. Nat. Commun. 2020:5430–5511. doi: 10.1038/s41467-020-19249-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Han Y., Zhuang Q., Sun B., Lv W., Wang S., Xiao Q., Pang B., Zhou Y., Wang F., Chi P., et al. Crystal structure of steroid reductase SRD5A reveals conserved steroid reduction mechanism. Nat. Commun. 2021;12 doi: 10.1038/S41467-020-20675-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hori T., Yokomizo T., Ago H., Sugahara M., Ueno G., Yamamoto M., Kumasaka T., Shimizu T., Miyano M. Structural basis of leukotriene B4 12-hydroxydehydrogenase/15-Oxo-prostaglandin 13-reductase catalytic mechanism and a possible Src homology 3 domain binding loop. J. Biol. Chem. 2004;279:22615–22623. doi: 10.1074/JBC.M312655200. [DOI] [PubMed] [Google Scholar]
- 54.Pippione A.C., Boschi D., Pors K., Oliaro-Bosso S., Lolli M.L. Androgen-AR axis in primary and metastatic prostate cancer: chasing steroidogenic enzymes for therapeutic intervention. J. Cancer Metastasis Treat. 2017;3:328–361. doi: 10.20517/2394-4722.2017.44. [DOI] [Google Scholar]
- 55.Turner G.W., Croteau R. Organization of monoterpene biosynthesis in Mentha. Immunocytochemical localizations of geranyl diphosphate synthase, limonene-6-hydroxylase, isopiperitenol dehydrogenase, and pulegone reductase. Plant Physiol. 2004;136:4215–4227. doi: 10.1104/PP.104.050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Funabashi M., Grove T.L., Wang M., Varma Y., McFadden M.E., Brown L.C., Guo C., Higginbottom S., Almo S.C., Fischbach M.A. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature. 2020;582:566–570. doi: 10.1038/S41586-020-2396-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rottenberg H. The Thermodynamic Description of Enzyme-Catalyzed Reactions: The Linear Relation between the Reaction Rate and the Affinity. Biophys. J. 1973;13:503–511. doi: 10.1016/S0006-3495(73)86004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stoll J., Rosenwald A.G., Krag S.S. A Chinese hamster ovary cell mutant F2A8 utilizes polyprenol rather than dolichol for its lipid-dependent asparagine-linked glycosylation reactions. J. Biol. Chem. 1988;263:10774–10782. doi: 10.1016/s0021-9258(18)38038-4. [DOI] [PubMed] [Google Scholar]
- 59.Rosenwald A.G., Stanley P., Krag S.S. Control of carbohydrate processing: increased beta-1,6 branching in N-linked carbohydrates of Lec9 CHO mutants appears to arise from a defect in oligosaccharide-dolichol biosynthesis. Mol. Cell Biol. 1989;9:914–924. doi: 10.1128/MCB.9.3.914-924.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kean E.L., Rush J.S., Waechter C.J. Activation of GlcNAc-P-P-dolichol synthesis by mannosylphosphoryldolichol is stereospecific and requires a saturated alpha-isoprene unit. Biochemistry. 1994;33:10508–10512. doi: 10.1021/BI00200A036. [DOI] [PubMed] [Google Scholar]
- 61.Kousal B., Honzík T., Hansíková H., Ondrušková N., Čechová A., Tesařová M., Stránecký V., Meliška M., Michaelides M., Lišková P., et al. Review of SRD5A3 Disease-Causing Sequence Variants and Ocular Findings in Steroid 5α-Reductase Type 3 Congenital Disorder of Glycosylation, and a Detailed New Case (retinal dystrophy/SRD5A3-CDG/SRD5A3/novel variant) Folia Biol. 2019;65:134–141. [PubMed] [Google Scholar]
- 62.Rauniyar N., Prokai L. Detection and identification of 4-hydroxy-2-nonenal Schiff-base adducts along with products of Michael addition using data-dependent neutral loss-driven MS3 acquisition: method evaluation through an in vitro study on cytochrome c oxidase modifications. Proteomics. 2009;9:5188–5193. doi: 10.1002/PMIC.200900116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mohamed M., Cantagrel V., Al-Gazali L., Wevers R.A., Lefeber D.J., Morava E. Normal glycosylation screening does not rule out SRD5A3-CDG. Eur. J. Hum. Genet. 2011;19:1019. doi: 10.1038/EJHG.2010.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quellhorst G.J., Hall C.W., Robbins A.R., Krag S.S. Synthesis of dolichol in a polyprenol reductase mutant is restored by elevation of cis-prenyl transferase activity. Arch. Biochem. Biophys. 1997;343:19–26. doi: 10.1006/ABBI.1997.0141. [DOI] [PubMed] [Google Scholar]
- 65.Bertorelli R., Capone L., Ambrosetti F., Garavelli L., Varriale L., Mazza V., Stanghellini I., Percesepe A., Forabosco A. The homozygous deletion of the 3’ enhancer of the SHOX gene causes Langer mesomelic dysplasia. Clin. Genet. 2007;72:490–491. doi: 10.1111/J.1399-0004.2007.00875.X. [DOI] [PubMed] [Google Scholar]
- 66.Martinez-Moczygemba M., Doan M.L., Elidemir O., Fan L.L., Cheung S.W., Lei J.T., Moore J.P., Tavana G., Lewis L.R., Zhu Y., et al. Pulmonary alveolar proteinosis caused by deletion of the GM-CSFRalpha gene in the X chromosome pseudoautosomal region 1. J. Exp. Med. 2008;205:2711–2716. doi: 10.1084/JEM.20080759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Suzuki T., Sakagami T., Rubin B.K., Nogee L.M., Wood R.E., Zimmerman S.L., Smolarek T., Dishop M.K., Wert S.E., Whitsett J.A., et al. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J. Exp. Med. 2008;205:2703–2710. doi: 10.1084/JEM.20080990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Berletch J.B., Yang F., Xu J., Carrel L., Disteche C.M. Genes that escape from X inactivation. Hum. Genet. 2011;130:237–245. doi: 10.1007/S00439-011-1011-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wainer Katsir K., Linial M. Human genes escaping X-inactivation revealed by single cell expression data. BMC Genom. 2019;20 doi: 10.1186/S12864-019-5507-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Viuff M., Skakkebæk A., Johannsen E.B., Chang S., Pedersen S.B., Lauritsen K.M., Pedersen M.G.B., Trolle C., Just J., Gravholt C.H. X chromosome dosage and the genetic impact across human tissues. Genome Med. 2023;15:21. doi: 10.1186/S13073-023-01169-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tartaglia N., Ayari N., Howell S., D’Epagnier C., Zeitler P. 48,XXYY, 48,XXXY and 49,XXXXY syndromes: not just variants of Klinefelter syndrome. Acta Paediatr. 2011;100:851–860. doi: 10.1111/J.1651-2227.2011.02235.X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Astro V., Alowaysi M., Fiacco E., Saera-Vila A., Cardona-Londoño K.J., Aiese Cigliano R., Adamo A. Pseudoautosomal Region 1 Overdosage Affects the Global Transcriptome in iPSCs From Patients With Klinefelter Syndrome and High-Grade X Chromosome Aneuploidies. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/FCELL.2021.801597/FULL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Skakkebæk A., Nielsen M.M., Trolle C., Vang S., Hornshøj H., Hedegaard J., Wallentin M., Bojesen A., Hertz J.M., Fedder J., et al. DNA hypermethylation and differential gene expression associated with Klinefelter syndrome. Sci. Rep. 2018;8 doi: 10.1038/S41598-018-31780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lefèvre N., Corazza F., Valsamis J., Delbaere A., De Maertelaer V., Duchateau J., Casimir G. The Number of X Chromosomes Influences Inflammatory Cytokine Production Following Toll-Like Receptor Stimulation. Front. Immunol. 2019;10:9560–9566. doi: 10.3389/FIMMU.2019.01052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Skuse D., Printzlau F., Wolstencroft J. Sex chromosome aneuploidies. Handb. Clin. Neurol. 2018;147:355–376. doi: 10.1016/B978-0-444-63233-3.00024-5. [DOI] [PubMed] [Google Scholar]
- 76.Costain G., Walker S., Marano M., Veenma D., Snell M., Curtis M., Luca S., Buera J., Arje D., Reuter M.S., et al. Genome Sequencing as a Diagnostic Test in Children With Unexplained Medical Complexity. JAMA Netw. Open. 2020;3 doi: 10.1001/jamanetworkopen.2020.18109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heremans I.P., Caligiore F., Gerin I., Bury M., Lutz M., Graff J., Stroobant V., Vertommen D., Teleman A.A., Van Schaftingen E., Bommer G.T. Parkinson’s disease protein PARK7 prevents metabolite and protein damage caused by a glycolytic metabolite. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2111338119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Corey E.J., Schmidt G. Useful procedures for the oxidation of alcohols involving pyridinium dichromate in aprotic media. Tetrahedron Lett. 1979;20:399–402. doi: 10.1016/S0040-4039(01)93515-4. [DOI] [Google Scholar]
- 79.Dewulf J.P., Gerin I., Rider M.H., Veiga-Da-Cunha M., Van Schaftingen E., Bommer G.T. The synthesis of branched-chain fatty acids is limited by enzymatic decarboxylation of ethyl- and methylmalonyl-CoA. Biochem. J. 2019;476:2427–2447. doi: 10.1042/BCJ20190500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD049174 and 10.6019/PXD049174. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.